

Abstract
Background and purpose:
AZ11645373 and N-{2-methyl-5-[(1R, 5S)-9-oxa-3,7-diazabicyclo[3.3.1]non-3-ylcarbonyl]phenyl}-2-tricyclo[3.3.1.13,7]dec-1-ylacetamide hydrochloride (compound-22) are recently described P2X7 receptor antagonists. In this study we have further characterized these compounds to determine their mechanism of action and interaction with other species orthologues.
Experimental approach:
Antagonist effects at recombinant and chimeric P2X7 receptors were assessed by ethidium accumulation and radioligand-binding studies.
Key results:
AZ11645373 and compound-22 were confirmed as selective non-competitive antagonists of human or rat P2X7 receptors respectively. Both compounds were weak antagonists of the mouse and guinea-pig P2X7 receptors and, for each compound, their potency estimates at human and dog P2X7 receptors were similar. The potency of compound-22 was moderately temperature-dependent while that of AZ11645373 was not. The antagonist effects of both compounds were slowly reversible and were not prevented by decavanadate, suggesting that they were allosteric antagonists. Indeed, the compounds competed for binding sites labelled by an allosteric radio-labelled P2X7 receptor antagonist. The species selectivity of AZ11645373, but not compound-22, was influenced by the nature of the amino acid at position 95 of the P2X7 receptor. N2-(3,4-difluorophenyl)-N1-[2-methyl-5-(1-piperazinylmethyl)phenyl]glycinamide dihydrochloride, a positive allosteric modulator of the rat receptor, reduced the potency of compound-22 at the rat receptor but had little effect on the actions of AZ11645373.
Conclusions:
AZ11645373 and compound-22 are allosteric antagonists of human and rat P2X7 receptors respectively. The differential interaction of the two compounds with the receptor suggests there may be more than one allosteric regulatory site on the P2X7 receptor at which antagonists can bind and affect receptor function.
Keywords: P2X7, ATP, BzATP, AZ11645373, GW791343, species selectivity
Introduction
The P2X7 receptor is a ligand-gated cation channel gated by extracellular ATP (North, 2002). The receptor plays a key role in the release of the pro-inflammatory cytokine interleukin-1β from immune cells (Ferrari et al., 2006) and has been implicated in inflammatory disease and pain disorders (Chessell et al., 2005; Honore et al., 2006; Donnelly-Roberts and Jarvis, 2007). This has generated considerable interest in developing antagonists of the P2X7 receptor and recently the properties of several of these have been described. These include AZ11645373 (Stokes et al., 2006), A740003 (Honore et al., 2006), compound-17 (Michel et al., 2008a), N-{2-methyl-5-[(1R, 5S)-9-oxa-3,7-diazabicyclo[3.3.1]non-3-ylcarbonyl]phenyl}-2-tricyclo[3.3.1.13,7]dec-1-ylacetamide hydrochloride (compound-22, Furber et al., 2007), N2-(3,4-difluorophenyl)-N1-[2-methyl-5-(1-piperazinylmethyl)phenyl]glycinamide dihydrochloride (GW791343) (Michel et al., 2008a) as well as numerous compounds described in patents (Baraldi et al., 2004; Romagnoli et al., 2005; Donnelly-Roberts and Jarvis, 2007).
A key requirement for the development of such antagonists as therapeutic agents is selectivity, which AZ11645373 and A-740003 exhibit to a high degree (Honore et al., 2006; Stokes et al., 2006). This is somewhat surprising as agents that block the effects of ATP might have been expected to suffer from limited selectivity or specificity. However, the mechanisms of action of AZ11645373 and compound-22 were not determined in the preliminary reports using these compounds and this may be important as a number of P2X7 receptor antagonists have recently been shown to be allosteric antagonists that do not bind at the ATP-binding site. Thus, in receptor protection studies using the rapidly reversible competitive P2X7 receptor antagonist, decavanadate, compounds, such as SB203580 (AD Michel, unpubl. obs.), 1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-L-tyrosyl]-4-phenylpiperazine (KN62) (Michel et al., 2006a) and compound-17 (Michel et al., 2008a), produced a long-lasting blockade of the receptor that was minimally affected by pretreatment with decavanadate. In contrast, decavanadate competitively protected the P2X7 receptor from the long-lasting blockade produced by agents that interact at the ATP-binding site such as pyridoxal phosphate-6-azophenyl-2′,4′-disulphonic acid (PPADS) and periodate oxidized ATP (Michel et al., 2006a).
Many antagonists are also selective for human, as compared with the rat, P2X7 receptor (Humphreys et al., 1998; Hibell et al., 2001; Stokes et al., 2006, Donnelly-Roberts and Jarvis, 2007) and we have identified the amino acid at position 95 of the receptor as a key determinant of the species selectivity of the allosteric antagonists GW791343, KN62 and SB203580 (Michel et al., 2008b). This residue does not affect the species selectivity of PPADS, which is instead determined by the amino acid at position 126 and residues between amino acids 264 and 304 (Michel et al., 2008b).
The initial reports describing AZ11645373 and compound-22 did not examine their mechanism of action in any detail but AZ11645373 did appear to function as a non-competitive inhibitor as it reduced maximal responses to ATP (Stokes et al., 2006). As slowly reversible antagonists such as PPADS that act at the ATP-binding site (Michel et al., 2006a), as well as negative allosteric antagonists (Michel et al., 2008a), decrease maximal responses, it is not possible from the published data to determine if AZ11645373 is a slowly reversible competitive antagonist or a negative allosteric antagonist.
In this study we have carried out further studies using AZ11645373 and compound-22 both to determine their mechanism of action and to further elucidate the factors determining their species selectivity. We have found that both are negative allosteric antagonists of the P2X7 receptor and that they appear to interact with the receptor in different ways.
Methods
Recombinant receptors
The nomenclature used for describing the P2X7 receptor conforms to the Journal's guidelines (Alexander et al., 2008). Studies on the human (Rassendren et al., 1997), dog (Accession Number, EU334661.1; Roman et al., 2007) and rat (Surprenant et al., 1996) P2X7 receptors were performed using HEK293 cells expressing the recombinant receptors (Fonfria et al., 2008) whereas studies on guinea-pig (Fonfria et al., 2008) and mouse (Young et al., 2006) P2X7 receptors were performed using the BacMam expression system previously described (Fonfria et al., 2008).
The generation of HEK293 cells expressing chimeric rat-human P2X7 and human-rat P2X7 receptors has been described previously (Michel et al., 2006b). Briefly, the human-rat P2X7 receptor comprises amino acids 1–255 of the human P2X7 receptor and amino acids 256–595 of the rat P2X7 receptor while the rat-human P2X7 receptor comprises amino acids 1–255 of the rat P2X7 receptor and amino acids 256–595 of the human P2X7 receptor.
The generation of the other chimeric P2X7 receptors has been described previously (Michel et al., 2008b). Briefly, the rat extracellular domain (ECD) P2X7 receptor (rat ECD) is a human receptor in which the entire ECD between amino acids 48 and 332 has been replaced with the corresponding rat residues. We also examined chimeric P2X7 receptors in which small sequences (25–42 amino acids) of the human P2X7 receptor were replaced with the corresponding residues from the rat P2X7 receptor. These were termed the domain 1 (rat residues 72–95), domain 2 (rat residues 108–136), domain 3 (rat residues 154–183), domain 4 (rat residues 212–244) and domain 5 (rat residues 264–304) P2X7 receptors. Several single point mutant receptors were used in which a single residue in the human P2X7 receptor was replaced with the corresponding residue from the rat P2X7 receptor. These were the lysine 72 to threonine (K72T), asparate 74 to asparagine (E74N), asparagine 78 to glycine (N78G), lysine 81 to threonine (K81T), serine 86 to glycine (S86G), phenylalanine 95 to leucine (F95L) and arginine 126 to glycine (R126G) P2X7 receptors. The effects of compounds were also evaluated at a histidine 155 to tyrosine (H155Y) receptor but this was produced in a human P2X7 receptor that also contained a histidine 270 to arginine mutation (H270R).
Cell culture and viral transductions
Studies on the human, rat, dog, human-rat, rat-human, rat L95F and domain 3 P2X7 receptors were performed using HEK293 cells stably transfected with the recombinant receptor. The guinea-pig, mouse, rat ECD, other chimeric domain swaps and single point mutant P2X7 receptors were transiently expressed in human osteosarcoma U-2 OS cells using the BacMam viral transduction method (Fonfria et al., 2008). For the later studies wild-type human and rat P2X7 receptors were also expressed in U-2 OS cells for comparative purposes. The BacMam viral transductions for ethidium accumulation studies were conducted as described previously (Fonfria et al., 2008). Briefly, U-2 OS cells were maintained in adherent culture conditions in the presence of Dulbecco's modified Eagle's medium: nutrient mixture F-12 supplemented with Glutamax (DMEM: F12 + Glutamax, Invitrogen) and 10% foetal bovine serum (PAA laboratories GmBH, Austria) at 37°C, 5% CO2. One day prior to assay, cells were harvested from the culture flasks using 0.05% trypsin/EDTA (Invitrogen, Paisley, UK) and resuspended at a concentration of ∼ 750 × 103 cells·mL−1 in culture media in the presence of the various BacMam viruses (1–2 × 108 plaque forming units·mL−1 of the BacMam virus stock in culture media). Cells (70–80 000) were plated into individual wells of poly-L-lysine pretreated 96-well plates (Costar, High Wycombe, UK) and the plates were incubated at 37°C, 5% CO2 overnight. HEK293 cells stably expressing the human, rat, dog, human-rat, rat-human or domain 3 recombinant P2X7 receptors were prepared in a similar manner except that the BacMam virus was omitted.
In studies to measure compound effects in radioligand-binding studies, U-2 OS cells were grown to confluence in T175 cm2 flasks and the media replaced with fresh growth media containing BacMam virus (1–2 × 108 plaque forming units·mL−1). The cells were incubated overnight, harvested using Versene (Invitrogen, Paisley, UK) and membranes prepared as described previously (Michel et al., 2007). Similar studies to determine the effects of agonists in radioligand-binding studies were performed using membranes prepared from HEK293 cells stably expressing the human, rat or domain 3 chimeric recombinant P2X7 receptors, using the same methods as described above for the U-2 OS cells.
Cellular ethidium accumulation measurements
Studies were performed as described previously (Michel et al., 2006a; Fonfria et al., 2008) using assay buffers comprising (in mmol·L−1): HEPES 10, N-methyl-D-glucamine 5, KCl 5.6, D-glucose 10, CaCl2 0.5 (pH 7.4) and supplemented with either 280 mmol·L−1 sucrose (sucrose buffer) or 140 mmol·L−1 NaCl (NaCl buffer). Before use, growth media was completely removed from the cells and they were rinsed with 350 µL of the appropriate assay buffer which was also removed before performing assay additions. In all studies the final assay volume was 100 µL and studies were performed at room temperature of 19–21°C.
Cells were incubated with antagonist for 40 min before addition of a mixture containing the agonists, ATP or 2′-& 3′-O-(4benzoylbenzoyl) ATP (BzATP), and ethidium bromide (100 µmol·L−1 final assay concentration). After agonist addition, incubations were continued until approximately 10–30% of maximal agonist-stimulated dye accumulation occurred. Reactions were rapidly terminated by addition of 25 µL of 1.3 mol·L−1 sucrose assay buffer containing 5 mmol·L−1 reactive black 5 and cellular accumulation of ethidium was determined by immediately measuring fluorescence (excitation wavelength of 530 nm and emission wavelength of 620 nm) from below the plate with a 96 well plate fluorescence reader (FlexStation, Molecular Devices, Wokingham, UK).
Protection of P2X7 receptors from the slowly reversible blockade by P2X7 receptor antagonists (receptor protection studies)
These studies were performed exactly as described previously (Michel et al., 2008b). Briefly, cells expressing rat or human P2X7 receptors were incubated with decavanadate for 10 min prior to addition of AZ11645373, compound-22 or PPADS. After a 40 min co-incubation, the antagonists were extensively washed over a 15 min period before measuring ATP-stimulated ethidium accumulation as described above. The studies were performed using NaCl buffer and for studies on the rat and human P2X7 receptors, the ATP concentrations were 0.5 mmol·L−1 and 2 mmol·L−1 respectively.
Interaction studies with GW791343
These studies were performed exactly as described previously (Michel et al., 2008b). Briefly, cells expressing rat P2X7 receptors were incubated with 30 µmol·L−1 GW791343 for 10 min prior to addition of AZ11645373 or compound-22. After a 40 min co-incubation period, ATP-stimulated ethidium accumulation was measured as described above.
Radioligand-binding studies
The radioligand-binding studies using [3H]-compound-17 were performed as described previously (Michel et al., 2007). Briefly, membranes prepared from U-2 OS cells transduced with human, rat or chimeric recombinant P2X7 receptors were incubated with the compounds and the radioligand, [3H]-compound-17 (2–3 nmol·L−1) for 60 min at room temperature in a final assay volume of 200 µL of 50 mmol·L−1 Tris HCl buffer containing 0.01% bovine serum albumen (pH 7.4 at RT). Reactions were terminated by vacuum filtration. Non-specific binding was defined using 10 µmol·L−1 compound-17.
Data analysis
Individual concentration-effect or inhibition curves from each experiment were fitted to a four-parameter logistic function to determine the maximum and minimum responses and to calculate the EC50 or IC50 values and the Hill slope. For graphical purposes, most concentration-effect and inhibition curves are presented as a percentage of the maximal response obtained in the control group.
As the compounds produced non-competitive antagonist effects in the Schild studies (see Figure 1A,D), the data from the Schild studies were also analysed to calculate antagonist pIC50 values at each agonist concentration as this provided some quantitative estimate of antagonist potency. To graphically represent the effect of agonist concentration on antagonist pIC50, the agonist concentration was expressed relative to its EC50 at the various receptors [logarithm (agonist concentration/agonist EC50)]. This enabled a simpler comparison of antagonist pIC50 values between the species orthologues and chimeric receptors (see Figure 1F). In addition, the relationship between pIC50 and [logarithm (agonist concentration/agonist EC50)] was linear (Figures 1F and 2F) and so, in order to provide a statistical comparison of antagonist potency between the various receptors, the data for each experiment were analysed by linear regression and the antagonist pIC50 at the agonist EC50 was calculated from the fit. In graphical terms, this corresponded to the pIC50 at 0 on the X-axis (Figure 1F) and this value is referred to as the normalized pIC50.
Figure 1.

The effect of AZ11645373 at the human P2X7 receptor in ethidium accumulation studies. HEK293 cells expressing the human recombinant P2X7 receptor were pre-incubated for 40 min with the indicated concentrations of AZ11645373 before measuring agonist-stimulated ethidium accumulation. (A) The effect of AZ11645373 on ATP responses in NaCl buffer at room temperature. (B) Transposition of the data from panel (A) to illustrate the effect of AZ11645373 on responses to ATP. (C) The effect of AZ11645373 on responses in NaCl buffer at 37°C. (D) The effect of AZ11645373 on BzATP responses in sucrose buffer at room temperature. (E) Transposition of the data from panel (D) to illustrate the effect of AZ11645373 on responses to BzATP. (F) The data from panels A, C and D were analysed to calculate the pIC50 of AZ11645373 at each concentration of agonist (RT, room temperature). Agonist concentration is expressed relative to agonist EC50 at each receptor such that Log (fold EC50) represents logarithm (agonist concentration/agonist EC50). Basal ethidium accumulation in the absence or presence of AZ11645373 is indicated on the X-ordinate as C in (A,C,D). In (B,E) the response to agonist in the absence of AZ11645373 is indicated on the X-ordinate as C. The data are the mean ± SEM of three to four separate experiments. BzATP, 2′-& 3′-O-(4benzoylbenzoyl) ATP.
Figure 2.

The effect of AZ11645373 at (A) dog, (B) guinea-pig (GP), (C) mouse and (D,E) rat P2X7 receptors in ethidium accumulation studies. HEK293 cells expressing the dog (A) or rat (D) recombinant receptors or U-2 OS cells transduced with guinea-pig (B) or mouse (C) P2X7 receptors were pre-incubated for 40 min with the indicated concentrations of AZ11645373 before measuring ATP-induced ethidium accumulation. Studies were performed in NaCl buffer at RT. (E) Transposition of the data from panel (D) to illustrate effect of AZ11645373 on responses to ATP at the rat receptor. (F) The data from (A–D) were analysed to calculate the pIC50 of AZ11645373 at each concentration of agonist. Agonist concentration is expressed relative to agonist EC50 at each receptor such that Log (fold EC50) represents logarithm (agonist concentration/agonist EC50). Basal ethidium accumulation in the absence or presence of AZ11645373 is indicated on the X-ordinate as C in (A–D). In (E) the response to agonist in the absence of AZ11645373 is indicated on the X-ordinate as C. The data are the mean ± SEM of three to four separate experiments.
The data for the decavanadate protection experiments were analysed as described previously (Michel et al., 2006a). Briefly, the IC50 of the antagonists to block ATP responses was determined in control cells and in cells pretreated with the various concentrations of decavanadate. For each concentration of decavanadate, a dose-ratio (DR) was calculated as the ratio of the IC50 of the compound determined in presence of decavanadate and in its absence. The log (DR-1) estimates from these studies were plotted against the log of the decavanadate concentration in order to construct a form of Schild plot to represent the effects of decavanadate on the antagonist IC50.
Statistical comparisons were made using Student's t-test or one-way anova followed by Tukey's post-hoc test. Differences were assessed as significant when P < 0.05. In these studies the data are the mean ± SEM of three to five experiments. All curve fitting and statistical analysis was performed using GraphPad Prism 3 (San Diego, CA, USA).
Materials
ATP, BzATP, ethidium bromide, PPADS, sodium orthovanadate and reactive black 5 were obtained from Sigma (Poole, UK). AZ11645373, compound-22, GW791343 and SB203580 were synthesized in the Chemistry Department of GSK, Harlow, UK. All culture media were obtained from Invitrogen (Paisley, Scotland) while other reagents were obtained from VWR (Loughborough, UK). [3H]-Compound-17 was from Tritec, Switzerland (specific activity was 2.1TBq·mmol−1 and purity was >99% by HPLC). Decavandate solutions were prepared as described previously (Michel et al., 2006a).
Results
AZ11645373 is a selective, non-competitive, antagonist of human and dog P2X7 receptors
Antagonist effects were studied in both sucrose and NaCl buffer as some receptors could only be studied in sucrose buffer and some antagonists have different potency in these two buffers (Hibell et al., 2001). AZ11645373 was a potent antagonist of the human P2X7 receptor whether studied in sucrose or NaCl buffer (Figure 1). In both buffers, AZ11645373 was a non-competitive antagonist reducing the maximal response to ATP and BzATP. The pIC50 varied slightly with ATP or BzATP concentration, significantly (P < 0.05, F-test) decreasing as agonist concentration increased (Figure 1F), and there was an approximately linear relationship between pIC50 and the logarithm of the agonist concentration. In order to provide a statistical comparison of potency between the various studies, we analysed the data in Figure 1F using linear regression and calculated the antagonist pIC50 at the agonist EC50. In graphical terms, this corresponded to the pIC50 at 0 on the X-axis and is termed the normalized pIC50. The potency of AZ11645373 at the human P2X7 receptor in NaCl buffer when using ATP as agonist (normalized pIC50= 7.46 ± 0.04) was significantly lower (P < 0.05, one-way anova followed by Tukey's post-hoc test) than in sucrose buffer using BzATP as agonist (normalized pIC50= 7.87 ± 0.02) (Figure 1F). In NaCl buffer using ATP as agonist, there was no significant difference (P < 0.05, one-way anova followed by Tukey's post-hoc test) in the potency of AZ11645373 at room temperature (normalized pIC50= 7.46 ± 0.04) and at 37°C (normalized pIC50= 7.31 ± 0.04) (Figure 1).
AZ11645373 was also a potent antagonist at the dog receptor (Figure 2A) where its normalized pIC50 of 7.40 ± 0.13 (Figure 2F) was similar to that at the human receptor (7.46 ± 0.04). AZ11645373 was also an antagonist of guinea-pig and mouse receptors producing almost complete inhibition of responses at 10 µmol·L−1 (Figure 2B,C). However, it was less potent than at the human or dog receptors and the normalized pIC50 values at mouse and guinea-pig receptors were 5.81 ± 0.13 and 5.94 ± 0.06 respectively (Figure 2F).
AZ11645373 was a low potency antagonist at the rat P2X7 receptor producing very little shift in the ATP (Figure 2D) or BzATP (data not shown but see Figure 3C) concentration-effect curves in NaCl buffer or that of BzATP in sucrose buffer (data not shown but see Figure 3D). AZ11645373 only appeared to inhibit responses at intermediate agonist concentrations in both NaCl and sucrose buffer (Figure 3) and at these intermediate agonist concentrations the inhibition of responses appeared to be incomplete with saturation of effect at the higher concentrations of AZ11645373 although we only examined the compound at concentrations up to 30 µmol·L−1. The inhibition of agonist effects produced by AZ11645373 was modest but reproducible in two separate studies (Figure 3A,B) although the normalized pIC50 determined using ATP as agonist in NaCl buffer varied between the studies (5.28 ± 0.05 and 5.90 ± 0.05) probably reflecting the difficulty in calculating pIC50 values with modest and incomplete inhibition of responses.
Figure 3.
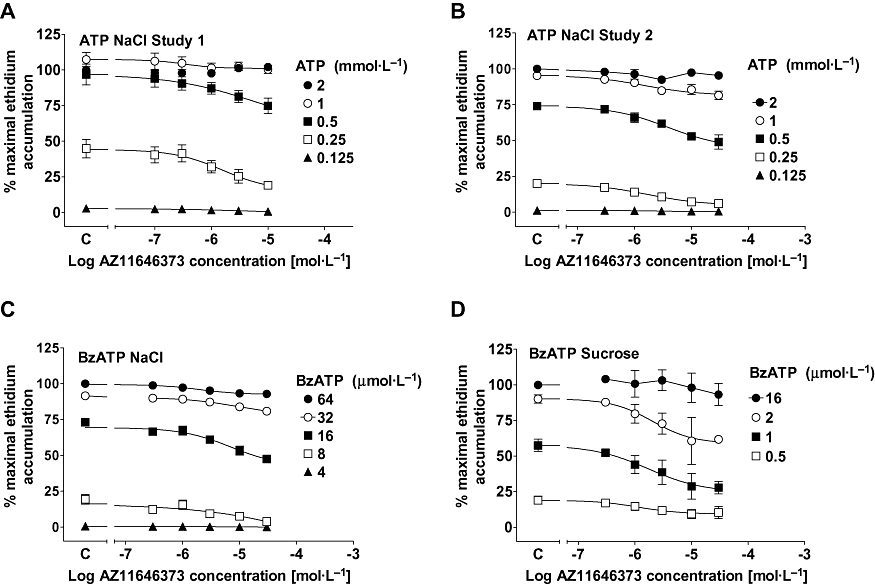
The effect of AZ11645373 at the rat P2X7 receptor in ethidium accumulation studies. HEK293 cells expressing the rat recombinant receptor were pre-incubated for 40 min with AZ11645373 before measuring agonist stimulated ethidium accumulation. (A) The effect of AZ11645373 on responses to ATP in NaCl buffer in study 1. (B) The effect of AZ11645373 on responses to ATP in NaCl buffer in study 2. (C) The effect of AZ11645373 on responses to BzATP in NaCl buffer. (D) The effect of AZ11645373 on responses to BzATP in sucrose buffer. The response to agonist in the absence of AZ11645373 is indicated on the X-ordinate as C. The data are the mean ± SEM of three to four separate experiments. BzATP, 2′-& 3′-O-(4benzoylbenzoyl) ATP.
AZ11645373 does not interact at the ATP-binding site
AZ11645373 produced a long-lasting inhibition of responses, with the inhibition of responses at 15 min after washout being the same as without washout (data not shown). This enabled AZ11645373 to be used in receptor protection experiments to determine if the rapidly reversible competitive antagonist decavanadate could affect the persistent antagonist effects of AZ11645373. Decavanadate had very little effect on the long-lasting inhibitory effects of AZ11645373 although it did produce a significant decrease in the pIC50 of AZ116435373 at concentrations of 30, 100 and 300 µmol·L−1 (P < 0.05, one-way anova followed by Tukey's post-hoc test) although this was no more than twofold and the effects at these three doses were identical (P > 0.05, one-way anova followed by Tukey's post-hoc test) (Figure 4A,C). These effects contrasted markedly with those observed with PPADS where decavanadate produced a more competitive shift in the PPADS inhibition curve (Figure 4B) and the resultant Schild plot of the data exhibited a slope of unity (Figure 4C, slope = 1.03 ± 0.03).
Figure 4.

The interaction of AZ11645373 or PPADS with decavanadate or GW791343 in ethidium accumulation studies. (A–C) HEK293 cells expressing the human recombinant P2X7 receptor were pre-incubated with the indicated concentrations of decavanadate (Dec) for 10 min prior to addition of AZ11645373 or PPADS. Following a further 40 min co-incubation the cells were washed before measuring 2 mmol·L−1 ATP stimulated ethidium accumulation. (C) Schild plot for the interaction between decavanadate and AZ11645373 or PPADS. (D,E) HEK293 cells expressing the rat recombinant P2X7 receptor were pre-incubated with 30 µmol·L−1 GW791343 for 10 min and then co-incubated with GW791343 and AZ11645373 for a further 40 min before measuring ATP responses. The effect of 30 µmol·L−1 GW791343 on AZ11645373 inhibition of responses to (D) a threshold or (E) a near maximal concentration of ATP is shown. The response to agonist in the absence of AZ11645373 is indicated on the X-ordinate as C in panels A, B, D and E. The data are the mean ± SEM of three to four separate experiments. GW791343, N2-(3,4-difluorophenyl)-N1-[2-methyl-5-(1-piperazinylmethyl)phenyl]glycinamide dihydrochloride; PPADS, pyridoxal phosphate-6-azophenyl-2′,4′-disulphonic acid.
GW791343 is a positive allosteric modulator of the rat P2X7 receptor that increases the potency and effect of ATP (Michel et al., 2008a). In the presence of GW791343, the potency of the negative allosteric P2X7 receptor modulator, compound-17, was reduced but the potency of compounds that act at the ATP-binding site, such as PPADS and decavanadate, was not affected (Michel et al., 2008a). The ability of AZ11645373 to inhibit responses to equi-effective concentrations of ATP at the rat P2X7 receptor was not detectably affected by 30 µmol·L−1 GW791343 (Figure 4D,E) although the small magnitude of the AZ11645373 effect clearly complicated these studies.
Effects of AZ11645373 at human-rat chimeric receptors
Previous studies have identified amino acid 95 as a key determinant in the species selectivity of several P2X7 receptor antagonists (see Introduction). To determine if this was also the case with AZ11645373, we examined its effects at chimeric human-rat receptors.
At the rat ECD receptor, as at the wild-type rat receptor, AZ11645373 had very little effect on responses to ATP (data not shown) suggesting its species selectivity was determined through an interaction with the ECD of the P2X7 receptor. The compound was a relatively potent antagonist of the human-rat receptor although these studies were complicated by the biphasic nature of the ATP concentration effect curve at this receptor (Figure 5A). To allow for this, the normalized pIC50 was calculated at ATP concentrations below 0.125 mmol·L−1, corresponding to the high affinity component of the ATP concentration-effect curve, and at concentrations of 0.25 mmol·L−1 and above, corresponding to the low affinity component of the ATP concentration-effect curve. However, the normalized pIC50 values were similar over each concentration range of ATP and were intermediate between the potency at the rat and the human receptors (Figure 5F). AZ11645373 was a very weak antagonist of BzATP effects at rat-human receptors (Figure 5B) where its normalized pIC50 value was less than at the rat receptor (Figure 5F). Taken together, this suggested that residues in the N-terminal 255 amino acids of the human receptor were more responsible for the species selectivity of AZ11645373.
Figure 5.

The effect of AZ11645373 on human rat chimeric receptors in ethidium accumulation studies. (A,B) HEK293 cells expressing chimeric human rat receptors or (C–E) U-2 OS cells transduced with chimeric human rat receptors were pre-incubated for 40 min with AZ11645373 before measuring agonist-induced ethidium accumulation. Studies were performed in (A,C,D,E) NaCl buffer using ATP as agonist or (B) sucrose buffer using BzATP as agonist. (E) The dependence of AZ11645373 pIC50 on agonist concentration. The data from (A–D), and other studies, were analysed to calculate the pIC50 of AZ11645373 at each concentration of agonist for each experiment. Agonist concentration is expressed relative to agonist EC50 at each receptor such that Log (fold EC50) represents logarithm (agonist concentration/agonist EC50). (F) AZ11645373 potency at chimeric and wild-type P2X7 receptors. The normalized pIC50 for AZ11645373, which corresponds to the extrapolated pIC50 for AZ11645373 at the agonist EC50, was calculated from (E), and other studies, as described in the methods. *Significantly different to normalized pIC50 determined at the human receptor using the same assay buffer, P < 0.05, one-way anova followed by Tukey's post hoc test. #Significantly different to normalized pIC50 determined at the rat receptor using the same assay buffer, P < 0.05, one-way anova followed by Tukey's post hoc test. Basal ethidium accumulation in the absence or presence of AZ11645373 is indicated on the X-ordinate as C in (A–D). The data are the mean ± SEM of three to four separate experiments. BzATP, 2′-& 3′-O-(4benzoylbenzoyl) ATP.
The normalized pIC50 of AZ11645373 at the domain 1 receptor (6.44 ± 0.03) was significantly less than the value of 7.87 ± 0.02 at the human receptor (Figure 5C,F; P < 0.05, one-way anova followed by Tukey's post-hoc test). However, the normalized pIC50 values at domain 2 (7.84 ± 0.05), domain 4 (7.57 ± 0.02) and domain 5 (7.59 ± 0.15) mutant receptors were not significantly different from the value of 7.87 ± 0.02 at the human receptor (Figure 5D–F, P > 0.05, one-way anova followed by Tukey's post-hoc test).
When AZ11645373 was evaluated at P2X7 receptors with single point mutations within domain 1 (Figure 6), its normalized pIC50 was only significantly reduced at the F95L mutant compared with the human receptor, although the normalized pIC50 of 6.91 ± 0.06 was significantly higher than the value of 6.44 ± 0.03 at the domain 1 mutant (P < 0.05, one-way anova followed by Tukey's post-hoc test).
Figure 6.

The effect of AZ11645373 on single point mutant receptors from within domain 1 of the P2X7 receptor measured in ethidium accumulation studies. U-2 OS cells transduced with mutant receptors were pre-incubated for 40 min with AZ11645373 before measuring BzATP-induced ethidium accumulation. Studies were performed in sucrose buffer. The effect of AZ11645373 on BzATP responses in cells expressing (A) the K72T or (B) the F95L P2X7 receptor. (C) The dependence of AZ11645373 pIC50 on agonist concentration. The data from (A,B) and other studies were analysed to calculate the pIC50 of AZ11645373 at each concentration of agonist. Agonist concentration is expressed relative to agonist EC50 at each receptor such that Log (fold EC50) represents logarithm (agonist concentration/agonist EC50). (D) AZ11645373 potency at chimeric and wild-type P2X7 receptors. The normalized pIC50 for AZ11645373, which corresponds to the extrapolated pIC50 for AZ11645373 at the agonist EC50, was calculated from the studies in (C). *Significantly different to normalized pIC50 determined at the human receptor, P < 0.05, one-way anova followed by Tukey's post hoc test. Basal ethidium accumulation in the absence or presence of AZ11645373 is indicated on the X-ordinate as C in (A,B). The data are the mean ± SEM of three to four separate experiments. BzATP, 2′-& 3′-O-(4benzoylbenzoyl) ATP.
Effect of AZ11645373 in radioligand-binding studies
AZ11645373 was a potent inhibitor of compound-17 binding to the human P2X7 receptor but had no detectable inhibitory effect at the rat P2X7 receptor and even slightly, but significantly (P < 0.05, one-way anova followed by Tukey's post-hoc test), increased binding at 1 and 3 µmol·L−1 (Figure 7). At the rat ECD chimeric receptor, the effects of AZ11645373 were similar to those at the rat receptor but the effects of AZ11645373 were reduced considerably at the domain 1 chimeric receptor and similarly reduced at the F95L mutant (Figure 7A–C). The effects of AZ11645373 at the domain 2 and 4 receptors were not significantly different from those at the human P2X7 receptor (Figure 7D,F) although at the domain 4 receptor the inhibition of binding produced at 10 µmol·L−1 was less than at 1 and 3 µmol·L−1 (Figure 7F). At the domain 3 receptor (Figure 7E) and domain 5 receptor (data not shown), AZ11645373 potency was slightly, but significantly, reduced when compared with the potency at the human receptor (P < 0.05, one-way anova followed by Tukey's post-hoc test).
Figure 7.

The ability of AZ11645373 to inhibit [3H]-compound-17 binding to membranes prepared from cells expressing human-rat recombinant chimeric P2X7 receptors. Membranes were prepared from U-2 OS cells transiently transduced with recombinant P2X7 receptors using BacMam virus. The radioligand concentration was 2 nmol·L−1 and specific binding was defined with 10 µmol·L−1 compound-17. The effect of AZ11645373 at wild-type rat and human receptors is compared with effects at (A) rat-ECD, (B) domain 1, (C) F95L, (D) domain 2, (E) domain 3 or (F) domain 4 P2X7 receptors. The data are the mean ± SEM of three separate experiments. ECD, extracellular domain.
Compound-22 is a selective non-competitive antagonist of the rat P2X7 receptor
In contrast to AZ11645373, compound-22 was a more potent antagonist of rat than of human receptors (Figure 8A,B,F). The compound was a weak antagonist of the dog receptor and even weaker antagonist of the mouse and guinea-pig receptors (Figure 8C–E). The effects of compound-22 at human and dog receptors were clearly non-competitive as the maximal response was reduced (Figure 8A,C). At the rat receptor, compound-22 appeared to act in a more competitive manner (Figure 8B) as it reduced the ATP EC50 and a pA2 of 8.12 ± 0.20 could be calculated. However, compound-22 reduced the maximal response to ATP and the slope of the Schild plot was significantly less than unity (0.36 ± 0.06).
Figure 8.

The effect of compound-22 at P2X7 receptor species orthologues in ethidium accumulation studies. Studies were performed using HEK293 cells (A–C) or in U-2 OS cells transduced with P2X7 receptors (D, E). Cells expressing (A) human, (B) rat, (C) dog, (D) mouse or (E) guinea-pig P2X7 receptors were pre-incubated for 40 min with compound-22 in NaCl buffer before measuring ATP-induced ethidium accumulation in NaCl buffer at room temperature. (F) The data from (A–C) were analysed to calculate the pIC50 of compound-22 at each concentration of agonist. Agonist concentration is expressed relative to agonist EC50 at each receptor such that Log (fold EC50) represents logarithm (agonist concentration/agonist EC50). Basal ethidium accumulation in the absence or presence of compound-22 is indicated on the X-ordinate as C in (A–E). The data are the mean ± SEM of three to four separate experiments. Compound-22, N-{2-methyl-5-[(1R, 5S)-9-oxa-3,7-diazabicyclo[3.3.1]non-3-ylcarbonyl]phenyl}-2-tricyclo[3.3.1.13,7]dec-1-ylacetamide hydrochloride.
Antagonist interaction studies with compound-22
Compound-22 produced a long-lasting inhibition of responses at the rat receptor with the inhibition of responses 15 min after washout being the same as without washout (data not shown). This enabled compound-22 to be used in receptor protection experiments with decavanadate as performed with AZ11645373. Decavanadate had no significant effect on the long-lasting inhibitory effects of compound-22 (Figure 9A, P > 0.05, one-way anova followed by Tukey's post-hoc test). These effects contrast markedly with those observed with PPADS (Figure 2B,C).
Figure 9.

The effect of decavanadate, GW791343 or temperature on the antagonist effect of compound-22 at the rat P2X7 receptor measured in ethidium accumulation studies. Studies were performed using HEK293 cells expressing the rat recombinant P2X7 receptor. (A) Cells were pre-incubated with decavanadate for 10 min prior to addition of compound-22. After a 40 min co-incubation the cells were washed over a 15 min period before measuring the response to 0.5 mmol·L−1 ATP. (B,C) Cells were incubated with compound-22 for 40 min before measuring ATP responses. Studies were performed at room temperature (B) without or (C) following pre-incubation with 30 µmol·L−1 GW791343 for 10 min prior to addition of compound-22. (D) Studies were performed at 37°C. (E) The effect of temperature and GW791343 on the potency of compound-22. The data from (B–D) were analysed to calculate the pIC50 of compound-22 at each concentration of agonist. Agonist concentration is expressed relative to agonist EC50 under each condition such that Log (fold EC50) represents logarithm (agonist concentration/agonist EC50). The response to agonist in the absence of decavanadate (A) or compound-22 (B–D) is indicated on the X-ordinate as C. The data are the mean ± SEM of three to four separate experiments. Compound-22, N-{2-methyl-5-[(1R, 5S)-9-oxa-3,7-diazabicyclo[3.3.1]non-3-ylcarbonyl]phenyl}-2-tricyclo[3.3.1.13,7]dec-1-ylacetamide hydrochloride.
In contrast to the data obtained with AZ11645373, the potency of compound-22 was reduced in the presence of GW791343 (Figure 9B,C,E). Thus, the normalized pIC50 values in the absence or presence of 10 µmol·L−1 GW791343 were 8.19 ± 0.02 and 7.22 ± 0.08 respectively.
Temperature and buffer sensitivity of effects of compound-22 at rat receptors
The antagonist potency of compound-22 at rat receptors against responses to ATP in NaCl buffer was moderately temperature sensitive with normalized pIC50 values at 37°C and room temperature being 7.41 ± 0.02 and 8.19 ± 0.02 respectively (Figure 9B,D,E).
The potency and effect of compound-22 at rat receptors were modestly affected by assay conditions as the normalized pIC50 against BzATP in sucrose buffer in U-2 OS cells (7.45 ± 0.10 see Figure 10) was lower than against ATP in NaCl buffer in HEK cells (8.19 ± 0.02 and 8.06 ± 0.05 in two separate studies, see Figures 8F and 9F).
Figure 10.

The effect of compound-22 on human rat chimeric P2X7 receptors measured in ethidium accumulation studies. U-2 OS cells transduced with chimeric receptors were pre-incubated for 40 min with the indicated concentrations of compound-22 before measuring BzATP responses. Studies were performed in sucrose buffer. The effect of compound-22 is shown in cells expressing (A) human, (B) rat, (C) domain 1, (D) domain 2 or (E) F95L P2X7 receptors. (F) The potency of compound-22 at chimeric and wild-type receptors. The normalized pIC50 for compound-22, which corresponds to the extrapolated pIC50 at the agonist EC50, was calculated as described in the methods. *Significantly different to the normalized pIC50 at the human P2X7 receptor, P < 0.05, one-way anova followed by Tukey's post hoc test. Basal ethidium accumulation in the absence or presence of compound-22 is indicated on the X-ordinate as C in (A–E). The data are the mean ± SEM of three to four separate experiments. Compound-22, N-{2-methyl-5-[(1R, 5S)-9-oxa-3,7-diazabicyclo[3.3.1]non-3-ylcarbonyl]phenyl}-2-tricyclo[3.3.1.13,7]dec-1-ylacetamide hydrochloride.
Furthermore, compound-22 did not reduce responses to BzATP at rat P2X7 receptors in sucrose buffer (Figure 10B) and even appeared to act in a more competitive manner as the slope of the Schild plot of the data (0.87 ± 0.14) was not significantly different to unity (P > 0.05, One sample t-test). Additional more direct comparisons would be required to determine if the compounds potency and effect was affected specifically by ionic conditions, agonist used or varied depending on expression system. Nevertheless, it is clear that compound-22 did not act as a simple competitive antagonist.
Effect of compound-22 at chimeric receptors
Compound-22 was an antagonist of the rat-human P2X7 receptor but was much less potent than at the rat, and even the human receptor, as it only inhibited responses at 3 and 10 µmol·L−1 (data not shown). At the human-rat P2X7 receptor, compound-22 was an even weaker antagonist and only blocked responses at 10 µmol·L−1 (data not shown).
Compound-22 potency at the domain 1, domain 2, domain 4, F95L and R126G chimeric P2X7 receptors was similar to that at the human P2X7 receptor while its potency at the domain 5 P2X7 receptor was significantly less than at the human receptor (Figure 10). The effects at the domain 3 P2X7 receptor (normalized pIC50 of 6.62 ± 0.05) were slightly more pronounced than at the human, wild-type, P2X7 receptor examined in HEK cells (normalized pIC50 of 6.36 ± 0.01). We only examined one mutant within domain 3. This was the His155Tyr P2X7 receptor where the normalized pIC50 of 6.16 ± 0.02 was similar to the value of 6.20 ± 0.02 at the wild-type human P2X7 receptor expressed in U-2 OS cells. This mutant also contains an Arg270His mutation but the normalized pIC50 values of 6.17 ± 0.01 and 6.20 ± 0.02 at the Arg270 and His270 P2X7 receptors, respectively, were not significantly different.
The effect of compound-22 was examined in radioligand-binding studies. These studies confirmed the rat P2X7 receptor selectivity of compound-22 (Figure 11). The potency of compound-22 was identical at the rat ECD and rat receptor suggesting residues in the ECD were responsible for its species difference in potency. Compared with its effect at the human wild-type receptor, the effects of compound-22 were significantly increased at the domain 1, domain 2 and F95L mutant P2X7 receptor (Figure 11B–D) and reduced at the domain 5 mutant receptor (Figure 11F). However, in each case the IC50 values were only 1.5-to twofold different to those at the wild-type human P2X7 receptor. There were no significant changes in pIC50 at the domain 3 (Figure 11E) or the domain 4 (data not shown) P2X7 receptors.
Figure 11.

The ability of compound-22 to inhibit [3H]-compound-17 binding to membranes prepared from cells expressing human-rat recombinant chimeric P2X7 receptors. Membranes were prepared from U-2 OS cells transiently transduced with recombinant P2X7 receptors using BacMam virus. The radioligand concentration was 2 nmol·L−1 and specific binding was defined with 10 µmol·L−1 compound-17. The effect of compound-22 at wild-type rat and human receptors is compared with effects at (A) rat-ECD, (B) domain 1, (C) F95L, (D) domain 2, (E) domain 3 or (F) domain 5 P2X7 receptors. The data are the mean ± SEM of three separate experiments. Compound-22, N-{2-methyl-5-[(1R, 5S)-9-oxa-3,7-diazabicyclo[3.3.1]non-3-ylcarbonyl]phenyl}-2-tricyclo[3.3.1.13,7]dec-1-ylacetamide hydrochloride; ECD, extracellular domain.
Discussion
In this study we have presented evidence that two previously described P2X7 antagonists are negative allosteric modulators of the P2X7 receptors and confirmed their species selectivity. We have also extended the information on their orthologue selectivity and shown that these two molecules may interact with different sites on the P2X7 receptor.
AZ11645373 was recently described as a selective antagonist of the human P2X7 receptor and its effects were examined in detail in electrophysiology, ion influx and cytokine release assays (Stokes et al., 2006). However, it was a non-competitive antagonist and its mechanism of action was not identified (see Introduction). In this study, we have studied the effects of this compound in ethidium accumulation and radioligand-binding studies in order to better understand its mechanism of action. Our studies confirm the non-competitive mechanism of action at the human receptor and also demonstrate that its effects are only slightly affected by assay buffer composition or assay temperature. The latter point may be important as several P2X7 antagonists (Fonfria et al., 2005; A.D. Michel, unpubl. obs.), as well as compound-22 (see below), display between three-and 10-fold lower affinities at the more physiologically relevant temperature of 37°C than at room temperature where most in vitro studies are performed.
AZ11645373 was confirmed as a highly selective antagonist of human, as opposed to rat, P2X7 receptors in both ethidium accumulation and radioligand-binding studies. The compound only partially blocked responses at the rat receptor but produced a much more pronounced blockade of the guinea-pig and mouse receptors although the pIC50 values were not very different between the three rodent orthologues. At the rat receptor, AZ11645373 produced partial and incomplete inhibition of responses which may suggest that it is acting in an allosteric manner (see below) and that the main difference between its effects at the rodent orthologues is in the magnitude of its efficacy to inhibit responses to ATP.
AZ11645373, like several other P2X7 receptor antagonists (Chessell et al., 1998; Michel et al., 2008a), appeared to have a very slow offset of action when studied using the ethidium accumulation technique, with effects persisting unchanged for at least 15 min after washout. The compound appeared to have faster offset kinetics in electrophysiology studies where its offset was described as slow but with responses returning to 10% of control within 15–25 min (Stokes et al., 2006). We do not know the reason for this difference in kinetics between studies but the slow offset of action did allow the compound to be used in receptor protection studies to explore its mechanism of action. In those studies, decavanadate had very little effect on the long-lasting inhibition produced by AZ11645373 and even the minimal effect it had saturated at concentrations of 30–300 µmol·L−1. This contrasted markedly with the pronounced and apparently competitive protection of the receptor by decavanadate from the persistent blockade produced by PPADS and was similar to the very limited effects of decavanadate on the persistent blockade of the receptor produced by the allosteric antagonist compound-17 in studies on the human receptor (Michel et al., 2008a). This suggested that, like compound-17, AZ11645373 is an allosteric antagonist of the receptor. In this respect, site-directed mutagenesis studies also provided indirect evidence that AZ11645373 was acting as an allosteric antagonist as the potency of AZ11645373 was affected by the same residues that affected the positive allosteric modulator, GW791343, and the negative allosteric modulators SB203580 and KN62 (Michel et al., 2008b). Thus, amino acid 95 was found to be a key residue in determining the allosteric effects of these allosteric agents and we found that this residue also contributed to the human/rat species selectivity of AZ11645373. Interestingly, although compound-17 and AZ11645373 both appear to be allosteric antagonists of the human P2X7 receptor, they may interact with the receptor in a different manner, as GW791343 was able to reduce the potency of compound-17 as an antagonist at the rat receptor (Michel et al., 2008a) but had little effect on the actions of AZ11645373.
The pharmacological properties of compound-22 have not previously been presented in any detail but it represented a unique compound in being selective for rat over human P2X7 receptors (Furber et al., 2007). We were able to confirm this selectivity and, additionally, found that compound-22 had little or no detectable affinity for mouse or guinea-pig P2X7 receptors and so also discriminated between rodent receptors. KN62 also discriminates between rat and mouse P2X7 receptors (Humphreys et al., 1998) and rat and guinea-pig P2X7 receptors (Fonfria et al., 2008) but compound-22 appears to be the most selective tool to date in this regard. Compound-22 also possessed low affinity for the dog P2X7 receptor where its potency was similar to that at the human receptor. Indeed, the effects of both AZ11645373 and compound-22 at human and dog P2X7 receptors were similar and contrasted with the marked species differences observed with the compounds between human and rodent P2X7 receptors. We have recently characterized the dog recombinant P2X7 receptor (Roman et al., 2007) and found that its antagonist sensitivity was similar to the human P2X7 receptor and so the present observations strengthen this correlation.
Compound-22, like AZ11645373, was a non-competitive P2X7 receptor antagonist and also appeared to be acting in an allosteric manner. The allosteric nature of its effect was evident in receptor protection experiments where there was no detectable interaction between compound-22 and decavanadate which appears to bind at, or close to, the ATP-binding site (Michel et al., 2006a). Furthermore, in radioligand-binding studies, compound-22 interacted with the sites labelled by the allosteric antagonist compound-17. In contrast, competitive P2X7 receptor antagonists such as PPADS and decavanadate have little effect on the binding of compound-17 (Michel et al., 2007). Finally, like compound-17 (Michel et al., 2008a), the potency of compound-22 at rat P2X7 receptors was reduced by GW791343.
Although AZ11645373 and compound-22 were both allosteric P2X7 receptor antagonists, they may bind to the receptor in different ways as the compounds differed in several respects. First, compound-22 potency was temperature-dependent but this was not observed with AZ11645373. Second, in compound interaction studies, GW791343 had little detectable effect on the potency of AZ11645373 but reduced the potency of compound-22 10-fold. Third, the potency of AZ11645373 was affected in the domain 1 chimera and the human F95L single point mutant receptors. In contrast, the potency of compound-22 was not affected in functional studies on the human F95L receptor and only slightly affected in the binding studies (twofold change in potency). Interestingly, compound-22 had little effect in rat-human or human-rat receptors suggesting that multiple sites may be required for its binding to the P2X7 receptor and that loss of either site results in a substantial loss of affinity for the receptor. This was not observed with AZ11645373. Taken together, it seems likely that compound-22 and AZ11645373 are both allosteric inhibitors of the P2X7 receptor but that each compound has a different mode of binding.
In conclusion, we have confirmed the species selectivity of the P2X7 receptor antagonists, AZ11645373 and compound-22, and characterized their interaction with three other species orthologues. The latter studies provided further evidence for pharmacological similarities in the human and dog P2X7 receptors and also identified quite marked species differences among rodent receptors in terms of their antagonist sensitivity. Furthermore, we have demonstrated that both AZ11645373 and compound-22 are allosteric antagonists, although they appear to bind at separate sites or induce their allosteric effects in different ways. It remains to be seen if the different mechanism of action will affect their potential therapeutic effects.
Glossary
Abbreviations:
- BzATP
2′-& 3′-O-(4benzoylbenzoyl) ATP
- compound-22
N-{2-methyl-5-[(1R, 5S)-9-oxa-3,7-diazabicyclo[3.3.1]non-3-ylcarbonyl]phenyl}-2-tricyclo[3.3.1.13,7]dec-1-ylacetamide hydrochloride
- DR
dose-ratio
- ECD
extracellular domain
- GW791343
N2-(3,4-difluorophenyl)-N1-[2-methyl-5-(1-piperazinylmethyl)phenyl]glycinamide dihydrochloride
- KN62
1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-L-tyrosyl]-4-phenylpiperazine
- PPADS
pyridoxal phosphate-6-azophenyl-2′,4′-disulphonic acid
Conflict of interest
The authors are employed by GlaxoSmithKline.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 3rd edition (2008 revision) Br J Pharmacol. 2008;153(2):S1–S209. doi: 10.1038/sj.bjp.0707746. Suppl. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baraldi PG, Di Virgilio F, Romagnoli R. Agonists and antagonists acting at P2X7 receptor. Curr Top Med Chem. 2004;4:1707–1717. doi: 10.2174/1568026043387223. [DOI] [PubMed] [Google Scholar]
- Chessell IP, Michel AD, Humphrey PP. Effect of antagonists at the human recombinant P2X7 receptor. Br J Pharmacol. 1998;124:1314–1320. doi: 10.1038/sj.bjp.0701958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chessell IP, Hatcher J, Bountra C, Michel AD, Hughes JB, Green P, et al. Disruption of the P2X7 purinoceptor gene abolishes chronic inflammatory and neuropathic pain. Pain. 2005;114:386–396. doi: 10.1016/j.pain.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Donnelly-Roberts DL, Jarvis MF. Discovery of P2X7 receptor-selective antagonists offers new insights into P2X7 receptor function and indicates a role in chronic pain states. Br J Pharmacol. 2007;151:571–579. doi: 10.1038/sj.bjp.0707265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, et al. The P2X7 receptor: a key player in IL-1β processing and release. J Immunol. 2006;176:3877–3883. doi: 10.4049/jimmunol.176.7.3877. [DOI] [PubMed] [Google Scholar]
- Fonfria E, Chambers LJ, Demont EH, Roman SA, Skaper SD, Michel AD. Species and temperature dependent effects of a novel P2X7 receptor antagonist on recombinant and native P2X7 receptors Soc Neurosci. Abstr no. 958.1.
- Fonfria E, Clay WC, Levy DS, Goodwin JA, Roman S, Smith GD, et al. Cloning and pharmacological characterisation of the guinea-pig P2X7 receptor orthologue. Br J Pharmacol. 2008;153:544–556. doi: 10.1038/sj.bjp.0707596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furber M, Alcar AZ, Bent JE, Beyerbach A, Bowers K, Braddock M, et al. Discovery of potent and selective adamantane-based small-molecule P2X7 receptor antagonists/interleukin-1beta inhibitors. J Med Chem. 2007;50:5882–5885. doi: 10.1021/jm700949w. [DOI] [PubMed] [Google Scholar]
- Hibell AD, Thompson KM, Xing M, Humphrey PP, Michel AD. Complexities of measuring antagonist potency at P2X7 receptor orthologs. J Pharmacol Exp Ther. 2001;296:947–957. [PubMed] [Google Scholar]
- Honore P, Donnelly-Roberts D, Namovic MT, Hsieh G, Zhu CZ, Mikusa JP, et al. A-740003 [N-(1-{[(cyanoimino)(5-quinolinylamino) methyl]amino}-2,2-dimethylpropyl)-2-(3,4-dimethoxyphenyl)acetamide], a novel and selective P2X7 receptor antagonist, dose-dependently reduces neuropathic pain in the rat. J Pharmacol Exp Ther. 2006;319:1376–1385. doi: 10.1124/jpet.106.111559. [DOI] [PubMed] [Google Scholar]
- Humphreys BD, Virginio C, Surprenant A, Rice J, Dubyak GR. Isolquinolines as antagonists of the P2X7 nucleotide receptor: high selectivity for the human versus the rat receptor homologues. Mol Pharmacol. 1998;54:22–32. doi: 10.1124/mol.54.1.22. [DOI] [PubMed] [Google Scholar]
- Michel AD, Xing M, Thompson KM, Jones CA, Humphrey PP. Decavanadate, a P2X receptor antagonist, and its use to study ligand interactions with P2X7 receptors. Eur J Pharmacol. 2006a;534:19–29. doi: 10.1016/j.ejphar.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Michel AD, Thompson KM, Simon J, Boyfield I, Fonfria E, Humphrey Patrick PA. Species and response dependent differences in the effects of MAPK inhibitors on P2X7 receptor function. Br J Pharmacol. 2006b;149:948–957. doi: 10.1038/sj.bjp.0706938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AD, Chambers LJ, Clay WC, Condreay JP, Walter DS, Chessell IP. Direct labelling of the human P2X7 receptor and identification of positive and negative cooperativity of binding. Br J Pharmacol. 2007;151:103–114. doi: 10.1038/sj.bjp.0707196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AD, Chambers LJ, Walter DS. Negative and positive allosteric modulators of the P2X7 receptor. Br J Pharmacol. 2008a;153:737–750. doi: 10.1038/sj.bjp.0707625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel AD, Clay WC, Ng SW, Roman S, Thompson K, Condreay JP, et al. Identification of regions of the P2X7 receptor that contribute to human and rat species differences in antagonist effects. Br J Pharmacol. 2008b;155:738–751. doi: 10.1038/bjp.2008.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RA. Molecular physiology of P2X receptors. Physiol Rev. 2002;82:1013–1067. doi: 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- Rassendren F, Buell GN, Virginio C, Collo G, North RA, Surprenant A. The permeabilizing ATP receptor, P2X7. Cloning and expression of a human cDNA. J Biol Chem. 1997;272:5482–5486. doi: 10.1074/jbc.272.9.5482. [DOI] [PubMed] [Google Scholar]
- Romagnoli R, Baraldi PG, Di Virgilio F. Recent progress in the discovery of antagonists acting at P2X7 receptor. Expert Opin Ther Patents. 2005;15:271–287. [Google Scholar]
- Roman S, Cusdin FS, Fonfria E, Goodwin JA, Clay WC, Michel AD. Cloning and pharmacological characterization of the dog recombinant P2X7 receptor. Proc Br Pharmacol Soc. 2007;5 doi: 10.1111/j.1476-5381.2009.00425.x. Abstract 0077 http://www.pA2online.org/abstracts/Vol5Issue2abst077P.pdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes L, Jiang LH, Alcaraz L, Bent J, Bowers K, Fagura M, et al. Characterization of a selective and potent antagonist of human P2X7 receptors, AZ11645373. Br J Pharmacol. 2006;149:880–887. doi: 10.1038/sj.bjp.0706933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surprenant A, Rassendren F, Kawashima E, North RA, Buell G. The cytolytic P2Z receptor for extracellular ATP identified as a P2X receptor (P2X7) Science. 1996;272:735–738. doi: 10.1126/science.272.5262.735. [DOI] [PubMed] [Google Scholar]
- Young MT, Pelegrin P, Surprenant A. Identification of Thr283 as a key determinant of P2X7 receptor function. Br J Pharmacol. 2006;149:261–268. doi: 10.1038/sj.bjp.0706880. [DOI] [PMC free article] [PubMed] [Google Scholar]