

Abstract
Background and purpose:
We showed previously that cisplatin inititates a signalling pathway mediated by PKC-δ/extracellular signal-regulated kinase (ERK), important for maintaining viability in PC Cl3 thyroid cells. The studies described herein examined whether c-fos was associated with cisplatin resistance and the signalling link between c-fos and PKC-δ/ERK.
Experimental approach:
Cells were treated with various pharmacological inhibitors of PKCs and ERK, or were depleted of c-fos, PKC-δ, PKC-ε and caspase-3 by small interfering RNA (siRNA), then incubated with cisplatin and cytotoxicity assessed.
Key results:
Cisplatin provokes the induction of c-fos and the activation of conventional PKC-β, and novel PKC-δ and -ε. The cisplatin-provoked c-fos induction was decreased by Gö6976, a PKC-β inhibitor; by siRNA for PKC-δ- but not that for PKC-ε or by PD98059, a mitogen-activated protein kinase/ERK kinase inhibitor. Expression of c-fos was abolished by GF109203X, an inhibitor of all PKC isoforms, or by PD98059 plus Gö6976 or by PKC-δ-siRNA plus Gö6976. When c-fos expression was blocked by siRNA, cisplatin cytotoxicity was strongly enhanced with increased caspase-3 activation. In PKC-δ-depleted cells treated with cisplatin, caspase-3 activation was increased and cell viability decreased. In these PKC-δ-depleted cells, PD98059 did not affect caspase-3 activation.
Conclusions and implications:
In PC Cl3 cells, in the cell signalling pathways that lead to cisplatin resistance, PKC-δ controls ERK activity and, together with PKC-β, also the induction of c-fos. Hence, the protective role of c-fos in thyroid cells has the potential to provide new opportunities for therapeutic intervention.
Keywords: PKC-δ, c-fos, ERK, cisplatin, thyroid, PC Cl3
Introduction
Thyroid cancer is the most common endocrine malignancy and is responsible for about 60% of deaths from endocrine cancer (Gimm, 2001; Vini and Harmer, 2002). Thyroid tumours exhibit a wide spectrum of neoplastic pathology, varying from well-differentiated benign tumours to highly malignant anaplastic carcinomas. Malignant transformation has been demonstrated to be caused by several factors, including the activation of proto-oncogene and the inactivation of tumour suppressor genes (Aasland et al., 1988; Terrier et al., 1988; Karga et al., 1991; Zou et al., 1993). Clinical trials with chemotherapeutic drugs have produced only rare and limited positive responses in thyroid cancer (Hanna et al., 1999; Haigh, 2000; Haigh et al., 2001). The expression of the multidrug resistance gene is altered in a small subset of thyroid carcinomas (Sugawara et al., 1995; Asakawa et al., 1997), but the molecular basis for the failure of chemotherapy-based regimens has not been defined in the vast majority of thyroid carcinomas.
Several studies (Kashani-Sabet et al., 1990; Scanlon et al., 1990; Moorehead and Singh, 2000) indicate that cisplatin resistance is associated with over-expression of the proto-oncogene c-fos in tumours from patients that did not respond to cisplatin-based chemotherapy and in established cisplatin-resistant cell lines (Jiao et al., 1991; Dempke et al., 2000).
The c-fos gene encodes a nuclear transcription factor, c-fos, that induces transcription of a number of other genes involved in the regulation of cell replication, cell cycle progression and differentiation through its interactions with members of the c-Jun and ATF/CREB families (Chiu et al., 1988; Kovary and Bravo, 1992; Schuermann, 1994). c-fos expression can be induced by physical stresses such as reactive oxygen species, heat shock and several DNA-damaging agents, including cisplatin (Hollander and Fornace, 1989; Amstad et al., 1992). Cells lacking the c-fos gene are hypersensitive to a number of DNA-damaging agents (Kaina et al., 1997). Transfection of a human ovarian carcinoma cell line with a c-fos expression vector induces cisplatin resistance (Moorehead and Singh, 2000). Treatment of cisplatin- resistant human ovarian carcinoma cells with cisplatin induces the expression of the c-fos gene as well as a number of genes involved in DNA repair (Kashani-Sabet et al., 1990). Finally, a retroviral vector that expresses c-fos antisense RNA is effective at reducing MCF-7 tumour size and incidence, and these findings have laid the foundation for a clinical trial investigating the effectiveness of this c-fos antisense retroviral vector against breast tumours (Arteaga and Holt, 1996; Bonovich et al., 2002). Although exciting in prospect, the effect of c-fos on tumour growth and cisplatin resistance has been examined in only a limited number of systems, and thus it is difficult to determine whether the ability of c-fos to regulate tumour growth and/or cisplatin sensitivity is specific for these systems or whether the actions of c-fos extend to other systems. Therefore, the studies described here examined whether c-fos expression was associated with cisplatin resistance in thyroid cells and whether reducing c-fos expression was an effective method for increasing cisplatin sensitivity in cells resistant to cisplatin. In this study we used the fully differentiated thyroid PC Cl3 cells, which express the typical markers of thyroid differentiation and are sensitive to thyrotropin (TSH) stimulation for their growth (Fusco et al., 1987). We have shown previously that, in PC Cl3 cells, cisplatin activated the extracellular signal-regulated kinase (ERK) cascade, which is important for maintaining the cell viability after drug treatments, through a signalling pathway mediated by PKC-δ (Muscella et al., 2005; Urso et al., 2005). Thus PC Cl3 cells may represent a helpful model in the investigation of the mechanisms by which ERK and other signal transduction pathways modulate the response to cisplatin and promote cell survival in response to cisplatin treatment.
Methods
Cell lines
PC Cl3, a rat differentiated thyroid cell line, was grown in Coon's modified Ham's F-12 medium (Celbio, Pero Milan, Italy) supplemented with 5% calf serum (Sigma, Milan, Italy) and a mixture of hormones and growth factors (insulin 1 µg·mL−1; TSH 1 mIU·mL−1; glycylhistidyl-L-lysine 10 ng·mL−1; human transferrin 5 µg·mL−1; cortisone 10 nmol·L−1; somatostatin 10 ng·mL−1; all from Sigma).
Reverse transcription and polymerase chain reaction (RT-PCR)
Total RNA was extracted from PC Cl3 cells using an SV Total RNA isolation kit and performed according to the manufacturer's protocols (Promega Corporation, USA). The RNA concentration was determined by measuring the absorbance at 260 nm. Samples were then stored at −20°C for subsequent RT-PCR analysis. 1 µg of total RNA was used to synthesize a complementary DNA (cDNA) using the RT kit (Promega Corporation, USA). The RT reaction was carried out at 25°C for 5 min followed by 42°C for 60 min and then at 95°C for 5 min. The samples were then placed on ice for 5 min and stored at −20°C for PCR amplification. PCR was performed using a BioRad iQ iCycler Detection System (BioRad Laboratories, Ltd) with SYBR green fluorophore (SyberGreen Supermix; Biorad Laboratories, USA) in the presence of 2 µM of specific primers, for each PKC isoforms and β-actin. A melt curve analysis was performed following every run to ensure a single amplified product for every reaction. For each gene, relative expression was determined using the 2−ΔΔCT method and normalized to β-actin expression (Livak and Schmittgen, 2001).
Preparation of subcellular fractions
To obtain protein cell extracts, cells were washed twice in ice-cold phosphate-buffered saline (PBS) and harvested in 1 mL of PBS. The samples were centrifuged for 30 s at 10 000× g, and cell pellets were resuspended in the following buffer (in mmol·L−1): 20 Tris–HCl, pH 8, containing 420 NaCl, 2 EDTA, 2 Na3VO4 and 0.2% Nonidet P-40 10% glycerol, supplemented with a cocktail of protease inhibitors. After a 10 min incubation on ice, cells were passed several times through a 20 gauge syringe needle and then centrifuged at 13 000× g for 10 min at 4°C. Other samples were centrifuged at 100 000× g for 20 min at 4°C. The resultant supernatant is referred to as the cytosolic fraction. The pellet was solubilized in buffer B (in mmol·L−1, 20 Tris-HCl, pH 7.5, 150 NaCl, 1 EGTA, 1 EDTA and protease inhibitors) containing 1% Nonidet P-40. We evaluated the Na+/K+-ATPase activity using a coupled enzyme assay method (Norby, 1988) to determine the purity of the cell membrane fraction used for immunoblotting. The enrichment factor (enzyme activities of final purified membrane pellet and cytosol compared with those of the initial homogenate) were 35 ± 2.2 and not determined. Lactate dehydrogenase activity (a marker enzyme for the cytoplasm) was determined by measuring the decrease at 340 nm due to the oxidation of NADH (Kochhar et al., 1992). The reaction buffer contained 50 mmol·L−1 HEPES, pH 7.5, 8 mmol·L−1 sodium pyruvate, 0.2 mmol·L−1 NADH and the protein sample in a 1 mL volume. Sodium pyruvate was added last to minimize non-specific NADH oxidation. The specific activity of LDH in the cytosol was 13.1 times higher than that in the homogenate.
Nuclei were pelleted by centrifugation at 2000× g for 15 min at 4°C and resuspended in high salt buffer (in mmol·L−1, 20 Tris-HCl, pH 7.9, 420 NaCl, 10 KCl, 0.1 Na3VO4, 1 EDTA, 1 EGTA, 20% glycerol, supplemented with a cocktail of protease inhibitors) and sonicated until no nuclei remained intact. The purity of fractions was tested by immunoblotting with anti-α subunit of Na+/K+-ATPase monoclonal antibody (membrane protein) or anti-histone-3/4 polyclonal antibody (nuclear proteins).
Western blot analysis
Proteins in homogenates and cellular fraction were determined using the Bio-Rad protein assay kit 1 (Milan, Italy). Lyophilized bovine serum albumin was used as a standard. Total cell proteins or proteins of the distinct sub cellular fractions were dissolved in sodium dodecyl sulphate (SDS) sample buffer and separated on 10% or 15% SDS gels. Separated proteins were transferred electrophoretically onto polyvinylidene difluoride membrane (Amersham International). Equal protein loading was confirmed by Ponceau S staining. Blots were incubated with specific primary antibodies and the immune complexes were detected using appropriate peroxidase-conjugated secondary antibodies and enhanced chemiluminescent detection reagent (Amersham International). Blots were stripped and used for several sequential incubations with control antibodies. Densitometric analysis was carried out on the Western blots using the NIH Image 1.62 software (National Institutes of Health, Bethesda, MD, USA). The pixel intensity for each region was analysed, the background was subtracted, and the c-fos protein expressions were normalized to β actin loading control for each lane.
Design and preparation of siRNAs
Small interfering RNAs (siRNAs) were prepared by an in vitro transcription method. For each siRNA, target sites specific to rat c-fos, PKC-δ, PKC-ε, caspase 3 mRNA, sense and antisense templates were designed based on each target sequence and partial T7 promoter sequence. The mRNA targets were: caspase-3 target sequence 5′-CCUCAGAGAGACAUUCAUG-3′, PKC-δ target sequence 5′-AACUGUUUGUGAAUUUG CCUU-3′, PKC-ε target sequence 5′-GCCCCUAAAGACA AUGAAGTT-3′; c-fos target sequence 5′-UCACAGGGCUAG CAGUGUGGGU-3′ In addition, a nonsense (scrambled) sequence 5′-AAUCGCAUAGCGUAUGCCGUU-3′ was used as a control.
All template oligonucleotides were chemically synthesized and polyacrylamide gel electrophoresis purified. In vitro transcription, annealing and purification of siRNA duplexes were performed using the protocol supplied with the T7 RiboMAX Express RNAi System (Promega). Briefly, approximately 2 µg of each single-strand (ss) transcription template was first annealed with the T7 promoter to form double-strand transcription templates. For preparation of each siRNA duplex, transcription reactions were first performed with separated antisense and sense templates using the T7 RNA polymerase provided with the kit and then annealed to form siRNA duplexes. Then, the siRNA duplex was treated with DNase and RNase to remove the extra nucleotides of transcribed siRNA to meet the structural 3′UU overhang and 5′ phosphate requirement.
Fos, PKCs and caspase immunoblottig were performed 24 h post transfection to determine the efficiency of siRNA incorporation in PC Cl3 and to measure proteins expression. Quantitative analysis of c-fos, PKC-δ, -ε and caspase 3 expressions, as measured by intensity of immunoreactivity, in siRNA or siRNA-NS-transfected PC Cl3 revealed a higher reduction in protein expression. Experiments were also performed by pharmacological inhibition of PKC-δ and PKC-ε (rottlerin and εV1 respectively).
siRNA transfection
PC Cl3 cells (50–70% confluence) were transfected with siRNA duplexes using the protocol supplied with the CodeBreaker siRNA transfection reagent (Promega). Briefly, the transfection reagent was first diluted into Coon's modified Ham's F12 medium without serum and antibiotics for about 15 min, and then the siRNA duplex was added to the medium to form a lipid–siRNA complex. Following additional 15 min incubation, transfection was initiated by adding the lipid–siRNA complex to 6-well plates. The final concentration of siRNA was 30 nmol·L−1.
Cytotoxicity assays
Cells at 70–80% confluency were trypsinized (0.25% trypsin with 1 mmol·L−1 EDTA), washed and resuspended in growth medium. 100 µL of a cell suspension (105 cells·mL−1) was added to each well of a 96 well plate. After overnight incubation, cells were treated with specific reagents for different incubation periods.
MTT assay
The conversion of MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenol tetrazolium bromide] by cells was used as an indicator of cell number as previously described (Muscella et al., 2005). This method measures the reduction of MTT by active mitochondria, which results in a colorimetric change measured at 550 nm wavelength. Experiments were performed to define the linear range of the assay. A good correlation was observed up to 50 000 cells per well (data not shown).
Increasing the concentration of heat-killed cells per well (killed by incubating at 70°C for 15 min) caused no significant change in the absorbance; thus, this spectrophotometric method was a valid technique for measuring the number of viable cells. All subsequent experiments performed were within the linear range of the assay.
The percentage cell survival was calculated as the absorbance ratio of treated to untreated cells. The data presented are means ± standard deviation (SD) from eight replicate wells per microtitre plate, repeated four times.
Sulforhodamine B (SRB) assay
The SRB assay was carried out as previously described (Skehan et al., 1990). Briefly, 70 µL 0.4% (w·v−1) SRB in 1% acetic acid solution was added to each well and left at room temperature for 20 min. SRB was removed and the plates washed five times with 1% acetic acid before air drying. Bound SRB was solubilized with 200 µL of 10 mmol·L−1 unbuffered Tris-base solution and plates were left on a plate shaker for at least 10 min. Absorbance was measured in a 96 well plate reader at 492 nm. The test optical density (OD) value was defined as the absorbance of each individual well, minus the blank value (‘blank’ is the mean optical density of the background control wells, n = 8). The percentage survival was calculated as the absorbance ratio of treated to untreated cells. The data presented are means ± SD from eight replicate wells per microtitre plate, repeated four times.
Trypan Blue dye exclusion assay
The cells were seeded in 60 mm tissue culture dishes (100 000 cells·mL−1). After overnight incubation, the cells were treated with the concentrations of cisplatin. Cell viability was estimated using the trypan blue exclusion assay and light microscopy.
Apoptosis analysis
For 4,6-diamine-2-phenylindole (DAPI) staining, cells treated with cisplatin were fixed with 3% formalin and stained with 1 µg·mL−1 DAPI in PBS for 10 min. Cells were mounted on glass slides, covered and analysed using fluorescence microscopy. For statistical analysis of each experiment, 5–10 fields (magnification ×400) were counted (between 400 and 700 cells in total) and the mean ± SD was calculated.
Statistical analysis
Experimental points represent means ± SD of replicates. Statistical analysis was carried out using the anova. When indicated, post hoc tests (Bonferroni/Dunne) were also performed. A P-value less than 0.05 was considered to achieve statistical significance.
Reagents
Glutamine, gentamicin, the mitogen-activated protein kinase (MAPK)/ERK kinase (MEK) inhibitor PD098059, the phosphoinositide 3-kinase (PI3K) inhibitors LY294002 and wortmannin, the PKC inhibitors GF109203X, Gö6976, rottlerin and the PKC-ε-selective translocation inhibitor εV1 were obtained from Sigma Chemical Co. (Milan, Italy). PKC isoforms, phospho-specific ERK1 and ERK2, anti-histone-3/4 antibodies, goat anti-rabbit IgG conjugated with peroxidase, as well as control antibodies, were obtained from Santa Cruz Biotechnology, Santa Cruz, CA, USA.
Results
Cisplatin induces c-fos gene expression in PC Cl3 cells
Incubation of PC Cl3 cells with cisplatin (1–100 µmol·L−1) led to a dose-dependent induction of c-fos mRNA (Figure 1A), as demonstrated by real time PCR. No further c-fos induction was shown at higher doses (200 µmol·L−1) of cisplatin (data not shown). c-fos induction peaked at 6 h (Figure 1B). No effect on the level of β-actin mRNA, used as an internal control, was found over the 24 h incubation with cisplatin.
Figure 1.

Cisplatin induces c-fos gene expression in PC Cl3 cells. Thyroid cells were treated without or with various concentrations of cisplatin, for 24 h (A), or with 100 µmol·L−1 cisplatin, for the indicated times (B) and then RNA was extracted. Upper figures: agarose gel electrophoresis of reverse transcription and polymerase chain reaction (RT-PCR) products revealed the specific transcripts for c-fos; β-actin was used to normalized the amount of DNA template in each PCR reaction. Lower figures: the samples were also analysed using real-time PCR. mRNA levels were normalized to β-actin and calculated as fold change values relative to positive control. Data were expressed as the mean ± SD of three different experiments. Values with shared letters are not significantly different according to Bonferroni/Dunne post hoc tests.
Western blot analysis using an antibody recognising the c-fos protein showed that in PC Cl3 cells cisplatin stimulated the expression of this protein in a dose- and time-dependent way (1–100 µmol·L−1; 0–24 h Figure 2A,B). Sequential incubation of the sheet with anti-actin antibody confirmed the equal protein loading. On the basis of these results further experiments were carried out using 6 and 12 h incubation time for c-fos mRNA and protein detection respectively.
Figure 2.

Dose- and time-dependent induction of c-fos by cisplatin in thyroid cell lines. Thyroid cells were treated without or with various concentrations of cisplatin, for 24 h (A), or with 100 µmol·L−1 cisplatin, for the indicated times (B). Lysates from thyroid cells were separated by 10% SDS-PAGE and analysed by Western blotting using the antibody against c-fos. Sequential incubation of the sheet with anti-actin antibody confirmed equal protein loading. The figures are representative of three independent experiments and results from densitometry are expressed as mean ± SD (n = 3) of sum of the grey level values. Values with shared letters are not significantly different according to Bonferroni/Dunne post hoc tests. PAGE, polyacrylamide gel electrophoresis; SDS, sodium dodecyl sulphate.
In addition, a translocation of c-fos from the cytosol to the nucleus of cisplatin-treated cells was also detected in Western blot assays (Figure 3).
Figure 3.
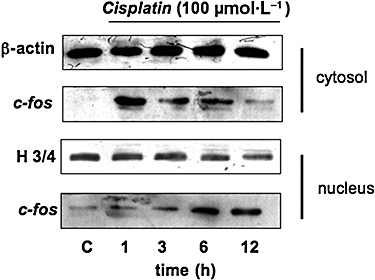
Time-dependent induction of c-fos by cisplatin in thyroid cell lines. Thyroid cells were treated without or with 100 µmol·L−1 cisplatin, for the indicated times. c-fos and its cytosol-to-nucleus translocation were detected by Western blotting. Cytosol (upper) and nuclear (lower) fractions from thyroid cells were separated by 10% SDS-PAGE and analysed by Western blotting using specific antibodies against c-fos. The purity of fractions was tested by immunoblotting with anti-histone-3/4 (H3/4) polyclonal and anti β-actin monoclonal antibodies. The figures are representative of three independent experiments. PAGE, polyacrylamide gel electrophoresis; SDS, sodium dodecyl sulphate.
Effect of cisplatin on expression and activation of PKC isoforms
PC Cl3 cells express PKC-α, -β1, -δ, -ε, -ζ (Marsigliante et al., 2003). PKC-δ plays a central role in the upstream activation of the ERK pathway after cisplatin treatment of PC Cl3 cells; furthermore, cisplatin increased PKC-δ expression and induced its proteolytic activation starting after 12 h (Urso et al., 2005). We here extended the examination of the relationship between cisplatin and PKC isoforms by using both RT-PCR and Western blot analyses. We found that cisplatin treatment caused a time-dependent decrease of mRNA level of PKC-βI, PKC-δ and PKC-ε and a time-dependent increase of PKC-ι for up to 12 h of treatment. No variation in the mRNA levels of PKC-α and PKC-ζ was found (Figure 4A). As expected, because cisplatin makes covalent DNA adducts blocking replication and transcription, no mRNAs of PKCs and β-actin were found in PC Cl3 cells incubated for more than 24 h (data not shown).
Figure 4.
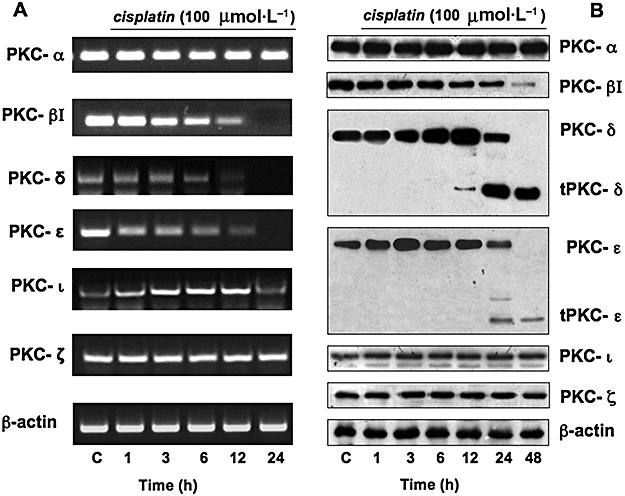
Effect of cisplatin on expression of PKC isoforms. Thyroid cells were treated without or with 100 µmol·L−1 cisplatin, for the indicated times. (A) Agarose gel electrophoresis of reverse transcription and polymerase chain reaction (RT-PCR) products revealed the specific transcripts for different PKC isozymes and β-actin used to normalize the amount of DNA template in each PCR reaction. The figures are representative of three independent experiments. (B) Cell lysates were separated by 10% SDS-PAGE and analysed by Western blotting using specific antibodies against the PKC isozymes. The figures are representative of four independent experiments. β-actin was used as a loading control. PAGE, polyacrylamide gel electrophoresis; SDS, sodium dodecyl sulphate.
Figure 4B shows Western blot analyses after cisplatin treatment of PC Cl3 cells; cisplatin caused a time-dependent decrease in the expression of PKC-βI, without generation of catalytic fragments; the expression of PKC-α showed no significant changes. Cisplatin increased the expression of novel PKC-δ after 6 h treatment and also induced its proteolytic activation after 12 h incubation, as previously shown (Urso et al., 2005). Cisplatin provoked a transient rise in PKC-ε expression, with maximal increase occurring at 3 h, followed by a progressively decreasing level (over 12 h) to the initial, pre-stimulated, level; cisplatin induced also PKC-ε cleavage after 24 h incubation. In summary, these results demonstrate that the increase in novel PKC expression is due to post-transcriptional regulation. No variation in the atypical PKC-ζ and -ι expression was found after cisplatin incubation. Sequential incubation of the sheet with anti-β-actin antibody confirmed the equal protein loading.
As activated PKCs translocate from the cytosol to the cellular membranes, we analysed by Western blot the distribution of PKC isozymes in PC Cl3 treated with cisplatin (100 µmol·L−1) for different incubation time (1, 3, 6, 12 and 24 h). Translocation of PKC-βI to the nuclear and membrane fractions was observed, with corresponding losses from the cytosolic fraction; conversely, the distribution of PKC-α was not altered by cisplatin treatment (Figure 5A). Regarding the novel PKCs, we observed the translocation of PKC-ε to the membranes (Figure 5A) and the translocation of PKC-δ to both membranes and nuclei (Figure 5B).
Figure 5.
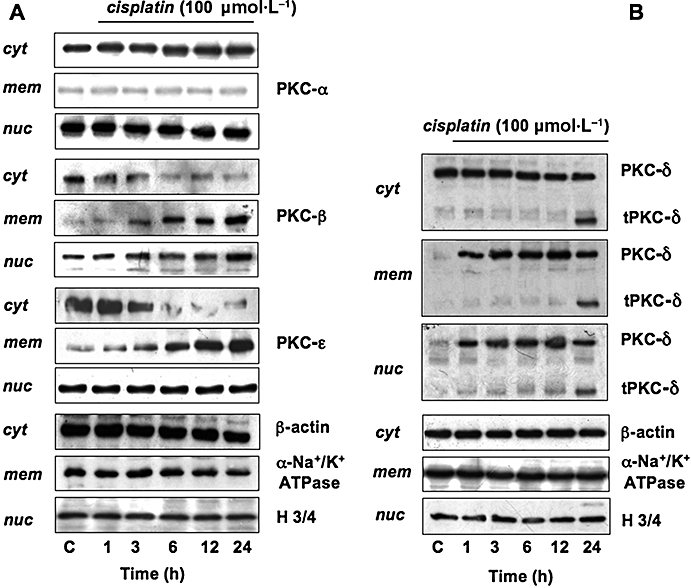
Effect of cisplatin on activation of PKCs. Thyroid cells were treated without or with 100 µmol·L−1 cisplatin, for the indicated times. Cell fractions – cytosol (cyt), membrane (mem), nuclear (nuc) – from thyroid cells were separated by 10% SDS-PAGE and analysed by Western blotting, using specific antibodies against the PKC isozymes. The purity of fractions was tested by immunoblotting with anti β-actin, anti-α subunit of Na+/K+ ATPase monoclonal or anti-histone-3/4 (H3/4) polyclonal antibodies. The figures are representative of four independent experiments. PAGE, polyacrylamide gel electrophoresis; SDS, sodium dodecyl sulphate.
Mechanism of cisplatin-induced c-fos expression
Role of PKC
The role of PKC in the cisplatin-induced expression of c-fos protein was examined by incubating the cells with GF109203X (0.1–10 µmol·L−1), which interferes with the catalytic domain of all PKCs, or with Gö6976 (0.1–10 µmol·L−1), an inhibitor of conventional, Ca2+-dependent PKC, or with rottlerin (1–20 µmol·L−1), a PKC-δ inhibitor, for 60 min and then with 100 µmol·L−1 cisplatin (Martiny-Baron et al., 1993). The cisplatin-induced expression of c-fos protein was inhibited, in a dose-dependent way, by GF109203X (Figure 6A) and it was only partly decreased by Gö6976 (Figure 6B), or by rottlerin (Figure 6C), indicating that both conventional and non-conventional PKCs are implicated in the cisplatin-induced c-fos expression. When Gö6976 and rottlerin were combined, there was complete inhibition of the cisplatin-induced c-fos expression (Figure 6D).
Figure 6.

Mechanism of cisplatin-induced c-fos expression. Roles of PKC and MAPKs. Thyroid cells were treated without or with 100 µmol·L−1 cisplatin, after preincubation with GF109203X (A), Gö6976 (B), PD98059 and rottlerin (C) or Gö6976 plus PD98059, Gö6976 plus rottlerin or pretreated with siRNA-PKC-ε or siRNA-NS (D). Lysates from thyroid cell were separated by 10% SDS-PAGE and analysed by Western blotting using the antibody against c-fos (or the antibody against PKC-ε, in set). Sequential incubation of the sheet with anti β-actin antibody confirmed equal protein loading. The figures are representative of three independent experiments and results from densitometry are expressed as mean ± SD (n = 3) of sum of the grey level values. Values with shared letters are not significantly different according to Bonferroni/Dunne post hoc tests. PAGE, polyacrylamide gel electrophoresis; SDS, sodium dodecyl sulphate siRNA, small interfering RNA.
PC Cl3 cells were also pre-incubated with 10 µmol·L−1 of the PKC-ε translocation inhibitor peptide εV1 (that selectively inhibits the translocation of PKC-ε to sub cellular sites (Yedovitzky et al., 1997) for 60 min. Such preincubation did not affect the induction of c-fos evoked by cisplatin (data not shown). PC Cl3 cells were then transfected with 30 nmol·L−1 of siRNA-PKC-ε, or with nonsilencing siRNA (siRNA-NS) using the Code Breaker transfection reagent (Promega). Depletion of PKC-ε by siRNA efficiently reduced PKC-ε protein level but not cisplatin-induced c-fos expression (Figure 6D).
Role of MAPK
To determine whether the MAPK cascade mediates cisplatin-evoked c-fos protein induction, PD098059, a selective inhibitor of the ERK/MAPK activator MEK (Alessi et al., 1995) was used. Pretreatment of the cells for 30 min with PD98059 (15–30 µmol·L−1) decreased the cisplatin-induced expression of c-fos (Figure 6C). When Gö6976 and PD098059 or Gö6976 and rottlerin were combined, there was total inhibition of the cisplatin-induced c-fos expression (Figure 6D); conversely, when rottlerin and PD098059 were combined, no additive effects on the cisplatin-induced c-fos expression was found (data not shown).
The effects of GF109203X, Gö6976, rottlerin and PD098059 were also studied by using RT-PCR; Figure 7A shows that the expression of c-fos mRNA was reduced by both Gö6976 and PD098059; co-incubation of cells with Gö6976 and PD098059 or with Gö6976 and rottlerin provoked a drastic reduction of c-fos mRNA level. GF109203X provoked a complete inhibition of the cisplatin-induced c-fos mRNA (Figure 7B).
Figure 7.

Mechanism of cisplatin-induced c-fos induction: roles of PKC and MAPKs. Thyroid cells were treated without or with 100 µmol·L−1 cisplatin, after preincubation with Gö6976, PD98059, GF109203X, or with Gö6976 plus rottlerin, then RNA was extracted. Analysis by (A) electrophoresis of reverse transcription and polymerase chain reaction (RT-PCR) products or (B) agarose gel (GF109203X; 0.1–10 µmol·L−1) revealed the specific transcripts for c-fos and β-actin used to normalized the amount of DNA template in each PCR reaction. The figures are representative of three independent experiments.
Role of PI3K
Pre-incubation of PC Cl3 cells with LY294002 (1–100 µmol·L−1), or with wortmannin (5–50 µmol·L−1), two PI3K inhibitors that inhibited the phosphorylation of PKB/Akt, did not have any effect on the cisplatin-induced c-fos expression in PC-Cl3 cells, suggesting that its induction was not related to the activity of PI3K (data not shown).
c-fos expression and cisplatin resistance in PC Cl3 cells
To examine whether cisplatin sensitivity could be restored by the inhibition of c-fos expression, PC Cl3 cells were exposed to cisplatin in the presence and absence of PD98059 or Gö6976 or rottlerin, able to repress c-fos expression. Viable cell number was determined after 24 h by MTT assay and confirmed by SRB assay to rule out potential effects of cisplatin on mitochondrial enzymes. Indeed, comparable results were obtained when cell number was directly determined by cell counting; consequently, we used the MTT assay in all the cytotoxicity experiments described herein. As previously demonstrated, pretreatment with PD98059 a MEK inhibitor or rottlerin, a PKC-δ inhibitor, both able to block cisplatin-provoked ERK phosphorylation, resulted in enhanced sensitivity to cisplatin inasmuch as a significant decrease in cell survival after treatment was observed in PC Cl3 cells (Urso et al., 2005). Here, to confirm the specificity of data obtained by the use of rottlerin, we examined the effects of PKC-δ depletion by siRNA on cisplatin-provoked ERK phosphorylation, c-fos expression and cell sensitivity.
PC Cl3 cells were transfected with 30 nmol·L−1 of siRNA-PKC-δ or with nonsilencing siRNA (siRNA-NS) using the Code Breaker transfection reagent (Promega). Cell lysates were extracted at 24 and 48 h post transfection and the levels of PKC-δ protein expression were determined by Western blotting. Depletion of PKC-δ by siRNA reduced PKC-δ protein level, cisplatin-induced ERK phosphorylation and also c-fos induction, but not total ERK expression (Figure 8A). Figure 8B shows the effects of PKC-δ depletion on cell viability in cisplatin-treated PC Cl3 cells; it can be seen that si-RNA-PKC-δ increased the cytotoxic effects of cisplatin, reducing the viable cells from 50 to 23%. siRNA-NS did not have any effect (Figure 8A,B).
Figure 8.
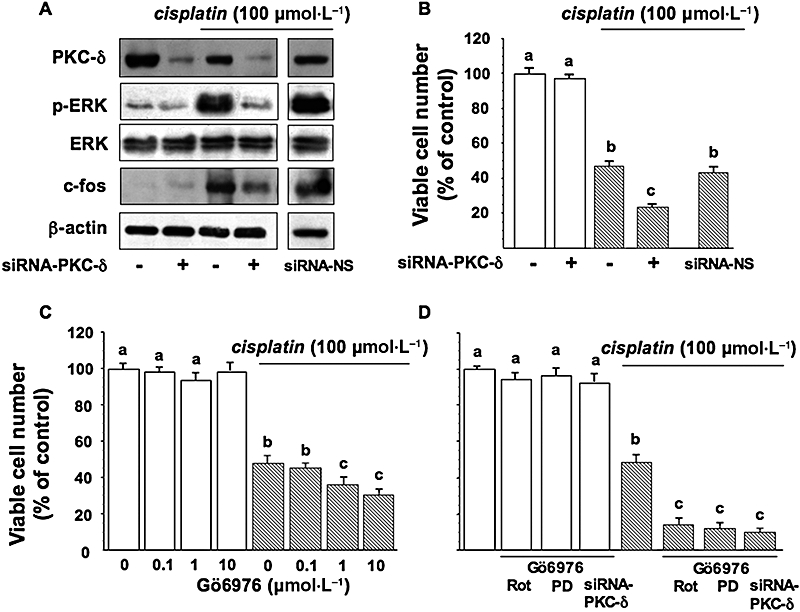
c-fos expression and cisplatin resistance in PC Cl3 cells. Cells pretreated with siRNA-PKC-δ were treated with cisplatin. (A) Lysates from cell were separated by 10% SDS-PAGE and analysed by Western blotting using the antibodies against PKC-δ, p-ERK and total ERK. Sequential incubation of the sheet with anti β-actin antibody confirmed equal protein loading. (B) Viable cell numbers assessed by a MTT assay, as described in Material and Methods. siRNA-NS, nonsilencing siRNA. Thyroid cells were treated without or with 100 µmol·L−1 cisplatin, after preincubation with Gö6976 (C), or with Gö6976 combined with rottlerin/siRNA-PKC-δ or to PD98059 (D). Viable cell numbers were assessed by the MTT assay. The data are means ± SD of four different experiments run in eight replicates and are presented as percent of control. Values with shared letters are not significantly different according to Bonferroni/Dunne post hoc tests. ERK, extracellular signal-regulated kinase; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenol tetrazolium bromide; PAGE, polyacrylamide gel electrophoresis; SDS, sodium dodecyl sulphate; siRNA, small interfering RNA.
Similarly, pre-incubation with Gö6976 (0.1–10 µmol·L−1) for 30 min enhanced the sensitivity to cisplatin in PC Cl3 (anova: P < 0.05; Figure 8C). When PC Cl3 cells were pre-incubated with both Gö6976 (10 µmol·L−1) and PD98059 (30 µmol·L−1) for 30 min, a condition in which c-fos was barely visible, the sensitivity to cisplatin was also enhanced, with less than 15% of cells surviving after 24 h (anova: P < 0.001; Figure 8D). The combination of Gö6976 and rottlerin or siRNA-PKC-δ had an additional effect on PC-Cl3 cell survival compared to inhibitors alone (anova: P < 0.001; Figure 8D).
The expression of c-fos protein was also down-regulated by siRNA. Preliminary experiments, using Western blotting, demonstrated that maximal down-regulation of c-fos protein levels, was apparent after 24 h (Figure 9A). siRNA-NS had no silencing effect c-fos protein expression at either 24 or 48 h posttransfection (Figure 9A).
Figure 9.
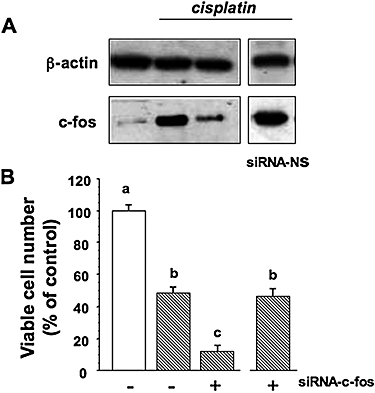
c-fos inhibition and cisplatin resistance in PC Cl3 cells. Cells pretreated with siRNA-c-fos were treated with cisplatin. (A) lysates were separated by 10% SDS-PAGE and analysed by Western blotting using the antibody against c-fos. Sequential incubation of the sheet with anti β-actin antibody confirmed the equal protein loading. (B) Viable cell number was determined 24 h later by the MTT assay. The data are means ± SD of four different experiments run in eight replicates and are presented as percent of control. Values with shared letters are not significantly different according to Bonferroni/Dunne post hoc tests. MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenol tetrazolium bromide; PAGE, polyacrylamide gel electrophoresis; SDS, sodium dodecyl sulphate; siRNA, small interfering RNA.
Cells treated with siRNA-c-fos were then treated with cisplatin, and viable cell number was determined 24 h later. We found that cisplatin was more toxic in cells treated with the siRNA c-fos than in cells treated with cisplatin alone, from 50% (cisplatin alone) to 10% (cisplatin plus c-fos antisense; P = 0.00012) (Figure 9B).
Effects of the inhibition of cisplatin-induced c-fos expression on apoptosis
Previous studies from our laboratory have demonstrated nuclear condensation and fragmentation and caspase-3 cleavage in PC Cl3 cells treated with cisplatin (Muscella et al., 2005; Urso et al., 2005). Since novel PKC isozymes are substrates for caspase-3 and cisplatin induced cleavage of PKC isozymes, we examined the effects of caspase-3 depletion, by siRNA, on the proteolytic activation of PKC-δ. PC Cl3 cells were transfected with 30 nmol·L−1 of siRNA-caspase-3 and siRNA-NS. Cell lysates were extracted at 24 and 48 h post transfection and the caspase-3 expression was determined by Western blotting. siRNA-caspase-3 entirely suppressed caspase-3 expression after 24 h, while siRNA-NS had no silencing effect (Figure 10A). Depletion of caspase-3 by siRNA did not have any effect on PKC-δ cleavage (Figure 10A). On the other hand, depletion of PKC-δ by siRNA, enhanced the conversion of the 32 kDa pro-caspase-3 to the 17 kDa active fragment and increased the cleavage of poly(ADP-ribose) polymerase (PARP) induced by cisplatin (Figure 10B).
Figure 10.
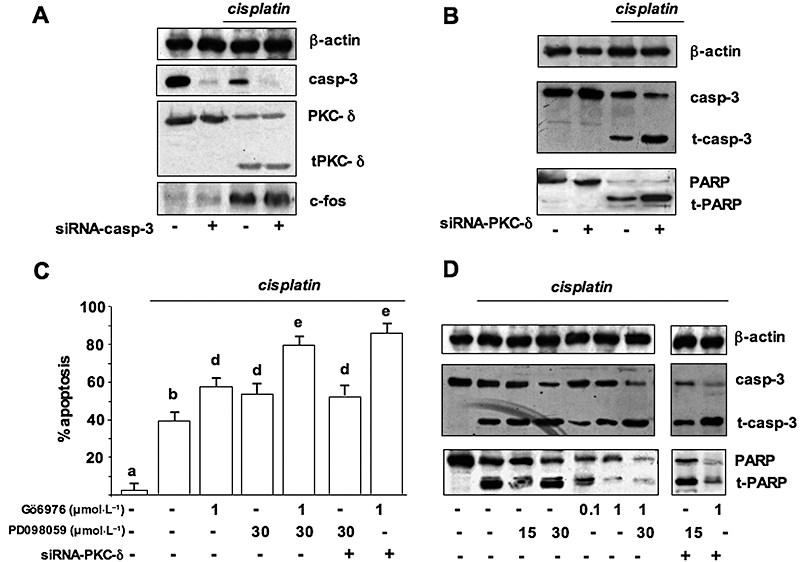
Effects of the inhibition of cisplatin-induced c-fos expression on apoptosis. (A) Cells pretreated with siRNA-caspase-3 were treated or not with cisplatin. Cell lysates were separated by 10% SDS-PAGE and analysed by Western blotting using the antibodies against c-fos, PKC-δ and caspase-3. (B) Cells pretreated with siRNA-PKC-δ were treated or not with cisplatin. Cell lysates were separated by 10% SDS-PAGE and analysed by Western blotting using the antibodies against caspase-3 and PARP. (C and D) Cells, pretreated or not with siRNA-PKC-δ, were pre-incubated with Gö6976, PD98059, or with Gö6976 plus PD98059 and then incubated with 100 µmol·L−1 cisplatin; cells were stained with DAPI (C) or lysed for Western blot analysis (D). (C) Quantification of the percentage of apoptotic nuclei obtained from cells stained with DAPI (mean ± SD; n = 4). Values with shared letters are not significantly different according to Bonferroni/Dunne post hoc tests. (D) Cell lysates separated by 15% (caspase-3) or 10% SDS-PAGE (PARP) were analysed by Western blotting. Sequential incubation of the sheet with anti β-actin antibody confirmed equal protein loading. The figures are representative of three independent experiments. MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenol tetrazolium bromide; DAPI, 4,6-diamine-2-phenylindole; PAGE, polyacrylamide gel electrophoresis; PARP, poly(ADP-ribose) polymerase; SDS, sodium dodecyl sulphate; siRNA, small interfering RNA.
We examined the effects of the inhibition of c-fos by Gö6976 (1 µmol·L−1) and PD98059 (30 µmol·L−1) on condensation of nuclei and caspase-3 activation, both early markers of apoptosis. Figure 10C shows the results of a quantification of nuclear changes obtained by DAPI staining; it can be seen that the percentage of cisplatin-induced apoptotic cells (39%) was significantly increased in both Gö6976 (59%) and PD98059-pretreated (53%) PC Cl3 cells (Figure 10C). The combination of Gö6976 and PD98059 had a higher effect on PC-Cl3 cell nuclei condensation (80%; anova: P < 0.001; Figure 10C). Moreover, pretreatment of cells with Gö6976 caused a significant decrease in pro-caspase-3 level, and an increase in the activated forms of low molecular mass. A similar effect was found in cells pre-incubated with PD98059; when cells were pretreated simultaneously with Gö6976 plus PD98059 the effects on caspase-3 activation was highest (Figure 10D), suggesting that conventional PKC pathway is separated from the MAPK pathway ending in the control of caspase-3 activity. On the contrary, in PKC-δ-depleted cells by siRNA, pre-incubation with PD98059 did not alter the caspase-3 active level while pre-incubation with Gö6976 further enhanced caspase-3 activation. This observation suggests the existence of one pathway combining PKC-δ and ERK and of another, independent, pathway relying on the activity of conventional PKCs. We also examined PARP cleavage by Western blotting of proteins obtained from isolated nuclei. As shown in Figure 10D, pretreatment with Gö6976 or with PD98059 increased cleaved PARP with respect to the fragmentation obtained with cisplatin alone. The greatest effect on PARP cleavage resulted from pretreatment with Gö6976 plus PD98059 (Figure 10D). Sequential incubation of the blot with anti-β-actin antibody confirmed equal protein loading.
Discussion
Cisplatin is one of the most effective cytotoxic agents against a number of solid tumours, but its use is limited by the frequent emergence of cisplatin-resistant cell populations (Scanlon et al., 1989; Eastman, 1990). Resistance to cisplatin is particularly apparent in thyroid tumours (Sugawara et al., 1995; Asakawa et al., 1997; Haigh et al., 2001); therefore, prediction of chemo-sensitivity may be important for improving the efficacy of treatment for thyroid cancer. Cisplatin resistance has been associated with a number of cellular mechanisms, several of which have been extensively investigated. For example, ERK is activated by cisplatin and it mediates a physiological response to DNA damage such as induction of one or more DNA repair enzymes (Scanlon et al., 1989; Sweeney et al., 1999). In the fully differentiated thyroid PC Cl3 cells, the activation of ERK is important for induction of cisplatin resistance inasmuch as the inhibition of ERK activity enhanced cisplatin-induced cell death (Muscella et al., 2005; Urso et al., 2005). Generally speaking, the activation of ERK (Matsuda et al., 1994), ultimately leads to the activation of downstream transcription factors, such as c-fos, which in turn regulate expression of diverse proteins and play a critical role in cellular defence mechanisms. The elevated expression of c-fos gene has been associated with cisplatin resistance in established cisplatin-resistant cell lines (Kashani-Sabet et al., 1990; Jiao et al., 1991; Moorehead and Singh, 2000). In agreement with these earlier reports, our studies described here showed that c-fos expression was associated with cisplatin resistance in thyroid PC Cl3 cells, and that the decrease of c-fos expression was an effective method for increasing sensitivity of PC Cl3 cells. In these cells, cisplatin was responsible for a dose- and time-dependent induction of c-fos mRNA (Figure 1) and protein (Figures 2 and 3); such induction appeared to be due to the activity of PKCs, as it decreased greatly in the presence of GF109203X, an inhibitor of all PKC isoforms (Figure 7B). Indeed, several PKC isoforms are involved in the transduction of a number of signals important for the regulation of cell growth, differentiation, apoptosis and other cellular functions. As cisplatin provokes the activation by cytosol-to-membrane translocation of conventional PKC-β and novel PKC-ε and PKC-δ, and a late proteolytic activation of PKC-δ (Figure 5), it was necessary to distinguish between the effects of these three PKC isoforms on c-fos induction. To this end, we used pharmacological and molecular techniques in order to inhibit conventional PKC-β (by the use of Gö6976) and novel PKC-δ (by the use of rottlerin and siRNA) and PKC-ε (using the PKC-ε translocation inhibitor peptide and siRNA). Results obtained showed that PKC-ε was not implicated in the control of c-fos expression, while both PKC-β and PKC-δ were involved (Figure 6). Indeed, PKC-β acted as a mediator of cell survival in response to apoptosis induced by chemotherapeutic agents in gastric cancer (Zhu et al., 2000; Jiang et al., 2002); furthermore, inhibition of PKC-α or PKC-β resulted in enhanced sensitivity to chemotherapeutic drugs in gastric cancer MKN-45 cells (Jiang et al., 2004). Also over-expression of PKC-α decreased the sensitivity to cisplatin-induced apoptosis in NIH3T3 cells (Spitaler et al., 1999) and down-regulation of this PKC isoform occurs during drug-induced apoptosis in Cos cells (Whelan and Parker, 1998). Thus, conventional, calcium-dependent PKCs play an important role in regulating cell survival and cell death and overall plays an important role in influencing cisplatin-induced apoptosis (Basu and Akkaraju, 1999; Basu, 2003).
In a previous study (Urso et al., 2005) we found that cisplatin brought about a signalling pathway mediated by PKC-δ, which appeared to be a crucial element of the pathway linking cisplatin to the ERK cascade in PC Cl3 cells. Here, when cells were preincubated with PD98059, decreased c-fos protein (Figure 6) and mRNA (Figure 7) were found. This inhibition was complete when PD98059 and Gö6976 were used simultaneously thus indicating the existence of two different pathways linking cisplatin to c-fos, namely PKC-β and PKC-δ/ERK. The effects of PD98059 plus Gö6976 and rottlerin plus Gö6976 were practically the same and completely inhibited the induction of c-fos (Figure 6).
Next, we aimed to understand the role of c-fos on the cytotoxic effects of cisplatin. To this end we measured the viable cell numbers after cisplatin administration in cells in which PKCs and ERK pathways were inhibited and therefore c-fos expression blocked. Indeed, the cytotoxicity of cisplatin was always enhanced reaching a maximum when Gö6976 plus rottlerin/PKC-δ siRNA or Gö6976 plus PD98059 were used (Figure 8), conditions in which the expression of c-fos was barely visible, thus suggesting that c-fos has protective roles. This effect was also evident when the expression of c-fos was knocked-down by siRNA (Figure 9). In order to assess whether c-fos has anti-apoptotic roles in PC Cl3 cells, the effects of Gö6976 and PD98059 on apoptotic cell number and caspase-3 activation (Figure 10) were studied. The marked increase in the number of apoptotic cells, caspase-3 activation and PARP cleavage found under conditions in which c-fos expression was inhibited, suggested an anti-apoptotic role of c-fos in PC Cl3 cells treated with cisplatin.
As it has been shown that PKC-δ is a substrate for caspase-3 and that proteolytic activation of PKC-δ is associated with DNA damage-induced apoptosis (Emoto et al., 1995; Ghayur et al., 1996), we examined the existence of a physiological link between PKC-δ and caspase-3. We found that depletion of caspase-3 did not affect the cleavage of PKC-δ in cisplatin-treated cells (Figure 10B), thus excluding the possibility that proteolysis of PKC-δ was due to caspase-3 activity. Conversely, when PKC-δ was depleted we found a decrease of the 32 kDa pro-enzyme caspase-3 and an increase of the active subunit, thus showing an anti-apoptotic function of PKC-δ after cisplatin administration (Figure 10C). This PKC-δ function was confirmed by assessing the viable cell number of PC Cl3 cells treated with cisplatin and expressing or not PKC-δ (Figure 8B). Thus, PKC-δ has, in PC Cl3 cells, anti-apoptotic roles related to the control of ERK activation and c-fos induction. Similarly, in some other cell lines, the cytotoxic effect of cisplatin was enhanced by ERK inhibition (Persons et al., 1999; Mandic et al., 2001). On the other hand, opposing effects of ERK have been demonstrated in human melanoma cell line AA (Mandic et al., 2001), in PC12 pheochromocytoma cells (Xia et al., 1995) and in HeLa endometrial carcinoma cells (Wang et al., 2000; Yeh et al., 2002). In addition, there are reports that inhibition of ERK and PKC-δ prevents cisplatin-induced cell death (Huang et al., 2004; Basu and Tu, 2005). Thus, in response to cisplatin, activation of ERK can have either survival or cell death as its outcome. This divergence indicates that the relationship between the activity of ERK and the cellular response to cisplatin might depend on the individual cellular context such as the extent of MAPK activity and the signalling pathways downstream of ERK that will eventually determine cell fate, and also the levels of stress.
PKC-δ has been shown to be activated by multiple apoptotic stimuli. Activation of caspase-3 during apoptosis cleaves the full-length PKC-δ and produces a PKC-δ catalytic domain; alternatively, PKC-δ may also be an activator of caspase-3 (Anantharam et al., 2002). Furthermore, inhibition of PKC-δ has been shown to reduce the effects of apoptosis caused by external stimuli (Xia et al., 2007). Although the majority of the studies indicate that PKC-δ is involved in the induction of apoptosis, there are also studies pointing to a role of PKC-δ in anti-apoptotic responses and in cell survival. For instance, PKC-δ plays a role in the anti-apoptotic effect of tumour necrosis factor-α in human neutrophils (Kilpatrick et al., 2002) and in the anti-apoptotic effect of basic fibroblast growth factor in granulosa cells (Peluso et al., 2001) and in serum-deprived PC-12 cells (Wert and Palfrey, 2000). PKC-δ protects RAW264.7 macrophages from nitric oxide-induced apoptosis (Jun et al., 1999) glioma cells from infection with a virulent strain of Sindbis virus (Zrachia et al., 2002) and the melanoma cells from docetaxel (Mhaidat et al., 2007).
This discrepancy of function in apoptosis attributed to PKC-δ can be explained by the presence of distinct PKC-δ isoforms, generated by alternative splicing. PKC-δI is pro-apoptotic and is a mediator of apoptosis (Patel et al., 2006) whereas the mouse specific PKC-δII isoform and human PKC-δVIII isoform (Jiang et al., 2008) function as anti-apoptotic proteins.
In conclusion, cisplatin resistance is associated with a number of cellular mechanisms; of these, in PC Cl3 cells ERK and c-fos appeared to have crucial roles. In the cell signalling pathways ending to cisplatin resistance, novel PKC-δ operates to control ERK activity and, together with conventional PKC-β, also the induction of c-fos. PKC-δ/ERK and conventional PKC pathways separately enhanced the activation of caspase-3.
Glossary
Abbreviations:
- DAPI
4,6-diamine-2-phenylindole
- ERK
extracellular signal-regulated kinase
- MAPK
mitogen-activated protein kinase
- MEK
MAPK/ERK kinase
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenol tetrazolium bromide
- PARP
poly(ADP-ribose) polymerase
- PBS
phosphate-buffered saline
- PI3K
phosphoinositide 3-kinase
- SDS
sodium dodecyl sulphate
- siRNA
small interfering RNA
- SRB
sulforhodamine B
Conflict of interest
The authors state no conflict of interest.
References
- Aasland R, Lillehaug JR, Male R, Jøsendal O, Varhaug JE, Kleppe K. Expression of oncogenes in thyroid tumours: coexpression of c-erb B2/neu and c-erb B. Br J Cancer. 1988;57:358–363. doi: 10.1038/bjc.1988.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi A, Cuenda P, Cohen DT, Dudley AR, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- Amstad PA, Krupitza G, Cerutti PA. Mechanism of c-fos induction by active oxygen. Cancer Res. 1992;52:3952–3960. [PubMed] [Google Scholar]
- Anantharam V, Kitazawa M, Wagner J, Kaul S, Kanthasamy AG. Caspase-3-dependent proteolytic cleavage of protein kinase Cdelta is essential for oxidative stress-mediated dopaminergic cell death after exposure to methylcyclopentadienyl manganese tricarbonyl. J Neurosci. 2002;22:1738–1751. doi: 10.1523/JNEUROSCI.22-05-01738.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arteaga CL, Holt JT. Tissue-targeted antisense c-fos retroviral vector inhibits established breast cancer xenografts in nude mice. Cancer Res. 1996;56:1098–1103. [PubMed] [Google Scholar]
- Asakawa H, Kobayashi T, Komoike Y, Maruyama H, Nakano Y, Tamaki Y, et al. Chemosensitivity of anaplastic thyroid carcinoma and poorly differentiated thyroid carcinoma. Anticancer Res. 1997;17:2757–2762. [PubMed] [Google Scholar]
- Basu A. Involvement of protein kinase C-delta in DNA damage-induced apoptosis. J Cell Mol Med. 2003;7:341–350. doi: 10.1111/j.1582-4934.2003.tb00237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A, Akkaraju GR. Regulation of caspase activation and cisdiamminedichloroplatinum(II)-induced cell death by protein kinase C. Biochemistry. 1999;38:4245–4251. doi: 10.1021/bi982854q. [DOI] [PubMed] [Google Scholar]
- Basu A, Tu H. Activation of ERK during DNA damage-induced apoptosis involves protein kinase C. Biochem Biophys Res Commun. 2005;334:1068–1073. doi: 10.1016/j.bbrc.2005.06.199. [DOI] [PubMed] [Google Scholar]
- Bonovich M, Olive M, Reed E, O'Connell B, Vinson C. Adenoviral delivery of A-FOS, an AP-1 dominant negative, selectively inhibits drug resistance in two human cancer cell lines. Cancer Gene Ther. 2002;9:62–70. doi: 10.1038/sj.cgt.7700409. [DOI] [PubMed] [Google Scholar]
- Chiu R, Boyle WJ, Meek J, Smeal T, Hunter T, Karin M. The c-Fos protein interacts with c-Jun/AP-1 to stimulate transcription of AP-1 responsive genes. Cell. 1988;54:541–552. doi: 10.1016/0092-8674(88)90076-1. [DOI] [PubMed] [Google Scholar]
- Dempke W, Voigt W, Grothey A, Hill BT, Schmoll HJ. Cisplatin resistance and oncogenes – a review. Anticancer Drugs. 2000;11:225–236. doi: 10.1097/00001813-200004000-00001. [DOI] [PubMed] [Google Scholar]
- Eastman A. Activation of programmed cell death by anticancer agents: cisplatin as a model system. Cancer Cells. 1990;2:275–280. [PubMed] [Google Scholar]
- Emoto Y, Manome Y, Meinhardt G, Kisaki H, Kharbanda S, Robertson M, et al. Proteolytic activation of protein kinase C d by an ICE-like protease in apoptosis cells. EMBO J. 1995;14:6148–6156. doi: 10.1002/j.1460-2075.1995.tb00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco A, Berlingieri MT, Di Fiore PP, Portella G, Grieco M, Vecchio G. One- and two-step transformations of rat thyroid epithelial cells by retroviral oncogenes. Mol Cell Biol. 1987;7:3365–3370. doi: 10.1128/mcb.7.9.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghayur T, Hugunin M, Talanian RV, Ratnofsky S, Quinlan C, Emoto Y, et al. Proteolytic activation of protein kinase C d by an ICE/CED 3 like protease induces characteristics of apoptosis. J Exp Med. 1996;184:2399–2404. doi: 10.1084/jem.184.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimm O. Thyroid cancer. Cancer Lett. 2001;163:143–156. doi: 10.1016/s0304-3835(00)00697-2. [DOI] [PubMed] [Google Scholar]
- Haigh PI. Anaplastic thyroid carcinoma. Curr Treat Options Oncol. 2000;1:353–357. doi: 10.1007/s11864-000-0051-8. [DOI] [PubMed] [Google Scholar]
- Haigh PI, Ituarte PH, Wu HS, Treseler PA, Posner MD, Quivey JM, et al. Completely resected anaplastic thyroid carcinoma combined with adjuvant chemotherapy and irradiation is associated with prolonged survival. Cancer. 2001;91:2335–2342. [PubMed] [Google Scholar]
- Hanna NN, McGrath PC, Sloan DA, Kenady DE. Advances in the pathogenesis and treatment of thyroid cancer. Curr Opin Oncol. 1999;11:42–47. doi: 10.1097/00001622-199901000-00010. [DOI] [PubMed] [Google Scholar]
- Hollander MC, Fornace AJ., Jr Induction of fos RNA by DNA-damaging agents. Cancer Res. 1989;49:1687–1692. [PubMed] [Google Scholar]
- Huang J, Mohanty S, Basu A. Cisplatin resistance is associated with deregulation in protein kinase C-δ. Biochem Biophys Res Commun. 2004;316:1002–1008. doi: 10.1016/j.bbrc.2004.02.149. [DOI] [PubMed] [Google Scholar]
- Jiang K, Apostolatos AH, Ghansah T, Watson JE, Vickers T, Cooper DR, et al. Identification of a novel antiapoptotic human protein kinase C delta isoform, PKCdeltaVIII in NT2 cells. Biochemistry. 2008;47:787–797. doi: 10.1021/bi7019782. [DOI] [PubMed] [Google Scholar]
- Jiang XH, Lam SK, Lin MC, Jiang SH, Kung HF, Slosberg ED, et al. Novel target for induction of apoptosis by cyclo-oxygenase-2 inhibitor SC-236 through a protein kinase C-beta(1)-dependent pathway. Oncogene. 2002;21:6113–6122. doi: 10.1038/sj.onc.1205778. [DOI] [PubMed] [Google Scholar]
- Jiang XH, Tu SP, Cui JT, Lin MC, Xia HH, Wong WM, et al. Antisense targeting protein kinase C alpha and beta1 inhibits gastric carcinogenesis. Cancer Res. 2004;64:5787–5794. doi: 10.1158/0008-5472.CAN-03-1172. [DOI] [PubMed] [Google Scholar]
- Jiao L, Funato T, Wang W, Tone T, Kashani-Sabet M, Scanlon KJ. The role of the c-fos oncogene in cisplatin resistance. In: Howell SB, editor. Platinum and Other Metal Coordination Compounds in Cancer Chemotherapy. New York: Plenum Press; 1991. pp. 303–313. [Google Scholar]
- Jun CD, Oh CD, Kwak HJ, Pae HO, Yoo JC, Choi BM, et al. Overexpression of protein kinase C isoforms protectsRAW264.7 macrophages from nitric oxide-induced apoptosis: involvement of c-Jun N-terminal kinase/stress-activated protein kinase, p38 kinase, and CPP-32 protease pathways. J Immunol. 1999;162:3395–3401. [PubMed] [Google Scholar]
- Kaina B, Haas S, Kappes H. A general role for c-fos in cellular protection against DNA-damaging carcinogens and cytostatic drugs. Cancer Res. 1997;57:2721–2731. [PubMed] [Google Scholar]
- Karga H, Lee JK, Vickery AL, Thor A, Gaz RD, Jameson JL. ras oncogene mutations in benign and malignant thyroid neoplasms. J Clin Endocrinol Metab. 1991;73:832–836. doi: 10.1210/jcem-73-4-832. [DOI] [PubMed] [Google Scholar]
- Kashani-Sabet M, Wang W, Scanlon KJ. Cyclosporin A suppresses cisplatin induced c-fos gene expression in ovarian carcinoma cells. J Biol Chem. 1990;265:11285–11288. [PubMed] [Google Scholar]
- Kilpatrick LE, Lee JY, Haines KM, Campbell DE, Sullivan KE, Korchak HM. A role for PKC-δ and PI 3-kinase in TNFalpha-mediated antiapoptotic signaling in the human neutrophil. Am J Physiol Cell Physiol. 2002;283:48–57. doi: 10.1152/ajpcell.00385.2001. [DOI] [PubMed] [Google Scholar]
- Kochhar S, Hunziker PE, Leong-Morgenthaler P, Hovius R. Primary structure, physicochemical properties, and chemical modification of NAD(+)-dependent D-lactate dehydrogenase. Evidence for the presence of Arg-235, His-303, Tyr-101, and Trp-19 at or near the active site. J Biol Chem. 1992;267:8499–8513. [PubMed] [Google Scholar]
- Kovary K, Bravo R. Existence of different Fos/Jun complexes during the G0-to-G1 transition and during exponential growth in mouse fibroblasts: differential role of Fos proteins. Mol Cell Biol. 1992;12:5015–5023. doi: 10.1128/mcb.12.11.5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TH. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Mandic A, Viktorsson K, Heiden T, Hansson J, Shoshan MC. The MEK1 inhibitor PD98059 sensitizes C8161 melanoma cells to cisplatin-induced apoptosis. Melanoma Res. 2001;11:11–19. doi: 10.1097/00008390-200102000-00002. [DOI] [PubMed] [Google Scholar]
- Marsigliante S, Muscella A, Elia MG, Greco S, Storelli C. Angiotensin II AT1 receptor stimulates Na+ -K+ATPase activity through a pathway involving PKC-zeta in rat thyroid cells. J Physiol. 2003;54:461–470. doi: 10.1113/jphysiol.2002.027466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martiny-Baron G, Kazanietz H, Mischak PM, Blumberg G, Kochs H, Hug D, et al. Selective inhibition of protein kinase C isozymes by the indolocarbazole Gö 6976. J Biol Chem. 1993;268:9194–9197. [PubMed] [Google Scholar]
- Matsuda S, Gotoh Y, Nishida E. Signaling pathways mediated by the mitogen-activated protein (MAP) kinase/MAP kinase cascade. J Leukoc Biol. 1994;56:548–553. doi: 10.1002/jlb.56.5.548. [DOI] [PubMed] [Google Scholar]
- Mhaidat NM, Thorne RF, Zhang XD, Hersey P. Regulation of docetaxel-induced apoptosis of human melanoma cells by different isoforms of protein kinase C. Mol Cancer Res. 2007;5:1073–1081. doi: 10.1158/1541-7786.MCR-07-0059. [DOI] [PubMed] [Google Scholar]
- Moorehead RA, Singh G. Influence of the proto-oncogene c-fos on cisplatin sensitivity. Biochem Pharmacol. 2000;59:337–345. doi: 10.1016/s0006-2952(99)00333-0. [DOI] [PubMed] [Google Scholar]
- Muscella A, Urso L, Calabriso N, Ciccarese A, Migoni D, Fanizzi FP, et al. Differential response of normal, dedifferentiated and transformed thyroid cell lines to cisplatin treatment. Biochem Pharmacol. 2005;19:50–60. doi: 10.1016/j.bcp.2005.10.022. [DOI] [PubMed] [Google Scholar]
- Norby N. Methods in Enzymology. New York: Academic Press; 1988. Coupled assay of Na+/K+ATPase activity; p. 116. Vol. 156. p. [DOI] [PubMed] [Google Scholar]
- Patel NA, Song S, Cooper DR. PKCdelta alternatively spliced isoforms modulate cellular apoptosis in retinoic-induced differentiation of human NT2 cells and mouse embryonic stem cells. Gene Expr. 2006;13:73–84. doi: 10.3727/000000006783991890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peluso JJ, Pappalardo A, Fernandez G. Basic fibroblast growth factor maintains calcium homeostasis and granulosa cell viability by stimulating calcium efflux via a PKC δ-dependent pathway. Endocrinology. 2001;142:4203–4211. doi: 10.1210/endo.142.10.8460. [DOI] [PubMed] [Google Scholar]
- Persons DL, Yazlovitskaya EM, Cui W, Pelling JC. Cisplatin induced activation of mitogen-activated protein kinases in ovarian carcinoma cells: inhibition of extracellular signal-egulated kinase activity increases sensitivity to cisplatin. Clin Cancer Res. 1999;5:1007–1014. [PubMed] [Google Scholar]
- Scanlon KJ, Kashani-Sabet M, Miyachi H, Sowers LC, Rossi J. Molecular basis of cisplatin resistance in human carcinomas: model systems and patients. Anticancer Res. 1989;9:1301–1312. [PubMed] [Google Scholar]
- Scanlon KJ, Wang W, Han H. Cyclosporin A suppresses cisplatin-induced oncogene expression in human cancer cells. Cancer Treat Rev. 1990;17:27–35. doi: 10.1016/0305-7372(90)90013-6. [DOI] [PubMed] [Google Scholar]
- Schuermann M. The Fos family: gene and protein structure, homologies, and differences. In: Angel PE, Herrlich PA, editors. The FOS and JUN Families of Transcription Factors. Boca Raton: CRC Press; 1994. pp. 15–35. [Google Scholar]
- Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- Spitaler M, Wiesenhofer B, Biedermann V, Seppi T, Zimmermann J, Grunicke H, et al. The involvement of protein kinase C isoenzymes alpha, epsilon and zeta in the sensitivity to antitumor treatment and apoptosis induction. Anticancer Res. 1999;19:3969–3976. [PubMed] [Google Scholar]
- Sugawara I, Masunaga A, Itoyama S, Sumizawa T, Akiyama S, Yamashita T. Expression of multidrug resistance-associated protein (MRP) in thyroid cancers. Cancer Lett. 1995;95:135–138. doi: 10.1016/0304-3835(95)03878-z. [DOI] [PubMed] [Google Scholar]
- Sweeney JF, Nguyen PK, Omann G, Hinshaw DB. Granulocyte-macrophage colony-stimulating factor rescues human polymorphonuclear leukocytes from ultraviolet irradiation-accelerated apoptosis. J Surg Res. 1999;81:108–112. doi: 10.1006/jsre.1998.5471. [DOI] [PubMed] [Google Scholar]
- Terrier P, Sheng ZM, Schlumberger M, Tubiana M, Caillou B, Travagli JP, et al. Structure and expression of c-myc and c-fos proto-oncogenes in thyroid carcinomas. Br J Cancer. 1988;57:43–47. doi: 10.1038/bjc.1988.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urso L, Muscella A, Calabriso N, Ciccarese A, Fanizzi FP, Migoni D, et al. Differential functions of PKC-delta and PKC-zeta in cisplatin response of normal and transformed thyroid cells. Biochem Biophys Res Commun. 2005;337:297–305. doi: 10.1016/j.bbrc.2005.09.046. [DOI] [PubMed] [Google Scholar]
- Vini L, Harmer C. Management of thyroid cancer. Lancet Oncol. 2002;3:407–414. doi: 10.1016/s1470-2045(02)00787-8. [DOI] [PubMed] [Google Scholar]
- Wang X, Martindale JL, Holbrook NJ. Requirement for ERK activation in cisplatin-induced apoptosis. J Biol Chem. 2000;275:39435–39443. doi: 10.1074/jbc.M004583200. [DOI] [PubMed] [Google Scholar]
- Wert MM, Palfrey HC. Divergence in the anti-apoptotic signaling pathways used by nerve growth factor and basic fibroblast growth factor (bFGF) in PC12 cells: rescue by bFGF involves protein kinase Cδ. Biochem J. 2000;352:175–182. [PMC free article] [PubMed] [Google Scholar]
- Whelan RD, Parker PJ. Loss of protein kinase C function induces an apoptotic response. Oncogene. 1998;16:1939–1944. doi: 10.1038/sj.onc.1201725. [DOI] [PubMed] [Google Scholar]
- Xia S, Forman LW, Faller DV. PKC-delta is required for survival of cells expressing activated p21Ras. J Biol Chem. 2007;282:13199–13210. doi: 10.1074/jbc.M610225200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Yedovitzky M, Mochly-Rosen D, Johnson JA, Gray MO, Ron D, Abramovitch E, et al. Translocation inhibitors define specificity of protein kinase C isoenzymes in pancreatic beta-cells. J Biol Chem. 1997;272:1417–1420. doi: 10.1074/jbc.272.3.1417. [DOI] [PubMed] [Google Scholar]
- Yeh PY, Chuang SE, Yeh KH, Song YC, Ea CK, Cheng AL. Increase of the resistance of human cervical carcinoma cells to cisplatin by inhibition of the MEK to ERK signaling pathway partly via enhancement of anticancer druginduced NF-kB activation. Biochem Pharmacol. 2002;63:1423–1430. doi: 10.1016/s0006-2952(02)00908-5. [DOI] [PubMed] [Google Scholar]
- Zhu GH, Wong BC, Slosberg E, Eggo M, Ching CK, Yuen S, et al. Overexpression of protein kinase C-beta1 isoenzyme suppresses indomethacin-induced apoptosis in gastric epithelial cells. Gastroenterology. 2000;118:507–514. doi: 10.1016/s0016-5085(00)70256-3. [DOI] [PubMed] [Google Scholar]
- Zou M, Shi Y, Farid NR. p53 mutations in all stages of thyroid carcinomas. J Clin Endocrinol Metab. 1993;77:1054–1058. doi: 10.1210/jcem.77.4.8408453. [DOI] [PubMed] [Google Scholar]
- Zrachia A, Dobroslav M, Blass M, Kazimirsky G, Kronfeld I, Blumberg PM, et al. Infection of glioma cells with Sindbis virus induces selective activation and tyrosine phosphorylation of protein kinase Cδ. Implications for Sindbis virus-induced apoptosis. J Biol Chem. 2002;277:23693–23701. doi: 10.1074/jbc.M111658200. [DOI] [PubMed] [Google Scholar]