

Abstract
Background and purpose
Allopurinol is a potent inhibitor of the enzyme xanthine oxidase, used primarily in the treatment of hyperuricemia and gout. It is well known that purines exert multiple effects on pain transmission. We hypothesized that the inhibition of xanthine oxidase by allopurinol, thereby reducing purine degradation, could be a valid strategy to enhance purinergic activity. The aim of this study was to investigate the anti-nociceptive profile of allopurinol on chemical and thermal pain models in mice.
Experimental approach
Mice received an intraperitoneal (i.p.) injection of vehicle (Tween 10%) or allopurinol (10–400 mg kg−1). Anti-nociceptive effects were measured with intraplantar capsaicin, intraplantar glutamate, tail-flick or hot-plate tests.
Key results
Allopurinol presented dose-dependent anti-nociceptive effects in all models. The opioid antagonist naloxone did not affect these anti-nociceptive effects. The non-selective adenosine-receptor antagonist caffeine and the selective A1 adenosine-receptor antagonist, DPCPX, but not the selective A2A adenosine-receptor antagonist, SCH58261, completely prevented allopurinol-induced anti-nociception. No obvious motor deficits were produced by allopurinol, at doses up to 200 mg kg−1. Allopurinol also caused an increase in cerebrospinal fluid levels of purines, including the nucleosides adenosine and guanosine, and decreased cerebrospinal fluid concentration of uric acid.
Conclusions and implications
Allopurinol-induced anti-nociception may be related to adenosine accumulation. Allopurinol is an old and extensively used compound and seems to be well tolerated with no obvious central nervous system toxic effects at high doses. This drug may be useful to treat pain syndromes in humans.
Keywords: allopurinol, purines, pain, adenosine, guanosine, xanthine oxidase, anti-nociception
Introduction
Allopurinol [1,5-dihydro-4H-pyrazolo(3,4-d)pyrimidin-4-one] is a structural analogue of hypoxanthine and a potent inhibitor of the enzyme xanthine oxidase that catalyses the transformation of hypoxanthine to xanthine and uric acid, reducing both uric acid formation and purine degradation (Pacher et al., 2006; Day et al., 2007). Allopurinol is used primarily in the treatment of hyperuricemia and gout (Rundles, 1982). Besides its hypouricemic effects, allopurinol has been studied for several other indications, including treatment of seizures, psychiatric disorders, ischaemia-reperfusion injury, protozoal diseases and as a measure of liver impairment (Das et al., 1987; Van Waeg et al., 1988; Garcia Garcia et al., 1990; Day et al., 1994; Akhondzadeh et al., 2005).
Both healthy and hyperuricemic patients exhibit a reduction of uric acid levels after allopurinol, probably leading to accumulation of purines, including the neuromodulator adenosine, which may explain its beneficial anticonvulsant and antipsychotic effects (Tada et al., 1991; Wada et al., 1992; Zagnoni et al., 1994; Lara et al., 2000; 2003; Machado-Vieira et al., 2001). Of note, positive effects of allopurinol in refractory epilepsy (Tada et al., 1991), aggressive behaviour (Lara et al., 2000; 2003), mania (Machado-Vieira et al., 2001) and schizophrenia (Lara et al., 2001a; Brunstein et al., 2005) have been suggested to be secondary to its inhibitory effect on purine degradation and thus enhancing activities of adenosine, despite the lack of direct data to support this hypothesis.
The purinergic system involves adenosine and adenosine triphosphate (ATP) as major endogenous effectors, acting on P1 and P2 receptors respectively (Ralevic and Burnstock, 1998). Adenosine is mainly an inhibitory neuromodulator, regulating synaptic activity and release of several neurotransmitters, such as noradrenaline, dopamine, serotonin, acetylcholine and glutamate (Brundege and Dunwiddie, 1997; Ralevic and Burnstock, 1998). It is well known that adenosine and its analogues exert multiple effects on pain transmission at peripheral and central sites (Sawynok, 1998; Sawynok and Liu, 2003). Anti-nociceptive effects of adenosine may be related to the inhibition of intrinsic neurons by an increase in K+ conductance and pre-synaptic inhibition of sensory nerve terminals, decreasing the release of substance P and glutamate (Sollevi, 1997); attenuation by NMDA-induced production of nitric oxide also may be involved (Bhardwaj et al., 1995). Adenosine has been shown to mediate opioid analgesia (Bennett, 2000).
Caffeine and theophylline are the classic P1 adenosine antagonists currently used in humans, but adenosine agonists for human use are still lacking. We hypothesized that the inhibition of xanthine oxidase by allopurinol, thereby reducing purine degradation, could be a valid strategy to enhance purinergic activity, which is in line with the anti-convulsant and neuropsychiatric effects observed with allopurinol treatment (Tada et al., 1991; Wada et al., 1992; Zagnoni et al., 1994; Lara et al., 2000; 2001a; 2003; Machado-Vieira et al., 2001; Brunstein et al., 2005). Based on the considerations above, the aims of the present study were: (i) to investigate the anti-nociceptive activity induced by allopurinol in chemical and thermal pain models in mice; (ii) to identify, by means of pharmacological as well as neurochemical approaches, possible mechanisms by which allopurinol causes anti-nociception in mice; and (iii) to evaluate the acute toxicity induced by allopurinol using behavioural paradigms.
Methods
Animals
All animal procedures and studies followed the ethical guidelines for investigations of experimental pain in conscious animals (Zimmermann, 1983) and our institutional protocols for experiments with animals, designed to avoid suffering and limit the number of animals used. The number of animals and intensities of noxious stimuli used were the minimum necessary to demonstrate the consistent effects of the drug treatments. Male adult Swiss albino mice (2–3 months of age, 30–40 g) were kept on a 12 h light/dark cycle (light on at 7:00 a.m.) at temperature of 22 ± 1°C, housed in plastic cages (five per cage) with tap water and commercial food pellets ad libitum. All behavioural procedures were conducted between 8:00 and 10:00 a.m. In all experiments of nociceptive behavioural, the animals were acclimatized to the laboratory for at least 1 h before testing and were used only once throughout the experiments.
Drug administration
Experiments were performed according to the method described by Schmidt et al. (2000): 20 min before the experiment, animals were placed individually in acrylic boxes, which also served as observation chambers. After this adaptation period, treatments were performed. Animals were given an intraperitoneal (i.p.) injection (10 mL kg−1) of vehicle (saline or 10% Tween) or allopurinol (10–400 mg kg−1). In order to investigate the mechanism of action of allopurinol, some animals were also pre-treated (15 min in advance) with an i.p. injection of the non-selective (A1 and A2A) adenosine receptor antagonist caffeine (30 mg kg−1), the selective A1 adenosine receptor antagonist DPCPX (0.1 mg kg−1), the selective A2A adenosine receptor antagonist SCH58261 (0.5 mg kg−1) or the non-selective opioid receptor antagonist naloxone (1 mg kg−1). Adenosine (100 mg kg−1) and morphine sulphate (6 mg kg−1) were used as positive controls for those experiments. Caffeine, adenosine, DPCPX and SCH58261 doses were based on earlier work (Lara et al., 2001b; Peana et al., 2006; Dall'Igna et al., 2007).
Capsaicin-induced nociception
The method used for capsaicin-induced licking was similar to that described by Sakurada et al. (1993). Thirty minutes after i.p. treatments, 20 µL of capsaicin (1.6 µg per paw) was injected intraplantarly (i.pl.), under the plantar skin of the right hind paw (Hamilton microsyringe with a 26-gauge needle). Animals were observed individually for 5 min after capsaicin administration for the time spent licking the injected paw, which was recorded and considered a measure of nociception.
Glutamate-induced nociception
The procedure used was similar to that described previously (Beirith et al., 2002). Thirty minutes after i.p. treatments, a volume of 20 µL of glutamate solution (10 µmol per paw prepared in saline) was injected i.pl., as described above. Mice were observed individually for 15 min following glutamate injection and the amount of time spent licking the injected paw was recorded and considered as indicative of nociception.
Tail-flick test
Nociception was assessed with a tail-flick apparatus (Albrasch Electronic Equipments, Brazil), as described in detail elsewhere (D'Amour and Smith, 1941). A source of light was positioned below the tail, focused on a point 2.3 cm rostral to the tip of the tail and the time that the mouse took to withdraw its tail from the noxious stimulus was recorded. Deflection of the tail activates a photocell and automatically terminates the trial. A cut-off time of 10 s was employed in order to prevent tissue damage (a mouse that did not flick its tail by 10 s was considered as fully analgesic). On day one, animals were first habituated with the tail-flick apparatus through three separate measures (data not shown). On day two, baseline tail-flick latency (TFL) was measured for each mouse prior to the treatments. Animals displaying at least two TFL of 10 s on the baseline were excluded from the study. Immediately after the third TFL measurement, animals received i.p. treatments and 30 min thereafter were submitted to the tail-flick apparatus. Data for tail-flick are expressed as mean per cent of maximum possible effect (% MPE) ± SEM, according to the following formula (Calcagnetti et al., 1990): % MPE: 100 × (post-drug latency − baseline latency) × (cut-off time − baseline latency)−1.
Hot-plate test
The hot-plate test was used to measure the response latencies according to the method described by Eddy and Leimbach (1953), with minor modifications. In these experiments, the hot-plate apparatus (Ugo Basile, model-DS 37, Italy) was maintained at 55 ± 0.5°C. Animals were placed into a glass cylinder of 24 cm diameter on the heated surface, and the time between placing of the animal on the hot-plate and the occurrence of licking of hind paws or jumping off the surface was recorded as response latency. On day one, the animals were first habituated to the apparatus. On day two, mice were tested and animals displaying baseline latencies of more than 15 s were excluded from the study. An automatic 20 s cut-off was used to prevent tissue damage. Each animal was tested before administration of drugs in order to obtain the baseline. Thirty minutes after i.p. treatments, animals were placed on the heated surface and response latency recorded as described above. Data for hot-plate are expressed as mean % MPE ± SEM.
Hole-board test
The hole-board apparatus (Ugo Basile, Italy) consisted of grey Perspex panels (40 cm × 40 cm, 2.2 cm thick) with 16 equidistant holes 3 cm in diameter in the floor. Photocells below the surface of the holes measured the number of head-dips. The board was positioned 15 cm above the table and divided into nine squares of 10 cm × 10 cm with a water-resistant marker. Thirty minutes after i.p. treatments, each animal was placed singly in the centre of the board facing away from the observer and its behaviour recorded for 5 min. The number of head-dips, crossings (number of squares crossed with all four paws), rearings, groomings and defecations was recorded, as well as the latency to start locomotion (Vinadéet al., 2003).
Measurement of motor performance
In order to evaluate non-specific muscle relaxant or neurotoxic effects, we evaluated the effects of allopurinol in the rotarod test and in spontaneous locomotor activity test. The rotarod apparatus (Ugo Basile, Italy) consists of a rotating (18 r.p.m.) bar (2.5 cm diameter), subdivided by disks into six compartments. As previously described (Leal et al., 2000), mice were initially trained to remain on the rotarod apparatus for 120 s. Those not remaining on the bar for at least two out of three consecutive trials were discarded. On the day after training, the latency to fall from the rotarod (one trial with a maximum of 60 s) was determined 30 min after i.p. treatments. The method to evaluate spontaneous locomotor activity was adapted from Creese et al. (1976). Activity cages (45 cm × 25 cm × 20 cm, Albarsch Electronic Equipment, Brazil), equipped with three parallel photocells, automatically recorded the number of crossings. Animals were individually habituated to the activity cage for 10 min before receiving the i.p. treatments. Animals were placed again in the activity cages 30 min after treatments, and the crossings were recorded for 15 min.
Potentiation of barbiturate sleeping time in mice
In order to investigate sedative properties of allopurinol, mice pre-treated with allopurinol (50, 100 or 200 mg kg−1) or vehicle (30 min in advance) received an i.p. injection of sodium pentobarbital (30 mg kg−1). After the barbiturate injection, the sleeping time (time elapsed between loss and recuperation of righting reflex) was recorded. Criterion for recuperation of righting reflex is that animals have to regain their normal posture for three consecutive times when challenged to remain on their backs (Yamamoto et al., 1987).
Rectal temperature
This was measured by using a flexible probe before and 30 min after allopurinol (50, 100 or 200 mg kg−1) or vehicle treatment.
Cerebrospinal fluid sampling
Groups of mice were treated similarly with i.p. administration of allopurinol (200 mg kg−1) or vehicle. After 30 min, mice were anaesthetized with sodium thiopental (60 mg kg−1, 10 mL kg−1, i.p.) and placed in a stereotaxic apparatus, where the cerebrospinal fluid (CSF) was drawn (10–20 µL per mouse) by direct puncture of the cisterna magna, with an insulin syringe (27 gauge × 1/2 in length), with the help of a magnifying glass. All samples were centrifuged at 10 000× g in an Eppendorf centrifuge for 5 min to obtain cell-free supernatants and stored in separate tubes in −70°C until analysis.
High-performance liquid chromatography procedure
High-performance liquid chromatography was performed with aliquots obtained from the CSF cell-free supernatants. The following purines were measured according to Domanski et al. (2006): ATP, adenosine diphosphate, adenosine monophosphate, adenosine, guanosine triphosphate, guanosine diphosphate, guanosine monophosphate, guanosine, inosine monophosphate (IMP), inosine, hypoxanthine, xanthine and uric acid. Analyses were performed with Shimadzu Class-VP chromatography system consisting of a quaternary gradient pump with vacuum degassing and piston desalting modules, Shimadzu SIL-10AF auto-injector valve with 50 µL loop, and an UV detector. Separations were achieved on a Supelco C18 250 mm × 4.6 mm, 5 µm particle size column. The mobile phase flow rate was 1.2 mL min−1 and column temperature was 24°C. Buffer composition remained unchanged (A: 150 mmol L−1 phosphate buffer, pH 6.0, containing 150 mmol L−1 potassium chloride; B: 15% acetonitrile in buffer A). The gradient profile was modified to the following content of buffer B in the mobile phase: 0% at 0.00 min, 2% at 0.05 min, 7% at 2.45 min, 50% at 10.00 min, 100% at 11.00 min, 100% at 12.30 min and 0% at 12.40 min. Samples of 10 µL were injected every 18 min into the injection valve loop. Absorbance was read at 254 nm.
Statistical analysis
Data are expressed as mean ± SEM, except the ID50 values (i.e. the dose of allopurinol necessary to reduce the nociceptive response by 50% relative to the control value), which are reported as geometric means accompanied by their respective 95% confidence limits. The ID50 value was determined by linear regression from individual experiments using linear regression GraphPad software (GraphPad software, San Diego, CA, USA). CSF concentrations of purines are expressed as mean ± SEM in µmol·L−1. Data were submitted to Kolmogorov–Smirnov, Levene and Bartlett tests for normality evaluation. Statistical analysis between groups was performed using one-way anova plus the post hoc Student–Newman–Keuls multiple comparisons test when necessary. All results with P < 0.05 were considered statistically significant.
Drugs
Allopurinol, capsaicin, adenosine, caffeine, naloxone and glutamate were purchased from Sigma Chemicals (St Louis, MO, USA). DPCPX (8-cyclopentyl-1,3-dipropylxanthine) was purchased from Tocris (Northpoint, UK). SCH58261 [5-amino-2-(2-furyl)-7-phenylethyl-pyrazolo-(4,3-e)-1,2,4-triazolo(1,5c)pyrimidine] was provided by S. Weiss (Vernalis, UK). Sodium thiopental and morphine sulphate were obtained from Cristália (SP, Brazil). Allopurinol was dissolved in a 10% Tween solution. The dose of Tween (10%) did not cause any detectable effect. Capsaicin was diluted in 5% DMSO (dimethyl sulphoxide); DPCPX and SCH58261 were diluted in 10% DMSO. All other drugs were dissolved in saline (NaCl 0.9%) and buffered with 0.1 N NaOH or 0.1 N HCl to pH 7.4 when necessary. All other chemicals were purchased from local suppliers. Drug and molecular target nomenclature used in this manuscript conforms to the Guide to Receptors and Channels (Alexander et al., 2008).
Results
Figures 1–4 (panel A) show that i.p. administration of allopurinol produced anti-nociception in the tail-flick, hot-plate, i.pl. glutamate and i.pl. capsaicin tests in mice. Mean ID50 values (and their respective 95% confidence limits) in the glutamate and capsaicin tests were 102.5 (61.9–169.8) and 119.1 (54.0–262.7 mg kg−1) respectively, and maximal inhibitions were 63 ± 12% and 58 ± 8% respectively. Vehicle (10% Tween) did not affect nociception as compared with control (sham) animals (data not shown). Morphine (6 mg kg−1– positive control) produced anti-nociception in all models.
Figure 1.
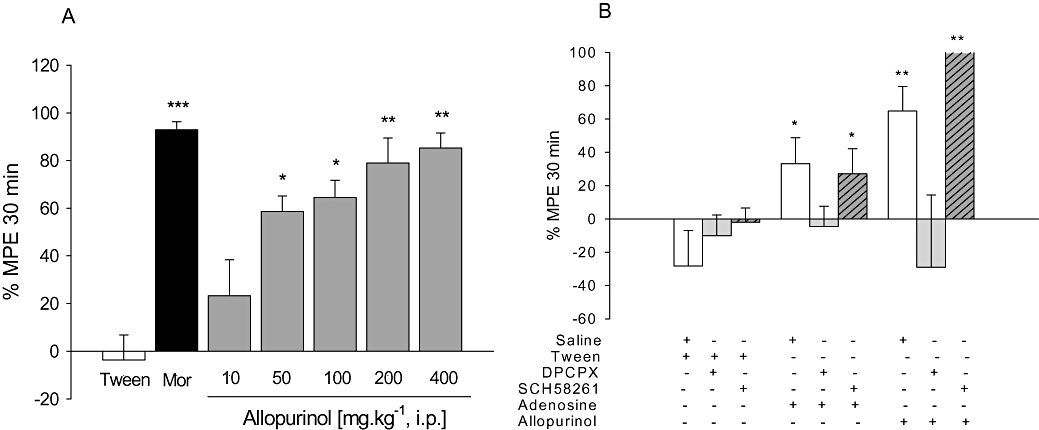
A. Anti-nociceptive effects of allopurinol (10–400 mg kg−1, i.p.) or morphine (6 mg kg−1; Mor) on tail-flick test; mean baseline latencies (s) were: Tween – 6.5 ± 0.4; morphine – 5.4 ± 0.3; allopurinol 10 to 400 mg kg−1– 7.4 ± 0.4, 6.4 ± 0.4, 6.8 ± 0.4, 6.9 ± 0.5 and 6.9 ± 0.5 s respectively. B. Effects of DPCPX (0.1 mg kg−1, i.p.) or SCH58261 (0.5 mg kg−1, i.p.) on anti-nociceptive effects of adenosine (100 mg kg−1, i.p.) or allopurinol (200 mg kg−1, i.p.) on tail-flick test. The columns represent mean values of % of maximum possible effect (% MPE) and vertical bars represent SEM. n = 8–12 animals per group. *P < 0.05, **P < 0.01 and ***P < 0.001 compared with control (10% Tween or saline + Tween), one-way anova followed by Student–Newman–Keuls test.
Figure 4.
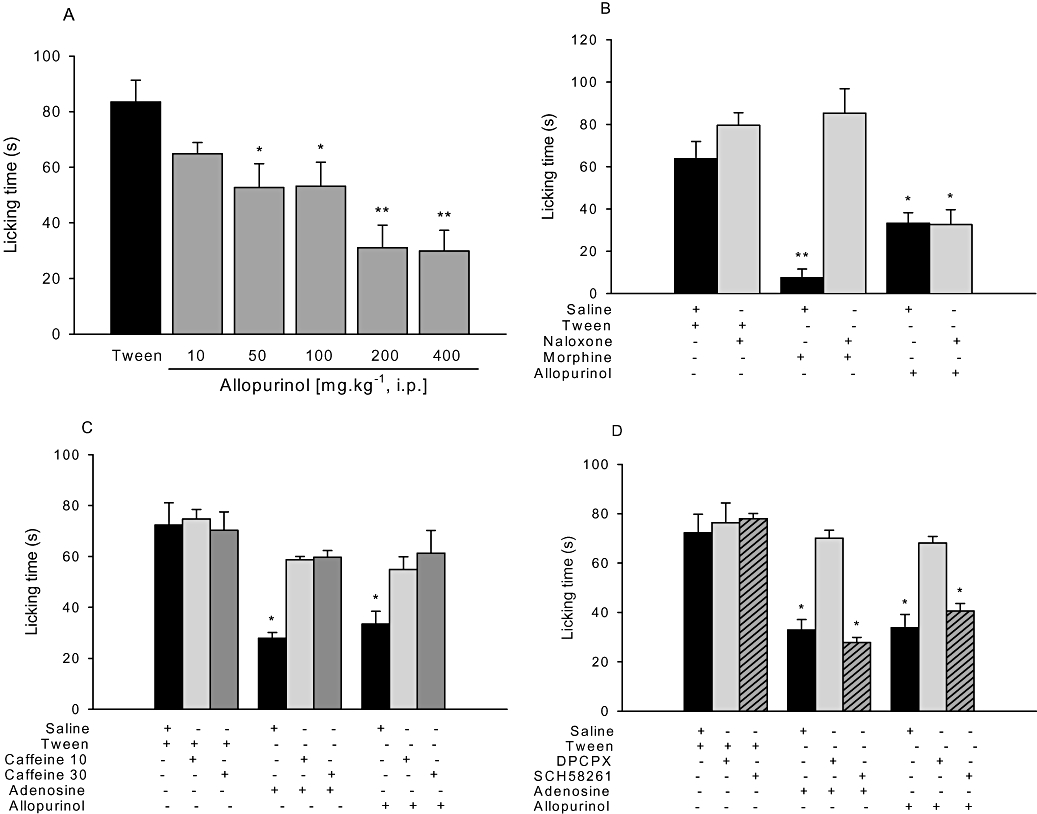
A. Anti-nociceptive effects of allopurinol (10–400 mg kg−1, i.p.) on capsaicin-induced pain. B. Effects of naloxone (1 mg kg−1, i.p.) on the anti-nociceptive effects of morphine (6 mg kg−1, i.p.) or allopurinol (200 mg kg−1, i.p.) on capsaicin-induced pain. C. Effects of caffeine (10 or 30 mg kg−1, i.p.) on adenosine (100 mg kg−1, i.p.) or allopurinol (200 mg kg−1, i.p.) anti-nociception on capsaicin-induced pain. D. Effects of DPCPX (0.1 mg kg−1, i.p.) or SCH58261 (0.5 mg kg−1, i.p.) on anti-nociceptive effects of adenosine (100 mg kg−1, i.p.) or allopurinol (200 mg kg−1, i.p.) on capsaicin-induced pain. The columns represent mean time spent licking the injected hind paw and vertical bars represent SEM. n = 8–12 animals per group. *P < 0.05, **P < 0.01 and ***P < 0.001 compared with control (10% Tween or saline + Tween), one-way anova followed by Student–Newman–Keuls test.
Figure 2.
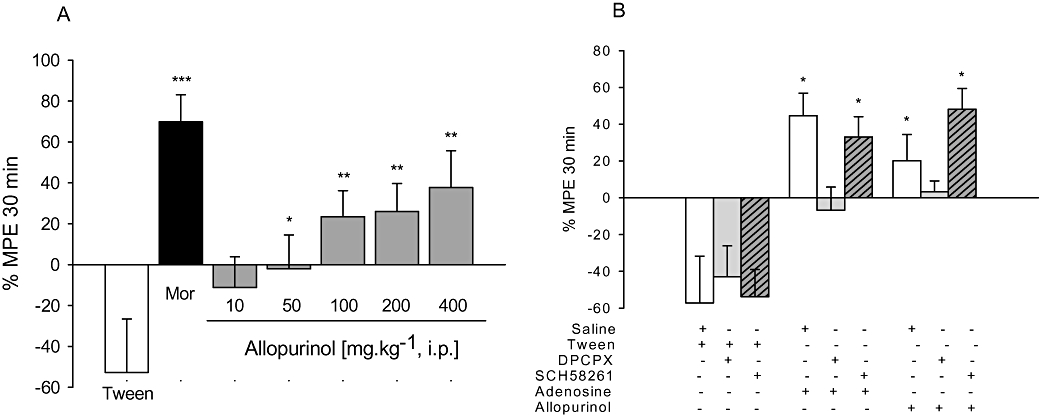
A. Anti-nociceptive effects of allopurinol (10–400 mg kg−1, i.p.) or morphine (6 mg kg−1; Mor) on the hot-plate test; mean baseline latencies (s) were: Tween – 10.4 ± 0.7; morphine – 7.4 ± 0.4; allopurinol 10 to 400 mg kg−1– 8.5 ± 0.4, 9.0 ± 0.7, 10.2 ± 0.6, 8.8 ± 0.7 and 10.2 ± 0.9 s respectively. B. Effects of DPCPX (0.1 mg kg−1, i.p.) or SCH58261 (0.5 mg kg−1, i.p.) on anti-nociceptive effects of adenosine (100 mg kg−1, i.p.) or allopurinol (200 mg kg−1, i.p.) on hot-plate test. The columns represent mean values of % of maximum possible effect (% MPE) and vertical bars represent SEM. n = 8–12 animals per group. *P < 0.05, **P < 0.01 and ***P < 0.001 compared with control (10% Tween or saline + Tween), one-way anova followed by Student–Newman–Keuls test.
Figure 4B shows that the non-selective opioid-receptor antagonist naloxone completely prevented morphine-induced anti-nociception, without affecting anti-nociception induced by allopurinol. As shown in Figure 4C, i.p. adenosine (100 mg kg−1), as well as allopurinol, produced anti-nociceptive effects against capsaicin-induced pain, an effect prevented by pre-treatment with the non-selective adenosine receptor antagonist caffeine (30 mg kg−1). The results depicted in Figures 1–3 (panel B) and Figure 4D show that the selective A1 adenosine receptor antagonist DPCPX (0.1 mg kg−1), but not the selective A2A adenosine receptor antagonist SCH58261 (0.5 mg kg−1), had no effect per se, but prevented anti-nociception induced by allopurinol and adenosine in the tail-flick, hot-plate, i.pl. glutamate and i.pl. capsaicin pain tests.
Figure 3.
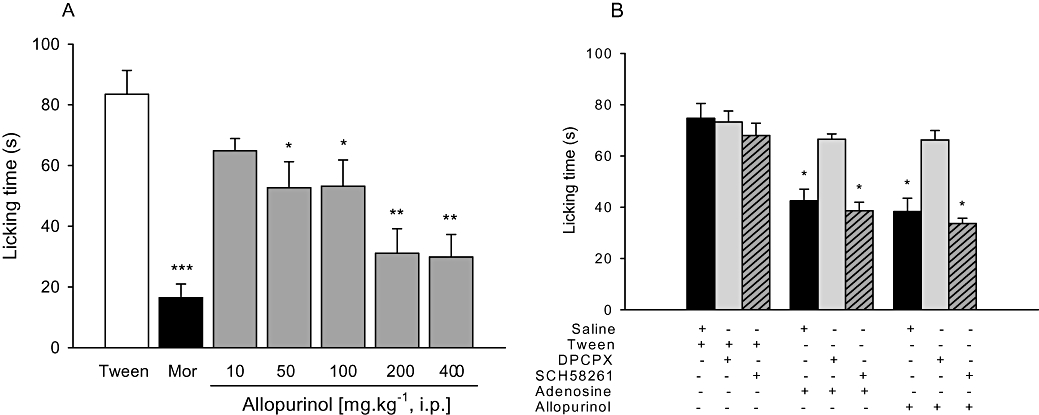
A. Anti-nociceptive effects of allopurinol (10–400 mg kg−1, i.p.) or morphine (6 mg kg−1; Mor) on glutamate-induced pain. B. Effects of DPCPX (0.1 mg kg−1, i.p.) or SCH58261 (0.5 mg kg−1, i.p.) on anti-nociceptive effects of adenosine (100 mg kg−1, i.p.) or allopurinol (200 mg kg−1, i.p.) on glutamate-induced pain. The columns represent mean time spent licking the injected hind paw and vertical bars represent SEM. n = 8–12 animals per group. *P < 0.05, **P < 0.01 and ***P < 0.001 compared with control (10% Tween or saline + Tween), one-way anova followed by Student–Newman–Keuls test.
In the hole-board model (Table 1), neither i.p. allopurinol (10–200 mg kg−1) nor vehicle affected latency to first head-dip, number of head-dips, groomings and defecations. However, allopurinol, at the highest dose (400 mg kg−1), induced a significant decrease in the number of crossings and rearings, compared with vehicle.
Table 1.
Effects of allopurinol on the hole-board, rotarod and spontaneous locomotor activity tests, used in mice
| Treatment |
Allopurinol (mg kg−1) |
|||||
|---|---|---|---|---|---|---|
| Tween 10% | 10 | 50 | 100 | 200 | 400 | |
| Latency to head-dip (s) | 6.0 (0.9) | 6.4 (0.8) | 5.5 (0.9) | 7.5 (0.2) | 5.3 (0.7) | 8.5 (0.7) |
| Head-dips (n) | 50.1 (5.7) | 56.0 (2.2) | 49.7 (8.6) | 43.5 (6.2) | 47.0 (7.5) | 35.0 (6.3) |
| Squares crossed (n) | 42.1 (5.4) | 40.7 (5.9) | 44.0 (7.2) | 47.7 (5.9) | 46.0 (9.4) | 21.7 (9.4)* |
| Rearings (n) | 2.0 (0.8) | 2.3 (0.8) | 2.6 (0.9) | 1.8 (1.0) | 1.7 (0.8) | 0.7 (0.3)* |
| Groomings (n) | 1.6 (0.3) | 1.5 (0.3) | 0.8 (0.4) | 1.2 (0.4) | 1.6 (0.6) | 1.3 (0.4) |
| Fecal boli (n) | 0.8 (0.4) | 1.2 (0.4) | 1.2 (0.5) | 1.0 (0.3) | 1.5 (0.5) | 0.8 (0.5) |
| Latency to fall (s) | 56.4 (1.6) | 50.6 (7.0) | 58.0 (1.5) | 50.8 (6.8) | 56.2 (2.5) | 34.5 (5.8)* |
| Crossings (n) | 234 (28) | 250 (37) | 222 (10) | 213 (15) | 212 (19) | 140 (14)* |
Vehicle (10% Tween) or allopurinol was given i.p., 30 min prior to the behaviour measurements: latency to the first head-dip; head-dips; squares crossed; rearings; groomings; fecal boli; latency to fall (rotarod); number of crossings (spontaneous locomotor activity). n = 8 animals per group.
P < 0.05 compared with control (10% Tween), one-way anova followed by Student–Newman–Keuls test.
Allopurinol (10–200 mg kg−1) did not induce motor deficits, ataxia or affected spontaneous locomotor activity, as evaluated by the performance in the rotarod test and in the activity cages. However, allopurinol (400 mg kg−1) decreased latency to fall in the rotarod test and spontaneous locomotor activity measured by activity cages as shown in Table 1. Allopurinol had no effect on pentobarbital-induced sleeping time (n = 8 for all groups; Tween – 33.5 ± 5.8 min; allopurinol 50 mg kg−1– 33.9 ± 10.4 min; allopurinol 100 mg kg−1– 13.4 ± 8.7 min; and allopurinol 200 mg kg−1– 21.2 ± 8.1 min; P = 0.36) and rectal temperature (n = 8 for all groups; Tween – 35.8 ± 0.2°C; allopurinol 50 mg kg−1– 36.1 ± 0.3°C; allopurinol 100 mg kg−1– 35.5 ± 0.3°C; and allopurinol 200 mg kg−1– 36.1 ± 0.4°C; P = 0.72).
Figure 5 shows that the CSF concentration of uric acid was significantly reduced 30 min after treatment with allopurinol (200 mg kg−1). Conversely, the CSF concentrations of xanthine, hypoxanthine, guanosine, adenosine, inosine and adenosine monophosphate were significantly increased in mice treated with allopurinol compared with mice receiving vehicle. The most significant changes were observed for adenosine (sevenfold increase) and guanosine (14-fold increase). Intraperitoneal administration of allopurinol did not affect CSF levels of ATP, adenosine diphosphate, guanosine triphosphate, guanosine diphosphate, guanosine monophosphate and IMP, compared with vehicle (data not shown). CSF allopurinol concentration was estimated to be 57.7 ± 5.9 µmol·L−1, 30 min after a single i.p. dose of allopurinol (200 mg kg−1). Allopurinol was not detected in the CSF of control animals.
Figure 5.
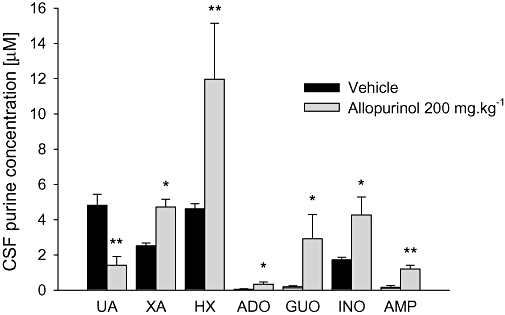
Effects of allopurinol (200 mg kg−1, i.p.) on cerebrospinal fluid (CSF) concentration of purines. The columns represent mean (µmol·L−1) and vertical bars represent SEM. Vehicle was 10% Tween. Vehicle or allopurinol was given i.p. 30 min prior to the CSF sampling. UA, uric acid; XA, xanthine; HX, hypoxanthine; ADO, adenosine; GUO, guanosine; INO, inosine; AMP, adenosine monophosphate. n = 8 animals per group. *P < 0.05 and **P < 0.01 compared with vehicle (control), one-way anova followed by Student–Newman–Keuls test.
Discussion and conclusions
In this study, i.p. administration of the xanthine oxidase inhibitor, allopurinol, produced dose-dependent anti-nociceptive effects in the i.pl. capsaicin, i.pl. glutamate, tail-flick and hot-plate pain models in mice. The opioid antagonist naloxone did not alter the anti-nociceptive effects of allopurinol. However, the non-selective adenosine receptor antagonist caffeine and the selective A1 adenosine receptor antagonist DPCPX, but not the selective A2A adenosine receptor antagonist SCH58261, prevented allopurinol-induced anti-nociception. No obvious motor deficits were produced by allopurinol at doses up to 200 mg kg−1. This study also demonstrated that i.p. administration of allopurinol significantly increased CSF levels of purines, including the nucleosides adenosine and guanosine, and decreased CSF concentration of uric acid.
Although allopurinol has been traditionally used in the treatment of gout and its related symptoms (including pain), only anecdotal reports investigating the effects of allopurinol per se on pain are found in the literature (Pinelli et al., 1991; Daskalopoulou et al., 2005; Hacimuftuoglu et al., 2006; Inkster et al., 2007). Interestingly, the present study demonstrated that allopurinol produced anti-nociception in four different animal pain models. Although these animal models are essentially based on acute, short-lasting noxious stimuli, some differences between tests can be found. Tail-flick and hot-plate tests are thermal models of pain but the tail-flick refers predominantly to a spinal reflex with modest control by supraspinal structures, while the hot-plate test is a more complex pain model, producing two behavioural components (i.e. paw licking and jumping) considered to be supraspinally integrated responses (Le Bars et al., 2001). Intraplantar injection of algogenic chemical agents (capsaicin or glutamate) usually produces similar nociceptive responses and represents a longer-lasting stimulus (tonic pain). However, i.pl. administration of glutamate produces a nociceptive response and paw oedema that are mainly mediated by non-NMDA receptors (Beirith et al., 2002), while the capsaicin test involves a more complex mechanism, predominantly mediated by tachykinin and NMDA receptors (Sakurada et al., 1993).
The rationale to administer allopurinol for pain is derived from evidence in basic and clinical research on the purinergic system. Purines and their analogues have been considered important targets for the development of new drugs for pain management, as the nucleoside adenosine and its analogues present anti-nociceptive effects at spinal, supraspinal and peripheral sites (Sawynok, 1998; Sawynok and Liu, 2003), and P1 and P2 receptors are closely involved in the mechanisms of pain transmission (Sawynok, 1998; Sawynok and Liu, 2003). Adenosine can alter pain transmission by acting on both nociceptive afferent and transmission neurons, and these actions are mediated primarily by adenosine A1 receptors (Sawynok, 1998). Additional effects on inflammatory cells at peripheral sites (Fredholm, 1997) and on glia in the central nervous system (CNS) (Gebicke-Haerter et al., 1996; Ogata and Schubert, 1996) mediated by adenosine A2A, A2B and A3 receptors also occur, and these potentially can produce indirect effects on pain transmission. Endogenous adenosine can be released in the CNS and peripheral tissues, and the regulation of its levels by various pharmacological agents can alter pain processing through activation of adenosine A1 receptors on neurons, and perhaps other receptors on adjacent structures (Sawynok and Liu, 2003). Anti-nociception induced by adenine-derived purines seems to be related to adenosine receptors, probably A1 receptors, as adenosine antagonists such as caffeine and theophylline block their effect and adenosine uptake blockers and adenosine deaminase inhibitors enhance anti-nociception (McGaraughty et al., 2001; Donnelly-Roberts et al., 2008). Therefore, allopurinol, by inhibiting xanthine oxidase and production of uric acid, may produce accumulation of other purines (for example adenosine), which may account for its anti-nociceptive properties.
With regard to the mechanism of action of allopurinol, our findings demonstrated that the activation of a analoxone-sensitive opioid pathway is unlikely to be involved in the anti-nociception caused by allopurinol, as naloxone, under conditions where it fully reversed morphine-induced anti-nociception, had no effect against anti-nociception after allopurinol. However, caffeine and DPCPX, but not SCH58261, prevented allopurinol-induced anti-nociception; these results indicate that A1 adenosine receptors and adenosine are involved in these effects. Importantly, there is no evidence that allopurinol presents any direct agonist or antagonist effect on adenosine receptors (Day et al., 2007).
The basic mechanism of action of allopurinol and its metabolite oxypurinol is inhibition of xanthine oxidase (they bind strongly to the reduced form of xanthine oxidase and inhibit the enzyme). This leads to a decrease in the systemic concentration of uric acid and an increase in the concentration of the precursors, hypoxanthine and xanthine (Day et al., 2007). In addition, hypoxanthine can be converted to inosine, IMP and consequently, to adenosine and guanosine (Day et al., 2007). Thus, the primary effect of both allopurinol and oxypurinol is inhibition of uric acid production, and the overall result is the inhibition of the metabolism of xanthine and hypoxanthine leading to greater salvage of these purines by their conversion to inosine, adenosine and guanosine. These findings, both in CNS and periphery, have been extensively demonstrated after systemic administration of allopurinol in several studies in animals and humans (Kim et al., 1987a, b; Ceballos et al., 1994; Marro et al., 2006). In fact, a significant concentration of allopurinol has been demonstrated in CSF after its systemic administration and a remarkable suppression of CSF uric acid levels has been observed (Kim et al., 1987a; Enrico et al., 1997; Akdemir et al., 2001). Accordingly, in this study, we demonstrated a marked increase in the CSF concentrations of allopurinol (approximately 58 µmol·L−1) and of the nucleosides adenosine and guanosine and their metabolites, 30 min after an i.p. administration of allopurinol (200 mg kg−1). Therefore, allopurinol-induced CSF adenosine accumulation may play a role in the anti-nociceptive action of allopurinol.
Although our findings indicate a role for adenosine in allopurinol-induced anti-nociception, we cannot rule out the influence of other purines. This study also demonstrated a significant increase in the CSF concentration of the nucleoside guanosine. Our group and others (Dobolyi et al., 2000; Oses et al., 2004; 2007; Cunha, 2005) have demonstrated that the nucleosides guanosine and adenosine closely interact in the CNS. More recently, we have proposed a specific guanine-based purinergic system with relevant physiological and pathological implications to the CNS, in addition to the well-characterized adenine-based purinergic system (Schmidt et al., 2007). Of note, we have demonstrated that guanosine, as well as adenosine, may modulate pain transmission (Schmidt et al., 2008). Therefore, it is not possible to exclude that the nucleoside guanosine may also influence allopurinol-induced anti-nociception. Unfortunately, a limitation of our study is that guanosine receptor antagonists are not available, which would more directly assess the role of guanosine in the anti-nociceptive effects of allopurinol.
Allopurinol was developed and has been extensively used as an inhibitor of the enzyme xanthine oxidase (Day et al., 2007). Xanthine oxidase is a highly versatile flavoprotein enzyme that catalyses the oxidative hydroxylation of purine substrates and generates reactive oxygen species (ROS) (Borges et al., 2002). ROS have been proposed to contribute to and/or maintain conditions of chronic pain (Kim et al., 2006). More recently, some data indicated that ROS may also mediate acute pain transmission (Hacimuftuoglu et al., 2006). Notably, there is overwhelming acceptance that xanthine oxidase activity is significantly increased in various pathological states, including some pain states (Khalil and Khodr, 2001). Therefore, the inhibition of this enzymatic pathway may be beneficial for treating pain (Lee et al., 2007).
The administration of allopurinol has been shown to decrease tissue injury following ischaemia/reperfusion in a variety of in vitro and in vivo models (Garcia Garcia et al., 1990; Reilly et al., 1991). Recently, Inkster et al. (2007) showed that allopurinol treatment (50 and 250 mg kg−1) had marked beneficial effects on nerve and vascular function in diabetic rats. That same study also demonstrated that allopurinol (150 mg kg−1) attenuated diabetes-induced tactile allodynia, thermal and mechanical hyperalgesia. These effects may be related to a reduction on xanthine oxidase activity and consequently on aspects of ROS-mediated nerve dysfunction via adverse vascular effects (Inkster et al., 2007).
Central and systemic administration of the nucleoside adenosine has been traditionally related to significant side effects, such as hypotension, sedation and impaired motor function (Sawynok and Liu, 2003). Therefore, if allopurinol induces anti-nociception by increasing levels of adenosine, the reduction in pain scores could be related to these alterations. However, our results show that allopurinol up to 200 mg kg−1 produced no obvious behavioural disturbances (hole-board) and did not alter motor coordination (rotarod) and spontaneous ambulation (activity cages). However, at a higher dose (400 mg kg−1), some CNS side effects were observed (decreased locomotor activity as shown in the hole-board test and activity cages and impaired motor coordination as observed in the rotarod test).
In summary, to the best of our knowledge, this is the first study reporting the anti-nociceptive profile of allopurinol in well-established pain models. Allopurinol-induced anti-nociception may be related to an accumulation of adenosine, and perhaps guanosine, in the CNS. Although it is too early to propose the use of adenine- or guanine-based purines for clinical research, an interesting approach to investigate their role clinically is the investigation of purine derivatives already used in humans, such as allopurinol. Moreover, because allopurinol is a well-known and extensively used compound and seems to be well tolerated, with no obvious CNS toxic effects, except at high doses, this drug may be useful to treat pain syndromes in humans.
Acknowledgments
This research was supported by the FINEP research Grant ‘Rede Instituto Brasileiro de Neurociência (IBN-Net)’ # 01.06.0842-00, CNPq, CAPES, FAPERGS, UFRGS.
Glossary
Abbreviations
- P1
purinergic receptor type 1
- P2
purinergic receptor type 2
- A1
adenosine receptor type 1
- A2A
adenosine receptor type 2A
- A2B
adenosine receptor type 2B
- A3
adenosine receptor type 3
- DMSO
dimethyl sulphoxide
- TFL
tail-flick latency
- MPE
maximum possible effect
- CSF
cerebrospinal fluid
- HPLC
high-performance liquid chromatography
- ADA
adenosine deaminase
- ROS
reactive oxygen species
- DPCPX
8-cyclopentyl-1,3-dipropylxanthine
- SCH58261
5-amino-2-(2-furyl)-7-phenylethyl-pyrazolo-[4,3-e]-1,2,4-triazolo[1,5c]pyrimidine
Conflicts of interest
None.
References
- Akdemir H, Asik Z, Pasaoğlu H, Karaküçük I, Oktem IS, Koç RK. The effect of allopurinol on focal cerebral ischaemia: an experimental study in rabbits. Neurosurg Rev. 2001;24:131–135. doi: 10.1007/pl00012397. [DOI] [PubMed] [Google Scholar]
- Akhondzadeh S, Safarcherati A, Amini H. Beneficial antipsychotic effects of allopurinol as add-on therapy for schizophrenia: a double blind, randomized and placebo controlled trial. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:253–259. doi: 10.1016/j.pnpbp.2004.11.008. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153:S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirith A, Santos ARS, Calixto JB. Mechanisms underlying the nociception and paw oedema caused by injection of glutamate into the mouse paw. Brain Res. 2002;924:219–228. doi: 10.1016/s0006-8993(01)03240-1. [DOI] [PubMed] [Google Scholar]
- Bennett GJ. Update on the neurophysiology of pain transmission and modulation: focus on the NMDA-receptor. J Pain Symptom Manage. 2000;19:S2–S6. doi: 10.1016/s0885-3924(99)00120-7. [DOI] [PubMed] [Google Scholar]
- Bhardwaj A, Northington FJ, Koehler RC, Stiefel T, Hanley DF, Traystman RJ. Adenosine modulates N-methyl-D-aspartate-stimulated hippocampal nitric oxide production in vivo. Stroke. 1995;26:1627–1633. doi: 10.1161/01.str.26.9.1627. [DOI] [PubMed] [Google Scholar]
- Borges F, Fernandes E, Roleira F. Progress towards the discovery of xanthine oxidase inhibitors. Curr Med Chem. 2002;9:195–217. doi: 10.2174/0929867023371229. [DOI] [PubMed] [Google Scholar]
- Brundege JM, Dunwiddie TV. Role of adenosine as a modulator of synaptic activity in the central nervous system. Adv Pharmacol. 1997;39:353–391. doi: 10.1016/s1054-3589(08)60076-9. [DOI] [PubMed] [Google Scholar]
- Brunstein MG, Ghisolfi ES, Ramos FL, Lara DR. A clinical trial of adjuvant allopurinol therapy for moderately refractory schizophrenia. J Clin Psychiatry. 2005;66:213–219. doi: 10.4088/jcp.v66n0209. [DOI] [PubMed] [Google Scholar]
- Calcagnetti DJ, Fleetwood SW, Holtzman SG. Pharmacological profile of the potentiation of opioid analgesia by restraint stress. Pharmacol Biochem Behav. 1990;37:193–199. doi: 10.1016/0091-3057(90)90061-l. [DOI] [PubMed] [Google Scholar]
- Ceballos G, Tuttle JB, Rubio R. Differential distribution of purine metabolizing enzymes between glia and neurons. J Neurochem. 1994;62:1144–1153. doi: 10.1046/j.1471-4159.1994.62031144.x. [DOI] [PubMed] [Google Scholar]
- Creese I, Burt DR, Snyder SH. DA receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science. 1976;192:481–483. doi: 10.1126/science.3854. [DOI] [PubMed] [Google Scholar]
- Cunha RA. Neuroprotection by adenosine in the brain: from A1 receptor activation to A2A receptor blockade. Purinergic Signal. 2005;1:111–134. doi: 10.1007/s11302-005-0649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amour FE, Smith DL. A method for determining loss of pain sensation. J Pharmacol Exp Ther. 1941;72:74–79. [Google Scholar]
- Dall'Igna OP, Fett P, Gomes MW, Souza DO, Cunha RA, Lara DR. Caffeine and adenosine A(2a) receptor antagonists prevent beta-amyloid (25–35)-induced cognitive deficits in mice. Exp Neurol. 2007;203:241–245. doi: 10.1016/j.expneurol.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Das DK, Engelman RM, Clement R, Otani H, Prasad MR, Rao PS. Role of xanthine oxidase inhibitor as free radical scavenger. A novel mechanism of action of allopurinol and oxypurinol in myocardial salvage. Biochem Biophys Res Commun. 1987;148:314–319. doi: 10.1016/0006-291x(87)91112-0. [DOI] [PubMed] [Google Scholar]
- Daskalopoulou SS, Tzovaras V, Mikhailidis DP, Elisaf M. Effect on serum uric acid levels of drugs prescribed for indications other than treating hyperuricaemia. Curr Pharm Des. 2005;11:4161–4175. doi: 10.2174/138161205774913309. [DOI] [PubMed] [Google Scholar]
- Day R, Birkett DJ, Hicks M, Miners JO, Graham GG, Brooks PM. Allopurinol: new uses. Drugs. 1994;48:339–344. doi: 10.2165/00003495-199448030-00002. [DOI] [PubMed] [Google Scholar]
- Day RO, Graham GG, Hicks M, McLachlan AJ, Stocker SL, Williams KM. Clinical pharmacokinetics and pharmacodynamics of allopurinol and oxypurinol. Clin Pharmacokinet. 2007;46:623–644. doi: 10.2165/00003088-200746080-00001. [DOI] [PubMed] [Google Scholar]
- Dobolyi A, Reichart A, Szikra T, Nyitrai G, Kekesi KA, Juhasz G. Sustained depolarisation induces changes in the extracellular concentrations of purine and pyrimidine nucleosides in the rat thalamus. Neurochem Int. 2000;37:71–79. doi: 10.1016/s0197-0186(99)00162-x. [DOI] [PubMed] [Google Scholar]
- Domanski L, Sulikowski T, Safranow K, Pawlik A, Olszewska M, Chlubek D, et al. Effect of trimetazidine on the nucleotide profile in rat kidney with ischemia-reperfusion injury. Eur J Pharm Sci. 2006;27:320–327. doi: 10.1016/j.ejps.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Donnelly-Roberts D, McGaraughty S, Shieh CC, Honore P, Jarvis MF. Painful purinergic receptors. J Pharmacol Exp Ther. 2008;324:409–415. doi: 10.1124/jpet.106.105890. [DOI] [PubMed] [Google Scholar]
- Eddy NB, Leimbach D. Synthetic analgesics. II. Dithienylbutenyl- and dithienylbutylamines. J Pharmacol Exp Ther. 1953;107:385–393. [PubMed] [Google Scholar]
- Enrico P, Esposito G, Mura MA, Migheli R, Serra PA, Desole MS, et al. Effects of allopurinol on striatal dopamine, ascorbate and uric acid during an acute morphine challenge: ex vivo and in vivo studies. Pharmacol Res. 1997;35(6):577–585. doi: 10.1006/phrs.1997.0193. [DOI] [PubMed] [Google Scholar]
- Fredholm BB. Purines and neutrophil leukocytes. Gen Pharmacol. 1997;28:345–350. doi: 10.1016/s0306-3623(96)00169-3. [DOI] [PubMed] [Google Scholar]
- Garcia Garcia J, Martin Rollan C, Refoyo Enrinquez MA, Holgado Madruga M, Marino Hernandez E, Marcias Nuñez JF, et al. Improved survival in intestinal ischemia by allopurinol not related to xanthine-oxidase inhibition. J Surg Res. 1990;48:144–146. doi: 10.1016/0022-4804(90)90206-h. [DOI] [PubMed] [Google Scholar]
- Gebicke-Haerter PJ, Christoffel F, Timmer J, Northoff H, Berger M, Van Calker D. Both adenosine A1- and A2-receptors are required to stimulate microglial proliferation. Neurochem Int. 1996;29:37–42. [PubMed] [Google Scholar]
- Hacimuftuoglu A, Handy CR, Goettl VM, Lin CG, Dane S, Stephens RL., Jr Antioxidants attenuate multiple phases of formalin-induced nociceptive response in mice. Behav Brain Res. 2006;173:211–216. doi: 10.1016/j.bbr.2006.06.030. [DOI] [PubMed] [Google Scholar]
- Inkster ME, Cotter MA, Cameron NE. Treatment with the xanthine oxidase inhibitor, allopurinol, improves nerve and vascular function in diabetic rats. Eur J Pharmacol. 2007;561:63–71. doi: 10.1016/j.ejphar.2006.12.029. [DOI] [PubMed] [Google Scholar]
- Khalil Z, Khodr B. A role for free radicals and nitric oxide in delayed recovery in aged rats with chronic constriction nerve injury. Free Radic Biol Med. 2001;31:430–439. doi: 10.1016/s0891-5849(01)00597-4. [DOI] [PubMed] [Google Scholar]
- Kim HK, Kim JH, Gao X, Zhou J-L, Lee I, Chung K, et al. Analgesic effect of vitamin E is mediated by reducing central sensitization in neuropathic pain. Pain. 2006;122:53–62. doi: 10.1016/j.pain.2006.01.013. [DOI] [PubMed] [Google Scholar]
- Kim P, Yaksh TL, Burnett PC, Blum MR, Sundt TM., Jr Cerebrospinal fluid levels of uric acid in dogs and the effect of allopurinol. Brain Res. 1987a;402:87–92. doi: 10.1016/0006-8993(87)91050-x. [DOI] [PubMed] [Google Scholar]
- Kim P, Yaksh TL, Romero SD, Sundt TM., Jr Production of uric acid in cerebrospinal fluid after subarachnoid hemorrhage in dogs: investigation of the possible role of xanthine oxidase in chronic vasospasm. Neurosurgery. 1987b;21:39–44. doi: 10.1227/00006123-198707000-00008. [DOI] [PubMed] [Google Scholar]
- Lara DR, Belmonte-de-Abreu P, Souza DO. Allopurinol for refractory aggression and self-inflicted behaviour. J Psychopharmacol. 2000;14:81–83. doi: 10.1177/026988110001400112. [DOI] [PubMed] [Google Scholar]
- Lara DR, Brunstein MG, Ghisolfi ES, Lobato MI, Belmonte-de-Abreu P, Souza DO. Allopurinol augmentation for poorly responsive schizophrenia. Int Clin Psychopharmacol. 2001a;16:235–237. doi: 10.1097/00004850-200107000-00008. [DOI] [PubMed] [Google Scholar]
- Lara DR, Schmidt AP, Frizzo ME, Burgos JS, Ramírez G, Souza DO. Effect of orally administered guanosine on seizures and death induced by glutamatergic agents. Brain Res. 2001b;912:176–180. doi: 10.1016/s0006-8993(01)02734-2. [DOI] [PubMed] [Google Scholar]
- Lara DR, Cruz MR, Xavier F, Souza DO, Moriguchi EH. Allopurinol for the treatment of aggressive behaviour in patients with dementia. Int Clin Psychopharmacol. 2003;18:53–55. doi: 10.1097/00004850-200301000-00009. [DOI] [PubMed] [Google Scholar]
- Le Bars D, Gozariu M, Cadden SW. Animal models of nociception. Pharmacol Rev. 2001;53:597–652. [PubMed] [Google Scholar]
- Leal MB, Souza DO, Elisabetsky E. Long-lasting ibogaine protection against NMDA-induced convulsions in mice. Neurochem Res. 2000;25:1083–1087. doi: 10.1023/a:1007665911622. [DOI] [PubMed] [Google Scholar]
- Lee I, Kim HK, Kim JH, Chung K, Chung JM. The role of reactive oxygen species in capsaicin-induced mechanical hyperalgesia and in the activities of dorsal horn neurons. Pain. 2007;133:9–17. doi: 10.1016/j.pain.2007.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaraughty S, Cowart M, Jarvis MF. Recent developments in the discovery of novel adenosine kinase inhibitors: mechanism of action and therapeutic potential. CNS Drug Rev. 2001;7(4):415–432. doi: 10.1111/j.1527-3458.2001.tb00208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado-Vieira R, Lara DR, Souza DO, Kapczinski F. Therapeutic efficacy of allopurinol in mania associated with hyperuricemia. J Clin Psychopharmacol. 2001;21:621–622. doi: 10.1097/00004714-200112000-00017. [DOI] [PubMed] [Google Scholar]
- Marro PJ, Mishra OP, Delivoria-Papadopoulos M. Effect of allopurinol on brain adenosine levels during hypoxia in newborn piglets. Brain Res. 2006;1073–1074:444–450. doi: 10.1016/j.brainres.2005.11.061. [DOI] [PubMed] [Google Scholar]
- Ogata T, Schubert P. Programmed cell death in rat microglia is controlled by extracellular adenosine. Neurosci Lett. 1996;218:91–94. doi: 10.1016/s0304-3940(96)13118-9. [DOI] [PubMed] [Google Scholar]
- Oses JP, Leke R, Portela LV, Lara DR, Schmidt AP, Casali EA, et al. Biochemical brain markers and purinergic parameters in rat CSF after seizure induced by pentylenetetrazol. Brain Res Bull. 2004;64:237–242. doi: 10.1016/j.brainresbull.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Oses JP, Viola GG, de Paula Cognato G, Júnior VH, Hansel G, Böhmer AE, et al. Pentylenetetrazol kindling alters adenine and guanine nucleotide catabolism in rat hippocampal slices and cerebrospinal fluid. Epilepsy Res. 2007;75:104–111. doi: 10.1016/j.eplepsyres.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Pacher P, Nivorozhkin A, Szabó C. Therapeutic effects of xanthine oxidase inhibitors: renaissance half a century after the discovery of allopurinol. Pharmacol Rev. 2006;58:87–114. doi: 10.1124/pr.58.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peana AT, Rubattu P, Piga GG, Fumagalli S, Boatto G, Pippia P, et al. Involvement of adenosine A1 and A2A receptors in (−)-linalool-induced anti-nociception. Life Sci. 2006;78:2471–2474. doi: 10.1016/j.lfs.2005.10.025. [DOI] [PubMed] [Google Scholar]
- Pinelli A, Trivulzio S, Malvezzi L, Zecca L. Potentiation of the analgesic effects of tryptophan by allopurinol in rats. Arzneimittelforschung. 1991;41:809–811. [PubMed] [Google Scholar]
- Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev. 1998;50:413–492. [PubMed] [Google Scholar]
- Reilly PM, Schiller HJ, Bulkley GB. Pharmacologic approach to tissue injury mediated by free radicals and other reactive oxygen metabolites. Am J Surg. 1991;161:488–503. doi: 10.1016/0002-9610(91)91120-8. [DOI] [PubMed] [Google Scholar]
- Rundles RW. The development of allopurinol. Arch Intern Med. 1982;145:89–94. [PubMed] [Google Scholar]
- Sakurada T, Katsumata K, Yogo H, Tan-No K, Sakurada S, Kisara K. Anti-nociception induced by CP 96345, a non-peptide NK-1 receptor antagonist, in the formalin and capsaicin tests. Neurosci Lett. 1993;151:142–145. doi: 10.1016/0304-3940(93)90006-7. [DOI] [PubMed] [Google Scholar]
- Sawynok J. Adenosine receptor activation and nociception. Eur J Pharmacol. 1998;347:1–11. doi: 10.1016/s0014-2999(97)01605-1. [DOI] [PubMed] [Google Scholar]
- Sawynok J, Liu XJ. Adenosine in the spinal cord and periphery: release and regulation of pain. Prog Neurobiol. 2003;69:313–340. doi: 10.1016/s0301-0082(03)00050-9. [DOI] [PubMed] [Google Scholar]
- Schmidt AP, Lara DR, Maraschin JF, Perla AS, Souza DO. Guanosine and GMP prevent seizures induced by quinolinic acid in mice. Brain Res. 2000;864:40–43. doi: 10.1016/s0006-8993(00)02106-5. [DOI] [PubMed] [Google Scholar]
- Schmidt AP, Lara DR, Souza DO. Proposal of a guanine-based purinergic system in the mammalian central nervous system. Pharmacol Ther. 2007;116:401–416. doi: 10.1016/j.pharmthera.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Schmidt AP, Böhmer AE, Leke R, Schallenberger C, Antunes C, Pereira ML, et al. Anti-nociceptive effects of intracerebroventricular administration of guanine-based purines in mice: evidences for the mechanism of action. Brain Res. 2008;1239:50–58. doi: 10.1016/j.brainres.2008.07.091. [DOI] [PubMed] [Google Scholar]
- Sollevi A. Adenosine for pain control. Acta Anaesthesiol Scand Suppl. 1997;110:135–136. doi: 10.1111/j.1399-6576.1997.tb05532.x. [DOI] [PubMed] [Google Scholar]
- Tada H, Morooka K, Arimoto K, Matsuo T. Clinical effects of allopurinol on intractable epilepsy. Epilepsia. 1991;32:279–283. doi: 10.1111/j.1528-1157.1991.tb05256.x. [DOI] [PubMed] [Google Scholar]
- Van Waeg G, Loof L, Groth T, Niklasson F. Allopurinol kinetics in humans as a means to assess liver function: evaluation of allopurinol loading test. Scand J Clin Lab Med. 1988;48:45–57. doi: 10.3109/00365518809085393. [DOI] [PubMed] [Google Scholar]
- Vinadé ER, Schmidt AP, Frizzo MES, Izquierdo I, Elizabetsky E, Souza DO. Chronically administered guanosine is anticonvulsant, amnesic and anxiolytic in mice. Brain Res. 2003;977:97–102. doi: 10.1016/s0006-8993(03)02769-0. [DOI] [PubMed] [Google Scholar]
- Wada Y, Hasegawa H, Nakamura M, Yamaguchi N. Anticonvulsant effect of allopurinol on hippocampal-kindled seizures. Pharmacol Biochem Behav. 1992;42:899–901. doi: 10.1016/0091-3057(92)90046-i. [DOI] [PubMed] [Google Scholar]
- Yamamoto I, Kimura T, Tateoka Y, Watanabe K, Kang Ho I. Hypnotic activity of N3-benzilthymidine on mice. Life Sci. 1987;41:2791–2797. doi: 10.1016/0024-3205(87)90424-3. [DOI] [PubMed] [Google Scholar]
- Zagnoni PG, Bianchi A, Zolo P, Canger R, Cornaggia C, D'Alessandro P, et al. Allopurinol as add-on therapy in refractory epilepsy: a double-blind placebo-controlled randomized study. Epilepsia. 1994;35:107–112. doi: 10.1111/j.1528-1157.1994.tb02919.x. [DOI] [PubMed] [Google Scholar]
- Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]