

Abstract
KCNQ genes encode five Kv7 K+ channel subunits (Kv7.1–Kv7.5). Four of these (Kv7.2–Kv7.5) are expressed in the nervous system. Kv7.2 and Kv7.3 are the principal molecular components of the slow voltage-gated M-channel, which widely regulates neuronal excitability, although other subunits may contribute to M-like currents in some locations. M-channels are closed by receptors coupled to Gq such as M1 and M3 muscarinic receptors; this increases neuronal excitability and underlies some forms of cholinergic excitation. Muscarinic closure results from activation of phospholipase C and consequent hydrolysis and depletion of membrane phosphatidylinositol-4,5-bisphosphate, which is required for channel opening. Some effects of M-channel closure, determined from transmitter action, selective blocking drugs (linopirdine and XE991) and KCNQ2 gene disruption or manipulation, are as follows: (i) in sympathetic neurons: facilitation of repetitive discharges and conversion from phasic to tonic firing; (ii) in sensory nociceptive systems: facilitation of A-delta peripheral sensory fibre responses to noxious heat; and (iii) in hippocampal pyramidal neurons: facilitation of repetitive discharges, enhanced after-depolarization and burst-firing, and induction of spontaneous firing through a reduction of action potential threshold at the axon initial segment. Several drugs including flupirtine and retigabine enhance neural Kv7/M-channel activity, principally through a hyperpolarizing shift in their voltage gating. In consequence they reduce neural excitability and can inhibit nociceptive stimulation and transmission. Flupirtine is in use as a central analgesic; retigabine is under clinical trial as a broad-spectrum anticonvulsant and is an effective analgesic in animal models of chronic inflammatory and neuropathic pain.
Keywords: potassium channels, M-channels, sympathetic neurons, sensory systems, hippocampal neurons, muscarinic receptors, excitability, action potentials, anticonvulsant, analgesic
Introduction
KCNQ genes encode members of the Kv7 family of K+ channel subunits. There are five members of this family – Kv7.1 to Kv7.5; of these, four (Kv7.2–Kv7.5) are expressed in the nervous system (see Jentsch, 2000; Robbins, 2001; Table 1). There they form subunits of the low-threshold voltage-gated K+ channel originally termed the ‘M-channel’ (Brown and Adams, 1980; Brown, 1988). While all members of the family generate M-like currents when expressed in oocytes or cell lines, in most neurons native M-channels are composed of Kv7.2 and Kv7.3 subunits (Wang et al., 1998) or sometimes of homomeric Kv7.2 subunits (Hadley et al., 2003; Schwarz et al., 2006), although probably with a contribution by Kv7.5 in some neurons(e.g. Shah et al., 2002); Kv7.4 subunits are predominantly expressed in the auditory and vestibular systems, but also probably contribute to M-channels in central dopaminergic neurons (Hansen et al., 2008).
Table 1.
Kv7 subunits: location, channel, function and channelopathy
| Subunit | Main location | Channel/current | Function(s) | Disease mutation |
|---|---|---|---|---|
| Kv7.1 | Heart | Delayed rectifier IKs | Repolarizes action potential | Long QT-syndrome (RWS) |
| Cochlea | Potassium transport | Congenital deafness (JLNS) | ||
| Kv7.2 | Brain, ganglia | M-current IK(M) | Controls excitability | Epilepsy (BFNC) |
| Kv7.3 | Brain, ganglia | M-current IK(M) | Controls excitability | Epilepsy (BFNC) |
| Kv7.4 | Cochlear, vestibular hair cells | OHC K+ current IK,s | Potassium transport | Deafness (DFNA2) |
| Kv7.5 | Brain, ganglia | M-current (?) | Controls excitability (?) | None |
BFNC, Benign Familial Neonatal Convulsions; DFNA2, dominant progressive hearing loss; JLNS, Jervill and Lange-Nielsen Syndrome; OHC, outer hair cell; RWS, Romano–Ward Syndrome.
Properties of neural M-channels
M-channels activate at subthreshold potentials, from about −60 mV (Figure 1). They do not inactivate so generate a steady voltage-dependent outward current. This assists in stabilizing the membrane potential in the presence of depolarizing currents and may (variably) contribute to the resting potential. Because their activation is relatively slow (tens of milliseconds) they do not contribute materially to the repolarization of individual action potentials. However, they can exert a profound dampening effect on repetitive or burst-firing and on the general excitability of neurons (Brown, 1988).
Figure 1.

Kv7/M-current in a rat sympathetic neuron. Records in (a) show M-currents recorded from a dissociated rat superior cervical sympathetic neuron with a perforated patch electrode on depolarizing the cell from −60 mV to −30 mV in 10 mV steps. Records in (b) show corresponding steady-state M-channel activity recorded through a cell-attached patch electrode on depolarizing the patch to the equivalent potentials to those in (a). (Adapted from figure 1 in Selyanko et al., 1992).
A signal (although not unique) property of M-channels is that they are inhibited by a variety of transmitters and hormones acting on G protein-coupled receptors, principally those coupling to Gq and/or G11. This was first observed by using muscarinic acetylcholine receptor (mAChR) agonists (hence the name ‘M-channels’; Brown and Adams, 1980), resulting (in this case) from activation of M1 (or at some sites M3 or possibly M5) mAChRs; their closure underlies part or all of the widespread slow excitatory effect of synaptically released ACh in both peripheral and central nervous systems, and hence for some of the physiological consequences of cholinergic activation. However, they can also be closed by many other similarly coupled receptors, including mGluR1 and mGluR5 metabotropic glutamate receptors, histamine H1, 5-hydroxytryptamine 5HT2C and P2Y1, 2, 4 and 6 nucleotide receptors, and also by several peptide receptors (e.g. for GnRH, substance P, bradykinin and angiotensin) (Marrion, 1997).
How does stimulating mAChRs close M-channels?
It has been clear for some time that M-channel inhibition is a rather indirect response to mAChR stimulation (Figure 2), and that it was likely to be associated with the well-known effect of Gq activation, to stimulate phospholipase Cβ and catalyse the hydrolysis of membrane phosphatidylinositol-4,5-bisphosphate (PIP2) (Marrion, 1997). Earlier work concentrated (not surprisingly) on the actions of the products of this hydrolysis, inositol-1,4,5-trisphosphate (IP3) and diacylglycerol as potential ‘diffusible messengers’. However, the more recent demonstration that – like many other membrane proteins – Kv7 (and native M) channels actually required PIP2 in order to enter the open state and close when membrane PIP2 levels are reduced or its polar head groups neutralized (Suh and Hille, 2002; Zhang et al., 2003; Robbins et al., 2006; Suh et al., 2006) has led to a complete volte-face – namely, that closure results, not from the accumulation of hydrolysis products but instead from the reduction in membrane PIP2 levels that results from its hydrolysis. Perhaps the most convincing evidence for this is that mAChR-induced inhibition of both expressed Kv7.2/7.3 channels and native neuronal M-channels is actually reduced or prevented when membrane PIP2 levels are increased by over-expressing the PI5-kinase (Winks et al., 2005; Suh et al., 2006), rather than increased as would be anticipated were closure to depend on hydrolysis products. This in turn has highlighted the very large and rapid changes in membrane PIP2 levels following mAChR stimulation, the concentration apparently falling by 90% plus within a few seconds (Winks et al., 2005; Suh et al., 2004; see also Willars et al., 1998). Direct evidence for this rapid depletion has recently been obtained by using a fluorescent probe that binds to PIP2 in the membrane then moves into the cytosol as PIP2 levels fall (Hughes et al., 2007; Quinn et al., 2008). The primary PIP2-binding site in the Kv7.2/7.3 channel has recently been identified as a cluster of basic amino acids in the carboxy-terminus (Hernandez et al., 2008): homology screening suggests that this region forms an interaction site with membrane lipids similar to that formed by Kir channels, which are also activated by PIP2 (see Logothetis et al., 2007).
Figure 2.

Inhibition of heteromeric Kv7.2/7.3 channels by the muscarinic agonist oxotremorine-M (Oxo-M). The channels were expressed in Chinese Hamster Ovary cells by KCNQ2 and KCNQ3 cDNA transfection. (A) Inhibition of whole-cell current recorded with a perforated patch electrode. Currents were generated by depolarizing the cell to −20 mV; records show deactivation of this steady outward current by 1 s hyperpolarizing steps to −50 mV. (B) Equivalent multi-channel activity recorded within a cell-attached membrane patch is inhibited by applying Oxo-M to the solution bathing the cell outside the patch, thereby activating muscarinic receptors remote from the Kv7.2/7.3 channels (‘remote inhibition’). (From Selyanko et al., 2000).
Physiology and pharmacology of M/Kv7 channels
While confirming the general properties of the current, considerably more detailed information about some of their specific properties at different neural loci has accrued over the last 10 years, from three main sources: the development of drugs that selectively block (e.g. linopirdine, XE991) or enhance (e.g. retigabine) M-channel activity (Miceli et al., 2008); and, following their molecular identification, the development of specific antibodies against the channel subunits that has provided more precise information about their localization; and from human genetic mutations (see Jentsch, 2000; Maljevic et al., 2008) and other genetic manipulations. In this report I concentrate on three types of neuron – sympathetic neurons, sensory neurons and hippocampal pyramidal neurons.
M/Kv7 channels in sympathetic neurons
Early inferences about M-channel function in sympathetic neurons depended partly, at least, on the effects of muscarinic agonists (Adams et al., 1986; Brown, 1988). Because (not surprisingly) these also affect other sympathetic neuron currents (e.g. Cassell and McLachlan, 1987), these inferences were always subject to some constraints. Notwithstanding, the principal conclusions so derived have been well substantiated by the effects of M-channel-specific drugs, both qualitatively (see Figure 3) and quantitatively (Zaika et al., 2006), and also from induced truncation of the KCNQ2 gene (Passmore et al., 2006). Thus, as shown in Figure 3, rat sympathetic neurons only fire one (or a very few) action potentials in response to long depolarizing steps; enhancement of M-current with retigabine both hyperpolarizes the neuron and (when this is corrected) completely inhibits spike generation, while channel blockade with XE991 allows a much more prolonged spike discharge – in essence converting the neuron from phasic firing to tonic firing (see also Brown and Selyanko, 1985; Zaika et al., 2006). As originally suggested (Brown, 1983) and recently confirmed by Zaika et al. (2006), the reason why M-current abbreviates the spike discharge is that the increased membrane conductance and enhanced outward current following the initial depolarization and spike-burst raises the threshold for subsequent spikes. Indeed, it is the absence or presence of an M-current that substantially determines whether sympathetic neurons naturally fire tonically or phasically (Wang and McKinnon, 1995). [An additional apamin-sensitive Ca2+-activated K+ current also contributes to spike-frequency adaptation in sympathetic neurons (Kawai and Watanabe, 1986); indeed, this current is enhanced when M-current is blocked, because the additional spikes increase Ca2+ influx. Hence, the full tonic-firing capacity of these neurons is only revealed when both M and KCa channels are blocked (see Brown, 1986; Yamada et al., 1998).]
Figure 3.

Effects of enhancing Kv/M-channel activity with retigabine and blocking Kv7/M-channels with XE991 on (left) membrane currents and (right) action potential generation in a dissociated rat sympathetic neuron. Records on the left show membrane currents recorded by depolarizing the cell to −20 mV (to activate Kv7/M-channels) then hyperpolarizing it to −50 mV (to deactivate the Kv7/M-current). Records in the presence of the drug are in red. Retigabine increases the outward Kv7/M-current at −20 mV and slows deactivation during hyperpolarization but accelerates reactivation. XE991 reduces the outward current and suppresses the slow deactivation relaxation, showing that Kv7/M-channels are blocked. Records on the right show responses to 4 s depolarizing currents up to 200 pA in 20 pA steps. Under normal conditions current injections ≥40 pA generated one (or sometimes two) action potentials at the beginning of the step. Retigabine suppressed spike generation whereas XE991 promoted repetitive spike discharges. Note: the starting membrane potential was held at −55 mV by constant current injection, offsetting the hyperpolarization and depolarization induced by retigabine and XE991 respectively (G.M. Passmore, unpublished).
Kv7/M-channels in sensory neurons
Many sensory neurons in rat dorsal root ganglia, including small neurons that respond to capsaicin (and hence are presumably nociceptive) express Kv7.2, Kv7.3 and/or Kv7.5 immunoreactivity and exhibit clear M-currents under voltage-clamp (Passmore et al., 2003). Further, retigabine enhances these currents, reduces the transmission of Aδ and C-fibre responses into the dorsal horn of the spinal cord (Passmore et al., 2003) and also hyperpolarizes primary afferent fibres (Rivera-Arconada and Lopez-Garcia, 2006). More recently, retigabine has been found to attenuate sensory Aδ and C-fibre discharges induced by heat stimulation when applied to the peripheral endings of sensory fibres in the isolated rat skin nerve preparation (Passmore and Brown, 2007), and to raise the threshold for C-fibre stimulation in human sural nerves (Lang et al., 2008). In (presumed) consequence of some or all of these effects, retigabine has been reported to exert an analgesic action in some models of chronic inflammatory or neuropathic pain (e.g. Blackburn-Munro and Jensen, 2003; Dost et al., 2004; Passmore et al., 2003).
These experiments clearly demonstrate the presence of Kv7/M-channels at various sites along the sensory neuraxis and also indicate the therapeutic potential of enhancing their activity (see following). However, they do not show whether these channels affect normal sensory processing: this requires tests with blocking agents. For example, while XE991 antagonizes the primary afferent hyperpolarization produced by retigabine, it did not itself change the primary afferent potential (Rivera-Arconada and Lopez-Garcia, 2006), implying a minimal contribution of Kv7/M-channels to primary afferent membrane potential. In contrast, XE991 strongly enhances peripheral Aδ discharges produced by thermal or mechanical stimulation in the isolated skin nerve preparation (Passmore and Brown, 2007; Figure 4). Likewise, Kv7.2, Kv7.3 and Kv7.5 immunoreactivity is present in peripheral baroceptor afferents, and here also XE991 strongly enhances baroceptor afferent discharges in response to increasing intra-arterial pressure in the aortic arch (Wladyka et al., 2008). Thus, it appears that Kv7/M-channels play a significant role in regulating some forms of peripheral sensory activity at least.
Figure 4.

Enhanced sensitivity of Aδ fibres to thermal stimulation by local application of XE991 in the rat isolated skin-nerve preparation. The preparation and recordings were obtained as described by Reeh (1986). Drugs were applied to the receptive field via the corium. Heat ramps from 30 to 46–47°C were applied for 90 s at 5 min intervals as shown. Single unit responses were recorded from a central portion of the saphenous nerve. Conduction velocity was 6.2 m·s−1. Records show second (control), fifth (+XE991 3 µmol·L−1) and eighth (wash) responses. Discharges per stimulus were 10, 52 and 10 respectively. (G.M. Passmore, unpublished).
The hippocampal CA1 pyramidal neuron: an example of a central neuron
M-current was first identified in these neurons by Halliwell and Adams (1982). As in ganglion cells, the channels are probably composed principally of Kv7.2 and Kv7.3 subunits, but with a possible contribution of Kv7.5 subunits (Shah et al., 2002). At least three ‘functions’ of these channels have been demonstrated.
Control of repetitive firing
As in sympathetic neurons, activation of Kv7/M-channels during the initial stages of an action potential discharge serves to suppress later action potentials and abbreviate the duration and frequency of the spike train induced by sustained depolarization. Thus, inhibition of channel activity, by either a blocking drug such as linopirdine (Aiken et al., 1995) or XE991, or expression of a dominant-negative Kv7.2 construct (Peters et al., 2005), strongly enhances repetitive firing (Figure 5). Also as in sympathetic neurons (but to an even greater extent), this form of excitability regulation by Kv7/M-channels is supplemented by a calcium-activated K+ current (Madison and Nicoll, 1984), carried in this case by a set of apamin-insensitive K+ channels of as yet unknown identity (Stocker et al., 2004).
Figure 5.

Blocking Kv7/M-channels or suppressing Kv7.2 channel expression facilitates repetitive spike discharges in mouse CA1 pyramidal neurons. Records show spike discharges in response to 500 ms depolarizing currents, up to 200 pA, in hippocampal slices from (A–C) normal mice and (D–F) transgenic mice expressing a dominant-negative (pore-defective) Kv7.2 mutant. Neurons from normal mice (A) show strong spike-frequency adaptation; XE991 (B) greatly reduces adaptation, facilitating repetitive discharge. Neurons from the transgenic mice (D) show much less spike adaptation and already fire repetitively; this is not significantly enhanced by XE991 (E). (Adapted from Peters et al., 2005, with permission of Nature Publishing Group).
Control of endogenous burst-firing
Another function of Kv7/M-channels in CA1 neurons is to suppress their intrinsic capacity for burst-firing. In these cells, an action potential may be followed by an after-depolarization, generated by a persistent Na+ current. This is normally opposed by the outward current carried by Kv7/M-channels. However, progressive blockade of these channels with linopirdine (Figure 6) or XE991 enhances the after-depolarization and eventually leads to a brief spike-burst (Yue and Yaari, 2004). Some CA1 neurons exhibit burst activity even in the absence of overt Kv7/M-channel block. This arises through an influx of Ca2+ ions through voltage-gated Ca2+ channels during the initial depolarization and spiking, which has the effect of temporarily inhibiting the Kv7/M-channels (Selyanko and Sim, 1998); Figure 7), thereby unmasking the effect of the persistent Na+ current and generating an after-depolarization (Chen and Yaari, 2008).
Figure 6.

Blocking Kv7/M-channels enhances a spike after-depolarization and induces burst-firing in rat CA1 hippocampal pyramidal neurons. Normally a brief current injection induces a single action potential (Aa1,b1). In a2–a4, XE991 gradually depolarized the cell and induced burst-firing. This resulted from an enhanced after-depolarization (B). Row Ab1–b4 and panel C show that XE991 had the same effects even when the depolarization was prevented. (Adapted from Yue and Yaari, 2004, with permission of the Journal of Neuroscience).
Figure 7.
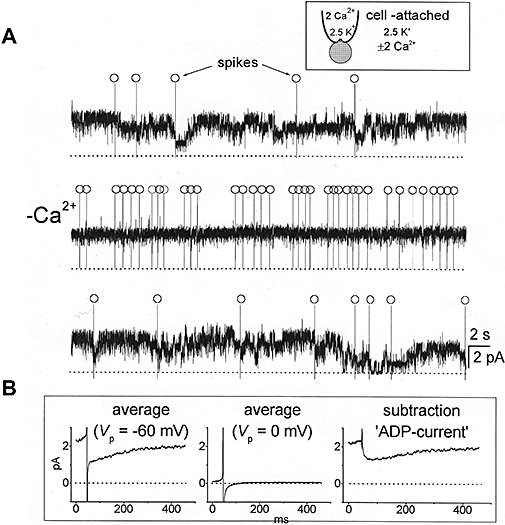
Transient Ca2+-dependent inhibition of Kv7/M-channels in dissociated hippocampal pyramidal neurons following spontaneous action potentials. (A) Cell-attached pipette recordings of multi-M channel activity obtained in 2 (upper and lower traces) and 0 (middle trace) mmole.L−1[Ca2+]. Spontaneous action potentials (‘spikes’) are marked (○). Note that action potentials are associated with intermittent channel closures in Ca2+-containing solution but not in Ca2+-free solution. (B) Ensembles of post-spike channel activity in Ca2+-containing solution recorded at −60 mV patch-potential (Vp), i.e., near-zero membrane potential (94 individual records) and 0 mV Vp, i.e., rest potential where most M-channels are shut (68 records). The latter was then subtracted from the former to yield (on the right) the net after-depolarizing current (ADP-current) resulting from spike-induced M-channel inhibition. (A.A.Selyanko, unpublished; data from Figures 11 and 12 in Selyanko & Sim, 1998, with permission of the Journal of Physiology.)
Control of action potential threshold and suppression of spontaneous firing
The highest density of Kv7.2 and Kv7.3 immunoreactivity in the CA1 region is not in the neuron somata (or dendrites) but in the axon initial segments (AIS), where the action potential is generated; there the Kv7 channels co-localize with the Na+ channels through binding to ankyrin G (Chung et al., 2006; Pan et al., 2006; Rasmussen et al., 2007) and serve to regulate the action potential threshold. Thus, inhibiting all CA1 channels in the neuron with XE991 both depolarizes the neuron by some 9 mV and reduces the action potential threshold by around 7–8 mV: as a result the action potential threshold is now set at or near the resting potential, so that many neurons now fire spontaneously, even in the total absence of synaptic activity (Shah et al., 2008; Figures 8A and 9). This change in action potential threshold is due to block of AIS Kv7 channels, not somatic channels, as it was replicated by intracellular application of a peptide designed to inhibit Kv7 channel binding to ankyrin G, which had no effect on somatic M-conductance (Figures 8B and 9). Computer simulations indicated that the observed control of AIS action potential threshold required an axon: somatic Kv7 density ratio of between 3:1 and 5:1 (Shah et al., 2008).
Figure 8.

Kv7/M-channel inhibition or disruption of Kv7/M-channel binding to ankyrin at the axon initial segment induces spontaneous action potential firing in hippocampal CA1 pyramidal neurons. Note: synaptic transmission was blocked by using a mixture of ionotropic and metabotropic glutamate and GABA antagonists. (A) Bath-applied XE991 (3 µmol·L−1) (a) depolarized the neuron, increased input resistance and facilitated repetitive firing in response to depolarizing current injections (a) and (b) induced spontaneous firing. (B) These effects were partly replicated by intracellular application of a peptide (ABP, ankyrin-binding peptide), designed to disrupt Kv7 channel binding to ankyrin at the axon initial segment, except that the peptide did not increase somatic input resistance (indicating that it did not affect somatic Kv7/M-channels). (Adapted from Shah et al., 2008).
Figure 9.
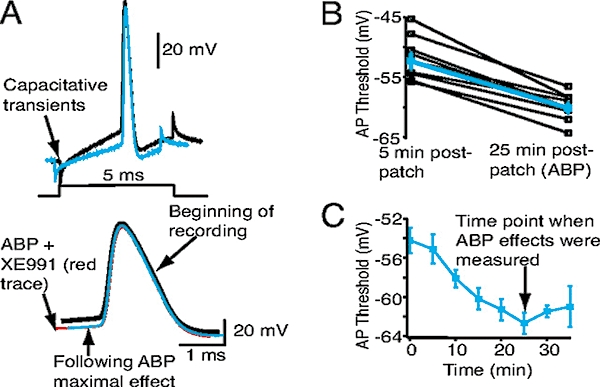
Kv7/M-channels in the axon initial segment control action potential (AP) threshold in hippocampal CA1 pyramidal neurons: inclusion of ankyrin-binding peptide (ABP; see Figure 8) in the patch-pipette progressively reduces AP threshold. (A) Superimposed examples of single APs 5 (black trace) and 25 min (blue trace) post patching with ABP in the patch-pipette solution. Each AP was generated by a 5 ms depolarizing current injection from −70 mV, as shown in the schematic. The magnitude of the current injection was adjusted to ensure that the step produced only a single AP. The APs are also shown on an enhanced time scale below. Note that subsequent bath addition of XE991 (red trace) did not further reduce the AP threshold. (B) Average (blue squares) AP thresholds at the beginning and 25 min post patching with ABP in the internal pipette solution. Open black squares represent the effects in individual neurons. (C) Time course of the reduction in AP threshold with ABP. (From Shah et al., 2008).
Interestingly, Kv7.2 subunits are also co-localized with Na+ channels at nodes of Ranvier in both central and peripheral nerves (Devaux et al., 2004; Schwarz et al., 2006). At the latter (peripheral) nodes they are responsible for the nodal K+ current previously termed IKs (DuBois, 1981) and – as at the AIS – regulate the action potential threshold and suppress repetitive firing (Schwarz et al., 2006). Thus, in some respects, the Kv7/M-channel can be regarded as a device to both stabilize the neuron's resting potential and constrain over-excitability by regulating the action potential threshold.
Pharmacological implications
Unlike most voltage-gated K+ channels, Kv7 channels have a rather unique pharmacology, which offers scope for selective intervention. These have recently been well reviewed by Miceli et al. (2008) (see also Gribkoff 2008).
The first selective blocking agent, linopirdine, was originally introduced as a cognition enhancer (Aiken et al., 1995; 1996), for potential application in Alzheimer's disease. However, it did not prove very effective in clinical trials (e.g. Rockwood et al., 1997), probably partly because pro-epileptic side effects such as tremors (not surprising in view of the effect of Kv7.2 and Kv7.3 mutations) limited the attainable dosage. Another problem with linopirdine (and its successor XE991) is that they are not selective for neural Kv7 channels but can also block cardiac Kv7.1 channels, albeit with lower potency when associated with their KCNE1 subunit as the native cardiac delayed rectifier (Wang et al., 2000). The widespread effects of Kv7/M-channel block may preclude any well-targeted use of blocking drugs.
M-channel enhancers offer more scope for therapeutic advances. Thus, flupirtine has been in clinical use as an analgesic (with some muscle-relaxant properties) for some 20 years – long before its effect on Kv7 channels was known (Friedel and Fitton, 1993). Flupirtine also showed some anticonvulsant properties; exploitation of this led to the development of retigabine, which shows an unusually broad spectrum of anticonvulsant properties in animal tests (Rostock et al., 1996) and shows efficacy in patients with partial onset seizures that are refractory to other anticonvulsants (Porter et al., 2007). Retigabine also shows analgesic activity, especially in animal models of chronic inflammatory and neuropathic pain (Blackburn-Munro and Jensen, 2003; Dost et al., 2004; Passmore et al., 2006; Munro et al., 2007)
Retigabine enhances Kv7 currents primarily (but not exclusively) through a hyperpolarizing shift of the voltage–conductance curve (Main et al., 2000; Wickenden et al., 2000; Tatulian et al., 2001; see Figure 10). This results from an increase in channel open times and a large (10-fold) reduction in the longest closed time in the steady-state open-closed time distributions, thereby effectively stabilizing the open state and increasing open probability (Tatulian and Brown, 2003). Importantly, retigabine does not enhance Kv7.1 currents. This difference was exploited to identify the binding site, as a hydrophobic pocket at the cytoplasmic ends of the S5–S6 domains of the Kv7 channel protein, with the obligatory presence of a tryptophan in the S5 domain (Schenzer et al., 2005; Wuttke et al., 2005). Thus, although retigabine shifts the voltage sensitivity of the Kv7 channel, it does not act on the voltage sensor (S4 domain) as such but rather by stabilizing the S5–S6 domain hinge in the open state.
Figure 10.
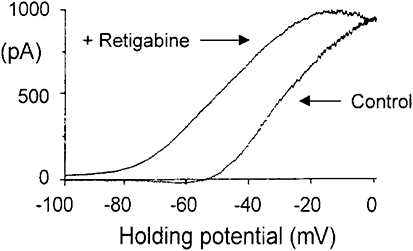
Retigabine (10 µmol·L−1) produces a 20 mV shift of the Kv7/M-current in a sympathetic neuron. Ordinates: current, abscissae: membrane voltage. Currents were evoked by using −100–0 mV ramped voltage commands and Kv7/M-current measured as that current blocked by 10 µmol·L−1 linopirdine. As a result of the voltage shift, an outward current is generated at the resting potential (around −60 mV), leading to an average hyperpolarization of 9 mV. (From Tatulian et al., 2001).
Retigabine shows no strong selectivity among neural Kv7 subunits (Kv7.2–Kv7.5) and is not totally specific for Kv7 channels (for example, it can also enhance GABAA currents: Otto et al., 2002). Hence there has been a search for other Kv7 enhancers with greater Kv7 specificity and/or subunit selectivity. Other, or more recent, enhancers include: (i) BMS-204352, originally developed as a KCa enhancer, which interacts with the retigabine site but with a noticeably strong action on Kv7.4 channels (Schrøder et al., 2001;Korsgaard et al., 2005); (ii) acrylamides with an enantiomer-specific effect, also interacting at the retigabine site and with a strong effect on Kv7.4 and Kv7.5 subunits (Bentzen et al., 2006); (iii) the non-steroidal analgesics diclofenac and meclofenamate (Peretz et al., 2005) and other compounds derived from these (Peretz et al., 2007), which may act at a different site from retigabine; (iv) a benzamide derivative ICA-27242, selective for Kv7.2/7.3 over Kv7.4 or Kv7.3/7.5 channels (Wickenden et al., 2008), and which is effective against a broad range of epileptic protocols in animal experiments, with no apparent effect on the water-maze cognition test (Roeloffs et al., 2008); and (v) zinc pyrithione, which also interacts with the S5–S6 domains but at a different (extracellular) site from retigabine that does not require the S5-tryptophan, and so enhances Kv7.1 currents as well as the neural currents (Xiong et al., 2007; 2008).
Potential therapeutic applications of these Kv7 enhancers, as deduced from animal experimentation, have expanded from anti-nociceptive and anticonvulsant to treatment of migraine, anxiety (Korsgaard et al., 2005; Munro et al., 2007), mania (Dencker et al., 2008) and addiction to psychostimulants (Hansen et al., 2007). This should not be taken to imply that Kv7/M-channel dysfunction necessarily plays any part in such disorders, merely that the widespread distribution of these channels provides a means of reducing overall neural excitability when Kv7 channel activity is enhanced. Thus, very few types of epilepsies can be attributed to Kv7 channel dysfunction, but retigabine can still reduce epileptiform discharges of whatever origin, for two reasons. First, the shift in Kv7 channel voltage activation means that many more channels are now open at the cell's resting potential and this both increases resting membrane conductance and hyperpolarizes the neuron, thereby reducing excitability (as seen in Figure 3); and second, because the accelerated channel opening during depolarization or following an action potential (which is partly but not entirely, a consequence of the voltage shift) enhances the natural effectiveness of Kv7 currents in suppressing repetitive firing.
Acknowledgments
The work from our laboratory referred to in this review was supported by grants from the UK Medical Research Council, the Wellcome Trust and the European Union.
Glossary
Abbreviations:
- AIS
axon initial segment
- IP3
inositol-1,4,5-trisphosphate
- mAChR
muscarinic acetylcholine receptor
- PIP2
phosphatidylinositol-4,5-bisphosphate
Conflict of interest
There are no conflict of interest.
References
- Adams PR, Jones SW, Pennefather P, Brown DA, Kock C, Lancaster B. Slow synaptic transmission in frog sympathetic ganglia. J Exp Biol. 1986;124:259–285. doi: 10.1242/jeb.124.1.259. [DOI] [PubMed] [Google Scholar]
- Aiken SP, Lampe BJ, Murphy PA, Brown BS. Reduction of spike frequency adaptation and blockade of M-current in rat CA1 pyramidal neurones by linopirdine (DuP 996), a neurotransmitter release enhancer. Br J Pharmacol. 1995;115:1163–1168. doi: 10.1111/j.1476-5381.1995.tb15019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiken SP, Zaczek R, Brown BS. Pharmacology of the neurotransmitter release enhancer linopirdine (DuP 996), and insights into its mechanism of action. Adv Pharmacol. 1996;35:349–384. doi: 10.1016/s1054-3589(08)60281-1. [DOI] [PubMed] [Google Scholar]
- Bentzen BH, Schmitt N, Calloe K, Dalby Brown W, Grunnet M, Olesen SP. The acrylamide (S)-1 differentially affects Kv7 (KCNQ) potassium channels. Neuropharmacology. 2006;51:1068–1077. doi: 10.1016/j.neuropharm.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Blackburn-Munro G, Jensen BS. The anticonvulsant retigabine attenuates nociceptive behaviours in rat models of persistent and neuropathic pain. Eur J Pharmacol. 2003;460:109–116. doi: 10.1016/s0014-2999(02)02924-2. [DOI] [PubMed] [Google Scholar]
- Brown DA. Slow cholinergic excitation – a mechanism for increasing neuronal excitability. Trends Neurosci. 1983;6:302–306. [Google Scholar]
- Brown DA. Voltage-sensitive ion channels mediating modulatory effects of acetylcholine, amines and peptides. In: Iversen LL, Goodman E, editors. Fast and Slow Chemical Signalling in the Nervous System. Oxford: Oxford University Press; 1986. pp. 130–150. [Google Scholar]
- Brown DA. M currents. In: Narahashi T, editor. Ion Channels. New York: Plenum Press; 1988. pp. 55–99. Vol. 1. [DOI] [PubMed] [Google Scholar]
- Brown DA, Adams PR. Muscarinic suppression of a novel voltage-sensitive K+-current in a vertebrate neurone. Nature. 1980;283:673–676. doi: 10.1038/283673a0. [DOI] [PubMed] [Google Scholar]
- Brown DA, Selyanko AA. Membrane currents underlying the slow excitatpry post-synaptic potential in the rat sympathetic ganglion. J Physiol. 1985;365:335–364. doi: 10.1113/jphysiol.1985.sp015777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassell JF, McLachlan EM. Muscarinic agonists block five different potassium conductances in guinea-pig sympathetic neurones. Br J Pharmacol. 1987;91:259–261. doi: 10.1111/j.1476-5381.1987.tb10279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Yaari Y. Spike Ca2+ influx upmodulates the spike afterdepolarization and bursting via intracellular inhibition of KV7/M channels. J Physiol. 2008;586:1351–1363. doi: 10.1113/jphysiol.2007.148171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HJ, Jan YN, Jan LY. Polarized axonal surface expression of neuronal KCNQ channels is mediated by multiple signals in theKCNQ2andKCNQ3C-terminal domains. Proc Natl Acad Sci USA. 2006;103:8870–8875. doi: 10.1073/pnas.0603376103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dencker D, Dias R, Pedersen ML, Husum H. Effect of the new antiepileptic drug retigabine in a rodent model of mania. Epilepsy Behav. 2008;12:49–53. doi: 10.1016/j.yebeh.2007.09.023. [DOI] [PubMed] [Google Scholar]
- Devaux JJ, Kleopa KA, Cooper EC, Scherer SS. KCNQ2 is a nodal K+ channel. J Neurosci. 2004;24:1236–1244. doi: 10.1523/JNEUROSCI.4512-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dost R, Rostock A, Rundfeldt C. The anti-hyperalgesic activity of retigabine is mediated by KCNQ potassium channel activation. Naunyn Schmiedebergs Arch Pharmacol. 2004;369:382–390. doi: 10.1007/s00210-004-0881-1. [DOI] [PubMed] [Google Scholar]
- Dubois JM. Evidence for the existence of three types of potassium channels in the frog Ranvier node membrane. J Physiol. 1981;318:297–316. doi: 10.1113/jphysiol.1981.sp013865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedel HA, Fitton A. Flupirtine. A review of its pharmacological properties, and therapeutic efficacy in pain states. Drugs. 1993;45:548–569. doi: 10.2165/00003495-199345040-00007. [DOI] [PubMed] [Google Scholar]
- Gribkoff VK. The therapeutic potential of neuronal Kv7 (KCNQ) channel modulators: an update. Expert Opin Ther Targets. 2008;12:565–581. doi: 10.1517/14728222.12.5.565. [DOI] [PubMed] [Google Scholar]
- Hadley JK, Passmore GM, Tatulian L, Al-Qatari M, Ye F, Wickenden AD, et al. Stoichiometry of expressed KCNQ2/KCNQ3 channels and subunit composition of native ganglionic M-channels deduced from block by tetraethylammonium (TEA) J Neurosci. 2003;23:5012–5019. doi: 10.1523/JNEUROSCI.23-12-05012.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell JV, Adams PR. Voltage-clamp analysis of muscarinic excitation in hippocampal neurons. Brain Res. 1982;250:71–92. doi: 10.1016/0006-8993(82)90954-4. [DOI] [PubMed] [Google Scholar]
- Hansen HH, Andreasen JT, Weikop P, Mirza N, Scheel-Krüger J, Mikkelsen JD. The neuronal KCNQ channel opener retigabine inhibits locomotor activity and reduces forebrain excitatory responses to the psychostimulants cocaine, methylphenidate and phencyclidine. Eur J Pharmacol. 2007;570:77–88. doi: 10.1016/j.ejphar.2007.05.029. [DOI] [PubMed] [Google Scholar]
- Hansen HH, Waroux O, Seutin V, Jentsch TJ, Aznar S, Mikkelsen JD. Kv7 channels: interaction with dopaminergic and serotonergic neurotransmission in the CNS. J Physiol. 2008;586:1823–1832. doi: 10.1113/jphysiol.2007.149450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez CC, Zaika O, Shapiro MS. A carboxy-terminal inter-helix linker as the site of phosphatidylinositol 4,5-bisphosphate action on Kv7 (M-type) K+ channels. J Gen Physiol. 2008;132:361–381. doi: 10.1085/jgp.200810007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes S, Marsh SJ, Tinker A, Brown DA. PIP(2)-dependent inhibition of M-type (Kv7.2/7.3) potassium channels: direct on-line assessment of PIP(2) depletion by Gq-coupled receptors in single living neurons. Pflugers Arch. 2007;455:115–124. doi: 10.1007/s00424-007-0259-6. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci. 2000;1:21–30. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- Kawai T, Watanabe M. Blockade of Ca-activated K conductance by apamin in rat sympathetic neurones. Br J Pharmacol. 1986;87:225–232. doi: 10.1111/j.1476-5381.1986.tb10175.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsgaard MP, Hartz BP, Brown WD, Ahring PK, Strøbaek D, Mirza NR. Anxiolytic effects of Maxipost (BMS-204352) and retigabine via activation of neuronal Kv7 channels. J Pharmacol Exp Ther. 2005;314:282–292. doi: 10.1124/jpet.105.083923. [DOI] [PubMed] [Google Scholar]
- Lang PM, Fleckenstein J, Passmore GM, Brown DA, Grafe P. Retigabine reduces the excitability of unmyelinated peripheral human axons. Neuropharmacology. 2008;54:1271–1278. doi: 10.1016/j.neuropharm.2008.04.006. [DOI] [PubMed] [Google Scholar]
- Logothetis DE, Jin T, Lupyan D, Rosenhouse-Dantsker A. Phosphoinositide-mediated gating of inwardly rectifying K(+) channels. Pflugers Arch. 2007;455:83–95. doi: 10.1007/s00424-007-0276-5. [DOI] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA. Control of the repetitive discharge of rat CA 1 pyramidal neurones in vitro. J Physiol. 1984;354:319–331. doi: 10.1113/jphysiol.1984.sp015378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Main MJ, Cryan JE, Dupere JRB, Cox B, Clare JJ, Burbidge SA. Modulation of KCNQ2/3 potassium channels by the novel anticonvulsant retigabine. Mol Pharmacol. 2000;58:253–262. doi: 10.1124/mol.58.2.253. [DOI] [PubMed] [Google Scholar]
- Maljevic S, Wuttke TV, Lerche H. Nervous system KV7 disorders: breakdown of a subthreshold brake. J Physiol. 2008;586:1791–1801. doi: 10.1113/jphysiol.2008.150656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrion NV. Control of M-current. Ann Rev Physiol. 1997;59:483–504. doi: 10.1146/annurev.physiol.59.1.483. [DOI] [PubMed] [Google Scholar]
- Miceli F, Soloveri MV, Martire M, Taglialatela M. Molecular pharmacology and therapeutic potential of neuronal Kv7-modulating drugs. Curr Opin Pharmacol. 2008;8:65–74. doi: 10.1016/j.coph.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Munro G, Erichsen HK, Mirza NR. Pharmacological comparison of anticonvulsant drugs in animal models of persistent pain and anxiety. Neuropharmacology. 2007;53:609–618. doi: 10.1016/j.neuropharm.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Otto JF, Kimball MM, Wilcox KS. Effects of the anticonvulsant retigabine on cultured cortical neurons: changes in electroresponsive properties and synaptic transmission. Mol Pharmacol. 2002;61:921–927. doi: 10.1124/mol.61.4.921. [DOI] [PubMed] [Google Scholar]
- Pan Z, Kao T, Horvath Z, Lemos J, Sul JY, Cranstoun SD, et al. A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J Neurosci. 2006;26:2599–2613. doi: 10.1523/JNEUROSCI.4314-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passmore GM, Brown DA. Effects of M-channel modulators on peripheral excitability in rat hairy skin. Abstr Soc Neurosci. 2007;681:8. [Google Scholar]
- Passmore GM, Selyanko AA, Mistry M, Al-Qatari M, Marsh SJ, Matthews EA, Dickenson AH, Brown TA, Burbidge SA, Main M, Brown DA. KCNQ/M currents in sensory neurons: significance for pain therapy. J Neurosci. 2003;23:7236–7727. doi: 10.1523/JNEUROSCI.23-18-07227.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passmore GM, Robbins J, Abogadie F, Brown DA. The KCNQ2 (Kv7.2) gene is required for functional M-channels in embryonic mouse superior cervical ganglion (SCG) neurons. Proc Physiol Soc. 2006;3:C106. [Google Scholar]
- Peretz A, Degani N, Nachman R, Uziyel Y, Gibor G, Shabat D, et al. Meclofenamic acid and diclofenac, novel templates of KCNQ2/Q3 potassium channel openers, depress cortical neuron activity and exhibit anticonvulsant properties. Mol Pharmacol. 2005;67:1053–1066. doi: 10.1124/mol.104.007112. [DOI] [PubMed] [Google Scholar]
- Peretz A, Degani-Katzav N, Talmon M, Danieli E, Gopin A, Malka E, Nachman R, Raz A, Shabat O, Attali B. A tale of switched functions: from cyclooxygenase inhibition to M-channel modulation in new diphenylamine derivatives. PLoSONE. 2007;2:e1332. doi: 10.1371/journal.pone.0001332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters HC, Hu H, Pongs O, Storm JF, Isbrandt D. Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nat Neurosci. 2005;8:51–60. doi: 10.1038/nn1375. [DOI] [PubMed] [Google Scholar]
- Porter RJ, Partiot A, Sachdeo R, Nohria V, Alves WM. Randomized, multicenter, dose-ranging trial of retigabine for partial-onset seizures. Neurology. 2007;68:1197–1204. doi: 10.1212/01.wnl.0000259034.45049.00. [DOI] [PubMed] [Google Scholar]
- Quinn KV, Behe P, Tinker A. Monitoring changes in membrane phosphatidylinositol 4,5-bisphosphate in living cells using a domain from the transcription factor tubby. J Physiol. 2008;586:2855–2871. doi: 10.1113/jphysiol.2008.153791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen HB, Frøkjaer-Jensen C, Jensen CS, Jensen HS, Jørgensen NK, Misonou H, et al. Requirement of subunit co-assembly and ankyrin-G for M-channel localization at the axon initial segment. J Cell Sci. 2007;120:953–963. doi: 10.1242/jcs.03396. [DOI] [PubMed] [Google Scholar]
- Reeh PW. Sensory receptors in mammalian skin in an in vitro preparation. Neurosci Letts. 1986;66:141–146. doi: 10.1016/0304-3940(86)90180-1. [DOI] [PubMed] [Google Scholar]
- Rivera-Arconada I, Lopez-Garcia JA. Retigabine-induced population primary afferent hyperpolarisation in vitro. Neuropharmacology. 2006;51:756–763. doi: 10.1016/j.neuropharm.2006.05.015. [DOI] [PubMed] [Google Scholar]
- Robbins J. KCNQ potassium channels: physiology, pathophysiology, and pharmacology. Pharmacol Ther. 2001;90:1–19. doi: 10.1016/s0163-7258(01)00116-4. [DOI] [PubMed] [Google Scholar]
- Robbins J, Marsh SJ, Brown DA. Probing the regulation of M(Kv7) channels in intact neurons with membrane-targeted peptides. J Neurosci. 2006;26:7950–7961. doi: 10.1523/JNEUROSCI.2138-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockwood K, Beattie BL, Eastwood MR, Feldman H, Mohr E, Pryse-Phillips W, et al. A randomized, controlled trial of linopirdine in the treatment of Alzheimer's disease. Can J Neurol Sci. 1997;24:140–145. doi: 10.1017/s031716710002148x. [DOI] [PubMed] [Google Scholar]
- Roeloffs R, Wickenden AD, Crean C, Werness S, McNaughton-Smith G, Stables J, et al. In vivo profile of ICA-27243 [N-(6-chloro-pyridin-3-yl)-3,4-difluoro-benzamide], a potent and selective KCNQ2/Q3 (Kv7.2/Kv7.3) activator in rodent anticonvulsant models. J Pharmacol Exp Ther. 2008;326:818–828. doi: 10.1124/jpet.108.137794. [DOI] [PubMed] [Google Scholar]
- Rostock A, Tober C, Rundfeldt C, Bartsch R, Engel J, Polymeropoulos EE, et al. D-23129: a new anticonvulsant with a broad spectrum activity in animal models of epileptic seizures. Epilepsy Res. 1996;23:211–223. doi: 10.1016/0920-1211(95)00101-8. [DOI] [PubMed] [Google Scholar]
- Schenzer A, Friedrich T, Pusch M, Saftig P, Jentsch TJ, Grotzinger J, et al. Molecular determinants of KCNQ (Kv7) K+ channel sensitivity to the anticonvulsant retigabine. J Neurosci. 2005;25:5051–5060. doi: 10.1523/JNEUROSCI.0128-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrøder RL, Jespersen T, Christophersen P, Strøbaek D, Jensen BS, Olesen SP. KCNQ4 channel activation by BMS-204352 and retigabine. Neuropharmacology. 2001;40:888–898. doi: 10.1016/s0028-3908(01)00029-6. [DOI] [PubMed] [Google Scholar]
- Schwarz JR, Glassmeier G, Cooper E, Kao T, Nodera H, Tabuena D, et al. KCNQ channels mediate IKs, a slow K+ current regulating excitability in the node of Ranvier. J Physiol. 2006;573:17–34. doi: 10.1113/jphysiol.2006.106815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selyanko AA, Sim JA. Ca2+-inhibited non-inactivating K+ channels in cultured rat hippocampal pyramidal neurones. J Physiol. 1998;510:71–91. doi: 10.1111/j.1469-7793.1998.071bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selyanko AA, Stansfeld CE, Brown DA. Closure of potassium M-channels by muscarinic acetylcholine-receptor stimulants requires a diffusible messenger. Proc Biol Sci. 1992;250:119–125. doi: 10.1098/rspb.1992.0139. [DOI] [PubMed] [Google Scholar]
- Selyanko AA, Hadley JK, Wood IC, Abogadie FC, Jentsch TJ, Brown DA. Inhibition of KCNQ1-4 potassium channels expressed in mammalian cells via M1 muscarinic acetylcholine receptors. J Physiol. 2000;522:349–355. doi: 10.1111/j.1469-7793.2000.t01-2-00349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Mistry M, Marsh SJ, Brown DA, Delmas P. Molecular correlates of the M-current in cultured rat hippocampal neurons. J Physiol. 2002;544:29–37. doi: 10.1113/jphysiol.2002.028571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Migliore M, Valencia I, Cooper EC, Brown DA. Functional significance of axonal Kv7 channels in hippocampal pyramidal neurons. Proc Natl Acad Sci USA. 2008;105:7869–7874. doi: 10.1073/pnas.0802805105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker M, Hirzel K, D'hoedt D, Pedarzani P. Matching molecules to function: neuronal Ca2+-activated K+ channels and afterhyperpolarizations. Toxicon. 2004;43:933–949. doi: 10.1016/j.toxicon.2003.12.009. [DOI] [PubMed] [Google Scholar]
- Suh BC, Hille B. Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron. 2002;35:507–520. doi: 10.1016/s0896-6273(02)00790-0. [DOI] [PubMed] [Google Scholar]
- Suh BC, Horowitz LF, Hirdes W, Mackie K, Hille B. Regulation of KCNQ21KCNQ3 current by G protein cycling: the kinetics of receptor-mediated signaling by Gq. J Gen Physiol. 2004;123:663–683. doi: 10.1085/jgp.200409029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh BC, Inoue T, Meyer T, Hille B. Rapid chemically induced changes of PtdIns(4,5)P2 gate KCNQ ion channels. Science. 2006;314:1454–1457. doi: 10.1126/science.1131163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatulian L, Brown DA. Effect of the KCNQ potassium channel opener retigabine on single KCNQ2/3 channels expressed in CHO cells. J Physiol. 2003;549:57–63. doi: 10.1113/jphysiol.2003.039842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatulian L, Delmas P, Abogadie FC, Brown DA. Activation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabine. J Neurosci. 2001;21:5535–5545. doi: 10.1523/JNEUROSCI.21-15-05535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HS, McKinnon D. Potassium currents in rat prevertebral and paravertebral sympathetic neurones. J Physiol. 1995;485:319–337. doi: 10.1113/jphysiol.1995.sp020732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, et al. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- Wang HS, Brown BS, McKinnon D, Cohen IS. Molecular basis for differential sensitivity of KCNQ and I(Ks) channels to the cognitive enhancer XE991. Mol Pharmacol. 2000;57:1218–1223. [PubMed] [Google Scholar]
- Wickenden AD, Yu W, Zou A, Jegla T, Wagoner PK. Retigabine, a novel anti-convulsant, enhances activation of KCNQ2/Q3 potassium channels. Mol Pharmacol. 2000;58:591–600. doi: 10.1124/mol.58.3.591. [DOI] [PubMed] [Google Scholar]
- Wickenden AD, Krajewski JL, London B, Wagoner PK, Wilson WA, Clark S, et al. N-(6-chloro-pyridin-3-yl)-3,4-difluoro-benzamide (ICA-27243): a novel, selective KCNQ2/Q3 potassium channel activator. Mol Pharmacol. 2008;73:977–986. doi: 10.1124/mol.107.043216. [DOI] [PubMed] [Google Scholar]
- Willars GB, Nahorski SR, Challis RAJ. Differential regulation of muscarinic acetylcholine receptor-sensitive polyphosphoinositide pools and consequences for signaling in human neuroblastoma cells. J Biol Chem. 1998;273:5037–5046. doi: 10.1074/jbc.273.9.5037. [DOI] [PubMed] [Google Scholar]
- Winks JS, Hughes S, Filippov AK, Tatulian L, Abogadie FC, Brown DA, et al. Relationship between membrane phosphatidylinositol-4,5-bisphosphate and receptor-mediated inhibition of native neuronal M channels. J Neurosci. 2005;25:3400–3413. doi: 10.1523/JNEUROSCI.3231-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wladyka CL, Feng B, Glazebrook PA, Schild JH, Kunze DL. The KCNQ/M-current modulates arterial baroreceptor function at the sensory terminal in rats. J Physiol. 2008;586:795–802. doi: 10.1113/jphysiol.2007.145284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuttke TV, Seebohm G, Bail S, Maljevic S, Lerche H. The new anticonvulsant retigabine favors voltage-dependent opening of the Kv7.2 (KCNQ2) channel by binding to its activation gate. Mol Pharmacol. 2005;67:1009–1017. doi: 10.1124/mol.104.010793. [DOI] [PubMed] [Google Scholar]
- Xiong Q, Sun H, Li M. Zinc pyrithione-mediated activation of voltage-gated KCNQ potassium channels rescues epileptogenic mutants. Nat Chem Biol. 2007;3:287–296. doi: 10.1038/nchembio874. [DOI] [PubMed] [Google Scholar]
- Xiong Q, Sun H, Zhang Y, Nan F, Li M. Combinatorial augmentation of voltage-gated KCNQ potassium channels by chemical openers. Proc Natl Acad Sci USA. 2008;105:3128–3133. doi: 10.1073/pnas.0712256105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada W, Koch C, Adams P. Multiple channels and calcium dynamics. In: Koch C, Segev L, editors. Methods in Neuronal Modelling: from Synapse to Networks. 2nd. Cambridge, MA: MIT Press; 1998. pp. 137–170. [Google Scholar]
- Yue C, Yaari Y. KCNQ/M channels control spike afterdepolarization and burst generation in hippocampal neurons. J Neurosci. 2004;24:4614–4624. doi: 10.1523/JNEUROSCI.0765-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaika O, Lara LS, Gamper N, Hilgemann DW, Jaffe DB, Shapiro MS. Angiotensin-II regulates neuronal excitability via PIP2-dependent modulation of Kv7 (M-type) potassium channels. J Physiol. 2006;575:49–67. doi: 10.1113/jphysiol.2006.114074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Craciun LC, Mirshahi T, Rohacs T, Lopes CM, Jin T, et al. PIP(2) activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron. 2003;37:963–975. doi: 10.1016/s0896-6273(03)00125-9. [DOI] [PubMed] [Google Scholar]