

Abstract
Background and purpose:
Protein kinase (PK) A and the ε isoform of PKC (PKCε) are involved in the development of hypernociception (increased sensitivity to noxious or innocuous stimuli) in several animal models of acute and persistent inflammatory pain. The present study evaluated the contribution of PKA and PKCε to the development of prostaglandin E2 (PGE2)-induced mechanical hypernociception.
Experimental approach:
Prostaglandin E2-induced mechanical hypernociception was assessed by constant pressure rat paw test. The activation of PKA or PKCε was evaluated by radioactive enzymic assay in the dorsal root ganglia (DRG) of sensory neurons from the hind paws.
Key results:
Hypernociception induced by PGE2 (100 ng) by intraplantar (i.pl.) injection, was reduced by i.pl. treatment with inhibitors of PKA [A-kinase-anchoring protein St-Ht31 inhibitor peptide (AKAPI)], PKCε (PKCεI) or adenylyl cyclase. PKA activity was essential in the early phase of the induction of hypernociception, whereas PKC activity was involved in the maintenance of the later phase of hypernociception. In the DRG (L4-L5), activity of PKA increased at 30 min after injection of PGE2 but PKC activity increased only after 180 min. Moreover, i.pl. injection of the catalytic subunit of PKA induced hypernociception which was markedly reduced by pretreatment with an inhibitor of PKCε, while the hypernociception induced by paw injection of PKCε agonist was not affected by an inhibitor of PKA (AKAPI).
Conclusions and implications:
Taken together, these findings are consistent with the suggestion that PKA activates PKCε, which is a novel mechanism of interaction between these kinases during the development of PGE2-induced mechanical hypernociception.
Keywords: PKA, PKC, cAMP, PGE2 and rat mechanical hypernociception
Introduction
Tissue injury and inflammation are associated with increased prostanoid synthesis which sensitizes primary sensory neurons (Martin et al., 1987; Schaible and Schmidt, 1988; Rueff and Dray, 1993). This sensitization of the nociceptive neurons in humans results in hyperalgesia (Ferreira, 1972) (an increased response to a stimulus which is normally painful) or allodynia (pain from stimuli that are not normally painful). However, in animal behaviour models of mechanical nociception, hyperalgesia and allodynia can be distinguished by the use of apparently different mechanical stimuli, as in the Randall-Selitto and the electronic von Frey tests. Moreover, the terms hyperalgesia and allodynia have been developed for use in clinical practice rather than for experimental work, physiology or anatomical purposes (see IASP Pain Terminology). Therefore, we have used the term hypernociception to describe the decrease of behavioural nociceptive threshold in experimental animals.
Mechanical hypernociception induced by prostaglandin E2 (PGE2), assessed by modification of the Randall-Selitto mechanical test, was linked with increased adenosine 3′,5′-cyclic monophosphate (cAMP) in neurons (Ferreira and Nakamura, 1979). This conclusion was later supported and extended (Taiwo et al., 1989; Taiwo and Levine, 1991; Ouseph et al., 1995; Aley and Levine, 1999; Cunha et al., 1999). The adenylyl cyclase (AC) activator, forskolin or inhibitors of the phosphodiesterases enhanced the mechanical hypernociception induced by PGE2 (Taiwo et al., 1989; Taiwo and Levine, 1991; Ouseph et al., 1995; Cunha et al., 1999; Kassuya et al., 2007). In primary nociceptive neurons, increased cAMP is associated with the activation of protein kinase A (PKA) in vitro and in vivo (Scott, 1991; Beebe, 1994; England et al., 1996; Liao et al., 1999; Smith et al., 2000; Distler et al., 2003; Wang et al., 2007). In this context, PKA participates in the inflammatory hypernociception induced by PGE2 (Malmberg et al., 1997; Aley and Levine, 1999; Cunha et al., 1999; Kassuya et al., 2007).
Besides PKA, there is an extensive literature documenting a role for PKC in nociceptor activation as well as sensitization. Studies in vitro (Barber and Vasko, 1996; Leng et al., 1996; Di Castro et al., 2006) demonstrated that phorbol-activated PKC sensitizes primary nociceptive neurons. In this context, the hypernociception caused by inflammatory nociceptive mediators, such as adrenaline, endothelins and bradykinin, is related to the PKC pathway (Cesare et al. 1999; Khasar et al., 1999a; Souza et al., 2002; Cunha et al., 2004). In primary afferent neurons, the ε isoform of PKC (PKCε) participates in inflammatory hypernociception, as selective PKCε inhibitors reduced hypernociception induced by a variety of nociceptive stimuli (Khasar et al., 1999b; Dina et al., 2000; 2006; Hucho et al., 2005; Parada et al., 2005; Summer et al., 2006; Yamamoto et al., 2006). Furthermore, PKCε was up-regulated in dorsal root ganglia (DRG) after peripheral administration of carrageenan (Zhou et al., 2003).
Although the second messenger cascade associated with PGE2-induced hypernociception involves the participation of PKA downstream from cAMP, this evidence does not exclude the participation of PKC in this pathway. Thus, the aim of the present study was to investigate whether, in mechanical hypernociception, PKC participates in the PGE2/cAMP/PKA intracellular signalling pathway in the primary nociceptive neurons.
Methods
Animals
Animal care and handling procedures were in accordance with International Association for the Study of Pain guidelines for the use of animals in pain research and with the approval of the Ethics Committee of the School of Medicine of Ribeirão Preto (University of São Paulo). All efforts were made to minimize discomfort for the animals. Experiments were performed with 180 to 200 g male Wistar rats kept in a 12 h light–dark cycle, with controlled humidity (60–80%) and temperature (22–25°C). Food and water were available ad libitum. The animals were taken to the testing area at least 1 h before testing. Each experiment used five rats per group, previously found to be the minimum number of animals necessary to detect significant variations of the paw nociceptive sensitivity. All behavioural testing was performed between 9:00 am and 4:00 pm.
Nociceptive test: constant pressure rat paw test
Nociception was measured in the present study by applying a constant pressure via a ring-shaped syringe piston (15 mm2) to the dorsal surface of the hind paw. The piston pressure (20 mm Hg) was measured by a sphygmomanometer connected to the source of compressed air (Ferreira et al., 1978). The end point of the mechanical nociception is a typical behavioural reaction (freezing) characterized by a combination of signs: a brief apnoea, retraction of the head and forepaws and reduction in the escape movements from the position imposed by the experimental situation. Usually, the apnoea is associated with successive waves of muscular tremor. The latency of the freezing reaction is measured before (control reaction time at zero time) and at different times after intraplantar (i.pl.) administration of the tested substance in the same paws. The reduction of the latency obtained by subtracting the value of latency at a given time point from the value at zero time (31.5 ± 0.1 s, n = 50) quantified the intensity of mechanical hypernociception (Ferreira et al., 1978). In this test, local or systemic pretreatment with steroidal and non-steroidal anti-inflammatory drugs reduces the intensity of hypernociception by inflammatory stimuli (carrageenan, endotoxin, cytokines/chemokines) but does not affect hypernociception induced by directly sensitizing mediators like PGE2 or dopamine (Nakamura and Ferreira, 1987; Cunha et al., 1991; 1992; Lorenzetti et al., 2002). Thus this test, using a constant pressure as mechanical nociceptive stimulus, allows the measurement of variations of nociceptive behaviour.
Determination of PKA and PKC activity in DRG
Rats were given a lethal overdose of sodium pentobarbital and the L4 and L5 DRG removed. These samples were frozen on dry ice and homogenized (Homogeneizador Kont). The PKA and PKC activity was measured using a commercial kit (protein kinase A assay kit, protein kinase C assay kit, Calbiochem) according to the manufacturer's instructions. Assays were only carried out using the DRG (L4-L5) ipsilateral to the hind paws receiving the i.pl. injection of PGE2 (100 ng per paw) or N6,2′-O-dibutyryladenosine 3′:5′-cyclic monophosphate (db-cAMP) (100 µg per paw), removed at 30 or 180 min after injection. The assay is based on the reaction of the enzyme sample with a biotinylated peptide substrate and γ32P-ATP, and is expressed as pmoles of phosphate incorporated per minute.
Effect of drug treatments
To investigate the involvement of cAMP, PKA or PKCε in hypernociception induced by PGE2 (100 ng; Ferreira and Nakamura, 1979) or db-cAMP (100 µg; Ferreira and Nakamura, 1979), the animals were pretreated with subcutaneous i.pl. injections into the same (ipsilateral) paw. Note all i.pl. treatments are shown as the dose per paw. Pretreatment with inhibitors of AC (SQ22536; 3-27 µg), of PKA [A-kinase-anchoring protein St-Ht31 inhibitor peptide (AKAPI); 0.03-0.3 µg] or of PKCε (PKCεI, 1-9 µg) was given 5 min before the i.pl. injection of PGE2 or db-cAMP. The participation of PKA or PKCε in later phases of PGE2 or db-cAMP-induced hypernociception was assessed by post-treatment (30 or 90 min) with AKAPI (0.3 µg) or PKCεI (9 µg). To investigate if PKA or PKCε had intrinsic hypernociceptive effects, the animals were injected with the catalytic subunit of PKA (PKAcs 0.3–9 U) or pseudo receptor octapeptide for activated PKCε, a specific agonist of PKCε (ΨεRACK 0.3-9 µg; Dorn et al., 1999). To determine if PKA was able to activate PKCε, the animals were pretreated with inhibitor AKAPI (0.3 µg) or PKCε (PKCεI, 9 µg) 30 min before the PKAcs (1 U) or the specific agonist of PKCε (ΨεRACK 1 µg). In the present study, all hypernociceptive stimuli (PGE2, db-cAMP, ΨεRACK and PKAcs) or antagonists used were given by subcutaneous injection into the hind paw.
Data analysis
Results are presented as means ± SEM for groups of five animals. Analysis of variance (one-way anova) was used, followed by the Bonferroni test. The level of significance was set at P < 0.05. The dose–response relationships for SQ22536, AKAPI and PKCεI were analysed by non-linear regression.
Drugs and reagents
The pseudo receptor for activated PKCε octapeptide (ΨεRACK; Dorn et al., 1999) and PKCεV1–2 peptide (a selective PKCε inhibitor; Gray et al., 1997) were obtained from SynPep Corp (Dublin, CA, USA). SQ22536 was obtained from Biomol (Aley and Levine, 1999). The PKAcs was obtained from Calbiochem (La Jolla, CA, USA). PGE2 and db-cAMP (Taiwo et al., 1989; Taiwo and Levine, 1991; Ouseph et al., 1995) were obtained from Sigma (St Louis, MO, USA). InCELLlect® AKAPI (Moita et al., 2002; Parada et al., 2005) was obtained from Promega Corp (Madison, WI, USA). The stock solution of PGE2 (1 µg·µL−1) was prepared in 10% ethanol, and additional dilutions were made in physiological saline (0.9% NaCl), yielding a final concentration of ethanol of less than 1%. All other drugs were dissolved directly in saline. Protein kinase A assay kit and protein kinase C assay kit were from Calbiochem.
Results
PKA and PKCε participate in the PGE2-activated, cAMP second messenger cascade in primary afferent neurons
The results presented in Figure 1 show that subcutaneous i.pl. pretreatment with an inhibitor of AC (SQ22536; 3, 9 and 27 µg; ID50: 4.01 µg; Figure 1A) or of PKA (AKAPI; 0.03, 0.1 and 0.3 µg; ID50: 0.22 µg; Figure 1B) prevented PGE2-induced hypernociception in a dose-dependent manner. The PKCε inhibitor (PKCεI; 1, 3 and 9 µg; ID50: 2.6 µg; Figure 1C) prevented in a dose-dependent manner the hypernociception induced by PGE2. Pretreatment with PKCεI (9 µg), or AKAPI (0.3 µg), but not with the AC inhibitor SQ22536 (27 µg), inhibited the hypernociception induced by db-cAMP (100 µg, Figure 1D). It should be stressed that SQ22536 (27 µg, Figure 1A), AKAPI (0.3 µg, Figure 1B) or PKCεI (9 µg, Figure 1C) at the highest doses used here had no nociceptive effect when given alone into normal paws.
Figure 1.
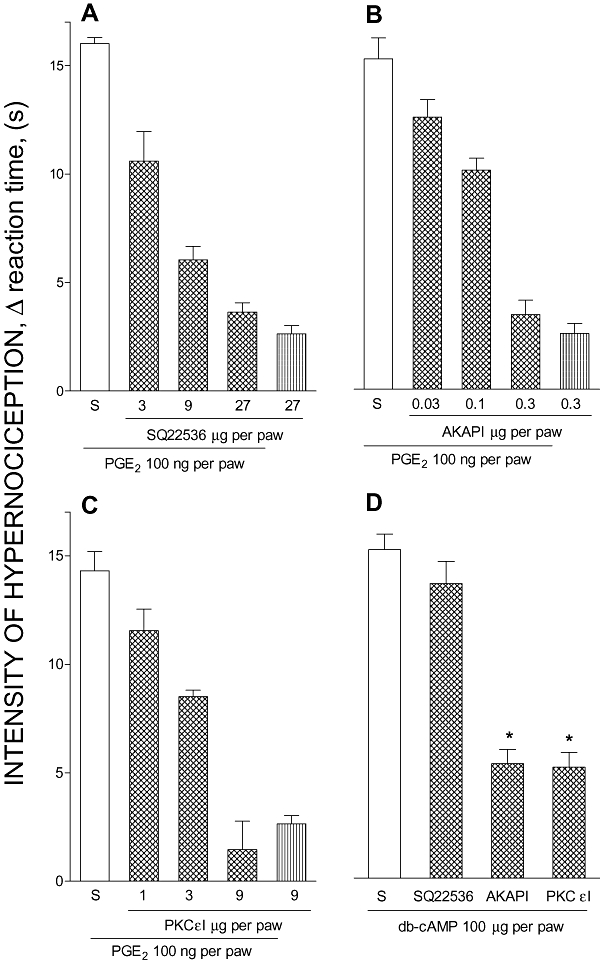
Effect of adenylyl cyclase, PKA or PKC inhibitors upon PGE2- or db-cAMP-evoked hypernociception. All inhibitors were given i.pl. and doses are shown as the dose per paw. In panel (A) adenylyl cyclase (SQ22536, 3, 9 or 27 µg), (B) PKA (AKAPI, 0.03, 0.1 or 0.3 µg), (C) PKCε (PKCεI, 1, 3 or 9 µg) inhibitors or saline (S, 50 µL) were injected 5 min before PGE2 (100 ng, i.pl.) injection. In (D) SQ22536 (27 µg), AKAPI (0.3 µg), PKCεI (9 µg) or saline (S, 50 µL) was injected 5 min before db-cAMP (100 µg; i.pl.) injection. The last bar of panels (A), (B) and (C) represents the effects of the inhibitors SQ22536 (27 µg), AKAPI (0.3 µg) or PKCεI (9 µg) injected alone into the paw respectively. The intensity of hypernociception was determined 3 h after i.pl. injection of PGE2 (100 ng), db-cAMP (100 µg) or saline (50 µL). The data are the means ± SEM of five animals per group. There were dose-dependent effects for SQ22536 (A; non-linear regression, R2 = 0.98), AKAPI (B; non-linear regression, R2 = 0.92) and PKCεI (C; non-linear regression, R2 = 0.96) pretreatments. *P < 0.05 compared with rats pretreated with saline and injected with db-cAMP (one-way anova followed by Bonferroni test). AKAPI, A-kinase anchoring protein St-Ht31 inhibitor peptide; db-cAMP, N6,2′-O-dibutyryladenosine 3′:5′-cyclic monophosphate; i.pl., intraplantar; PGE2, prostaglandin E2; PKA, protein kinase A; PKC, protein kinase C; PKCε, ε isoform of protein kinase C; PKCεI, PKCεV1–2 peptide, a selective PKCε inhibitor.
Differential time effects of PKA and PKC inhibitors on the hypernociception induced by PGE2 or db-cAMP
Treatment with inhibitors of PKA (AKAPI, 0.3 µg) or PKCε (PKCεI, 9 µg), either before or 30 min after the i.pl. injection of PGE2 (100 ng) or db-cAMP (100 µg), reduced the mechanical hypernociception. However, later post-treatment (90 min after PGE2 or db-cAMP i.pl. injection) with AKAPI was ineffective but similar treatment with PKCεI clearly reduced mechanical hypernociception. Together, these results suggest that PKA activity is essential only in the early phase of the establishment of hypernociception, whereas PKC activity is involved in the maintenance of later phases of hypernociception. In support of this suggestion, the radioactive assay for PKA activity in ipsilateral DRG (L4-L5) was increased at 30 min, but not 180 min after paw injection of PGE2 (Figure 2B) or db-cAMP (Figure 3B). However, the PKC activity was enhanced much later, mainly at 180 min after i.pl. injection of PGE2 (Figure 2D) or db-cAMP (Figure 3D).
Figure 2.
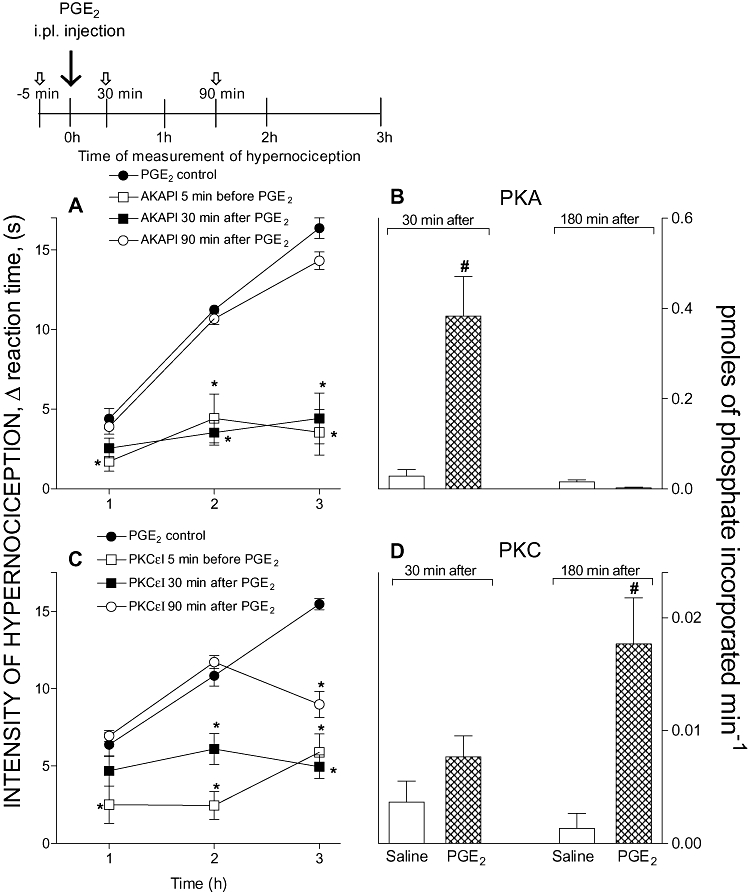
Time-dependence of the inhibitory effect of pre- or post-treatments with PKA (A) or PKC (C) inhibitors upon PGE2-induced hypernociception. Note all i.pl. treatments are shown as the dose per paw. Panels (B) and (D) show the PKA and PKC activities in DRG (L4-L5) of rats injected i.pl. with PGE2 (100 ng). (A) AKAPI (0.3 µg) or (C) PKCεI (9 µg) was administered 5 min before or 30 or 90 min after i.pl. injection of PGE2 (100 ng). Inhibitors of PKA or PKC were given at the times indicated by the short arrows. The intensity of hypernociception was determined 1, 2 or 3 h after i.pl. injection of PGE2. Inserted above panel (A) is a diagram showing the schedule of treatments and hypernociception determinations. The activities of PKA (B) and PKC (D) were evaluated in DRG (L4-L5) of the rats 30 or 180 min after i.pl. injection of saline (50 µL) or PGE2 (100 ng) and expressed as pmoles of phosphate incorporated min−1. The data are the means ± SEM of five animals per group in panels (A) and (C) and mean ± SEM of three animals per group in panels (B) and (D). *P < 0.05 compared with PGE2-control rats. #P < 0.05 compared with rats injected with saline (one-way anova followed by Bonferroni). AKAPI, A-kinase anchoring protein St-Ht31 inhibitor peptide; DRG, dorsal root ganglia; i.pl., intraplantar; PGE2, prostaglandin E2; PKA, protein kinase A; PKC, protein kinase C; PKCε, ε isoform of protein kinase C; PKCεI, PKCεV1–2 peptide, a selective PKCε inhibitor.
Figure 3.
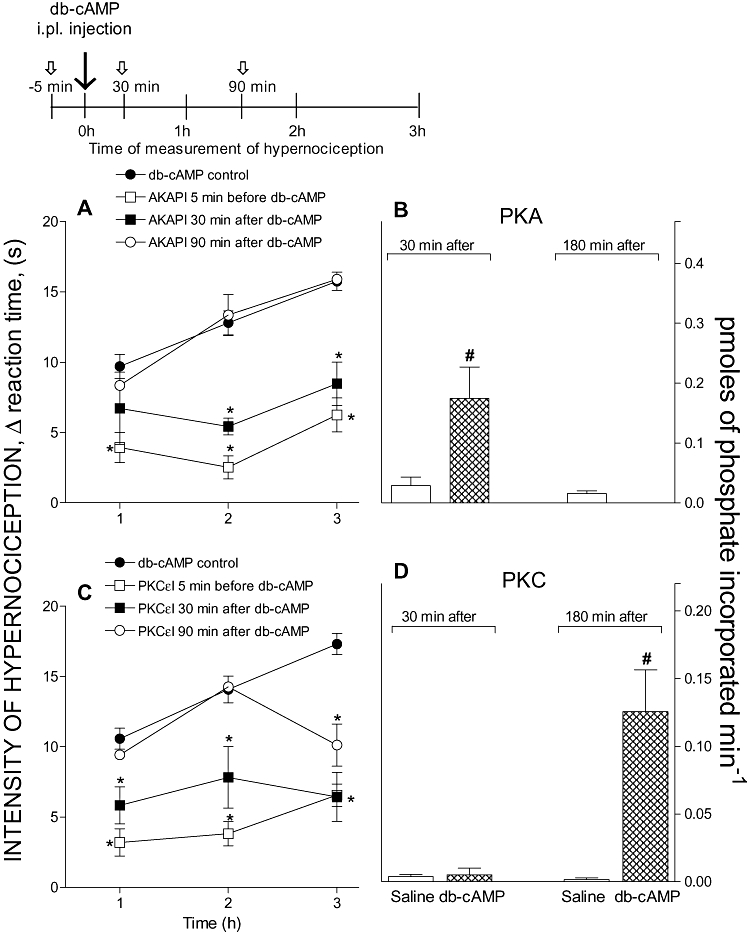
Time-dependence of the inhibitory effect of pre- or post-treatments with PKA (A) or PKC (C) inhibitors upon db-cAMP-induced hypernociception. All inhibitors were given i.pl. and doses are shown as the dose per paw. Panels (B) and (D) show the PKA and PKC activities in DRG (L4-L5) of rats injected i.pl. with db-cAMP (100 µg). (A) AKAPI (0.3 µg) or (C) PKCεI (9 µg) was administered 5 min before or 30 or 90 min after i.pl. injection of db-cAMP (100 µg). Inhibitors of PKA or PKC were given at the times indicated by the short arrows. The intensity of hypernociception was determined 1, 2 or 3 h after i.pl. injection of db-cAMP (100 µg). Inserted above panel (A) is a diagram showing the schedule of treatments and hypernociception determinations. The activities of PKA (B) and PKC (D) were evaluated in DRG (L4-L5) of the rats 30 or 180 min after intraplantar injection of saline (50 µL) or db-cAMP (100 µg) and expressed as pmoles of phosphate incorporated min−1. The data are the means ± SEM of five animals per group in panels (A) and (C) and means ± SEM of three animals per group in panels (B) and (D). *P < 0.05 compared with db-cAMP-control rats treated with saline. #P < 0.05 compared with rats injected with saline (one-way anova followed by Bonferroni). AKAPI, A-kinase anchoring protein St-Ht31 inhibitor peptide; db-cAMP, N6,2′-O-dibutyryladenosine 3′: 5′-cyclic monophosphate; DRG, dorsal root ganglia; i.pl., intraplantar; PKA, protein kinase A; PKC, protein kinase C; PKCε, ε isoform of protein kinase C; PKCεI, PKCεV1–2 peptide, a selective PKCε inhibitor.
PKA activates PKCε during PGE2-induced hypernociception
We also investigated whether the hypernociception induced by a PKAcs depends on PKCε activation, and whether the hypernociceptive activity of ψεRACK depends on PKA activation. Both PKAcs (0.3, 1, 3 and 9 U; ED50: 1.31 U; Figure 4A) and ψεRACK (0.3, 1, 3 and 9 µg; ED50: 1.67 µg; Figure 4B) given alone induced dose-dependent mechanical hypernociception. Pretreatment (30 min before) with the inhibitor of PKCε (9 µg) significantly inhibited the hypernociception induced by PKAcs (1 U; Figure 4C). In contrast, the PKA inhibitor AKAPI (0.3 µg) given 30 min before the administration of ψεRACK (1 µg) did not change the hypernociceptive effect (Figure 4D). These results are compatible with the proposition that PKA activates PKCε during PGE2-induced hypernociception.
Figure 4.
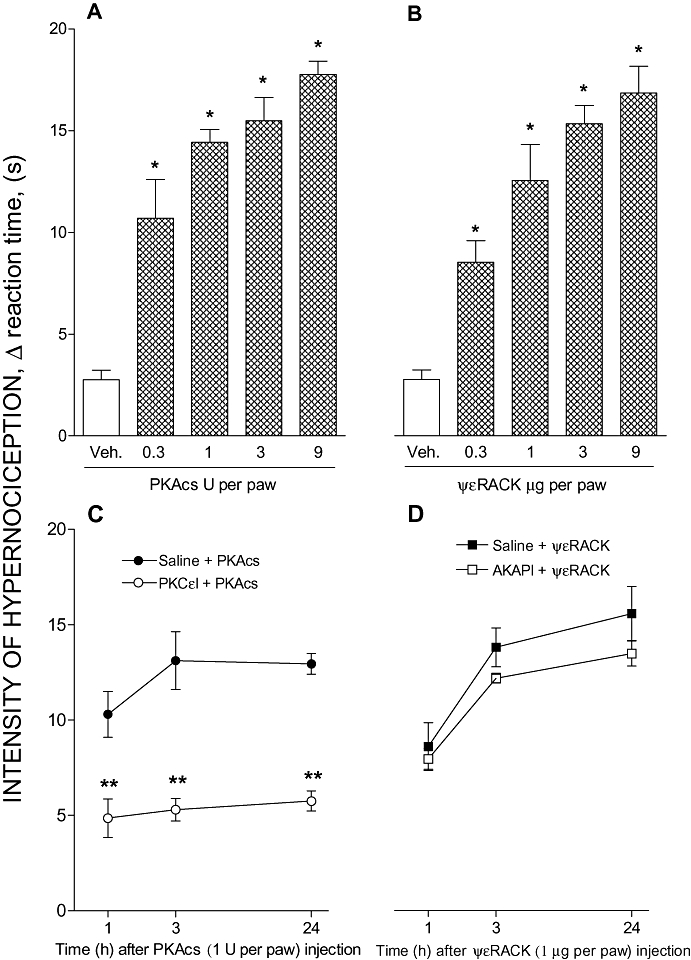
Crosstalk between PKA and PKCε in paw hypernociception. All i.pl. treatments are shown as the dose per paw. Panels (A) and (B): Hypernociceptive effect of the catalytic subunit of PKA (PKAcs) or a specific agonist of PKC (ΨεRACK). PKAcs (0.3, 1, 3 or 9 U) or ψεRACK (0.3, 1, 3 or 9 µg) was injected i.pl. and the intensity of hypernociception was determined 1 h after PKAcs (A) or ψεRACK (B) administration respectively. Panels (C) and (D): The effect of PKC inhibitor (PKCεI) upon PKAcs-evoked hypernociception and of PKA inhibitor (AKAP) upon ΨεRACK-evoked hypernociception. The pretreatment with PKCεI [9 µg; i.pl., panel (C)] or AKAPI [0.3 µg; i.pl., panel (D)] was performed 30 min before PKAcs (1 U; i.pl.) or ψεRACK (1 µg; i.pl.) injections respectively. The intensity of hypernociception was determined 1, 3 and 24 h after PKAcs (C) or ψεRACK (D) injection. The data are the means ± SEM of five animals per group. *P < 0.05 compared with control group i.pl. injected with vehicle (saline, 50 µL). **P < 0.05 compared with PKAcs-control rats treated with saline (50 µL). AKAPI, A-kinase anchoring protein St-Ht31 inhibitor peptide; i.pl., intraplantar; PKA, protein kinase A; PKAcs, catalytic subunit of PKA; PKCε, ε isoform of protein kinase C; ΨεRACK, pseudo receptor octapeptide for activated PKCε, a specific agonist of PKCε; PKCεI, PKCεV1–2 peptide, a selective PKCε inhibitor.
Discussion
Enhanced sensitivity to nociceptive stimuli is one of the characteristics of the inflammatory response and results from an increase in the excitability of primary nociceptive neurons. There is substantial experimental evidence demonstrating that PGs, among other mediators (sympathetic amines, endothelins, 5-hydroxytryptamine (5-HT), adenosine and cytokines) sensitize the primary nociceptive neurons to innocuous mechanical (Handwerker and Neher, 1976; Pateromichelakis and Rood, 1982; Schaible and Schmidt, 1988; Mizumura et al., 1993; Moriyama et al., 2005) and chemical stimuli (Nicol and Cui, 1994; Smith et al., 2000). This sensitization is referred in the present study as hypernociception, thus avoiding the use of terms like hyperalgesia or allodynia, because they describe an aggregate of characteristic human pathological nociceptive symptoms (see IASP Pain Terminology).
It is now well established that inflammatory hypernociception, such as induced by PGE2, depends on activation of signalling pathways, which require neuronal activation of PKA in neurons, downstream of the second messenger cAMP (Ferreira and Nakamura, 1979; Taiwo and Levine, 1990; 1991; 1992; Taiwo et al., 1992; Wang et al., 1996; Aley and Levine, 1999; Cunha et al., 1999; Kassuya et al., 2007). Moreover, there are a great number of studies suggesting that the PKC pathway also participates in nociceptor sensitization, induced by nociceptive mediators, such as adrenaline, endothelins and bradykinin, as well as by more general inflammatory stimuli such as carrageenan and formalin (Cesare et al. 1999; Khasar et al., 1999b; Souza et al., 2002; Cunha et al., 2004). Furthermore, PKCε is the isoform involved in nociceptor sensitization, as the mechanical nociception induced by carrageenan or formalin-induced nociceptive behaviour were reduced by treatment with inhibitors of PKCε (Khasar et al., 1999a; Sweitzer et al., 2004). Further, long-lasting hypernociception induced by PGE2, in paws previously primed by carrageenan or TNFα, was associated with activation of neuronal PKCε (Aley et al., 2000; Parada et al., 2003; 2005).
In the present study, we confirmed that the treatment with cAMP or PKA inhibitors (Figure 1) prevents PGE2-induced hypernociception, as already described (Taiwo and Levine, 1990; 1992; Aley and Levine, 1999; Parada et al., 2005; Kassuya et al., 2007). In addition, we found that inhibition of the PKCε prevented PGE2 or db-cAMP-induced hypernociception. These results suggest that an increase in cAMP levels is necessary for the activation of PKCε (directly or indirectly) for the induction of PGE2 hypernociception. Further, the activation of PKA downstream from cAMP was involved in the onset of hypernociception, mainly in the initial 30 min. After that, a continuous PKCε activity was required for the development of hypernociception induced by PGE2. As we have shown, in contrast with PKA, PKCε inhibitors significantly reduced the hypernociception induced by PGE2 (Figure 2A,C) or db-cAMP (Figure 3A,C), by either early (30 min after) or later (90 min after) post-treatment. Together, these results suggest that PKA activity is essential in the early phase of the establishment of hypernociception, whereas PKC activity is involved in the maintenance of later phases of hypernociception. To support this suggestion, we investigated the activities of PKA and PKCε in the cell bodies of the DRG neurons after PGE2 and db-cAMP injection in the rat paws, assuming that they reflect biochemical events occurring at peripheral terminations of sensory neurons. In fact, PKA activity increased in ipsilateral DRG (L4-L5) mainly in the early phase (30 min after the PGE or db-cAMP injection) and returned to basal levels 180 min after (Figures 2B and 3B). Although the kinetics of PKA activity induced by PGE2 and db-cAMP were similar, the enzyme activity at 30 min after PGE2 injection was higher that that induced by db-cAMP. Experiments using different doses of these mediators might explain this difference. The increase in PKC activity, however, was low when PKA levels were maximal and maximal when the PKA levels returned to the base line (Figures 2D and 3D). These results are in line with the assumption that PKC activation is downstream to the activation of PKA. Zhou et al. (2003) demonstrated that in paw inflammation induced by carrageenan, in which there is a significant activation of PKCε in the DRG, these kinases are synthesized and transcribed in the soma and delivered to the nerve terminals by axonal transport. Finally, the PKCε inhibitor significantly reduced hypernociception induced by the PKAcs (Figure 4C). Nevertheless, the PKA inhibitor AKAPI did not change the hypernociceptive effect of ψεRACK (Figure 4D), thus supporting the suggestion that hypernociceptive effect induced by a PKAcs depends on PKCε activity. Hypernociception induced by PGE2 did not cause variations in PKA and PKC activities in the paw tissues, probably because the nociceptive neurons constitute a relatively small portion of the total tissue of the paw (data not shown).
Besides, PKA and PKC and other kinases or factors such as (c-Jun N-terminalkinase (JNK), extracellular-regulated kinase, mitogen-activated protein kinases or cAMP-activated guanine exchange factor (Epac), all participate in the inflammatory response. Nevertheless, in acute mechanical hyperalgesia, PKCε and PKA (PGE2)-mediated hyperalgesia was independent of extracellular signal-regulated kinase (MEK) activity (Aley et al., 2001). Further, JNK inhibition did not inhibit PGE2-induced mechanical allodynia (Kassuya et al., 2007). Additionally, p38 and JNK, are activated by the peripheral injection of PGE2 and cause paw oedema in mice but no nociceptive behaviours (Claudino et al., 2006). For this reason, we also did not evaluate the participation of those intracellular pathways in the inflammatory sensitization. There is an alternative mechanism by which cAMP might lead to activation of PKCε. The Epac can induce PKCε translocation via phospholipase C or D in DRG neurons (Hucho et al., 2005). However, the Epac activator (CPTOMe) did not induce mechanical hypernociception in our behavioural test (data not shown).
We did not address the mechanism underlying PKA/PKC crosstalk in the present study. However, there is evidence pointing to a role for PKA in the activation and translocation of PKC. In fact, activation of dopamine D1 receptors, known to couple to Gαs, increases PKC activity and translocation in LTK cells (Yu et al., 1996). In addiction, Huang et al. (2001) demonstrated that PKCζ is a key downstream component of a PKA-dependent, anti-apoptotic signalling pathway activated by a G protein-coupled receptor. Recently, Yao et al. (2008) demonstrated that activation of PKA by Sp-cAMPS (PKA activator) was sufficient to induce activity and translocation of PKCε.
In conclusion, the present study shows that both PKA and PKCε participate in acute mechanical hypernociception downstream from PGE2 receptor activation and suggest that PKA may activate PKCε. Thus, our results describe a novel signalling pathway, in addition to the usual cAMP/PKA pathway, in mechanical hypernociception induced by PGE2.
Acknowledgments
This study was supported by grants from the CNPq and FAPESP, Brazil. We thank IRS Schivo, SR Rosa and FL Mestriner for excellent technical assistance.
Glossary
Abbreviations:
- AC
adenylyl cyclase
- AKAPI
A-kinase-anchoring protein St-Ht31 inhibitor peptide
- i.pl.
intraplantar
- PGE2
prostaglandin E2
- PKA
protein kinase A
- PKAcs
catalytic subunit of PKA
- PKC
protein kinase C
- PKCε
ε isoform of protein kinase C
- ΨεRACK
pseudo receptor octapeptide for activated PKCε, a specific agonist of PKCε
- PKCεI
PKCεV1–2 peptide, a selective PKCε inhibitor
Conflict of interest
The authors state no conflict of interest.
References
- Aley KO, Levine JD. Role of protein kinase A in the maintenance of inflammatory pain. J Neurosci. 1999;19:2181–2186. doi: 10.1523/JNEUROSCI.19-06-02181.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aley KO, Messing RO, Mochly-Rosen D, Levine JD. Chronic hypersensitivity for inflammatory nociceptor sensitization mediated by the epsilon isozyme of protein kinase C. J Neurosci. 2000;20:4680–4685. doi: 10.1523/JNEUROSCI.20-12-04680.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aley KO, Martin A, McMahon T, Mok J, Levine JD, Messing RO. Nociceptor sensitization by extracellular signal-regulated kinases. J Neurosci. 2001;17:6933–6939. doi: 10.1523/JNEUROSCI.21-17-06933.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber LA, Vasko MR. Activation of protein kinase C augments peptide release from rat sensory neurons. J Neurochem. 1996;67:72–80. doi: 10.1046/j.1471-4159.1996.67010072.x. [DOI] [PubMed] [Google Scholar]
- Beebe SJ. The cAMP-dependent protein kinases and cAMP signal transduction. Semin Cancer Biol. 1994;5:285–294. [PubMed] [Google Scholar]
- Cesare P, Dekker LV, Sardini A, Parker PJ, McNaughton PA. Specific involvement of PKC-e in sensitization of the neuronal response to painful heat. Neuron. 1999;23:617–624. doi: 10.1016/s0896-6273(00)80813-2. [DOI] [PubMed] [Google Scholar]
- Claudino RF, Kassuya CAL, Ferreira J, Calixto JB. Pharmacological and molecular characterization of the mechanisms involved in the prostaglandin E2 (PGE2)-induced edematogenic responses in mice. J Pharmacol Exp Ther. 2006;318:611–618. doi: 10.1124/jpet.106.102806. [DOI] [PubMed] [Google Scholar]
- Cunha FQ, Lorenzetti BB, Poole S, Ferreira SH. Interleukin-8 as a mediator of sympathetic pain. Br J Pharmacol. 1991;104:765–767. doi: 10.1111/j.1476-5381.1991.tb12502.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha FQ, Poole S, Lorenzetti BB, Ferreira SH. The pivotal role of tumour necrosis factor alpha in the development of inflammatory hyperalgesia. Br J Pharmacol. 1992;107:660–664. doi: 10.1111/j.1476-5381.1992.tb14503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha FQ, Teixeira MM, Ferreira SH. Pharmacological modulation of secondary mediator systems – cyclic AMP and cyclic GMP – on inflammatory hyperalgesia. Br J Pharmacol. 1999;127:671–678. doi: 10.1038/sj.bjp.0702601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha JM, Rae GA, Ferreira SH, Cunha FQ. Endothelins induce ETB receptor-mediated mechanical hypernociception in rat hindpaw: roles of cAMP and protein kinase C. Eur J Pharmacol. 2004;501:87–94. doi: 10.1016/j.ejphar.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Di Castro A, Drew LJ, Wood JN, Cesare P. Modulation of sensory neuron mechanotransduction by PKC- and nerve growth factor-dependent pathways. Proc Nat Acad Sci USA. 2006;103:4699–4704. doi: 10.1073/pnas.0508005103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dina OA, Barletta J, Chen X, Mutero A, Martin A, Messing RO, et al. Key role for the epsilon isoform of protein kinase C in painful alcoholic neuropathy in the rat. J Neurosci. 2000;20:8614–8619. doi: 10.1523/JNEUROSCI.20-22-08614.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dina OA, Messing RO, Levine JD. Ethanol withdrawal induces hyperalgesia mediated by PKCepsilon. Eur J Neurosci. 2006;24:197–204. doi: 10.1111/j.1460-9568.2006.04886.x. [DOI] [PubMed] [Google Scholar]
- Distler C, Rathee PK, Lips KS, Obreja O, Neuhuber W, Kress M. Fast Ca2+-induced potentiation of heat-activated ionic currents requires cAMP/PKA signaling and functional AKAP anchoring. J Neurophysiol. 2003;89:2499–2505. doi: 10.1152/jn.00713.2002. [DOI] [PubMed] [Google Scholar]
- Dorn GW, II, Souroujon MC, Liron T, Chen CH, Gray MO, Zhou HZ, et al. Sustained in vivo cardiac protection by a rationally designed peptide that causes epsilon protein kinase C translocation. Proc Natl Acad Sci USA. 1999;96:12798–12803. doi: 10.1073/pnas.96.22.12798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England S, Bevan S, Docherty RJ. PGE2 modulates the tetrodotoxin-resistant sodium current in neonatal rat dorsal root ganglion neurones via the cyclic AMP-protein kinase A cascade. J Physiol. 1996;495:429–440. doi: 10.1113/jphysiol.1996.sp021604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira SH. Prostaglandins, aspirin-like drugs and analgesia. Nat New Biol. 1972;240(102):200–203. doi: 10.1038/newbio240200a0. [DOI] [PubMed] [Google Scholar]
- Ferreira SH, Nakamura M. I-Prostaglandin hyperalgesia, a cAMP/Ca2+ dependent process. Prostaglandins. 1979;18:179–190. doi: 10.1016/0090-6980(79)90103-5. [DOI] [PubMed] [Google Scholar]
- Ferreira SH, Lorenzetti BB, Correa FM. Central and peripheral antialgesic action of aspirin-like drugs. Eur J Pharmacol. 1978;53:39–48. doi: 10.1016/0014-2999(78)90265-0. [DOI] [PubMed] [Google Scholar]
- Gray MO, Karliner JS, Mochly-Rosen D. A selective epsilon-protein kinaseC antagonist inhibits protection of cardiac myocytes from hypoxiainduced cell death. J Biol Chem. 1997;272:30945–30951. doi: 10.1074/jbc.272.49.30945. [DOI] [PubMed] [Google Scholar]
- Handwerker HO, Neher KD. Characteristics of C-fibre receptors in the cat's foot responding to stepwise increase of skin temperature to noxious levels. Pflugers Arch. 1976;365:221–229. doi: 10.1007/BF01067022. [DOI] [PubMed] [Google Scholar]
- Huang NK, Lin YW, Huang CL, Messing RO, Chern Y. Activation of protein kinase A and atypical protein kinase C by A(2A) adenosine receptors antagonizes apoptosis due to serum deprivation in PC12 cells. J Biol Chem. 2001;276:13838–13846. doi: 10.1074/jbc.M008589200. [DOI] [PubMed] [Google Scholar]
- Hucho TB, Dina OA, Levine JD. Epac mediates a cAMP-to-PKC signaling in inflammatory pain: an isolectin B4(+) neuron-specific mechanism. J Neurosci. 2005;25:6119–6126. doi: 10.1523/JNEUROSCI.0285-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassuya CA, Ferreira J, Claudino RF, Calixto JB. Intraplantar PGE2 causes nociceptive behaviour and mechanical allodynia: the role of prostanoid E receptors and protein kinases. Br J Pharmacol. 2007;150:727–737. doi: 10.1038/sj.bjp.0707149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D, et al. A novel nociceptor signaling pathway revealed in protein kinase C epsilon mutant mice. Neuron. 1999a;24:253–260. doi: 10.1016/s0896-6273(00)80837-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasar SG, McCarter G, Levine JD. Epinephrine produces a beta-adrenergic receptor-mediated mechanical hyperalgesia and in vitro sensitization of rat nociceptors. J Neurophysiol. 1999b;81:1104–1112. doi: 10.1152/jn.1999.81.3.1104. [DOI] [PubMed] [Google Scholar]
- Leng S, Mizumura K, Koda H, Kumazawa T. Excitation and sensitization of the heat response induced by a phorbol ester in canine visceral polymodal receptors studied in vitro. Neurosci Lett. 1996;206:13–16. doi: 10.1016/0304-3940(96)12414-9. [DOI] [PubMed] [Google Scholar]
- Liao X, Gunstream JD, Lewin MR, Ambron RT, Walters ET. Activation of protein kinase A contributes to the expression but not the induction of long-term hyperexcitability caused by axotomy of Aplysia sensory neurons. J Neurosci. 1999;19:1247–1256. doi: 10.1523/JNEUROSCI.19-04-01247.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzetti BB, Veiga FH, Canetti CA, Poole S, Cunha FQ, Ferreira SH. Cytokine-induced neutrophil chemoattractant 1 (CINC-1) mediates the sympathetic component of inflammatory mechanical hypersensitivitiy in rats. Eur Cytokine Netw. 2002;13:456–461. [PubMed] [Google Scholar]
- Malmberg AB, Brandon EP, Idzerda RL, Liu H, McKnight GS, Basbaum AI. Diminished inflammation and nociceptive pain with preservation of neuropathic pain in mice with a targeted mutation of the type I regulatory subunit of cAMP-dependent protein kinase. J Neurosci. 1997;17:7462–7470. doi: 10.1523/JNEUROSCI.17-19-07462.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin HA, Basbaum AI, Kwiat GC, Goetzl EJ, Levine JD. Leukotriene and prostaglandin sensitization of cutaneous highthreshold C-mechanonociceptors in the rat. Neuroscience. 1987;22:651–659. doi: 10.1016/0306-4522(87)90360-5. [DOI] [PubMed] [Google Scholar]
- Mizumura K, Minagawa M, Tsujii Y, Kumazawa T. Prostaglandin E2-induced sensitization of the heat response of canine visceral polymodal receptors in vitro. Neurosci Lett. 1993;161:117–119. doi: 10.1016/0304-3940(93)90154-d. [DOI] [PubMed] [Google Scholar]
- Moita MA, Lamprecht R, Nader K, LeDoux JE. A-kinase anchoring proteins in amygdala are involved in auditory fear memory. Nat Neurosci. 2002;5:837–838. doi: 10.1038/nn901. [DOI] [PubMed] [Google Scholar]
- Moriyama T, Higashi T, Togashi K, Lida T, Segi E, Sugimoto Y, et al. Sensitization of TRPV1 by EP1 and IP reveals peripheral nociceptive mechanism of prostaglandins. Mol Pain. 2005;1:1–3. doi: 10.1186/1744-8069-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Ferreira SH. A peripheral sympathetic component in inflammatory hyperalgesia. Eur J Pharmacol. 1987;135:145–153. doi: 10.1016/0014-2999(87)90606-6. [DOI] [PubMed] [Google Scholar]
- Nicol GD, Cui M. Enhancement by prostaglandin E2 of bradykinin activation of embryonic rat sensory neurones. J Physiol. 1994;480:485–492. doi: 10.1113/jphysiol.1994.sp020377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouseph AK, Khasar SG, Levine JD. Multiple second messenger systems act sequentially to mediate rolipram-induced prolongation of prostaglandin E2-induced mechanical hyperalgesia in the rat. Neuroscience. 1995;64:769–776. doi: 10.1016/0306-4522(94)00397-n. [DOI] [PubMed] [Google Scholar]
- Parada CA, Yeh JJ, Joseph EK, Levine JD. Tumor necrosis factor receptor type-1 in sensory neurons contributes to induction of chronic enhancement of inflammatory hyperalgesia in rat. Eur J Neurosci. 2003;17:1847–1852. doi: 10.1046/j.1460-9568.2003.02626.x. [DOI] [PubMed] [Google Scholar]
- Parada CA, Reichling DB, Levine JD. Chronic hyperalgesic priming in the rat involves a novel interaction between cAMP and PKCepsilon second messenger pathways. Pain. 2005;113:185–190. doi: 10.1016/j.pain.2004.10.021. [DOI] [PubMed] [Google Scholar]
- Pateromichelakis S, Rood JP. Prostaglandin E1-induced sensitization of A delta moderate pressure mechanoreceptors. Brain Res. 1982;232:89–96. doi: 10.1016/0006-8993(82)90612-6. [DOI] [PubMed] [Google Scholar]
- Rueff A, Dray A. Sensitization of peripheral afferent fibers in the in vitro neonatal rat spinal cord-tail by bradykinin and prostaglandins. Neuroscience. 1993;54:527–535. doi: 10.1016/0306-4522(93)90272-h. [DOI] [PubMed] [Google Scholar]
- Schaible HG, Schmidt RF. Excitation and sensitization of fine articular afferents from cat's knee joint by prostaglandin E2. J Physiol. 1988;403:91–104. doi: 10.1113/jphysiol.1988.sp017240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott JD. Cyclic nucleotide-dependent protein kinases. Pharmacol Ther. 1991;50:123–145. doi: 10.1016/0163-7258(91)90075-w. [DOI] [PubMed] [Google Scholar]
- Smith JA, Davis CL, Burgess GM. Prostaglandin E2-induced sensitization of bradykinin-evoked responses in rat dorsal root ganglion neurons is mediated by cAMP-dependent protein kinase A. Eur J Neurosci. 2000;12:3250–3258. doi: 10.1046/j.1460-9568.2000.00218.x. [DOI] [PubMed] [Google Scholar]
- Souza AL, Moreira FA, Almeida KR, Bertollo CM, Costa KA, Coelho MM. In vivo evidence for a role of protein kinase C in peripheral nociceptive processing. Br J Pharmacol. 2002;135:239–247. doi: 10.1038/sj.bjp.0704434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summer GJ, Puntillo KA, Miaskowski C, Dina OA, Green PG, Levine JD. TrkA and PKC-epsilon in thermal burn-induced mechanical hyperalgesia in the rat. J Pain. 2006;12:884–891. doi: 10.1016/j.jpain.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Sweitzer SM, Wong SM, Peters MC, Mochly-Rosen D, Yeomans DC, Kendig JJ. Protein kinase C epsilon and gamma: involvement in formalin-induced nociception in neonatal rats. J Pharmacol Exp Ther. 2004;309:616–625. doi: 10.1124/jpet.103.060350. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Levine JD. Direct cutaneous hyperalgesia induced by adenosine. Neuroscience. 1990;38:757–762. doi: 10.1016/0306-4522(90)90068-f. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Levine JD. Further confirmation of the role of adenyl cyclase and of cAMP-dependent protein kinase in primary afferent hyperalgesia. Neuroscience. 1991;44:131–135. doi: 10.1016/0306-4522(91)90255-m. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Levine JD. Serotonin is a directly-acting hyperalgesic agent in the rat. Neuroscience. 1992;48:485–490. doi: 10.1016/0306-4522(92)90508-y. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Bjerknes LK, Goetzl EJ, Levine JD. Mediation of primary afferent peripheral hyperalgesia by the cAMP second messenger system. Neuroscience. 1989;32:577–580. doi: 10.1016/0306-4522(89)90280-7. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Heller PH, Levine JD. Mediation of serotonin hyperalgesia by the cAMP second messenger system. Neuroscience. 1992;48:479–483. doi: 10.1016/0306-4522(92)90507-x. [DOI] [PubMed] [Google Scholar]
- Wang C, Li GW, Huang LY. Prostaglandin E2 potentiation of P2X3 receptor mediated currents in dorsal root ganglion neurons. Mol Pain. 2007;3:22. doi: 10.1186/1744-8069-3-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JF, Khasar SG, Ahlgren SC, Levine JD. Sensitization of C-fibres by prostaglandin E2 in the rat is inhibited by guanosine 5′-O-(2-thiodiphosphate), 2′,5′-dideoxyadenosine and Walsh inhibitor peptide. Neuroscience. 1996;71:259–263. doi: 10.1016/0306-4522(95)00429-7. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Kawamata T, Ninomiya T, Omote K, Namiki A. Endothelin-1 enhances capsaicin-evoked intracellular Ca2+ response via activation of endothelin a receptor in a protein kinase Cepsilon-dependent manner in dorsal root ganglion neurons. Neuroscience. 2006;137:949–960. doi: 10.1016/j.neuroscience.2005.09.036. [DOI] [PubMed] [Google Scholar]
- Yao L, Fan P, Jiang Z, Gordon A, Mochly-Rosen D, Diamond I. Dopamine and ethanol cause translocation of εPKC associated with εRACK: cross-talk between cAMP-Dependent Protein Kinase A and Protein Kinase C Signaling Pathways. Mol Pharmacol. 2008;73:1105–1112. doi: 10.1124/mol.107.042580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu PY, Eisner GM, Yamaguchi I, Mouradian MM, Felder RA, Jose PA. Dopamine D1A receptor regulation of phospholipase C isoform. J Biol Chem. 1996;271:19503–19508. doi: 10.1074/jbc.271.32.19503. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Li GD, Zhao ZQ. State-dependent phosphorylation of epsilon-isozyme of protein kinase C in adult rat dorsal root ganglia after inflammation and nerve injury. J Neurochem. 2003;85:571–580. doi: 10.1046/j.1471-4159.2003.01675.x. [DOI] [PubMed] [Google Scholar]