

Abstract
Background and purpose
The objective of this study was to characterize the effects of the cysteinyl leukotriene receptor antagonist, montelukast (0.1–2 µmol·L−1), on Ca2+-dependent pro-inflammatory activities, cytosolic Ca2+ fluxes and intracellular cAMP in isolated human neutrophils activated with the chemoattractants, N-formyl-L-methionyl-L-leucyl-L-phenylalanine (1 µmol·L−1) and platelet-activating factor (200 nmol·L−1).
Experimental approach
Generation of reactive oxygen species was measured by lucigenin- and luminol-enhanced chemiluminescence, elastase release by a colourimetric assay, leukotriene B4 and cAMP by competitive binding ELISA procedures, and Ca2+ fluxes by fura-2/AM-based spectrofluorimetric and radiometric (45Ca2+) procedures.
Key results
Pre-incubation of neutrophils with montelukast resulted in dose-related inhibition of the generation of reactive oxygen species and leukotriene B4 by chemoattractant-activated neutrophils, as well as release of elastase, all of which were maximal at 2 µmol·L−1 (mean percentages of the control values of 30 ± 1, 12 ± 3 and 21 ± 3 respectively; P < 0.05). From a mechanistic perspective, treatment of chemoattractant-activated neutrophils with montelukast resulted in significant reductions in both post-peak cytosolic Ca2+ concentrations and store-operated Ca2+ influx. These montelukast-mediated alterations in Ca2+ handling by the cells were associated with a significant elevation in basal cAMP levels, which resulted from inhibition of cyclic nucleotide phosphodiesterases.
Conclusions and implications
Montelukast, primarily a cysteinyl leukotriene (CysLT1) receptor antagonist, exhibited previously undocumented, secondary, neutrophil-directed anti-inflammatory properties, which appeared to be cAMP-dependent.
Keywords: Adenosine 3′,5′-cyclic monophosphate; calcium; elastase; leukotriene B4; montelukast; neutrophils; phosphodiesterases; reactive oxygen species
Introduction
Montelukast, a highly selective antagonist of cysteinyl leukotriene (CysLT) receptors, is widely used in the treatment of bronchial asthma, primarily as an adjunct to corticosteroids (Anonymous, 2004; Currie et al., 2005; Diamant and van der Molen, 2005; Riccioni et al., 2007). In this setting, the therapeutic activity of montelukast is achieved through antagonism of CysLT-mediated bronchoconstriction, increased vascular permeability and mucus secretion, following release of these mediators, mainly from monocytes/macrophages, eosinophils, mast cells and basophils, as well as by anti-inflammatory actions targeting type 2 helper CD4+ T-lymphocytes (Peters-Golden and Henderson, 2007). Unlike corticosteroids, montelukast has been reported to modulate airway remodelling in patients with chronic asthma, compatible with an extended spectrum of anti-inflammatory activity (Henderson et al. 2006; Muz et al., 2006).
Montelukast has also been reported to possess therapeutic activity in other diseases such as chronic obstructive pulmonary disease, a disorder that is believed to be of neutrophilic aetiology (Rubinstein et al., 2004; Celik et al., 2005). Although they do not produce CysLTs, neutrophils do possess receptors for LTC4 and LTD4, activation of which triggers relatively modest pro-inflammatory responses in these cells (Lärfars et al., 1999; Zhu et al., 2005). Interference with neutrophil activation by CysLTs released from other cell types, such as monocytes/macrophages, mast cells or eosinophils, may therefore underlie the neutrophil-directed therapeutic efficacy of montelukast. Alternatively, montelukast may possess secondary anti-inflammatory properties that are distinct from conventional antagonism of CysLT receptors. These include interference with activation of the transcription factor, nuclear factor kappa B in immune and inflammatory cells, promotion of sustained production of interleukin-10 in inflamed airways or by inhibition of signalling pathways triggered by P2Y receptors (Mamedova et al., 2005; Wu et al., 2006). However, the contribution of these mechanisms to the possible neutrophil-targeted anti-inflammatory activity of montelukast is unclear.
In the current study, we have investigated the effects of montelukast, at therapeutically relevant concentrations, on the mobilization of stored and extracellular Ca2+ by chemoattractant-activated human neutrophils, as well as on several Ca2+-dependent, pro-inflammatory activities of the cells. Our results demonstrate that montelukast antagonizes the pro-inflammatory activities of neutrophils by a mechanism involving inhibition of cyclic nucleotide phosphodiesterases (PDE) and cAMP-mediated attenuation of Ca2+ influx.
Methods
Preparation of neutrophils
The study was approved by the Faculty of Health Sciences Research Ethics Committee of the University of Pretoria, and prior informed consent was obtained from all blood donors.
These cells were isolated from heparinized venous blood (5 units of preservative-free heparin per ml of blood) from healthy adult volunteers. Neutrophils were separated from mononuclear leucocytes by centrifugation on Histopaque-1077 (Sigma Diagnostics) cushions at 400× g for 25 min at room temperature. The resultant pellets were suspended in phosphate-buffered saline (PBS, 0.15 M, pH 7.4) and sedimented with 3% gelatine to remove most of the erythrocytes. Following centrifugation (280× g at 10°C for 10 min), residual erythrocytes were removed by selective lysis with 0.83% ammonium chloride at 4°C for 10 min. The neutrophils, which were routinely of high purity (>90%) and viability (>95%), determined by flowcytometric procedures, were re-suspended to 1 × 107 ml−1 in PBS and held on ice until used.
Measurement of reactive oxygen species
These were measured using lucigenin (bis-N-methyl-acridinium nitrate)- and luminol (5-amino-2,3-dihydro-1,4-phthalazine dione)-enhanced chemiluminescence (CL) procedures that predominantly detect superoxide and reactive oxygen species (ROS) generated by the myeloperoxidase/H2O2/halide system respectively (Minkenberg and Ferber, 1984). Briefly, neutrophils (106 cells) were pre-incubated for 10 min at 37°C, without and with montelukast (0.1–2 µmol·L−1) in 900 µl of Hanks’ balanced salt solution (HBSS) containing either lucigenin (0.2 mmol·L−1) or luminol (0.1 mmol·L−1), followed by addition of either 100 µl of HBSS (unstimulated control systems) or the chemoattractant, N-formyl-L-methionyl-L-leucyl-L-phenylalanine (FMLP, 1 µmol·L−1) and CL responses recorded using a Lumac Biocounter (Model 2010, Lumac Systems Inc., Titusville, FL, USA). The final volume in each vial was 1 ml, and the results, which are expressed in relative light units (rlu), are the peak values for FMLP-activated systems that were reached 40–50 s after addition of the stimulant.
MK886, an inhibitor of 5-lipoxygenase-activating protein, was used to investigate the possible contribution of LTs generated by neutrophils, as well as by contaminating cells in the neutrophil suspensions, to superoxide generation by FMLP-activated cells, especially the involvement of LTC4 and LTD4. Neutrophils were pre-incubated with MK886 (0.5 µmol·L−1, final) for 5 min at 37°C followed by addition of montelukast (0.5 µmol·L−1) and a further pre-incubation of 5 min followed by addition of FMLP (1 µmol·L−1) and measurement of lucigenin-enhanced CL. Control systems included neutrophils only, as well as cells treated with either MK886 or montelukast only. The efficacy of MK886 as an inhibitor of 5-lipoxygenase in FMLP-activated neutrophils was measured according to the magnitude of inhibition of production of LTB4 by the cells using the method described below.
The superoxide-scavenging potential of montelukast (2 µmol·L−1) was measured using a cell-free xanthine (1 mmol·L−1)/xanthine oxidase (130 mU ml−1) lucigenin-dependent CL procedure.
NADPH oxidase in isolated neutrophil membranes
Neutrophils (1 × 106 ml−1) were pre-incubated for 10 min at 37°C without or with montelukast at a fixed concentration of 2 µmol·L−1, followed by addition of FMLP (1 µmol·L−1). After 3 min of incubation at 37°C, the reactions were terminated by addition of a large volume of ice-cold HBSS and the tubes transferred to an ice bath. The cells were then pelleted by centrifugation at 4°C and the pellets pooled and re-suspended to 5 × 106 ml−1 in 0.34 M sucrose supplemented with 0.5 mmol·L−1 phenylmethylsulphonyl fluoride (PMSF, Calbiochem Corp., La Jolla, CA, USA) and disrupted by sonication. Cellular debris was removed by centrifugation and the membrane fractions in the supernatants were harvested after centrifugation at 70 000× g for 30 min. The membrane pellets were dispersed in 1 ml of sucrose and assayed for NADPH oxidase activity using lucigenin-enhanced CL. Reaction mixtures (1 ml) contained lucigenin, membrane fractions (200 µl) and NADPH (2 mmol·L−1), which was added last to initiate superoxide generation.
Oxygen consumption
This was measured using a three-channel oxygen electrode (Model DW1, Hansatech Ltd, King's Lynn, Norfolk, UK). Neutrophils (2 × 106 ml−1) were pre-incubated for 10 min at 37°C in HBSS without or with montelukast at a fixed concentration of 1 µmol·L−1 followed by addition of FMLP (1 µmol·L−1) and measurement of PO2 over a 5 min time course.
Elastase release
Neutrophil degranulation was measured according to the extent of release of the primary granule enzyme, elastase. Neutrophils were incubated at a concentration of 2 × 106 ml−1 in HBSS with and without montelukast (0.1–2 µmol·L−1) for 10 min at 37°C. FMLP (1 µmol·L−1) in combination with a submaximal concentration of cytochalasin B (0.5 µmol·L−1, final) was then added to the cells that were incubated for 15 min at 37°C. The tubes were then transferred to an ice bath, followed by centrifugation at 400× g for 5 min to pellet the cells. The neutrophil-free supernatants were then decanted and assayed for elastase using a micromodification of a standard colourimetric procedure (Beatty et al., 1982). Briefly, 125 µl of supernatant were added to the elastase substrate, N-succinyl-L-alanyl-L-alanyl-L-alanine-p-nitroanilide [3 mmol·L−1 in dimethylsulphoxide (DMSO)] in 0.05 M Tris-HCl (pH 8.0), and elastase activity was monitored spectrophotometrically at a wavelength of 405 nmol·L−1.
Spectrofluorimetric measurement of cytosolic Ca2+
Fura-2/AM was used as the fluorescent, Ca2+-sensitive indicator for these experiments (Grynkiewicz et al., 1985). Neutrophils (1 × 107 ml−1) were incubated with fura-2/AM (2 µmol·L−1) for 30 min at 37°C in PBS, washed and re-suspended in indicator-free HBSS (pH 7.4), containing 1.25 mmol·L−1 CaCl2. The fura-2-loaded cells (2 × 106 ml−1) were then pre-incubated for 5 min at 37°C with montelukast (0.25–2 µmol·L−1) or an equivalent volume of DMSO in control systems, after which they were transferred to disposable reaction cuvettes, which were maintained at 37°C in a Hitachi 650 10S fluorescence spectrophotometer with excitation and emission wavelengths set at 340 and 500 nm respectively. After a stable baseline was obtained (±1 min), the neutrophils were activated by the addition of the chemoattractants FMLP (1 µmol·L−1, final), or platelet-activating factor (PAF, 200 nmol·L−1, final) and alterations in fluorescence intensity monitored over a 5–10 min time course. Cytosolic calcium concentrations were calculated as described previously (Grynkiewicz et al., 1985).
Radiometric assessment of Ca2+ influx
A radiometric procedure was also used to measure the net influx of 45Ca2+ into FMLP (1 µmol·L−1)- or PAF (200 nmol·L−1)-activated neutrophils uncomplicated by concomitant efflux of the radiolabelled cation. The cells were pre-incubated for 10 min at 37°C in Ca2+-replete (1.25 mmol·L−1) HBSS to ensure that intracellular Ca2+ stores were full to minimize spontaneous uptake of 45Ca2+ (unrelated to activation with FMLP or PAF) in the influx assay. The cells were then pelleted by centrifugation and re-suspended to a concentration of 1 × 107 ml−1 in HBSS containing 25 µmol·L−1 cold, carrier CaCl2. The Ca2+-loaded neutrophils (2 × 106 ml−1) were then incubated for 5 min at 37°C in HBSS containing 25 µmol·L−1 CaCl2 in the absence or presence of montelukast (2 µmol·L−1) followed by simultaneous addition of FMLP or PAF and 2 µCi ml−145Ca2+ (as 45[Ca]Cl2, specific activity 24.3 mCi mg−1, Perkin Elmer Life and Analytical Sciences, Boston, MA, USA), or 45Ca2+ only to control, unstimulated systems. The cells, in a final volume of 5 ml, were then incubated for 5 min at 37°C, after which chemoattractant-activated, store-operated uptake of Ca2+ is complete (Steel and Anderson, 2002), and the reactions stopped by the addition of 10 ml of ice-cold, Ca2+-replete HBSS to the tubes, which were transferred immediately to an ice bath. The cells were then pelleted by centrifugation at 400× g for 5 min followed by washing with 15 ml of ice-cold, Ca2+-replete HBSS and the cell pellets dissolved in 0.5 ml 0.1% Triton X-100/0.1 M NaOH and the radioactivity measured in a liquid scintillation spectrometer. The results are presented as the amount of cell associated radioactivity (pmol 45Ca2+·107 cells−1).
Measurement of LTB4 and cyclic AMP
Competitive binding enzyme immunoassay procedures (Correlate-EIA™, Assay Designs Inc., Ann Arbor, MI, USA) were used to measure LTB4 in the supernatants of neutrophils activated with PAF (200 nmol·L−1), while cAMP was measured in the extracts of unstimulated neutrophils, in the absence and presence of montelukast (0.25–2 µmol·L−1). In the case of LTB4, neutrophils (2 × 106 ml−1, final) in HBSS were pre-incubated for 10 min at 37°C with montelukast after which PAF was added to the cells and the reactions stopped after 3 min incubation at 37°C (predetermined in preliminary time course experiments) by the addition of an equal volume of ice-cold HBSS to the tubes, which were then held in an ice bath prior to pelletting the cells by centrifugation. The cell-free supernatants were then assayed for LTB4 using the enzyme immunoassay procedure. Supernatants from cells activated with PAF were diluted 1:4 prior to assay. These results are expressed as pg 107 cells−1.
In the case of cAMP, neutrophils (2 × 106 ml−1, final) were pre-incubated for 10 min at 37°C followed by the addition of montelukast (0.25–2 µmol·L−1) after which the cells were incubated for a further period of 5 min at 37°C and the reactions were stopped by the addition of an equal volume of ice-cold HBSS to the tubes, which were then held on ice prior to pelletting the cells by centrifugation. Following centrifugation, the supernatants were discarded and cAMP extracted from the cell pellets by addition of 1 ml of 0.1 M HCl for 10–15 min followed by centrifugation to remove cell debris and the supernatants decanted and assayed for cAMP. These results are expressed as pmol cAMP 107 cells−1.
In an additional series of experiments, the cells were exposed to montelukast (2 µmol·L−1) or vehicle (0.05% DMSO) for 5 min at 37°C followed by the addition of salbutamol (β2-adrenoreceptor agonist, 5 µmol·L−1), CGS21680 (adenosine A2A receptor agonist, 1 µmol·L−1) or rolipram (type 4 PDE inhibitor, 0.1 µmol·L−1) for 3–5 min at 37°C after which cAMP was assayed in the cell extracts.
PDE activity
To prepare neutrophil cytosol, the cells (5 × 106 ml−1) in PBS were pelleted by centrifugation, then re-suspended in 0.34 M sucrose and 0.5 mmol·L−1 PMSF. The cells were then disrupted by sonication and cellular debris removed by centrifugation. The sonicates were then fractionated by ultracentrifugation at 70 000× g for 30 min and the supernatants harvested for assessment of PDE activity using a scintillation proximity assay (SPA, Amersham Biosciences, UK). Briefly, assays were performed at 30°C for 10 min in buffer containing 50 mmol·L−1 Tris-HCl (pH 7.5), 8.3 mmol·L−1 MgCl2, 17 mmol·L−1 EGTA and 0.3 mg ml−1 bovine serum albumin. Each assay was performed in a reaction volume of 200 µl containing neutrophil cytosol (20 µl) as a source of PDE and approximately 0.05 µCi [3H]cAMP or [3H]cGMP in the absence and presence of montelukast (0.25–20 µmol·L−1), as well as rolipram (20 µmol·L−1), or the non-specific PDE inhibitor, 3-isobutyl-1-methylxanthine (50 µmol·L−1) in control systems. Reactions were terminated by the addition of 75 µl of PDE SPA beads suspended in 18 mmol·L−1 zinc sulphate and PDE-mediated hydrolysis of [3H]cAMP or [3H]cGMP determined by liquid scintillation spectrometry.
The effects of montelukast on the activity of PDE in a preparation isolated from bovine heart (Sigma Chemical Co.) were also investigated, using the enzyme preparation at a fixed, final concentration of 1 mU ml−1.
In an additional series of experiments, the effects of pre-treatment of neutrophils with montelukast (2 µmol·L−1) on the activities of cAMP PDE in matched, isolated membranes and cytosol fractions prepared from both unstimulated and FMLP (1 µmol·L−1)-activated cells were investigated. Briefly, neutrophils were pre-incubated for 10 min at 37°C in the absence and presence of montelukast, followed by the addition of FMLP (or an equal volume of HBSS to control cells) and termination of reaction 1 min later by addition of ice-cold HBSS. The cells were then pelleted by centrifugation, re-suspended in 0.34 M sucrose/0.5 mmol·L−1 PMSF, sonicated, and membrane and cytosol fractions prepared as described above and assayed for cAMP PDE activity by SPA. For purposes of comparison, the membrane and cytosol fractions were assayed for protein content and the results expressed as enzyme activity min−1 mg protein−1.
Inositol triphosphate (inositol-1,4,5-triphosphate)
Neutrophils at a concentration of 4 × 106 ml−1 were pre-incubated for 5 min at 37°C in HBSS without or with montelukast (2 µmol·L−1) after which the cells were activated with PAF (200 nmol·L−1) in a final volume of 1 ml. The reactions were terminated and the inositol-1,4,5-triphosphate (IP3) extracted by the addition of 1 ml of 20% perchloric acid at 5 and 10 s after the addition of PAF. Following a 20 min incubation on ice, the tubes were centrifuged at 2000× g for 15 min and the supernatants decanted and titrated to pH 7.5 with 5 M KOH followed by centrifugation at 2000× g for 15 min to remove precipitated KClO4. The supernatants were assayed for IP3 using the inositol-1,4,5-triphosphate (3H) radioreceptor assay kit (Perkin Elmer Life and Analytical Sciences), which is a competitive ligand binding assay, and the results expressed as pmol 107 cells−1.
Cellular ATP levels
To determine the effects of montelukast (2 µmol·L−1) on neutrophil viability, intracellular ATP concentrations were measured in cell lysates (1 × 106 cells ml−1) following exposure of the cells to the drug for 15 min at 37°C, using a luciferin/luciferase CL procedure (Holmsen et al., 1972). These results are expressed as nmol ATP 107 cells−1.
Statistical analysis
With the exception of the results of the fura-2 fluorescence experiments, some of which are presented as representative traces, the results of each series of experiments are presented as the mean values ± SEM, either as the absolute values or as mean percentages of the corresponding drug-free control systems where n = the number of different donors used in each series of experiments, with the number of replicates for each drug concentration and drug-free control system for each experiment shown in the figure legends and table footnotes. Levels of statistical significance were determined by comparing the absolute values for each drug-treated system with the corresponding values for the relevant drug-free control systems for each assay using the Friedman repeated measures anova with Dunn's multiple comparisons post-test, or the Wilcoxon matched pairs signed-ranks test where appropriate, while a two-way repeated measures anova with the Bonferroni post-test was used to analyse the data shown in Table 1.
Table 1.
Effects of MK886 and montelukast, separately and in combination, on the lucigenin-enhanced chemiluminescence (LECL) responses of N-formyl-L-methionyl-L-leucyl-L-phenylalanine (FMLP)-activated neutrophils
| System | LECL | P-values (relative light units) |
|---|---|---|
| a.FMLP only | 6401 ± 496 | |
| b.FMLP + MK886 (0.5 µmol·L−1) | 6448 ± 571 | NSa |
| c.FMLP + Montelukast (0.5 µmol·L−1) | 4707 ± 354 | <0.01 to <0.001a |
| d.FMLP + MK886 + Montelukast | 4741 ± 349 | NSb |
The results of four experiments are expressed as the mean peak LECL values ± SEM.
For comparison with the FMLP-activated, drug-free control system.
For comparison between systems c and d.
Chemicals and reagents
Montelukast sodium, 2-[1[[1-[3-[2-(7-chloroquinolin-2-yl)ethenyl]phenyl]-3-2[1-hydroxy-1-methyl-ethyl)phenyl]-propyl]sulphanyl-methyl]-propyl]cyclopropyl]ethanoic acid, was kindly provided by Merck Research Laboratories, Rahway, NJ, USA, and dissolved in DMSO to a stock concentration of 10 mmol·L−1. Unless indicated, all other chemicals and reagents were purchased from Sigma-Aldrich, St Louis, MO, USA. In the various assay systems described below, montelukast was used at final concentrations of 0.1, 0.25, 0.5, 1 and 2 µmol·L−1. Peak serum concentrations of 0.5–1 µmol·L−1 are attainable during oral administration of montelukast (Cheng et al., 1996; Knorr et al., 2001). The final DMSO concentration was 0.05%, and DMSO control systems were included in each assay.
Results
Production of ROS
The effects of montelukast (0.1–2 µmol·L−1) on the FMLP-activated generation of ROS using the lucigenin- and luminol-enhanced CL procedures are shown in Figure 1. Treatment of the cells with montelukast resulted in dose-related inhibition of the generation of ROS, which was evident using both procedures and which achieved statistical significance at concentrations of 0.5 µmol·L−1 (lucigenin, P < 0.001) or 1µmol·L−1 (luminol, P < 0.01). Maximal inhibition was observed at 2 µmol·L−1 montelukast, resulting in 70% and 60% mean inhibition of the generation of ROS by FMLP-activated neutrophils with the lucigenin- and luminol-enhanced CL procedures respectively, the IC50 for the latter being 1.5 µmol·L−1 (confidence intervals 1.1–1.9).
Figure 1.
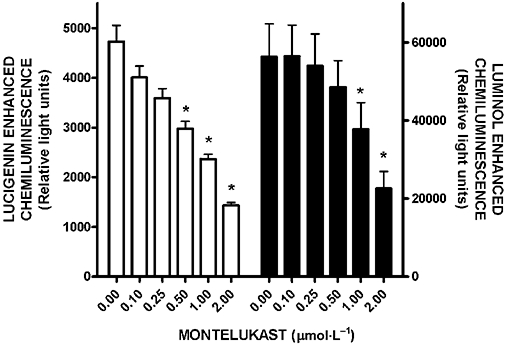
Effects of montelukast (0.1–2 µmol·L−1) on the lucigenin- and luminol-enhanced chemiluminescence responses of neutrophils activated by N-formyl-L-methionyl-L-leucyl-L-phenylalanine (FMLP) (1 µmol·L−1). The results are expressed as the mean peak chemiluminescence values in relative light units measured 30–50 s after the addition of FMLP and vertical lines show SEM. In the case of lucigenin-enhanced chemiluminescence (left graph, n = 3 with three to four replicates for each drug concentration and control system in each experiment), the absolute values for unstimulated neutrophils and for cells activated with FMLP in the absence of montelukast were 1086 ± 147 and 4729 ± 325 respectively, while the corresponding values for luminol-enhanced chemiluminescence (right graph, n = 5 with two to three replicates for each drug concentration and control system in each experiment) were 3047 ± 127 and 56 397 ± 8394 relative light units. *P < 0.01 to P < 0.001 for comparison with the FMLP-activated, montelukast-free control system.
As shown in Table 1, pre-treatment of neutrophils with MK886 (0.5 µmol·L−1) did not affect the generation of superoxide by FMLP-activated neutrophils in either the absence or presence of montelukast (0.5 µmol·L−1). Treatment of neutrophils with MK886 resulted in almost complete inhibition of the FMLP-activated production of LTB4 by the cells, the values for unstimulated cells and for FMLP-activated cells in the absence and presence of MK886 being 59 ± 8, 332 ± 23 and 22 ± 3 pg LTB4 107 cells−1 (n = 5 with a minimum of two replicates for each system).
The activity of NADPH oxidase in isolated membranes prepared from neutrophils activated with FMLP was markedly attenuated by treatment of the cells with montelukast (2 µmol·L−1). The results for membrane fractions prepared from unstimulated neutrophils and those from neutrophils activated with FMLP in the absence and presence of montelukast were 2852 ± 291, 11 543 ± 698 and 6518 ± 407 rlu respectively (n = 5 with two replicates for each system in each experiment; P < 0.05 for comparison of FMLP-activated systems without and with montelukast).
At the maximum concentration of montelukast used in these studies (2 µmol·L−1), the drug did not possess detectable superoxide-scavenging activity, with the lucigenin-enhanced CL values of the xanthine oxidase/hypoxanthine superoxide-generating system in the absence and presence of montelukast being 22 939 ± 4850 and 22 271 ± 5413 rlu respectively (data from three separate experiments with three to four replicates for the control and drug-treated systems).
Activation of neutrophils with FMLP (1 µmol·L−1) resulted in increased oxygen consumption by the cells that was linear over a 1 min period and was significantly attenuated by pre-treatment of the cells with 1 µmol·L−1 montelukast (65 ± 11% of control; n = 6 with one to three replicates for each system; P < 0.05 for comparison of FMLP-activated systems without and with montelukast).
Elastase release
The effects of montelukast on the release of elastase from neutrophils activated with FMLP/cytochalasin B are shown in Figure 2. Treatment of the cells with montelukast resulted in dose-related inhibition of the release of elastase, which achieved statistical significance (P < 0.001) at concentrations of 0.5 µmol·L−1 and greater, with maximal inhibition (79%) observed at 2 µmol·L−1 of this agent. The IC50 value for montelukast-mediated inhibition of elastase release was 1.2 µmol·L−1 (95% confidence intervals 0.9–1.4).
Figure 2.
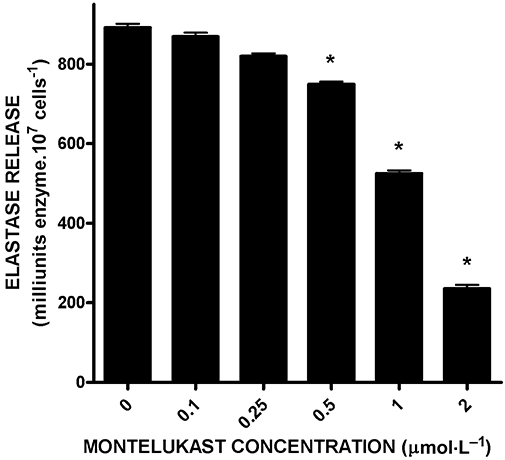
Effects of montelukast (0.1–2 µmol·L−1) on the release of elastase from neutrophils activated with N-formyl-L-methionyl-L-leucyl-L-phenylalanine (1 µmol·L−1)/cytochalasin B (0.5 µmol·L−1). The results (n = 4 with duplicate data sets for each experiment with 10 replicates for each drug concentration and control system in each experiment) are expressed as the mean values for total extracellular elastase (milliunits 107 cells−1) and vertical lines show SEM. The absolute values for the unstimulated control system and for cells activated with N-formyl-L-methionyl-L-leucyl-L-phenylalanine/cytochalasin B in the absence of montelukast were 221 ± 44 and 892 ± 9.6 milliunits elastase 107 cells−1 respectively. *P < 0.001 for comparison with the drug-free control systems.
Leukotriene B4
The effects of montelukast on the production of LTB4 by PAF (200 nmol·L−1)-activated neutrophils are shown in Figure 3. Treatment of neutrophils with this agent resulted in dose-related inhibition of the generation of LTB4, which achieved statistical significance (P < 0.001) at concentrations of 1 µmol·L−1 and greater, with maximal inhibition (89 ± 3%) observed at 2 µmol·L−1 montelukast. The IC50 value for montelukast-mediated inhibition of LTB4 production was 1.2 µmol·L−1 (95% confidence intervals 0.7–1.6).
Figure 3.
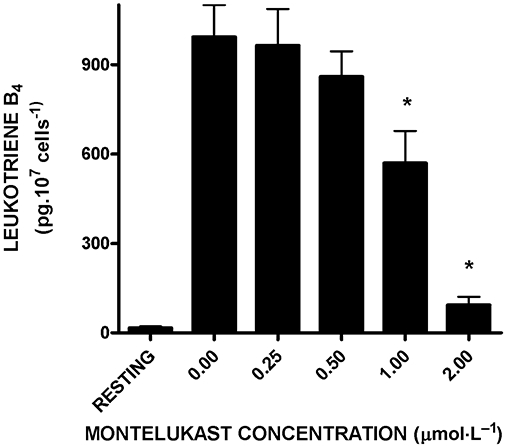
Effects of montelukast (0.25–2 µmol·L−1) on the production of leukotriene B4 (LTB4) by neutrophils activated with platelet-activating factor (200 nmol·L−1). The results are presented as the mean values for total extracellular LTB4 (pg 107 cells−1) and vertical lines show SEM (n = 8, with two to three replicates for each drug concentration and control system in each experiment). The absolute values for the unstimulated control system and for cells activated with platelet-activating factor in the absence of montelukast were 16 ± 6 and 993 ± 107 pg LTB4 107 cells−1 respectively. *P < 0.001 for comparison with the drug-free control system.
Fura-2 fluorescence responses of activated neutrophils
The results shown in Figure 4 are typical traces of the FMLP- and PAF-activated fluorescence responses of neutrophils in the absence and presence of montelukast at 2 µmol·L−1. Addition of FMLP to neutrophils was accompanied by the characteristic, abrupt increase in fura-2 fluorescence intensity, which accompanies increased cytosolic concentrations of Ca2+, rising from a basal value of 83 ± 8 nmol·L−1 to a peak value of 419 ± 60 nmol·L−1. This was followed by a rapid decrease in fluorescence intensity, which slowed after 1–2 min, coincident with influx of Ca2+. Although the peak cytosolic Ca2+ concentrations were equivalent in control and montelukast-treated neutrophils, the rate of decline in fluorescence intensity was faster in montelukast-treated cells. The time taken for fluorescence intensity to decline to half peak values was 1.3 ± 0.1, 0.9 ± 0.1 and 1.0 ± 0.1 min for FMLP-activated cells in the absence (control system) and presence of 1 and 2 µmol·L−1 montelukast respectively (n = 8, P < 0.05 to P < 0.001 for comparison of the control system with each drug-treated system). These observations are compatible with increased efficiency of sequestration/resequestration of cytosolic Ca2+ into stores and/or decreased store-operated influx of the cation.
Figure 4.
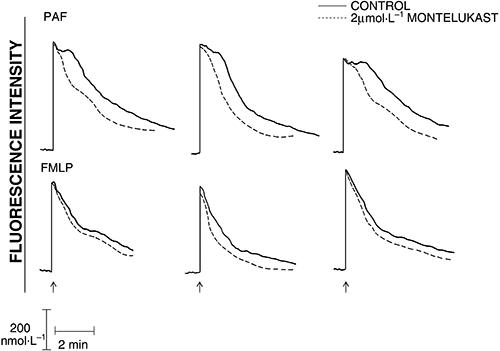
Chemoattractant-activated fura-2 fluorescence responses of control and montelukast (2 µmol·L−1)-treated neutrophils. Platelet-activating factor (PAF) (200 nmol·L−1) or N-formyl-L-methionyl-L-leucyl-L-phenylalanine (FMLP) (1 µmol·L−1) were added as indicated (↑) when a stable baseline was obtained (±1 min). The traces shown are from three different representative experiments (8 for FMLP and 12 for PAF in the series).
In the case of PAF-activated cells, the peak increases in cytosolic Ca2+ were sustained for about 1 min (Fig. 4) as described previously (Steel and Anderson, 2002). Treatment of neutrophils with montelukast (2 µmol·L−1) markedly attenuated the duration of the sustained peak elevation in cytosolic Ca2+, without affecting the magnitude of the peak response. The mean duration of the peak plateau elevation in cytosolic Ca2+ for PAF-activated control cells was 1.13 ± 0.1 min, while the corresponding values for systems treated with 2 µmol·L−1 montelukast was 0.3 ± 0.1 min (n = 12, P < 0.001 for comparison of the control system with each drug-treated system).
Ca2+ influx
The effects of varying concentrations of montelukast (0.25–2 µmol·L−1) on influx of 45Ca2+ following activation of the cells with the chemoattractants are shown in Figure 5. Treatment of the cells with montelukast resulted in a dose-related decrease in the influx of Ca2+ activated by both FMLP and PAF, which was statistically significant at concentrations of 1 and 2 µmol·L−1.
Figure 5.
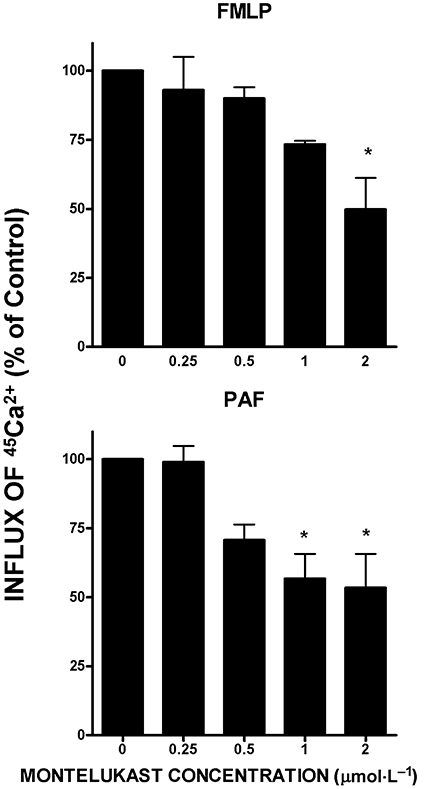
Effects of montelukast (0.25–2 µmol·L−1) on the influx of 45Ca2+ into the neutrophils activated with either N-formyl-L-methionyl-L-leucyl-L-phenylalanine (FMLP) (1 µmol·L−1, upper graph) or platelet-activating factor (PAF) (200 nmol·L−1, lower graph). The results are expressed as the mean percentages of the drug-free control systems and vertical lines show SEM (n = 4–8 with two to four replicates for each drug concentration and control system). The absolute values for uptake of 45Ca2+ by unstimulated neutrophils and for cells activated with FMLP or PAF were 47 ± 25, 150 ± 34 and 148 ± 14 pmol 45Ca2+ 107 cells−1 respectively. *P < 0.05 to P < 0.01 for comparison with the corresponding chemoattractant-activated montelukast-free control systems (according to the repeated measures anova, there were significant effects of montelukast at both 1 and 2 µmol·L−1 for the FMLP system; on post hoc testing significance remained at 2 µmol·L−1).
Cyclic AMP
Exposure of neutrophils to montelukast caused a dose-related increase in intracellular cAMP, which achieved statistical significance (P < 0.05) at 1 µmol·L−1, the values for the control system and systems treated with 0.5, 1 and 2 µmol·L−1 montelukast being 4.8 ± 0.3, 6.2 ± 0.2, 7.7 ± 0.3 and 7.3 ± 0.2 pmol cAMP 107 cells−1 respectively. The effects of montelukast alone or in combination with CGS21680, rolipram or salbutamol are shown in Figure 6. Treatment of neutrophils with montelukast (2 µmol·L−1) in combination with either CGS21680, rolipram or salbutamol resulted in elevations in cAMP, which were significantly (P < 0.05) greater than those observed with the individual agents.
Figure 6.
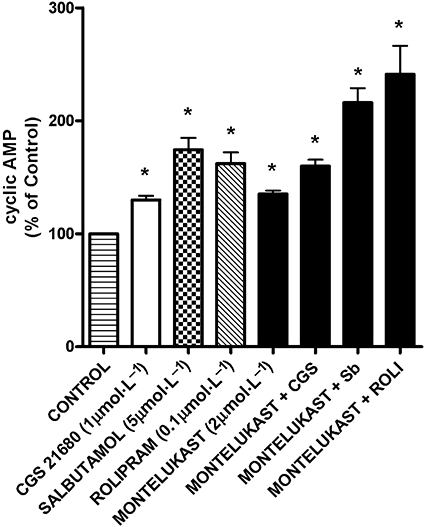
Effects of CGS21680 (CGS; 1 µmol·L−1), salbutamol (Sb; 5 µmol·L−1), rolipram (ROLI; 0.1 µmol·L−1) and montelukast (2 µmol·L−1) individually, as well as those of montelukast in combination with the other agents on neutrophil intracellular cAMP. The results are presented as the mean percentages of the drug-free control system and vertical lines show SEM (n = 6, with two to three replicates for each drug concentration and control system in each experiment). The absolute value for the drug-free control system was 4.3 ± 0.3 pmol cAMP 107 cells−1. *P < 0.05 for comparison with the drug-free control systems.
PDE activity
The effects of montelukast, relative to those of rolipram and 3-isobutyl-1-methylxanthine, on cAMP and cGMP PDE activity, when added directly to neutrophil cytosolic extracts, are shown in Figure 7. Montelukast caused dose-related inhibition of both cAMP and cGMP PDE activity, which achieved statistical significance (P < 0.05) at concentrations of 1 µmol·L−1 and higher. Although not shown, similar effects of montelukast were observed using the PDE preparation from bovine heart. Using neutrophil cytosol as the source of PDE activity, the IC50 value for montelukast-mediated inhibition of cAMP PDE activity was 3.4 µmol·L−1 (95% confidence intervals 2.9–3.9).
Figure 7.
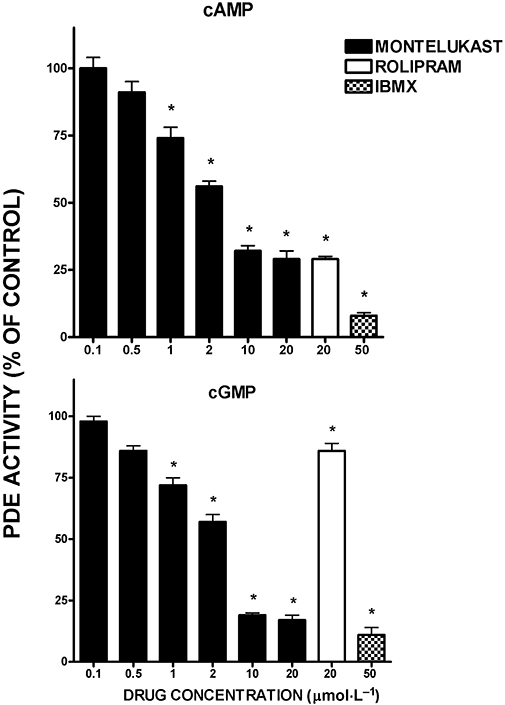
Effects of montelukast (0.5–20 µmol·L−1), rolipram (20 µmol·L−1) and 3-isobutyl-1-methylxanthine (50 µmol·L−1) on cAMP (upper graph) and cGMP (lower graph) phosphodiesterase (PDE) activities in neutrophil cytosol. The results of four to eight and two to four experiments for cAMP and cGMP PDE activity respectively are presented as the mean percentages of the drug-free control systems and vertical lines show SEM. In the case of the cAMP/PDE experiments, the absolute values for the cytosol-free background system and for the cytosol-containing systems in the absence of the drugs were 1213 ± 53 and 14 525 ± 232 counts per minute respectively. The corresponding values for the cGMP/PDE experiments were 2031 ± 206 and 21 381 ± 501 counts per minute. *P < 0.05 for comparison with the corresponding drug-free control system.
The cAMP PDE activities of matched cytosol and membrane fractions prepared from montelukast (2 µmol·L−1)-treated and untreated, unstimulated and FMLP-activated neutrophils are shown in Table 2. Enzyme activity was considerably lower in the membrane fractions, while no redistribution of enzyme activity between the cytosol and membrane compartments was evident following activation of the cells with FMLP. Pre-treatment of the cells with montelukast was accompanied by decreased cAMP PDE activity in the cytosol, and especially the membrane fractions. Given that the cell pellets were diluted approximately 20-fold in sucrose/PMSF following exposure to montelukast ± FMLP, it is likely that the inhibitory effects of montelukast on cAMP PDE were underestimated using this experimental design.
Table 2.
Cyclic AMP phosphodiesterase (PDE) activities in cytosol and membrane fractions prepared from matched control and montelukast-treated unstimulated and N-formyl-L-methionyl-L-leucyl-L-phenylalanine (FMLP)-activated neutrophils
| System | PDE activity (cpm × 10−2 min−1 mg protein−1) | |
|---|---|---|
| Membranes | Cytosol | |
| Control, unstimulated cells | 108 ± 2 | 895 ± 56 |
| Unstimulated cells + 2 µmol·L−1 montelukast | 34 ± 10* | 718 ± 57* |
| FMLP-activated control cells | 60 ± 10 | 938 ± 38 |
| FMLP-activated cells + 2 µmol·L−1 montelukast | 21 ± 6* | 609 ± 34* |
Results are expressed as the mean values ± SEM (n = 4, 2 replicates for each system in each experiment).
P < 0.05 for comparison with the corresponding drug-free control systems.
Inositol triphosphate
The basal IP3 value for unstimulated cells was 45 ± 2 pmol 107 cells−1, increasing to 63 ± 2 pmol 107 cells−1 at 10 s following the addition of PAF (200 nmol·L−1) to control neutrophils (P < 0.05 for comparison with the basal value), while the corresponding value for PAF-activated, montelukast (2 µmol·L−1)-treated neutrophils was 60 ± 3 pmol IP3 107 cells−1, which did not differ significantly from the control system (n = 12, with two to five replicates for each drug concentration and control system in each experiment).
ATP levels
Treatment of neutrophils with montelukast (2 µmol·L−1) did not affect neutrophil ATP levels; the values for control and drug-treated cells following a 15 min exposure at 37°C were 62 ± 2 and 58 ± 3 pmol ATP 107 cells−1 respectively (n = 2, with seven replicates for each system in each experiment).
Discussion and conclusions
Montelukast, a selective antagonist of CysLT1 receptors, is used primarily in the treatment of allergic conditions such as bronchial asthma and allergic rhinitis (Fox-Spencer, 2006; Nayak and Langdon, 2007; Peters-Golden and Henderson, 2007). The reported pA2 value for montelukast antagonism of LTD4-mediated contraction of guinea pig trachea is 9.3 (Jones et al., 1995). Interestingly, beneficial therapeutic effects of this agent have been reported for diverse diseases in which neutrophils play a pathogenetic role, including chronic obstructive pulmonary disease, respiratory bronchiolitis, cystic fibrosis and atherosclerosis (Anonymous, 2004; Rubinstein et al., 2004; Celik et al., 2005; Fitzgerald and Mellis, 2006; Riccioni et al., 2007). The current study was designed to probe potential anti-inflammatory interactions of montelukast with activated human neutrophils in vitro.
Montelukast, at concentrations within the therapeutic range (Cheng et al., 1996; Knorr et al., 2001) and above, caused significant dose-related inhibition of superoxide (lucigenin CL) and hypochlorous acid (luminol CL) generation, as well as production of LTB4 and release of elastase, by activated neutrophils. In the case of superoxide production, the inhibitory effects of montelukast were found to result from interference with the activation of NADPH oxidase. This conclusion is based on observations that montelukast, at concentrations of up to 2 µmol·L−1, did not possess superoxide-scavenging activity, while treatment of the cells with this agent resulted in decreased oxygen consumption following activation with FMLP, as well as markedly reduced activity of NADPH oxidase in membrane fractions prepared from these cells. MK886, an inhibitor of 5-lipoxygenase-activating protein, was used to probe the possible involvement of LTC4 and LTD4 generated by contaminating cells in the neutrophil preparations, in the production of superoxide by these cells. The failure of MK886 to affect the production of superoxide by activated control neutrophils demonstrates that LTC4 and LTD4 were not present at high enough concentrations in the cell suspensions to affect neutrophil NADPH oxidase activity. More importantly, however, the failure of MK886 to attenuate montelukast-mediated inhibition of superoxide production by FMLP-activated neutrophils clearly demonstrates that the observed anti-inflammatory effects of montelukast, in this experimental design, are directed primarily at neutrophils and not at contaminating cells in the cell suspension and, further, the effects were not mediated via antagonism of CysLT1 receptors.
Considering that all the pro-inflammatory activities of neutrophils mentioned above are dependent on elevations in cytosolic Ca2+, we also investigated the effects of montelukast on Ca2+ fluxes in FMLP/PAF-activated neutrophils. Peak cytosolic Ca2+ concentrations in PAF-activated neutrophils were sustained for 60–90 s, followed by a gradual subsidence over a time course of several minutes. The prolonged peak cytosolic Ca2+ response observed in PAF-activated neutrophils results from the failure of this chemoattractant to activate both NADPH oxidase and adenylyl cyclase (Nick et al., 1997; Steel and Anderson, 2002), resulting in early store-operated influx of Ca2+ and failure of cAMP-dependent protein kinase (PKA)-mediated restoration of Ca2+ homeostasis (as described below) respectively. In the case of FMLP-activated cells, NADPH oxidase-mediated membrane depolarization limits influx of Ca2+, while activation of adenylyl cyclase favours rapid clearance of cytosolic Ca2+ (Iannone et al., 1989; Tintinger et al., 2001). Consequently, the peak cytosolic Ca2+ response observed in FMLP-activated neutrophils is of brief duration, declining rapidly for 1–2 min, followed by a levelling-off, coincident with store-dependent influx of Ca2+ (Geiszt et al., 1997; Tintinger et al., 2001).
Treatment of neutrophils with montelukast did not affect the magnitudes of the immediate peak increase in cytosolic Ca2+ in neutrophils activated with either FMLP or PAF. Taken together with the absence of effects of montelukast on IP3 production by PAF-activated neutrophils, these observations demonstrate that neither phospholipase C nor the Ca2+-mobilizing interactions of IP3 with its receptor on intracellular Ca2+ stores are affected by this agent. Treatment of neutrophils with montelukast did, however, significantly attenuate the duration of the prolonged peak cytosolic Ca2+ response of PAF-activated neutrophils, while hastening the rate of decline in cytosolic Ca2+ concentrations in FMLP-activated neutrophils, compatible with decreased store-operated influx of Ca2+ in drug-treated cells. Using procedures that selectively measure the influx of Ca2+ into chemoattractant-activated neutrophils, we observed that montelukast did indeed cause significant, dose-related inhibition of the uptake of Ca2+ by cells activated by both FMLP and PAF, with mean values for inhibition of uptake of 50% and 66%, respectively, for cells treated with 2 µmol·L−1 montelukast. Importantly, Ca2+ influx is necessary to sustain the Ca2+-dependent pro-inflammatory activities of neutrophils (Bréchard and Tschirhart, 2008).
Treatment of neutrophils with montelukast, at the same concentrations that suppressed the Ca2+-dependent pro-inflammatory activities of the cells, was found to cause a significant increase in basal cAMP. Basal cAMP is probably maintained by the autocrine interactions of secreted adenosine with adenosine A2A receptors (Mundell et al., 2001). From a mechanistic perspective, the elevation in intracellular cAMP observed in montelukast-treated neutrophils represented the most likely explanation for the Ca2+ handling-targeted, anti-inflammatory interactions of this agent with activated neutrophils. Interestingly, pre-treatment of neutrophils with montelukast followed by addition of CGS21680, rolipram or salbutamol resulted in elevations in neutrophil cAMP, which were significantly greater than those observed with the individual agents. With respect to CGS21680 and salbutamol, these agents were used at concentrations likely to cause saturation of adenosine A2A and β2-adrenoceptors respectively, compatible with lack of agonist interactions of montelukast with either of these G protein/adenylyl cyclase-coupled receptor types. This observation, taken together with the interactive effects of montelukast and rolipram on raising basal cAMP in neutrophils, as well as the findings of a limited series of experiments that revealed an increase in basal cGMP in montelukast-treated cells (data not included), suggested that the drug possessed non-specific PDE inhibitory activity.
The effects of montelukast on cAMP and cGMP PDE activity were measured using cytosolic fractions from isolated neutrophils, as well as a PDE preparation from bovine heart. Addition of montelukast to either of these resulted in striking, dose-related inhibition of the activities of both cAMP and cGMP PDEs in neutrophil cytosol with an IC50 value of 3.4 µmol·L−1 for the former. The concentrations of montelukast that were found to possess non-specific PDE inhibitory activity therefore closely paralleled those that elevated cAMP and inhibited the Ca2+-dependent pro-inflammatory activities of neutrophils, compatible with a causal association between these events. Although the IC50 values for montelukast-mediated inhibition of neutrophil PDEs are somewhat higher than those for inhibition of superoxide and LTB4 production and elastase release (1, 1.2 and 1.3 µmol·L−1 respectively), this difference may be due to intracellular accumulation of lipophilic montelukast by intact cells, as the drug has an oil : water partition coefficient of LogKD = 2.3 ± 0.2 (data on file, Merck Research Laboratories).
We also attempted to measure the effects of addition of montelukast to intact neutrophils on cAMP PDE activity in cytosol and membrane fractions prepared from unstimulated and FMLP-activated cells. Activation of neutrophils with FMLP did not result in either increased activity of cytosolic cAMP PDE, or redistribution of enzyme to the membrane, with activity in the membrane fraction being low relative to the cytosol. Treatment of intact neutrophils with montelukast resulted in decreased cAMP PDE activity in the cytosolic and especially the membrane fractions of unstimulated and FMLP-activated cells. In the case of the cytosol, however, this was of lesser magnitude than that observed following direct addition of the drug to the cytosol, due, presumably, to loss and dilution of the drug during cell processing.
As a consequence of activation of PKA, cAMP promotes restoration of Ca2+ homeostasis in neutrophils and other cell types by multiple mechanisms, including phosphorylative inactivation of PLCγ (Ali et al., 1998); inactivation of IP3-receptors (Bai and Sanderson, 2006); phosphorylative up-regulation of the Ca2+ sequestering/resequestering endomembrane Ca2+-ATPase (Anderson et al., 1998; Anderson et al., 2000); inactivation of store-operated Ca2+ channels (Binnaz et al., 2006); and inhibition of p38 MAP kinase and consequent interference with the activation of 5-lipoxygenase (Flamand et al., 2002), thereby attenuating an autocrine, LTB4-mediated secondary wave of Ca2+ uptake by the cells (Steel et al., 2007). While the first of these mechanisms does not appear to contribute significantly to the effects of montelukast on Ca2+ handling by activated neutrophils observed in the current study, all the other mechanisms may be operative. Moreover, cross-activation of PKA and PKG by cAMP and cGMP may also contribute to restoration of Ca2+ homeostasis as PKG has also been reported to restrict store-operated uptake of Ca2+ (Ruiz-Velasco et al., 1998).
Although PDE4 subtype B2 appears to be the predominant PDE in human neutrophils (Wang et al., 1999), it is noteworthy that cilostazol, a PDE3 inhibitor, has been reported to attenuate Ca2+ fluxes in activated human neutrophils, as well as superoxide generation (Yang et al., 2006), demonstrating, albeit indirectly, the presence of PDE3 in these cells. Using selective inhibitors of PDE3 (cilostamide) and PDE5 (MY5445), we have observed that neutrophils exhibit appreciable activities of both of these PDEs in addition to PDE4 (R. Anderson et al., unpubl. obs.). Given the ability of PDE3 to hydrolyze cAMP, as well as cross-activation of PKAs A and G by cAMP and cGMP, non-specific PDE inhibitors that target PDEs 3, 4 and 5 in neutrophils may be more effective anti-inflammatory agents than those that selectively target PDE4, by preventing compensatory, counteracting increases in the activities of PDEs 3 and 5. The apparent benefits of a combination of PDE3 and PDE4 inhibitors, as opposed to either category of inhibitor alone, have been already described in an animal model of allergen-induced bronchospasm (Underwood et al., 1994).
Although the non-specific PDE inhibitory effects of montelukast described here have not, to our knowledge, been reported previously, several of the early, experimental CysLT1 receptor antagonists such as FPL55712 and LY171883 were documented to possess this property (Fleisch et al., 1984; Hay et al., 1987). More recently, CR3465, a novel CysLT1 receptor antagonist, was reported to possess PDE inhibitory activity (Ferrari et al., 2004). In the case of FPL55712 and LY171883, PDE inhibitory activity appeared to represent a limitation in respect of specificity of pharmacological mode of action and clinical development (Fleisch et al., 1984; Hay et al., 1987), whereas for CR3465 the combination of CysLT1 receptor antagonism and PDE inhibitory activity was considered to be beneficial, because the latter property conferred additional protection by targeting spasmogenic and inflammatory mediators other than CysLTs (Ferrari et al., 2004).
It is noteworthy that PKA also possesses anti-inflammatory activities that are distinct from its effects on Ca2+ handling by activated immune and inflammatory cells. These include interference with the activation of NADPH oxidase, and inhibition of p38 MAP kinase (as mentioned above) and phosphatidylinositol 3-kinase (Bengis-Garber and Gruener, 1996; Flamand et al., 2002; Burelout et al., 2007). In addition, montelukast has also been reported to inhibit human recombinant 5-lipoxygenase with a relatively high IC50 of 30–50 µmol·L−1, while synthesis of LTB4 by activated neutrophils was inhibited at drug concentrations of >1 µmol·L−1 (Ramires et al.,2004). However, effects on Ca2+ fluxes and cAMP, which may explain the greater sensitivity of intact cells to the inhibitory effects of montelukast on LTB4 production, were not investigated in this study. Nevertheless, inhibition of 5-lipoxygenase, either directly or indirectly by the mechanisms described in the current study, together with reported inhibition of signalling via P2Y receptors (Mamedova et al., 2005), suggests that montelukast may be particularly effective in attenuating both the generation and action of autocrine inflammatory mediators.
Antagonism of CysLT1 receptors is clearly the primary mechanism of therapeutic activity of montelukast. However, the PDE-targeted anti-inflammatory activity of this agent described in the current study may contribute to the beneficial effects of this agent, used in addition to inhaled corticosteroids, in some categories of patients with bronchial asthma (Laviolette et al., 1999), possibly by enabling control of the corticosteroid-resistant neutrophil (Barnes, 2007), as well as by countering bronchospasm via direct cyclic nucleotide-mediated effects on airway smooth muscle (Binnaz et al., 2006).
Acknowledgments
This study was supported in part by a research grant awarded to the University of Pretoria by MSD (Pty) Ltd, Johannesburg, South Africa.
Glossary
Abbreviations
- CB
cytochalasin B
- CL
chemiluminescence
- CysLT
cysteinyl leukotriene
- DMSO
dimethylsulphoxide
- FLAP
5-lipoxygenase activating protein
- FMLP
N-formyl-L-methionyl-L-leucyl-L-phenylalanine
- HBSS
Hanks’ balanced salt solution
- IBMX
3-isobutyl-1-methylxanthine
- IP3
inositol-1,4,5-triphosphate
- PAF
platelet-activating factor
- PDE
phosphodiesterase
- ROS
reactive oxygen species
Conflicts of interest
CF acts on the Speakers’ Bureau and Advisory Board of MSD (Pty) Ltd, Johannesburg, South Africa and has received congress travel support from MSD. CG acts on the Speakers’ Bureau of MSD (Pty) Ltd, Johannesburg, South Africa.
References
- Ali H, Sozzani S, Fisher I, Barr AJ, Richardson RM, Haribabu B, et al. Differential regulation of formyl peptide and platelet-activating factor receptors: role of phospholipase Cβ3 phosphorylation by protein kinase A. J Biol Chem. 1998;273:11012–11016. doi: 10.1074/jbc.273.18.11012. [DOI] [PubMed] [Google Scholar]
- Anderson R, Goolam Mahomed A, Theron AJ, Ramafi G, Feldman C. Effects of rolipram and dibutyryl cyclic AMP on resequestration of cytosolic calcium in FMLP-activated human neutrophils. Br J Pharmacol. 1998;124:547–555. doi: 10.1038/sj.bjp.0701849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson R, Visser SS, Ramafi G, Theron AJ. Accelerated resequestration of cytosolic calcium and suppression of the pro-inflammatory activities of human neutrophils by CGS 21680. Br J Pharmacol. 2000;130:717–724. doi: 10.1038/sj.bjp.0703344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anonymous. Drug Review. Asthma. Montelukast, drugs in context. Part E. Respir Med Infect. 2004;1:1–40. [Google Scholar]
- Bai Y, Sanderson MJ. Airway smooth muscle relaxation results from a reduction in the frequency of Ca2+ oscillations induced by a cAMP-mediated inhibition of the IP3 receptor. Respir Res. 2006;7:34. doi: 10.1186/1465-9921-7-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ. New molecular targets for the treatment of neutrophilic diseases. J Allergy Clin Immunol. 2007;119:1055–1062. doi: 10.1016/j.jaci.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Beatty K, Robertie P, Senior RM, Travis J. Determination of oxidized alpha-1-proteinase inhibitor in serum. J Lab Clin Med. 1982;100:186–192. [PubMed] [Google Scholar]
- Bengis-Garber C, Gruener N. Protein kinase A downregulates the phosphorylation of p47 phox in human neutrophils: a possible pathway for inhibition of the respiratory burst. Cell Signal. 1996;8:291–296. doi: 10.1016/0898-6568(96)00052-6. [DOI] [PubMed] [Google Scholar]
- Binnaz A, Iyanoye A, Sieck G, Prakash YS, Pabelick CM. Cyclic nucleotide regulation of store-operated Ca2+ influx in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2006;290:L278–L283. doi: 10.1152/ajplung.00188.2005. [DOI] [PubMed] [Google Scholar]
- Bréchard S, Tschirhart EJ. Regulation of superoxide production in neutrophils: role of calcium influx. J Leuk Biol. 2008;84:1–15. doi: 10.1189/jlb.0807553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burelout C, Thibault N, Harbour D, Naccache PH, Bourgoin SG. The PGE2-induced inhibition of the PLD activation pathway stimulated by fMLP in human neutrophils is mediated by PKA at the PI3-Kgamma level. Biochem Pharmacol. 2007;74:730–741. doi: 10.1016/j.bcp.2007.06.013. [DOI] [PubMed] [Google Scholar]
- Celik P, Sakar A, Havlucu Y, Yuksel H, Turkdogan P, Yorganioglu A. Short-term effects of montelukast in stable patients with moderate to severe COPD. Respir Med. 2005;99:444–450. doi: 10.1016/j.rmed.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Cheng H, Leff JA, Amin R, Gertz BJ, De Smet M, Noonan N, et al. Pharmacokinetics, bioavailability, and safety of montelukast sodium (MK-0476) in healthy males and females. Pharm Res. 1996;13:445–448. doi: 10.1023/a:1016056912698. [DOI] [PubMed] [Google Scholar]
- Currie GP, Srivastava P, Dempsey OJ, Lee DKC. Therapeutic modulation of allergic airways disease with leukotriene receptor antagonists. Q J Med. 2005;98:171–182. doi: 10.1093/qjmed/hci024. [DOI] [PubMed] [Google Scholar]
- Diamant Z, van der Molen T. Treating asthma: is there a place for leukotriene receptor antagonists? Respir Med. 2005;99:655–662. doi: 10.1016/j.rmed.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Ferrari F, Mennuni L, Caselli G, Zanelli T, Makovec F. Pharmacological profile of CR3465, a new leukotriene CysLT1 receptor antagonist with broad anti-inflammatory activity. Eur J Pharmacol. 2004;504:223–233. doi: 10.1016/j.ejphar.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Fitzgerald DA, Mellis CM. Leukotriene receptor antagonists in virus-induced wheezing: evidence to date. Treat Respir Med. 2006;5:407–417. doi: 10.2165/00151829-200605060-00006. [DOI] [PubMed] [Google Scholar]
- Flamand N, Surette ME, Picard S, Bourgoin S, Borgeat P. Cyclic AMP-mediated inhibition of 5-lipoxygenase translocation and leukotriene biosynthesis in human neutrophils. Mol Pharmacol. 2002;62:250–256. doi: 10.1124/mol.62.2.250. [DOI] [PubMed] [Google Scholar]
- Fleisch JH, Rinkema LE, Marshall WS. Pharmacologic receptors for the leukotrienes. Biochem Pharmacol. 1984;33:3919–3922. doi: 10.1016/0006-2952(84)90001-7. [DOI] [PubMed] [Google Scholar]
- Fox-Spencer R. Drug review. Allergic rhinitis and asthma. Montelukast. Drugs Context. 2006;2:413–456. [Google Scholar]
- Geiszt M, Kapus A, Nemet K, Farkas L, Ligeti F. Regulation of capacitative calcium influx in neutrophil granulocytes: alterations in chronic granulomatous disease. J Biol Chem. 1997;272:26471–26478. doi: 10.1074/jbc.272.42.26471. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescent properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hay DWP, Muccitelli RM, Tucker SS, Vickery-Clark LM, Wilson KA, Gleason JG, et al. Pharmacologic profile of SK&F 104353: a novel potent and selective peptidoleukotriene receptor antagonist in guinea pig and human airways. J Pharmacol Exp Ther. 1987;243:474–481. [PubMed] [Google Scholar]
- Henderson WR, Jr, Chiang GK, Tien YT, Chi EY. Reversal of allergen-induced airway remodeling by CysLT1 receptor blockade. Am J Respir Crit Care Med. 2006;173:718–728. doi: 10.1164/rccm.200501-088OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmsen H, Storm E, Day HJ. Determination of ATP and ADP in blood platelets: a modification of the firefly luciferase assay for plasma. Anal Biochem. 1972;46:489–501. doi: 10.1016/0003-2697(72)90323-5. [DOI] [PubMed] [Google Scholar]
- Iannone MA, Wolberg G, Zimmerman TP. Chemotactic peptide induces cAMP elevation in human neutrophils by amplification of the adenylate cyclase response to endogenously produced adenosine. J Biol Chem. 1989;264:20177–20180. [PubMed] [Google Scholar]
- Jones TR, Labelle M, Belley M, Champion E, Charette L, Ford-Hutchinson AW, et al. Pharmacology of montelukast sodium (Singulair), a potent and selective leukotriene D4 receptor antagonist. Can J Physiol Pharmacol. 1995;73:191–201. doi: 10.1139/y95-028. [DOI] [PubMed] [Google Scholar]
- Knorr B, Holland S, Schwartz J, Douglas Rogers J, Reiss TF. Clinical pharmacology of montelukast. Clin Exp Allergy Rev. 2001;1:254–260. [Google Scholar]
- Lärfars G, Lantoine F, Devynck MA, Palmblad J, Gyllenhammar H. Activation of nitric oxide release and oxidative metabolism by leukotrienes B4, C4 and D4 in human polymorphonuclear leukocytes. Blood. 1999;93:1399–1405. [PubMed] [Google Scholar]
- Laviolette M, Malmstrom K, Lu S, Chervinsky P, Pujet JC, Peszek I, et al. Montelukast added to inhaled beclamethasone in treatment of asthma. Am J Respir Crit Care Med. 1999;160:1862–1868. doi: 10.1164/ajrccm.160.6.9803042. [DOI] [PubMed] [Google Scholar]
- Mamedova L, Capra V, Accomazzo MR, Gao ZG, Ferrario S, Fumagalli M, et al. CysLT1 leukotriene receptor antagonists inhibit the effects of nucleotides acting at P2Y receptors. Biochem Pharmaol. 2005;71:115–125. doi: 10.1016/j.bcp.2005.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkenberg I, Ferber E. Lucigenin-dependent chemiluminescence as a new assay for NADPH-oxidase activity in particulate fractions of human polymorphonuclear leukocytes. J Immunol Methods. 1984;71:61–67. doi: 10.1016/0022-1759(84)90206-0. [DOI] [PubMed] [Google Scholar]
- Mundell SJ, Olah ME, Panettieri RA, Benovic JL, Penn RB. Regulation of G-protein-coupled receptor-adenyl cyclase responsiveness in human airway smooth muscle by exogenous and autocrine adenosine. Am J Respir Cell Mol Biol. 2001;24:155–163. doi: 10.1165/ajrcmb.24.2.4243. [DOI] [PubMed] [Google Scholar]
- Muz MH, Deveci F, Bulut Y, Illhan N, Yekeler H, Turgut T. The effects of low dose leukotriene receptor antagonist therapy on airway remodeling and cysteinyl leukotriene expression in a mouse asthma model. Exp Mol Med. 2006;38:109–118. doi: 10.1038/emm.2006.14. [DOI] [PubMed] [Google Scholar]
- Nayak A, Langdon RB. Montelukast in the treatment of allergic rhinitis. An evidence-based review. Drugs. 2007;67:887–901. doi: 10.2165/00003495-200767060-00005. [DOI] [PubMed] [Google Scholar]
- Nick JA, Avdi NJ, Young SK, Knall C, Gerwins P, Johnson GL, et al. Common and distinct intracellular signaling pathways in human neutrophils utilized by platelet activating factor and FMLP. J Clin Invest. 1997;99:975–986. doi: 10.1172/JCI119263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters-Golden M, Henderson WR. Leukotrienes. N Engl J Med. 2007;357:1841–1854. doi: 10.1056/NEJMra071371. [DOI] [PubMed] [Google Scholar]
- Ramires R, Caiaffa ME, Tursi A, Haeggström JZ, Macchia L. Novel inhibitory effects on 5-lipoxygenase activity by the anti-asthma drug montelukast. Biochem Biophys Res Commun. 2004;324:815–821. doi: 10.1016/j.bbrc.2004.09.125. [DOI] [PubMed] [Google Scholar]
- Riccioni G, Bucciarelli T, Mancini B, Di Ilio C, D'Orazio N. Antileukotriene drugs: clinical application, effectiveness and safety. Curr Med Chem. 2007;14:1966–1977. doi: 10.2174/092986707781368522. [DOI] [PubMed] [Google Scholar]
- Rubinstein I, Kumar B, Schriever C. Long-term montelukast therapy in moderate to severe COPD- a preliminary observation. Respir Med. 2004;98:134–138. doi: 10.1016/j.rmed.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Ruiz-Velasco V, Zhong J, Hume JR, Keef KD. Modulation of Ca2+ channels by cyclic nucleotide cross activation of opposing protein kinases in rabbit portal vein. Circ Res. 1998;82:557–565. doi: 10.1161/01.res.82.5.557. [DOI] [PubMed] [Google Scholar]
- Steel HC, Anderson R. Dissociation of the PAF-receptor from NADPH oxidase and adenylate cyclase in human neutrophils results in accelerated influx and delayed clearance of cytosolic calcium. Br J Pharmacol. 2002;136:81–89. doi: 10.1038/sj.bjp.0704685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steel HC, Tintinger GR, Theron AJ, Anderson R. Itraconazole-mediated inhibition of calcium entry into platelet-activating factor-stimulated human neutrophils is due to interference with production of leukotriene B4. Clin Exp Immunol. 2007;150:144–150. doi: 10.1111/j.1365-2249.2007.03470.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tintinger GR, Theron AJ, Steel HC, Anderson R. Accelerated calcium influx and hyperactivation of neutrophils in chronic granulomatous disease. Clin Exp Immunol. 2001;123:254–263. doi: 10.1046/j.1365-2249.2001.01447.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underwood DC, Kotzer CJ, Bochnowicz S, Osborn RR, Luttman MA, Hay DW, et al. Comparison of phosphodiesterase III, IV and dual III/IV inhibitors on bronchospasm and pulmonary eosinophils influx in guinea pigs. J Pharmacol Exp Ther. 1994;270:250–259. [PubMed] [Google Scholar]
- Wang P, Wu P, Ohleth KM, Egan RW, Billah MM. Phosphodiesterase 4B2 is the predominant phosphodiesterase species and undergoes differential regulation of gene expression in human monocytes and neutrophils. Mol Pharmacol. 1999;56:170–174. doi: 10.1124/mol.56.1.170. [DOI] [PubMed] [Google Scholar]
- Wu Y, Zhou C, Tao J, Li S. Montelukast prevents the decrease of interleukin-10 and inhibits NF-kappa B activation in inflammatory airway of asthmatic guinea pigs. Can J Physiol Pharmacol. 2006;84:531–537. doi: 10.1139/y06-003. [DOI] [PubMed] [Google Scholar]
- Yang Y, Luo J, Kazumura K, Takeuchi K, Inui N, Hayashi H, et al. Cilostazol suppresses adhesion of human neutrophils to HUVECs stimulated by FMLP and its mechanisms. Life Sci. 2006;79:629–636. doi: 10.1016/j.lfs.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Zhu J, Qiu YS, Figueroa DJ, Bandi V, Galczenski H, Hamada K, et al. Localization and upregulation of cysteinyl leukotriene-1 receptor in asthmatic bronchial mucosa. Am J Respir Cell Mol Biol. 2005;33:531–540. doi: 10.1165/rcmb.2005-0124OC. [DOI] [PubMed] [Google Scholar]