

Abstract
Background and purpose:
Highly selective M3 muscarinic receptor antagonists may represent a better treatment for overactive bladder syndrome, diminishing side effects. Cardiac side effects of non-selective antimuscarinics have been associated with activity at M2 receptors as these receptors are mainly responsible for muscarinic receptor-dependent bradycardia. We have investigated a novel antimuscarinic, SVT-40776, highly selective for M3 over M2 receptors (Ki = 0.19 nmol·L−1 for M3 receptor affinity). This study reports the functional activity of SVT-40776 in the bladder, relative to its activity in atria.
Experimental approach:
In vitro and ex vivo (oral dosing) inhibition of mouse detrusor and atrial contractile responses to carbachol were used to study the functional activity of SVT-40776. The in vivo efficacy of SVT-40776 was characterized by suppression of isovolumetric spontaneous bladder contractions in anaesthetized guinea pigs after intravenous administration.
Key results:
SVT-40776 was the most potent in inhibiting carbachol-induced bladder contractions of the anti-cholinergic agents tested, without affecting atrial contractions over the same range of concentrations. SVT-40776 exhibited the highest urinary versus cardiac selectivity (199-fold). In the guinea pig in vivo model, SVT-40776 inhibited 25% of spontaneous bladder contractions at a very low dose (6.97 µg·kg−1 i.v), without affecting arterial blood pressure.
Conclusions and implications:
SVT-40776 is a potent inhibitor of M3 receptor-related detrusor contractile activity. The absence of effects on isolated atria preparations represents an interesting characteristic and suggests that SVT-40776 may lack unwanted cardiac effects; a feature especially relevant in a compound intended to treat mainly elderly patients.
British Journal of Pharmacology (2009) doi:10.1111/j.1476-5381.2008.00082.x
Keywords: muscarinic antagonist, urinary bladder, overactive bladder, SVT-40776, carbachol, isolated bladder tissue, atria contractions
Introduction
Overactive bladder (OAB) is a widely prevalent condition characterized by urgency, with or without urge incontinence, usually accompanied by frequency and nocturia (Abrams et al., 2003). It arises from involuntary contractions of the detrusor muscle during bladder filling (Wein and Rovner, 2002). In an attempt to clarify the etiology of this bladder dysfunction, different theories endorse its either myogenic (Brading, 1997) or neural (de Groat, 1997) origin. However, in most cases, OAB is idiopathic in nature.
Muscarinic acetylcholine receptors are the predominant receptor system controlling bladder contractility (Andersson and Yoshida, 2003, Abrams et al., 2006) and its pharmacological characterization mediating detrusor smooth muscle contraction has been well established in humans (Fetscher et al., 2002; Tyagi et al., 2006), mice (Choppin and Eglen, 2001; Choppin, 2002), rats (Longhurst et al., 1995; Hegde et al., 1997; Longhurst and Levendusky, 2000), guinea pigs (Wang et al., 1995), rabbits (Tobin and Sjogren, 1995; Choppin et al., 1998) and monkeys (Lai et al., 1998). M2 and M3 muscarinic receptors coexist in the bladder of different mammalian species, including humans, with a major presence of the M2 receptor (M2 receptors account for 70–80% of the receptor population whereas the M3 receptors comprise only 20–30%) (Eglen et al., 1996). However, although M2 receptors predominate in number, numerous studies have suggested that the M3 receptor represents the main subtype controlling bladder contractility in humans (Hegde and Eglen, 1999, Chess-Williams et al., 2001; Andersson, 2002a; Fetscher et al., 2002; Stevens et al., 2007), through a mechanism involving activation of phosphoinositide breakdown followed by an increase in intracellular Ca2+ levels (Harriss et al., 1995; Tran et al., 2006), in addition to activation of the Rho-kinase pathway (Schneider et al., 2004). Further evidence comes from studies using M3 receptor knockout mice, in which bladder contractile response to carbachol is virtually abolished (Matsui et al., 2000). The residual (∼5%) contractions that persist in those animals are completely lost in M2/M3 double knockout mice, revealing the occurrence of minor M2 receptor-mediated contractions (Matsui et al., 2002). Moreover, M2 receptors are capable of mediating bladder contractions by enhancing the contractile response to M3 receptor activation, indicating a contribution of M2 receptors in M3 receptor-mediated contractile responses (Ehlert et al., 2005). Although this contribution has been suggested to become more important in diseased states (Pontari et al., 2004; Stevens et al., 2007), in both normal and overactive human bladders, direct contractile response to carbachol is mediated mainly by M3 receptors.
Muscarinic M2 receptors, the major muscarinic subtype present in mammalian heart (Caulfield, 1993; Hoover et al., 1994), have been reported to be essential for muscarinic receptor-dependent bradycardia, modulating pacemaker activity and atrioventricular conduction (Stengel et al., 2000; Dhein et al, 2001; Andersson and Olshansky, 2007). Although expression of M1, M3 and M4 muscarinic receptors have been reported in guinea pig and rat intrinsic intracardiac neurons (Hassall et al., 1993) as well as in canine atria (Shi et al., 1999), their functional role is not completely understood. Also in human heart, M1, M3 and M5 receptors have been localized (Hellgren et al., 2000; Wang et al., 2001; Andersson and Olshansky, 2007). However, to date, no data indicate that any other muscarinic receptor subtype, apart from M2 receptors, mediate effects on heart rate (Andersson and Olshansky, 2007).
This study reports the functional characterization of SVT-40776, a novel quinuclidine derivative (Farrerons et al., 2002), highly selective for M3 over M2 receptors, on mouse detrusor and isolated atrial tissues, as well as in a guinea pig bladder contraction model in vivo. We have compared its pharmacological activity with those of the currently marketed anticholinergic agents, tolterodine, darifenacin and solifenacin. Our results demonstrated that SVT-40776 was the most potent in inhibiting carbachol-induced bladder contractions, compared with the anticholinergic agents tested, without affecting atrial contractions at the same range of concentrations. SVT-40776 exhibited the highest urinary versus cardiac selectivity (199-fold) in the animal models studied. These data may suggest that SVT-40776 could provide improved tolerability over currently available treatments for OAB.
Methods
Animal experiments
All animal procedures were according to the published guidelines on the use of animals in research (EU Directive 86/609/ECC). CD-1 male mice (25–30 g) and DH female guinea pigs (250–300 g) were purchased from Harlan Iberica (Spain). They were housed in a temperature-controlled environment (20 ± 2°C, 12 h light/dark cycle) with standard laboratory food and water freely available. All animals were fasted for 18 h prior to the experiment.
In vitro contractile study
Bladder detrusor preparations
Mice were killed by CO2 and the urinary bladder was isolated and placed in Krebs' solution (composition in mmol·L−1: NaCl 118, KCl 4.6, CaCl2 1.5, MgCl2 1.5, KH2PO4 1.15, NaHCO3 25 and glucose 11). Indomethacin (30 µmol·L−1) and hexamethonium (1 mmol·L−1) were routinely included in the Krebs' solution to abolish prostaglandin-induced spontaneous activity and any possible nicotinic receptormediated activity respectively. One strip of tissue per animal (4 × 2 mm) was cut from the posterior region of bladder body, parallel to the longitudinal axis. Tissues were mounted in 25 mL organ baths containing Krebs' solution, maintained at 37°C and constantly aerated with 95% O2 and 5% CO2 (pH = 7.4). Isometric tension generated by the tissue was measured by pure isometric transducers (Cibertec) and recorded using the PowerLab® software (ADInstruments, Bella Vista, Australia). Tissues were maintained at a resting tension of 5 mN during an equilibration period of 60 min. Tension adjustments were made as necessary. Tissues were washed every 15 min. The viability of each tissue was assessed by determining the contractile response to KCl (120 mmol·L−1) at the start of the experimental protocol. After washing, tissues were re-equilibrated for 15 min and allowed to regain baseline tension. Repeated contractions to carbachol (3 µmol·L−1) were then induced, in order to obtain two consecutive contractions with less than 10% difference. After tissue equilibration, cumulative consecutive concentration-effect curves to carbachol were then constructed in each bladder preparation. Antagonist was incubated for a 60 min period between curves.
Atrial preparations
Mice were killed by CO2 and both atria were isolated, placed in aerated Krebs' solution (without either indomethacin or hexamethonium) with ligatures placed on the right and left atrium. Tissues were maintained at a resting tension of 1.47 mN during an equilibration period of 10 min to obtain spontaneous contractions in order to measure the beating frequency. After tissue equilibration, cumulative consecutive concentration-effect curves to carbachol were then constructed in each atrial preparation. Antagonist was incubated for a 60 min period between the first and second curve.
Ex vivo contractile study
Bladder and atria preparations
Groups of animals (n = 4–6 per dose) received a single oral dose (0.3 to 50 mg·kg−1) of antagonists or vehicle (three to five doses per compound). Mice were killed 3 h later and the urinary bladder and atria were excised and prepared as described before. After tissue equilibration, the viability of each strip was assessed by determining the contractile response to KCl at the start of the experimental protocol. Repeated KCl contractions were used to warm up the tissue. A unique cumulative concentration-effect curve to carbachol was then constructed in each tissue and referred to KCl effect.
In vivo animal study
Guinea pigs were anaesthetized with urethane 1.5 g·kg−1 i.p. (n = 4–6 animals per compound). A polyethylene catheter (PE-50) was implanted in the bladder via the urethra and the bladder was emptied. Carotid artery and jugular vein were cannulated to register arterial pressure (AP) and to administer drugs respectively. Bladder and carotid catheters were connected to pressure transducers (Transpac IV) and analysed using PowerLab® Software.
A baseline AP of 59.8 ± 1.7 mmHg was registered. The bladder was filled with 2.8 mL of saline to obtain a mean pressure of 58.5 ± 2.8 mmHg, which induced regular spontaneous contractions. After obtaining a stable response, the compound was administered by intravenous bolus followed by a cumulative consecutive dose–response protocol (15 min between doses or when stable contractions were obtained). Mean AP was measured during the first 5 min post-dose periods. Amplitude from all bladder contractions (intravesical pressure) was measured during the 15 min period between doses and a mean amplitude was calculated for baseline and for each dose. Per cent change per dose was calculated, relative to baseline effects and an ED25 (effective dose 25% of maximum response) was obtained for each compound.
Data analysis
In the in vitro study, bladder contractions were recorded as changes in tension from baseline and expressed as a percentage of the maximum response of the first agonist concentration-effect curve. Atrial beating frequency was recorded and expressed as a percentage of the initial frequency before the first agonist concentration-effect curve. Carbachol concentration-effect curves were analysed by using a non-linear fitting program (GraphPad Prism software) and pEC50 values were calculated. Concentration ratio (CR) was determined from pEC50 values in the presence and absence of different antagonist concentrations. pA2 values were obtained from the x-axis intercept from the Schild plot and expressed as means with 95% confidence interval. The Schild plots of all antagonists were linear, with slopes close to unity. All other data are expressed as mean ± SEM.
In the ex vivo protocol, carbachol concentration-effect curves were fitted as described above. CR was determined from pEC50 values, considering curves obtained from vehicle-treated groups as ‘control pEC50’ and curves from antagonist-treated animals as ‘treated animals pEC50’. A pA2-equivalent dose (pA2-ED) value was obtained from the Schild plot, using the oral doses instead of bath concentrations for each compound and given as means with 95% confidence interval. All other data are shown as mean ± SEM.
In the in vivo study, ED25 values were obtained fitting the percentage of variation to a cubic spline curve (GraphPad Prism software). These values represent the doses needed to inhibit 25% bladder contractions. For the results of in vitro assays, a non-paired Student's t-test was used for statistical analysis.
Materials
SVT-40776, darifenacin, solifenacin and tolterodine were synthesized by the Medicinal Chemistry Department (SALVAT). Carbachol, indomethacin and hexamethonium were purchased from Sigma Chemical Co. (St. Louis, MO, USA). For the in vitro protocol, compounds tested were prepared at 1 mmol·L−1 in dimethylsulfoxide (DMSO) and dilutions made in deionized water. For the ex vivo protocol, compounds tested were freshly suspended in vehicle (methylcellulose 0.5% and Tween-80 0.1%) 1 h before oral administration (10 mL·kg−1). For the anaesthetized animal study, stock solutions of SVT-40776 and solifenacin were dissolved in saline. Tolterodine was dissolved in deionized water. Darifenacin stock solution was prepared in 10% DMSO in deionized water. Subsequent dilutions for all compounds were made in saline.
Results
In vitro functional characterization of SVT-40776 on mouse isolated bladder and atria preparations
Carbachol induced concentration-dependent contractions of mouse urinary bladder smooth muscle. The first curve yielded a mean pEC50 = 5.66 ± 0.04 with a maximum response (Emax) of 11.7 ± 2.2 mN. A consecutive additional carbachol concentration-response curve within this preparation in the absence of antagonist yielded a mean pEC50 = 5.66 ± 0.08 with an Emax of 11.9 ± 1.9 mN (n = 4). Thus, two consecutive concentration-effect curves to carbachol could be constructed in the same tissue with no significant change in the agonist potency and maximum response (Figure 1A). All the compounds tested antagonized cumulative agonist concentration-response curves, in a concentration-dependent fashion, with parallel right-ward shifts. While tolterodine and solifenacin did not significantly alter maximum carbachol response (Figure 1B,C), darifenacin exposure clearly reduced Emax (from 66% at 3 nmol·L−1 to 58% at 100 nmol·L−1) (Figure 1D). SVT-40776 slightly reduced Emax at 10 nmol·L−1 and 100 nmol·L−1 concentrations (to 71% and 80% respectively) (Figure 1E). All four antagonists yielded Schild regression lines, with slopes close to unity.
Figure 1.
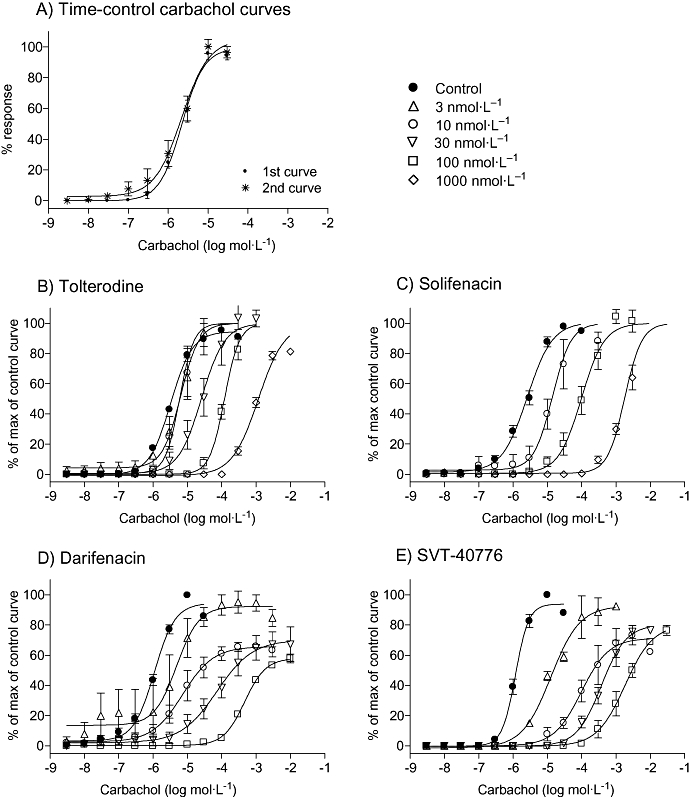
Effects of muscarinic receptor antagonists on the cumulative consecutive concentration-response curves to carbachol on mouse urinary bladder. In (A), two repeated control concentration-response curves show the reproducibility and stability of the preparation. In (B)–(E), the effects of anatagonists (3–1000 nmol·L−1) on the carbachol concentration-response curves are shown. Direct contractile effects were expressed as percentages of the maximum response of the control curve. Data are expressed as mean ± SEM, n = 4–8 animals per concentration.
In mouse atrial preparations, carbachol induced concentration-dependent negative chronotropism of spontaneous beating right atria. Two consecutive concentration-effect curves to carbachol could be constructed in the same tissue with no significant change in the agonist potency and maximum response. First and second curves yielded pEC50 of 6.23 ± 0.05 and 6.89 ± 0.08 (n = 10) respectively. Maximum effect, which was the complete inhibition of beating, was also maintained in the second curve (Figure 2A). The four antagonists tested shifted the carbachol curve dose-dependently to the right (Figure 2B–E).
Figure 2.
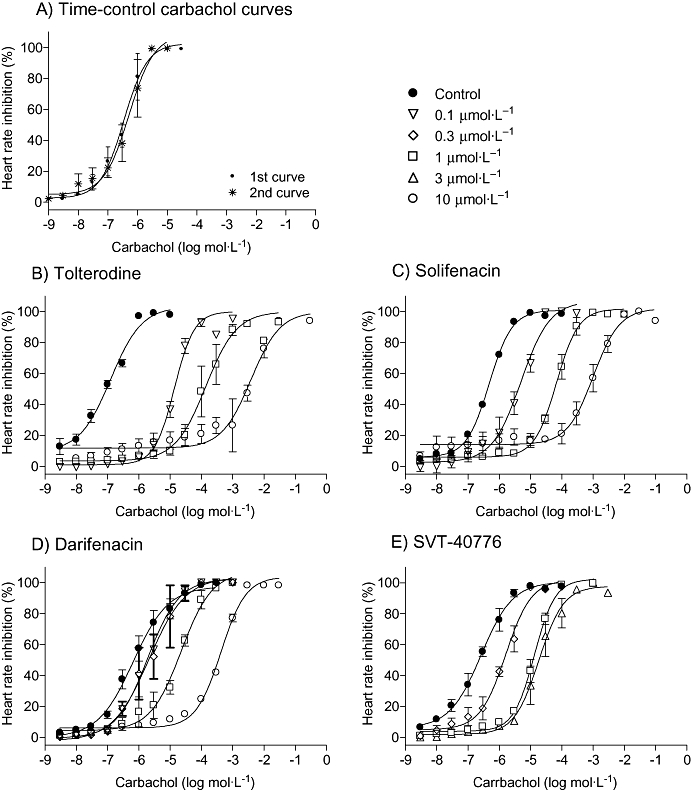
Effect of tolterodine (B), solifenacin (C), darifenacin (D) and SVT-40776 (E) treatment on the cumulative consecutive concentration-response curves to carbachol (A) on mouse atria. As in Figure 1, the reproducibility of the control curves is shown in (A) and the effects of the antagonists (0.1–10 µmol·L−1) in (B)–(E). Direct contractile effects were expressed as percentages of the maximum response of the control curve. Data are expressed as mean ± SEM, n = 4–8 animals per concentration.
Antagonist affinities (pA2) on carbachol-induced responses on bladder and heart isolated tissues in vitro are summarized in Table 1. SVT-40776 was clearly the most potent antagonist in the bladder, lacking any relevant effect in atria at the same range of concentrations. Furthermore, as shown in Table 1, SVT-40776 exhibited the highest urinary versus cardiac selectivity (199-fold).
Table 1.
In vitro affinities (pA2) of muscarinic M3 receptor antagonists on carbachol-induced responses in isolated bladder and atrial tissues
| Compound |
pA2(CI) |
Selectivity ratio | |||
|---|---|---|---|---|---|
| Bladder | Slope | Atria | Slope | ||
| Tolterodine | 8.4 | 1.09 ± 0.10 | 8.5 | 1.24 ± 0.17 | 0.79 |
| (8.2–8.6) | (7.8–9.2) | ||||
| Darifenacin | 8.8 | 1.27 ± 0.52 | 7.3 | 1.26 ± 0.02 | 31.6 |
| (8.2–9.4) | (7.2–7.4) | ||||
| Solifenacin | 8.6 | 1.11 ± 0.06 | 7.8 | 1.20 ± 0.02 | 6.3 |
| (8.3–8.9) | (7.7–7.8) | ||||
| SVT-40776 | 9.5 | 1.36 ± 0.12 | 7.2 | 1.15 ± 0.45 | 199 |
| (9.2–9.8) | (7.0–8.1) | ||||
pA2 values are expressed as mean (95% CI); the slope shown is from Schild plot analysis, mean ± SEM, n = 4–8 animals for each antagonist concentration.
Ex vivo functional characterization of SVT-40776 on mouse isolated bladder and atria preparations
Detrusor smooth muscle from control animals killed 3 h after receiving an oral dose of vehicle did not behave differently from that of non-treated animals. Carbachol induced concentration-response curves, yielding a pEC50 = 5.16 ± 0.06 (n = 71) (Figure 3A). This value was assigned as control, in order to compare it with pEC50 from antagonist-treated animals. KCl produced a maximum effect similar to carbachol (11.4 ± 1.8 mN). Right-ward shifts of the carbachol response curves for tolterodine, solifenacin, darifenacin and SVT-40776 were obtained (Figure 3B–E). While tolterodine, solifenacin and SVT-40776 did not significantly affect the maximum response to carbachol, darifenacin at 50 mg·kg−1 significantly reduced the Emax. All four antagonists yielded Schild regression lines with slopes close to unity.
Figure 3.
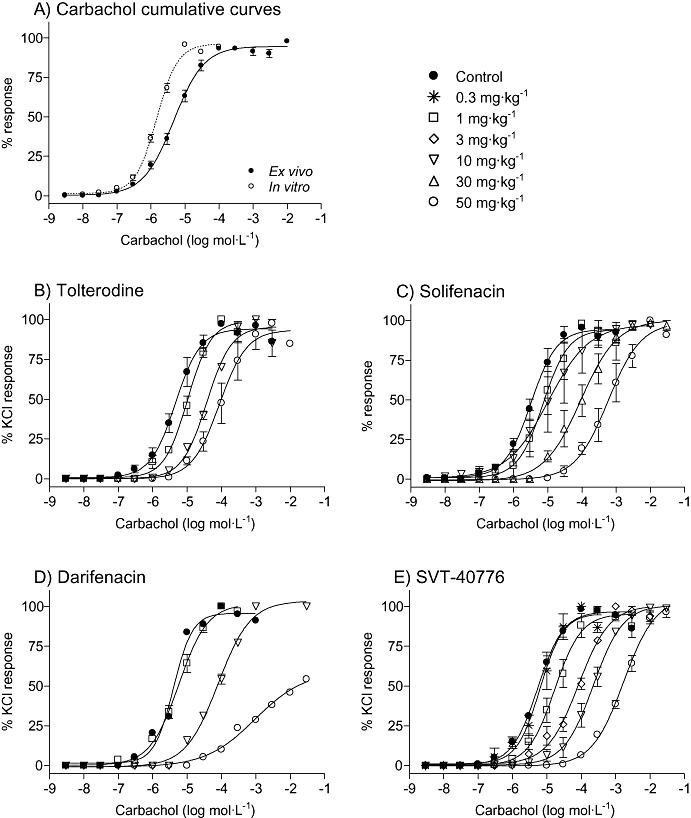
Effect of oral administration of the M3 receptor antagonists on the contractile response to carbachol in mouse urinary bladder ex vivo. Data are expressed as mean ± SEM, n = 4–6 animals per dose.
Atria from control animals killed 3 h after receiving an oral dose of vehicle showed the same behaviour as those from non-treated animals. Carbachol induced concentration-response curves, yielding pEC50 = 6.55 ± 0.09 (n = 27) (Figure 4A). This value was assigned as control, in order to compare it with the pEC50 from antagonist-treated animals. In this protocol, as in the in vitro atria, maximum effect was seen when complete inhibition of beating was obtained. Tolterodine and solifenacin dose-dependently shifted carbachol curves to the right. Darifenacin exhibited less potency than tolterodine and solifenacin. SVT-40776 did not induce any relevant displacement of carbachol curves to the right, up to a dose of 30 mg·kg−1 (Figure 4B–E). Only at the very high dose of 50 mg·kg−1, it was able to shift carbachol curves to the right, to yield pED50 values twice that of control.
Figure 4.
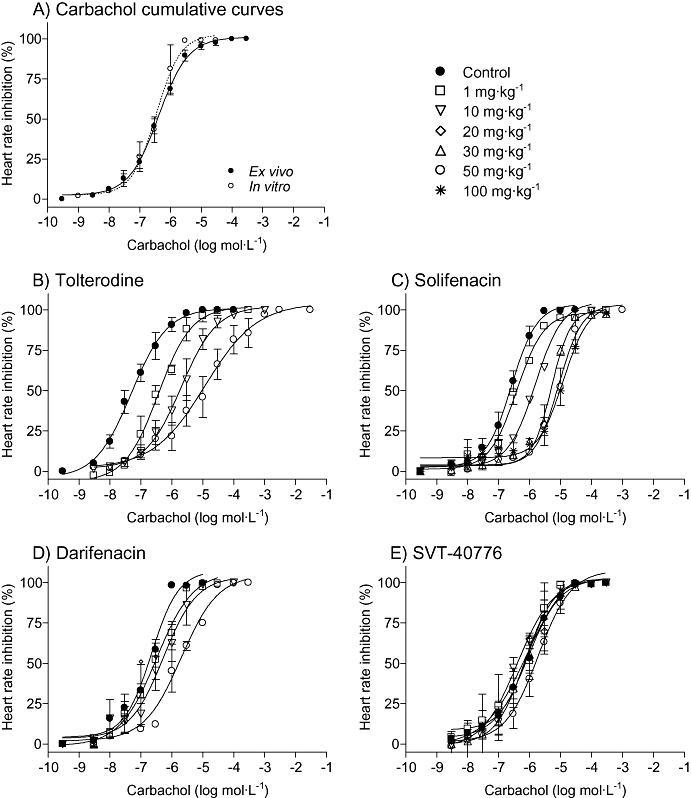
Effect of oral administration of the M3 receptor antagonists on the contractile response to carbachol in mouse atria ex vivo. Data are expressed as mean ± SEM, n = 4–6 animals per dose.
Antagonist activities (expressed as pA2-ED) at carbachol-induced responses on bladder and heart isolated tissues ex vivo are reported in Table 2. In accordance with results obtained in the in vitro study, SVT-40776 exhibited the highest urinary versus cardiac selectivity (58-fold), whereas darifenacin, solifenacin and tolterodine showed much lesser selectivity (2.4-, 1.5- and 0.21-fold respectively).
Table 2.
Ex vivo affinities, shown as pA2-equivalent dose (pA2-ED) of muscarinic receptor antagonists on carbachol-induced responses in isolated bladder and atrial tissues
| Compound |
pA2-ED (mg·kg−1oral) |
Selectivity ratio | |
|---|---|---|---|
| Bladder | Atria | ||
| Tolterodine | 0.7 | 0.14 | 0.21 |
| (0.4–1.0) | (0.07–0.3) | ||
| Darifenacin | 1.6 | 3.9 | 2.4 |
| (0.35–6.7) | (2.1–7.5) | ||
| Solifenacin | 1.3 | 2.0 | 1.5 |
| (0.46–4.2) | (1.6–2.6) | ||
| SVT-40776 | 0.7 | 40.3 | 58 |
| (0.4–1.1) | (32.6–51.6) | ||
Mice were killed 3 h after receiving a single oral dose of antagonists or vehicle. Values are expressed as mean (95% CI) n = 4–6 animals for each antagonist dose, three to five doses for each compound.
Table 3 shows carbachol EC50 values from detrusor and atria preparations, comparing in vitro and ex vivo experiments. These values are very similar, which demonstrates the reliability of the ex vivo technique.
Table 3.
Comparative in vitro and ex vivo carbachol pEC50 obtained in isolated detrusor and in atria from mice
| In vitro pEC50 (n) | Ex vivo pEC50 (n) | |
|---|---|---|
| Detrusor | 5.66 ± 0.04 | 5.16 ± 0.06 |
| (4) | (71) | |
| Atria | 6.23 ± 0.05 | 6.55 ± 0.09 |
| (10) | (27) |
pEC50 values are expressed as mean ± SEM, n = number of animals used.
In vivo functional characterization of SVT-40776 on guinea pig bladder
Figure 5 shows a representative trace of spontaneous contractions before and after consecutive saline (A) or antagonist (B) bolus. Amplitudes of bladder contraction were measured during the 15 min period between doses and the per cent change in amplitude was calculated, relative to baseline values. Intravenous administration of SVT-40776, tolterodine, darifenacin and solifenacin changed bladder contraction amplitude in a dose-dependent manner (Figure 6). SVT-40776 was the most potent compared with the antagonists tested in the guinea pig in vivo model, inhibiting 25% of spontaneous bladder contractions at a very low dose of 17.1 nmol·kg−1 i.v. (6.97 µg·kg−1). Calculated effective doses of darifenacin, solifenacin and tolterodine were 3-, 12- and 17-fold higher than that of SVT-40776 respectively (Table 4). In addition, the bladder versus vascular selectivity of SVT-40776 was more than 175-fold. SVT-40776, darifenacin and solifenacin showed higher urinary selectivity than tolterodine in this assay (Table 4).
Figure 5.
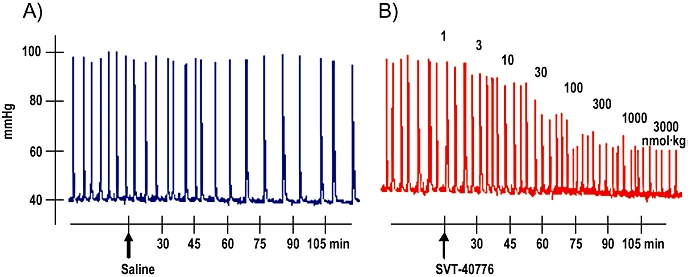
Representative trace of spontaneous contractions of guinea pig bladder in vivo before and after consecutive saline (a) or antagonist (b) bolus. Amplitude of bladder contraction was measured during the 15 min period between doses and the per cent change in bladder contraction amplitude was calculated, relative to baseline values.
Figure 6.
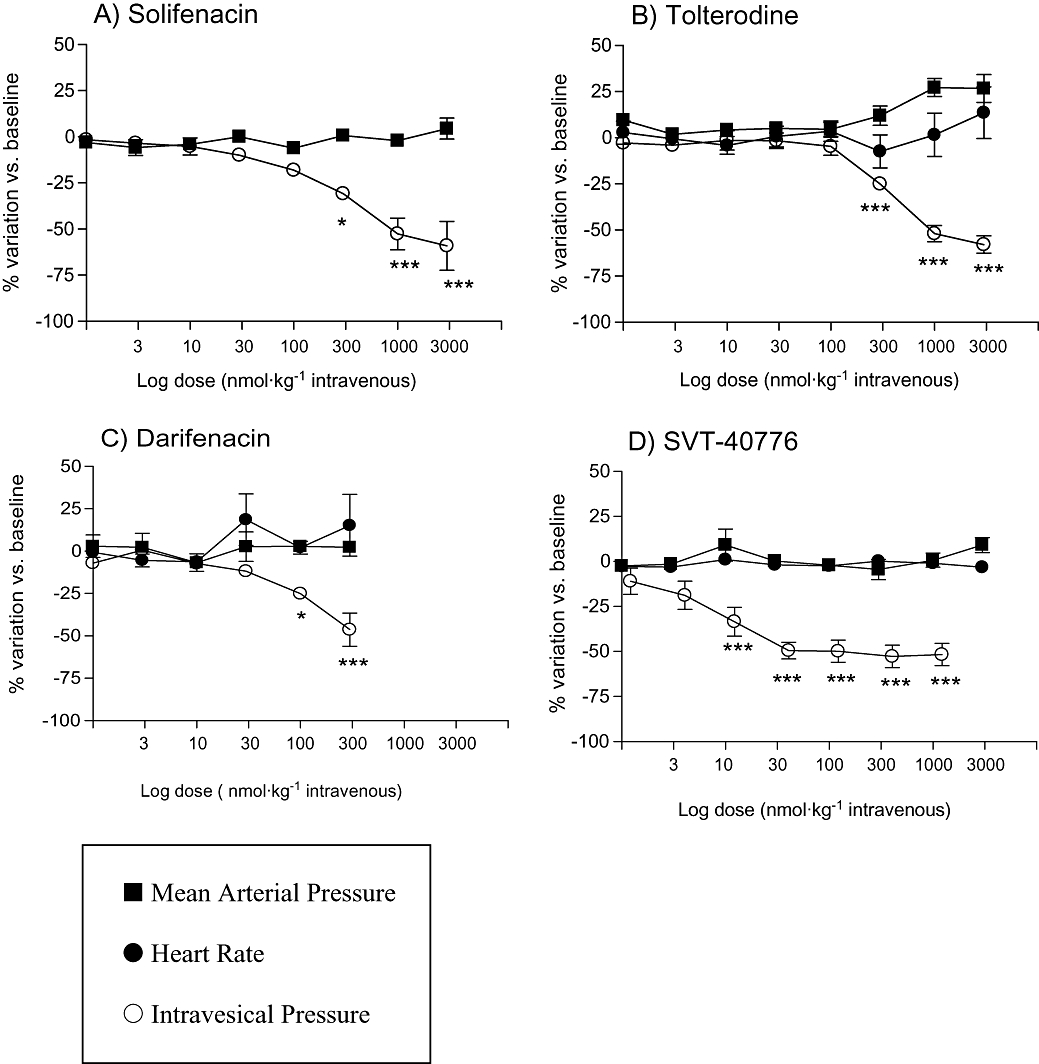
Effect of intravenous administration of four muscarinic receptor antagonists on bladder contraction, blood pressure and heart rate in guinea pigs. Amplitude of bladder contraction, mean arterial blood pressure and heart rate were measured at the same times (see Methods). The per cent change in each variable was calculated relative to baseline values. *P < 0.05, **P < 0.01 and ***P < 0.001 versus control; Student's t-test: n = 4–6 animals per compound.
Table 4.
In viv o effects of treatment with M3 receptor antagonists on bladder intravesical pressure and arterial blood pressure
| Compound |
ED25 (nmol·kg−1, i.v.) |
|
|---|---|---|
| IVP inhibition | MAP increase | |
| Tolterodine | 299.2 | 820 |
| Darifenacin | 53.2 | >1000 |
| Solifenacin | 200.9 | >3000 |
| SVT-40776 | 17.1 | >3000 |
Values are calculated from n = 4–6 animals per dose. Darifenacin was not given at higher doses because of its solubility problems.
IVP, intravesical pressure; MAP, mean arterial pressure.
Discussion and conclusions
Antimuscarinic agents (muscarinic receptor antagonists) are commonly the first line of treatment for OAB. However, their side effects, stemming from a lack of selectivity, compromise their clinical use (Andersson, 2002b; 2004; Andersson and Olshansky, 2007). Because of this, there are clear potential benefits in terms of efficacy and tolerability to be provided by selective antagonists of muscarinic M3 receptors (Andersson, 2002b).
Efficacy of antimuscarinic drugs for the treatment of OAB has been evaluated in several clinical trials. A systematic review of 32 randomized controlled trials conducted by Herbison et al. (2003) concluded that antimuscarinic agents produced significant improvements in OAB symptoms compared with placebo, even though the clinical relevance of these differences was uncertain. A recent update of a Cochrane systematic review has corroborated the efficacy of anticholinergic medication, suggesting also that improvements in symptoms may be associated with modest improvement in quality of life (Nabi et al., 2006). The overall concept of improvement in quality of life was introduced to support a reported efficacy that did not reflect the real limited effectiveness, a handicap that no recently launched treatment has been able to overcome.
From a rational point of view, two logical questions are: is efficacy compromised because the dose level is limited and is the dose limited because of the probability of adverse events? The incidence of typical muscarinic adverse events such as constipation or dry mouth has been shown to increase with dose (Chapple et al., 2005; Hay-Smith et al., 2005). However, cardiac effects due to blockade of M2, receptors which would be unacceptable for a non-life-threatening condition, have been clearly under-reported. Increase in heart rate is an adverse effect exhibited by non-selective anticholinergic agents, which may become prominent when used at high doses (Howell and Kovalsky, 1995; Andersson and Olshansky, 2007). The last two antimuscarinic agents launched for OAB, solifenacin and darifenacin, were developed with the intention of obtaining a safer cardiovascular profile. It should be noted that cardiovascular disorders, including hypertension, ischaemic heart disease and arrhythmias, have a prevalence of 47% in OAB patients treated with anitmuscarinics (Andersson and Olshansky, 2007). The effect of darifenacin and tolterodine treatment on heart rate have been recently evaluated in patients with OAB (Romanzi et al., 2005; Olshansky et al., 2006; 2008). Tolterodine significantly increased heart rate in comparison with darifenacin and placebo. The percentage of patients with an increase in heart rate of ≥5 bpm from baseline to last observation was significantly greater with immediate-release tolterodine (2 mg twice daily) (39.3%) than placebo (23.2%) or darifenacin (15 mg·kg−1 once daily) (23%) (Olshansky et al., 2006). Very similar results were obtained in a second study where extended release-tolterodine (4 mg once daily) and darifenacin (15 mg·kg−1 once daily) were compared with placebo (Olshansky et al, 2008). Tiotropium, a well-known non-selective muscarinic antagonist developed for the treatment of chronic obstructive pulmonary disease (COPD), showed in a pooled clinical trial analysis, a slightly elevated risk of tachycardia when compared with placebo (Kesten et al., 2005). Moreover, Barr et al. (2006) published a meta-analysis of available randomized trials, in which, among the adverse events reported, the authors pointed out that the frequency of arrhythmias was significantly higher with tiotropium than with placebo.
In order to increase both the efficacy and tolerability of antimuscarinics in the treatment of OAB, new antagonists with greater selectivity for M3 receptors are being developed. The present study reports the functional activity of a novel antimuscarinic, SVT-40776, with M3 receptor antagonist properties. Previous binding studies performed in our laboratory have characterized the binding properties of SVT-40776 (data not shown). These studies demonstrated the high affinity and selectivity for binding to M3 receptors over M2 of SVT-40776 (Ki of 0.19 nmol·L−1 for human M3 receptor affinity and 203-fold for M3 vs. M2 affinity) (Farrerons et al., 2002 SALVAT S.A., PCT Patent application WO02/00652; Salcedo et al., 2003; Balsa et al., 2004; Fernández et al., 2005). In the present study, we have characterized the functional activity of SVT-40776 on bladder contraction and compared it with its activity on atrial contractions in order to assess the functional selectivity of the compound.
Functional in vitro studies in mouse urinary bladder smooth muscle have shown that SVT-40776 was more potent in inhibiting carbachol-induced bladder contractions than the marketed antimuscarinic agents tolterodine, solifenacin and darifenacin. SVT-40776 was able to produce a right-ward parallel shift of the cumulative agonist concentration-response curves, obtaining a pA2 of 9.5, while pA2 values for tolterodine, solifenacin and darifenacin were 8.4, 8.6 and 8.8 respectively. Tolterodine and solifenacin did not significantly alter maximum carbachol response. However, darifenacin reduced Emax (from 66% at 3 nmol·L−1 to 58% at 100 nmol·L−1), which means that darifenacin behaves insurmountably in the mouse bladder. SVT-40776 slightly reduced the Emax to 71% at 10 nmol·L−1 and to 80% at 100 nmol·L−1, concentrations that are 25 and 250-fold higher than its intrinsic activity. Although the pharmacological meaning of these results has not been elucidated so far and would require further investigation, one explanation can be that at these high concentrations a depletion of the antagonist from the medium can occur as a consequence of binding to other receptors or other structures, which would lead to a slope higher than 1. However, it should be noted that at concentrations in the order of its affinity for M3 receptors, SVT-40776 behaves as a competitive antagonist, as do tolterodine and solifenacin, but not darifenacin. The results from darifenacin were compatible with an insurmountable blockade and are consistent with previous findings in rat (Hegde et al., 1997), dog (Choppin and Eglen, 2001), mouse (Yamada, 2006) and human bladder (Fetscher et al., 2002).
In mouse atrial preparations, carbachol curves were antagonized by all compounds tested, in a concentration-dependent fashion, with parallel right-ward displacements at lower potencies than in bladder tissue. The rank order of antagonist activities (pA2) was tolterodine (8.5), solifenacin (7.8), darifenacin (7.3) and SVT-40776 (7.2).
Having in mind that the M3 receptors represent the main receptor system controlling bladder contractility and that the M2 receptor is mainly responsible for muscarinic cardiac effects, the high urinary versus cardiac functional selectivity (199-fold) exhibited by SVT-40776 is in agreement with the results previously found in binding studies where SVT-40776 showed a high affinity for the M3 receptor (Ki = 0.19 nmol·L−1) and a clear selectivity (203-fold) for M3 versus M2 receptor subtypes (Salcedo et al. 2003, Balsa et al. 2004, Fernández et al., 2005). As a result of the pharmacological profile exhibited by SVT-40776 in binding as well as in functional studies, it is therefore not expected for the compound to cause any M2 receptor-related unwanted cardiac effects.
The ex vivo protocol was intended to get a closer approach to the in vivo situation, as it integrates the pharmacokinetic and distribution pattern of the compounds. SVT-40776 inhibited carbachol-induced bladder contractions in a concentration-dependent manner 3 h after oral administration. Neither tolterodine, solifenacin nor SVT-40776 significantly altered the maximum response to carbachol. On the contrary, darifenacin significantly reduced the Emax at 50 mg·kg−1, verifying the antagonist profile shown in the in vitro protocol. Likewise, darifenacin exhibited less potency than tolterodine and solifenacin in the atria, which accounts for the M3 versus M2 receptor selectivity attributed to darifenacin (Gillberg et al., 1998). Interestingly, SVT-40776 did not induce any significant right-ward shifts of the carbachol curves at doses up to 30 mg·kg−1. As the ex vivo model reflects more accurately the physiological conditions, these data support the fact that SVT-40776 is a M3 receptor antagonist clearly devoid of any relevant M2 receptor affinity.
In line with the above observations, in the anaesthetized guinea pig model, SVT-40776 inhibited isovolumetric-induced contractions in a dose-dependent fashion, without changing cardiovascular parameters. Tolterodine, darifenacin and solifenacin also changed bladder contraction amplitude in a dose-dependent manner. Nonetheless, SVT-40776 revealed itself as the most potent muscarinic antagonist of the compounds tested in this in vivo model, with an ED25 of 17.1 nmol·kg−1 (6.97 µg·kg−1). These experiments provide in vivo functional data that may indicate potential advantages for an M3 selective drug, in terms of cardiovascular safety.
The present study has shown that SVT-40776, a novel substituted quinuclidine derivative, is a potent inhibitor of M3 receptor-related detrusor contractile activity. Its functional selectivity for urinary bladder over cardiac tissues is in the order of 200-fold, a value not reached with any of the current agents used in the treatment of OAB. Studies performed in our laboratory have shown that SVT-40776 exhibits also a relevant selectivity for mouse bladder tissue over salivary glands, a tissue known to contain M1 and M3 receptors and whose activation is necessary for salivary secretion (Balsa et al. 2005). This is an important characteristic of the compound as it may predict a lack of dry mouth, a side effect exhibited by most of the antimuscarinic agents used for OAB. In summary, the wide experimental selectivity ratio for bladder over cardiac function (and salivary glands) exhibited by SVT-40776 may predict a good tolerability profile regarding to antimuscarinic adverse effects. The compound has successfully completed Phase I clinical trials and is currently undergoing Phase II clinical trials for the treatment of OAB.
Glossary
Abbreviations:
- AP
arterial pressure
- OAB
overactive bladder
Conflict of interest
All the authors were employees of Laboratorios Salvat at the time of these experiments.
References
- Abrams P, Cardozo L, Fall M, Griffiths D, Rosier P, Ulmsten U, et al. The standardisation of terminology in lower urinary tract function: report from the standardisation sub-committee of the international continence society. Urology. 2003;61:37–49. doi: 10.1016/s0090-4295(02)02243-4. [DOI] [PubMed] [Google Scholar]
- Abrams P, Andersson KE, Buccafusco JJ, Chapple C, de Groat WC, Fryer AD, et al. Muscarinic receptors: their distribution and function in body systems, and the implications for treating overactive bladder. Br J Pharmacol. 2006;148:565–578. doi: 10.1038/sj.bjp.0706780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson KE. Overactive bladder – pharmacological aspects. Scand J Urol Nephrol Suppl. 2002a:72–81. doi: 10.1080/003655902320766006. [DOI] [PubMed] [Google Scholar]
- Andersson KE. Potential benefits of muscarinic M3 receptor selectivity. Eur Urol Suppl. 2002b;1:23–28. [Google Scholar]
- Andersson KE. Antimuscarinics for treatment of overactive bladder. Lancet Neurol. 2004;3:46–53. doi: 10.1016/s1474-4422(03)00622-7. [DOI] [PubMed] [Google Scholar]
- Andersson KE, Olshansky B. Treating patients with overactive blabber syndrome with antimuscarinics: heart rate considerations. BJU Int. 2007;100:1007–1014. doi: 10.1111/j.1464-410X.2007.07100.x. [DOI] [PubMed] [Google Scholar]
- Andersson KE, Yoshida M. Antimuscarinics and the overactive detrusor – which is the main mechanism of action? Eur Urol. 2003;43:1–5. doi: 10.1016/s0302-2838(02)00540-7. [DOI] [PubMed] [Google Scholar]
- Balsa D, Salcedo C, Enrich A, Davalillo S, Cabellos J, Pellicer T, et al. SVT-40776: a selective, competitive and reversible antagonist of the human recombinant muscarinic M3 receptor. International Continence Society 34th Annual Meeting 2004 (Abstract 260.
- Balsa D, Salcedo C, Enrich A, Davalillo S, Cabellos J, Fernández AG. Functional bladder selectivity of SVT-40776: a comparative study. 35th Annual Meeting of the International Continence Society. Montreal. P 284.
- Barr RG, Bourbeau J, Camargo CA, Ram FS. Tiotropium for stable chronic obstructive pulmonary disease: a meta-analysis. Thorax. 2006;61:854–862. doi: 10.1136/thx.2006.063271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brading AF. A myogenic basis for the overactive bladder. Urology. 1997;50:57–67. doi: 10.1016/s0090-4295(97)00591-8. [DOI] [PubMed] [Google Scholar]
- Caulfield MP. Muscarinic receptors – characterization, coupling and function. Pharmacol Ther. 1993;58:319–379. doi: 10.1016/0163-7258(93)90027-b. [DOI] [PubMed] [Google Scholar]
- Chapple C, Steers W, Norton P, Millard R, Kralidis G, Glavind K, et al. A pooled analysis of three phase III studies to investigate the efficacy, tolerability and safety of darifenacin, a muscarinic M3 selective receptor antagonist, in the treatment of overactive bladder. BJU Int. 2005;95:993–1001. doi: 10.1111/j.1464-410X.2005.05454.x. [DOI] [PubMed] [Google Scholar]
- Chess-Williams R, Chapple CR, Yamanishi T, Yasuda K, Sellers DJ. The minor population of M3-receptors mediate contraction of human detrusor muscle in vitro. J Auton Pharmacol. 2001;21:243–248. doi: 10.1046/j.1365-2680.2001.00231.x. [DOI] [PubMed] [Google Scholar]
- Choppin A. Muscarinic receptors in isolated urinary bladder smooth muscle from different mouse strains. Br J Pharmacol. 2002;137:522–528. doi: 10.1038/sj.bjp.0704897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choppin A, Eglen RM. Pharmacological characterization of muscarinic receptors in mouse isolated urinary bladder smooth muscle. Br J Pharmacol. 2001;133:1035–1040. doi: 10.1038/sj.bjp.0704165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choppin A, Eglen RM, Hegde SS. Pharmacological characterization of muscarinic receptors in rabbit isolated iris sphincter muscle and urinary bladder smooth muscle. Br J Pharmacol. 1998;124:883–888. doi: 10.1038/sj.bjp.0701920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groat WC. A neurologic basis for the overactive bladder. Urology. 1997;50:36–52. doi: 10.1016/s0090-4295(97)00587-6. [DOI] [PubMed] [Google Scholar]
- Dhein S, van Koppen CJ, Brodde OE. Muscarinic receptors in the mammalian heart. Pharmacol Res. 2001;44:161–182. doi: 10.1006/phrs.2001.0835. [DOI] [PubMed] [Google Scholar]
- Eglen RM, Hegde SS, Watson N. Muscarinic receptor subtypes and smooth muscle function. Pharmacol Rev. 1996;48:531–565. [PubMed] [Google Scholar]
- Ehlert FJ, Griffin MT, Abe DM, Vo TH, Taketo MM, Manabe T, et al. The M2 muscarinic receptor mediates contraction through indirect mechanisms in mouse urinary bladder. J Pharmacol Exp Ther. 2005;313:368–378. doi: 10.1124/jpet.104.077909. [DOI] [PubMed] [Google Scholar]
- Farrerons C, Catena J, Fernandez-Serrat A, Miquel I, Balsa D, Bonilla JI, et al. Carbamates derived from arylalkylamines. PCT/ES01/00252 [WO 02/00652]
- Fernández AG, Viayna C, Salcedo C, Davalillo S, Sanagustín J, Balsa D, et al. Bladder selectivity of SVT-40776: a pharmacokinetic/pharmacodynamic (PK/PD) analysis based on preclinical and clinical studies. International Continence Society 35th Annual Meeting 2005.
- Fetscher C, Fleichman M, Schmidt M, Krege S, Michel MC. M(3) muscarinic receptors mediate contraction of human urinary bladder. Br J Pharmacol. 2002;136:641–643. doi: 10.1038/sj.bjp.0704781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillberg PG, Sundquist S, Nilvebrant L. Comparison of the in vitro and in vivo profiles of tolterodine with those of subtype-selective muscarinic receptor antagonists. Eur J Pharmacol. 1998;349:285–292. doi: 10.1016/s0014-2999(98)00214-3. [DOI] [PubMed] [Google Scholar]
- Harriss DR, Marsh KA, Birmingham AT, Hill SJ. Expression of muscarinic M3-receptors coupled to inositol phospholipid hydrolysis in human detrusor cultured smooth muscle cells. J Urol. 1995;154:1241–1245. [PubMed] [Google Scholar]
- Hassall CJ, Stanford SC, Burnstock G, Buckley NJ. Co-expression of four muscarinic receptor genes by the intrinsic neurons of the rat and guinea-pig heart. Neuroscience. 1993;56:1041–1048. doi: 10.1016/0306-4522(93)90149-a. [DOI] [PubMed] [Google Scholar]
- Hay-Smith J, Herbison P, Ellis G, Morris A. Which anticholinergic drug for overactive bladder symptoms in adults. Cochrane Database Syst Rev. 2005;20:CD005429. doi: 10.1002/14651858.CD005429. [DOI] [PubMed] [Google Scholar]
- Hegde SS, Eglen RM. Muscarinic receptor subtypes modulating smooth muscle contractility in the urinary bladder. Life Sci. 1999;64:419–428. doi: 10.1016/s0024-3205(98)00581-5. [DOI] [PubMed] [Google Scholar]
- Hegde SS, Choppin A, Bonhaus D, Briaud S, Loeb M, Moy TM, et al. Functional role of M2 and M3 muscarinic receptors in the urinary bladder of rats in vitro and in vivo. Br J Pharmacol. 1997;120:1409–1418. doi: 10.1038/sj.bjp.0701048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellgren I, Mustafa A, Riazi M, Suliman I, Sylven C, Adem A. Muscarinic M3 receptor subtype gene expression in the human heart. Cell Mol Life Sci. 2000;57:175–180. doi: 10.1007/s000180050507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbison P, Hay-Smith J, Ellis G, Moore K. Effectiveness of anticholinergic drugs compared with placebo in the treatment of overactive bladder: systematic review. British Medical Journal. 2003;326:841–844. doi: 10.1136/bmj.326.7394.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover DB, Baisden RH, Xi-Moy SX. Localization of muscarinic receptor MRNAs in rat heart and intrinsic cardiac ganglia by in situ hybridization. Circ Res. 1994;75:813–820. doi: 10.1161/01.res.75.5.813. [DOI] [PubMed] [Google Scholar]
- Howell RE, Kovalsky MP. Hypotensive effect of an M2-selective muscarinic antagonist in anaesthetized guinea-pigs. J Auton Pharmacol. 1995;15:19–26. doi: 10.1111/j.1474-8673.1995.tb00344.x. [DOI] [PubMed] [Google Scholar]
- Kesten S, Jara M, Wentworth C, Lanes S. Pooled clinical trial analysis of the safety of tiotropium. Chest. 2005;128:257S. doi: 10.1378/chest.130.6.1695. [DOI] [PubMed] [Google Scholar]
- Lai FM, Cobuzzi A, Spinelli W. Characterization of muscarinic receptors mediating the contraction of the urinary detrusor muscle in cynomolgus monkeys and guinea pigs. Life Sci. 1998;62:1179–1186. doi: 10.1016/s0024-3205(98)00044-7. [DOI] [PubMed] [Google Scholar]
- Longhurst PA, Levendusky M. Influence of gender and the oestrous cycle on in vitro contractile responses of the rat urinary bladder to cholinergic stimulation. Br J Pharmacol. 2000;131:177–184. doi: 10.1038/sj.bjp.0703551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longhurst PA, Leggett RE, Briscoe JA. Characterization of the functional muscarinic receptors in the rat urinary bladder. Br J Pharmacol. 1995;116:2279–2285. doi: 10.1111/j.1476-5381.1995.tb15065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui M, Motomura D, Karasawa H, Fujikawa T, Jiang J, Komiya Y, et al. Multiple functional defects in peripheral autonomic organs in mice lacking muscarinic acetylcholine receptor gene for the M3 subtype. Proc Natl Acad Sci USA. 2000;97:9579–9584. doi: 10.1073/pnas.97.17.9579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui M, Motomura D, Fujikawa T, Jiang J, Takahashi S, Manabe T, et al. Mice lacking M2 and M3 muscarinic acetylcholine receptors are devoid of cholinergic smooth muscle contractions but still viable. J Neurosci. 2002;22:10627–10632. doi: 10.1523/JNEUROSCI.22-24-10627.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabi G, Cody JD, Ellis G, Herbison P, Hay-Smith J. Anticholinergic drugs versus placebo for overactive bladder syndrome in adults. Cochrane Database Syst Rev. 2006;4:CD003781. doi: 10.1002/14651858.CD003781.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshansky B, Foote J, Arguinzoniz M, Lheritier K, Quebe-Fehling E, Steel M. The effects of darifenacin and tolterodine on Heart Rate (HR) in patients with overactive bladder. Neurourol Urodyn. 2006;25:A124. [Google Scholar]
- Olshansky B, Ebinger U, Egermark M, Luthra A, Rekedac A, Brum J. Antimuscarinics and heart rate: results from a randomized, double-blind, placebo-controlled, 3 way crossover study in subjects over age 50. ICI 2008 July 5–8, Paris, Abst 32.
- Pontari MA, Braverman AS, Ruggieri MR., Sr The M2 muscarinic receptor mediates in vitro bladder contractions from patients with neurogenic bladder dysfunction. Am J Physiol Regul Integr Comp Physiol. 2004;286:R874–R880. doi: 10.1152/ajpregu.00391.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanzi LJ, Delconte A, Kralidis G. Impact of darifenacin compared with tolterodine on incontinence episodes in patients with overactive bladder. Obstet Gynecol. 2005;105:88S. [Google Scholar]
- Salcedo C, Balsa D, Enrich A, Davalillo S, Pellicer T, Lagunas C, et al. SVT-40776, a new selective M3 muscarinic antagonist: human receptor binding profile and bladder effects in the guinea-pig. Neurourol Urodyn. 2003;22:382–384. [Google Scholar]
- Schneider T, Fetscher C, Krege S, Michel MC. Signal transduction underlying carbachol-induced contraction of human urinary bladder. J Pharmacol Exp Ther. 2004;309:1148–1153. doi: 10.1124/jpet.103.063735. [DOI] [PubMed] [Google Scholar]
- Shi H, Wang H, Wang Z. Identification and characterization of multiple subtypes of muscarinic acetylcholine receptors and their physiological functions in canine hearts. Mol Pharmacol. 1999;55:497–507. [PubMed] [Google Scholar]
- Stengel PW, Gomeza J, Wess J, Cohen ML. M(2) and M(4) receptor knockout mice: muscarinic receptor function in cardiac and smooth muscle in vitro. J Pharmacol Exp Ther. 2000;292:877–885. [PubMed] [Google Scholar]
- Stevens LA, Chapple CR, Chess-Williams R. Human idiopathic and neurogenic overactive bladders and the role of M2 muscarinic receptors in contraction. Eur Urol. 2007;52:531–538. doi: 10.1016/j.eururo.2006.11.016. [DOI] [PubMed] [Google Scholar]
- Tobin G, Sjogren C. In vivo and in vitro effects of muscarinic receptor antagonists on contractions and release of [3H]Acetylcholine in the rabbit urinary bladder. Eur J Pharmacol. 1995;281:1–8. doi: 10.1016/0014-2999(95)00221-6. [DOI] [PubMed] [Google Scholar]
- Tran JA, Matsui M, Ehlert FJ. Differential coupling of muscarinic M-1, M-2, and M-3 receptors to phosphoinositide hydrolysis in urinary bladder and longitudinal muscle of the ileum of the Mouse. J Pharmacol Exp Ther. 2006;318:649–656. doi: 10.1124/jpet.106.103093. [DOI] [PubMed] [Google Scholar]
- Tyagi S, Tyagi P, Van le S, Yoshimura N, Chancellor MB, de Miguel F. Qualitative and quantitative expression profile of muscarinic receptors in human urothelium and detrusor. J Urol. 2006;176:1673–1678. doi: 10.1016/j.juro.2006.06.088. [DOI] [PubMed] [Google Scholar]
- Wang H, Han H, Zhang L, Shi H, Schram G, Nattel S, et al. Expression of multiple subtypes of muscarinic receptors and cellular distribution in the human heart. Mol Pharmacol. 2001;59:1029–1036. doi: 10.1124/mol.59.5.1029. [DOI] [PubMed] [Google Scholar]
- Wang P, Luthin GR, Ruggieri MR. Muscarinic acetylcholine receptor subtypes mediating urinary bladder contractility and coupling to GTP binding proteins. J Pharmacol Exp Ther. 1995;273:959–966. [PMC free article] [PubMed] [Google Scholar]
- Wein AJ, Rovner ES. Definition and epidemiology of overactive bladder. Urology. 2002;60:7–12. doi: 10.1016/s0090-4295(02)01784-3. [DOI] [PubMed] [Google Scholar]
- Yamada S, Maruyama S, Takagi Y, Uchida S, Oki T. In vivo demonstration of M3 muscarinic receptor subtype selectivity of darifenacin in mice. Life Sci. 2006;80:127–132. doi: 10.1016/j.lfs.2006.08.028. [DOI] [PubMed] [Google Scholar]