

Abstract
The therapeutic potential for manipulation of glucocorticoid metabolism in cardiovascular disease was revolutionized by the recognition that access of glucocorticoids to their receptors is regulated in a tissue-specific manner by the isozymes of 11β-hydroxysteroid dehydrogenase. Selective inhibitors of 11β-hydroxysteroid dehydrogenase type 1 have been shown recently to ameliorate cardiovascular risk factors and inhibit the development of atherosclerosis. This article addresses the possibility that inhibition of 11β-hydroxsteroid dehydrogenase type 1 activity in cells of the cardiovascular system contributes to this beneficial action. The link between glucocorticoids and cardiovascular disease is complex as glucocorticoid excess is linked with increased cardiovascular events but glucocorticoid administration can reduce atherogenesis and restenosis in animal models. There is considerable evidence that glucocorticoids can interact directly with cells of the cardiovascular system to alter their function and structure and the inflammatory response to injury. These actions may be regulated by glucocorticoid and/or mineralocorticoid receptors but are also dependent on the 11β-hydroxysteroid dehydrogenases which may be expressed in cardiac, vascular (endothelial, smooth muscle) and inflammatory (macrophages, neutrophils) cells. The activity of 11β-hydroxysteroid dehydrogenases in these cells is dependent upon differentiation state, the action of pro-inflammaotory cytokines and the influence of endogenous inhibitors (oxysterols, bile acids). Further investigations are required to clarify the link between glucocorticoid excess and cardiovascular events and to determine the mechanism through which glucocorticoid treatment inhibits atherosclerosis/restenosis. This will provide greater insights into the potential benefit of selective 11β-hydroxysteroid dehydrogenase inhibitors in treatment of cardiovascular disease.
Keywords: glucocorticoids, cardiovascular disease, 11β-hydroxysteroid dehydrogenase, selective inhibition
Introduction
Glucocorticoids have complex, and often contradictory, influences on cardiovascular disease and cardiovascular risk (Walker, 2007a). Systemic glucocorticoid excess, caused by either increased secretion of endogenous steroid or by chronic exogenous treatment, is associated with increased cardiovascular risk. In contrast, the well-established anti-inflammatory, anti-proliferative and anti-migratory properties of glucocorticoids have led to their investigation as possible therapeutic inhibitors of atherosclerosis and restenosis following percutaneous coronary intervention (Hadoke et al., 2006).
Research performed over the past 20 years has considerably improved understanding of the physiological regulation of glucocorticoid activity. Of key importance has been the demonstration that receptor activation in target tissues is not determined solely by circulating glucocorticoids but also by intra-cellular, pre-receptor inter-conversion of active and inactive forms of the steroid. This pre-receptor metabolism is catalysed by the two isozymes of 11β-hydroxysteroid dehydrogenase (11-HSD). Identification of this mechanism for local regulation of glucocorticoid action has prompted the concept that tissue-specific glucocorticoid excess and deficiency, in the face of normal circulating concentrations, may contribute to disease pathogenesis (Seckl and Walker, 2001). Furthermore, the presence of tissue-specific mechanisms for regulation of glucocorticoid activity suggests promising new therapeutic targets in a variety of conditions, and has prompted a drive by pharmaceutical companies to produce selective inhibitors of the 11-HSD isozymes (Webster and Pallin, 2007). There is already considerable evidence that selective inhibitors of 11-HSD1 reductase can ameliorate risk factors strongly associated with cardiovascular disease (type 2 diabetes mellitus, obesity, high blood pressure, dyslipidaemia) and this has been the subject of recent reviews (Walker, 2007a; Wamil and Seckl, 2007; Webster and Pallin, 2007). In addition to these systemic effects, it is possible that manipulation of glucocorticoid generation has direct effects in target tissues as both isozymes of 11-HSD are expressed in the heart and blood vessel wall (Walker et al., 1991; Hadoke et al., 2001). Indeed, recent data suggest that selective 11-HSD1 inhibition may reduce atherosclerotic lesion formation by direct interaction with the arterial wall (Hermanowski-Vosatka et al., 2005) although the mechanisms responsible for this action remain obscure. The role of interactions between 11-HSDs, glucocorticoids and cells of the heart and vascular wall in the development of cardiovascular disease (in humans or animals) has not been clearly established. Therefore, this article will review the evidence that glucocorticoids influence cardiovascular disease by direct interaction with the heart and vascular wall and will evaluate the potential for systemic and targeted manipulation of glucocorticoid activity for the treatment of cardiovascular disease.
Glucocorticoids: systemic generation, regulation and action
Generation and metabolism
Glucocorticoids are stress hormones with a vital role in regulation of metabolic and defence responses. Their generation from cholesterol (Figure 1A), which occurs in the zonae fasciculata and reticularis of the adrenal cortex, is tightly regulated by the hypothalamic-pituitary-adrenal (HPA) axis with glucocorticoids regulating their own generation by negative feedback inhibition on several components of the axis. Under this control, glucocorticoids are produced de novo and released into the blood as required, with a clear circadian rhythm producing peak blood concentrations in the early morning diminishing to a nadir in the evening (Dallman et al., 1993). Cortisol is the major glucocorticoid in man, whereas in rodents, which lack the enzyme 17α-hydroxylase in the adrenal, corticosterone predominates. On secretion into the blood, most (90–95%) glucocorticoids are sequestered to corticosteroid-binding globulin and albumin with only the unbound fraction available to interact with their receptors (Hammond et al., 1990). Metabolic inactivation of glucocorticoids occurs predominantly in the liver, and also in the kidney, with inactive metabolites excreted in the urine. This involves a complex modification process (Figure 1B) in which glucocorticoids [and their 11-keto-metabolites (cortisone, 11-dehydrocorticosterone)] are reduced in a pathway involving 5α- and 5β-reductases, 3α-hydroxysteroid dehydrogenase, 20α- and 20β-hydroxysteroid dehydrogenases and 21-oxidase followed by conjugation with glucuronic acid or sulphates. The 11-keto metabolites are biologically inert at the glucocorticoid receptor (GR) but have been shown to attenuate the response to aldosterone (Odermatt et al., 2001). In addition, some products of glucocorticoid metabolism (e.g. 5α-terahydrocorticosterone) can activate GRs (McInnes et al., 2004).
Figure 1.

Strategies for pharmacological manipulation of glucocorticoid activity. (A) Therapeutic manipulation of glucocorticoid generation and action. Synthesis of active glucocorticoids (predominantly cortisol in man and corticosterone in rodents) can be targeted by inhibiting key enzymes in the pathway. Action of glucocorticoids on corticosteroid receptors can be blocked using selective antagonists of glucocorticoid (GR) and mineralocorticoid (MR) receptors. (B) Manipulation of glucocorticoid metabolism. Glucocorticoid generation in target tissues can be targeted using inhibitors of 11β-hydroxysteroid dehydrogenase (11-HSD) which interconverts active steroid and its inert 11-keto metabolite. Clearance of glucocorticoids by 5α-reductase is also inhibited by compounds used to inhibit conversion of testosterone into dihydrotestosterone.
Receptor activation
Glucocorticoids are ligands both for high affinity type I [or mineralocorticoid receptors (MR)] and for low affinity type II (or GRs) corticosteroid receptors (Figure 1A) which are members of the nuclear receptor superfamily of ligand-activated transcription factors [nomenclature conforms with the BJP's guide to Receptors and Channels (Alexander et al., 2008)]. These are predominantly intra-cellular receptors: although there is increasing evidence for membrane-bound versions on the cell surface (Bartholome et al., 2004) their physiological relevance has not been established. GR may exist in two forms – GRα, which has a high affinity for glucocorticoids and is expressed throughout the body, and GRβ (Solakidi et al., 2007) – which does not bind traditional GR agonists, acts as a dominant-negative inhibitor of GRα, is expressed in humans [but not animals (Otto et al., 1997) – except perhaps the zebrafish (Schaaf et al., 2008)] and has no known function in vivo. In contrast to GRα, MR are expressed in relatively few tissues. MR in extra-renal rat tissues bind aldosterone and corticosterone with similar affinities (Krozowski and Funder, 1983) while human MR (Kd ∼1 nmol·L−1) has (10–40 fold) higher affinity for cortisol than GR (Arriza et al., 1987). Thus, the selectivity shown by MR for aldosterone over cortisol (which is present in the plasma in concentrations 100–1000 times higher than aldosterone) in vivo is largely dependent on pre-receptor metabolism of glucocorticoids by 11-HSD type 2 [(Stewart and Krozowski, 1999); see below], although other processes also have a role (Funder and Myles, 1996). Consequently, the cellular response to glucocorticoids will depend upon whether the target tissue expresses GR and/or MR and/or the isozymes of 11-HSD [discussed in (Walker, 2007b)]. Glucocorticoids bind to cytoplasmic GR after entering the cell (probably via passive diffusion), prompting dissociation of key heat shock proteins, receptor dimerization and translocation to the nucleus. Receptor dimers then bind to glucocorticoid response elements in target genes leading to alterations (induction or inhibition) in transcription which ultimately result in the appropriate physiological response. In addition, GR may interact with other factors which modify gene transcription and rapid, receptor-mediated, non-genomic actions of glucocorticoids have also been reported, resulting from initiation of signal transduction within the cytosol (Hafezi-Moghadam et al., 2002).
The main actions of glucocorticoids mediated by GR stimulation are: regulation of carbohydrate and protein metabolism, negative feedback on the HPA axis, and anti-inflammatory and immunosuppressive effects. In addition, it is recognized that glucocorticoid activity also influences the cardiovascular system (Figure 2). In healthy individuals, glucocorticoids are required for blood pressure maintenance (Ullian, 1999) although the mechanisms involved are complex and incompletely understood. It is likely that several distinct interactions contribute to this activity, including regulation of: renal electrolyte and water homeostasis [by effects on glomerular filtration rate, proximal tubular epithelial sodium transport and free water clearance (Montrella-Waybill et al., 1991)]; intravascular volume [by increased generation of angiotensinogen, arginine vasopressin (Raff, 1987) and atrial natriuretic peptide from the liver, hypothalamus and cardiac myocytes (Shields et al., 1988), respectively]; and vascular contractility (Ullian, 1999).
Figure 2.
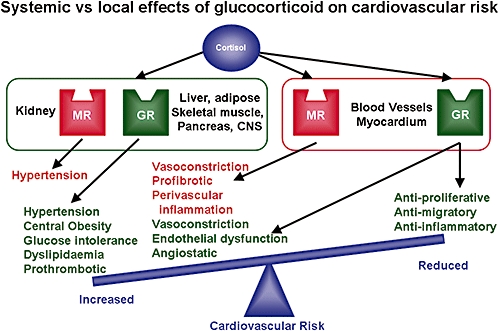
Systemic vs. local effects of glucocorticoids on the cardiovascular system. Systemic actions of glucocorticoids are associated with increased cardiovascular risk and are likely to promote cardiovascular disease development. Local effects on cells of the cardiovascular system may be mediated by glucocorticoid (GR) and/or mineralocorticoid (MR) receptors and could be predicted either to promote or oppose lesion development.
Tissue-specific metabolism of glucocorticoids by 11β-hydroxysteroid dehydrogenases
A major shift in understanding of the physiological regulation of glucocorticoid activity came with the demonstration that 11-HSD, which was originally described more than 50 years ago (Amelung et al., 1953; Hubener et al., 1956), did not simply provide yet another mechanism for glucocorticoid clearance. Rather, it was shown that 11-HSD activity was essential for maintaining aldosterone selectivity of MR (Edwards et al., 1988; Funder et al., 1988). This observation has prompted a re-evaluation of the processes regulating glucocorticoid activity and changed the concept of glucocorticoid excess (Seckl and Walker, 2001).
The isozymes of 11-HSD
Two isozymes of 11-HSD, type 1 and type 2 (Figure 1B), have now been identified, both of which are microsomal enzymes of the short-chain alcohol dehydrogenase superfamily (Stewart and Krozowski, 1999) and catalyse the inter-conversion of active glucocorticoids and their inert 11-keto forms (Amelung et al., 1953). 11-HSD type 1 (11-HSD1) is a low affinity NADP(H)-dependent enzyme which acts predominantly as a reductase in vivo converting cortisone to cortisol (or 11-dehydrocorticosterone to corticosterone). Intact cells or organs [including liver (Jamieson et al., 1995; 2000; Ricketts et al., 1998), adipose tissue (Bujalska et al., 1997), neurones (Rajan et al., 1996) and vascular smooth muscle (Hatakeyama et al., 1999)] generally do not exhibit the dehydrogenase activity of this isozyme [although some tissues, such as testis, do exibit 11-HSD1 which functions as a dehydrogenase (Gao et al., 1997)]. Indeed, it seems likely that early suggestions of 11-HSD1 dehydrogenase activity in vascular smooth muscle (Brem et al., 1995) are attributable to 11-HSD2 (Hatakeyama et al., 1999) while in other in vitro preparations dehydrogenase activity may be explained by release of the enzyme from damaged or dying cells (Monder and Lakshmi, 1989). The latter would result in release of 11-HSD1 from the intra-cellular environment, alteration of co-factor and substrate availability and change in redox potential: all of which may be important in driving the enzyme in the reductase direction. For example, dissociation from hexose-6-phosphate dehydrogenase may be important as this enzyme is thought to generate the high nicotinamide adenine dinucleotide phosphate (NADPH) concentrations required for reductase activity (Atanasov et al., 2004). 11-HSD1, which has a Km in the µmol·L−1 range for both cortisol and corticosterone (Lakshmi and Monder, 1988), is widely expressed in many glucocorticoid-target tissues [including: liver, lung, adipose tissue, brain, vascular smooth muscle, skeletal muscle, anterior pituitary, gonads and adrenal cortex (Stewart and Krozowski, 1999)] where it amplifies local glucocorticoid concentrations (Seckl and Walker, 2001). Its synthesis and activity are regulated by a complex interaction of many factors, including: glucocorticoids (Hammami and Siiteri, 1991; Low et al., 1994a; Takeda et al., 1994), stress (Walker et al., 1994; Jamieson et al., 1997), sex steroids (Low et al., 1994b), growth hormone (Painson et al., 1992), cytokines (Cai et al., 2001) and peroxisome proliferator activated receptor agonists (PPAR) (Tomlinson et al., 2004).
In contrast to 11-HSD1, 11-HSD2 is a high affinity nicotinamide adenine dinucleotide dependent, exclusive dehydrogenase and converts active glucocorticoids into inactive 11-ketosteroids. It has a Km for cortisol and corticosterone in the nmol·L−1 range and is expressed constitutively, mainly in mineralocorticoid target tissues [kidney, sweat glands, salivary glands and colon (Stewart and Krozowski, 1999)] where it protects MR from illicit occupation by glucocorticoids. 11-HSD2 inhibition (using liquorice or its derivatives) [reviewed in (Walker and Edwards, 1994)], transgenic disruption in mice (Kotelevtsev et al., 1999) or congenital deficiency in man (Walker et al., 1992), produces the glucocorticoid-dependent syndrome of ‘apparent’ mineralocorticoid excess (SAME), in which inappropriate activation of MR by glucocorticoids results in characteristic sodium retention, hypokalaemia and hypertension (Walker and Edwards, 1994). It has also been noted that 11-HSD2 is expressed in tissues (such as lung, lymph nodes, heart, blood vessel wall and placenta) which are not classic MR targets (Stewart et al., 1995; Slight et al., 1996; Waddell et al., 1998). In the placenta 11-HSD2 protects the fetus from exposure to maternal glucocorticoids (Murphy et al., 1974; Brown et al., 1996) while in the heart it may be important in preventing glucocorticoid-dependent, MR-mediated fibrosis (Konishi et al., 2003).
Tissue-specific glucocorticoid excess/deficiency
The presence of 11-HSD-mediated pre-receptor metabolism of glucocorticoids in target tissues has highlighted the possibility of tissue-specific glucocorticoid excess (or deficiency) in the presence of normal circulating concentrations of the enzyme (Wamil and Seckl, 2007). Consequently, altered activity of 11-HSD isozymes in adipose tissue, liver, skeletal muscle and the brain have been linked to diabetes mellitus, the metabolic syndrome and cognitive dysfunction [reviewed in (Wamil and Seckl, 2007)]. The association of some of these conditions with hypertension and atherosclerosis has suggested a role for 11-HSD activity in the development of cardiovascular disease (Walker, 2007a). Whether this role is linked to regulation of systemic risk factors or to direct influence of 11-HSD activity on regulation of the cells of the heart and vascular wall has yet to be established.
Pharmacological manipulation of systemic glucocorticoid activity
Therapeutic glucocorticoid administration
Therapeutic approaches to manipulating glucocorticoids include both direct steroid administration and pharmacological manipulation of synthetic (Figure 1A) and excretory (Figure 1B) pathways. Clinically, glucocorticoid administration (often using hydrocortisone, the synthetic form of cortisol) is used predominantly to provide physiologic replacement in glucocorticoid deficiency and, in higher doses, as an anti-inflammatory (to suppress various inflammatory, allergic and autoimmune disorders) or immunosuppressant (to prevent graft rejection following transplant). The use of endogenous glucocorticoids, or their metabolites, has been supplemented by the development of synthetic compounds with greater potency, a higher degree of receptor selectivity and improved bioavailability. For example, prednisolone and methyl prednisolone have higher (3–8 fold) selectivity than cortisol for GR and have longer biological half-lives (16–40 h compared with 2–8 h for cortisol). Dexamethasone and betamethasone have even better selectivity (25–80 times) and longer biological half-lives (36–54 h). Synthetic steroids also vary in the susceptibility to metabolism by 11-HSDs for, while both prednisolone and dexamethasone are substrates for these isozymes, dexamethasone is relatively protected from dehydrogenation (Best et al., 1997). It should be noted that treatments often use inactive precursor molecules (such as cortisone or prednisone) which require conversion to the active steroid. Replacement therapy, which usually uses glucocorticoids with both GR and MR activity (cortisol), is limited by their high bioavailability and short half-life (∼90 min) and, consequently, cannot replicate physiological circulating concentrations (and cannot replicate the diurnal rhythm of cortisol secretion which peaks before waking). As a result, doses used tend to produce supra-physiological concentrations in the first 1–2 h after administration. In contrast to physiologic replacement, anti-inflammatory therapy tends to use GR selective compounds (such as prednisolone). The anti-angiogenic properties of glucocorticoids are also harnessed clinically in the treatment of vascular lesions such as proliferating capillary haemangiomas (Hasan et al., 2000; 2003). The ubiquitous expression of GR limits the therapeutic use of glucocorticoids by mediating a variety of serious systemic side effects (including: immunosuppression; osteoporosis; pubertal delay; central obesity; hypertension; anovulation). These can be reduced by targeting therapy (e.g. topical administration for skin conditions; inhalation for treatment of asthma). Finally, dexamethasone is also used clinically, in the dexamethasone suppression test, to assess HPA axis function (Swade et al., 1987).
Pharmacological manipulation of glucocorticoid activity
In addition to therapeutic administration, pharmacological approaches have been used clinically to regulate the action of endogenous glucocorticoids. These have involved two main strategies: (i) modulation of glucocorticoid synthesis; and (ii) corticosteroid receptor antagonism (Figure 1A). Generation of glucocorticoids can be manipulated pharmacologically by inhibiting different steps in the synthetic pathway using aminoglutethimide [which inhibits 11β-hydroxylase, 17α-hydroxylase and conversion of cholesterol to pregnenalone (Brodie, 1993)]; or high dose ketoconazole [which has similar actions to aminoglutethimide and is also a GR antagonist (Sonino, 1987)]. More selective approaches include inhibition of 3β-dehydrogenase (using trilostane) or 11β-hydroxylase [metyrapone (Miller and Crapo, 1993)]. Receptor blockade can be achieved using the potent GR (and progesterone receptor) antagonist, Mifepristone [RU38486 (Spitz and Bardin, 1993); which is used primarily to induce termination of pregnancy] or the MR antagonists spironolactone and (the more selective) eplerenone (Rabasseda et al., 1999). Inhibition of glucocorticoid synthesis and GR blockade have been used clinically in the treatment of Cushing's syndrome (Engelhardt and Weber, 1994) but are limited by their tendency to alter beneficial, as well as deleterious actions of glucocorticoid; leading to major side effects and compensatory changes in cortisol generation (returning cortisol to pre-treatment levels). Systemic regulation of cortisol synthesis remains a pharmacological target, however, with phase IIb trials scheduled for this year involving DiObex 2S, 4R ketoconazole (DIO-902; one of two enantiomers contained in ketoconazole) which may be safer and more effective than racemic ketoconazole (http://www.diobex.com/product902.html). Finally, glucocorticoid availability can be influenced by manipulation of steroid breakdown. For example, non-selective (dutasteride) and type 2 selective (finasteride) 5α-reductase inhibitors (Figure 1B), which are used in the treatment of benign prostatic hyperplasia and male pattern baldness (Metcalf et al., 1989), may also inhibit metabolic clearance of glucocorticoids. The most significant development in this area, however, has been the recent drive to produce selective inhibitors of the 11-HSD isozymes (see below), which has considerable potential in the treatment of cardiovascular disease pathogenesis.
Influence of glucocorticoids on the cardiovascular system
For glucocorticoids to contribute to cardiovascular disease they must directly influence the function of the heart and vasculature and/or increase cardiovascular risk factors (Figure 2). Evidence that this is indeed the case is perhaps most clearly indicated by the increased cardiovascular risk factors (elevated blood pressure, central obesity, dyslipidaemia, insulin resistance) in patients with excessive production of these steroids (Cushing's syndrome). It seems likely that much of the impact of glucocorticoids on cardiovascular risk is due to interaction with the kidney, liver, adipose and central nervous system (Bjorntorp, 1991). However, while the influence of glucocorticoids on homeostasis is probably due predominantly to renal sodium retention and intravascular volume overload there is also evidence for additional, non-renal mechanisms. Notably, glucocorticoids increase peripheral vascular resistance in animals devoid of renal mass (Langford and Snavely, 1959). This is consistent with the observation that glucocorticoids can interact directly with the cells of the heart and vascular wall to alter their structure and function.
Cardiac and vascular function
Direct modulation of the heart and vasculature by glucocorticoids is, obviously, dependent on the presence of the appropriate receptors and enzymes. MR and GR are present in intact arteries (Kornel et al., 1982; Christy et al., 2003), cultured vascular smooth muscle (VSMC) (Meyer and Nicholls, 1981; Scott et al., 1987) and endothelial (EC) (Inoue et al., 1999; Jun et al., 1999; Golestaneh et al., 2001; Newton et al., 2002; Oberleithner et al., 2003) cells from several different species. Their distribution may be territory-dependent, however, as MR have been detected in rabbit aortic and pulmonary VSMCs but not in small arteries (Lombes et al., 1992). Vascular GR and MR have both been shown to be active: dexamethasone-mediated induction of angiotensin converting enzyme (ACE) activity in rat aortic ECs (Sugiyama et al., 2005), cortisol-mediated inhibition of prostacyclin synthesis in rat aorta (Jeremy and Dandona, 1986) and dexamethasone or cortisol-mediated increases in protein kinase C in porcine coronary artery are all sensitive to GR antagonism (Maddali et al., 2005). Similarly, exposure to angiotensin II and aldosterone induces hypertrophy of VSMCs (Hatakeyama et al., 1994a) and swelling of ECs (Oberleithner et al., 2003) respectively. Whether membrane binding sites for corticosteroids are present, or have a role, in the vascular wall has not been established. The nature of the interaction between glucocorticoids and vascular cells may be complex as prolonged exposure inhibits proliferation of cultured vascular smooth muscle cells whereas short exposures (2 min-6 h) can stimulate a GR-dependent increase in proliferation [probably by stimulation of autocrine growth factor release (Kawai et al., 1998)]. GR (Pujols et al., 2002) and MR (Lombes et al., 1995) are both also expressed in the myocardium, with co-expression of MR with 11-HSD2 (Konishi et al., 2003) ensuring mineralocorticoid selectivity. Their relationship to cardiac function has been demonstrated by conditional GR over-expression in the heart which induces atrio-ventricular (AV) block (Sainte-Marie et al., 2007). Similarly, MR knockdown induces severe heart failure and fibrosis (without hypertension or hyperaldosteronism) (Beggah et al., 2002) while mice over-expressing cardiac MR develop ventricular arrhythmias (Ouvrard-Pascaud et al., 2005).
The influence of glucocorticoid exposure on the heart and vascular wall is still controversial despite many reports of glucocorticoid-mediated changes in function and structure. Indeed, many early in vitro investigations must be discounted for using inappropriately high concentrations of steroid and short exposure times [reviewed in Walker and Williams (1992)]. In man, topical administration of glucocorticoids induces dermal vasoconstriction (Walker et al., 1992) although the precise mechanism of this response remains unclear. It is widely accepted that glucocorticoid exposure potentiates contractile responses to noradrenaline and angiotensin II, although whether this is due to alterations within the VSMC or EC has not been established [reviewed in Ullian (1999) and Hadoke et al. (2006)]. In VSMCs glucocorticoids have been shown to up-regulate contractile receptors, alter intracellular second messenger activation and modulate the activity and synthesis of vasoactive substances leading to a direct enhancement of contraction. Increased contractility has also been attributed to changes in the endothelium but it is not clear whether this is due to: (i) increased release of endothelium-derived vasoconstrictors [such as angiotensin II or endothelin-1 (Mendelsohn et al., 1982; Morin et al., 1998)] and/or, (ii) impaired endothelium-dependent relaxation (Mangos et al., 2000) due to impaired vasodilator (e.g. prostaglandins, nitric oxide) activity (Gerritsen and Rosenbaum, 1985; Simmons et al., 1996; Wallerath et al., 1999). Functional modulations of the vasculature may occur through stimulation of either GR or MR (Nagata and Hirata, 2007) as they have been reported with GR-(dexamethasone) and MR-(aldosterone) selective ligands.
In the heart, glucocorticoids may help maintain normal contractile function. Adrenalectomy results in a decreased contractile force generation in rat papillary muscles which can be prevented by treatment with dexamethasone (Lefer, 1968) which may act by modulating membrane Ca2+ transport (Whitehurst, Jr et al., 1999; Narayanan et al., 2004) and activity of K+ channels (Lefer, 1968; Penefsky and Kahn, 1971; Wang et al., 1999). Similarly, dexamethasone enhances the development of contractile tension and increases contraction and relaxation velocities in cardiac muscles (Penefsky and Kahn, 1971) although short-term administration of this compound has also been shown to decrease resting heart rate in healthy human volunteers (Brotman et al., 2005). This influence of glucocorticoids on cardiac function is supported by observations from mice with over-expression of cardiac GR which have reduced heart rate and depressed cardiac conduction with AV block (Sainte-Marie et al., 2007). The role of MR in regulating cardiac function is more ambiguous although recent evidence suggests MR are necessary for mediating corticosteroid-induced up-regulation of the cardiac calcium current (Rougier et al., 2008).
Cardiovascular remodelling
While their ability to influence cardiovascular function is imperfectly understood, glucocorticoid-induced changes in vascular structure and growth appear more straightforward. In general glucocorticoids inhibit tube formation by endothelial cells (Nicosia and Ottinetti, 1990) and migration and proliferation of vascular smooth muscle cells (Longenecker et al., 1982; 1984; Berk et al., 1988); a characteristic that has been exploited in attempts to inhibit neointimal proliferation (see below). To complicate matters, however, the direct inhibition of smooth muscle cell growth may be countered by the ability of both glucocorticoids and mineralocorticoids to stimulate proliferation in these cells by potentiating the action of other hormones [see Ullian (1999)]. MR may have a role in this process as MR antagonism attenuated angiotensin II mediated hypertrophy of smooth muscle cells (Hatakeyama et al., 1994b). Thus, the impact of glucocorticoids in vivo may reflect a balance between direct inhibition of hypertrophy, hyperplasia and migration of smooth muscle cells countered by indirect stimulation of hypertrophy and hyperplasia mediated through other factors. This process may involve both MR and GR but surprisingly few studies have directly addressed the role of these receptors in mediating corticosteroid-mediated changes in migration and proliferation of vascular smooth muscle.
The ability of glucocorticoids to alter vascular remodelling is exemplified in their inhibition of angiogenesis; a property first demonstrated by Folkman 25 years ago (Folkman et al., 1983) and extended to include several steroids without classical glucocorticoid activity (Crum et al., 1985; Folkman and Ingber, 1987). This work suggested that GR activation is not required for inhibition of new vessel growth but our own recent investigations have shown that corticosterone-mediated inhibition of angiogenesis in mouse aorta and sub-cutaneous sponge implants (Small et al., 2005) is abolished by GR-receptor antagonism. The mechanism responsible for inhibition of angiogenesis has not been established, despite extensive research over many years. As inflammation plays a significant role in stimulation of angiogenesis (Risau, 1997), it is often difficult to distinguish immunosuppressive and anti-inflammatory effects of glucocorticoids from direct effects on remodelling of the vascular wall. However, the ability of glucocorticoids to inhibit the formation of tube-like structures in vitro, in isolation from the immune system (Nicosia and Ottinetti, 1990; Small et al., 2005), suggests that direct interaction with the vascular wall does have a role. The role of glucocorticoid-mediated inhibition of angiogenesis in man has not been established and may be complex as endocrine [glucocorticoid- (Cushing's syndrome) and mineralocorticoid (primary aldosteronism)-induced] hypertension is associated with increased circulating levels of the pro-angiogenic vascular endothelial growth factor (Zacharieva et al., 2004).
In the heart, there is evidence from experimental and human studies indicating that glucocorticoid treatment is harmful, leading to cardiomyopathy (Zecca et al., 2001; Mitsuya et al., 2004) characterized by an accumulation of lipid droplets, cardiomyocyte hypertrophy and dissolution of myofibrils (Clark et al., 1982; de Vries et al., 2002). In the adult, dexamethasone treatment can induce hypertrophy and precocious degeneration of cardiomyocytes (de Vries et al., 2002) while glucocorticoid exposure in neonates can induce myocardial hypertrophy and changes in contractile proteins (Werner et al., 1992; La Mear et al., 1997). The mechanisms involved are not entirely understood but it has been suggested that cardiomyocyte hypertrophy up-regulates GR and MR expression and allows corticosteroid-mediated potentiation of α-adrenoceptor-mediated signalling (Lister et al., 2006). Dexamethasone also increases ACE activity (Barreto-Chaves et al., 2000) which can induce myocardial fibrosis and heart failure via direct and indirect mechanisms. The role of GR in modulating cardiac remodelling is questioned, however, by the demonstration that over-expression of cardiac GR does not induce major ventricular hypertrophy or ventricular arrhythmias (Sainte-Marie et al., 2007). MR activation, in contrast, is clearly associated with cardiac remodelling. Mineralocorticoids have potent pro-fibrotic effects, in vitro (Neumann et al., 2002; Stockand and Meszaros, 2003) and in vivo (Brilla et al., 1990a; 1993), and can promote oxidative stress (Sun et al., 2002). Aldosterone can also amplify the action of angiotensin II by increasing AT1 receptor density (Robert et al., 1999) and ACE activity (Harada et al., 2001) leading to cardiac fibrosis (Brilla et al., 1990b; McEwan et al., 1998; Ramires et al., 1998; Lijnen and Petrov, 2000; Lijnen et al., 2000). Blockade of MR (eplerenone) attenuates ventricular hypertrophy, ventricular fibrosis, myocardial stiffening and relaxation abnormalities (Ohtani et al., 2007) while clinical studies report beneficial effects of MR antagonists on mortality in heart failure patients (Pitt et al., 1999; 2003). In addition, mice with over-expression of cardiac MR die in the embryonic and perinatal period secondary to severe ventricular arrhythmia without high-degree AV block (Ouvrard-Pascaud et al., 2005).
In addition to direct modulation of remodelling by cardiac and vascular cells, glucocorticoids may also alter structural changes in the cardiovascular system by regulating the inflammatory response to injury. It is becoming apparent, however, that the model of glucocorticoids as inhibitors of inflammation may be simplistic as, depending on circumstances [including glucocorticoid concentration (Lim et al., 2007)], they may inhibit or stimulate the inflammatory response [reviewed in Yeager et al. (2004)]. Much of this anti-inflammatory activity may be due to direct interaction with inflammatory cells. Glucocorticoids can influence a variety of GR-dependent functions of macrophages, lymphocytes, eosinophils and neutrophils (Heasman et al., 2003), including apoptosis, phagocytosis, adhesion molecule expression and expression of inflammatory genes [reviewed in Valledor and Ricote (2004)]. For example, glucocorticoids inhibit up-regulation of adhesion molecules on lymphocytes by stimulation of GR (blocked by RU38486) and also by non-genomic mechanisms [reviewed in Pitzalis et al. (2002)]. In macrophages both GR and MR are expressed but the action of glucocorticoids on these cells appears to be solely dependent on GR stimulation (Lim et al., 2007). In addition, GR- (Wheller and Perretti, 1997) and MR- (Caprio et al., 2008) dependent mechanisms also regulate induction of adhesion molecules in the vascular endothelium and, thus, suppress passage of neutrophils into the vessel wall. The role of these interactions is discussed further in relation to cardiovascular disease (below).
Finally, glucocorticoids may also influence the cardiovascular system by modulating coagulation of the blood both by direct modulation of clotting factors and by inhibition of anti-thrombotic pathways in the endothelium (Yamamoto et al., 2004). In vitro evidence suggests that glucocorticoids may activate haemostasis and inhibit thrombolysis, thereby increasing the likelihood of clot formation. Whether this is significant in vivo may depend on dose and duration of treatment (Brotman et al., 2006).
Prenatal programming of the cardiovascular system
A more esoteric mechanism through which glucocorticoids may alter the function and structure of the cardiovascular system is the process of prenatal ‘programming’. This is based on the increasing evidence that exposure to glucocorticoid excess in utero can induce low birth weight and programme the development of increased cardiovascular risk in later life (Seckl and Meaney, 2004). Experimental fetal exposure to excess maternal glucocorticoid (by direct administration or by inhibition of placental 11-HSD2) leads to reduced birth weight (Benediktsson et al., 1993) which is associated with increased risk of cardiovascular and metabolic disease in adulthood (Barker et al., 1990). Indeed, maternal dietary restriction and maternal stress, both major causes of low birth weight, may act by modulation of glucocorticoid activity (Woodall et al., 1996; Lesage et al., 2001). Whether development of cardiovascular risk factors, such as hypertension, in adulthood (Dodic et al., 1998) is due to programmed changes in the vasculature itself has not been established but may be related to the ability of glucocorticoids to increase blood pressure and alter vascular function in the fetus (Gao et al., 1996). However, although altered vascular structure and enhanced contractility are evident in rats (Lamireau et al., 2002; Khan et al., 2005) and sheep (Roghair et al., 2005) with programmed hypertension [as is left ventricular hypertrophy and reduced cardiac functional reserve (Dodic et al., 2001)], it is not clear whether this contributes to (or is a consequence of) elevated blood pressure.
Cardiovascular 11-HSDs
Whatever the influence of glucocorticoids on the heart and vascular wall, these interactions are liable to modification by pre-receptor metabolism as both isozymes of 11-HSD are expressed in cardiac (Walker et al., 1991; Lombes et al., 1995; Slight et al., 1996) and vascular cells (Ullian, 1999; Alzamora et al., 2000). Evidence suggests that 11-HSD2 may be the predominant isozyme in cardiac cells (Slight et al., 1996) whereas 11-HSD1 is the most active isozyme in the arterial wall (Figure 3; Walker et al., 1991; Christy et al., 2003). The cellular localization of 11-HSD isozymes in cardiovascular tissues is not clear. There are several reports of both enzymes in the VSMC (Hatakeyama et al., 1999; Cai et al., 2001) and also in the EC (Brem et al., 1998): our own studies have suggested that (in the mouse and rat aorta) 11-HSD2 is localized to ECs whereas 11-HSD1 is predominantly in the VSMC (Walker et al., 1991; Christy et al., 2003). It is often difficult to compare studies, however, as there is variation in arteries studied (species, anatomical origin) and analytical techniques employed [e.g. in contrast to our work in intact arteries, 11-HSD2 activity was not detected in human umbilical vein ECs (Schleimer, 1991)]. This is significant as the cellular distribution of 11-HSDs differs between vascular territories and 11-HSD activity may increase as arterial diameter decreases (Walker et al., 1991). Interpretation of studies performed in culture is also difficult as the expression and activity of 11-HSDs appears to be regulated by cell proliferation and passage number (Dover et al., 2007). Despite these uncertainties, it is becoming increasingly evident that 11-HSD activity is an important determinant of glucocorticoid-mediated modulation of vascular function, structure, growth and inflammation.
Figure 3.
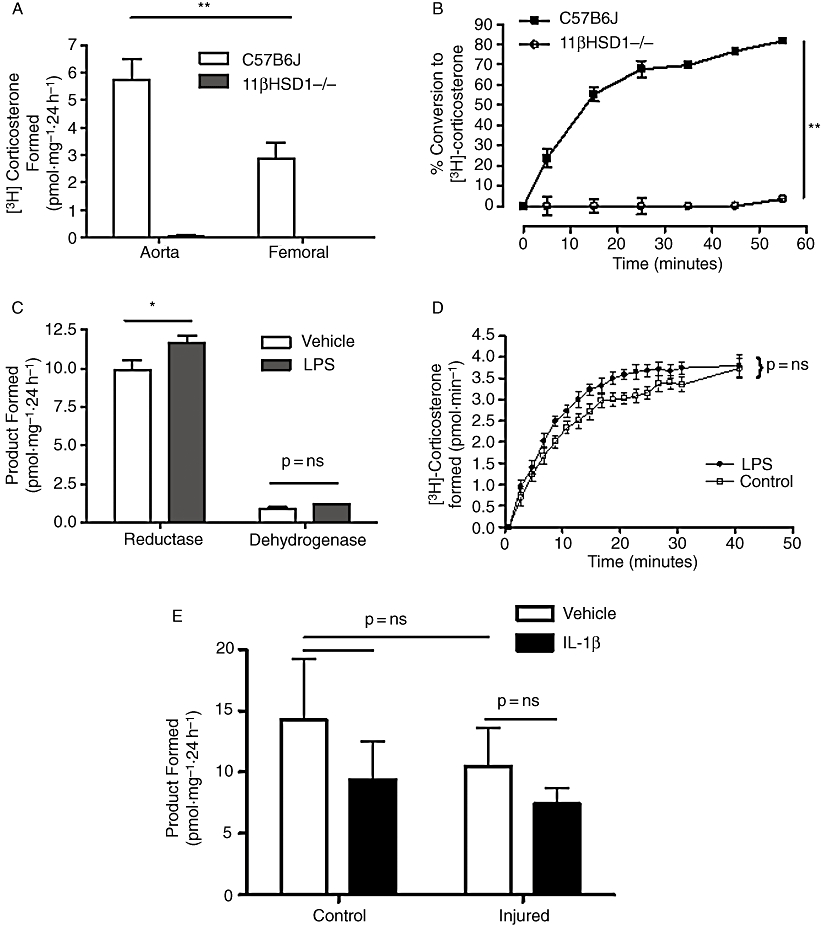
The influence of inflammation on 11β-hydroxysteroid dehydrogenase activity in murine arteries. Glucocorticoid generation in (A) isolated mouse aorta and femoral artery and (B) perfused mouse hindlimb is catalysed exclusively by 11-HSD1 type 1 as deletion of this isozyme completely prevents generation of corticosterone. Exposure of mice to a pro-inflammatory stimulus [lipopolysaccharide (LPS), 6 h] produced a small increase in reductase activity that achieved significance in isolated arteries (C) but not in the perfused hindlimb (D). Similarly, induction of an inflammatory response to intravascular injury in the mouse femoral artery (E) did not increase 11-HSD1 reductase activity in either the presence or absence of pro-inflammatory cytokines (Interleukin 1β; IL-1β) in vitro. Adapted with permission from Dover et al., 2007Endocrinology, 148, 166–172.
The influence of 11-HSD activity on vascular structure has been addressed using mice with selective 11-HSD isozyme deletion. No evidence has been found for altered cardiovascular structure in 11-HSD1−/− mice (Kotelevtsev et al., 1997) but 11-HSD2 deletion results in cardiac hypertrophy and occasional aortic rupture (particularly in pregnant females) (Kotelevtsev et al., 1999). However, as with determining the effects of glucocorticoids on cardiovascular structure, it is difficult to establish the direct effects of 11-HSD deletion from secondary effects mediated by changes in blood pressure. The role of 11-HSDs in regulating angiogenesis can, however, be addressed directly using isolated vascular rings (Small et al., 2005). Work by our own group has shown that 11-HSD1 in the vascular wall regulates angiogenesis by generation of active glucocorticoids from their 11-keto forms (Small et al., 2005). Similar results were obtained in a model of in vivo angiogenesis although this effect may be influenced by alterations in inflammatory response.
More recently, 11-HSD activity has been described in inflammatory cells from mice (macrophages, lymphocytes) and humans (macrophages, neutrophils). In both species, 11-HSD activity in these cells appears to be exclusively reductase activity of the type 1 isozyme (Thieringer et al., 2001; Zhang et al., 2005a; Ishii et al., 2007; Kardon et al., 2008). 11-HSD1 expression is induced on cellular differentiation: for example from monocytes to macrophages (Thieringer et al., 2001), and in polarization of lymphocytes into Th1 or Th2 cells (Zhang et al., 2005a). In addition, 11-HSD1 activity in inflammatory cells is increased by exposure to pro-inflammatory cytokines (Thieringer et al., 2001; Zhang et al., 2005a) and regulates both GR activation (Zhang et al., 2005a) and cellular function [such as neutrophil apoptosis (Kardon et al., 2008) and phagocytosis of apoptotic neutrophils by macrophages (Gilmour et al., 2006)]. These data clearly suggest, therefore, that 11-HSD1 activity in inflammatory cells has a role in regulating the inflammatory response (Chapman et al., 2006).
Glucocorticoids and cardiovascular pathophysiology
The link between glucocorticoids and cardiovascular disease has been evident since the work of Adlersberg and colleagues in the 1950s suggesting that the impact of cortisone treatment on serum lipids might cause premature atherosclerosis (Adlersberg et al., 1950a,b,c). Pathological evidence associating elevated cortisol with atherosclerosis emerged in the same decade (Etheridge and Hochligeti, 1952). It is evident that glucocorticoids have the potential to regulate the cardiovascular system both indirectly, by systemic modulation of risk factors [hypertension and obesity; see Walker et al. (1998)], and directly by interaction with cells of the heart and vascular wall. Most of the indirect systemic effects of glucocorticoids lead to an increase in cardiovascular risk factors and, therefore, are linked to the prognosis of increased cardiovascular events (Dallman et al., 1993; Andrews and Walker, 1999; Walker, 2007b). In contrast, direct interaction of glucocorticoids with cardiovascular cells can induce changes that could variously promote or inhibit development of cardiovascular disease and it is not clear which predominate (Figure 2). Since they induce profound inhibition of inflammation, proliferation and migration, it seems logical that, in the absence of systemic effects, glucocorticoid administration would inhibit the remodelling processes that lead to lesion formation in atherosclerosis, restenosis and chronic graft rejection. In addition, glucocorticoid-mediated regulation of inflammation and angiogenesis may have a beneficial impact on myocardial remodelling following ischaemia. With the demonstration that MR activation can have a profound influence cardiovascular disease development [reviewed in Young (2008)], it is becoming increasingly evident that, in addition to their classical actions on GR, stimulation of MR by glucocorticoids may also have a significant role in these disease pathophysiology.
The major remodelling processes associated with cardiovascular disease all have their basis in the process of wound healing in response to injury. Atherosclerosis is widely accepted to be the result of an unfettered inflammatory response to chronic insult (Ross, 1999; Libby et al., 2002), while restenosis following revascularization is mediated predominantly by inflammation, combined with proliferation and migration of VSMCs, in response to acute injury (Wainwright et al., 2001). Similarly, chronic graft rejection is caused by a progressive thickening of the vascular wall associated with inflammation and EC dysfunction (Brazelton et al., 1999) while, in the heart, myocardial ischaemia prompts a wound healing response characterized by inflammation, angiogenesis and fibrosis (Michael et al., 1999). In atherosclerosis, symptoms are caused by the interruption of blood supply to vulnerable tissues as a result either of a progressive increase in lesion size or, more significantly, of rupture and thrombosis of vulnerable lesions (Weissberg, 2000). The ability of HMGCoA reductase inhibitors (‘statins’) to reduce cardiovascular events without appreciable changes in lesion size has been attributed to increased plaque stability (Son et al., 2003). This has important implications for anti-atherosclerotic interventions as agents that inhibit VSMC proliferation might inhibit development of restenotic lesions but, by degrading the protective fibrous cap, increase the vulnerability of atheroma.
Based on our understanding of cardiovascular remodelling processes, the direct action of glucocorticoids could be predicted to increase or decrease risk, depending on disease pathology and on which action predominates (Figure 2). Inhibition of inflammation, in the heart and in the vasculature (and by actions on circulating inflammatory cells) would be expected to inhibit the development of fibro-fatty lesions in atherosclerosis, remove the stimulus for VSMC migration and proliferation in restenosis (Miller et al., 2001; Wainwright et al., 2001) and graft rejection and also attenuate scarring and fibrosis in the healing myocardium. However, the influence of glucocorticoids on adhesion molecules, which mediate the inflammatory response, is complex. In healthy men, for example, dexamethasone inhibits circulating E-selectin and intercellular adhesion molecule (ICAM)-1 (Jilma et al., 2000) but, while glucocorticoids inhibit NFκB – induced endothelial vascular cell adhesion molecule (VCAM)-1 expression, they also stabilize VCAM-1 mRNA (Simoncini et al., 2000). Similarly, in restenosis/graft rejection, glucocorticoid-mediated inhibition of VSMC migration (Goncharova et al., 2003) and proliferation (Longenecker et al., 1982; 1984), plus their ability to inhibit thrombin-induced expression of growth factors [platelet-derived growth factor A chain and heparin binding epidermal growth factor like growth factors] by VSMCs (Nakano et al., 1993), would be predicted to be beneficial, reducing neointimal lesion formation. In contrast to these beneficial effects, however, inhibition of these processes, combined with stimulation of glucocorticoid-induced tumour necrosis factor (TNF) receptor family-related protein-mediated activation of macrophages (Kim et al., 2006b), is likely to contribute to destabilization of atherosclerotic lesions. Glucocorticoids may also exacerbate the consequences of lesion rupture by modulating factors involved in coagulation and fibrinolysis (clotting factors and fibrinogen) to produce a prothrombotic state (Brotman et al., 2006). Other processes may also favour lesion development, including: glucocorticoid-mediated inhibition of endothelial nitric oxide generation [possibly by production of superoxide (Iuchi et al., 2003) or inhibition of tetrahydrobiopterin (BH4) generation (Johns et al., 2001)], leading to increased VSMC proliferation and migration (Radomski et al., 1990); stimulation of ACE activity (Mendelsohn et al., 1982; Fishel et al., 1995) contributing to increased VSMC proliferation [since ACE inhibition limits neointimal proliferation following balloon injury (Powell et al., 1989; Capron et al., 1991)]; stimulating release of the vasoconstrictor and growth factor, endothelin-1 (Kato et al., 1995); increasing vasoconstriction leading to reduced vascular lumen diameter (Ullian, 1999); impairment of cholesterol removal from the arterial wall [by stimulating cholesteryl ester formation in smooth muscle cells (Petrichenko et al., 1997)]; and, increasing vascular calcification [due to promotion of osteoblastic differentiation of VSMCs (Mori et al., 1999)]. Finally, some studies from the 1970s and 1980s suggested that inhibition of inflammation and VSMC proliferation and atherosclerotic plaque formation (possibly by inhibition of heat shock proteins) in the heart is likely to be beneficial following infarction (Alisky, 2006). These effects may be counter-balanced, however, by a detrimental inhibition of angiogenesis during wound healing (Small et al., 2005).
In the context of inflammatory cardiovascular remodelling, the direct interaction of glucocorticoids with inflammatory cells may have an influence on the wound healing response. For example, dexamethasone inhibits interleukin-6 (IL-6) release (Wirtz et al., 2004a), but stimulates TNF-α secretion (Wirtz et al., 2004b), from monocytes. These processes may be linked [since dexamethasone-mediated inhibition of IL-6 production is blocked in the presence of high TNF-α (Wirtz et al., 2004a)] and may also be influenced by systemic risk factors [as dexamethasone-induced TNF-α release is increased in monocytes from patients with essential hypertension (Wirtz et al., 2004b)]. Given the role of macrophages in the formation of atherosclerotic and neointimal lesions, the ability of glucocorticoids to increase phagocytic activity (McColl et al., 2007), inhibit scavenger receptor activity (Roberts et al., 1998) and inhibit endothelin-1-mediated release of TNF-α from these cells (Ruetten and Thiemermann, 1997) may be important. In addition, glucocorticoids may promote foam cell formation by increasing the accumulation of cholesterol esters, by enhancing acyl coenzyme A: cholesterol acyltransferase 1 (ACAT-1) expression (Yang et al., 2004). The ability of glucocorticoids to influence inflammatory cells is not limited to monocyte/macrophage/foam cells as dexamethasone can also attenuate serum amyloid-A-induced release of TNF-α from neutrophils (Hatanaka et al., 2004).
Pathophysiological role of cardiovascular 11-HSDs
Positive and negative effects of glucocorticoids on the heart, blood vessel wall and inflammatory response are likely to be modulated by pre-receptor metabolism in cells containing 11-HSD1 and/or 11-HSD2. Indeed, a key role of these enzymes may be to regulate access of glucocorticoids to MR in the heart and vasculature (Molnar et al., 2008). The induction of 11-HSD1 activity in monocytes as they differentiate into macrophages (Thieringer et al., 2001) suggests a mechanism for amplification of phagocytosis (McColl et al., 2007) and, hence, accelerated resolution of inflammation. Perhaps more striking is the demonstration that pro-inflammatory cytokines up-regulate 11-HSD1, but down-regulate 11-HSD2, in human smooth muscle cells, thereby favouring the generation of active glucocorticoid (Cai et al., 2001). This suggests a mechanism for local feedback regulation of inflammation in the vascular wall which would be expected to protect against the excessive inflammatory response central to atherogenesis. In our own studies (Dover et al., 2007) with intact murine arteries, however, in vitro exposure to various inflammatory stimuli or in vivo exposure to lipopolysaccharide had no effect on (or produced only small increases in) 11-HSD1 reducatse activity (Figure 3C,D). In addition, our work in mice (Small et al., 2005) indicates that 11-HSD isozymes regulate the angiogenic growth of blood vessels. This may influence both cardiac remodelling and atherogenesis in which angiogenesis provides an important mechanism for supplying oxygen to vascular and cardiac cells.
Glucocorticoid-mediated regulation of atherosclerosis
The various actions of glucocorticoids on the heart, vascular wall and inflammatory cells, combined with their ability to influence cardiovascular risk factors, make it difficult to predict their effect on cardiovascular disease in vivo. Endogenous glucocorticoid excess in humans appears to be predictive of cardiovascular morbidity and mortality (Colao et al., 1999): predominantly epidemiological studies have shown correlation of endogenous plasma corticosteroid levels with disease severity (Table 1). Retrospective analysis of the association between plasma cortisol concentrations and atherosclerosis demonstrated a significant correlation of elevated morning plasma cortisol levels with angiographically determined moderate-to-severe coronary atherosclerosis (Troxler et al., 1977) and an increase, independent of age or sex, in the number of coronary vessels with severe stenosis (Alevizaki et al., 2007). These results are consistent with studies of endogenous corticosteroid excess which can be caused by a variety of conditions (e.g. Cushing's syndrome, primary hyperaldosteronism, renal artery stenosis). For example, patients with untreated or non-remitting Cushing's disease exhibit a fourfold increase in mortality, compared with the general population, mainly as a result of vascular disease (Etxabe and Vazquez, 1994) while the combination of chronic HPA axis hyper-reactivity with tissue hypersensitivity to glucocorticoids has been linked to more severe atherosclerosis (Alevizaki et al., 2007). This is supported by the demonstration that normalization of glucocorticoid levels in patients with Cushing's disease improves a variety of vascular parameters, including: the distensibility coefficient, systolic lumen diameter and intima-media thickness (Faggiano et al., 2003). In addition to Cushing's syndrome, congenital adrenal hyperplasia (due to 21-hydroxylase deficiency) which causes a tendency to obesity, high insulin and hypertension in children and adolescents, is linked with increased intima-media thickness in the aorta and other major conduit arteries (common carotid A, carotid bulb, femoral A) (Sartorato et al., 2007). It has also been proposed that aggressive treatment to lower low-density lipoprotein (LDL) levels in atherosclerosis may affect the synthesis of steroid hormones (Kanat et al., 2007). When evaluating these studies, however, it should be noted that, given the diurnal fluctuations in secretion and the importance of tissue-specific regulation, simple measurement of steroid concentrations in biological fluids (such as blood, saliva and urine) will give only a very crude indication of glucocorticoid activity.
Table 1.
The relationship between glucocorticoid excess and atherosclerosis in human studies
| Patient group | Design and intervention | Outcome | Reference |
|---|---|---|---|
| Endogenous glucocorticoid excess | |||
| Cushing's syndrome | Epidemiological study | Increased mortality (4×) | (Etxabe and Vazquez, 1994) |
| Normalization of cortisol levels | Improved vascular parameters | (Faggiano et al., 2003) | |
| Normalization of cortisol levels | Cardiovascular risk remains elevated | (Colao et al., 1999) | |
| CAH | Ultrasound analysis | Increased intima/media thickness | (Sartorato et al., 2007) |
| Requiring angiography | Elective cortisol measurement | Increased cortisol correlated with increased atherosclerosis | (Troxler et al., 1977), (Alevizaki et al., 2007) |
| Chronic glucocorticoid administration | |||
| Glucocorticoid users | Cohort study | Increased risk of cardiovascular events | (Wei et al., 2004) |
| Cohort study | Increased risk of heart failure | (Souverein et al., 2004) | |
| Cohort study | Increased risk of acute myocardial infarction | (Varas-Lorenzo et al., 2007) | |
| Rheumatoid arthritis | Cohort study | Increased cardiovascular mortality | (Sihvonen et al., 2006) |
| Cohort study | Increased risk of cardiovascular events | (Davis et al., 2007) | |
| Cohort study | Increased risk of cardiovascular events | (Solomon et al., 2006) | |
| Cohort study | Increased carotid artery plaque and arterial stiffness | (del Rincon et al., 2004) | |
| Cohort study | Increased cholesterol but no effect on atherosclerosis or EC function | (Hafstrom et al., 2007) | |
| Polymyalgia rheumatica | Cohort study | No increase in cardiovascular events | (Kremers et al., 2007) |
| Giant cell arteritis | Cohort study | No increase in atherosclerosis | (Gonzales-Juanatey et al., 2007) |
| Cohort study | Increased blood pressure | (Fardet et al., 2007) | |
| SLE | Cohort study | Increased carotid and femoral lesions | (Vlachoyiannopoulos et al., 2003) |
CAH, congenital adrenal hyperplasia; EC, endothelial cell; SLE, systemic lupus erythromatosis.
Exogenous glucocorticoid therapy is also associated with cardiovascular disease, as judged by assessment of the effect on cardiovascular risk factors in patients receiving glucocorticoid replacement, and by pharmacoepidemiological investigations of patients receiving chronic administration of anti-inflammatory doses of glucocorticoid for treatment of disease (e.g. Lupus, rheumatoid arthritis) (Table 1). This increased risk is cumulative and dose-dependent, is mainly observed during the first month of treatment (Wei et al., 2004; Davis et al., 2007) and is reduced if treatment is discontinued (Souverein et al., 2004; Varas-Lorenzo et al., 2007). Daily administration of prednisolone in dose equivalents exceeding 10 mg, for example, was associated with a twofold increased risk of acute myocardial infarction (Varas-Lorenzo et al., 2007). The relationship between chronic glucocorticoid therapy and cardiovascular disease has been addressed most consistently in patients with progressive inflammatory conditions, such as rheumatoid arthritis and systemic lupus erythromatosis (SLE). In rheumatoid arthritis the risk of mortality (predominantly from cardiovascular disease) increases by 14% after 1 year, rising to 69% after 10 years, in patients treated with low-dose, oral glucocorticoids (Sihvonen et al., 2006). These outcomes are consistent with detection of increased risk of cardiovascular risk in similar groups of patients (del Rincon et al., 2004; Solomon et al., 2006; Davis et al., 2007) and have been associated with carotid plaque and arterial distensibility independent of cardiovascular risk factors and clinical manifestations (del Rincon et al., 2004). It should be noted, however, that, in one study, oral low-dose prednisolone had no effect on endothelial cell function, atherosclerosis or atherosclerotic risk factors in a cohort of patients with rheumatoid arthritis (Hafstrom et al., 2007). Chronic glucocorticoid administration also increased cardiovascular risk in patients with SLE (Vlachoyiannopoulos et al., 2003; Fischer-Betz et al., 2005). Of some concern is a recent study on patients with SLE which suggested that glucocorticoid administration decreased the effectiveness of the anti-atherosclerotic drug pravastatin (Costenbader et al., 2007). This pro-atheromatous effect of glucocorticoids seems most likely to be caused by increasing systemic risk factors (e.g. hypertension) and, indeed, has been linked to: increased cholesterol levels [in patients with SLE (Sarkissian et al., 2006; 2007); impaired metabolism of atheroprotective high density lipoproteins (Beentjes et al., 2000); and increased insulin resistance following transplant (Armstrong et al., 2005; Oterdoom et al., 2007)]. Impaired EC function (flow-mediated dilatation) in SLE could not, however, be attributed to the cumulative dose of prednisolone (Lima et al., 2002). A limitation of these studies is that it is difficult to differentiate the effect of treatment from that of the underlying inflammatory condition, a factor which may contribute to investigations in patients with different forms of inflammatory disease (e.g. polymyalgia rheumatica) that found no association between glucocorticoid therapy and cardiovascular risk.
In addition to these associations between glucocorticoid excess and systemic risk factors, there is some data to suggest that the interaction of glucocorticoids with the vascular wall is impaired in cardiovascular disease. This mainly revolves around alteration of arterial GR activity with GR expression, which is high in the media of human carotid arteries, reduced in cells from human vascular lesions (Bray et al., 1999). This may be a consequence of increased lipid deposition as both LDL and VLDL can inhibit glucocorticoid binding by reducing the number of GR in human cells (Shakhov et al., 1989; 1993).
The effects of elevated glucocorticoids on atherosclerosis in animal models contrast strikingly with their adverse effects in clinical investigations (Table 2). Glucocorticoids, and other anti-inflammatory steroids, prevent or arrest atherosclerosis in fat-fed rabbits, despite increasing hyperlipidaemia (Jain et al., 1965; Bailey and Butler, 1985; Naito et al., 1992; Asai et al., 1993). This effect was first demonstrated over 50 years ago (Oppenheim and Bruger, 1952; Gordon et al., 1954; Stumpf and Wilens, 1954; Constantinides et al., 1962) and has subsequently proved to be reproducible in this model (Cavallero et al., 1976; Bailey and Butler, 1985; Naito et al., 1992; Asai et al., 1993) and in rabbits with inherited hypercholesterolaemia [Watanabe heritable hyperlipidaemic (WHHL); Makheja et al., 1989]. There is also an indication that social stress, which increases cortisol and corticosterone secretion in New Zealand white and WHHL rabbits (Szeto et al., 2004) increases atherogenesis in the latter (Mccabe et al., 2002). In female cynomologus monkeys, however, while depression increased sensitivity to negative feedback regulation of the HPA axis, it had no effect on atherogenesis or vascular function after prolonged exposure to a high-fat diet (Shively et al., 2002). A single study in atherosclerosis-susceptible pigeons suggested that glucocorticoid administration in early life influences aortic prostaglandin synthesis and morphology (Deitemeyer et al., 1985). In cockerels, cortisone has been shown to both increase (Stamler et al., 1954) and reduce atheroma (Jain et al., 1965) (while cortisol had no effect). There is insufficient information to determine the reason for these contradictions.
Table 2.
The influence of endogenous glucocorticoid excess or glucocorticoid treatment on atherosclerosis in animal models
| Sample | Design and intervention | Outcome | Reference |
|---|---|---|---|
| Monkey (stress) | Depression; impaired HPA feedback | No effect on atherosclerosis | (Shively et al., 2002) |
| Pigeon | Dexamethasone | Reduced abnormalities in aortic ECs | (Deitemeyer et al., 1985) |
| Cockerel (fat-fed), | Cortisone (1–15 mg·day−1) | Moderate increase in aortic and coronary atherogenesis | (Stamler et al., 1954) |
| Hydrocortisone (1–2 mg·day−1) | No effect on aortic or coronary atherogenesis | (Stamler et al., 1954) | |
| Hydrocortisone (1 mg·day−1) | Reduced coronary atheroma | (Jain et al., 1965) | |
| Rabbit (fat-fed) | Cortisone (10 mg 3× week−1) | Reduced lesion development | (Oppenheim and Bruger, 1952) |
| Cortisone (5 mg·day−1) | No effect on atherosclerosis | (Ashton and Cook, 1952) | |
| Cortisone (1–3 mg·kg−1) | Reduced aortic lesion development | (Gordon et al., 1954) | |
| Cortisone | Reduced arterial lipid deposition | (Stumpf and Wilens, 1954) | |
| Prednisolone | (Constantinides et al., 1962) | ||
| Cortisol | Reduced proliferation in lesions | (Cavallero et al., 1976) | |
| Cortisone 5–10 mg | Reduction in lesion formation | (Bailey and Butler, 1985) | |
| Dex (125 mg·day−1) | Reduced lesion development | (Naito et al., 1992) | |
| Dex (125 mg·day−1) | Reduced lesion development | (Asai et al., 1993) | |
| Rabbit (WHHL) | Cortisone (5 mg·day−1) | 60% reduction in atherogenesis | (Makheja et al., 1989) |
| Social stress | Increased lesion formation and severity | (Mccabe et al., 2002) |
EC, endothelial cell; Dex, dexamethasone; HPA, hypothalamic-pituitary adrenal axis; WHHL, Watanabe heritable hyperlipidaemic.
The mechanism(s) underlying this anti-atherosclerotic effect of glucocorticoids is (are) incompletely understood. Suggestions include: inhibition of DNA synthesis in the cellular component of lesions (in fat-fed rabbits; Cavallero et al., 1976); inhibition of inflammatory cell proliferation (Asai et al., 1993); inhibition of intimal VSMC proliferation (Voisard et al., 1994) and migration (Van Put et al., 1995); reduced chemotaxis of circulating monocytes and macrophages into the sub-endothelial space (Prescott et al., 1989; Yamada et al., 1993); inhibition of macrophage proliferation by GR-mediated-attenuation of oxidized-LDL-induced granulocyte/macrophage colony-stimulating factor production by (human and murine) inflammatory cells (Sakai et al., 1999); GR-mediated reduction of reactive oxygen species generation in (human) aortic VSMCs (Marumo et al., 1998); and GR-mediated inhibition of monocyte chemotactic protein (MCP)-1 (the dominant mediator of macrophage accumulation in atherosclerotic plaques) secretion by marked reduction in MCP-1 mRNA stability (Fasshauer et al., 2004; Dhawan et al., 2007). Whatever the mechanism(s) responsible for glucocorticoid-mediated inhibition of atherosclerosis in animal models, one of the most important questions has not been answered: why do these steroids apparently inhibit lesion development in animals but increase it in humans?
The ability of 11-HSD activity to influence atherosclerosis, presumably by regulating glucocorticoid generation in key metabolic and cardiovascular tissues, has been demonstrated in a small number of studies in which 11-HSD inhibitors were administered to dyslipidaemic mice (see below). However, while 11-HSD activity in inflammatory cells, cardiac myocytes, VSMCs and ECs (in animals and in man) suggests it has a role in regulating the inflammatory response to injury, there is little evidence that this is important in regulating cardiovascular disease.
Glucocorticoid-mediated regulation of neointimal proliferation
Several properties of glucocorticoids suggest that they may prevent the intense fibro-proliferative vascular remodelling that occurs following percutaneous revascularization (angioplasty, stenting), prompting a number of small-scale trials in patients and animal models (Table 3). Studies in animals have largely supported the hypothesis that glucocorticoid administration will reduce neointimal proliferation. Systemic dexamethasone administration inhibited neointimal lesion formation in rats (Villa et al., 1994; Guzman et al., 1996), rabbits (Van Put et al., 1995; Poon et al., 2001) and dogs (Strecker et al., 1998) with inhibition of macrophage accumulation proposed as a possible mechanism of action (Poon et al., 2001). Similar results were obtained with either oral or local prednisone in rabbit iliac artery (Ribichini et al., 2007a). This approach may also have potential in the prevention of chronic graft disease as short-term (7 days) oral dexamethasone (0.15 mg−1·kg−1·day−1) reduced vein graft thickening in ApoE3leiden mice (Schepers et al., 2006). Not all studies in animals have yielded positive results, however, as dexamethasone treatment did not reduce neointimal hyperplasia after angioplasty in the rabbit (Karim et al., 1997) or in the pig (Lincoff et al., 1997). Initial clinical trials were also disappointing: methylprednisolone did not inhibit restenosis after coronary angioplasty (Pepine et al., 1990) or stenting (Reimers et al., 1998) while dexamethasone-drug eluting stents (D-DES) have not reduced the incidence of restenosis (Hoffmann et al., 2004a; Ribichini et al., 2007b). The reasons for the differences between animals and humans remain unclear but there are several possible explanations, including: animal models providing a poor approximation of disease in humans; clinical trials using small numbers of patients, often with refractory or complex disease; differences in timing of drug administration in animal and clinical studies, and; detrimental systemic effects of glucocorticoids masking beneficial effects in the arterial wall. More recently, interest in the potential of glucocorticoids as anti-restenotics has been re-awakened (Liu et al., 2004; Radke et al., 2004; Ferrero et al., 2007a) with several trials suggesting a beneficial effect of glucocorticoids: oral prednisone produced a dose-dependent reduction in clinical events and angiographic restenosis rate after stenting (the IMPRESS trial) (Versaci et al., 2002; Ferrero et al., 2007b); low-dose dexamethasone was associated with low restenosis rate (the STRIDE trial) (Liu et al., 2003), D-DES produced a low rate of clinical events at 6 months (despite no inhibition of restenosis; the DESIRE trial) (Ribichini et al., 2007b). It should be noted, however, that the EMPEROR trial was discontinued before patient recruitment because of poor results from a pilot study (Hoffmann et al., 2004b). An intriguing addendum to this search for an anti-restenotic capability of glucocorticoids has been the recent announcement of a study designed to test the effect of glucocorticoid administration by encapsulation in erythrocytes (Versaci and Del Giudice; http://clinicaltrials.gov).
Table 3.
The effect of glucocorticoid administration on vascular remodeling
| Study sample | Study design & intervention | Outcome | Reference |
|---|---|---|---|
| Clinical Studies | |||
| PCI | Patients with ACS; D-DES implantation | Reduced clinical events (6 months); No antirestenotic effect | (Ribichini et al., 2007b) |
| PCI + stent | Oral prednisone (45 days) in patients with high CRP | Reduced clinical events and angiographic restenosis rate | (Versaci et al., 2002) |
| Oral prednisone, low dose vs high dose | Low dose less effective | (Ferrero et al., 2007b) | |
| Intra-wall delivery of methylprednisolone before elective stent implantation | No reduction in the incidence of restenosis | (Reimers et al., 1998) | |
| Stent | Dex | Increased Aneurysm | (Rab et al.,1991) |
| No effect | (Stone et al., 1989) | ||
| No effect | (Pepine et al., 1990) | ||
| Oral Methylprednisolone (1 g) | No effect | (Lee et al., 1999) | |
| High dose Dex | No effect | (Hoffmann et al., 2004b) | |
| Hydrocortisone (200 mg) | Reduced coronary restenosis | (Kakio et al., 2004) | |
| Dexamethasone (low dose) | Low restenosis rate | (Liu et al., 2003) | |
| Animal Studies | |||
| Dog | D-DES-coated stent | Reduced neointimal proliferation | (Strecker et al., 1998) |
| Pig | Stent (Dex) | No effect | (Lincoff et al., 1997) |
| Rat | Local Dex administration | Reduced neointimal proliferation | (Villa et al., 1994b) |
| Reduced neointimal proliferation | (Guzman et al., 1996) | ||
| Reduced neointimal proliferation | (Petrik et al., 1998) | ||
| Increased neointimal proliferation via increased ACE. | (Fishel et al., 1995) | ||
| Rabbit | Dex (1 mg·kg−1·day−1) oral or sc (2 weeks) after silicone collar induced injury to the carotid artery | Reduced neointimal proliferation | (Van Put et al., 1995) |
| Systemic (2.1 mg·kg−1·day−1) or local prednisone | Reduced neointimal proliferation | (Ribichini et al., 2007a) | |
| Rabbit (high fat) | Dex (1 mg·kg−1·day−1) (one week); bilateral iliac artery endothelial denudation, followed by angioplasty | No effect on intimal hyperplasia or fibrosis | (Karim et al., 1997) |
ACE, angiotensin converting enzyme; ACS, acute coronary syndrome; Dex, dexamethasone; D-DES, dexamethasone-drug eluting stent; PCI, Percutaneous coronary intervention.
Pharmacological inhibition of 11-HSDs in the treatment of cardiovascular disease
Plant-derived and endogenous 11-HSD inhibitors
Inhibition of 11-HSD activity in experimental and clinical studies depended, until relatively recently, upon the use of several naturally occurring compounds (Figure 1B). Principal among these were compounds derived from glycyrrhizic acid, the principal active component of the liquorice plant, Glycyrrhiza glabra. Indeed, the therapeutic activity of liquorice was well known to the Ancient Greeks and Romans, is exploited in traditional Chinese medicine and, until the introduction of H2 antagonists in the late 1970s, provided the most effective treatment for peptic ulcers (Davis and Morris, 1991). In addition, excessive liquorice ingestion causes SAME as a result of 11-HSD type 2 inhibition (Conn et al., 1968). Glycyrrhetinic acid (Adamson and Tillman, 1955), the hydrolytic product of glycyrrhizic acid, and its hemi-succinate derivative, carbenoxolone (Csonka and Murray, 1971), have anti-inflammatory properties in the skin and have also been used extensively for pharmacological inhibition of 11-HSD activity. The usefulness of these compounds is limited, however, as they inhibit both isozymes of 11-HSD [IC50s: Dehydrogenase activity: glycyrretinic acid; ∼2.5 × 10−8 mol·L−1; carbenoxolone ∼1.5 × 10−7 mol·L−1 (Schleimer, 1991)]; Oxo-reductase; glycyrretinic acid; ∼8 × 10−8 mol·L−1; carbenoxolone ∼1 × 10−7 mol·L−1 (Li et al., 1997) although it has been suggested that carbenoxolone is more active against the dehydrogenase activity (Ki 2 × 10−8 mol·L−1 compared with 4.1 × 10−7 mol·L−1 for the reductase) of 11-HSD (Brem et al., 1997; Morris et al., 2003). In addition, unwanted effects of carbenoxolone may be mediated by its ability to inhibit 3α-hydroxysteroid dehydrogenase (Akao et al., 1992) and to block myoendothelial gap junctions (Goldberg et al., 1996; Edwards et al., 1999).
In addition to these plant-derived compounds, several endogenous inhibitors of 11-HSD have also been identified. These include the cholesterol derivatives 11β-OH-progesterone and its metabolite 11-keto-progesterone, both of which inhibit 11-HSD; it has been suggested that 11β-OH-progesterone selectively inhibits 11-HSD dehydrogenase activity while 11-keto-progesterone is selective for 11-HSD reductase activity (Brem et al., 1997). Both compounds are relatively weak inhibitors (Ki = 5 × 10−7 mol·L−1 and 6.8 × 10−7 mol·L−1 for 11β-OH-progesterone and 11-keto-progesterone respectively), however, and the claim of selectivity for the latter is contentious. Endogenous bile acids, such as lithocholic acid and chenodeoxycholic acid, are also non-selective inhibitors of 11-HSD (Perschel et al., 1991; Latif et al., 1994) with the latter proposed to be selective for 11-HSD1 (Morris and Souness, 1996). More recently it has been demonstrated that 11-HSDs can metabolize 7-oxysterols in man and rodents (Hult et al., 2004a,b; Schweizer et al., 2004b) revealing the possibility that 7-oxysterols may inhibit 11-HSD-mediated metabolism of glucocorticoids by competing for the active site of the enzyme (this work also suggests that 11-HSDs could directly mediate the generation of atherogenic cholesterol metabolites). In general, however, inhibition of 11-HSD by compounds obtained from liquorice (Glycyrrhetinic acid, Glycyrrhizic acid, carbenoxolone), or generated endogenously (e.g. 11β-hydroxyprogesterone; chenodeoxycholic acid), tends to be non-selective or weakly selective.
The limited availability of isozyme-selective 11-HSD inhibitors has prompted the use of alternative approaches to differentiate their physiological roles. Transgenic deletion (Kotelevtsev et al., 1997; 1999) and over-expression (Masuzaki et al., 2001; 2003; Paterson et al., 2004) of 11-HSD isozymes has been invaluable in this respect. In addition, selective 11-HSD inhibition has been achieved using specific anti-sense oligonucleotide probes to 11-HSD1 and 11-HSD2 (Souness et al., 1995). The influence of tissue-specific regulation of 11-HSD1 on cardiovascular risk factors [with over-expression of 11-HSD1 in the adipose tissue (Masuzaki et al., 2001; 2003) or liver (Paterson et al., 2004) producing central obesity, hypertension and dyslipidaemia] and the presence of both isozymes in the heart and blood vessel wall has suggested that they may prove a useful therapeutic target in cardiovascular disease.
Selective 11-HSD inhibitors
The identification of 11-HSD1 as a potential target for the treatment of diabetes mellitus and metabolic syndrome has prompted considerable recent activity in the pharmaceutical industry; leading to the discovery and development of a multitude of novel non-steroidal 11-HSD1-selective inhibitors (>90 patents filed by ∼29 different companies and other organizations since 2002). These embrace a wide variety of compounds, based predominantly around: triazoles, aryl sulphonamide thiazoles, sulfonamides and adamantyl carboxamides (Webster and Pallin, 2007; Hughes et al., 2008). Development of these new compounds has not been trivial, with the challenge being to find a small molecule that is both potent and selective enough to exploit the extremely hydrophobic pocket in the active site of 11-HSD1 (Hosfield et al., 2005; Zhang et al., 2005b; Kim et al., 2006a). This requires the presence of lipophilic groups which, in most inhibitors, contributes to associated problems with stability and solubility. Both issues, however, have been successfully improved by appropriate chemical modification, producing compounds with good pharmacodynamic and pharmacokinetic profiles in target tissues.
Selective 11-HSD1 inhibitors potentially have a variety of clinical applications [reviewed in Wamil and Seckl (2007)], for example, in the treatment of wound healing and age-related cognitive impairment. Their application in the cardiovascular arena could include treatment of the metabolic syndrome, obesity, type 2 diabetes and prevention and treatment of atherosclerosis. It is recognized that pharmacological effects of these compounds on the metabolic syndrome require 11-HSD1 inhibition in the liver and adipose tissue (Webster and Pallin, 2007). Several companies have now produced orally available compounds that achieve inhibition in these tissues in rodents (rats, mice); with tissue inhibition in human adipose also recently demonstrated in phase IIa clinical trials in obese patients [reviewed in Webster and Pallin (2007)].
The therapeutic potential of 11-HSD1 inhibitors in metabolic syndrome (specifically, obesity, type 2 diabetes mellitus and dyslipidaemia) is very promising. For example, based on animal studies showing its ability to restore the glucose lowering action of insulin without influencing HPA axis function, INCB13739 (Incyte corp, Wilmington, Del) has been shown, in phase IIa clinical trials, to rapidly correct glucose levels, improve insulin resistance, fasting cholesterol and low-density lipoprotein levels in patients with type 2 diabetes (Press release Incyte web site; http://incyte.com/index.html). Additionally, phase I clinical trials of selective 11-HSD1 inhibitors have also been disclosed by Amgen and Biovitrum (AMG221) with a pre-clinical trial programme at Pfizer (Biocentury 2007, 15 (4), A1–A25).
11-HSD1 inhibition in cardiovascular disease
11-HSD1 knockout mice have an atheroprotective phenotype, including lower cholesterol and triglyceride levels and improved glucose tolerance (Morton et al., 2001) while non-selective 11-HSD1 inhibition with carbenoxolone (4 weeks) significantly reduced atherosclerosis in mice (Nuotio-Antar et al., 2007). In addition, it is possible that some anti-atherosclerotic interventions owe part of their effectiveness to regulation of 11-HSDs as PPAR-α agonists, such as fibrates (which are important agents for treating dyslipidaemia) reduce 11-HSD1 expression and activity in murine liver (Hermanowski-Vosatka et al., 2000). One concern is raised, however, by the ability of 11-HSD1 to metabolise 7-oxysterols, as this may be pro-atherogenic (Schroepfer, 2000). 7-Ketocholesterol which is present in very small quantities in the plasma is highly concentrated in human atherosclerotic lesions (Brown et al., 2000) and is linked with an increased risk of developing atherosclerotic plaques at an early age as demonstrated in patients with cerebrotendinous xanthomatosis (Fujiyama et al., 1991). 11-HSD1 inhibition in vivo results in increased 7-ketocholesterol levels in the liver and plasma in rats (Schweizer et al., 2004a,b).
Initial results from in vivo studies in mouse and rat models have produced promising evidence that selective 11-HSD1 inhibitors provide an effective treatment for systemic cardiovascular risk factors, including: obesity, type 2 diabetes and dyslipidaemia. It has been established that exposure to 11-dehydrocorticosterone can induce an 11-HSD1-dependent, GR-dependent increase in pancreatic 11-HSD1 activity, leading to reduced glucose-mediated insulin secretion from pancreatic islets (Wang et al., 2004; Ortsater et al., 2005). In addition, 11-HSD1 inhibition (with BVT: 2733) improved insulin sensitivity and dyslipidaemia [200 mg·kg−1 twice daily; Alberts et al., 2003] and decreased blood glucose [7 day administration by minipump; Alberts et al., 2002] in hyperglycaemic mice. Perhaps the most striking example of the benefit of selective 11-HSD1 inhibition in mouse models of disease is provided by a study with an inhibitor (Compound 544), produced by Merck Laboratories, that produces short-term selective inactivation of 11-HSD1 after oral administration (Hermanowski-Vosatka et al., 2005). This investigation confirmed that 11-HSD1 inhibition was beneficial in mice with diet-induced obesity and streptozotocin/high-fat diet-induced diabetes (reducing body weight, fasting glucose and serum lipids) after twice daily administration (20 or 30 mg·kg−1) for only 11 days. In addition, extended (8 week) administration of this compound (∼10 mg·kg−1·day−1) to atherosclerosis prone (Apolipoprotein E knockout) mice on a high-fat diet lowered serum cholesterol, triglycerides and free fatty acids and produced a dramatic (∼85%) reduction in atherosclerotic lesion development. Improvements in cardiovascular risk factors are not restricted to this compound or to mouse models as a low dose (3 mg·kg−1·day−1) of a 4-heteroarylbicyclo [222] octyl triazole compound improved fasting triglyceride levels and prevented lipid accumulation in tissues of rats with diet-induced obesity (Berthiaume et al., 2007a,b). These pre-clinical studies are now being augmented by the first clinical trials, for example the phase IIa trials with compound INCB13739 in patients with type 2 diabetes (see above: Incyte Press Release; http://incyte.com/index.html).
The mechanisms through which selective 11-HSD1 inhibitors reduce atherosclerosis have yet to be demonstrated unequivocally. It was notable that the dramatic reduction in lesion formation produced by Compound 544 in atherosclerotic mice was greater than predicted by the more modest improvements in systemic metabolic risk factors (Hermanowski-Vosatka et al., 2005). Similarly carbenoxolone-mediated inhibition of atherogenesis was independent of blood pressure (Nuotio-Antar et al., 2007). It was proposed, therefore, that 11-HSD1 inhibition may have a direct protective effect on the vascular wall (Hermanowski-Vosatka et al., 2005). If this is the case, the precise mechanisms have not been established although data were provided to suggest that 11-HSD1 inhibition was reducing concentrations of MCP-1 in the circulation and aortic wall [MCP-1 is not normally found in the arterial media or intima but has been found in human and rodent atherosclerotic plaques (Ylaherttuala et al., 1991)]. This is not consistent, however, with the reported GR-dependent inhibition of MCP-1 from arterial SMC by glucocorticoids (Dhawan et al., 2007). Furthermore, the role of 11-HSD1 in regulating the inflammatory response to injury or proliferation and migration of VSMCs in vivo has not been investigated in models of neointimal proliferation. We have shown recently (Dover et al., 2007), however, that intra-luminal injury followed by neointimal remodelling does not appear to produce the up-regulation of 11-HSD1 activity [or increased sensitivity to pro-inflammatory cytokines (Figure 3E)] reported in cultured VSMC (Cai et al., 2001). Finally, it should be noted that our recent demonstration that endogenous 11-HSD1 activity suppresses angiogenesis following myocardial infarction (Small et al., 2005) suggests that 11-HSD1 inhibition is likely to promote neovascularization following infarct with consequent improvements in cardiac function. Whether this will actually prove to be the case has yet to be determined.
Conclusions
While it is evident that glucocorticoids can influence the development of cardiovascular disease the processes involved are complex and incompletely understood. The ability of glucocorticoids to stimulate both GR and MR, to mediate opposing actions depending on steroid concentration, and to regulate systemic cardiovascular risk factors as well as functional and structural properties of cardiac, vascular and inflammatory cells makes it difficult to determine which mechanisms contribute to regulation of cardiovascular disease pathogenesis. Thus, it is still not clear why, despite the association between glucocorticoid excess and cardiovascular disease, glucocorticoid administration inhibits atherogenesis and restenosis in animal models. The situation is further complicated by the role of tissue-specific regulation of glucocorticoid activity by the 11-HSDs which introduces the possibility of local glucocorticoid deficiency/excess. The importance of 11-HSDs does, however, provide a potentially important novel therapeutic target. Traditional pharmacological approaches to regulation of glucocorticoid activity are limited by the varied systemic actions of these steroids and, thus, the multiplicity of complications associated with manipulation of this system. Targetting 11-HSD isozymes raises the possibility of tissue-specifc manipulation of 11-HSD activity without altering regulation of the HPA axis.
The potential implications of 11-HSD1 inhibition in treatment of cardiovascular disease have been difficult to predict given the complex relationship between glucocorticoids and this condition. Evidence from transgenically modified mice and a small number of studies using relevant inhibitors, have suggested that 11-HSD1 inhibition should reduce cardiovascular disease by reducing systemic cardiovascular risk factors. Theoretically, these inhibitors also have the potential to alter glucocorticoid activity in the heart, inflammatory cells and vascular wall: whether these actions are all desirable in prevention of atherosclerosis, and whether they contribute to the anti-atherosclerotic action of 11-HSD1 inhibitors has not been determined. Perhaps most importantly, however, much of the evidence that 11-HSD activity regulates cardiovascular disease comes from animal models. There is, as yet, only a superficial understanding of the role of 11-HSDs in regulating atherosclerosis and remodelling following infarction in humans. Given the current enthusiasm for generation and clinical testing of selective 11-HSD1 inhibitors for the treatment of metabolic conditions associated with cardiovascular disease, it is clearly imperative to develop an improved understanding of the action of glucocorticoids, and their endogenous metabolism by 11-HSD isozymes, on cardiovascular pathophysiology. The influence of endogenous glucocorticoid activity, and its regulation within the heart and vascular wall, on atherosclerosis, restenosis and cardiac infarction needs to be demonstrated. Understanding this relationship, particularly in man, will be key to confirming the mechanisms underlying the anti-atherosclerotic actions of selective 11-HSD1 inhibitors and predicting possible side effects.
Glossary
Abbreviations:
- ACAT
acyl coenzyme A, cholesterol acyltransferase
- ACE
angiotensin converting enzyme
- AV
atrio-ventricular
- BH4
tetrahydrobiopterin
- D-DES
Dexamethasone-drug eluting stents
- EC
endothelial cell
- GR
glucocorticoid receptor
- HMGCoA
3-Hydroxy-3-methyl-glutaryl co-enzyme A
- HPA
hypothalamic pituitary adrenal
- 11-HSD
11β-hyhroxysteroid dehydrogenase
- ICAM
intercellular adhesion molecule
- IL
interleukin
- MCP
monocyte chemotactic protein
- MR
mineralocorticoid receptor
- SAME
syndrome of ‘apparent’ mineralocorticoid excess
- SLE
systemic lupus erythromatosis
- TNF
tumour necrosis factor
- VCAM
vascular cell adhesion molecule
- (V)LDL
(very) low-density lipoprotein
- VSMC
vascular smooth muscle cell
- WHHL
Watanabe heritable hyperlipidaemic
Conflicts of interest
P.W.F.H. and B.R.W. are inventors on relevant patents owned by the University of Edinburgh. B.R.W. has received speaker's honoraria from Abbott, Bristol Myers Squibb, Merck and Novartis and consulted for Amgen, AstraZeneca, Biovitrum, Dianippon, Incyte, Ipsen, Johnson & Johnson, Merck, Roche, Syrrx, Vitae, Wyeth, and Zydus. J.I. has no conflicts of interest.
References
- Adamson AC, Tillman WG. Hydrocortisone. Br Med J. 1955;ii:1501. [Google Scholar]
- Adlersberg D, Schaefer L, Drachman SR. Development of hypercholesteremia during cortisone and Acth therapy. JAMA. 1950a;144:909–914. doi: 10.1001/jama.1950.02920110021006. [DOI] [PubMed] [Google Scholar]
- Adlersberg D, Schaefer LE, Dritch R. Effect of cortisone, adrenocorticotropic hormone (Acth), and desoxycorticosterone acetate (Doca) on serum lipids. J Clin Invest. 1950b;29:795. [PubMed] [Google Scholar]
- Adlersberg D, Schaefer LE, Dritch R. Studies on hormonal control of serum lipid partition in man. J Clin Endocrinol. 1950c;10:814–815. [PubMed] [Google Scholar]
- Akao T, Akao T, Hattori M, Namba T, Kobashi K. Inhibitory effects of glycyrrhetic acid and its related-compounds on 3-alpha-hydroxysteroid dehydrogenase of rat-liver cytosol. Chem Pharm Bull. 1992;40:1208–1210. doi: 10.1248/cpb.40.1208. [DOI] [PubMed] [Google Scholar]
- Alberts P, Engblom L, Edling N, Forsgren M, Klingstrom G, Larsson C, et al. Selective inhibition of 11beta-hydroxysteroid dehydrogenase type 1 decreases blood glucose concentrations in hyperglycaemic mice. Diabetologia. 2002;45:1528–1532. doi: 10.1007/s00125-002-0959-6. [DOI] [PubMed] [Google Scholar]
- Alberts P, Nilsson C, Selen G, Engblom LO, Edling NH, Norling S, et al. Selective inhibition of 11 beta-hydroxysteroid dehydrogenase type 1 improves hepatic insulin sensitivity in hyperglycemic mice strains. Endocrinology. 2003;144:4755–4762. doi: 10.1210/en.2003-0344. [DOI] [PubMed] [Google Scholar]
- Alevizaki M, Cimponeriu A, Lekakis J, Papamichael C, Chrousos GP. High anticipatory stress plasma cortisol levels and sensitivity to glucocorticoids predict severity of coronary artery disease in subjects undergoing coronary angiography. Metabolism. 2007;56:222–226. doi: 10.1016/j.metabol.2006.09.017. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition (2008 revision) Br J Pharmacol. 2008;153(Suppl.)(2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alisky JM. Dexamethasone could improve myocardial infarction outcomes and provide new therapeutic options for non-interventional patients. Med Hypotheses. 2006;67:53–56. doi: 10.1016/j.mehy.2005.12.034. [DOI] [PubMed] [Google Scholar]
- Alzamora R, Michea L, Marusic ET. Role of 11beta-hydroxysteroid dehydrogenase in nongenomic aldosterone effects in human arteries. Hypertension. 2000;35:1099–1104. doi: 10.1161/01.hyp.35.5.1099. [DOI] [PubMed] [Google Scholar]
- Amelung D, Huebner HJ, Roka L, Meyerheim G. Conversion of cortisone to compound F. J Clin Endocrinol Metab. 1953;13:1125. doi: 10.1210/jcem-13-9-1125. [DOI] [PubMed] [Google Scholar]
- Andrews RC, Walker BR. Glucocorticoids and insulin resistance: old hormones, new targets. Clin Sci. 1999;96:513–523. doi: 10.1042/cs0960513. [DOI] [PubMed] [Google Scholar]
- Armstrong KA, Hiremagalur B, Haluska BA, Campbell SB, Hawley CM, Marks L, et al. Free fatty acids are associated with obesity, insulin resistance, and atherosclerosis in renal transplant recipients. Transplantation. 2005;80:937–944. doi: 10.1097/01.tp.0000173792.53561.b6. [DOI] [PubMed] [Google Scholar]
- Arriza JL, Weinberger C, Cerelli G. Cloning of human mineralocorticoid receptor complementary DNA; structural and functional kinship with the glucocorticoid receptor. Science. 1987;237:268–275. doi: 10.1126/science.3037703. [DOI] [PubMed] [Google Scholar]
- Asai K, Funaki C, Hayashi T, Yamada K, Naito M, Kuzuya M, et al. Dexamethasone-induced suppression of aortic atherosclerosis in cholesterol-fed rabbits – possible mechanisms. Arterioscler Thromb. 1993;13:892–899. doi: 10.1161/01.atv.13.6.892. [DOI] [PubMed] [Google Scholar]
- Ashton N, Cook C. In vivo observations of the effects of cortisone upon blood vessels in rabbit ear chambers. Br J Exp Pathol. 1952;33:445–450. [PMC free article] [PubMed] [Google Scholar]
- Atanasov AG, Nashev LG, Schweizer RA, Frick C, Odermatt A. Hexose-6-phosphate dehydrogenase determines the reaction direction of 11beta-hydroxysteroid dehydrogenase type 1 as an oxoreductase. FEBS Lett. 2004;571:129–133. doi: 10.1016/j.febslet.2004.06.065. [DOI] [PubMed] [Google Scholar]
- Bailey JM, Butler J. Anti-inflammatory drugs in experimental atherosclerosis .6. combination therapy with steroid and non-steroid agents. Atherosclerosis. 1985;54:205–212. doi: 10.1016/0021-9150(85)90179-0. [DOI] [PubMed] [Google Scholar]
- Barker DJP, Bull AR, Osmond C, Simmonds SJ. Fetal and placental size and risk of hypertension in adult life. Br Med J. 1990;301:259–262. doi: 10.1136/bmj.301.6746.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreto-Chaves ML, Heimann A, Krieger JE. Stimulatory effect of dexamethasone on angiotensin-converting enzyme in neonatal rat cardiac myocytes. Braz J Med Biol Res. 2000;33:661–664. doi: 10.1590/s0100-879x2000000600007. [DOI] [PubMed] [Google Scholar]
- Bartholome B, Spies CM, Gaber T, Schuchmann S, Berki T, Kunkel D, et al. Membrane Glucocorticoid Receptors (MGCR) are expressed in normal human peripheral blood mononuclear cells and up-regulated after in vitro stimulation and in patients with rheumatoid arthritis. FASEB J. 2004;18:70–80. doi: 10.1096/fj.03-0328com. [DOI] [PubMed] [Google Scholar]
- Beentjes JAM, Van Tol A, Sluiter WJ, Dullaart RPF. Decreased plasma cholesterol esterification and cholesteryl ester transfer in hypopituitary patients on glucocorticoid replacement therapy. Scand J Clin Lab Invest. 2000;60:189–198. doi: 10.1080/003655100750044839. [DOI] [PubMed] [Google Scholar]
- Beggah AT, Escoubet B, Puttini S, Cailmail S, Delage V, Ouvrard-Pascaud A, et al. Reversible cardiac fibrosis and heart failure induced by conditional expression of an antisense MRNA of the mineralocorticoid receptor in cardiomyocytes. Proc Natl Acad Sci USA. 2002;99:7160–7165. doi: 10.1073/pnas.102673599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benediktsson R, Lindsay RS, Noble J, Seckl JR, Edwards CRW. Glucocorticoid exposure in utero: new model for adult hypertension. Lancet. 1993;341:339–341. doi: 10.1016/0140-6736(93)90138-7. [DOI] [PubMed] [Google Scholar]
- Berk BC, Vallega G, Griendling KK, Gordon JB, Cragoe EJ, Canessa M, et al. Effects of glucocorticoids on Na/H exchange and growth in cultured vascular smooth muscle cells. J Cell Physiol. 1988;137:391–401. doi: 10.1002/jcp.1041370302. [DOI] [PubMed] [Google Scholar]
- Berthiaume M, Laplante M, Festuccia W, Gelinas Y, Poulin S, Lalonde J, et al. Depot-specific modulation of rat intraabdominal adipose tissue lipid metabolism by pharmacological inhibition of 11 beta-hydroxysteroid dehydrogenase type 1. Endocrinology. 2007a;148:2391–2397. doi: 10.1210/en.2006-1199. [DOI] [PubMed] [Google Scholar]
- Berthiaume M, Laplante M, Festuccia WT, Cianflone K, Turcotte LP, Joanisse DR, et al. 11 Beta-HSD1 inhibition improves triglyceridemia through reduced liver VLDL secretion and partitions lipids toward oxidative tissues. Am J Physiol Endocrinol Metab. 2007b;293:E1045–E1052. doi: 10.1152/ajpendo.00276.2007. [DOI] [PubMed] [Google Scholar]
- Best R, Nelson SM, Walker BR. Dexamethasone and 11-dehydrodexamethasone as tools to investigate the isozymes of 11β-hydroxysteroid dehydrogenase in vitro and in vivo. J Endocrinol. 1997;153:41–48. doi: 10.1677/joe.0.1530041. [DOI] [PubMed] [Google Scholar]
- Bjorntorp P. Visceral fat accumulation: the missing link between psychosocial factors and cardiovascular disease? J Intern Med. 1991;230:195–201. doi: 10.1111/j.1365-2796.1991.tb00431.x. [DOI] [PubMed] [Google Scholar]
- Bray PJ, Du BH, Mejia VM, Hao SC, Deutsch E, Fu CZ, et al. Glucocorticoid resistance caused by reduced expression of the glucocorticoid receptor in cells from human vascular lesions. Arterioscler Thromb Vasc Biol. 1999;19:1180–1189. doi: 10.1161/01.atv.19.5.1180. [DOI] [PubMed] [Google Scholar]
- Brazelton TR, Adams B, Shorthouse R, Morris RE. Chronic rejection: the result of uncontrolled remodelling of graft tissue by recipient mesenchymal cells? data from two rodent models and the effects of immunosupressive therapies. Inflamm Res. 1999;48:S134–S135. doi: 10.1007/s000110050554. [DOI] [PubMed] [Google Scholar]
- Brem AS, Bina RB, King T, Morris DJ. Bidirectional activity of 11β-hydroxysteroid dehydrogenase in vascular smooth muscle cells. Steroids. 1995;60:406–410. doi: 10.1016/0039-128x(94)00074-m. [DOI] [PubMed] [Google Scholar]
- Brem AS, Bina RB, King T, Morris DJ. 11betaOH-progesterone affects vascular glucocorticoid metabolism and contractile response. Hypertension. 1997;30:449–454. doi: 10.1161/01.hyp.30.3.449. [DOI] [PubMed] [Google Scholar]
- Brem AS, Bina RB, King TC, Morris DJ. Localization of 2 11beta-OH steroid dehydrogenase isoforms in aortic endothelial cells. Hypertension. 1998;31:459–462. doi: 10.1161/01.hyp.31.1.459. [DOI] [PubMed] [Google Scholar]
- Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT. Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res. 1990a;67:1355–1364. doi: 10.1161/01.res.67.6.1355. [DOI] [PubMed] [Google Scholar]
- Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT. Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res. 1990b;67:1355–1364. doi: 10.1161/01.res.67.6.1355. [DOI] [PubMed] [Google Scholar]
- Brilla CG, Matsubara LS, Weber KT. Anti-aldosterone treatment and the prevention of myocardial fibrosis in primary and secondary hyperaldosteronism. J Mol Cell Cardiol. 1993;25:563–575. doi: 10.1006/jmcc.1993.1066. [DOI] [PubMed] [Google Scholar]
- Brodie AMH. Aromatase, its inhibitors and their use in breast-cancer-treatment. Pharmacol Ther. 1993;60:501–515. doi: 10.1016/0163-7258(93)90033-a. [DOI] [PubMed] [Google Scholar]
- Brotman DJ, Girod JP, Garcia MJ, Patel JV, Gupta M, Posch A, et al. Effects of short-term glucocorticoids on cardiovascular biomarkers. J Clin Endocrinol Metab. 2005;90:3202–3208. doi: 10.1210/jc.2004-2379. [DOI] [PubMed] [Google Scholar]
- Brotman DJ, Girod JP, Posch A, Jani JT, Patel JV, Gupta M, et al. Effects of short-term glucocorticoids on hemostatic factors in healthy volunteers. Thromb Res. 2006;118:247–252. doi: 10.1016/j.thromres.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Brown AJ, Watts GF, Burnett JR, Dean RT, Jessup W. Sterol 27-hydroxylase acts on 7-ketocholesterol in human atherosclerotic lesions and macrophages in culture. J Biol Chem. 2000;275:27627–27633. doi: 10.1074/jbc.M004060200. [DOI] [PubMed] [Google Scholar]
- Brown RW, Diaz R, Robson AC, Kotelevtsev YV, Mullins JJ, Kaufman MH, et al. The ontogeny of 11beta-hydroxysteroid dehydrogenase type 2 and mineralocorticoid receptor gene expression reveal intricate control of glucocorticoid action in development. Endocrinology. 1996;137:794–797. doi: 10.1210/endo.137.2.8593833. [DOI] [PubMed] [Google Scholar]
- Bujalska IJ, Kumar S, Stewart PM. Does central obesity reflect ‘Cushing's Disease of the Omentum’? Lancet. 1997;349:1210–1213. doi: 10.1016/S0140-6736(96)11222-8. [DOI] [PubMed] [Google Scholar]
- Cai TQ, Wong BM, Mundt SS, Thieringer R, Wright SD, Hermanowski-Vosatka A. Induction of 11beta-hydroxysteroid dehydrogenase type 1 but not type 2 in human aortic smooth muscle cells by inflammatory stimuli. J Steroid Biochem. 2001;77:117–122. doi: 10.1016/s0960-0760(01)00041-3. [DOI] [PubMed] [Google Scholar]
- Caprio M, Newfell BG, la Sala A, Baur W, Fabbri A, Rosano G, et al. Functional mineralocorticoid receptors in human vascular endothelial cells regulate intercellular adhesion molecule-1 expression and promote leukocyte adhesion. Circ Res. 2008;102:1359–1367. doi: 10.1161/CIRCRESAHA.108.174235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capron L, Heudes D, Chajara A, Bruneval P. Effect of ramipril, an inhibitor of angiotensin converting enzyme, on the response of rat thoracic aorta to injury with a balloon catheter. J Cardiovasc Pharmacol. 1991;18:207–211. doi: 10.1097/00005344-199108000-00005. [DOI] [PubMed] [Google Scholar]
- Cavallero C, Ditondo U, Mingazzini PL, Nicosia R, Pericoli MN, Sarti P, et al. Cell-proliferation in atherosclerotic plaques of cholesterol-fed rabbits .3. histological and autoradiographic observations on glucocorticoids-treated rabbits. Atherosclerosis. 1976;25:145–152. doi: 10.1016/0021-9150(76)90020-4. [DOI] [PubMed] [Google Scholar]
- Chapman KE, Coutinho A, Gray M, Gilmour JS, Savill JS, Seckl JR. Local amplification of glucocorticoids by 11 beta-hydroxysteroid dehydrogenase type 1 and its role in the inflammatory response. Ann NY Acad Sci. 2006;1088:265–273. doi: 10.1196/annals.1366.030. [DOI] [PubMed] [Google Scholar]
- Christy C, Hadoke PWF, Paterson JM, Mullins JJ, Seckl JR, Walker BR. 11 Beta-hydroxysteroid dehydrogenase type 2 in mouse aorta – localization and influence on response to glucocorticoids. Hypertension. 2003;42:580–587. doi: 10.1161/01.HYP.0000088855.06598.5B. [DOI] [PubMed] [Google Scholar]
- Clark AF, Tandler B, Vignos PJ., Jr Glucocorticoid-induced alterations in the rabbit heart. Lab Invest. 1982;47:603–610. [PubMed] [Google Scholar]
- Colao A, Pivonello R, Spiezia S, Faggiano A, Ferone D, Filippella M, et al. Persistence of increased cardiovascular risk in patients with Cushing's disease after five years of successful cure. J Clin Endocrinol Metab. 1999;84:2664–2672. doi: 10.1210/jcem.84.8.5896. [DOI] [PubMed] [Google Scholar]
- Conn JW, Rovner DR, Cohen EL. Licorice-induced pseudoaldosteronism. JAMA. 1968;205:495–496. doi: 10.1001/jama.205.7.492. [DOI] [PubMed] [Google Scholar]
- Constantinides P, Hospes D, Gutmannauersperg N, Williams K. Estriol and prednisolone in rabbit atherosclerosis. Arch Pathol. 1962;73:277. [PubMed] [Google Scholar]
- Costenbader KH, Liang MH, Chibnik LB, Aizer J, Kwon H, Gall V, et al. A pravastatin dose-escalation study in systemic lupus erythematosus. Rheumatol Int. 2007;27:1071–1077. doi: 10.1007/s00296-007-0341-6. [DOI] [PubMed] [Google Scholar]
- Crum R, Szabo S, Folkman J. A new class of steroids inhibits angiogenesis in the presence of heparin or a heparin fragment. Science. 1985;230:1375–1378. doi: 10.1126/science.2416056. [DOI] [PubMed] [Google Scholar]
- Csonka GW, Murray M. Clinical evaluation of carbenoxolone in balanitis. Br J Vener Dis. 1971;47:179–181. doi: 10.1136/sti.47.3.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallman MF, Strack AM, Akana SF, Bradbury MJ, Hanson ES, Scribner KA, et al. Feast and famine: critical role of glucocorticoids with insulin in daily energy flow. Front Neuroendocrinol. 1993;14:303–347. doi: 10.1006/frne.1993.1010. [DOI] [PubMed] [Google Scholar]
- Davis EA, Morris DJ. Medicinal uses of licorice through the millenia: the good and plenty of it. Mol Cell Endocrinol. 1991;78:1–6. doi: 10.1016/0303-7207(91)90179-v. [DOI] [PubMed] [Google Scholar]
- Davis JM, Kremers HM, Crowson CS, Nicola PJ, Ballman KV, Therneau TM, et al. Glucocorticoids and cardiovascular events in rheumatoid arthritis – a population-based cohort study. Arthritis Rheum. 2007;56:820–830. doi: 10.1002/art.22418. [DOI] [PubMed] [Google Scholar]
- Deitemeyer D, Yunker RL, Ashraf M, Subbiah MTR. Effect of glucocorticoid administration early in life on aortic prostaglandin synthesis and morphology in atherosclerosis-susceptible pigeons. Exp Clin Endocrinol. 1985;85:147–154. doi: 10.1055/s-0029-1210430. [DOI] [PubMed] [Google Scholar]
- Dhawan L, Liu B, Blaxall BC, Taubman MB. A novel role for the glucocorticoid receptor in the regulation of monocyte chemoattractant protein-1 MRNA stability. J Biol Chem. 2007;282:10146–10152. doi: 10.1074/jbc.M605925200. [DOI] [PubMed] [Google Scholar]
- Dodic M, May CN, Wintour EM, Coghlan JP. An early prenatal exposure to excess glucocorticoid leads to hypertensive offspring in sheep. Clin Sci. 1998;94:149–155. doi: 10.1042/cs0940149. [DOI] [PubMed] [Google Scholar]
- Dodic M, Samuel C, Moritz K, Wintour EM, Morgan J, Grigg L, et al. Impaired cardiac functional reserve and left ventricular hypertrophy in adult sheep after prenatal dexamethasone exposure. Circ Res. 2001;89:623–629. doi: 10.1161/hh1901.097086. [DOI] [PubMed] [Google Scholar]
- Dover AR, Hadoke PWF, Macdonald LJ, Miller E, Newby DE, Walker BR. Intravascular glucocorticoid metabolism during inflammation and injury in mice. Endocrinology. 2007;148:166–172. doi: 10.1210/en.2006-0996. [DOI] [PubMed] [Google Scholar]
- Edwards CRW, Stewart PM, Burt D, Brett L, McIntyre MA, Sutanto WS, et al. Localisation of 11β-hydroxysteroid dehydrogenase- tissue specific protector of the mineralocorticoid receptor. Lancet. 1988;ii:986–989. doi: 10.1016/s0140-6736(88)90742-8. [DOI] [PubMed] [Google Scholar]
- Edwards G, Feletou M, Gardener MJ, Thollon C, Vanhoutte PM, Weston AH. Role of gap junctions in the responses to EDHF in rat and guinea-pig small arteries. Br J Pharmacol. 1999;128:1788–1794. doi: 10.1038/sj.bjp.0703009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt D, Weber MM. Therapy of Cushings-syndrome with steroid-biosynthesis inhibitors. J Steroid Biochem Mol Biol. 1994;49:261–267. doi: 10.1016/0960-0760(94)90267-4. [DOI] [PubMed] [Google Scholar]
- Etheridge EM, Hochligeti C. Lipid deposition in aortas in younger age groups following cortisone and adrenocorticotrophic hormone. Am J Pathol. 1952;28:315–319. [PMC free article] [PubMed] [Google Scholar]
- Etxabe J, Vazquez JA. Morbidity and mortality in cushings-disease – an epidemiologic approach. Clin Endocrinol (Oxf) 1994;40:479–484. doi: 10.1111/j.1365-2265.1994.tb02486.x. [DOI] [PubMed] [Google Scholar]
- Faggiano A, Pivonello R, Spiezia S, De Martino MC, Filippella M, Di Somma C, et al. Cardiovascular risk factors and common carotid artery caliber and stiffness in patients with Cushing's disease during active disease and 1 year after disease remission. J Clin Endocrinol Metab. 2003;88:2527–2533. doi: 10.1210/jc.2002-021558. [DOI] [PubMed] [Google Scholar]
- Fardet L, Flahault A, Kettaneh A, Tiev KP, Généreau T, Tolédano C, Lebbé C, Cabane J. Corticosteroid-induced clinical adverse events: frequency, risk factors and patient's opinion. Brit J Dematol. 2007;157:142–148. doi: 10.1111/j.1365-2133.2007.07950.x. [DOI] [PubMed] [Google Scholar]
- Fasshauer M, Klein J, Kralisch S, Klier M, Lossner U, Bluher M, et al. Monocyte chemoattractant protein 1 expression is stimulated by growth hormone and interleukin-6 in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 2004;317:598–604. doi: 10.1016/j.bbrc.2004.03.090. [DOI] [PubMed] [Google Scholar]
- Ferrero V, Ribichini F, Pesarini G, Brunelleschi S, Vassanelli C. Glucocorticoids in the prevention of restenosis after coronary angioplasty – therapeutic potential. Drugs. 2007a;67:1243–1255. doi: 10.2165/00003495-200767090-00001. [DOI] [PubMed] [Google Scholar]
- Ferrero V, Ribichini F, Rognoni A, Marino P, Brunelleschi S, Vassanelli C. Comparison of efficacy and safety of lower-dose to higher-dose oral prednisone after percutaneous coronary interventions (the IMPRESS-LD Study) Am J Cardiol. 2007b;99:1082–1086. doi: 10.1016/j.amjcard.2006.11.064. [DOI] [PubMed] [Google Scholar]
- Fischer-Betz R, Beer S, Schneider M. Considering systemic lupus erythematosus as example for accelerated atherosclerosis in rheumatoid diseases – what is the consequence? Z Rheumatol. 2005;64:229–238. doi: 10.1007/s00393-005-0733-5. [DOI] [PubMed] [Google Scholar]
- Fishel RS, Eisenberg S, Shai SY, Redden RA, Bernstein KE, Berk BC. Glucocorticoids induce angiotensin-converting enzyme expression in vascular smooth-muscle. Hypertension. 1995;25:343–349. doi: 10.1161/01.hyp.25.3.343. [DOI] [PubMed] [Google Scholar]
- Folkman J, Ingber DE. Angiostatic steroids. Method of discovery and mechanism of action. Ann Surg. 1987;206:374–383. doi: 10.1097/00000658-198709000-00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkman J, Langer R, Linhardt RJ, Haudenschild C, Taylor S. Angiogenesis inhibition and tumor regression caused by heparin or a heparin fragment in the presence of cortisone. Science. 1983;221:719–725. doi: 10.1126/science.6192498. [DOI] [PubMed] [Google Scholar]
- Fujiyama J, Kuriyama M, Arima S, Shibata Y, Nagata K, Takenaga S, et al. Atherogenic risk-factors in cerebrotendinous xanthomatosis. Clin Chim Acta. 1991;200:1–11. doi: 10.1016/0009-8981(91)90328-a. [DOI] [PubMed] [Google Scholar]
- Funder J, Myles K. Exclusion of corticosterone from epithelial mineralocorticoid receptors is insufficient for selectivity of aldosterone action: in vivo binding studies. Endocrinol. 1996;137:5264–5268. doi: 10.1210/endo.137.12.8940344. [DOI] [PubMed] [Google Scholar]
- Funder JW, Pearce PT, Smith R, Smith AI. Mineralocorticoid action: target tissue specificity is enzyme, not receptor, mediated. Science. 1988;242:583–585. doi: 10.1126/science.2845584. [DOI] [PubMed] [Google Scholar]
- Gao H-B, Ge R-S, Lakshmi V, Marandici AN, Hardy P. Hormonal regulation of oxidative and reductive activities of 11beta-hydroxysteroid dehydrogenase in rat leydig cells. Endocrinology. 1997;138:156–161. doi: 10.1210/endo.138.1.4837. [DOI] [PubMed] [Google Scholar]
- Gao Y, Zhou H, Raj JU. Antenatal betamethasone therapy potentiates nitric oxide-mediated relaxation of preterm ovine coronary arteries. Am J Physiol Heart Circ Physiol. 1996;270:H538–H554. doi: 10.1152/ajpheart.1996.270.2.H538. [DOI] [PubMed] [Google Scholar]
- Gerritsen ME, Rosenbaum RM. Regulation of rabbit coronary microvessel endothelial cell (rcme) prostaglandin synthesis by glucocorticoids. Microvasc Res. 1985;29:222. [Google Scholar]
- Gilmour JS, Coutinho AE, Cailhier JF, Man TY, Clay M, Thomas G, et al. Local amplification of glucocorticoids by 11 beta-hydroxysteroid dehydrogenase type 1 promotes macrophage phagocytosis of apoptotic leukocytes. J Immunol. 2006;176:7605–7611. doi: 10.4049/jimmunol.176.12.7605. [DOI] [PubMed] [Google Scholar]
- Goldberg GS, Moreno AP, Bechberger JF, Hearn SS, Shivers RR, MacPhee DJ, et al. Evidence that disruption of connexon particle arrangements in gap junction plaques is associated with inhibition of gap junctional communication by a glycyrrhetinic acid derivative. Exp Cell Res. 1996;222:48–53. doi: 10.1006/excr.1996.0006. [DOI] [PubMed] [Google Scholar]
- Golestaneh N, Klein C, Valamanesh F, Suarez G, Agarwal MK, Mirshahi M. Mineralocorticoid receptor-mediated signaling regulates the ion gated sodium channel in vascular endothelial cells and requires an intact cytoskeleton. Biochem Biophys Res Commun. 2001;280:1300–1306. doi: 10.1006/bbrc.2001.4275. [DOI] [PubMed] [Google Scholar]
- Goncharova EA, Billington CK, Irani C, Vorotnikov AV, Tkachuk VA, Penn RB, et al. Cyclic AMP-mobilizing agents and glucocorticoids modulate human smooth muscle cell migration. Am J Respir Cell Mol Biol. 2003;29:19–27. doi: 10.1165/rcmb.2002-0254OC. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Juanatey C, Lorez Diaz MJ, Martin J, Llorca J, Gonzalez-Gay MA. Atherosclerosis in patients with biopsy-proven giant cell artertis. Arthritis Rheum. 2007;57:1481–1486. doi: 10.1002/art.23114. [DOI] [PubMed] [Google Scholar]
- Gordon D, Kobernick SD, Mcmillan GC, Duff GL. The effect of cortisone on the serum lipids and on the development of experimental cholesterol atherosclerosis in the rabbit. J Exp Med. 1954;99:371–386. doi: 10.1084/jem.99.4.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman LALV, Song CX, Jang YS, Lincoff AM, Levy R, Topol EJ. Local intraluminal infusion of biodegradable polymeric nanoparticles – a novel approach for prolonged drug delivery after balloon angioplasty. Circulation. 1996;94:1441–1448. doi: 10.1161/01.cir.94.6.1441. [DOI] [PubMed] [Google Scholar]
- Hadoke PW, Macdonald L, Logie JJ, Small GR, Dover AR, Walker BR. Intra-vascular glucocorticoid metabolism as a modulator of vascular structure and function. Cell Mol Life Sci. 2006;63:565–578. doi: 10.1007/s00018-005-5427-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadoke PWF, Christy C, Kotelevtsev YV, Williams BC, Kenyon CJ, Seckl JR, et al. Endothelial cell dysfunction in mice after transgenic knockout of type 2, but not type 1, 11β-hydroxysteroid dehydrogenase. Circulation. 2001;104:2832–2837. doi: 10.1161/hc4801.100077. [DOI] [PubMed] [Google Scholar]
- Hafezi-Moghadam A, Simoncini T, Yang E, Limbourg FP, Plumier JC, Rebsamen MC, et al. Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat Med. 2002;8:473–479. doi: 10.1038/nm0502-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafstrom I, Rohani M, Deneberg S, Wornert M, Jogestrand T, Frostegard J. Effects of low-dose prednisolone on endothelial function, atherosclerosis, and traditional risk factors for atherosclerosis in patients with rheumatoid arthritis – a randomized study. J Rheumatol. 2007;34:1810–1816. [PubMed] [Google Scholar]
- Hammami MM, Siiteri PK. Regulation of 11β-hydroxysteroid dehydrogenase activity in human skin fibroblasts: enzymatic modulation of glucocorticoid action. J Clin Endocrinol Metab. 1991;73:326–334. doi: 10.1210/jcem-73-2-326. [DOI] [PubMed] [Google Scholar]
- Hammond GL, Smith CL, Paterson NAM, Sibbald WJ. A role for corticosteroid-binding globulin in delivery of cortisol to activated neutrophils. J Clin Endocrinol Metab. 1990;71:34–39. doi: 10.1210/jcem-71-1-34. [DOI] [PubMed] [Google Scholar]
- Harada E, Yoshimura M, Yasue H, Nakagawa O, Nakagawa M, Harada M, et al. Aldosterone induces angiotensin-converting-enzyme gene expression in cultured neonatal rat cardiocytes. Circulation. 2001;104:137–139. doi: 10.1161/01.cir.104.2.137. [DOI] [PubMed] [Google Scholar]
- Hasan Q, Tan ST, Gush J, Peters SG, Davis PF. Steroid therapy of a proliferating hemangioma: histochemical and molecular changes. Pediatrics. 2000;105:117–120. doi: 10.1542/peds.105.1.117. [DOI] [PubMed] [Google Scholar]
- Hasan Q, Tan ST, Xu B, Davis PF. Effects of five commonly used glucocorticoids on haemangioma in vitro. Clin Exp Pharmacol Physiol. 2003;30:140–144. doi: 10.1046/j.1440-1681.2003.03815.x. [DOI] [PubMed] [Google Scholar]
- Hatakeyama H, Miyamori I, Fujita T, Takeda Y, Takeda R, Yamamoto H. Vascular aldosterone – biosynthesis and a link to angiotensin-ii-induced hypertrophy of vascular smooth-muscle cells. J Biol Chem. 1994a;269:24316–24320. [PubMed] [Google Scholar]
- Hatakeyama H, Miyamori I, Fujita T, Takeda Y, Takeda R, Yamamoto H. Vascular aldosterone – biosynthesis and a link to angiotensin-ii-induced hypertrophy of vascular smooth-muscle cells. J Biol Chem. 1994b;269:24316–24320. [PubMed] [Google Scholar]
- Hatakeyama H, Inaba S, Miyamori I. 11Beta-hydroxysteroid dehydrogenase in cultured human vascular cells: possible role in the development of hypertension. Hypertension. 1999;33:1179–1184. doi: 10.1161/01.hyp.33.5.1179. [DOI] [PubMed] [Google Scholar]
- Hatanaka E, Furlaneto CJ, Ribeiro FP, Souza GM, Campa A. Serum amyloid a-induced MRNA expression and release of tumor necrosis factor-alpha (TNF-alpha) in human neutrophils. Immunol Lett. 2004;91:33–37. doi: 10.1016/j.imlet.2003.09.011. [DOI] [PubMed] [Google Scholar]
- Heasman SJ, Giles KM, Ward C, Rossi AG, Haslett C, Dransfield I. Glucocorticoid-mediated regulation of granulocyte apoptosis and macrophage phagocytosis of apoptotic cells: implications for the resolution of inflammation. J Endocrinol. 2003;178:29–36. doi: 10.1677/joe.0.1780029. [DOI] [PubMed] [Google Scholar]
- Hermanowski-Vosatka A, Gerhold D, Mundt SS, Loving VA, Lu M, Chen Y, et al. PPARalpha agonists reduce 11beta-hydroxysteroid dehydrogenase type 1 in the liver. Biochem Biophys Res Commun. 2000;279:330–336. doi: 10.1006/bbrc.2000.3966. [DOI] [PubMed] [Google Scholar]
- Hermanowski-Vosatka A, Balkovec JM, Cheng K, Chen HY, Hernandez M, Koo GC, et al. 11 Beta-HSD1 inhibition ameliorates metabolic syndrome and prevents progression of atherosclerosis in mice. J Exp Med. 2005;202:517–527. doi: 10.1084/jem.20050119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann R, Radke P, Weber C, Ortlepp J, Blindt R, Haager P, et al. Failure of a high-dose dexamethasone-eluting stent to inhibit neointimal hyperplasia and restenosis. Eur Heart J. 2004a;25:525–526. [Google Scholar]
- Hoffmann R, Langenberg R, Radke P, Franke A, Blindt R, Ortlepp J, et al. Evaluation of a high-dose dexamethasone-eluting stent. Am J Cardiol. 2004b;94:193–195. doi: 10.1016/j.amjcard.2004.03.061. [DOI] [PubMed] [Google Scholar]
- Hosfield DJ, Wu YQ, Skene RJ, Hilgers M, Jennings A, Snell GP, et al. Conformational flexibility in crystal structures of human 11 beta-hydroxysteroid dehydrogenase type I provide insights into glucocorticoid interconversion and enzyme regulation. J Biol Chem. 2005;280:4639–4648. doi: 10.1074/jbc.M411104200. [DOI] [PubMed] [Google Scholar]
- Hubener HJ, Fukushima DK, Gallagher TF. Substrate specificity of enzymes reducing the 11-keto and 20-keto groups of steroids. J Biol Chem. 1956;220:499–511. [PubMed] [Google Scholar]
- Hughes KA, Webster SP, Walker BR. 11-Beta-hydroxysteroid dehydrogenase type 1 (11 beta-HSD1) inhibitors in type 2 diabetes mellitus and obesity. Expert Opin Investig Drugs. 2008;17:481–496. doi: 10.1517/13543784.17.4.481. [DOI] [PubMed] [Google Scholar]
- Hult M, Elleby B, Shafqat N, Svensson S, Rane A, Jornvall H, et al. Human and rodent type 1 11 beta-hydroxysteroid dehydrogenases are 7 beta-hydroxycholesterol dehydrogenases involved in oxysterol metabolism. Cell Mol Life Sci. 2004a;61:992–999. doi: 10.1007/s00018-003-3476-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hult M, Elleby B, Shafqat N, Svensson S, Rane A, Jornvall H, et al. Human and rodent type 1 11beta-hydroxysteroid dehydrogenases are 7beta-hydroxycholesterol dehydrogenases involved in oxysterol metabolism. Cell Mol Life Sci. 2004b;61:992–999. doi: 10.1007/s00018-003-3476-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H, Umesono K, Nishimori T, Hirata Y, Tanabe T. Glucocorticoid-mediated suppression of the promoter activity of the cyclooxygenase-2 gene is modulated by expression of its receptor in vascular endothelial cells. Biochem Biophys Res Commun. 1999;254:292–298. doi: 10.1006/bbrc.1998.9939. [DOI] [PubMed] [Google Scholar]
- Ishii T, Masuzaki H, Tanaka T, Arai N, Yasue S, Kobayashi N, et al. Augmentation of 11 beta-hydroxysteroid LPS-activated J774.1 macrophages dehydrogenase type 1 in – role of 11 beta-HSD1 in pro-inflammatory properties in macrophages. Febs Lett. 2007;581:349–354. doi: 10.1016/j.febslet.2006.11.032. [DOI] [PubMed] [Google Scholar]
- Iuchi T, Akaike M, Mitsui T, Ohshima Y, Shintani Y, Azuma H, et al. Glucocorticoid excess induces superoxide production in vascular endothelial cells and elicits vascular endothelial dysfunction. Circ Res. 2003;92:81–87. doi: 10.1161/01.res.0000050588.35034.3c. [DOI] [PubMed] [Google Scholar]
- Jain S, Pick R, Katz LN. Benefit from testosterone and hydrocortisone on coronary atherogenesis in cockerels on a low protein atherogenic diet. Circ Res. 1965;17:492. doi: 10.1161/01.res.17.6.492. [DOI] [PubMed] [Google Scholar]
- Jamieson PM, Chapman KE, Edwards CRW, Seckl JR. 11β-hydroxysteroid dehydrogenase is an exclusive 11β-reductase in primary cultures of rat hepatocytes: effect of physicochemical and hormonal manipulations. Endocrinology. 1995;136:4754–4761. doi: 10.1210/endo.136.11.7588203. [DOI] [PubMed] [Google Scholar]
- Jamieson PM, Fuchs E, Flugge G, Seckl JR. Attenuation of hippocampal 11beta-hydroxysteroid dehydrogenase type 1 by chronic psychosocial stress in the tree shrew. Stress. 1997;2:123–132. doi: 10.3109/10253899709014743. [DOI] [PubMed] [Google Scholar]
- Jamieson PM, Walker BR, Chapman KE, Rossiter S, Seckl JR. 11β-hydroxysteroid dehydrogenase type 1 is a predominant 11-reductase in the intact perfused rat liver. J Endocrinol. 2000;175:685–692. doi: 10.1677/joe.0.1650685. [DOI] [PubMed] [Google Scholar]
- Jeremy JY, Dandona P. Inhibition by hydrocortisone of prostacyclin synthesis by rat aorta and its reversal with Ru486. Endocrinology. 1986;119:661–665. doi: 10.1210/endo-119-2-661. [DOI] [PubMed] [Google Scholar]
- Jilma B, Blann AD, Stohlawetz P, Eichler HG, Kautzky-Willer A, Wagner OF. Dexamethasone lowers circulating E-selectin and ICAM-1 in healthy men. J Lab Clin Med. 2000;135:270–274. doi: 10.1067/mlc.2000.105214. [DOI] [PubMed] [Google Scholar]
- Johns DG, Dorrance AM, Tramontini NL, Webb RC. Glucocorticoids inhibit tetrahydrobiopterin-dependent endothelial function. Exp Biol Med. 2001;226:27–31. doi: 10.1177/153537020122600104. [DOI] [PubMed] [Google Scholar]
- Jun SS, Chen Z, Pace MC, Shaul PW. Glucocorticoids downregulate cyclooxygenase-1 gene expression and prostacyclin synthesis in fetal pulmonary artery endothelium. Circ Res. 1999;84:193–200. doi: 10.1161/01.res.84.2.193. [DOI] [PubMed] [Google Scholar]
- Kakio T, Matsumori A, Ohashi N, Yamada T, Saito T, Kawamoto A, et al. Hydrocortisone reduces restenosis after stenting of small coronary arteries. J Interv Cardiol. 2004;17:295–300. doi: 10.1111/j.1540-8183.2004.02999.x. [DOI] [PubMed] [Google Scholar]
- Kanat M, Sipahioglu M, Arinc H, Serin E, Yildiz O, Tunckale A, et al. Is lipid lowering treatment aiming for very low LDL levels safe in terms of the synthesis of steroid hormones? Med Hypotheses. 2007;69:104–112. doi: 10.1016/j.mehy.2006.10.058. [DOI] [PubMed] [Google Scholar]
- Kardon T, Senesi S, Marcolongo P, Legeza B, Banhegyi G, Mandl J, et al. Maintenance of luminal NADPH in the endoplasmic reticulum promotes the survival of human neutrophil granulocytes. Febs Lett. 2008;582:1809–1815. doi: 10.1016/j.febslet.2008.04.045. [DOI] [PubMed] [Google Scholar]
- Karim MA, Frizzell S, Inman S, Shinn L, Miller M. In vivo role of glucocorticoids in barotrauma, vascular repair and fibrosis. J Mol Cell Cardiol. 1997;29:1111–1122. doi: 10.1006/jmcc.1996.0330. [DOI] [PubMed] [Google Scholar]
- Kato H, Hayashi T, Koshino Y, Oida K, Kutsumi Y, Nakai T, et al. Autocrine production of endothelin-1 participates in the glucocorticoid-induced Ca2+ influx into vascular smooth-muscle cells. Biochem Biophys Res Commun. 1995;208:82–88. doi: 10.1006/bbrc.1995.1308. [DOI] [PubMed] [Google Scholar]
- Kawai Y, Hayashi T, Eguchi K, Asazuma K, Masamura K, Iwamuro A, et al. Effects of brief glucocorticoid exposure on growth of vascular smooth muscle cells in culture. Biochem Biophys Res Commun. 1998;245:493–496. doi: 10.1006/bbrc.1998.8462. [DOI] [PubMed] [Google Scholar]
- Khan IY, Dekou V, Douglas G, Jensen R, Hanson MA, Poston L, et al. A high-fat diet during rat pregnancy or suckling induces cardiovascular dysfunction in adult offspring. Am J Physiol Regul Integr Comp Physiol. 2005;288:R127–R133. doi: 10.1152/ajpregu.00354.2004. [DOI] [PubMed] [Google Scholar]
- Kim KW, Wang ZL, Busby J, Tsuruda T, Chen M, Hale C, et al. The role of tyrosine 177 in human 11 beta-hydroxysteroid dehydrogenase type 1 in substrate and inhibitor binding: an unlikely hydrogen bond donor for the substrate. Biochim Biophys Acta. 2006a;1764:824–830. doi: 10.1016/j.bbapap.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Kim WJ, Bae EM, Kang YJ, Bae HU, Hong SH, Lee JY, et al. Glucocorticoid-induced tumour necrosis factor receptor family related protein (GITR) mediates inflammatory activation of macrophages that can destabilize atherosclerotic plaques. Immunology. 2006b;119:421–429. doi: 10.1111/j.1365-2567.2006.02453.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi A, Tazawa C, Miki Y, Darnel AD, Suzuki T, Ohta Y, et al. The possible roles of mineralocorticoid receptor and 11 beta-hydroxy steroid dehydrogenase type 2 in cardiac fibrosis in the spontaneously hypertensive rat. J Steroid Biochem Mol Biol. 2003;85:439–442. doi: 10.1016/s0960-0760(03)00198-5. [DOI] [PubMed] [Google Scholar]
- Kornel L, Ramsay C, Kanamarlapudi N, Travers T, Packer W. Evidence for the presence in arterial-walls of intracellular-molecular mechanism for action of mineralocorticoids. Clin Exp Hypertens A. 1982;4:1561–1582. doi: 10.3109/10641968209061625. [DOI] [PubMed] [Google Scholar]
- Kotelevtsev YV, Holmes MC, Burchell A, Houston PM, Scholl D, Jamieson PM, et al. 11β-hydroxysteroid dehydrogenase type 1 knockout mice show attenuated glucocorticoid inducible responses and resist hyperglycaemia on obesity and stress. Proc Natl Acad Sci USA. 1997;94:14924–14929. doi: 10.1073/pnas.94.26.14924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotelevtsev YV, Brown RW, Fleming S, Edwards CRW, Seckl JR, Mullins JJ. Hypertension in mice caused by inactivation of 11β-hydroxysteroid dehydrogenase type 2. J Clin Invest. 1999;103:683–689. doi: 10.1172/JCI4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremers HM, Reinalda MS, Crowson CS, Davis JM, Hunder GG, Gabriel SE. Glucocorticoids and cardiovascular and cerebrovascular events in polymyalgia rheumatica. Arthritis Rheum. 2007;57:279–286. doi: 10.1002/art.22548. [DOI] [PubMed] [Google Scholar]
- Krozowski ZS, Funder JW. Renal mineralocorticoid receptors and hippocampal corticosterone-binding species have identical intrinsic steroid specificity. Proc Natl Acad Sci USA. 1983;80:6056–6060. doi: 10.1073/pnas.80.19.6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Mear NS, MacGilvray SS, Myers TF. Dexamethasone-induced myocardial hypertrophy in neonatal rats. Biol Neonate. 1997;72:175–180. doi: 10.1159/000244481. [DOI] [PubMed] [Google Scholar]
- Lakshmi V, Monder C. Purification and characterization of the corticosteroid 11β-dehydrogenase component of the rat liver 11β-hydroxysteroid dehydrogenase complex. Endocrinology. 1988;123:2390–2398. doi: 10.1210/endo-123-5-2390. [DOI] [PubMed] [Google Scholar]
- Lamireau D, Nuyt AM, Hou X, Bernier S, Beauchamp M, Gobeil F, et al. Altered vascular function in fetal programming of hypertension. Stroke. 2002;33:2992–2998. doi: 10.1161/01.str.0000039340.62995.f2. [DOI] [PubMed] [Google Scholar]
- Langford HG, Snavely JR. Effect of DCA on development of renoprival hypertension. Am J Physiol. 1959;196:449–450. doi: 10.1152/ajplegacy.1959.196.2.449. [DOI] [PubMed] [Google Scholar]
- Latif SA, Hartman LR, Souness GW, Morris DJ. Possible endogenous regulators of steroid inactivating enzymes and glucocorticoid-induced Na+ retention. Steroids. 1994;59:352–356. doi: 10.1016/0039-128x(94)90001-9. [DOI] [PubMed] [Google Scholar]
- Lee CW, Chae JK, Lim HY, Hong MK, Kim JJ, Park SW, et al. Prospective randomized trial of corticosteroids for the prevention of restenosis after intracoronary stent implantation. Am Heart J. 1999;138:60–63. doi: 10.1016/s0002-8703(99)70247-4. [DOI] [PubMed] [Google Scholar]
- Lefer AM. Influence of corticosteroids on mechanical performance of isolated rat papillary muscles. Am J Physiol. 1968;214:518–524. doi: 10.1152/ajplegacy.1968.214.3.518. [DOI] [PubMed] [Google Scholar]
- Lesage J, Blondeau B, Grino M, Breant B, Dupouy JP. Maternal undernutrition during late gestation induces foetal over-exposure to glucocorticoids and intra-iuterine growth retardation, and disturbs the hypothalamic-pituitary adrenal axis in the newborn rat. Endocrinology. 2001;142:1692–1702. doi: 10.1210/endo.142.5.8139. [DOI] [PubMed] [Google Scholar]
- Li KZ, Obeyesekere VR, Krozowski ZS, Ferrari P. Oxoreductase and dehydrogenase activities of the human and rat 11beta-hydroxysteroid dehydrogenase type 2 enzyme. Endocrinology. 1997;138:2948–2952. doi: 10.1210/endo.138.7.5232. [DOI] [PubMed] [Google Scholar]
- Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- Lijnen P, Petrov V. Induction of cardiac fibrosis by aldosterone. J Mol Cell Cardiol. 2000;32:865–879. doi: 10.1006/jmcc.2000.1129. [DOI] [PubMed] [Google Scholar]
- Lijnen PJ, Petrov VV, Fagard RH. Induction of cardiac fibrosis by angiotensin II. Methods Find Exp Clin Pharmacol. 2000;22:709–723. doi: 10.1358/mf.2000.22.10.802287. [DOI] [PubMed] [Google Scholar]
- Lim HY, Muller N, Herold MJ, van den Brandt J, Reichardt HM. Glucocorticoids exert opposing effects on macrophage function dependent on their concentration. Immunology. 2007;122:47–53. doi: 10.1111/j.1365-2567.2007.02611.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima DSN, Sato EI, Lima VC, Miranda F, Hatta FH. Brachial endothelial function is impaired in patients with systemic lupus erythematosus. Journal of Rheumatology. 2002;29:292–297. [PubMed] [Google Scholar]
- Lincoff AM, Furst JG, Ellis SG, Tuch RJ, Topol EJ. Sustained local delivery of dexamethasone by a novel intravascular eluting stent to prevent restenosis in the porcine coronary injury model. J Am Coll Cardiol. 1997;29:808–816. doi: 10.1016/s0735-1097(96)00584-0. [DOI] [PubMed] [Google Scholar]
- Lister K, Autelitano DJ, Jenkins A, Hannan RD, Sheppard KE. Cross talk between corticosteroids and alpha-adrenergic signalling augments cardiomyocyte hypertrophy: a possible role for SGK11. Cardiovasc Res. 2006;70:555–565. doi: 10.1016/j.cardiores.2006.02.010. [DOI] [PubMed] [Google Scholar]
- Liu X, De Scheerder I, Desmet W. Dexamethasone-eluting stent: an anti-inflammatory approach to inhibit coronary restenosis. Expert Rev Cardiovasc Ther. 2004;2:653–660. doi: 10.1586/14779072.2.5.653. [DOI] [PubMed] [Google Scholar]
- Liu XS, Huang YM, Hanet C, Vandormael M, Legrand V, Dens J, et al. Study of antirestenosis with the biodivysio dexamethasone-eluting stent (STRIDE): a first-in-human multicenter pilot trial. Catheter Cardiovasc Interv. 2003;60:172–178. doi: 10.1002/ccd.10636. [DOI] [PubMed] [Google Scholar]
- Lombes M, Oblin MF, Gasc JM, Baulieu FE, Farman N, Bonvalet JP. Immunohistochemical and biochemical evidence for a cardiovascular mineralocorticoid receptor. Circ Res. 1992;71:503–510. doi: 10.1161/01.res.71.3.503. [DOI] [PubMed] [Google Scholar]
- Lombes M, Alfaidy N, Eugene E, Lessana A, Farman N, Bonvalet J-P. Prerequisite for cardiac aldosterone action: mineralocorticoid receptor and 11beta-hydroxysteroid dehydrogenase in the human hear. Circulation. 1995;92:175–182. doi: 10.1161/01.cir.92.2.175. [DOI] [PubMed] [Google Scholar]
- Longenecker JP, Kilty LA, Johnson LK. Glucocorticoid influence on growth of vascular wall cells in culture. J Cell Physiol. 1982;113:197–202. doi: 10.1002/jcp.1041130203. [DOI] [PubMed] [Google Scholar]
- Longenecker JP, Kilty LA, Johnson LK. Glucocorticoid inhibition of vascular smooth muscle cell proliferation: influence of homologous extracellular matrix and serum mitogens. J Cell Biol. 1984;98:534–540. doi: 10.1083/jcb.98.2.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low SC, Moisan M-P, Edwards CRW, Seckl JR. Glucocorticoids regulate 11β-hydroxysteroid dehydrogenase activity and gene expression in vivo in the rat. J Neuroendocrinol. 1994a;6:285–290. doi: 10.1111/j.1365-2826.1994.tb00584.x. [DOI] [PubMed] [Google Scholar]
- Low SC, Chapman KE, Edwards CRW, Wells T, Robinson ICAF, Seckl JR. Sexual dimorphism of hepatic 11β-hydroxysteroid dehydrogenase in the rat: the role of growth hormone patterns. J Endocrinol. 1994b;143:541–548. doi: 10.1677/joe.0.1430541. [DOI] [PubMed] [Google Scholar]
- Mccabe PM, Gonzales JA, Zaias J, Szeto A, Kumar M, Herron AJ, et al. Social environment influences the progression of atherosclerosis in the Watanabe heritable hyperlipidemic rabbit. Circulation. 2002;105:354–359. doi: 10.1161/hc0302.102144. [DOI] [PubMed] [Google Scholar]
- McColl A, Michlewska S, Dransfield I, Rossi AG. Effects of glucocorticoids on apoptosis and clearance of apoptotic cells. ScientificWorldJournal. 2007;7:1165–1181. doi: 10.1100/tsw.2007.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan PE, Gray GA, Sherry L, Webb DJ, Kenyon CJ. Differential effects of angiotensin II on cardiac cell proliferation and intramyocardial perivascular fibrosis in vivo 3. Circulation. 1998;98:2765–2773. doi: 10.1161/01.cir.98.24.2765. [DOI] [PubMed] [Google Scholar]
- McInnes KJ, Kenyon CJ, Chapman KE, Livingstone DEW, Macdonald LJ, Walker BR, et al. 5 Alpha-reduced glucocorticoids, novel endogenous activators of the glucocorticoid receptor. J Biol Chem. 2004;279:22908–22912. doi: 10.1074/jbc.M402822200. [DOI] [PubMed] [Google Scholar]
- Maddali KK, Korzick DH, Turk JR, Bowles DK. Lsoform-specific modulation of coronary artery PKC by glucocorticoids. Vascul Pharmacol. 2005;42:153–162. doi: 10.1016/j.vph.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Makheja AN, Bloom S, Muesing R, Simon T, Bailey JM. Anti-inflammatory drugs in experimental atherosclerosis. VII. spontaneous atherosclerosis in WHHL rabbits and inhibition by cortisone acetate. Atherosclerosis. 1989;76:155–161. doi: 10.1016/0021-9150(89)90099-3. [DOI] [PubMed] [Google Scholar]
- Mangos G, Walker BR, Kelly JJ, Lawson J, Webb DJ, Whitworth JA. Cortisol inhibits cholinergic dilatation in the human forearm: towards an explanation for glucocorticoid-induced hypertension. Am J Hypertens. 2000;13:1155–1160. doi: 10.1016/s0895-7061(00)01201-2. [DOI] [PubMed] [Google Scholar]
- Marumo T, Schini-Kerth VB, Brandes RP, Busse R. Glucocorticoids inhibit superoxide anion production and p22 phox MRNA expression in human aortic smooth muscle cells. Hypertension. 1998;32:1083–1088. doi: 10.1161/01.hyp.32.6.1083. [DOI] [PubMed] [Google Scholar]
- Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR, et al. A transgenic model of visceral obesity and the metabolic syndrome. Science. 2001;294:2166–2170. doi: 10.1126/science.1066285. [DOI] [PubMed] [Google Scholar]
- Masuzaki H, Yamamoto H, Kenyon CJ, Elmquist JK, Morton NM, Paterson JM, et al. Transgenic amplification of glucocorticoid action in adipose tissue causes high blood pressure in mice. J Clin Invest. 2003;112:83–90. doi: 10.1172/JCI17845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn FAO, Lloyd CJ, Kachel C, Funder JW. Induction by glucocorticoids of angiotensin converting enzyme production from bovine endothelial cells in culture and rat lung in vivo. J Clin Invest. 1982;70:684–692. doi: 10.1172/JCI110663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf BW, Levy MA, Holt DA. Inhibitors of steroid 5 alpha-reductase in benign prostatic hyperplasia, male pattern baldness and acne. Trends Pharmacol Sci. 1989;10:491–495. doi: 10.1016/0165-6147(89)90048-5. [DOI] [PubMed] [Google Scholar]
- Meyer WJ, Nicholls NR. Mineralocorticoid binding on cultured smooth muscle cells and fibroblasts from rat aorta. J Steroid Biochem. 1981;14:1157–1168. doi: 10.1016/0022-4731(81)90046-7. [DOI] [PubMed] [Google Scholar]
- Michael LH, Ballantyne CM, Zachariah JP, Gould KE, Pocius JS, Taffet GE, et al. Myocardial infarction and remodeling in mice: effect of reperfusion. Am J Physiol. 1999;277:H660–H668. doi: 10.1152/ajpheart.1999.277.2.H660. [DOI] [PubMed] [Google Scholar]
- Miller AM, McPhaden AR, Wadsworth RM, Wainwright CL. Inhibition of leukocyte depletion of neointima formation after balloon angioplasty in a rabbit model of restenosis. Cardiovasc Res. 2001;49:838–850. doi: 10.1016/s0008-6363(00)00304-7. [DOI] [PubMed] [Google Scholar]
- Miller JW, Crapo L. The medical-treatment of Cushings-syndrome. Endocr Rev. 1993;14:443–458. doi: 10.1210/edrv-14-4-443. [DOI] [PubMed] [Google Scholar]
- Mitsuya N, Kishi R, Suzuki N, Tamura M, Imai Y, Tanaka O, et al. Efficacy of steroid therapy for pacing failure in a patient with chronic myocarditis. Intern Med. 2004;43:213–217. doi: 10.2169/internalmedicine.43.213. [DOI] [PubMed] [Google Scholar]
- Molnar GA, Lindschau C, Dubrovska G, Mertens PR, Kirsch T, Quinkler M, et al. Glucocorticoid-related signaling effects in vascular smooth muscle cells. Hypertension. 2008;51:1372–1378. doi: 10.1161/HYPERTENSIONAHA.107.105718. [DOI] [PubMed] [Google Scholar]
- Monder C, Lakshmi V. Evidence for kinetically distinct forms of corticosteroid 11β-dehydrogenase in rat liver microsomes. J Steroid Biochem. 1989;32:77–83. doi: 10.1016/0022-4731(89)90017-4. [DOI] [PubMed] [Google Scholar]
- Montrella-Waybill M, Clore JN, Schoolwerth AC, Watlington CO. Evidence that high dose cortisol-induced Na+ retention in man is not mediated by the mineralocorticoid receptor. J Clin Endocrinol Metab. 1991;72:1060–1066. doi: 10.1210/jcem-72-5-1060. [DOI] [PubMed] [Google Scholar]
- Mori K, Shioi A, Jono S, Nishizawa Y, Morii H. Dexamethasone enhances in vitro vascular calcification by promoting osteoblastic differentiation of vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1999;19:2112–2118. doi: 10.1161/01.atv.19.9.2112. [DOI] [PubMed] [Google Scholar]
- Morin C, Asselin C, Boudreau F, Provencher PH. Transcriptional regulation of pre-pro-endothelin-1 gene by glucocorticoids in vascular smooth muscle cells. Biochem Biophys Res Commun. 1998;244:583–587. doi: 10.1006/bbrc.1998.8300. [DOI] [PubMed] [Google Scholar]
- Morris DJ, Souness GW. Endogenous 11 beta-hydroxysteroid dehydrogenase inhibitors and their role in glucocorticoid Na+ retention and hypertension. Endocr Res. 1996;22:793–801. doi: 10.1080/07435809609043778. [DOI] [PubMed] [Google Scholar]
- Morris DJ, Brem AS, Ge RS, Jellinck PH, Sakai RR, Hardy MP. The functional roles of 11 beta-HSD1: vascular tissue, testis and brain. Mol Cell Endocrinol. 2003;203:1–12. doi: 10.1016/s0303-7207(03)00094-7. [DOI] [PubMed] [Google Scholar]
- Morton NM, Holmes MC, Fievet C, Staels B, Tailleux A, Mullins JJ, et al. Improved lipid and lipoprotein profile, hepatic insulin sensitivity, and glucose tolerance in 11β-hydroxysteroid dehydrogenase type 1 null mice. J Biol Chem. 2001;276:41293–41300. doi: 10.1074/jbc.M103676200. [DOI] [PubMed] [Google Scholar]
- Murphy BEP, Clark SJ, Donald IR, Pinsky M, Vedady D. Conversion of maternal cortisol to cortisone during placental transfer to human fetus. Am J Obstet Gynecol. 1974;118:538–541. doi: 10.1016/s0002-9378(16)33697-3. [DOI] [PubMed] [Google Scholar]
- Nagata D, Hirata Y. Molecular mechanism of cardiovascular damage induced by aldosterone. Yakugaku Zasshi. 2007;127:1339–1346. doi: 10.1248/yakushi.127.1339. [DOI] [PubMed] [Google Scholar]
- Naito M, Yasue M, Asai K, Yamada K, Hayashi T, Kuzuya M, et al. Effects of dexamethasone on experimental atherosclerosis in cholesterol-fed rabbits. J Nutr Sci Vitaminol. 1992;38:255–264. doi: 10.3177/jnsv.38.255. [DOI] [PubMed] [Google Scholar]
- Nakano T, Raines EW, Abraham JA, Wenzel FG, Higashiyama S, Klagsbrun M, et al. Glucocorticoid inhibits thrombin-induced expression of platelet-derived growth-factor a-chain and heparin-binding epidermal growth factor-like growth-factor in human aortic smooth-muscle cells. J Biol Chem. 1993;268:22941–22947. [PubMed] [Google Scholar]
- Narayanan N, Yang C, Xu A. Dexamethasone treatment improves sarcoplasmic reticulum function and contractile performance in aged myocardium. Mol Cell Biochem. 2004;266:31–36. doi: 10.1023/b:mcbi.0000049130.58074.73. [DOI] [PubMed] [Google Scholar]
- Neumann S, Huse K, Semrau R, Diegeler A, Gebhardt R, Buniatian GH, et al. Aldosterone and D-glucose stimulate the proliferation of human cardiac myofibroblasts in vitro. Hypertension. 2002;39:756–760. doi: 10.1161/hy0302.105295. [DOI] [PubMed] [Google Scholar]
- Newton CJ, Ran G, Xie YX, Bilko D, Burgoyne CH, Adams I, et al. Statin-induced apoptosis of vascular endothelial cells is blocked by dexamethasone. J Endocrinol. 2002;174:7–16. doi: 10.1677/joe.0.1740007. [DOI] [PubMed] [Google Scholar]
- Nicosia RF, Ottinetti A. Growth of microvessels in serum-free matrix culture of rat aorta. A quantitative assay of angiogenesis in vitro. Lab Invest. 1990;63:115–122. [PubMed] [Google Scholar]
- Nuotio-Antar AM, Hachey DL, Hasty AH. Carbenoxolone treatment attenuates symptoms of metabolic syndrome and atherogenesis in obese, hyperlipidemic mice. Am J Physiol Endocrinol Metab. 2007;293:E1517–E1528. doi: 10.1152/ajpendo.00522.2007. [DOI] [PubMed] [Google Scholar]
- Oberleithner H, Schneider SW, Albermann L, Hillebrand U, Ludwig T, Riethmuller C, et al. Endothelial cell swelling by aldosterone. J Membr Biol. 2003;196:163–172. doi: 10.1007/s00239-003-0635-6. [DOI] [PubMed] [Google Scholar]
- Odermatt A, Arnold P, Frey FJ. The intracellular localization of the mineralocorticoid receptor is regulated by 11β-hydroxysteroid dehydrogenase type 2. J Biol Chem. 2001;276:28484–28492. doi: 10.1074/jbc.M100374200. [DOI] [PubMed] [Google Scholar]
- Ohtani T, Ohta M, Yamamoto K, Mano T, Sakata Y, Nishio M, et al. Elevated cardiac tissue level of aldosterone and mineralocorticoid receptor in diastolic heart failure: beneficial effects of mineralocorticoid receptor blocker. Am J Physiol Regul Integr Comp Physiol. 2007;292:R946–R954. doi: 10.1152/ajpregu.00402.2006. [DOI] [PubMed] [Google Scholar]
- Oppenheim E, Bruger M. Experimental cholesterol atherosclerosis .11. studies with vitamin-A. AMA Arch Pathol. 1952;53:520–522. [PubMed] [Google Scholar]
- Ortsater H, Alberts P, Warpman U, Engblom LOM, Abrahmsen L, Bergsten P. Regulation of 11 beta-hydroxysteroid dehydrogenase type 1 and glucose-stimulated insulin secretion in pancreatic islets of langerhans. Diabetes Metab Res Rev. 2005;21:359–366. doi: 10.1002/dmrr.525. [DOI] [PubMed] [Google Scholar]
- Oterdoom LH, de Vries APJ, Gansevoort RT, van Son WJ, van der Heide JJH, Ploeg RJ, et al. Determinants of insulin resistance in renal transplant recipients. Transplantation. 2007;83:29–35. doi: 10.1097/01.tp.0000245844.27683.48. [DOI] [PubMed] [Google Scholar]
- Otto C, Reichardt HM, Schutz G. Absence of glucocorticoid receptor-beta in mice. J Biol Chem. 1997;272:26665–26668. doi: 10.1074/jbc.272.42.26665. [DOI] [PubMed] [Google Scholar]
- Ouvrard-Pascaud A, Sainte-Marie Y, Benitah JP, Perrier R, Soukaseum C, Cat AN, et al. Conditional mineralocorticoid receptor expression in the heart leads to life-threatening arrhythmias. Circulation. 2005;111:3025–3033. doi: 10.1161/CIRCULATIONAHA.104.503706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painson JC, Thorner MO, Krieg RJ, Tannenbaum GS. Short-term adult exposure to estradiol feminizes the male pattern of spontaneous and growth hormone-releasing factor-stimulated growth-hormone secretion in the rat. Endocrinology. 1992;130:511–519. doi: 10.1210/endo.130.1.1345780. [DOI] [PubMed] [Google Scholar]
- Paterson JM, Morton NM, Fievet C, Kenyon CJ, Holmes MC, Staels B, et al. Metabolic syndrome without obesity: hepatic overexpression of 11beta-hydroxysteroid dehydrogenase type 1 in transgenic mice. Proc Natl Acad Sci USA. 2004;101:7088–7093. doi: 10.1073/pnas.0305524101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penefsky ZJ, Kahn M. Inotropic effects of dexamethasone in mammalian heart muscle1. Eur J Pharmacol. 1971;15:259–266. doi: 10.1016/0014-2999(71)90091-4. [DOI] [PubMed] [Google Scholar]
- Pepine CJ, Hirshfeld JW, Macdonald RG, Henderson MA, Bass TA, Goldberg S, et al. A controlled trial of corticosteroids to prevent restenosis after coronary angioplasty. Circulation. 1990;81:1753–1761. doi: 10.1161/01.cir.81.6.1753. [DOI] [PubMed] [Google Scholar]
- Perschel FH, Buhler H, Hierholzer K. Bile acids and their amidates inhibit 11β-hydroxysteroid dehydrogenase obtained from rat kidney. Pflug Arch. 1991;418:538–543. doi: 10.1007/BF00370568. [DOI] [PubMed] [Google Scholar]
- Petrichenko IE, Daret D, Kolpakova GV, Shakov YA, Larrue J. Glucocorticoids stimulate cholesteryl ester formation in human smooth muscle cells. Arterioscler Thromb Vasc Biol. 1997;17:1143–1151. doi: 10.1161/01.atv.17.6.1143. [DOI] [PubMed] [Google Scholar]
- Petrik PV, Law MMMWSC, Quinones-Baldrich W, Gelabert HA. Dexamethasone and enalapril suppress intimal hyperplasia individually but have no synergistic effect. Ann Vasc Surg. 1998;12:216–220. doi: 10.1007/s100169900143. [DOI] [PubMed] [Google Scholar]
- Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, Perez A, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized aldactone evaluation study investigators. N Engl J Med. 1999;341:709–717. doi: 10.1056/NEJM199909023411001. [DOI] [PubMed] [Google Scholar]
- Pitt B, Remme W, Zannad F, Neaton J, Martinez F, Roniker B, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348:1309–1321. doi: 10.1056/NEJMoa030207. [DOI] [PubMed] [Google Scholar]
- Pitzalis C, Pipitone N, Perretti M. Regulation of leukocyte-endothelial interactions by glucocorticoids. Ann NY Acad Sci. 2002;966:108–118. doi: 10.1111/j.1749-6632.2002.tb04208.x. [DOI] [PubMed] [Google Scholar]
- Poon M, Gertz SD, Fallon JT, Wiegman P, Berman JW, Sarembock IJ, et al. Dexamethasoneinhibits macrophage accumulation after balloon arterial injury in cholesterol-fed rabbits. Atherosclerosis. 2001;155:371–380. doi: 10.1016/s0021-9150(00)00605-5. [DOI] [PubMed] [Google Scholar]
- Powell JS, Clozel JP, Muller RKM, Kuhn H, Hefti F, Hosang M, Baumgartner HR. Inhibitors of angiotensin-converting enzyme prevent myointimal proliferation after vascular injury. Science. 1989;245:186–188. doi: 10.1126/science.2526370. [DOI] [PubMed] [Google Scholar]
- Prescott MF, Mcbride CK, Court M. Development of intimal lesions after leukocyte migration into the vascular wall. Am J Pathol. 1989;135:835–846. [PMC free article] [PubMed] [Google Scholar]
- Pujols L, Mullol J, Roca-Ferrer J, Torrego A, Xaubet A, Cidlowski JA, et al. Expression of glucocorticoid receptor alpha- and beta-isoforms in human cells and tissues. Am J Physiol Cell Physiol. 2002;283:C1324–C1331. doi: 10.1152/ajpcell.00363.2001. [DOI] [PubMed] [Google Scholar]
- Rab ST, Roubin GS, Carlin S, Hearn JA, Douglas JS. Coronoary aneurysms after stent placement – a suggestion of altered vessel wall healing in the presence of anti-inflammatory agents. J Am Coll Cardiol. 1991;18:1525–1528. doi: 10.1016/0735-1097(91)90685-3. [DOI] [PubMed] [Google Scholar]
- Rabasseda X, Silvestre J, Castaner J. Eplerenone: antihypertensive treatment of heart failure aldosterone antagonist. Drugs Future. 1999;24:488–501. [Google Scholar]
- Radke PW, Weber C, Kaiser A, Schober A, Hoffmann R. Dexamethasone and restenosis after coronary stent implantation: new indication for an old drug? Curr Pharm Des. 2004;10:349–355. doi: 10.2174/1381612043453324. [DOI] [PubMed] [Google Scholar]
- Radomski MW, Palmer RMJ, Moncada S. Glucocorticoids inhibit the expression of an inducible, but not the constitutive, nitric oxide synthase in vascular endothelial cells. Proc Natl Acad Sci USA. 1990;87:10043–10047. doi: 10.1073/pnas.87.24.10043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raff H. Glucocorticoid inhibition of neurohypophysial vasopressin secretion. Am J Physiol. 1987;252:R635–R644. doi: 10.1152/ajpregu.1987.252.4.R635. [DOI] [PubMed] [Google Scholar]
- Rajan V, Edwards CRW, Seckl JR. 11β-hydroxysteroid dehydrogenase in cultured hippocampal cells reactivates inert 11-dehydrocorticosterone, potentiating neurotoxicity. J Neurosci. 1996;16:65–70. doi: 10.1523/JNEUROSCI.16-01-00065.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramires FJ, Sun Y, Weber KT. Myocardial fibrosis associated with aldosterone or angiotensin II administration: attenuation by calcium channel blockade. J Mol Cell Cardiol. 1998;30:475–483. doi: 10.1006/jmcc.1997.0612. [DOI] [PubMed] [Google Scholar]
- Reimers B, Moussa I, Akiyama T, Kobayashi Y, Albiero R, Di Francesco L, et al. Persistent high restenosis after local intrawall delivery of long-acting steroids before coronary stent implantation. J Invasive Cardiol. 1998;10:323–331. [PubMed] [Google Scholar]
- Ribichini F, Joner M, Ferrero V, Finn AV, Crimins J, Nakazawa G, et al. Effects of oral prednisone after stenting in a rabbit model of established atherosclerosis. J Am Coll Cardiol. 2007a;50:176–185. doi: 10.1016/j.jacc.2007.03.031. [DOI] [PubMed] [Google Scholar]
- Ribichini F, Tomai F, Paloscia L, Di Sciascio G, Carosio G, Romano M, et al. Steroid-eluting stents in patients with acute coronary syndrome: the dexamethasone eluting stent Italian registry. Heart. 2007b;93:598–600. doi: 10.1136/hrt.2006.098467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricketts ML, Shoesmith KJ, Hewison M, Strain A, Eggo MC, Stewart PM. Regulation of 11beta-hydroxysteroid dehydrogenase type 1 in primary cultures of rat and human hepatocytes. J Endocrinol. 1998;156:159–168. doi: 10.1677/joe.0.1560159. [DOI] [PubMed] [Google Scholar]
- del Rincon I, O'Leary DH, Haas RW, Escalante A. Effect of glucocorticoids on the arteries in rheumatoid arthritis. Arthritis Rheum. 2004;50:3813–3822. doi: 10.1002/art.20661. [DOI] [PubMed] [Google Scholar]
- Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- Robert V, Heymes C, Silvestre JS, Sabri A, Swynghedauw B, Delcayre C. Angiotensin AT1 receptor subtype as a cardiac target of aldosterone: role in aldosterone-salt-induced fibrosis. Hypertension. 1999;33:981–986. doi: 10.1161/01.hyp.33.4.981. [DOI] [PubMed] [Google Scholar]
- Roberts CP, Murphy AA, Santanam N, Parthasarathy S. Regulation of monocyte to macrophage differentiation by antiglucocorticoids and antioxidants. Am J Obstet Gynecol. 1998;179:354–361. doi: 10.1016/s0002-9378(98)70364-3. [DOI] [PubMed] [Google Scholar]
- Roghair RD, Segar JL, Sharma RV, Zimmerman MC, Jagadeesha DK, Segar EM, et al. Newborn lamb coronary artery reactivity is programmed by early gestation dexamethasone before the onset of systemic hypertension. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1169–R1176. doi: 10.1152/ajpregu.00369.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross R. Mechanisms of disease – atherosclerosis – an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- Rougier JS, Muller O, Berger S, Centeno G, Schutz G, Firsov D, et al. Mineralocorticoid receptor is essential for corticosteroid-induced up-regulation of L-type calcium currents in cultured neonatal cardiomyocytes. Pflugers Arch. 2008;456:407–412. doi: 10.1007/s00424-007-0387-z. [DOI] [PubMed] [Google Scholar]
- Ruetten H, Thiemermann C. Endothelin-1 stimulates the biosynthesis of tumour necrosis factor in macrophages: ET-receptors, signal transduction and inhibition by dexamethasone. J Physiol Pharmacol. 1997;48:675–688. [PubMed] [Google Scholar]
- Sainte-Marie Y, Cat AND, Perrier R, Mangin L, Soukaseum C, Peuchmaur M, et al. Conditional glucocorticoid receptor expression in the heart induces atrio-ventricular block. FASEB J. 2007;21:3133–3141. doi: 10.1096/fj.07-8357com. [DOI] [PubMed] [Google Scholar]
- Sakai M, Biwa T, Matsumura T, Takemura T, Matsuda H, Anami Y, et al. Glucocorticoid inhibits oxidized LDL-induced macrophage growth by suppressing the expression of granulocyte macrophage colony-stimulating factor. Arterioscler Thromb Vasc Biol. 1999;19:1726–1733. doi: 10.1161/01.atv.19.7.1726. [DOI] [PubMed] [Google Scholar]
- Sarkissian T, Beyenne J, Feldman B, Adeli K, Silverman E. The complex nature of the interaction between disease activity and therapy on the lipid profile in patients with pediatric systemic lupus erythematosus. Arthritis Rheum. 2006;54:1283–1290. doi: 10.1002/art.21748. [DOI] [PubMed] [Google Scholar]
- Sarkissian T, Beyene J, Feldman B, McCrindle B, Silverman ED. Longitudinal examination of lipid profiles in pediatric systemic lupus erythematosus. Arthritis Rheum. 2007;56:631–638. doi: 10.1002/art.22332. [DOI] [PubMed] [Google Scholar]
- Sartorato P, Zulian E, Benedini S, Mariniello B, Schiavi F, Bilora F, et al. Cardiovascular risk factors and ultrasound evaluation of intima-media thickness at common carotids, carotid bulbs, and femoral and abdominal aorta arteries in patients with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2007;92:1015–1018. doi: 10.1210/jc.2006-1711. [DOI] [PubMed] [Google Scholar]
- Schaaf MJM, Champagne D, van Laanen IHC, van Wijk DCWA, Meijer AH, Meijer OC, et al. Discovery of a functional glucocorticoid receptor beta-isoform in zebrafish. Endocrinology. 2008;149:1591–1599. doi: 10.1210/en.2007-1364. [DOI] [PubMed] [Google Scholar]
- Schepers A, Pires NMM, Eefting D, de Vries MR, van Bockel JH, Quax PHA. Short-term dexamethasone treatment inhibits vein graft thickening in hypcrcholcstcrolemic ApoE3Leiden transgenic mice. J Vasc Surg. 2006;43:809–815. doi: 10.1016/j.jvs.2005.11.019. [DOI] [PubMed] [Google Scholar]
- Schleimer RP. Potential regulation of inflammation in the lung by local metabolism of hydrocortisone. Am J Respir Cell Mol Biol. 1991;4:166–173. doi: 10.1165/ajrcmb/4.2.166. [DOI] [PubMed] [Google Scholar]
- Schroepfer GJ. Oxysterols: modulators of cholesterol metabolism and other processes. Physiol Rev. 2000;80:361–554. doi: 10.1152/physrev.2000.80.1.361. [DOI] [PubMed] [Google Scholar]
- Schweizer RA, Zurcher M, Balazs Z, Dick B, Odermatt A. Rapid metabolism of 7-ketocholesterol by 11 beta-hydroxysteroid dehydrogenase type 1 in the liver. FASEB J. 2004a;18:C264. doi: 10.1074/jbc.M313615200. [DOI] [PubMed] [Google Scholar]
- Schweizer RAS, Zurcher M, Balazs Z, Dick B, Odermatt A. Rapid hepatic metabolism of 7-ketocholesterol by 11 beta-hydroxysteroid dehydrogenase type 1 – species-specific differences between the rat, human, and hamster enzyme. J Biol Chem. 2004b;279:18415–18424. doi: 10.1074/jbc.M313615200. [DOI] [PubMed] [Google Scholar]
- Scott BA, Lawrence B, Nguyen HH, Meyer WJ. Aldosterone and dexamethasone binding in human arterial smooth muscle cells. J Hypertens. 1987;5:739–744. doi: 10.1097/00004872-198712000-00018. [DOI] [PubMed] [Google Scholar]
- Seckl JR, Meaney MJ. Glucocorticoid programming. Ann NY Acad Sci. 2004;1032:63–84. doi: 10.1196/annals.1314.006. [DOI] [PubMed] [Google Scholar]
- Seckl JR, Walker BR. 11β-hydroxysteroid dehydrogenase type 1 – a tissue-specific amplifier of glucocorticoid action. Endocrinology. 2001;142:1371–1376. doi: 10.1210/endo.142.4.8114. [DOI] [PubMed] [Google Scholar]
- Shakhov Y, Petrichenko I, Fuki I. Reduced sensitivity of cultured skin fibroblasts from familial hypercholesterolemic patients to glucocorticoids. Life Sci. 1993;53:1617–1624. doi: 10.1016/0024-3205(93)90185-6. [DOI] [PubMed] [Google Scholar]
- Shakhov YA, Petrichenko IE, Chepurnenko NV, Sokolova MA, Medvedeva LA. The hypercholesterolemia-associated decrease in cell sensitivity to glucocorticoid hormones. Biokhimiya. 1989;54:1772–1779. [PubMed] [Google Scholar]
- Shields PP, Dixon JE, Glembotski CC. The secretion of atrial natriuretic factor(99-126) by cultured cardiac myocytes is regulated by glucocorticoids. J Biol Chem. 1988;263:12619–12628. [PubMed] [Google Scholar]
- Shively CA, Williams JK, Laber-Laird K, Anton RF. Depression and coronary artery atherosclerosis and reactivity in female cynomolgus monkeys. Psychosom Med. 2002;64:699–706. doi: 10.1097/01.psy.0000021951.59258.c7. [DOI] [PubMed] [Google Scholar]
- Sihvonen S, Korpela M, Mustonen J, Huhtala H, Karstila K, Pasternack A. Mortality in patients with rheumatoid arthritis treated with low-dose oral glucocorticoids. A population-based cohort study. J Rheumatol. 2006;33:1740–1746. [PubMed] [Google Scholar]
- Simmons WW, Ungureanu-Longrois D, Smith GK, Smith TW, Kelly RA. Glucocorticoids regulate inducible nitric oxide synthase by inhibiting tetrahydrobiopterin synthesis and L-arginine transport. J Biol Chem. 1996;271:23928–23937. doi: 10.1074/jbc.271.39.23928. [DOI] [PubMed] [Google Scholar]
- Simoncini T, Maffei S, Basta G, Barsacchi G, Genazzani AR, Liao JK, et al. Estrogens and glucocorticoids inhibit endothelial vascular cell adhesion molecule-1 expression by different transcriptional mechanisms. Circ Res. 2000;87:19–25. doi: 10.1161/01.res.87.1.19. [DOI] [PubMed] [Google Scholar]
- Slight SH, Ganjam VK, Gomez-Sanchez CE, Zhou M-Y, Weber KT. High affinity NAD+-dependent 11beta-hydroxysteroid dehydrogenase in the human heart. J Mol Cell Cardiol. 1996;28:781–787. doi: 10.1006/jmcc.1996.0072. [DOI] [PubMed] [Google Scholar]
- Small GR, Hadoke PWF, Sharif I, Dover AR, Armour D, Kenyon CJ, et al. Preventing local regeneration of glucocorticoids by 11 beta-hydroxysteroid dehydrogenase type 1 enhances angiogenesis. Proc Natl Acad Sci USA. 2005;102:12165–12170. doi: 10.1073/pnas.0500641102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solakidi S, Psarra AM, Sekeris C. Differential distribution of glucocorticoid and estrogen receptor isoforms: localization of GR beta and ER alpha in nucleoli and GR alpha and ER beta in the mitochondria of human osteosarcoma SaOS-2 and hepatocarcinoma HepG2 cell lines. J Musculoskelet Neuronal Interact. 2007;7:240–245. [PubMed] [Google Scholar]
- Solomon DH, Avorn J, Katz JN, Weinblatt ME, Setoguchi S, Levin R, et al. Immunosuppressive medications and hospitalization for cardiovascular events in patients with rheumatoid arthritis. Arthritis Rheum. 2006;54:3790–3798. doi: 10.1002/art.22255. [DOI] [PubMed] [Google Scholar]
- Son JW, Koh KK, Ahn JY, Jin DK, Park GS, Kim DS, et al. Effects of statin on plaque stability and thrombogenicity in hypercholesterolemic patients with coronary artery disease. Int J Cardiol. 2003;88:77–82. doi: 10.1016/s0167-5273(02)00368-6. [DOI] [PubMed] [Google Scholar]
- Sonino N. Drug-therapy – the use of ketoconazole as an inhibitor of steroid-production. N Engl J Med. 1987;317:812–818. doi: 10.1056/NEJM198709243171307. [DOI] [PubMed] [Google Scholar]
- Souness GW, Latif SA, Laurenzo JL, Morris DJ. 11alpha- and 11beta-hydroxyprogesterone, potent inhibitors of 11beta-hydroxysteroid dehydrogenase (isoforms 1 and 2), confer marked mineralocorticoid activity on corticosterone in the ADX rat. Endocrinology. 1995;136:1809–1812. doi: 10.1210/endo.136.4.7895695. [DOI] [PubMed] [Google Scholar]
- Souverein PC, Berard A, van Staa TP, Cooper C, Egberts AC, Leufkens HG, et al. Use of oral glucocorticoids and risk of cardiovascular and cerebrovascular disease in a population based case-control study. Heart. 2004;90:859–865. doi: 10.1136/hrt.2003.020180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitz IM, Bardin CW. Drug-therapy – mifepristone (Ru-486) – a modulator of progestin and glucocorticoid action. N Engl J Med. 1993;329:404–412. doi: 10.1056/NEJM199308053290607. [DOI] [PubMed] [Google Scholar]
- Stamler J, Johnson P, Ellis A. Estrogen inhibition of corticoid hypertension in chickens. Circulation. 1954;10:896–901. doi: 10.1161/01.cir.10.6.896. [DOI] [PubMed] [Google Scholar]
- Stewart PM, Krozowski ZS. 11Beta hydroxysteroid dehydrogenase. Vitam Horm. 1999;57:249–324. [PubMed] [Google Scholar]
- Stewart PM, Rogerson FM, Mason JI. Type 2 11β-hydroxysteroid dehydrogenase messenger RNA and activity in human placenta and fetal membranes: its relationship to birth weight and putative role in fetal steroidogenesis. J Clin Endocrinol Metab. 1995;80:885–890. doi: 10.1210/jcem.80.3.7883847. Ref Type: Journal (Full. [DOI] [PubMed] [Google Scholar]
- Stockand JD, Meszaros JG. Aldosterone stimulates proliferation of cardiac fibroblasts by activating Ki-RasA and MAPK1/2 signaling. Am J Physiol Heart Circ Physiol. 2003;284:H176–H184. doi: 10.1152/ajpheart.00421.2002. [DOI] [PubMed] [Google Scholar]
- Stone GW, Rutherford BD, Mcconahay DR, Johnson WL, Giorgi LV, Ligon RW, et al. A randomized trial of corticosteroids for the prevention of restenosis in 102 patients undergoing repeat coronary angioplasty. Cathet Cardiovasc Diagn. 1989;18:227–231. doi: 10.1002/ccd.1810180407. [DOI] [PubMed] [Google Scholar]
- Strecker EP, Gabelmann A, Boos I, Lucas CXZY, Haberstroh J, Freudenberg N, et al. Effect on intimal hyperplasia of dexamethasone released from coated metal stents compared with non-coated stents in canine femoral arteries. Cardiovasc Intervent Radiol. 1998;21:487–496. doi: 10.1007/s002709900309. [DOI] [PubMed] [Google Scholar]
- Stumpf HH, Wilens SL. Inhibitory effect of cortisone hyperlipemia on arterial lipid deposition in cholesterol-fed rabbits. Proc Soc Exp Biol Med. 1954;86:219–223. doi: 10.3181/00379727-86-21055. [DOI] [PubMed] [Google Scholar]
- Sugiyama T, Yoshimoto T, Tsuchiya K, Gochou N, Hirono Y, Tateno T, et al. Aldosterone induces angiotensin converting enzyme gene expression via a JAK2-dependent pathway in rat endothelial cells. Endocrinology. 2005;146:3900–3906. doi: 10.1210/en.2004-1674. [DOI] [PubMed] [Google Scholar]
- Sun Y, Zhang J, Lu L, Chen SS, Quinn MT, Weber KT. Aldosterone-induced inflammation in the rat heart: role of oxidative stress1. Am J Pathol. 2002;161:1773–1781. doi: 10.1016/S0002-9440(10)64454-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swade C, Metcalfe M, Coppen A, Mendlewicz J, Linkowski P. Seasonal variations in the dexamethasone suppression test. J Affect Disord. 1987;13:9–11. doi: 10.1016/0165-0327(87)90068-1. [DOI] [PubMed] [Google Scholar]
- Szeto A, Gonzales JA, Spitzer SB, Levine JE, Zaias J, Saab PG, et al. Circulating levels of glucocorticoid hormones in WHHL and NZW rabbits: circadian cycle and response to repeated social encounter. Psychoneuroendocrinology. 2004;29:861–866. doi: 10.1016/S0306-4530(03)00153-7. [DOI] [PubMed] [Google Scholar]
- Takeda Y, Miyamori I, Yoneda T, Ito Y, Takeda R. Expression of 11β-hydroxysteroid dehydrogenase MRNA in rat vascular smooth muscle cells. Life Sci. 1994;54:281–285. doi: 10.1016/0024-3205(94)00818-3. [DOI] [PubMed] [Google Scholar]
- Thieringer R, Le Grand CB, Carbin L, Cai TQ, Wong B, Wright SD, et al. 11 Beta-hydroxysteroid dehydrogenase type 1 is induced in human monocytes upon differentiation to macrophages. J Immunol. 2001;167:30–35. doi: 10.4049/jimmunol.167.1.30. [DOI] [PubMed] [Google Scholar]
- Tomlinson JW, Walker EA, Bujalska IJ, Draper N, Lavery GG, Cooper MS, et al. 11Beta-hydroxysteroid dehydrogenase type 1: a tissue-specific regulator of glucocorticoid response. Endocr Rev. 2004;25:831–866. doi: 10.1210/er.2003-0031. [DOI] [PubMed] [Google Scholar]
- Troxler RG, Sprague EA, Albanese RA, Fuchs R, Thompson AJ. Association of elevated plasma cortisol and early atherosclerosis as demonstrated by coronary angiography. Atherosclerosis. 1977;26:151–162. doi: 10.1016/0021-9150(77)90098-3. [DOI] [PubMed] [Google Scholar]
- Ullian ME. The role of corticosteroids in the regulation of vascular tone. Cardiovasc Res. 1999;41:55–64. doi: 10.1016/s0008-6363(98)00230-2. [DOI] [PubMed] [Google Scholar]
- Valledor AF, Ricote M. Nuclear receptor signaling in macrophages. Biochem Pharmacol. 2004;67:201–212. doi: 10.1016/j.bcp.2003.10.016. [DOI] [PubMed] [Google Scholar]
- Van Put DJM, Van Hove CE, DeMeyer GRY, Wuyts F, Herman AG, Bult H. Dexamethasone influences intimal thickening and vascular reactivity in the rabbit collared carotid artery. Eur J Pharmacol. 1995;294:753–761. doi: 10.1016/0014-2999(95)00635-4. [DOI] [PubMed] [Google Scholar]
- Varas-Lorenzo C, Rodriguez LAG, Maguire A, Castellsague J, Perez-Gutthann S. Use of oral corticosteroids and the risk of acute myocardial infarction. Atherosclerosis. 2007;192:376–383. doi: 10.1016/j.atherosclerosis.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Versaci F, Gaspardone A, Tomai F, Ribichini F, Russo P, Proietti I, et al. Immunosuppressive therapy for the prevention of restenosis after coronary artery stent implantation (IMPRESS Study) J Am Coll Cardiol. 2002;40:1935–1942. doi: 10.1016/s0735-1097(02)02562-7. [DOI] [PubMed] [Google Scholar]
- Villa AE, Guzman LA, Chen W, Golomb G, Levy RJ, Topol EJ. Local delivery of dexamethasone for prevention of neointimal proliferation in a rat model of balloon angioplasty. J Clin Invest. 1994;93:1243–1249. doi: 10.1172/JCI117078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachoyiannopoulos PG, Kanellopoulos PG, Ioannidis JPA, Tektonidou MG, Mastorakou I, Moutsopoulos HM. Atherosclerosis in premenopausal women with antiphospholipid syndrome and systemic lupus erythematosus: a controlled study. Rheumatology. 2003;42:645–651. doi: 10.1093/rheumatology/keg182. [DOI] [PubMed] [Google Scholar]
- Voisard R, Seitzer U, Baur R, Dartsch PC, Osterhues H, Hoher M, et al. Corticosteroid agents inhibit proliferation of smooth-muscle cells from human atherosclerotic arteries in-vitro. Int J Cardiol. 1994;43:257–267. doi: 10.1016/0167-5273(94)90206-2. [DOI] [PubMed] [Google Scholar]
- de Vries WB, van der Leij FR, Bakker JM, Kamphuis PJ, van Oosterhout MF, Schipper ME, et al. Alterations in adult rat heart after neonatal dexamethasone therapy. Pediatr Res. 2002;52:900–906. doi: 10.1203/00006450-200212000-00015. [DOI] [PubMed] [Google Scholar]
- Waddell BJ, Benediktsson R, Brown RW, Seckl JR. Tissue-specific messenger ribonucleic acid expression of 11beta- hydroxysteroid dehydrogenase types 1 and 2 and the glucocorticoid receptor within rat placenta suggests exquisite local control of glucocorticoid action. Endocrinology. 1998;139:1517–1523. doi: 10.1210/endo.139.4.5900. [DOI] [PubMed] [Google Scholar]
- Wainwright CL, Miller AM, Wadsworth RM. Inflammation as a key event in the development of neointima formation following vascular balloon injury. Clin Exp Pharmacol Physiol. 2001;28:891–895. doi: 10.1046/j.1440-1681.2001.03543.x. [DOI] [PubMed] [Google Scholar]
- Walker BR. Glucocorticoids and cardiovascular disease. Eur J Endocrinol. 2007a;157:545–559. doi: 10.1530/EJE-07-0455. [DOI] [PubMed] [Google Scholar]
- Walker BR. Extra-adrenal regeneration of glucocorticoids by 11 beta-hydroxysteroid dehydrogenase type 1: physiological regulator and pharmacological target for energy partitioning. Proc Nutr Soc. 2007b;66:1–8. doi: 10.1017/S002966510700523X. [DOI] [PubMed] [Google Scholar]
- Walker BR, Edwards CRW. Licorice-induced hypertension and syndromes of apparent mineralocorticoid excess. Endocrinol Metab Clin N Amer. 1994;23(2):359–377. [PubMed] [Google Scholar]
- Walker BR, Williams BC. Corticosteroids and vascular tone: mapping the messenger maze. Clin Sci. 1992;82:597–605. doi: 10.1042/cs0820597. [DOI] [PubMed] [Google Scholar]
- Walker BR, Yau JL, Brett LP, Seckl JR, Monder C, Williams BC, et al. 11β-Hydroxysteroid dehydrogenase in vascular smooth muscle and heart: implications for cardiovascular responses to glucocorticoids. Endocrinology. 1991;129:3305–3312. doi: 10.1210/endo-129-6-3305. [DOI] [PubMed] [Google Scholar]
- Walker BR, Connacher AA, Webb DJ, Edwards CRW. Glucocorticoids and blood pressure: a role for the cortisol/cortisone shuttle in the control of vascular tone in man. Clin Sci. 1992;83:171–178. doi: 10.1042/cs0830171. [DOI] [PubMed] [Google Scholar]
- Walker BR, Williams BC, Edwards CRW. Regulation of 11β-hydroxysteroid dehydrogenase activity by the hypothalamic-pituitary-adrenal axis in the rat. J Endocrinol. 1994;141:467–472. doi: 10.1677/joe.0.1410467. [DOI] [PubMed] [Google Scholar]
- Walker BR, Phillips DIW, Noon JP, Panarelli M, Best R, Edwards HE, et al. Increased glucocorticoid activity in men with cardiovascular risk factors. Hypertension. 1998;31:891–895. doi: 10.1161/01.hyp.31.4.891. [DOI] [PubMed] [Google Scholar]
- Wallerath T, Witte K, Schafer SC, Schwartz PM, Prelwitz W, Wohlfart P, et al. Down-regulation of the expression of endothelial NO synthase is likely to contribute to glucocorticoid-mediated hypertension. Proc Natnl Acad Sci USA. 1999;92:13357–13362. doi: 10.1073/pnas.96.23.13357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamil M, Seckl JR. Inhibition of 11B-hydroxysteroid dehydrogenase type 1 as a promising therapeutic target. Drug Discov Today. 2007;12:504–520. doi: 10.1016/j.drudis.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Wang L, Feng ZP, Duff HJ. Glucocorticoid regulation of cardiac K+ currents and L-type Ca2+ current in neonatal mice. Circ Res. 1999;85:168–173. doi: 10.1161/01.res.85.2.168. [DOI] [PubMed] [Google Scholar]
- Wang XL, Herzog B, Waltner-Law M, Hall RK, Shiota M, Granner DK. The synergistic effect of dexamethasone and all-trans-retinoic acid on hepatic phosphoenolpyruvate carboxykinase gene expression involves the coactivator P300. J Biol Chem. 2004;279:34191–34200. doi: 10.1074/jbc.M403455200. [DOI] [PubMed] [Google Scholar]
- Webster SP, Pallin TD. 11 Beta-hydroxysteroid dehydrogenase type 1 inhibitors as therapeutic agents. Expert Opin Ther Pat. 2007;17:1407–1422. [Google Scholar]
- Wei L, MacDonald TM, Walker BR. Taking glucocorticoids by prescription is associated with subsequent cardiovascular disease. Ann Intern Med. 2004;141:764–770. doi: 10.7326/0003-4819-141-10-200411160-00007. [DOI] [PubMed] [Google Scholar]
- Weissberg PL. Atherogenesis: current understanding of the causes of atheroma. Heart. 2000;83:247–252. doi: 10.1136/heart.83.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner JC, Sicard RE, Hansen TW, Solomon E, Cowett RM, Oh W. Hypertrophic cardiomyopathy associated with dexamethasone therapy for bronchopulmonary dysplasia. J Pediatr. 1992;120:286–291. doi: 10.1016/s0022-3476(05)80446-9. [DOI] [PubMed] [Google Scholar]
- Wheller SK, Perretti M. Dexamethasone inhibits cytokine-induced intercellular adhesion molecule-1 up-regulation on endothelial cell lines. Eur J Pharmacol. 1997;331:65–71. doi: 10.1016/s0014-2999(97)01015-7. [DOI] [PubMed] [Google Scholar]
- Whitehurst RM, Jr, Zhang M, Bhattacharjee A, Li M. Dexamethasone-induced hypertrophy in rat neonatal cardiac myocytes involves an elevated L-type Ca(2+) current. J Mol Cell Cardiol. 1999;31:1551–1558. doi: 10.1006/jmcc.1999.0990. [DOI] [PubMed] [Google Scholar]
- Wirtz PH, von Kanel R, Kunz-Ebrecht S, Ehlert U, Fischer JE. Reduced glucocorticoid sensitivity of monocyte interleukin-6 release in male employees with high plasma levels of tumor necrosis factor-alpha. Life Sci. 2004a;75:1–10. doi: 10.1016/j.lfs.2003.11.024. [DOI] [PubMed] [Google Scholar]
- Wirtz PH, von Kanel R, Frey K, Ehlert U, Fischer JE. Glucocorticoid sensitivity of circulating monocytes in essential hypertension. Am J Hypertens. 2004b;17:489–494. doi: 10.1016/j.amjhyper.2004.01.010. [DOI] [PubMed] [Google Scholar]
- Woodall SM, Johnston BM, Breier BH, Gluckman PD. Chronic maternal undernutrition in the rat leads to delayed postnatal growth and elevated blood pressure of offspring. Pediatr Res. 1996;40:438–443. doi: 10.1203/00006450-199609000-00012. [DOI] [PubMed] [Google Scholar]
- Yamada K, Naito M, Hayashi T, Asai K, Yoshimine N, Iguchi A. Effects of dexamethasone on migration of human monocytes in response to oxidized beta-very low-density-lipoprotein. Artery. 1993;20:253–267. [PubMed] [Google Scholar]
- Yamamoto Y, Ishizu A, Ikeda H, Otsuka N, Yoshiki T. Dexamethasone increased plasminogen activator inhibitor-1 expression on human umbilical vein endothelial cells: an additive effect to tumor necrosis factor-alpha. Pathobiology. 2004;71:295–301. doi: 10.1159/000081724. [DOI] [PubMed] [Google Scholar]
- Yang L, Yang JB, Chen J, Yu GY, Zhou P, Lei L, et al. Enhancement of human ACAT1 gene expression to promote the macrophage-derived foam cell formation by dexamethasone. Cell Res. 2004;14:315–323. doi: 10.1038/sj.cr.7290231. [DOI] [PubMed] [Google Scholar]
- Yeager MP, Guyre PM, Munck AU. Glucocorticoid regulation of the inflammatory response to injury. Acta Anaesthesiol Scand. 2004;48:799–813. doi: 10.1111/j.1399-6576.2004.00434.x. [DOI] [PubMed] [Google Scholar]
- Ylaherttuala S, Lipton BA, Rosenfeld ME, Sarkioja T, Yoshimura T, Leonard EJ, et al. Expression of monocyte chemoattractant protein-1 in macrophage-rich areas of human and rabbit atherosclerotic lesions. Proc Natl Acad Sci USA. 1991;88:5252–5256. doi: 10.1073/pnas.88.12.5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MJ. Mechanisms of mineralocorticoid receptor-mediated cardiac fibrosis and vascular inflammation. Curr Opin Nephrol Hypertens. 2008;17:174–180. doi: 10.1097/MNH.0b013e3282f56854. [DOI] [PubMed] [Google Scholar]
- Zacharieva S, Atanassova I, Orbetzova M, Kirilov G, Nachev E, Kalinov K, et al. Vascular Endothelial Growth Factor (VEGF), prostaglandin E-2(PGE(2)) and active renin in hypertension of adrenal origin. J Endocrinol Invest. 2004;27:742–746. doi: 10.1007/BF03347516. [DOI] [PubMed] [Google Scholar]
- Zecca E, Papacci P, Maggio L, Gallini F, Elia S, De RG, et al. Cardiac adverse effects of early dexamethasone treatment in preterm infants: a randomized clinical trial. J Clin Pharmacol. 2001;41:1075–1081. doi: 10.1177/00912700122012670. [DOI] [PubMed] [Google Scholar]
- Zhang TY, Ding XH, nes RA. The expression of 11 beta-hydroxysteroid dehydrogenase type I by lymphocytes provides a novel means for intracrine regulation of glucocorticoid activities. J Immunol. 2005a;174:879–889. doi: 10.4049/jimmunol.174.2.879. [DOI] [PubMed] [Google Scholar]
- Zhang JD, Osslund TD, Plant MH, Clogston CL, Nybo RE, Xiong F, et al. Crystal structure of murine 11 beta-hydroxysteroid dehydrogenase 1: an important therapeutic target for diabetes. Biochemistry. 2005b;44:6948–6957. doi: 10.1021/bi047599q. [DOI] [PubMed] [Google Scholar]