

Abstract
Background and purpose:
Various complications consequent on disordered calcium and phosphate homeostasis occur frequently in chronic kidney disease (CKD) patients. Particularly, vascular calcification has high morbidity and mortality rates. There is a clear need for a better CKD model to examine various aspects of this disordered homeostasis.
Experimental approach:
Oral dosing with adenine induced CKD in rats in only 10 days. Serum calcium, phosphate and parathyroid hormone were measured and calcification in aorta was assessed histologically. The effects of varying phosphorus content of diet or treatment with phosphate binders or active vitamin D3 on these parameters were examined.
Key results:
After adenine dosing, significant hyperphosphatemia, hypocalcemia and secondary hyperparathyroidism (2HPT) were observed during the experimental period of 15 weeks. Aortic calcification was detected in only some of the animals even at 15 weeks (∼40%). Treatment with vitamin D3 for 18 days, even at a low dose (100 ng·kg−1, 3–4 times week−1, p.o), caused aortic calcification in all animals and increases in serum calcium levels up to the normal range. The vitamin D3-induced calcification was significantly inhibited by phosphate binders which lowered serum phosphate levels and the calcium × phosphate product, although serum calcium levels were elevated.
Conclusions:
These data suggest that rats dosed orally with adenine provide a more useful model for analysing calcium/phosphate homeostasis in severe CKD. Controlling serum calcium/phosphate levels with phosphate binders may be better than vitamin D3 treatment in hyperphosphatemia and 2HPT, to avoid vascular calcification.
Keywords: chronic kidney disease, renal failure, complications, hyperphosphatemia, cardiovascular calcification, phosphate-binders, secondary hyperparathyroidism, rats
Introduction
Patients with chronic kidney disease (CKD) commonly experience various complications including hyperphosphatemia, hypocalcemia, vascular calcification and secondary hyperparathyroidism (2HPT). Disordered metabolism of calcium and phosphorus and elevation of the serum calcium × phosphate (Ca × P) product are major risk factors (Delmez and Slatopolsky, 1992; Winchester et al., 1993). Cardiovascular calcification in these patients must be carefully monitored and reduced renal function is known to be an independent risk factor for cardiovascular disease (Manjunath et al., 2003). Cardiovascular events are responsible for almost 50% of the mortality in CKD patients receiving haemodialysis (Foley et al., 1998) and the presence and extent of vascular calcifications are strong predictors of cardiovascular mortality in these patients (Blacher et al., 2001).
The medical treatment of the complications is itself complex. In late stage CKD, hyperphosphatemia is evident, along with low levels of calcitriol (1,25(OH)2 vitamin D3) and worsening 2HPT (Delmez and Slatopolsky, 1992), indicating that controlling serum phosphate levels and recruitment of active vitamin D3 might inhibit the deteriorating complications. Major therapies for 2HPT and/or hyperphosphatemia involve use of active vitamin D analogues and a phosphate binding agent containing calcium (CaCO3), but such treatments are thought to elevate the Ca × P product and worsen vascular calcification because of excess Ca. A retrospective study demonstrated that ectopic calcification was detected in 60% of CKD patients with 2HPT, and active vitamin D treatment was strongly associated with this calcification (Milliner et al., 1990). Despite these findings, active vitamin D is frequently prescribed for dialysis patients and recommended for pre-dialysis CKD patients as well (Eknoyan et al., 2003). Many nephrologists, however, are debating the necessity and appropriate starting time for vitamin D treatment (Andress, 2005).
As animal models of CKD, 5/6 subtotally nephrectomized (5/6Nx) rats (Cozzolino et al., 2002; 2003; Terai et al., 2008) and adenine-fed rats (Katsumata et al., 2003; Nagano et al., 2006; Terai et al., 2008) are commonly used for exploring the association of various complications with progressive CKD. We demonstrated that 5/6Nx rats showed reproducible mild CKD, but the progression was slow and highly diverse (Terai et al., 2008). Cozzolino et al. (2003) have also reported that less than 20% of 5/6Nx rats can survive and demonstrate cardiovascular calcification after 24-week feeding with a high-phosphorus (Hi-P) diet. Adenine-fed rats show more severe CKD with hyperphosphatemia after 4 weeks of a diet containing 0.75% adenine (Katsumata et al., 2003; Terai et al., 2008). But continuous feeding with the adenine or the Hi-P diet often seems to be required to maintain significant hyperphosphatemia (Katsumata et al., 2003; Nagano et al., 2006). Although long-term feeding with a Hi-P diet is necessary to cause further complications in adenine-fed rats (Katsumata et al., 2003; Tamagaki et al., 2006), as well as in 5/6Nx rats, a few modifications have been reported to provide more useful models for research. Recently, we reported that giving adenine orally for 10–12 days caused a more stable hyperphosphatemia in rats fed a normal diet, than that in adenine-fed rats or 5/6Nx rats (Terai et al., 2008). In the present study, we have carefully characterized the orally adenine-dosed rats with respect to calcium and phosphate homeostasis to clarify their usefulness as a severe CKD model.
Some of the clinical outcomes of active vitamin D3 therapies for CKD patients have been noted in previous animal studies (Krog et al., 1984; Inagaki et al., 1995; Hirata et al., 2003). Hypercalcemic dosages of calcitriol and active vitamin D3 analogues were inhibitory for 2HPT in 5/6Nx rats. But relationships between the effective dosages and the appearance of vascular calcification have not been demonstrated. Despite such findings, it has been thought that vitamin D therapy is useful for 2HPT treatment in CKD patients and safe while serum Ca was maintained in the normal range. Recently, Goodman et al. (2004) suggested that treatments with active vitamin D metabolites for CKD patients could contribute to systemic and vascular calcifications, even in the absence of hypercalcemia. Thus, we have examined the effects of active vitamin D therapy for 2HPT and vascular calcification in the present studies using our orally adenine-dosed rat model of severe CKD.
Methods
Animals and chows
All experiments were approved by the Animal Ethical Committee of Astellas Pharma Inc. and were carried out in accordance with the guidelines of the US National Institutes of Health (NIH) on animal care. Every effort was made to minimize the number of animals used and their suffering. Six-week-old male Wistar rats (n= 103) were purchased (SLC, Tokyo, Japan) and were maintained in sterilized cages (4–6 rats cage−1) under a 12 h light/dark schedule (08:00–20:00). The animals were fed with a standard pellet chow for rodents containing 0.97% phosphorus and 1.03% calcium (normal chow, CE-2, Clea Japan Inc., Tokyo, Japan) and were given tap water ad libitum during an acclimation period of 1 week. For the following experimental periods, different diets were used: powder or pellet normal chow, a Hi-P (1.27% phosphorus and 1.12% calcium) or a low-phosphorus (Low-P; 0.90% phosphorus and 1.12% calcium) pellet chow (Oriental yeast industries, Tokyo, Japan). For model validation studies (Exp. 4), two phosphate binding agents were used: the non-calcium containing phosphate binder, sevelamer hydrochloride (Renagel, Chugai Pharmaceutical Co, Tokyo, Japan) or CaCO3 (Caltan, Fuso Pharmaceutical Industries Ltd, Osaka, Japan) in powdered form and mixed into powdered normal chow at a concentration of 3% (w/w). In order to maintain a constant phosphorus concentration in a control diet, methylcellulose (MC) (Wako, Osaka, Japan) was added to normal chow at a concentration of 3% (w/w).
Animal model preparation and experimental protocols
Orally adenine-dosed rats were prepared according to a previous report (Terai et al., 2008). Briefly, after the acclimatization period, an adenine suspension (100 mg·mL−1 adenine sulphate in a 0.5% MC solution, Wako, Osaka, Japan) was given orally to the animals once a day at 8:00–9:00 am for 10 days (d −10 to d −1).
Four separate experiments (Exps. 1–4) were then carried out using the adenine-dosed and age-matched normal rats. The experimental designs and feeding protocols are summarized in Figure 1. In all experiments, the body weight of each animal was recorded at 8:00–9:00 am. The amounts of chow eaten and water drunk from 8:00 am to 8:00 am the next day were recorded as the amounts of the second date. Blood was taken from the orbital vein under ether anesthesia at multiple time points (arrowheads, 1:00–2:00 pm, Figure 1). In Exp. 3 for determination of the glomerular filtration rate (GFR), functional excretion rates of P (FERP) and Ca, urine was collected for 24 h using metabolic cages in the second, fourth and eighth weeks. For tissue sampling, animals were killed by CO2 inhalation at time points indicated by round-bottomed arrows (1:00–3:00 pm) (Figure 1).
Figure 1.
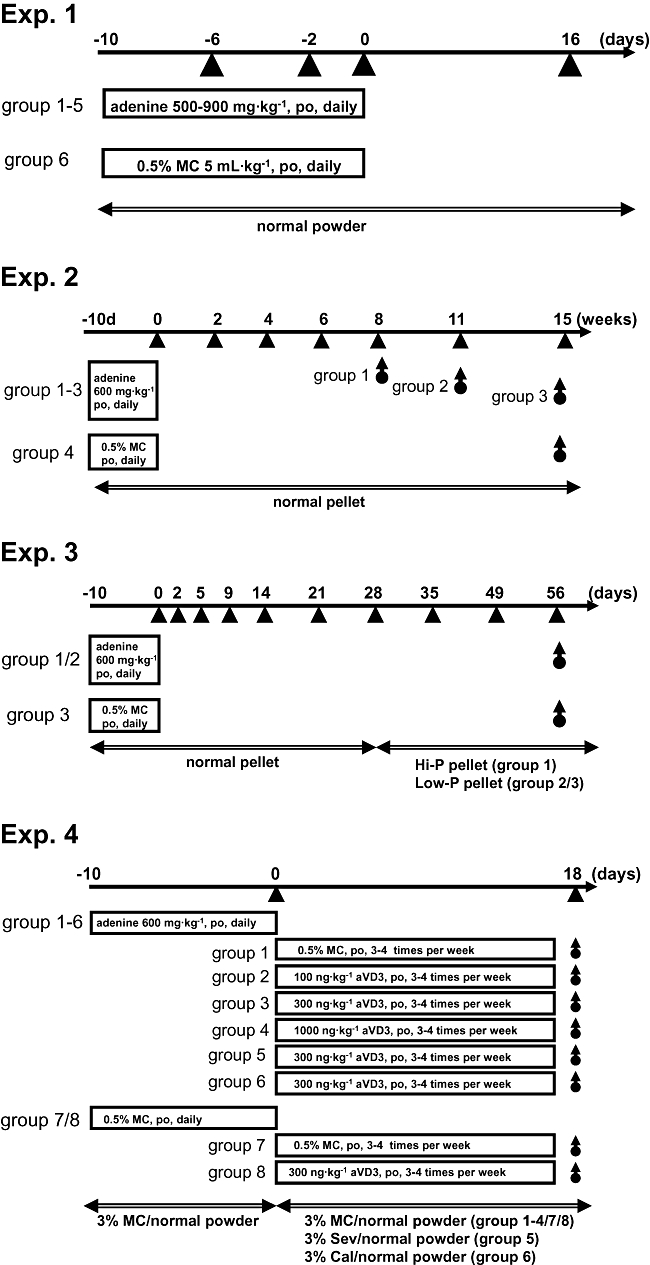
Experimental protocols. Four separate experiments were performed, and the feeding schedules and diets (double-head arrows) used are indicated for each experiment. Blood samples were taken at multiple time points (arrowheads) and used for serum parameter determinations. For biological and pathological analyses, animals in each group were sacrificed and tissue samples were taken at various time points (round-bottomed arrows). aVD3, active vitamin D3; Cal, CaCO3; MC, methylcellulose; Sev, sevelamer.
Exp. 1
A study to find an optimal dose of adenine for the induction of non-lethal, severe renal failure was performed. The adenine suspension was orally administered at dosages of 500 mg·kg−1 (5 mL·kg−1) to 900 mg·kg−1 (9 mL·kg−1) to rats (n= 4 each) for 10 days (d −10 to d −1). In control rats (n= 4), a MC solution (5 mL·kg−1) was given instead of the adenine suspension. For the following experiments (Exps. 2–4), an oral dose of 600 mg·kg−1 adenine (6 mL·kg−1) was selected.
Exp. 2
Long-term observations were made on the hyperphosphatemia, hypocalcemia, deficits of renal functions, vascular calcification and hyperparathyroidism in adenine-dosed rats. After 6 weeks of adenine dosing, animals were divided into three groups (n= 8 each) and used for tissue sampling at 8, 11 and 15 weeks. Serum parameters at 0 to 6 weeks were determined in all adenine-dosed rats (n= 24), and tissue at 8, 11 and 15 weeks was taken on eight animals in each of the three groups. Age-matched normal rats (n= 6) were used at all time points of serum parameter determination and their tissues were taken only at 15 weeks.
Exp. 3
The influence of the amount of dietary phosphorus on serum parameters was examined. At 28 days after adenine dosing, animals were divided into two groups, and were fed with a Hi-P diet (n= 5) or Low-P diet (n= 6) for the following 28 days. Age-matched normal rats (n= 4) were fed a Low-P diet from d 28 to d 56. Animals were put into metabolic cages to take urine at 2, 4 and 8 weeks.
Exp. 4
Vascular calcification induced by active vitamin D3 (1α-(OH) vitamin D3, Alfarol, Chugai, Tokyo, Japan) and the effect of phosphate binders on it were examined. At d 0 after adenine dosing, animals were divided into six groups (n= 4–5). Active vitamin D3 was orally administered at dosages of 0 (group 1), 100 (group 2), 300 (groups 3, 5 and 6) and 1000 ng·kg−1 (group 4) for 18 days (three to four times a week, timed on d 0, 1, 3, 5, 8, 10, 12, 15 and 17). During the 18th day of active vitamin D3 dosing, adenine-dosed rats were fed with normal chow containing 3% of either sevelamer (group 5), CaCO3 (group 6) or MC (other groups). Age-matched normal rats were given a MC solution (group 7) or active vitamin D3 (300 ng·kg−1, group 8) on the same experimental schedule. The tissues of the animals were taken on d 20.
Vascular calcification analyses
Vascular calcification was carefully examined using an atomic absorption method, histological staining and a combination of the two. The aortas, including arch to abdominal level just above the common iliac arteries, were taken and longitudinally incised, and were rinsed well to remove blood. After removing the arch, the major part of the aorta (thoracic to abdominal) was used for Ca measurement. The arch was used for alizarin staining to confirm Ca deposits in the tissue. Other large arteries, including the common carotid, renal and femoral arteries, were also prepared for Ca measurement. For Ca extraction, the tissue was immersed in 20 volumes of 2N HCl (v/w, wet tissue weight.) overnight at room temperature. Extracted Ca was measured using an atomic absorption spectrophotometer at 423 nm wavelength (ANA-182, Tokyo Photo-Electric Co., Ltd., Tokyo, Japan). For the histological detection of Ca deposits, some tissues were briefly fixed with 10% neutralized formalin overnight at room temperature. Coronal sections of 12 µm thickness were prepared with a cryostat (CM3050 S, Leica, Wetzlar, Germany).
In a control experiment, the aorta was fixed in formalin for 1 h and rinsed with distilled water, three times for 20 min each. The tissue was incubated with 1% Alizarin S solution for 5 min and washed three times with 70% ethanol for 20 min each. Aortas were cut into five to six segments (see Figure 5A). After taking photo-images under a microscope with a CCD camera (AX70, Olympus, Tokyo, Japan), the alizarin-positive area (Ca-burden area) was semi-quantitatively measured as a percentage of the total tissue area using a NIH image (Noda-Saita et al., 2004). After taking the images, segments were used for Ca measurements in an atomic absorption spectra photometer according to the method described above.
Figure 5.
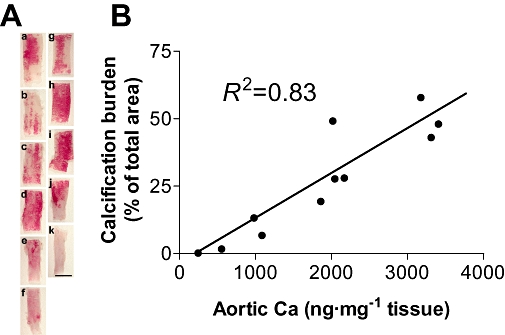
Validation of quantification methods for vascular calcification. The validation study was performed using separately prepared adenine-dosed rats fed with a normal diet for 22 weeks. Robust but heterogeneous alizarin-positive calcification is observed in the whole-mount aortas taken from two rats (A, a–f; six segments from one aorta, g–k; five from another aorta). After the histological measurement of calcified areas (% of total area), HCl-extracted Ca from each sample of tissue was quantified using the atomic absorption method (shown as ng·mg−1 tissue). The two measures of the Ca burden showed a significant correlation (B, R2= 0.83, n= 11). Bar is 5 mm (A).
Parathyroid glands
The parathyroid glands (PTG) were removed bilaterally under a binocular microscope and the total weight of the PTG from each animal was recorded. In a preliminary experiment, the localization of the PTG on the thyroidal organ was immunohistochemically examined (data not shown) using an antibody for parathyroid hormone (PTH), a marker of the PTG. This preliminary study facilitated the removal of the PTG using a microsurgery forceps.
Serum biochemistry
Serum phosphate and Ca levels were determined using a standard molybdate assay kit (Phospha C-test Wako, Wako, Osaka, Japan) and a methyl-xynol blue assay kit (Calcium E-test Wako, Wako, Osaka, Japan), respectively. Improved enzymatic assay kits were used for measurement of serum creatinine levels (Cre-EN Kinos, Kinos, Tokyo, Japan) and serum blood urea nitrogen (BUN) levels (BUN Kinos, Kinos, Tokyo, Japan). Serum PTH concentrations were determined using the rat intact PTH (iPTH) ELISA kit (Immunotopics, Inc., San Clemente, CA, USA).
Statistics
Data were expressed as the mean ± SEM. Statistical analyses were performed using the Statistical Analysis System ver. 8.2 (SAS Institute, Inc., Cary, NC, USA). Correlation analyses were performed with the use of Pearson's linear correlation. For parametric data analyses, the unpaired Student's t-test and Dunnett's test were used to compare the serum parameters between or among the animal groups. Statistical significance was defined as P < 0.05 (#) for the Student's t-test (two-sided), and P < 0.05 (*) or P < 0.01 (**) for Dunnett's test (two-sided).
Results
Optimizing adenine dosage for induction of CKD
To find an optimal dose of adenine for induction of severe CKD with hyperphosphatemia in rats, adenine doses ranging from 500 mg·kg−1 to 900 mg·kg−1 were administered for 10 days. Administration and feeding protocols are described in Exp. 1 in Figure 1. All doses of adenine induced hyperphosphatemia by the eleventh day (d 0 in the figure). Administration of 500–600 mg·kg−1 did not lead to animal deaths during the following 16 days, but higher doses resulted in deaths during the 10-day dosing period (Table 1). We therefore chose to use adenine at 600 mg·kg−1, given orally for 10 days, to prepare our models of rats with severe CKD.
Table 1.
Effects on serum phosphate levels and survival in rats orally dosed with different amounts of adenine
| Days | Serum phosphate, mg·mL−1 (survival rate, %) | |||||
|---|---|---|---|---|---|---|
| Adenine (mg·kg−1) | Normal | |||||
| 500 | 600 | 700 | 800 | 900 | ||
| −6 | 0.0766 ± 0.0010 | 0.0753 ± 0.0065 | 0.0823 ± 0.0064 | 0.0974 ± 0.0170 | 0.1300 ± 0.0146 | 0.0783 ± 0.0015 |
| (100) | (100) | (100) | (75) | (50) | (100) | |
| −2 | 0.1119 ± 0.0082 | 0.1393 ± 0.0212 | 0.1506 ± 0.0289 | 0.1353 ± 0.0310 | – | 0.0853 ± 0.0050 |
| (100) | (100) | (75) | (50) | (0) | (100) | |
| 0 | 0.1422 ± 0.0056 | 0.1502 ± 0.0204 | 0.1448 ± 0.0021 | – | – | 0.0727 ± 0.0059 |
| (100) | (100) | (50) | (0) | (0) | (100) | |
| 16 | 0.1008 ± 0.0073 | 0.1154 ± 0.0169 | 0.1173 ± 0.0091 | – | – | 0.0683 ± 0.0017 |
| (100) | (100) | (50) | (0) | (0) | (100) | |
Adenine sulphate (suspended in a 0.5% MC solution, 100 mg·mL−1) ranging from 500 mg·kg−1 to 900 mg·kg−1 or a 0.5% MC solution (5 mL·kg−1) was given orally to rats for 10 days (d −10 to d −1) (n= 4 each). Animals were fed with a normal diet during the experimental period of d −10 to d 16. Serum phosphate levels are expressed as the mean ± SD.
MC, methylcellulose.
Long-term observation of serum parameters
Serum parameters of adenine-dosed and normal rats fed normal chow were examined in a long-term experimental period of 15 weeks (Exp. 2). Serum creatinine and BUN levels in adenine-dosed rats were comparatively stable for 4 weeks after the induction of renal deficits, and then increased slowly over the experimental period of 15 weeks (Figure 2A,B). Serum levels of phosphate in adenine-dosed rats decreased slowly from 0.15 mg·mL−1 to 0.09–0.1 mg·mL−1 by 4–6 weeks after adenine dosing, and then increased again from 8 weeks (Figure 2C). The serum phosphate levels were significantly high even at the lowest time point of 6 weeks (0.088 mg·mL−1) compared with the levels in normal rats (0.059 mg·mL−1). Serum levels of Ca increased slowly to 6 weeks and then decreased (cf. Figure 2C,D). No animals died during the experimental 15 weeks.
Figure 2.
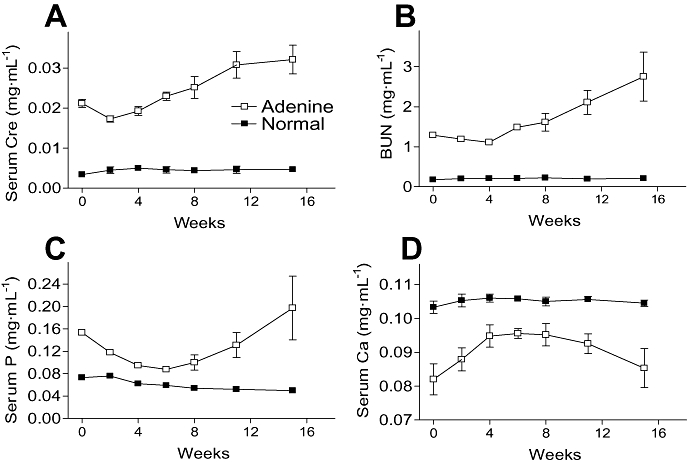
Long-term observation of serum parameters in adenine-dosed rats (Exp. 2). Serum creatinine (Cre; A), BUN (B), phosphate (P; C) and Ca (D) levels in adenine-dosed and normal rats (n= 6). Renal failure was induced with oral 10-day administration of 600 mg·kg−1 adenine, and normal animals were given 0.5% MC solution (6 mL·kg−1) instead of the adenine suspension. Serum parameters were measured at 0 (just after the end of adenine dosing), 2, 4, 6, 8, 11 and 15 weeks. At 6 weeks, adenine-dosed rats (n= 24) were divided into three groups (n= 8 each); thereafter, each adenine-dosed group was used at each time point of 8, 11 and 15 weeks for serum parameter measurements and tissue preparations (n= 8 each). Data are expressed as the mean ± SEM. BUN, blood urea nitrogen; MC, methylcellulose.
Effects of dietary content of phosphorus on serum parameters and renal function
Using adenine-dosed hyperphosphatemia rats maintained for a month, we studied whether the amount of phosphorus in the diet affected serum phosphate levels (Exp. 3). The serum phosphate levels decreased on the Low-P diet, and somewhat increased in rats on the Hi-P diet after 1 week of feeding, and both serum levels were stable during the 4 week feeding (Figure 3C). Serum Ca levels responded oppositely to the phosphate levels during the feeding period (Figure 3D). Serum creatinine and BUN were comparatively stable and were no different between Hi-P and Low-P groups after the 4 week feeding (Figure 3A,B). Values of other serum and urine parameters related to renal function at just before feeding with the Hi-P/Low-P diets (pre) and after the 4 week feeding (post) are summarized in Table 2. GFRs in adenine-dosed rats were approximately 13–20% of age-matched normal rats before the Hi-P/Low-P feeding, and those in adenine-dosed rats fed with Hi-P and Low-P diets did not differ before and after the feeding. The FERP of adenine-dosed rats fed the Hi-P diet was over 100%, but that of adenine-dosed rats fed the Low-P diet was approximately 70% (Table 2).
Figure 3.
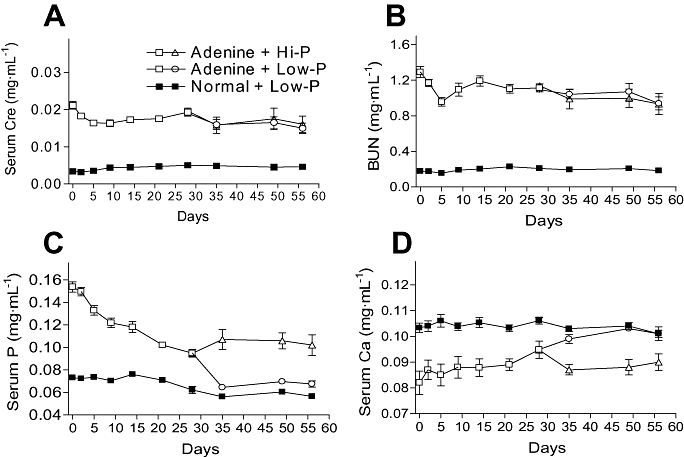
Influence of the phosphorus content of the diet on serum parameters in adenine-dosed rats (Exp. 3). Serum creatinine (Cre; A), BUN (B), phosphate (P; C) and Ca (D) levels were determined at multiple time points. Renal failure was induced with oral 10-day administration of 600 mg·kg−1 adenine, and normal animals were given 0.5% MC solution (6 mL·kg−1) instead of the adenine suspension. Adenine-dosed rats (n= 11) and normal rats (n= 4) were fed with a normal diet from d 0 to d 28. From d 28, adenine-dosed rats were split into two groups, one fed with a Hi-P diet (n= 5) and the other on a Low-P diet (n= 6), for the next 28 days. The normal rats were fed with a Low-P diet (n= 4) from d 28 to d 56. Data are expressed as the mean ± SEM. BUN, blood urea nitrogen; Hi-P, high-phosphorus; Low-P, low-phosphorus; MC, methylcellulose.
Table 2.
Urinary, functional and other parameters in adenine-dosed rats before and after feeding of Hi-P/Low-P diets
| Animal type | 2 weeks | 4 weeks | 8 weeks | ||||
|---|---|---|---|---|---|---|---|
| Adenine | Normal | Adenine | Normal | Adenine | Normal | ||
| Adenine chow | CE-2 | CE-2 | CE-2 | CE-2 | Hi-P | Low-P | Low-P |
| n | 11 | 4 | 11 | 4 | 5 | 6 | 4 |
| Urinary biochemistry | |||||||
| Creatinine (mg·day−1) | 2.7 ± 0.2 | 6.6 ± 0.3 | 3.8 ± 0.2 | 7.1 ± 0.3 | 5.7 ± 0.6 | 4.8 ± 0.3 | 7.6 ± 0.2 |
| Phosphate (mg·day−1) | 15.0 ± 1.2 | 22.6 ± 1.4 | 22.5 ± 1.5 | 22.6 ± 1.2 | 43.8 ± 4.9 | 15.4 ± 1.2 | 6.1 ± 0.8 |
| Ca (mg·day−1) | 0.5 ± 0.1 | 0.9 ± 0.1 | 1.3 ± 0.1 | 1.0 ± 0.1 | 1.0 ± 0.1 | 1.9 ± 0.3 | 1.2 ± 0.2 |
| Functional parameters | |||||||
| GFR (mL·day−1) (body weight 100 g) | 102 ± 6 | 751 ± 103 | 115 ± 6 | 610 ± 60 | 144 ± 15 | 122 ± 8 | 612 ± 39 |
| FERP (%) | 85.5 ± 6.5 | 19.9 ± 3.1 | 117.6 ± 4.3 | 25.9 ± 2.2 | 120.6 ± 6.8 | 70.8 ± 4.8 | 6.7 ± 1.0 |
| FERCa (%) | 4.4 ± 0.9 | 0.6 ± 0.1 | 6.1 ± 0.7 | 0.7 ± 0.1 | 3.6 ± 1.0 | 5.1 ± 0.5 | 0.7 ± 0.1 |
| Other parameters | |||||||
| Urine output (mL·day−1) | 37.6 ± 2.0 | 10.8 ± 3.8 | 44.0 ± 2.1 | 7.5 ± 0.7 | 45.1 ± 3.6 | 39.9 ± 2.8 | 5.2 ± 0.6 |
| Water drunk (mL·day−1) | 52.7 ± 2.0 | 30.7 ± 7.3 | 62.2 ± 3.4 | 21.4 ± 0.8 | 59.3 ± 4.8 | 50.1 ± 4.3 | 16.5 ± 1.7 |
| Food eaten (g·day−1) | 10.2 ± 0.5 | 14.3 ± 0.5 | 11.9 ± 0.3 | 18.3 ± 0.7 | 11.3 ± 0.5 | 12.4 ± 1.1 | 12.9 ± 0.4 |
| Body weight (g) | 152 ± 5 | 210 ± 5 | 177 ± 5 | 236 ± 4 | 213 ± 15 | 220 ± 10 | 270 ± 8 |
Adenine sulphate (600 mg·kg−1; suspended in 0.5% MC) (n= 11) or 0.5% MC (n= 4) was given orally for 10 days to rats, which were fed a normal diet (CE-2) for the following 4 weeks. At 4 weeks, the normal diet for adenine-dosed rats was changed to a Hi-P diet (n= 5) or a Low-P diet (n= 6), and that for normal rats to a Low-P diet (n= 4). These diets were maintained for another 4 weeks. Data are expressed as the mean ± SEM.
FERCa, functional excretion rates of Ca; FERP, functional excretion rates of P; GFR, glomerular filtration rate; Hi-P, high-phosphorus; Low-P, low-phosphorus; MC, methylcellulose.
Vascular calcification and 2HPT
In control experiments, we carefully validated quantitative methods for vascular calcification using separately prepared adenine-dosed rats fed a normal diet for 15–22 weeks. Heterogeneous and patchy alizarin-positive staining specific for Ca deposits was found in the aorta of adenine-dosed rats (Figure 4A,B) at 15 weeks after adenine dosing, but not in that of age-matched normal rats (Figure 4C). In coronal sections of the kidney artery, the positive staining was only found in the internal elastic lamina to the media (Figure 4D, E). Robust but heterogeneous alizarin-positive calcification was detected in aortas taken from adenine-dosed rats (n= 2) at 22 weeks (Figure 5A, a–f; six segments from one aorta, g–k; five from another aorta). After the histological measurement (% total area), Ca was extracted by HCl from each segmented tissue and quantified (ng·mg−1 tissue) using an atomic absorption method. After the extraction with HCl, alizarin did not show any staining of tissues (data not shown). The percentages of aorta area showing a histological Ca-burden and the Ca amounts (as ng·mg−1 tissue) extracted from each segment showed a significant correlation (Figure 5B, R2= 0.83, n= 11).
Figure 4.
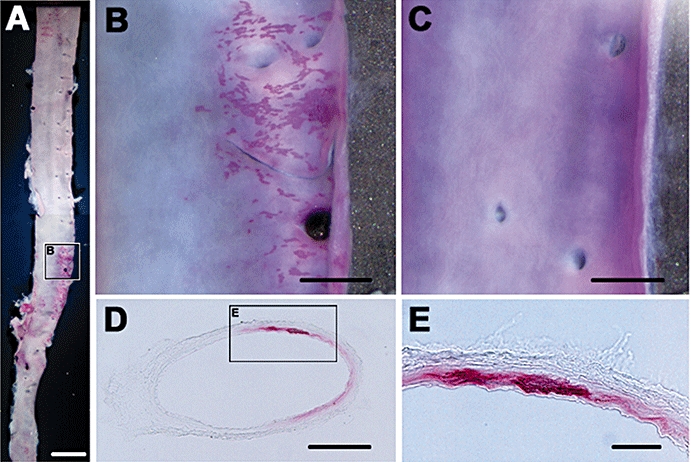
Vascular calcification in adenine-dosed rats (Exp. 2). Vascular calcification was demonstrated with alizarin staining specific for Ca deposits. The positive heterogeneous and patchy staining is localized in the whole-mount aorta only of rats (A, B) at 15 weeks after adenine dosing, but not in that of age-matched normal rats (C). In coronal sections of the kidney artery, the positive staining is only found in the internal elastic lamina to the media (D, E). Bars are 5 mm (A), 1 mm (B, C), 500 µm (D) and 100 µm (E).
In the long-term experimental study, quantification of Ca using the atomic absorption method revealed that 12.5% (1/8) of the animals showed aortic calcification at 8 weeks and 37.5% (3/8) at 15 weeks (Figure 6A). Only the animals with aortic calcification (>220 ng Ca mg−1 wet tissue) also showed alizarin-positive staining in the arch (data not shown), and the Ca × P product was comparatively higher (>0.012 mg2·mL−2) compared with animals not showing calcification (0.0067–0.0108 mg2·mL−2, Figure 6B). The amounts of Ca in the calcified aortas (n= 7) were significantly correlated with the Ca × P product (R2= 0.66, P= 0.02), but not with serum levels of phosphate (R2= 0.27, P= 0.23) or Ca (R2= 0.004, P= 0.90). At 15 weeks, the PTG weights (bilateral) in the adenine-dosed rats were five-fold higher (Figure 6C) than those in age-matched normal rats and serum iPTH levels were also very much higher (4229 ± 1308 pg·mL−1) more than those of the normal rats (11–12 pg·mL−1). The PTG weights (ranging 1.4–3.1 mg) and iPTH levels (ranging 370–11 960 pg·mL−1) varied widely among animals, but the PTG weights were significantly correlated with serum iPTH levels (Figure 6D, R2= 0.82, n= 8, P= 0.002).
Figure 6.
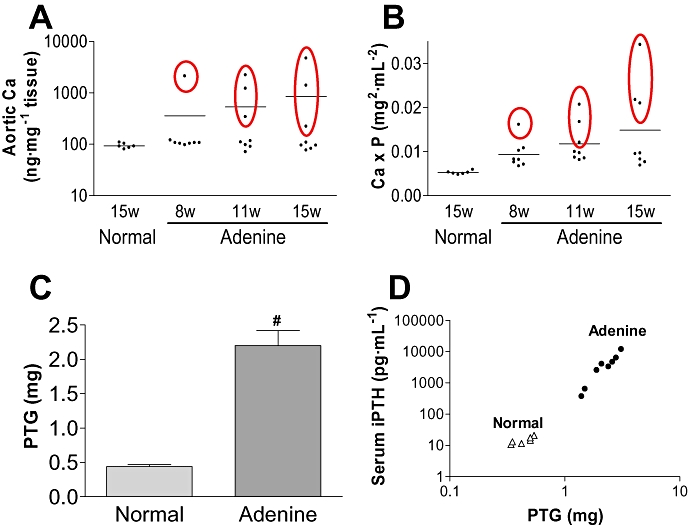
Aortic calcification and 2HPT in adenine-dosed rats. In a long-term experiment, quantification of Ca using the atomic absorption method was performed in adenine-dosed rats at multiple time points of 8 weeks (w), 11 w and 15 w (n= 8 each) and in normal rats at 15 w (n= 6) (A). At the same time points, the serum Ca × P product was determined (B). Data satisfying a criterion of significant calcification (more than 150 ng·mg−1 tissue) are surrounded by ovals and bars indicate the means (A, B). PTG weights of adenine-dosed rats (n= 8) and age-matched normal rats (n= 6) at 15 w (C). Bilateral PTG were removed and the total tissue weights recorded. The PTG weights and serum iPTH levels correlate when the data for adenine-dosed rats and age-matched normal rats are plotted (D). Data are expressed as the mean ± SEM. Statistical analyses were performed by the Student's t-test for comparison of PTG weights in adenine-dosed control rats and normal rats (#P < 0.05). 2HPT, secondary hyperparathyroidism; Ca × P, calcium × phosphate; iPTH, intact parathyroid hormone; PTG, parathyroid glands.
Vitamin D-induced vascular calcification and phosphate binders
To produce a hyperphosphatemic vascular calcification rat model, we used an active form of vitamin D3 (1α-hydroxy- vitamin D3, Alfarol) at doses from 100 to 1000 ng·kg−1 for 18 days (orally administered three to four times per week) after CKD induction with adenine. At the 18th day of active vitamin D3 administration (28 days after starting adenine), serum levels of phosphate, Ca and iPTH, aortic calcification and PTG weights were measured, and the results are summarized in Table 3. Serum Ca levels and the Ca × P product increased in the active vitamin D3 groups compared with the control group, but serum phosphate levels did not. Aortic calcification was not detected in the control group and normal rats, nor in normal rats treated with active vitamin D3, but there was an active vitamin D3-induced dose-dependent calcification in the adenine-dosed rats. Even at the lowest active vitamin D3 dose of 100 ng·kg−1, calcification was detected in all animals. Active vitamin D3 treatment did not affect PTG weights, but iPTH levels were significantly decreased. The amounts of food eaten and body weights did not differ between the adenine-dosed control group and active vitamin D3-treated adenine-dosed groups, during the experimental period of 18 days (shown at d 2 and d 16 in Table 3).
Table 3.
Effects of active vitamin D3 (aVD3) on serum biochemistry, vascular calcification and 2HPT in adenine-dosed and normal rats
| aVD3(ng.kg−) | Adenine | Normal | ||||
|---|---|---|---|---|---|---|
| 0 | 100 | 300 | 1000 | 0 | 300 | |
| n | 5 | 4 | 4 | 4 | 4 | 4 |
| Phospahte(mg.mL−) | 0.135±0.007# | 0.129±0.010 | 0.130±0.009 | 0.120±0.007 | 0.061±0.001 | 0.062±0.002 |
| Ca(mg.mL−) | 0.090±0.005# | 0.111±0.002** | 0.111±0.004** | 0.131±0.003** | 0.108±0.001 | 0.109±0.002 |
| Ca×(mg2.mL−2) | 0.0120±0.0005# | 0.0144±0.0008* | 0.0144±0.0005* | 0.0157±0.0007** | 0.0065±0.0001 | 0.0067±0.0003 |
| Calcification (ng.mg−1) | 89±6# | 580±231* | 2581±630** | 4970±492** | 114±3 | 112±4 |
| PTG (mg) | 0.98±0.13# | 1.11±0.15 | 0.99±0.24 | 0.97±0.13 | 0.38±0.06 | 0.38±0.09 |
| iPTH (pg.mL−1) | 3520±347# | 1680±288* | 2000±641* | 235±137** | 13±6 | 12±4 |
| Body weight(g) | ||||||
| d 2 | 181±3# | 168±8 | 175±7 | 175±2 | 219±7 | 214±5 |
| d 16 | 10.0±0.8# | 12.3±0.6 | 10.3±1.6 | 10.9±0.7 | 18.5±0.9 | 18.6±1.0 |
| d 16 | 174±9# | 183±6 | 176±2 | 179±4 | 265±6 | 264±6 |
| Food eaten(g.day−1) | ||||||
| d 2 | 10.8±0.8# | 9.7±1.2 | 10.3±1.0 | 10.4±1.3 | 17.8±0.5 | 17.1±1.0 |
| d 16 | 10.0±0.8# | 12.3±0.6 | 10.3±1.6 | 10.9±0.7 | 18.5±0.9 | 18.6±1.0 |
After a 10-day induction of renal failure with adenine (600 mg·kg−1, p.o), animals were administered aVD3 (suspended in 0.5% MC) at doses of 100, 300 or 1000 ng·kg−1 for 18 days (3–4 time week−1, p.o) and fed a control diet (normal diet containing 3% MC, w/w). For normal rats and adenine-dosed control rats, 0.5% MC was administered instead of adenine or the aVD3 suspension. At d 18, serum parameters were determined and tissue samples were taken. Data are expressed as the mean ± SEM. Statistical analyses were performed by Student's t-test for comparison of adenine-dosed control rats and normal rats without aVD3 dosing (#P<0.05), and among adenine-dosed rats with or without aVD3 dosing by Dunnett's test
(P<0.05.
P<0.01.)
2HPT, secondary hyperparathyroidism; Ca × P, calcium × phosphate; iPTH, intact parathyroid hormone; MC, methylcellulose; PTG, parathyroid glands.
In the same protocol of active vitamin D3-induced aortic calcification (300 ng·kg−1), feeding diets with 3% of sevelamer or 3% of CaCO3 decreased significantly the serum phosphate levels (Figure 7A) and the Ca × P product (Figure 7C), and increased serum Ca levels (Figure 7B). Sevelamer inhibited aortic calcification completely, but CaCO3 inhibited only partially (Figure 7D). Sevelamer also prevented hyperplasia of the PTG, but CaCO3 did not (Figure 7E). Both sevelamer and Ca CO3 decreased serum iPTH levels to the normal range (Figure 7F). There were differences in body weight and food intake among the active vitamin D3-treated adenine-dosed rats fed diets containing sevelamer, CaCO3 and MC during the experimental period of 18 days.
Figure 7.
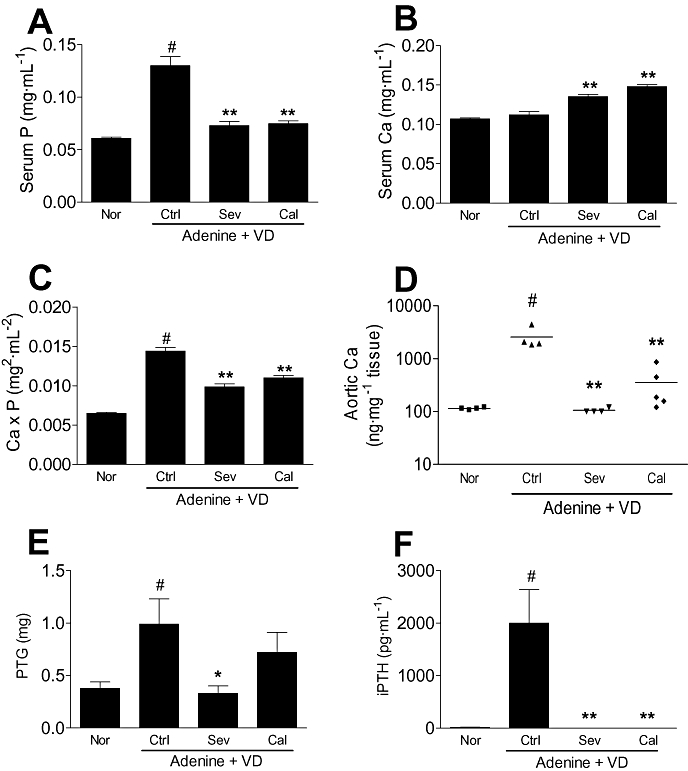
Effects of phosphate binders on active vitamin D3-induced vascular calcification in adenine-dosed rats and on the serum parameters (Exp. 4). Renal failure was induced by 10-day oral administration of adenine at a dose of 600 mg·kg−1, and normal animals were given an MC solution (6 mL·kg−1) instead of the adenine suspension. For 18 days after induction of renal failure, active vitamin D3 (VD; 300 ng·kg−1, 3–4 time week−1) was orally administered to adenine-dosed rats fed with normal powder diets containing 3% MC (n= 4), 3% sevelamer (n= 4) or 3% CaCO3 (n= 5). Normal rats were administered a MC solution instead of active vitamin D3 and fed with a 3% MC diet (n= 4). Sevelamer (Sev) and CaCO3 (Cal) significantly lowered serum phosphate levels (A) and Ca × P products (C), and increased serum Ca levels (B) in adenine-dosed rats given active vitamin D3 orally. Sevelamer inhibited aortic calcification completely and CaCO3 did partially (D). Sevelamer also prevented the hyperplasia of the parathyroid glands (PTG) (E). Both sevelamer and CaCO3 decreased serum iPTH levels to the normal concentration range (F). Data are expressed as the mean ± SEM (A, B, C, E, F) or plotted individually (D). Statistical analyses were performed by the Student's t-test for comparison of adenine-dosed control rats and normal rats without active vitamin D3 dosing (#P < 0.05), and among adenine-dosed rats with or without active vitamin D3 dosing by Dunnett's test (*P < 0.05, **P < 0.01). Ca × P, calcium × phosphate; MC, methylcellulose.
Discussion
In a previous study, we reported that short-term treatment of rats with oral adenine at a dose of 100 mg for 10–12 days caused CKD with hyperphosphatemia (Terai et al., 2008). The renal failure was manifested as elevation of serum creatinine and BUN to chronically stable levels during the experimental period of over 5 weeks. In the present dose-optimizing study, 600 mg·kg−1 of adenine for 10 days was deemed best for the later experiments because this dose induced significant hyperphosphatemia without death in the weeks following, while higher doses (700–900 mg·kg−1) did cause deaths. These data indicate that the amount of adenine is important for stable induction of severe CKD in rats and that it should be controlled by oral dosing. We have previously demonstrated that it is difficult to control the amounts of adenine ingested by rats given adenine in their diet (Terai et al., 2008).
In long-term observation of serum parameters, the data indicate that the adenine-induced CKD is irreversible, progressive and not diverse among the animals. At the stable phase 2–6 weeks after CKD induction, GFRs in adenine-dosed rats were approximately 13–20% of age-matched normal rats. Under the Kidney Disease Outcomes Quality Initiative (K/DOQI) guidelines, CKD is classified into five stages based on the decrements of GFR: stage 1 (GFR > 90 mL·min−1 1.73 m−2), stage 2 (GFR, 60–89 mL·min−1 1.73 m−2), stage 3 (GFR, 30–59 mL·min−1 1.73 m−2), stage 4 (GFR, 15–29 mL·min−1 1.73 m−2) and stage 5 (GFR < 15 mL·min−1 1.73 m−2 or dialysis) (Eknoyan et al., 2003). GFRs in healthy individuals have been estimated from 365 kidney donors (Rule et al., 2004). The mean GFR was 111 mL·min−1 1.73 m−2 in a 20-year-old population, and declined age-dependently by 4.9 mL·min−1 1.73 m−1·decade−1. In line with these guidelines and the normal GFR, adenine-dosed rats may be equivalent to stage 4 of CKD patients.
The K/DOQI guidelines (Eknoyan et al., 2003) recommend dietary restriction of phosphorus (less than 1000 mg·day−1, estimated 20–40% reduction of normal intake) for controlling the hyperphosphatemia in CKD patients (stage 4/5), mainly because of the lack of evidence of adverse effects (Klahr et al., 1994). But it is likely to be very difficult to provide the recommended diets because the patients are also advised to take high levels of proteins in processed foods that contain large amounts of uncounted phosphorus (Kestenbaum, 2007; Uribarri, 2007). The effectiveness of dietary phosphorus restriction for hyperphosphatemia in CKD patients has been analysed in 19 studies examining 2476 patients (Eknoyan et al., 2003), but no dramatic impact on serum phosphate levels was demonstrated (Klahr et al., 1994; Kestenbaum, 2007). In the present study, serum phosphate levels in adenine-dosed hyperphosphatemic rats responded to low-dietary-phosphorus intake. The serum phosphate levels were almost stable with a Hi-P diet (1.27% phosphorus) and decreased with a Low-P diet (0.90% phosphorus) to the normal range found in age-matched normal rats during 4 week feeding with the diets. These data clearly demonstrate that dietary restriction of about 30% (the difference between Hi-P and Low-P diets) phosphorus was effective for treatment of hyperphosphatemia in adenine-dosed rats, so such an effect may be expected in CKD patients.
Vascular calcification has been pathologically evaluated using von Kossa's silver nitrate staining specific for phosphate salts in 5/6Nx rats (Cozzolino et al., 2003; Hirata et al., 2003; Haffner et al., 2005; Henley et al., 2005; Neves et al., 2007), adenine-fed rats (Katsumata et al., 2003; Tamagaki et al., 2006) and patients (Milliner et al., 1990). The evaluations were mainly performed by scoring on staining degrees in representative tissue sections taken from limited parts of the aorta. In the present study, alizarin-positive staining specific for Ca deposits was found only in the internal elastic lamina to the media of the aorta and large arteries in adenine-dosed rats. The localization was compatible with the previous reports.
Alizarin staining on whole-mount aorta in the present study demonstrated heterogeneous and patchy deposits of Ca, and primitive Ca deposits looking pepper-and-salt (fat-marbled) throughout the aorta. This is a novel pathological finding, because the precise distribution throughout the aorta in whole-mount tissues has not been demonstrated by von Kossa staining. It may be difficult to apply von Kossa staining to whole-mount aorta because of the difficulty of detecting the specific staining against the high background in whole-mount aorta found in the present study (data not shown). Further study of whole-mount tissues stained with alizarin should be meaningful to demonstrate clearly high prevalence sites and the expanding patterns of calcification. Recently, Persy et al. (2006) demonstrated in vivo 3-demensional images of aortic calcification in adenine-fed rats using a X-ray micro-tomography mCT technique. Comparison of images at 2 week intervals revealed that calcified regions were heterogeneous and growing, and that newly formed calcified foci appeared during the interval.
Alternatively, biochemical Ca quantification using the atomic absorption method has been performed on HCl extracts from various tissues (Cozzolino et al., 2003; Hirata et al., 2003). However, the relationship of pathologically calcified areas and biologically quantified Ca amounts has not been clarified. In the present study, we carefully rated pathological Ca-burden areas detected by alizarin staining and compared them with biologically quantified amounts of Ca. The significant correlation between the calcified areas and the amounts of Ca in aortas indicates that the amounts of Ca measured with the atomic absorption method are excellent indicators of the degrees of pathological Ca deposits.
The amounts of aortic Ca in normal rats and in adenine-dosed rats without pathological calcification were around 100 ng·mg−1 and did not exceed 150 ng·mg−1 wet tissue weight. Hence concentrations of Ca in the aorta higher than 150 ng·mg−1 were regarded as indicating significant calcification in the present study. Aortic calcifications confirmed by the Ca quantification method were detected in some of the adenine-dosed rats (about 40%) even at 15 weeks after CKD induction. Interestingly, 40% of pre-dialysis patients are reported to have vascular calcification (Block, 2000; Raggi et al., 2002; Russo et al., 2004). Appearance of vascular calcification could be explained by the elevation of the Ca × P product. These values in the calcified rats were significantly higher (>0.012 mg2·mL−2) than in rats without calcification (0.0067–0.0108 mg2·mL−2), and the amounts of Ca in calcified aortas were significant correlated with the Ca × P product (R2= 0.66). Elevation of the serum Ca × P product has been identified as the most reliable risk factor for vascular calcification in CKD patients (Block, 2000).
Furthermore, adenine-dosed rats in the present study mimicked in detail a number of clinical features of CKD patients. For the diagnosis of 2HPT in CKD patients, elevation of serum iPTH levels and PTG hyperplasia are determinant findings (Eknoyan et al., 2003). Notably, the elevations in PTG weights were significantly correlated with serum iPTH levels in adenine-dosed rats. These findings suggest that adenine-dosed rats are an excellent 2HPT model which shows the clinical features as well as the vascular calcification. Another important aspect that has not been examined in the present study, but would certainly be worthwhile to study in the future in this model, is bone metabolism.
Orally administered active vitamin D3 (Alfarol) for 18 days (3–4 times per week) after CKD induction in adenine-dosed rats caused dose-dependent vascular calcification in the present study. Vascular calcification was detected in all animals even at the lowest dose of 100 ng·kg−1 of the active vitamin D3. The vascular calcification clearly demonstrated in the present study may depend on the CKD and hyperphosphatemia induced by oral adenine dosing. Notably, 100–300 ng·kg−1 of active vitamin D3 did not induce hypercalcemia, nor affect the amount of diet eaten (ingested amount of Ca) compared with adenine-dosed control rats. Those data indicate that active vitamin D3 treatment can definitely induce vascular calcification in adenine-dosed rats even while serum Ca levels remain in the normal range. The same may be true in CKD patients, as suggested by Goodman et al. (2004).
The active vitamin D3 treatment decreased serum iPTH levels partially even at the hypercalcemic dosage of 1000 ng·mL−1, compared with adenine-dosed control rats, and did not affect PTG hyperplasia. An effective dose of this active vitamin D3 (alfarol) for reduction of bone mineral density (BMD) in ovarectomized rats has been estimated to be 300 ng·kg−1·day−1 (Teramura et al., 2002). Oral treatment with this dose for 4 weeks significantly inhibited the trabecular BMD reduction accompanying significant serum Ca elevation. Taken together, it might be difficult in CKD patients to separate worsening vascular calcification from treatments for 2HPT and osteoporosis by active vitamin D3. However, new vitamin D analogues have been reported to suppress iPTH with less risk of cardiovascular calcification (Slatopolsky et al., 2002; Hirata et al., 2003; Slatopolsky et al., 2003), while some undisclosed analogs are still in preclinical (BXL-083, CTAP-201) and clinical developmental (CTA-018, LR-103) stages. It might be meaningful to evaluate the effects of such vitamin D analogs on vascular calcification, hypercalcemia and 2HPT treatment in adenine-dosed rats.
The phosphate binders, sevelamer and CaCO3, lowered serum phosphate to similar degrees in adenine-dosed rats, but the Ca elevation in the CaCO3 group was larger than that in the sevelamer group, so that the Ca × P products were greater in the CaCO3 group than in the sevelamer group. Complete inhibition of active vitamin D3-induced calcification by sevelamer, with only partial inhibition by CaCO3, suggests that the increase in Ca seen with CaCO3 may contribute to the vascular calcification. Moreover, phosphate binders inhibited the iPTH elevation and PTG hyperplasia as well as the aortic calcification, although active vitamin D3 failed to inhibit PTG hyperplasia even in a hypercalcemic dose. Those data suggest that phosphate binders may be more promising than vitamin D recruitment therapy for 2HPT as well as hyperphosphatemia. The problem is that both phosphate binders and vitamin D may elevate serum Ca levels. These data suggest that controlling serum levels of phosphate and Ca with phosphate binders should be tried in CKD patients before vitamin D recruitment, to avoid vascular calcification. Thereafter, if necessary, non-hypercalcemic dosage of vitamin D may be prescribed with serum Ca monitoring.
In conclusion, we established an adenine-induced stable hyperphosphatemia rat model which appeared comparable to stage 4 CKD patients under K/DOQI guidelines. The effects of vitamin D and phosphate binders on hyperphosphatemia, 2HPT and vascular calcification were clearly demonstratable in this rat CKD model. The data suggest that controlling serum phosphate and Ca levels with phosphate binders should be better than vitamin D recruitment in respect to vascular calcification and treatment for hyperphosphatemia and 2HPT in CKD patients. The rat CKD model may be useful to mimic clinical experiences in the treatment of CKD complications and be helpful for assessing the therapeutic potential of newly suggested agents. Finally, the novelty of this study is derives from the fact that animals were dosed orally with adenine in this model, in contrast to other studies, where adenine-enriched diets were used.
Acknowledgments
The authors thank Drs. Edith and Patrick McGeer for their revision of an early draft of this manuscript (Kinsmen Laboratory of Neurological Research, UBC, Vancouver, Canada).
Glossary
Abbreviations:
- 2HPT
secondary hyperparathyroidism
- 5/6Nx
5/6 subtotally nephrectomized
- BMD
bone mineral density
- BUN
blood urea nitrogen
- CKD
chronic kidney disease
- FERP
functional excretion rates of P
- GFR
glomerular filtration rate
- iPTH
intact parathyroid hormone
- MC
methylcellulose
- PTG
parathyroid glands
Conflict of interests
None.
References
- Andress DL. Vitamin D treatment in chronic kidney disease. Semin Dial. 2005;18:315–321. doi: 10.1111/j.1525-139X.2005.18408.x. [DOI] [PubMed] [Google Scholar]
- Blacher J, Guerin AP, Pannier B, Marchais SJ, London GM. Arterial calcifications, arterial stiffness, and cardiovascular risk in end-stage renal disease. Hypertension. 2001;38:938–942. doi: 10.1161/hy1001.096358. [DOI] [PubMed] [Google Scholar]
- Block GA. Prevalence and clinical consequences of elevated Ca × P product in hemodialysis patients. Clin Nephrol. 2000;54:318–324. [PubMed] [Google Scholar]
- Cozzolino M, Dusso AS, Liapis H, Finch J, Lu Y, Burke SK, et al. The effects of sevelamer hydrochloride and calcium carbonate on kidney calcification in uremic rats. J Am Soc Nephrol. 2002;13:2299–2308. doi: 10.1097/01.asn.0000025782.24383.0d. [DOI] [PubMed] [Google Scholar]
- Cozzolino M, Staniforth ME, Liapis H, Finch J, Burke SK, Dusso AS, et al. Sevelamer hydrochloride attenuates kidney and cardiovascular calcifications in long-term experimental uremia. Kidney Int. 2003;64:1653–1661. doi: 10.1046/j.1523-1755.2003.00284.x. [DOI] [PubMed] [Google Scholar]
- Delmez JA, Slatopolsky E. Hyperphosphatemia: its consequences and treatment in patients with chronic renal disease. Am J Kidney Dis. 1992;19:303–317. doi: 10.1016/s0272-6386(12)80446-x. [DOI] [PubMed] [Google Scholar]
- Eknoyan G, Levin A, Levin NW. K/DOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease – foreword. Am J Kidney Dis. 2003;42:S7–S201. [PubMed] [Google Scholar]
- Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am J Kidney Dis. 1998;32:S112–S119. doi: 10.1053/ajkd.1998.v32.pm9820470. [DOI] [PubMed] [Google Scholar]
- Goodman WG, London G, Amann K, Block GA, Giachelli C, Hruska KA, et al. Vascular calcification in chronic kidney disease. Am J Kidney Dis. 2004;43:572–579. doi: 10.1053/j.ajkd.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Haffner D, Hocher B, Muller D, Simon K, Konig K, Richter CM, et al. Systemic cardiovascular disease in uremic rats induced by 1,25(OH)2D3. J Hypertens. 2005;23:1067–1075. doi: 10.1097/01.hjh.0000166849.72721.1c. [DOI] [PubMed] [Google Scholar]
- Henley C, Colloton M, Cattley RC, Shatzen E, Towler DA, Lacey D, et al. 1,25-Dihydroxyvitamin D3 but not cinacalcet HCl (Sensipar/Mimpara) treatment mediates aortic calcification in a rat model of secondary hyperparathyroidism. Nephrol Dial Transplantat. 2005;20:1370–1377. doi: 10.1093/ndt/gfh834. [DOI] [PubMed] [Google Scholar]
- Hirata M, Katsumata K, Endo K, Fulcushima N, Ohkawa H, Fukagawa M. In subtotally nephrectomized rats 22-oxacalcitriol suppresses parathyroid hormone with less risk of cardiovascular calcification or deterioration of residual renal function than 1,25(OH)2 vitamin D3. Nephrol Dial Transplantat. 2003;18:1770–1776. doi: 10.1093/ndt/gfg296. [DOI] [PubMed] [Google Scholar]
- Inagaki O, Nakagawa K, Syono T, Nishian Y, Takenaka Y, Takamitsu Y, et al. Effect of 1,25-dihydroxyvitamin D3 and diltiazem on tissue calcium in uremic rat. Renal Fail. 1995;17:651–657. doi: 10.3109/08860229509037632. [DOI] [PubMed] [Google Scholar]
- Katsumata K, Kusano K, Hirata M, Tsunemi K, Nagano N, Burke SK, et al. Sevelamer hydrochloride prevents ectopic calcification and renal osteodystrophy in chronic renal failure rats. Kidney Int. 2003;64:441–450. doi: 10.1046/j.1523-1755.2003.00126.x. [DOI] [PubMed] [Google Scholar]
- Kestenbaum B. Phosphate metabolism in the setting of chronic kidney disease: significance and recommendations for treatment. Semin Dial. 2007;20:286–294. doi: 10.1111/j.1525-139X.2007.00303.x. [DOI] [PubMed] [Google Scholar]
- Klahr S, Levey AS, Beck GJ, Caggiula AW, Hunsicker L, Kusek JW, et al. The effects of dietary protein restriction and blood-pressure control on the progression of chronic renal disease. N Engl J Med. 1994;330:877–884. doi: 10.1056/NEJM199403313301301. [DOI] [PubMed] [Google Scholar]
- Krog M, Ejerblad S, Eriksson I, Johansson H, Krog M, Ejerblad S, et al. Arterial calcifications in uraemic rats treated with 1-α-hydroxycholecalciferol and parathyroidectomy. Scand J Urol Nephrol. 1984;18:227–239. doi: 10.3109/00365598409180188. [DOI] [PubMed] [Google Scholar]
- Manjunath G, Tighiouart H, Coresh J, MacLeod B, Salem DN, Griffith JL, et al. Level of kidney function as a risk factor for cardiovascular outcomes in the elderly. Kidney Int. 2003;63:1121–1129. doi: 10.1046/j.1523-1755.2003.00838.x. [DOI] [PubMed] [Google Scholar]
- Milliner DS, Zinsmeister AR, Lieberman E, Landing B, Milliner DS, Zinsmeister AR, et al. Soft tissue calcification in pediatric patients with end-stage renal disease. Kidney Int. 1990;38:931–936. doi: 10.1038/ki.1990.293. [DOI] [PubMed] [Google Scholar]
- Nagano N, Miyata S, Abe M, Kobayashi N, Wakita S, Yamashita T, et al. Effect of manipulating serum phosphorus with phosphate binder on circulating PTH and FGF23 in renal failure rats. Kidney Int. 2006;69:531–537. doi: 10.1038/sj.ki.5000020. [DOI] [PubMed] [Google Scholar]
- Neves KR, Graciolli FG, dos Reis LM, Graciolli RG, Neves CL, Magalhaes AO, et al. Vascular calcification: contribution of parathyroid hormone in renal failure. Kidney Int. 2007;71:1262–1270. doi: 10.1038/sj.ki.5002241. [DOI] [PubMed] [Google Scholar]
- Noda-Saita K, Terai K, Iwai A, Tsukamoto M, Shitaka Y, Kawabata S, et al. Exclusive association and simultaneous appearance of congophilic plaques and AT8-positive dystrophic neurites in Tg2576 mice suggest a mechanism of senile plaque formation and progression of neuritic dystrophy in Alzheimer's disease. Acta Neuropathol. 2004;108:435–442. doi: 10.1007/s00401-004-0907-2. [DOI] [PubMed] [Google Scholar]
- Persy V, Postnov A, Neven E, Dams G, De Broe M, D'Haese P, et al. High-resolution X-ray microtomography is a sensitive method to detect vascular calcification in living rats with chronic renal failure. Arterioscler Thromb Vasc Biol. 2006;26:2110–2116. doi: 10.1161/01.ATV.0000236200.02726.f7. [DOI] [PubMed] [Google Scholar]
- Raggi P, Boulay A, Chasan-Taber S, Amin N, Dillon M, Burke SK, et al. Cardiac calcification in adult Hemodialysis patients – A link between end-stage renal disease and cardiovascular disease? J Am Coll Cardiol. 2002;39:695–701. doi: 10.1016/s0735-1097(01)01781-8. [DOI] [PubMed] [Google Scholar]
- Rule AD, Gussak HM, Pond GR, Bergstralh EJ, Stegall MD, Cosio FG, et al. Measured and estimated GFR in healthy potential kidney donors. Am J Kidney Dis. 2004;43:112–119. doi: 10.1053/j.ajkd.2003.09.026. [DOI] [PubMed] [Google Scholar]
- Russo D, Palmiero G, De Blasio AP, Balletta MM, Andreucci VE. Coronary artery calcification in patients with CRF not undergoing dialysis. Am J Kidney Dis. 2004;44:1024–1030. doi: 10.1053/j.ajkd.2004.07.022. [DOI] [PubMed] [Google Scholar]
- Slatopolsky E, Cozzolino M, Finch JL. Differential effects of 19-nor-1,25-(OH) 2D2 and 1α-hydroxyvitamin D2 on calcium and phosphorus in normal and uremic rats. Kidney Int. 2002;62:1277–1284. doi: 10.1111/j.1523-1755.2002.kid573.x. [DOI] [PubMed] [Google Scholar]
- Slatopolsky E, Finch J, Brown A. New vitamin D analogs. Kidney Int Suppl. 2003;85:S83–S87. doi: 10.1046/j.1523-1755.63.s85.20.x. [DOI] [PubMed] [Google Scholar]
- Tamagaki K, Yuan Q, Ohkawa H, Imazeki I, Moriguchi Y, Imai N, et al. Severe hyperparathyroidism with bone abnormalities and metastatic calcification in rats with adenine-induced uraemia. Nephrol Dial Transplant. 2006;21:651–659. doi: 10.1093/ndt/gfi273. [DOI] [PubMed] [Google Scholar]
- Terai K, Mizukami K, Okada M. Comparison of chronic renal failure rats and modification of the preparation protocol as a hyperphosphataemia model. Nephrology. 2008;13:139–146. doi: 10.1111/j.1440-1797.2007.00844.x. [DOI] [PubMed] [Google Scholar]
- Teramura K, Fukushima S, Iwai T, Nozaki K, Kokubo S, Takahashi K, et al. Incadronate inhibits osteoporosis in ovariectomized rats. Eur J Pharmacol. 2002;457:51–56. doi: 10.1016/s0014-2999(02)02659-6. [DOI] [PubMed] [Google Scholar]
- Uribarri J. Phosphorus homeostasis in normal health and in chronic kidney disease patients with special emphasis on dietary phosphorus intake. Semin Dial. 2007;20:295–301. doi: 10.1111/j.1525-139X.2007.00309.x. [DOI] [PubMed] [Google Scholar]
- Winchester JF, Rotellar C, Goggins M, Robino D, Rakowski TA, Argy WP. Calcium and phosphate balance in dialysis patients. Kidney Int Suppl. 1993;41:S174–S178. [PubMed] [Google Scholar]