

Abstract
Background and purpose:
M2, M3 and/or M4 muscarinic acetylcholine receptors have been reported to mediate presynaptic inhibition in sympathetic neurons. M1 receptors mediate an inhibition of Kv7, CaV1 and CaV2.2 channels. These effects cause increases and decreases in transmitter release, respectively, but presynaptic M1 receptors are generally considered facilitatory. Here, we searched for inhibitory presynaptic M1 receptors.
Experimental approach:
In primary cultures of rat superior cervical ganglion neurons, Ca2+ currents were recorded via the perforated patch-clamp technique, and the release of [3H]-noradrenaline was determined.
Key results:
The muscarinic agonist oxotremorine M (OxoM) transiently enhanced 3H outflow and reduced electrically evoked release, once the stimulant effect had faded. The stimulant effect was enhanced by pertussis toxin (PTX) and was abolished by blocking M1 receptors, by opening Kv7 channels and by preventing action potential propagation. The inhibitory effect was not altered by preventing action potentials or by opening Kv7 channels, but was reduced by PTX and ω-conotoxin GVIA. The inhibition remaining after PTX treatment was abolished by blockage of M1 receptors or inhibition of phospholipase C. When [3H]-noradrenaline release was triggered independently of voltage-activated Ca2+ channels (VACCs), OxoM failed to cause any inhibition. The inhibition of Ca2+ currents by OxoM was also reduced by ω-conotoxin and PTX and was abolished by M1 antagonism in PTX-treated neurons.
Conclusions and implications:
These results demonstrate that M1, in addition to M2, M3 and M4, receptors mediate presynaptic inhibition in sympathetic neurons using phospholipase C to close VACCs.
Keywords: muscarinic receptors, noradrenaline release, presynaptic inhibition, phospholipase C, voltage-activated Ca2+ channels, Kv7 channels
Introduction
The reduction of noradrenaline release due to the activation of muscarinic acetylcholine receptors (mAChRs) located at sympathetic axon terminals was described for the first time at least four decades ago (Lindmar et al., 1968). Thus, this mechanism is, presumably, the best-documented example of presynaptic heteroinhibition in the sympathetic nervous system. Nevertheless, presynaptic modulation via muscarinic receptors remained controversial, as an enhancement of noradrenaline release via these receptors has also been observed (Fuder and Muscholl, 1995; Boehm and Kubista, 2002).
The family of mAChRs comprises at least five different subtypes (M1 through M5) which were originally characterized by the use of various antagonists that discriminate between them by up to 100-fold differences in affinities (Caulfield and Birdsall, 1998). More recently, toxins have been used instead which display more than 1000-fold differences in affinities (Onali et al., 2005). Complementary information on the roles of single mAChRs has been obtained in mutant mice (Wess et al., 2007). In the case of presynaptic mAChRs on sympathetic neurons, inhibitory effects have most commonly been reported to involve M2, whereas facilitatory effects are rarely observed and rather assigned to M1 receptors (Fuder and Muscholl, 1995; Boehm and Kubista, 2002). Results obtained with various sympathetically innervated tissues from mAChR knockout mice suggest that several combinations of presynaptic M2, M3 and M4 receptors control noradrenaline release (Trendelenburg et al., 2005). The different mAChRs are linked to various effector systems via one of the two following prototypic signalling cascades: M1, M3 and M5 via pertussis toxin (PTX)-insensitive Gq-type G-proteins and phospholipase Cβ, M2 and M4 via PTX-sensitive Gi-type G-proteins and adenylyl cyclase (Caulfield and Birdsall, 1998; Wess et al., 2007).
Inhibition of noradrenaline release via presynaptic G-protein-coupled receptors (GPCRs) is generally believed to rely on an inhibition of voltage-activated Ca2+ channels (VACCs) which in most, but not all, cases involves PTX-sensitive G-proteins (Kubista and Boehm, 2006). This mechanism has been investigated most thoroughly in sympathetic neurons derived from superior cervical ganglia (SCG) (Hille, 1994). With mAChRs, this effect involves several subtypes: the PTX-sensitive inhibition is mediated by M2 and M4, and the PTX-insensitive inhibition involves M1 (Shapiro et al., 1999; Liu and Rittenhouse, 2003b). This latter receptor not only controls the gating of VACCs, but also mediates an inhibition of Kv7 (KCNQ) channels which leads to a depolarization of SCG neurons and to increased action potential firing (Lechner et al., 2003; Hernandez et al., 2008). Moreover, the inhibition of Kv7 channels was shown to promote action potential-mediated noradrenaline release from sympathetic neurons (Kristufek et al., 1999; Lechner et al., 2003; Hernandez et al., 2008) and was suggested to mediate the facilitation of dopamine release by presynaptic mAChRs in striatal nerve terminals (Martire et al., 2007). Hence, in SCG neurons, M1 receptors can activate signalling cascades that mediate either an inhibition or a facilitation of action potential-evoked noradrenaline release.
In previous experiments, activation of M1 mAChRs has been demonstrated to trigger noradrenaline release from SCG neurons, but this effect was abolished by tetrodotoxin (TTX), that is, it required action potential propagation, and was thus not mediated by presynaptic receptors (Lechner et al., 2003). The role of presynaptic M1 mAChRs in SCG neurons, in contrast, remained elusive. Accordingly, this study was designed to clarify whether presynaptic M1 receptors can control noradrenaline release from rat SCG neurons in either a facilitatory or an inhibitory manner and whether either of these effects involves a modulation of Kv7 channels or VACCs respectively. The results reveal that activation of presynaptic M1 receptors reduces transmitter release due to an inhibition of VACCs via phospholipase C.
Methods
Primary cultures of rat superior cervical ganglion neurons
Primary cultures of dissociated SCG neurons from neonatal rats were prepared as described previously (Boehm, 1999). Newborn Sprague-Dawley rats were kept and killed 3 to 10 days after birth by decapitation in full accordance with all rules of the Austrian animal protection law (see http://ris1.bka.gv.at/Appl/findbgbl.aspx?name=entwurf&format=pdf&docid=COO_2026_100_2_72288) and the Austrian animal experiment by-laws (see http://www.ris2.bka.gv.at/Dokumente/BgblPdf/2000_198_2/2000_198_2.pdf). Ganglia were removed immediately after decapitation of the animals, cut into three to four pieces, and incubated in collagenase (1.5 mg·mL−1) and dispase (3.0 mg·mL−1) for 20 min at 36°C. Subsequently, they were further incubated in trypsin (0.25% trypsin) for 15 min at 36°C, dissociated by trituration and resuspended in Dulbeccos modified Eagle's medium containing 2.2 g·L−1 glucose, 10 mg·L−1 insulin, 25 000 iu·L−1 penicillin and 25 mg·L−1 streptomycin, 50 µg·L−1 nerve growth factor, and 5% foetal calf serum. Finally, all cells were seeded onto 5 mm plastic discs for radiotracer release experiments and onto 35 mm culture dishes for electrophysiological experiments. All tissue culture plastic was coated with rat tail collagen. The cultures were stored for 4 to 8 days in a humidified 5% CO2 atmosphere at 36°C. On days 1 and 4 after dissociation, the medium was exchanged entirely.
Determination of [3H]-noradrenaline release
[3H]-noradrenaline uptake and superfusion were performed as described previously (Boehm, 1999). The plastic discs with dissociated neurons were incubated in 0.05 µmol·L−1[3H]-noradrenaline (specific activity 37 Ci·mmol−1) in culture medium supplemented with 1 mmol·L−1 ascorbic acid at 36°C for 1 h. Thereafter, the culture discs were transferred to small chambers and superfused with a buffer containing (mmol·L−1): NaCl 120, KCl 6.0, CaCl2 2.0, MgCl2 2.0, glucose 20, HEPES 10, fumaric acid 0.5, Na-pyruvate 5.0 and ascorbic acid 0.57, adjusted to pH 7.4 with NaOH. Superfusion was performed at 25°C at a rate of about 1.0 mL·min−1. Collection of 4 min fractions of superfusate was started after a 60 min washout period during which excess radioactivity had been removed.
Depolarization-dependent tritium overflow was triggered either by electrical fields or by 30 mmol·L−1 KCl (NaCl was reduced accordingly to maintain isotonicity), both being applied for periods of 60 s. Stimulation by monophasic rectangular electrical pulses (0.5 ms, 60 mA, 50 V·cm−1) at 1.0 Hz was performed after 73 (S1) and 113 (S3) min of superfusion (see e.g. Figure 1A), stimulation by K+ after 73 (S1) and 93 (S3) min of superfusion (see e.g. Figure 4A). Tritium overflow independent of VACCs was elicited by 60 s applications of 100 µmol·L−1 adenosine–5′–triphosphate (ATP) in the presence of TTX to selectively activate Ca2+-permeable presynaptic P2X receptors (Boehm, 1999), again after 73 (S1) and 93 (S3) min of superfusion. Muscarinic agonists [oxotremorine M (OxoM) and bethanechol] were included in the buffer from minute 88 (see e.g. Figure 4A) onward. As OxoM is known to stimulate noradrenaline release from SCG neurons (Lechner et al., 2003), the accompanying transient rise in tritium outflow was defined as second stimulation period (S2). Modifying agents, such as TTX or retigabine, were included in the buffer after 50 min of superfusion (i.e. 10 min prior to the start of sample collection). The radioactivity remaining in the cells after the completion of experiments was extracted by immersion of the discs in 2% perchloric acid followed by sonication. Radioactivity in extracts and collected fractions was determined by liquid scintillation counting (Packard Tri-Carb 2800 TR). Radioactivity released in response to electrical field stimulation from rat sympathetic neurons after labelling with tritiated noradrenaline under conditions similar to those of the present study had previously been shown to consist predominantly of the authentic transmitter and to contain only small amounts (≤15%) of metabolites (Schwartz and Malik, 1993). Hence, the outflow of tritium measured in this study was assumed to reflect the release of noradrenaline and not that of metabolites.
Figure 1.
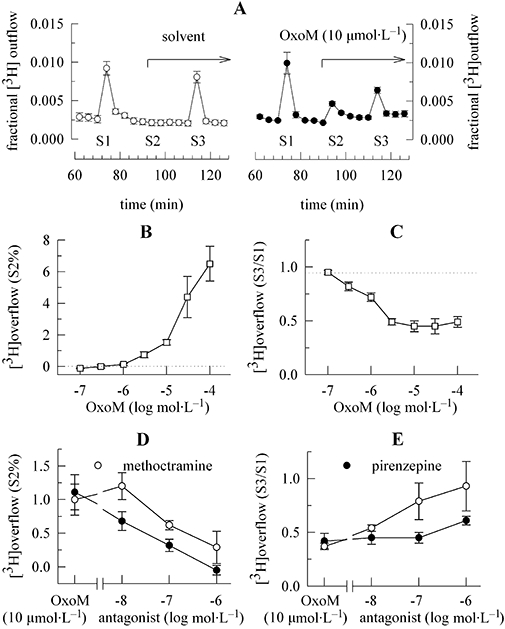
Enhancement of spontaneous and inhibition of electrically evoked noradrenaline release by OxoM via different sets of receptors. Primary cultures of rat SCG neurons were labelled with [3H]-noradrenaline for 1 h and then superfused. Subsequent to a 60 min washout period, 4 min fractions of superfusate were collected. Electrical field stimulation was applied at 73 (S1) and 113 (S3) min of superfusion. OxoM, either alone or together with pirenzepine or methoctramine, was present as indicated by the arrow on the right hand side in (A) and caused transient increases in the rate of spontaneous 3H outflow (S2); as a control for statistical comparison, solvent was applied instead of OxoM. (A) The time course of fractional 3H outflow·min−1 and its alteration by 10 µmol·L−1 OxoM on the right hand side (n= 3). For comparison, the effect of solvent is depicted on the left hand side (n= 3). (B) The concentration-dependence of OxoM-induced 3H overflow, as exemplified by S2% values. The indicated concentrations of OxoM were applied as shown in (A) (n= 5 to 6). (C) The concentration-dependence of the inhibition of electrically evoked tritium overflow by OxoM, as exemplified by S3/S1 ratios. The indicated concentrations of OxoM were applied as shown in (A) (n= 5 to 6). (D) The concentration-dependent attenuation of the 3H overflow induced by 10 µmol·L−1 OxoM by pirenzepine or methoctramine, as exemplified by S2% values. OxoM (10 µmol·L−1) was applied as shown in (A) either alone or together with the indicated concentrations of pirenzepine or methoctramine (n= 6 to 8). (E) The concentration-dependent attenuation of the inhibition of electrically evoked tritium overflow by 10 µmol·L−1 OxoM in the presence of pirenzepine or methoctramine, as exemplified by S3/S1 ratios. OxoM (10 µmol·L−1) was applied as shown in (A) either alone or together with the indicated concentrations of pirenzepine or methoctramine (n= 6 to 8). OxoM, oxotremorine M; SCG, superior cervical ganglia.
Figure 4.
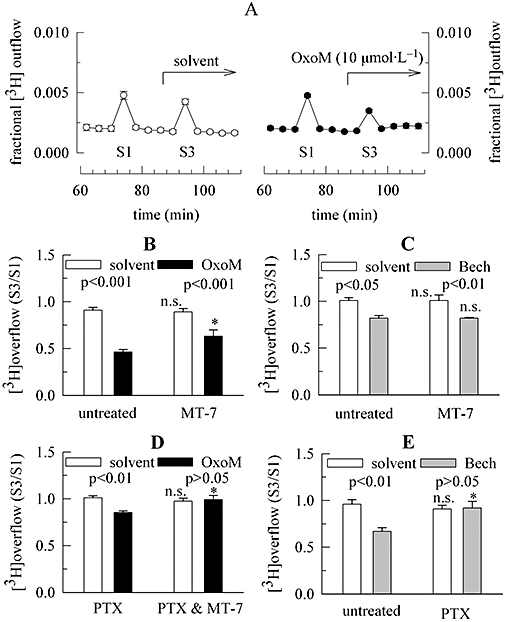
Characterization of the inhibition of K+-evoked noradrenaline release via mAChRs in the presence of TTX. Primary cultures of rat SCG neurons were either treated with pertussis toxin (PTX; 100 ng·mL−1 for 24 h) or remained untreated. Cultures were then labelled with [3H]-noradrenaline for 1 h in the absence or presence of 100 nmol·L−1 MT-7 and were superfused. From 50 min of superfusion onward, 0.1 µmol·L−1 TTX was present. Subsequent to a 60 min washout period, 4 min fractions of superfusate were collected. KCl 30 mmol·L−1 (NaCl was reduced accordingly) was present at 73 (S1) and 93 (S3) min of superfusion. OxoM 10 µmol·L−1 or 100 µmol·L−1 bethanechol (Bech) was present as indicated by the arrow; as a control for statistical comparison, solvent was applied instead of OxoM or bethanechol. (A) The time course of fractional 3H outflow·min−1 and its alteration by 10 µmol·L−1 OxoM on the right hand side (n= 3). For comparison, the effect of solvent is depicted on the left hand side (n= 3). (B) The inhibition of K+-evoked tritium overflow (as exemplified by S3/S1 ratios) by 10 µmol·L−1 OxoM in either untreated or MT-7-treated cultures; its effect is compared with that of solvent (n= 9). (C) The inhibition of K+-evoked tritium overflow (as exemplified by S3/S1 ratios) by 100 µmol·L−1 bethanechol in either untreated or MT-7-treated cultures; its effect is compared with that of solvent (n= 6). (D) The inhibition of K+-evoked tritium overflow (as exemplified by S3/S1 ratios) by 10 µmol·L−1 OxoM in cultures treated either with PTX or with PTX plus MT-7; the effect of OxoM is compared with that of solvent (n= 8 to 9). (E) The inhibition of K+-evoked tritium overflow (as exemplified by S3/S1 ratios) by 100 µmol·L−1 bethanechol in either untreated or PTX-treated cultures; the effect of bethanechol is compared with that of solvent (n= 6). In (B) to (E), significances of differences between pairs of bars are indicated above the corresponding pairs. * Indicates a significant difference versus the corresponding results obtained in either untreated (B, C and E) or PTX-treated (D) cultures at P < 0.05; n.s. indicates the lack of such a significance. mAChRs, muscarinic acetylcholine receptors; MT-7, muscarinic toxin 7; OxoM, oxotremorine M; PTX, pertussis toxin; SCG, superior cervical ganglia; TTX, tetrodotoxin.
The spontaneous (unstimulated) rate of 3H efflux was obtained by expressing the radioactivity retrieved during a collection period as a fraction of the total radioactivity in the cultures at the beginning of this period (fractional 3H outflow). Stimulation-evoked tritium overflow was calculated as the difference between the total tritium outflow during and after stimulation and the estimated basal outflow, which was assumed to follow a linear time course throughout experiments. Therefore, basal outflow during periods of stimulation was assumed to equate to the arithmetic mean of the samples preceding and those following stimulation, respectively. Differences between total and estimated basal outflow during periods of stimulation were expressed as percentages of total radioactivity in the cultures at the onset of stimulation (% of total radioactivity; S%). The amount of radioactivity in the cultures at the beginning of each collection period is calculated by summing up the radioactivity remaining in the cells at the end of experiments and that retrieved during the respective and all subsequent collection periods.
As the amount of depolarization-or drug-induced tritium overflow may vary considerably between different cultures (Scholze et al., 2002), the effects of mAChR ligands on depolarization-dependent release were evaluated by determining changes in the ratio of tritium overflow evoked during the two periods of electrical or K+ stimulation (S3/S1). When cultures had been subjected to a certain treatment, for example PTX or muscarinic toxin 7 (MT-7), control experiments were also performed in sister cultures that had not been exposed to that treatment (i.e. remained ‘untreated’).
Electrophysiology
Ca2+ currents of sympathetic neurons were determined using the perforated patch clamp technique as described previously (Lechner et al., 2005). Currents were recorded at room temperature (20–24°C) from single SCG neurons in vitro using an Axopatch 200B amplifier and the pCLAMP 8.0 hard-and software. Signals were low-pass filtered at 5 kHz, digitized at 10 to 50 kHz and stored on an IBM compatible computer. Traces were analysed off-line by the Clampfit 8.1 program (Axon). Patch electrodes were pulled (Flaming-Brown puller) from borosilicate glass capillaries, front-filled with a solution consisting of (mmol·L−1): CsCl 130, tetraethylammonium chloride 20, CaCl2 0.24, glucose 10, HEPES 10, EGTA 5, adjusted to pH 7.3 with KOH, and then back-filled with the same solution containing 200 µg·mL−1 amphotericin B (in 0.8% dimethyl sulfoxide (DMSO)) which yielded tip resistances of 2 to 3 MΩ. This combination of solutions results in small liquid junction potentials of about +2 mV, which, however, were ignored. Drugs were applied via a DAD-12 drug application device, which permits a complete exchange of solutions surrounding the cells being investigated within less than 100 ms (Boehm, 1999).
Ca2+ currents were elicited every 15 s by 30 ms depolarizations from a holding potential of −80 mV to +10 mV. Leakage currents were corrected for by an on-line leak subtraction protocol, which applies four hyperpolarizing pulses prior to the depolarization to +10 mV in order to determine the extent of leakage. The extent of current inhibition by muscarinic agonists or isradipine was quantified according to the equation: % inhibition = 100 − (200 * B/(A + C)), where B is peak current amplitudes determined 120 s after the start of drug application and A and C are the amplitudes of control currents measured directly before and 120 s after the end of the application of agonists. As ω-conotoxin GVIA causes an irreversible inhibition of voltage-gated Ca2+ currents in sympathetic neurons (Boehm and Huck, 1996), 1 µmol·L−1 of this toxin was applied until the reduction of current amplitudes reached a steady state. The amplitudes after this treatment were compared with those determined prior to the application of the toxin, and the effect was again expressed as % inhibition.
Statistics and nomenclature
All values are arithmetic means ± s.e.mean. Values of n reflect single cells in electrophysiological experiments and numbers of cultures in radiotracer release experiments. Statistical significance of differences between two groups was determined by the Mann–Whitney test. Statistical significance of differences between multiple groups was performed by one-way analyses of variances followed by Bonferroni's multiple comparison corrections; P values <0.05 were considered as indicating statistical significance. The nomenclature for ion channels and receptors follows the guide to receptors and channels (Alexander et al., 2008).
Materials
(-)-[Ring-2,5,6-3H]-noradrenaline was obtained from PerkinElmer (Vienna, Austria); collagenase, amphotericin B, OxoM, pirenzepine, methoctramine, bethanechol, UK 14304 (5-bromo-N-(4,5-dihydro-1H-imidazol-2-yl)-6-quinoxalinamine), U73122 (1-[6-[[(17b)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione), U73343 (1-[6-[[(17b)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-2,5-pyrollidinedione), isradipine and PTX from Sigma (Vienna, Austria); TTX from Latoxan (Rosans, France); MT-7 from the Peptide Institute Inc. (Osaka, Japan); ω-conotoxin GVIA from Alomone Labs Ltd. (Jerusalem, Israel), bulk chemicals were from Merck (Vienna, Austria). Retigabine was kindly donated by Dr B. Pechstein (Viatris, Frankfurt/Main, Germany). Water-insoluble drugs were first dissolved in DMSO and then diluted in buffer to yield final DMSO concentrations of up to 0.1% which were also included in control solutions. At these concentrations, DMSO did not affect any of the parameters investigated (Lechner et al., 2003).
Dispase was obtained from Boehringer Mannheim (Vienna, Austria); trypsin, Worthington (Lakewood, NJ, USA); Dulbeccos modified Eagle's medium, streptomycin and foetal calf serum, InVitrogen (Lofer, Austria); nerve growth factor, R&D Systems Inc. (Minneapolis, MN, USA); rat tail collagen, Biomedical Technologies Inc. (Stoughton, MA, USA). The pCLAMP 8.0 hard-and software was from Molecular Devices (Sunnyvale, CA, USA); the Flaming-Brown puller, Sutter Instruments (Novato, CA, USA); borosilicate glass capillaries, Science Products (Frankfurt/Main, Germany); DAD-12 drug application device, Adams & List (Westbury, NY, USA).
Results
Multiple effects of mAChR ligands on noradrenaline release from rat SCG neurons
Activation of M1 mAChRs in primary cultures of rat SCG neurons is known to trigger noradrenaline release (Lechner et al., 2003). This effect contrasts with the previously reported inhibition of sympathetic transmitter release via presynaptic mAChRs (Boehm and Kubista, 2002). When rat SCG neurons were exposed to 10 µmol·L−1 OxoM, there first was an increase in the rate of spontaneous tritium outflow, which despite the continuing presence of this muscarinic agonist returned to basal levels. No such effect was observed if solvent was applied instead of OxoM. The subsequent application of electrical pulses was able to trigger tritium overflow again, but the amount of stimulation-dependent overflow was smaller than that in the presence of solvent (Figure 1A). Accordingly, the S3/S1 ratio of electrically evoked 3H overflow was significantly smaller in the presence than in the absence of 10 µmol·L−1 OxoM (Figure 1C).
Both the stimulation of tritium overflow and the inhibition of electrically evoked overflow by OxoM were concentration-dependent, but the concentrations required to achieve a maximum inhibition were obviously lower than those required to result in a maximal stimulation (Figure 1B,C). In fact, even at 100 µmol·L−1, the stimulant effect of OxoM did not show an unequivocal sign of saturation (Figure 1B) as observed previously (Lechner et al., 2003). From this, one may conclude that different receptors and/or different signalling mechanisms are involved in the opposing effects of OxoM on noradrenaline release.
To further substantiate this conclusion, the effects of two muscarinic antagonists with widely differing rank orders of receptor subtype affinities were used: the rank order of subtype affinities is M2 > M4 > M1 > M3= M5 for methoctramine and M1 > M4 > M5 > M3 > M2 for pirenzepine (Caulfield and Birdsall, 1998; Eglen et al., 2001). Pirenzepine was approximately 10-fold more potent than methoctramine at attenuating the stimulation of tritium overflow caused by 10 µmol·L−1 OxoM (Figure 1D), whereas methoctramine was more than 10-fold more potent than pirenzepine at attenuating the inhibition of electrically evoked tritium overflow caused by 10 µmol·L−1 OxoM (Figure 1E). Thus, the opposing effects of OxoM involve different receptor populations.
Evidence for M1 receptors being involved in the inhibition of electrically evoked noradrenaline release by OxoM
In order to further characterize the receptors mediating the inhibition of electrically evoked noradrenaline release, we used the muscarinic agonist bethanechol which has been shown to selectively activate M2 mAChRs in rat SCG neurons and to achieve maximal effects at a concentration of 100 µmol·L−1 (Liu and Rittenhouse, 2003b). Bethanechol (100 µmol·L−1) failed to significantly alter spontaneous (unstimulated) tritium outflow (Figure 2A,B), but did reduce electrically evoked overflow (Figure 2A,C). This latter effect appeared to be concentration-dependent between 10 and 100 µmol·L−1, and the maximum amounted to 24.5 ± 3.7% inhibition (n= 9; Figure 2C). For comparison, the maximum inhibition achieved with OxoM amounted to 52.4 ± 7.1% (n= 6; Figure 1C). This may be taken as an indication that M2 mAChRs contribute to, but are not sufficient to entirely mediate, the inhibitory effects of OxoM.
Figure 2.
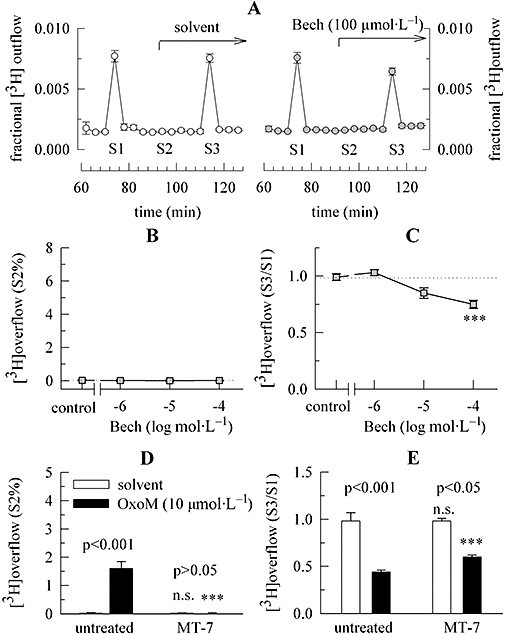
Characterization of the mAChRs mediating the inhibition of electrically evoked noradrenaline release. Primary cultures of rat SCG neurons were labelled with [3H]-noradrenaline for 1 h in the absence or presence of 100 nmol·L−1 MT-7 and were then superfused. Subsequent to a 60 min washout period, 4 min fractions of superfusate were collected. Electrical field stimulation was applied at 73 (S1) and 113 (S3) min of superfusion. Either OxoM or bethanechol (Bech) was present as indicated by the arrow on the right hand side in (A); OxoM caused transient increases in the rate of spontaneous 3H outflow (S2) as shown in Figure 1A; as a control for statistical comparison, solvent was applied instead of OxoM or bethanechol. (A) The time course of fractional 3H outflow·min−1 and its alteration by 100 µmol·L−1 bethanechol on the right hand side (n= 3). For comparison, the effect of solvent is depicted on the left hand side (n= 3). (B) Indicates a lack of bethanechol-induced 3H overflow, as exemplified by S2% values. The indicated concentrations of bethanechol were applied as shown in (A) (n= 6 to 9). (C) The concentration-dependence of the inhibition of electrically evoked tritium overflow by bethanechol, as exemplified by S3/S1 ratios. The indicated concentrations of bethanechol were applied as shown in (A) (n= 6 to 9); *** indicates a significant difference versus the result obtained in the presence of solvent at P < 0.001. (D) 3H overflow induced by 10 µmol·L−1 OxoM (as exemplified by S2% values) in either untreated or MT-7-treated cultures; its effect is compared with that of solvent (n= 9). (E) The inhibition of electrically evoked tritium overflow by 10 µmol·L−1 OxoM in either untreated cultures or in cultures treated with 100 nmol·L−1 MT-7 (as exemplified by S3/S1 ratios); the effect of OxoM is compared with that of solvent (n= 9). In (D) and (E) significances of differences between pairs of columns are indicated above the corresponding pairs. *** Indicates a significant difference versus the corresponding results obtained in untreated cultures at P < 0.001; n.s. indicates the lack of such a significance. mAChRs, muscarinic acetylcholine receptors; MT-7, muscarinic toxin 7; OxoM, oxotremorine M; SCG, superior cervical ganglia.
To specifically search for a role of M1 receptors in the inhibition of electrically evoked release, cultures were incubated in MT-7 (100 nmol·L−1 for 60 min) which selectively and irreversibly blocks M1 mAChRs (Olianas et al., 2000). In cultures treated with MT-7, OxoM (10 µmol·L−1) failed to cause significant changes in spontaneous tritium outflow when compared with the effects of solvent (Figure 2D), thus indicating that M1 receptors were sufficiently blocked. In parallel, the inhibition of electrically evoked 3H overflow was significantly attenuated from 55.4 ± 2.5% to 39.0 ± 2.3% (n= 9; Figure 2E). This indicates a role for M1 receptors in this latter effect.
Role of PTX-sensitive G-proteins in the inhibition of electrically evoked noradrenaline release by OxoM
Inhibition of noradrenaline release via presynaptic receptors involves in most, but not all, cases PTX-sensitive G-proteins (Kubista and Boehm, 2006). Therefore, cultures were treated with PTX (100 ng·mL−1 for 24 h). This treatment greatly enhanced the stimulation of tritium outflow by OxoM (Figure 3A,B), but left the amount of tritium overflow evoked by electrical field stimulation unaltered (S1% values: untreated: 2.47 ± 0.23, n= 17; PTX-treated: 2.63 ± 0.15, n= 17; P > 0.1). In parallel, the inhibitory effect of OxoM was reduced from 47.8 ± 3.9% inhibition (n= 9) to 16.7 ± 2.7% inhibition (n= 9; Figure 3C), but was not abolished. This indicates that another, pertussis-toxin-insensitive, signalling cascade is also involved in the inhibition of electrically evoked noradrenaline release via mAChRs.
Figure 3.
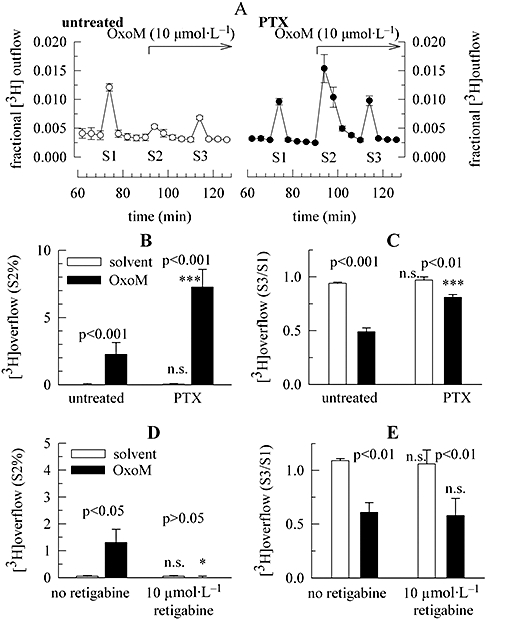
Characterization of signalling mechanisms involved in the inhibition of electrically evoked noradrenaline release via mAChRs. Primary cultures of rat SCG neurons were either treated with pertussis toxin (PTX; 100 ng·mL−1 for 24 h) or remained untreated. Cultures were then labelled with [3H]-noradrenaline for 1 h and were superfused. Subsequent to a 60 min washout period, 4 min fractions of superfusate were collected. Electrical field stimulation was applied at 73 (S1) and 113 (S3) min of superfusion. OxoM 10 µmol·L−1 was present as indicated by the arrows; OxoM caused transient increases in the rate of spontaneous 3H outflow (S2) as seen in (A); as a control for statistical comparison, solvent was applied instead of OxoM (see Figure 1A). (A) The time course of fractional 3H outflow·min−1 and its alteration by 10 µmol·L−1 OxoM in untreated (left) or PTX-treated cultures (right); n= 3. (B) 3H overflow induced by 10 µmol·L−1 OxoM (as exemplified by S2% values) in either untreated or PTX-treated cultures; its effect is compared with that of solvent (n= 8 to 9). (C) The inhibition of electrically evoked tritium overflow (as exemplified by S3/S1 ratios) by OxoM; its effect is compared with that of solvent (n= 8 to 9). (D) 3H overflow induced by 10 µmol·L−1 OxoM (as exemplified by S2% values) in either the absence or the presence of 10 µmol·L−1 retigabine; the effect of OxoM is compared with that of solvent (n= 9). (E) The inhibition of electrically evoked tritium overflow by 10 µmol·L−1 OxoM (as exemplified by S3/S1 ratios) in either the absence or the presence of 10 µmol·L−1 retigabine; the effect of OxoM is compared with that of solvent (n= 9). In (B) to (E), significances of differences between pairs of columns are indicated above the corresponding pairs. *** and * Indicate significant differences versus the corresponding results obtained in untreated cultures in the absence of retigabine at P < 0.001 and P < 0.05 respectively; n.s. indicates the lack of such a significance. mAChRs, muscarinic acetylcholine receptors; OxoM, oxotremorine M; PTX, pertussis toxin; SCG, superior cervical ganglia.
Role of Kv7 channels in the inhibition of electrically evoked noradrenaline release by OxoM
M1 mAChRs are generally linked to Gq-type G-proteins and thereby couple to phospholipase C (Wess et al., 2007). In SCG neurons, activation of phospholipase C leads to an inhibition of Kv7 channels via the depletion of membrane phosphatidylinositol-4,5-bisphosphate (PIP2) (Suh and Hille, 2002). The stimulation of noradrenaline release via M1 receptors is mediated by this Kv7 channel inhibition and is thus abolished by the Kv7 channel opener retigabine (Lechner et al., 2003). In line with our previous results, 10 µmol·L−1 retigabine entirely prevented the enhancement of the rate of spontaneous tritium outflow by OxoM (Figure 3D). However, neither electrically evoked 3H overflow itself, nor its inhibition by OxoM, was significantly affected by retigabine (Figure 3E). These results argue against a role of Kv7 channels in the inhibitory effect of OxoM.
Evidence for the presynaptic localization of inhibitory M1 receptors
The stimulation of noradrenaline release via M1 receptors is abolished when action potential propagation is blocked, thus indicating that these receptors are not directly localized at the presynaptic nerve terminals where exocytosis takes place (Lechner et al., 2003). To determine whether the muscarinic receptors that mediate the inhibition of electrically evoked noradrenaline release are located at presynaptic sites, experiments were also performed in the presence of 0.1 µmol·L−1 TTX. As expected, OxoM then failed to alter the rate of spontaneous 3H outflow (Figure 4A). As electrical field stimulation also fails to trigger noradrenaline release from SCG neurons when Na+ channels are blocked by TTX (Lechner et al., 2003), exposure to 30 mmol·L−1 K+ was used instead of electrical stimulation to elicit depolarization-dependent tritium overflow. This type of overflow was also reduced by 10 µmol·L−1 OxoM, and the inhibition amounted to 48.9 ± 3.1% (n= 9; Figure 4B). When cultures had been treated with MT-7 (100 nmol·L−1 for 60 min), this inhibition by OxoM was significantly reduced to 29.0 ± 7.4% (n= 8; Figure 4B). When 100 µmol·L−1 bethanechol was used instead of OxoM to reduce K+-evoked tritium overflow in the presence of TTX, the inhibition amounted to 18.5 ± 3.3% (n= 6; Figure 4C). Treatment of the cultures with MT-7 as above did not alter the effect of bethanechol, which then caused 19.1 ± 1.2% inhibition (n= 6; Figure 4C). Conversely, in cultures treated with PTX, the inhibition by 10 µmol·L−1 OxoM amounted to 15.4 ± 1.7% (n= 9; Figure 4D). When cultures were treated with both MT-7 and PTX the inhibition of K+-evoked tritium overflow by OxoM was abolished (Figure 4D). Likewise, in PTX-treated cultures, 100 µmol·L−1 bethanechol failed to alter K+-evoked overflow, whereas it did cause a clear-cut inhibition in untreated sister cultures (Figure 4E). Once again, the PTX treatment did not alter the amount (S1%) of K+-evoked overflow (not shown). These results verify that the inhibitory effects of mAChR agonists on noradrenaline release are the same whether action potential propagation is blocked or not. Thus, this inhibition is mediated via a presynaptic site of action.
Presynaptic inhibition of noradrenaline release by OxoM is mediated by an inhibition of VACCs
OxoM inhibits VACCs in rat SCG neurons via multiple signalling cascades (Shapiro et al., 1999), and the inhibition of VACCs is a predominant mechanism in the regulation of noradrenaline release via presynaptic GPCRs (Kubista and Boehm, 2006). To determine whether the inhibition of VACCs via muscarinic receptors displays the same pharmacological characteristics as the presynaptic inhibition of noradrenaline release, Ca2+ currents were measured in perforated patch-clamp recordings in order to maintain signalling cascades, including soluble components (Lechner et al., 2005). Under these experimental conditions, the Cav1 (L-type) channel blocker isradipine (3 µmol·L−1) reversibly reduced Ca2+ current amplitudes by 9.9 ± 2.6% (n= 9), whereas the Cav2.2 (N-type) channel blocker ω-conotoxin GVIA (1 µmol·L−1) caused an irreversible reduction of current amplitudes by 75.4 ± 3.1% (n= 8); 10 µmol·L−1 OxoM reduced overall Ca2+ current amplitudes by 59.0 ± 5.7% (n= 10; Figure 5A). In the presence of 3 µmol·L−1 isradipine, the inhibition by OxoM remained unchanged, but it was significantly attenuated when the cells had been exposed to ω-conotoxin GVIA (Figure 5B). Thus, OxoM affects primarily, but not exclusively, Cav2.2 (N-type) Ca2+ channels. To detemine whether the same holds true for the presynaptic inhibition, tritium overflow was triggered by 30 mmol·L−1 KCl in the presence of TTX in cultures that had either been treated with 1 µmol·L−1ω-conotoxin GVIA for 24 h or had remained untreated. Such a treatment minimizes the contribution of Cav2.2 channels to depolarization-induced noradrenaline release from sympathetic neurons (Boehm and Huck, 1996). This treatment with ω-conotoxin GVIA reduced the amount of K+-evoked tritium overflow (S1%) by 69.7 ± 2.5% (n= 17; P < 0.001). Furthermore, in ω-conotoxin-treated neurons the inhibition of tritium overflow by OxoM was significantly reduced when compared with that in untreated neurons (Figure 5C).
Figure 5.
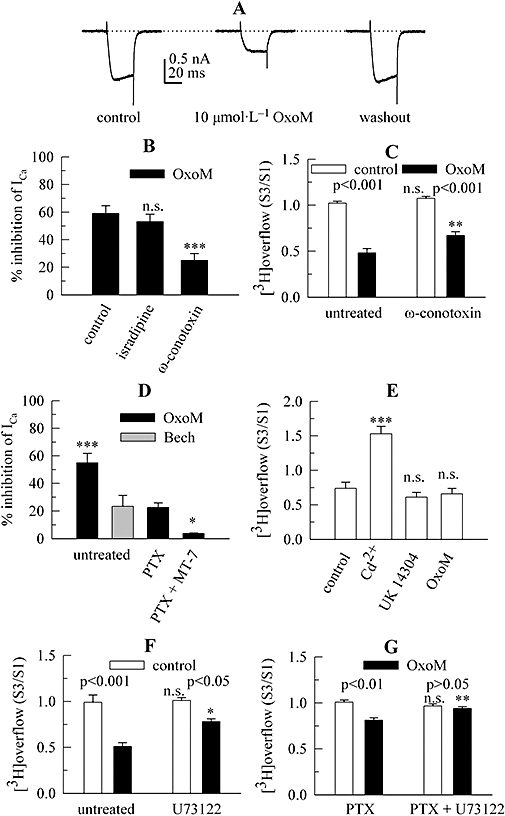
Role of VACCs in the inhibition of noradrenaline release via presynaptic mAChRs. Primary cultures of rat SCG neurons were treated with pertussis toxin (PTX; 100 ng·mL−1 for 24 h), with ω-conotoxin GVIA (1 µmol·L−1 for 24 h), or remained untreated. Using the perforated patch clamp technique, Ca2+ currents were evoked by 30 ms depolarizations from −80 to +10 mV. Alternatively, cultures were labelled with [3H]-noradrenaline for 1 h in the absence or presence of 3 µmol·L−1 U73122 and were superfused. From 50 min of superfusion onward, 0.1 µmol·L−1 TTX was present. Subsequent to a 60 min washout period, 4 min fractions of superfusate were collected. KCl 30 mmol·L−1 (NaCl was reduced accordingly) or 100 µmol·L−1 ATP were present at 73 (S1) and 93 (S3) min of superfusion. CdCl2 100 µmol·L−1, 10 µmol·L−1 UK 14304 or 10 µmol·L−1 OxoM was present from 88 min onward (see Figure 4A); as a control for statistical comparison, solvent was applied instead of these agents. (A) Original current traces of one neuron before during and after the presence of 10 µmol·L−1 OxoM. The dashed line indicates the zero current level. (B) The inhibition of Ca2+ currents by 10 µmol·L−1 OxoM under control conditions, in the continuous presence of 3 µmol·L−1 isradipine, and in cells that had been exposed to 1 µmol·L−1ω-conotoxin GVIA in order to irreversibly block Cav2.2 Ca2+ channels (n= 7 to 9). *** Indicates a significant difference versus the inhibition in the absence of Ca2+ channels blockers at P < 0.001; n.s. indicates the lack of such a significant difference. (C) Quantifies the effects of a treatment with 1 µmol·L−1ω-conotoxin GVIA for 24 h on the inhibition of K+-evoked tritium overflow (S3/S1) by 10 µmol·L−1 OxoM; n= 8 to 9. Significances of differences between pairs of columns are indicated above the corresponding results. ** Indicates a significant difference versus the corresponding result obtained in untreated cultures at P < 0.01; n.s. indicates the lack of such a significance; n= 6 to 9. (D) The effects of 10 µmol·L−1 OxoM or 100 µmol·L−1 bethanechol (Bech) on Ca2+ current amplitudes in untreated, pertussis toxin (PTX)-treated, and PTX-plus MT-7-treated (PTX + MT-7) neurons; n= 6 to 8. *** and * Indicate significant differences versus the inhibition by OxoM observed in PTX-treated neurons at P < 0.001 and P < 0.05 respectively. (E) Quantifies the effects of 100 µmol·L−1 CdCl2, 10 µmol·L−1 UK 14304 or 10 µmol·L−1 OxoM on ATP-evoked tritium overflow (S3/S1); n= 8 to 9. *** Indicates a significant difference versus the results obtained with solvent (control) at P < 0.001; n.s. indicates the lack of such a significance. (F) Quantifies the effects of a treatment with 3 µmol·L−1 U73122 on the inhibition of K+-evoked tritium overflow (S3/S1) by 10 µmol·L−1 OxoM; n= 5 to 6. Significances of differences between pairs of columns are indicated above the corresponding pairs. * Indicates a significant difference versus the corresponding results obtained in cultures that had not been exposed to U73122 at P < 0.05; n.s. indicates the lack of such a significance. (G) Quantifies the effects of a treatment with 3 µmol·L−1 U73122 on the inhibition of K+-evoked tritium overflow (S3/S1) by 10 µmol·L−1 OxoM in neurons treated with PTX; n= 6 to 9. Significances of differences between pairs of columns are indicated above the corresponding pairs. ** Indicates a significant difference versus the corresponding results obtained in cultures that had not been exposed to U73122 at P < 0.01; n.s. indicates the lack of such a significance. ATP, adenosine–5′–triphosphate; mAChRs, muscarinic acetylcholine receptors; OxoM, oxotremorine M; PTX, pertussis toxin; SCG, superior cervical ganglia; VACCs, voltage-activated Ca2+ channels.
Bethanechol (100 µmol·L−1) reduced Ca2+ current amplitudes significantly less than 10 µmol·L−1 OxoM (Figure 5D). When cultures had been treated with PTX (100 ng·mL−1 for 24 h), the inhibition of Ca2+ currents by OxoM was significantly reduced, and the remaining inhibition was abolished when neurons had also been exposed to 100 nmol·L−1 MT-7 (Figure 5D). This confirms that the PTX-insensitive component of Ca2+ current inhibition is mediated by M1 mAChRs.
To prove that the inhibition of VACCs is a prerequisite for the presynaptic inhibition of transmitter release, tritium overflow was induced by a stimulation paradigm that bypasses VACCs, namely the activation of presynaptic P2X receptors (Boehm, 1999). TTX was present throughout these experiments to prevent the stimulant effects of OxoM and the contribution of non-presynaptic P2X receptors (Boehm, 1999). ATP 100 µmol·L−1 was applied as a secretagogue stimulus. With this stimulation paradigm, the blockade of VACCs by 100 µmol·L−1 Cd2+ did not abolish stimulation-evoked tritium overflow (as it does with depolarizing stimuli; Lechner et al., 2003), but rather Cd2+ exerted an enhancing action on transmitter release (Figure 5E). A similar effect has been observed in PC12 cells and was shown to be caused by a direct allosteric potentiation of P2X receptors by Cd2+ ions (Ikeda et al., 1996). Moreover, the α2-adrenoceptor agonist UK 14304 (3 µmol·L−1), which inhibits noradrenaline release exclusively through a blockade of VACCs (Boehm and Huck, 1995), failed to cause a significant reduction in ATP-induced tritium overflow (Figure 5E). OxoM (10 µmol·L−1) also failed to inhibit this type of overflow (Figure 5E), thus indicating that its inhibitory action requires the contribution of VACCs and is not caused by actions on vesicle exocytosis downstream of Ca2+ influx.
Presynaptic inhibition of noradrenaline release by OxoM is mediated by phospholipase C
The inhibition of Ca2+ currents in rat SCG neurons via M1 mAChRs is mediated by phospholipase C and thus attenuated by U73122 (Liu and Rittenhouse, 2003a). This drug causes an irreversible inhibition of phospholipase C-dependent signalling cascades in SCG neurons (Bofill-Cardona et al., 2000). When cultures had been treated with 3 µmol·L−1 U73122, the inhibition of K+-evoked tritium overflow by OxoM was reduced from 48.6 ± 2.3% (n= 6) to 24.4 ± 1.6% (Figure 5F). The inactive analogue, U73343, however, left the inhibition unaltered (not shown, but see Lechner et al., 2005). Furthermore, the 19.5 ± 2.7% inhibition remaining in PTX-treated neurons was abolished by U73122 (Figure 5G). Thus, the PTX-insensitive presynaptic inhibition induced by OxoM involves phospholipase C.
Discussion
Presynaptic inhibition via GPCRs is generally believed to be mediated by activated G-protein βγ subunits that directly interact with VACCs and thereby reduce the Ca2+ influx required for vesicle exocytosis (Stevens, 2004). Although this also holds true for sympathetic neurons in a large number of cases (Boehm and Kubista, 2002), another signalling pathway of inhibitory presynaptic GPCRs has been identified recently: one that involves B2 bradykinin receptors, phospholipase C, depletion of membrane PIP2 and a resulting closure of VACCs (Lechner et al., 2005). This very signalling cascade is also linked to M1 mAChRs (Gamper et al., 2004; Liu et al., 2004), but evidence for inhibitory presynaptic M1 receptors has been lacking. The results shown above demonstrate that this receptor subtype contributes a small, but nevertheless significant, part to the presynaptic inhibition of noradrenaline release from rat SCG neurons elicited by mAChR agonists.
In primary cultures of rat SCG neurons, activation of M1 mAChRs is sufficient to trigger action potential-dependent noradrenaline release in the absence of any other secretagogue stimulus (Lechner et al., 2003). When OxoM was applied in the experiments shown above, a transient increase in the rate of noradrenaline release was also observed. In addition, OxoM caused a reduction of noradrenaline release evoked by electrical field stimulation once the secretagogue action of the muscarinic agonist had faded. This inhibitory effect was mediated by a different set of mAChRs than the secretagogue action as evidenced by the following results: (i) pirenzepine was more potent than methoctramine in antagonizing the stimulant action of OxoM, and the reverse was true for the inhibitory effect; (ii) the mAChR agonist bethanechol mimicked the inhibitory, but not the stimulant, effect of OxoM; and (iii) the selective and irreversible M1 receptor antagonist MT-7 (Olianas et al., 2000) abolished the stimulant effect, but only attenuated the inhibitory action of OxoM. As bethanechol is selective for M2 mAChRs in rat SCG neurons (Liu and Rittenhouse, 2003b), this receptor subtype can only be involved in the inhibition of noradrenaline release. The abolition of the secretagogue action of OxoM by MT-7 confirms that this latter effect is mediated by M1 receptors only. The fact that this toxin attenuated the reduction of noradrenaline release by OxoM was the first indication that M1 receptors may be involved in presynaptic inhibition.
One might argue that the MT-7-dependent attenuation of the inhibition of noradrenaline release by OxoM might be correlated with the loss of the secretagogue action of this agonist. However, the following results argue against such a conclusion. (i) Retigabine also abolished the secretagogue action of OxoM, but did not alter its inhibitory action. (ii) MT-7 attenuated the inhibition of depolarization-dependent release by OxoM even when the muscarinic agonist did not enhance the rate of otherwise unstimulated noradrenaline release, as was the case in the presence of TTX. Furthermore, the observation of an inhibition of depolarization-evoked transmitter release with action potential propagation being blocked by TTX demonstrates that all the mAChRs involved, including M1, must be located close to the sites of vesicle exocytosis, that is must be presynaptic receptors.
In rat SCG neurons, activation of mAChRs modulates the functions of Kv7 K+ channels and of Cav2.2 (N-type) as well as Cav1 (L-type) Ca2+ channels (Hille, 1994). The resulting changes in the accompanying conductances may impinge on the excitability of the neurons and on the amount of transmitter being released (Boehm and Kubista, 2002; Hernandez et al., 2008). To evaluate the role of these channels in the effects of OxoM on noradrenaline release, we first employed the Kv7 channel opener retigabine (Tatulian et al., 2001) which abolishes the secretagogue action of OxoM (see above and Lechner et al., 2003). However, 10 µmol·L−1 retigabine did not alter the inhibitory action of OxoM on electrically evoked release. Thus, the presynaptic inhibition via mAChRs is independent of Kv7 channels.
Activation of primarily M2, but also M4, receptors mediates an inhibition of Cav2.2 Ca2+ channels in rat SCG neurons through a direct interaction of βγ subunits of inhibitory G-proteins with the channel proteins (Bernheim et al., 1992; Liu and Rittenhouse, 2003b). This effect is abolished by PTX. Activation of M1 receptors, in contrast, inhibits VACCs in a PTX-insensitive manner (Hille, 1994). In accordance with these data, the 45% to 50% inhibition of depolarization-evoked noradrenaline release by OxoM was reduced by PTX treatment to 15% to 20% inhibition. Moreover, the 15% to 20% inhibition by the M2 selective agonist bethanechol was even abolished by PTX. And the 15% to 20% presynaptic inhibition by OxoM remaining in PTX-treated cultures was abolished by MT-7. These data concerning the inhibition of noradrenaline release were paralleled by electrophysiological results. OxoM caused a 55% to 60% reduction and bethanechol a 23% reduction of Ca2+ current amplitudes in untreated neurons. In PTX-treated neurons, however, the inhibition by OxoM was reduced to 23%, and this remainder was abolished by MT-7. The congruence of results obtained with the presynaptic inhibition of noradrenaline release, on the one hand, and with the inhibition of Ca2+ currents, on the other hand, suggests a causal association between these two phenomena. This causal association was challenged in experiments where tritium overflow was induced by a method that excludes VACCs from excitation-secretion coupling: the activation of presynaptic P2X receptors which mediate the transmembrane Ca2+ entry required for exocytosis themselves (Boehm, 1999). Under these conditions, OxoM failed to reduce noradrenaline release, as did the α2-adrenoceptor agonist UK 14304. Activation of presynaptic α2-adrenoceptors has been well documented to reduce noradrenaline release through an inhibition of VACCs (Kubista and Boehm, 2006). Accordingly, the present results demonstrate that the same holds true for the presynaptic inhibition via mAChRs.
Activation of M1 mAChRs controls the gating of Cav1 and Cav2.2 Ca2+ channels in rat SCG neurons via signalling cascades that include Gαq G-protein subunits and the activation of phospholipase C with ensuing depletion of membrane PIP2 and generation of arachidonic acid (Haley et al., 2000; Liu and Rittenhouse, 2003a; Gamper et al., 2004; Liu et al., 2006). With respect to the presynaptic inhibition via M1 receptors, the same signalling mechanism was found to operate as the inhibitory action of OxoM was attenuated by a phospholipase C inhibitor (and even abolished when neurons had also been treated with PTX). Both Ca2+ current amplitudes and noradrenaline release were reduced by ω-conotoxin GVIA by 70% to 75%. The remaining 25% to 30% of Ca2+ currents and evoked noradrenaline release were still reduced by OxoM, but the percentage of inhibition (about 30%) was significantly smaller than that in neurons with unblocked Cav2.2 channels. Moreover, one must not forget that this 30% inhibition of Ca2+ currents and noradrenaline release in ω-conotoxin-treated neurons equals no more than 10% of overall Ca2+ current amplitudes and evoked noradrenaline release. Thus, the inhibition of Cav2.2 Ca2+ channels is the most decisive, but not the only mechanism underlying the inhibition of noradrenaline release via presynaptic mAChRs.
The regulation of VACCs and Kv7 channels in SCG neurons via phospholipase C is initiated not only by M1 mAChRs, but also by bradykinin B2 receptors (Gamper et al., 2004; Lechner et al., 2005). As for M1 receptors here, B2 receptor activation has been found to trigger action potentials and thus noradrenaline release from SCG neurons (Boehm and Huck, 1997), on the one hand, and to simultaneously mediate presynaptic inhibition at the nerve terminals (Edelbauer et al., 2005), on the other hand. Furthermore, these opposing effects of bradykinin were mediated by the inhibition of Kv7 channels and VACCs respectively (Edelbauer et al., 2005). This leads to the question why one or the other effect triggered by these receptors predominates in presynaptic and non-presynaptic regions of SCG neurons respectively. One likely explanation is the diverging densities of the affected channels in different parts of a neuron. Cav2.2 Ca2+ channels are concentrated at the active zones of presynaptic nerve terminals (Khanna et al., 2007), whereas Kv7 channels are rather located close to neuronal somata (Hu et al., 2007; Rasmussen et al., 2007). In addition, certain components of the signalling cascades involved, such as phospholipase A2 (Liu et al., 2006) and diacylglycerol lipase (Liu et al., 2008) which mediate the inhibition of VACCs, but not that of Kv7 channels, may be differentially distributed throughout the SCG neurons. Moreover, the pattern of stimulating or inhibitory effects of Gq-coupled receptors on transmitter release may also vary between different types of neurons. Presynaptic M3 mAChRs, for instance, have been shown to enhance dopamine release from striatal neurons through an inhibition of Kv7 channels (Martire et al., 2007), and evidence for stimulating presynaptic M1 receptors has also been obtained in sympathetically innervated tissues (Boehm and Kubista, 2002). Here, we have demonstrated that presynaptic M1 mAChRs can mediate an inhibition of transmitter release. It will thus be of interesting to determine how signalling cascades are organized to mediate either inhibitory or stimulating actions of one type of presynaptic GPCR.
In conclusion, the present data reveal that M1 receptors contribute to the acetylcholine-dependent presynaptic inhibition in the sympathetic nervous system via a signal cascade that includes phospholipase C and VACCs.
Acknowledgments
This study was supported by grants P17611 (to SB) and P19710 (to HK) from the Austrian Science Funds (FWF) and by the ‘Virologiefonds’ of the Medical University of Vienna. The excellent technical assistance of Gabi Gaupmann and Martina Molin is gratefully acknowledged.
Glossary
Abbreviations:
- GPCR
G-protein-coupled receptor
- mAChRs
muscarinic acetylcholine receptors
- MT-7
muscarinic toxin 7
- OxoM
oxotremorine M
- PTX
pertussis toxin
- PIP2
phosphatidylinositol-4,5-bisphosphate
- SCG
superior cervical ganglia
- TTX
tetrodotoxin
- VACCs
voltage-activated Ca2+ channels
Conflict of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 3rd edition (2008 revision) Br J Pharmacol. 2008;153(2):S1–S209. doi: 10.1038/sj.bjp.0707746. Suppl. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernheim L, Mathie A, Hille B. Characterization of muscarinic receptor subtypes inhibiting Ca2+ current and M current in rat sympathetic neurons. Proc Natl Acad Sci USA. 1992;89:9544–9548. doi: 10.1073/pnas.89.20.9544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm S. ATP stimulates sympathetic transmitter release via presynaptic P2X purinoceptors. J Neurosci. 1999;19:737–746. doi: 10.1523/JNEUROSCI.19-02-00737.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm S, Huck S. alpha 2-Adrenoreceptor-mediated inhibition of acetylcholine-induced noradrenaline release from rat sympathetic neurons: an action at voltage-gated Ca2+ channels. Neuroscience. 1995;69:221–231. doi: 10.1016/0306-4522(95)00235-b. [DOI] [PubMed] [Google Scholar]
- Boehm S, Huck S. Inhibition of N-type calcium channels: the only mechanism by which presynaptic alpha 2-autoreceptors control sympathetic transmitter release. Eur J Neurosci. 1996;8:1924–1931. doi: 10.1111/j.1460-9568.1996.tb01336.x. [DOI] [PubMed] [Google Scholar]
- Boehm S, Huck S. Noradrenaline release from rat sympathetic neurones triggered by activation of B2 bradykinin receptors. Br J Pharmacol. 1997;122:455–462. doi: 10.1038/sj.bjp.0701404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm S, Kubista H. Fine tuning of sympathetic transmitter release via ionotropic and metabotropic presynaptic receptors. Pharmacol Rev. 2002;54:43–99. doi: 10.1124/pr.54.1.43. [DOI] [PubMed] [Google Scholar]
- Bofill-Cardona E, Vartian N, Nanoff C, Freissmuth M, Boehm S. Two different signaling mechanisms involved in the excitation of rat sympathetic neurons by uridine nucleotides. Mol Pharmacol. 2000;57:1165–1172. [PubMed] [Google Scholar]
- Caulfield MP, Birdsall NJ. International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol Rev. 1998;50:279–290. [PubMed] [Google Scholar]
- Edelbauer H, Lechner SG, Mayer M, Scholze T, Boehm S. Presynaptic inhibition of transmitter release from rat sympathetic neurons by bradykinin. J Neurochem. 2005;93:1110–1121. doi: 10.1111/j.1471-4159.2005.03084.x. [DOI] [PubMed] [Google Scholar]
- Eglen RM, Choppin A, Watson N. Therapeutic opportunities from muscarinic receptor research. Trends Pharmacol Sci. 2001;22:409–414. doi: 10.1016/s0165-6147(00)01737-5. [DOI] [PubMed] [Google Scholar]
- Fuder H, Muscholl E. Heteroreceptor-mediated modulation of noradrenaline and acetylcholine release from peripheral nerves. Rev Physiol Biochem Pharmacol. 1995;126:265–412. doi: 10.1007/BFb0049778. [DOI] [PubMed] [Google Scholar]
- Gamper N, Reznikov V, Yamada Y, Yang J, Shapiro MS. Phosphatidylinositol 4,5-bisphosphate signals underlie receptor-specific Gq/11-mediated modulation of N-type Ca2+ channels. J Neurosci. 2004;24:10980–10992. doi: 10.1523/JNEUROSCI.3869-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley JE, Delmas P, Offermanns S, Abogadie FC, Simon MI, Buckley NJ, et al. Muscarinic inhibition of calcium current and M current in Galpha q-deficient mice. J Neurosci. 2000;20:3973–3979. doi: 10.1523/JNEUROSCI.20-11-03973.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez CC, Zaika O, Tolstykh GP, Shapiro MS. Regulation of neural KCNQ channels: signalling pathways, structural motifs and functional implications. J Physiol. 2008;586:1811–1821. doi: 10.1113/jphysiol.2007.148304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- Hu H, Vervaeke K, Storm JF. M-channels (Kv7/KCNQ channels) that regulate synaptic integration, excitability, and spike pattern of CA1 pyramidal cells are located in the perisomatic region. J Neurosci. 2007;27:1853–1867. doi: 10.1523/JNEUROSCI.4463-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda M, Koizumi S, Nakazawa K, Inoue K, Ito K, Inoue K. Potentiation by cadmium ion of ATP-evoked dopamine release in rat phaeochromocytoma cells. Br J Pharmacol. 1996;117:950–954. doi: 10.1111/j.1476-5381.1996.tb15286.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna R, Li Q, Bewersdorf J, Stanley EF. The presynaptic Cav2.2 channel-transmitter release site core complex. Eur J Neurosci. 2007;26:547–559. doi: 10.1111/j.1460-9568.2007.05680.x. [DOI] [PubMed] [Google Scholar]
- Kristufek D, Koth G, Motejlek A, Schwarz K, Huck S, Boehm S. Modulation of spontaneous and stimulation-evoked transmitter release from rat sympathetic neurons by the cognition enhancer linopirdine: insights into its mechanisms of action. J Neurochem. 1999;72:2083–2091. doi: 10.1046/j.1471-4159.1999.0722083.x. [DOI] [PubMed] [Google Scholar]
- Kubista H, Boehm S. Molecular mechanisms underlying the modulation of exocytotic noradrenaline release via presynaptic receptors. Pharmacol Ther. 2006;112:213–242. doi: 10.1016/j.pharmthera.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Lechner SG, Mayer M, Boehm S. Activation of M1 muscarinic receptors triggers transmitter release from rat sympathetic neurons through an inhibition of M-type K+ channels. J Physiol. 2003;553:789–802. doi: 10.1113/jphysiol.2003.052449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner SG, Hussl S, Schicker KW, Drobny H, Boehm S. Presynaptic inhibition via a phospholipase C-and phosphatidylinositol bisphosphate-dependent regulation of neuronal Ca2+ channels. Mol Pharmacol. 2005;68:1387–1396. doi: 10.1124/mol.105.014886. [DOI] [PubMed] [Google Scholar]
- Lindmar R, Loffelholz K, Muscholl E. A muscarinic mechanism inhibiting the release of noradrenaline from peripheral adrenergic nerve fibres by nicotinic agents. Br J Pharmacol Chemother. 1968;32:280–294. doi: 10.1111/j.1476-5381.1968.tb00972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Heneghan JF, Michael GJ, Stanish LF, Egertova M, Rittenhouse AR. L-and N-current but not M-current inhibition by M1 muscarinic receptors requires DAG lipase activity. J Cell Physiol. 2008;216:91–100. doi: 10.1002/jcp.21378. [DOI] [PubMed] [Google Scholar]
- Liu L, Rittenhouse AR. Arachidonic acid mediates muscarinic inhibition and enhancement of N-type Ca2+ current in sympathetic neurons. Proc Natl Acad Sci USA. 2003a;100:295–300. doi: 10.1073/pnas.0136826100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Rittenhouse AR. Pharmacological discrimination between muscarinic receptor signal transduction cascades with bethanechol chloride. Br J Pharmacol. 2003b;138:1259–1270. doi: 10.1038/sj.bjp.0705157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Roberts ML, Rittenhouse AR. Phospholipid metabolism is required for M1 muscarinic inhibition of N-type calcium current in sympathetic neurons. Eur Biophys J. 2004;33:255–264. doi: 10.1007/s00249-003-0387-7. [DOI] [PubMed] [Google Scholar]
- Liu L, Zhao R, Bai Y, Stanish LF, Evans JE, Sanderson MJ, et al. M1 muscarinic receptors inhibit L-type Ca2+ current and M-current by divergent signal transduction cascades. J Neurosci. 2006;26:11588–11598. doi: 10.1523/JNEUROSCI.2102-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martire M, D'Amico M, Panza E, Miceli F, Viggiano D, Lavergata F, et al. Involvement of KCNQ2 subunits in [3H]dopamine release triggered by depolarization and pre-synaptic muscarinic receptor activation from rat striatal synaptosomes. J Neurochem. 2007;102:179–193. doi: 10.1111/j.1471-4159.2007.04562.x. [DOI] [PubMed] [Google Scholar]
- Olianas MC, Maullu C, Adem A, Mulugeta E, Karlsson E, Onali P. Inhibition of acetylcholine muscarinic M1 receptor function by the M1-selective ligand muscarinic toxin 7 (MT-7) Br J Pharmacol. 2000;131:447–452. doi: 10.1038/sj.bjp.0703606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onali P, Adem A, Karlsson E, Olianas MC. The pharmacological action of MT-7. Life Sci. 2005;76:1547–1552. doi: 10.1016/j.lfs.2004.10.029. [DOI] [PubMed] [Google Scholar]
- Rasmussen HB, Frokjaer-Jensen C, Jensen CS, Jensen HS, Jorgensen NK, Misonou H, et al. Requirement of subunit co-assembly and ankyrin-G for M-channel localization at the axon initial segment. J Cell Sci. 2007;120:953–963. doi: 10.1242/jcs.03396. [DOI] [PubMed] [Google Scholar]
- Scholze T, Moskvina E, Mayer M, Just H, Kubista H, Boehm S. Sympathoexcitation by bradykinin involves Ca2+–independent protein kinase C. J Neurosci. 2002;22:5823–5832. doi: 10.1523/JNEUROSCI.22-14-05823.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz DD, Malik KU. Cyclic AMP modulates but does not mediate the inhibition of [3H]norepinephrine release by activation of alpha-2 adrenergic receptors in cultured rat ganglion cells. Neuroscience. 1993;52:107–113. doi: 10.1016/0306-4522(93)90186-j. [DOI] [PubMed] [Google Scholar]
- Shapiro MS, Loose MD, Hamilton SE, Nathanson NM, Gomeza J, Wess J, et al. Assignment of muscarinic receptor subtypes mediating G-protein modulation of Ca2+ channels by using knockout mice. Proc Natl Acad Sci USA. 1999;96:10899–10904. doi: 10.1073/pnas.96.19.10899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF. Presynaptic function. Curr Opin Neurobiol. 2004;14:341–345. doi: 10.1016/j.conb.2004.04.004. [DOI] [PubMed] [Google Scholar]
- Suh BC, Hille B. Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron. 2002;35:507–520. doi: 10.1016/s0896-6273(02)00790-0. [DOI] [PubMed] [Google Scholar]
- Tatulian L, Delmas P, Abogadie FC, Brown DA. Activation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabine. J Neurosci. 2001;21:5535–5545. doi: 10.1523/JNEUROSCI.21-15-05535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trendelenburg AU, Meyer A, Wess J, Starke K. Distinct mixtures of muscarinic receptor subtypes mediate inhibition of noradrenaline release in different mouse peripheral tissues, as studied with receptor knockout mice. Br J Pharmacol. 2005;145:1153–1159. doi: 10.1038/sj.bjp.0706297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wess J, Eglen RM, Gautam D. Muscarinic acetylcholine receptors: mutant mice provide new insights for drug development. Nat Rev Drug Discov. 2007;6:721–733. doi: 10.1038/nrd2379. [DOI] [PubMed] [Google Scholar]