

Abstract
Background and purpose
The anti-cancer agent [Arg6, D-Trp7,9, NmePhe8]-substance P (6-11) (SP-G) modulates gastrin releasing peptide (GRP) and arginine vasopressin signalling in small cell lung cancer cells leading to growth arrest and apoptosis. We have shown that SP-G acts as a biased agonist at GRP receptors. This work examines the hypothesis that SP-G acts as a biased agonist at the V1A vasopressin receptor.
Experimental approach
The human V1A receptor was expressed in CHO-K1 cells. Extracellular regulated kinase (ERK) activation and intracellular Ca2+ were measured using activation state-specific antibodies and Fura-2-AM respectively. The effect of SP-G on tumourigenicity was assessed by colony assay.
Key results
In V1A receptor expressing cells, SP-G caused a sustained activation of ERK via a stimulation of V1A receptor coupling to Gi. Inhibition of Gi with Pertussis toxin attenuated the inhibition by SP-G of the growth of CHO-K1 cells stably expressing the V1A receptor. Chimeric V1A receptors containing the second or third intracellular loop of the V2 receptor were capable of binding vasopressin and SP-G but had altered ability to activate phospholipase C (PLC) and ERK. The second intracellular loop of the V1A receptor was essential for vasopressin-stimulated PLC and ERK activation but not for SP-G-induced ERK activation.
Conclusions and implications
This work provides mechanistic insight, for biased agonists at V1A receptors and highlights a potential role for such agents as anti-cancer agents.
Keywords: [Arg6, D-Trp7,9, NmePhe8]-substance P (6-11); vasopressin; V1A receptor; cancer; directed signalling; biased agonist
Introduction
Studies on neuroendocrine tumours such as small cell lung cancer (SCLC) have shown that these tumours show altered expression and sensitivity to neuropeptides which act as mitogens and promote SCLC cell growth via their G-protein coupled receptors (GPCRs) (Sethi and Rozengurt, 1991; Sethi et al., 1992). Gastrin releasing peptide (GRP) receptors are frequently aberrantly expressed in human neuroendocrine lung tumours (Cuttitta et al., 1985; Moody et al., 2003) and 2A11 a monoclonal antibody that binds GRP, thus preventing receptor interaction, has been shown to inhibit the growth of SCLC in vitro and as xenografts in nude mice (Cuttitta et al., 1985). SCLC cells also secrete arginine vasopressin (AVP) and express V1A receptors implying the existence of an autocrine growth loop (North et al., 1998a,b). SCLC patients frequently display symptoms of inappropriate AVP secretion such as hyponatremia and urinary hyperosmolality (Johnson et al., 1997). Independent studies have shown expression of V1A receptors in 5/5 SCLC lines and 0/4 non-small cell lung cancer (NSCLC) lines (Ocejo-Garcia et al., 2001) while we showed expression of V1A receptors in 4/4 SCLC lines (Waters et al., 2003). Expression of V1A receptors and AVP is the most useful diagnostic tool for differentiating SCLC from NSCLC and other cancers (Coulson et al., 2003). SCLC cells can also express the AVP gene as pro-vasopressin, which remains attached to the cell membrane and could possibly contribute to the autocrine-driven mitogenesis (Friedmann et al., 1994). Antibodies recognising this cell surface antigen have been developed as a potential diagnostic and therapeutic tool that targets SCLC tumours in vivo (Keegan et al., 2002). Taken together, these findings demonstrate that the AVP/V1A axis represents a novel target for new therapies for SCLC.
Gastrin releasing peptide and V1A receptors couple predominantly to Gq causing an increase in phospholipase C (PLC) activation, generation of inositol trisphosphates (IP3) and an increase of intracellular free calcium (Woll and Rozengurt, 1989; Sethi and Rozengurt, 1991) and increase activation of the extracellular signal regulated kinase (ERK) cascade through both protein kinase C (PKC) dependent and independent mechanisms (Ghosh et al., 2004; Sinnett-Smith et al., 2004). Stimulation of these pathways by GRP and AVP leads to an enhanced overall transcriptional activity, which controls cell proliferation and increases cell survival (Gutkind et al., 1997; Rozengurt, 1998).
Analogues of substance P such as [Arg6, D-Trp7,9, NmePhe8]-substance P (6-11) (SP-G), [D-Arg1, D-Phe5, DTrp7,9, Leu11-]-substance P (SP-D) and [D-Arg1, DTrp5,7,9, Leu11-]-substance P (SP-A) while being relatively poor tachykinin antagonists, block the mitogenic effects of AVP and GRP in fibroblasts and SCLC cells (Woll and Rozengurt, 1988; Langdon et al., 1992; Sethi et al., 1992). These analogues have been termed ‘broad spectrum neuropeptide antagonists’ and block the proliferation of SCLC cells in liquid culture and in vivo as xenografts in nude mice (Langdon et al., 1992; Sethi et al., 1992). SP-A has also been demonstrated to block angiogenesis in pancreatic cancer xenografts in vivo (Guha et al., 2005). SP-G has been taken into phase I clinical studies for SCLC where therapeutic plasma levels were achieved without dose-limiting toxicity (Clive et al., 2001).
Our work has focused on the mechanism of action of these analogues. SP-G and SP-D block GRP and AVP-induced calcium elevations but can also activate the receptors independently leading to a protracted increase in ERK and c-jun-N-terminal kinase (JNK) activation leading to apoptosis (Jarpe et al., 1998; MacKinnon et al., 2001; Waters et al., 2003). The results suggest that substance P analogues induce ligand-specific receptor conformations which result in opposing efficacy for two sets of responses; a property called ‘biased agonism’ (Jarpe et al., 1998; MacKinnon et al., 2001). Given that this pharmacological activity may have a clinical utility for cancer therapy, the molecular mechanisms of this biased agonism required more rigorous investigation.
Using CHO-K1 cells expressing V1A receptors we sought to dissect the mechanisms of SP-G biased agonism at V1A receptors by examining the effects of AVP and SP-G on intracellular Ca2+ and ERK activation and the subsequent effects on cell growth. We show that SP-G activates a Gi-dependent pathway to stimulate sustained ERK activation, but blocks AVP/Gq-stimulated increase in intracellular Ca2+. Blocking Gi with Pertussis toxin (PTX) inhibits SP-G-induced inhibition of cell growth in transfected CHO-K1 cells and SCLC cells. Using V1A/V2 receptor chimeras we show that the second intracellular loop of V1AR is essential for PLC activation and increased intracellular Ca2+ but not for SP-G-induced ERK activation. This study provides experimental evidence for agonist selective V1A receptor conformations and gives mechanistic insight into the therapeutic utility for V1A receptor biased agonists as anti-cancer agents in SCLC.
Methods
Cell culture and transfections
H69-SCLC cells were cultured in RPMI-1640 medium supplemented with 10% (v/v) foetal calf serum (FCS) 50 U mL−1 penicillin, 50 µg mL−1 streptomycin and 5 µg mL−1 L-glutamine. For experimental purposes, H69-SCLC cells were cultured in SITA medium consisting of RPMI-1640 medium supplemented with 30 nmol·L−1 selenium, 5 µg mL−1 insulin, 10 µg mL−1 transferrin and 0.25% (w/v) bovine serum albumin (BSA). CHO-K1 cells were maintained in Dulbecco's modified Eagles medium (DMEM) supplemented with 10% (v/v) FCS, 50 U mL−1 penicillin, 50 µg mL−1 streptomycin and 5 µg mL−1 L-glutamine in a humidified atmosphere of 5% CO2/95% air at 37°C. CHO-K1 cells were transfected with full-length V1A receptor or V1A receptor chimeras using lipofectamine plus (Invitrogen) according to the manufacturer's instructions. Stable cell cultures were maintained in the presence of 400 µg mL−1 G418-sulphate.
Liquid growth
Exponentially growing H69-SCLC or CHO-K1 cells were suspended in SITA medium (H69-SCLC cells) or DMEM with 5% FCS (CHO-K1 cells) at a density of 5 × 104 cells per plate in the presence or absence of mediators in triplicate. Cells were grown for 1–9 days and cell number determined using a Coulter Counter (model Z1, Coulter).
MTT assay
In some assays MTT (3-[4,5-dimethylthiazol-2yl]-2,5-diphenyl tetrazolium bromide) formazan production (Sigma) was used to measure proliferation according to the manufacturer's instructions.
Clonogenic assay
H69-SCLC cells or CHO-K1 (2 × 104) cells were suspended in SITA (H69-SCLC) or DMEM with 5% FCS (CHO-K1) containing 0.3% agarose in the presence or absence of mediators and layered over a solid base of 0.5% agarose in 35 mm plastic dishes. The cultures were incubated at 37°C for 1–10 days, and then stained with 1 mg mL−1 MTT overnight at 37°C. Colonies from 10 separate fields were counted using a microscope with a ×4 objective. Cloning efficiency was calculated as the % of original number of seeded cells forming colonies of >6 cells.
Aggregation assay
CHO-K1 cells (2 × 104) were suspended in DMEM in the presence of 5% FCS and seeded into low adhesion tissue culture plates on top of a layer (1 mL) of 0.5% agar. Under these conditions the cells did not adhere. Cells were maintained in culture for 7 days briefly trypsinized to disaggregate clusters and viable cells counted.
Receptor binding
Confluent cultures of CHO-K1 cells expressing V1A receptor were washed twice in ice-cold phosphate buffered saline (PBS). The cells were lysed in ice-cold lysis buffer (10 mmol·L−1 Tris HCl pH 7.4, 5 mmol·L−1 EDTA, 5 mmol·L−1 EGTA, 1 mmol·L−1 phenyl methyl sulphonyl fluoride) and briefly homogenised using a Polytron tissue homogeniser. After centrifugation at 500× g for 4 min, the supernatant was centrifuged at 49 000× g for 15 min at 4°C and the pellet washed twice by repeated homogenisation and centrifugation in lysis buffer. The final pellet was suspended in 50 mmol·L−1 Tris HCl (pH 7.4), adjusted to 1 mg mL−1 protein, and stored at −80°C. Protein was determined using Pierce BCA protein assay reagent (Pierce UK). Membranes (150–250 µg protein) were incubated with 1 nmol·L−1[3H]-AVP and test agents for 30 min at 37°C in 50 mmol·L−1 Tris HCl (pH 7.4), 0.5% BSA, 3 mmol·L−1 MgCl2. The assay was terminated by immediate filtration over GFB glass fibre filters using a Tomtec 96 cell harvester. The filters were washed with 2 × 3 mL binding buffer, dried and counted for 3H in a Wallac Betaplate scintillation counter. Non-specific binding was determined with 1 µmol·L−1 AVP. The binding parameters Kd and Bmax were calculated from competition binding isotherms with unlabelled ligand (DeBlasi et al., 1989). The IC50 (concentration of drug displacing 50% specific binding) was converted to the inhibitory constant (Ki), where Ki = IC50/(1 + [ligand]/Kd) (Cheng and Prusoff, 1973).
PLC activation
[3H]-inositol phosphate formation was measured by labelling cells overnight in serum-free and inositol-free medium containing 1 µCi mL−1 myo-[2-3H]-inositol. Cells (2 × 106 per assay point) were washed and incubated in Hank's balanced salt solution containing 20 mmol·L−1 HEPES, 1.8% glucose, 0.2% BSA and 20 mmol·L−1 LiCl, for 30 min at 37°C prior to addition of agonist for a further 30 min. Reactions were extracted by removal of assay buffer and addition of ice-cold 10 mmol·L−1 formic acid for 60 min on ice. [3H]-inositol phosphates were separated by anion exchange chromatography on Dowex (200–400 mesh, formate form) columns, and [3H] determined by scintillation counting.
[35S]-GTPγS binding and immunoprecipitation of Gα subunits
[35S]-Guanosine 5'-O-(γ-thio)triphosphate ([35S]-GTPγS) binding was carried out essentially as previously described (Weiland and Jakobs, 1994). Briefly, cell membranes (∼10 µg protein) were incubated in a final volume of 100 µL binding buffer (20 mmol·L−1 HEPES, pH 7.4, 100 mmol·L−1 NaCl, 3 mmol·L−1 MgCl2, 10 µmol·L−1 GDP, 0.2 mmol·L−1 ascorbic acid) with 0.2 nmol·L−1[35S]-GTPγS for 60 min at 4°C in the presence or absence of test compounds. Bound radioactivity was determined by filtration onto GF-B glass fibre filters and scintillation counting. Non-specific binding was determined in the presence of 100 µmol·L−1 unlabelled GTPγS. [35S]-GTPγS binding to Gαi or Gαq/11 was determined as above and membrane pellets solubilised in 50 µL 1.5% Triton, 0.2% SDS. Pellets were diluted in 1 mL immunoprecipitation buffer [50 mmol·L−1 Tris HCl pH 7.4, 150 mmol·L−1 NaCl. 2 mmol·L−1 KCl, 1 mmol·L−1 EDTA, 20 mmol·L−1 MgCl2, 1% Triton and protease inhibitors (Complete inhibitor tablet, Roche Belgium)] and subunits immunoprecipitated overnight at 4°C with 1 µg rabbit anti-Gαi or Gαq/11. Complexes were captured with protein A agarose and washed ×4 with immunoprecipitation buffer. Bound [35S]-GTPγS was eluted in SDS-PAGE buffer and counted by scintillation counting.
Determination of intracellular Ca2+ concentration
CHO-K1 cells stably expressing the V1A receptor chimeras were grown to confluence on 10 cm plates and rested overnight in DMEM containing 0.1% FCS. Cells (5 × 106 cells per data point) were trypsinised and loaded with FURA-2-tetraacetoxymethylester (Fura-2-AME 1 µmol·L−1) in calcium-free Hank's balanced salt solution for 10 min at 37°C. The cells were pelleted and resuspended in 2 mL of Hank's balanced salt solution containing 1.8 mmol·L−1 CaCl2. Fluorescence was recorded in a Model F2000 fluorescence spectrophotometer (Hitachi). Alternate dual wavelength excitation at 380 nm and 410 nm allowed ratiometric analysis of bound and unbound Fura-2 when measured at 505 nm. [Ca2+] was calculated according to the equation [Ca2+] = K(F − Fmin)/(Fmax − F), where F is the ratio of the unknown sample, Fmax is the ratio after the addition of 0.1% triton X-100 and Fmin is the ratio after Ca2+ chelation with 10 mmol·L−1 EGTA. K is the dissociation constant for Fura-2, which is 224 nmol·L−1.
Western blotting/ERK phosphorylation
Quiescent cell cultures (1 × 106 cells) were treated as described in figure legends and lysed at 4°C in lysis buffer containing; 25 mmol·L−1 HEPES pH 7.4, 0.3 M NaCl, 1.5 mmol·L−1 MgCl2, 0.2 mmol·L−1 EDTA, 0.5% Triton X-100, 20 mmol·L−1β-glycerophosphate, 0.5 mmol·L−1 dithiothreitol, 1 mmol·L−1 sodium orthovanadate and protease inhibitors [protease inhibitor cocktail (Boehringer Mannheim, Sussex, UK)]. Lysates were clarified by centrifugation, equalised for protein content using Pierce BCA protein assay reagent and denatured by boiling (5 min) in SDS-PAGE loading buffer. A total of 20 µg lysate per lane was resolved on 12% SDS-PAGE gels and electroblotted onto nitrocellulose membranes. Membranes were blocked in 3% BSA in PBS containing 0.05% tween-20. Blots were probed with primary antibody followed by the appropriate horseradish peroxidase-labelled goat IgG (DAKO, UK). Bands were visualised using enhanced chemiluminescence (ECL plus, Amersham) and quantified by Image J.
Statistical analysis
Results are presented as means ± SEM. Significance of the differences between means was assessed using Student's t-test or by ANOVA for comparison between groups. When ANOVA showed a significant treatment effect, Dunnet's post hoc test was used to compare individual means. Differences were considered statistically significant at P < 0.05. Unless stated otherwise, studies were performed on 3–6 independent occasions.
Materials
CHO-K1 cells and H69-SCLC cells were purchased for the European Cell Culture Collection; DMEM, RPMI-1640, AVP, d(CH2)-5-TyrMe-AVP and monoclonal antibody to diphosphorylated ERK 1 and 2 (M 8159) were from Sigma (Poole, UK); polyclonal antibodies to Gαi (sc-26761) and Gαq/11 (sc-392) and ERK2 were from Insight Biotechnology; Rabbit anti-caspase-3 (Asp-175) from Cell Signalling Technology; [Arg6, D-Trp7,9, NmePhe8]-substance P (6-11) (SP-G) was synthesized by Cancer Research UK (London, UK). PTX was from Alexis Biochemicals (Nottingham, UK). The human V1A receptor construct in pcDNA3.1 was a kind gift from M. Thibonnier (Case Western Reserve University School of Medicine, OH, USA). The V1A receptor mutants V1i2 and V1i3 were kindly provided by Dr Wess (National Institute of Health, Bethesda, MD, USA). The following sequences were exchanged between the rat V1A and the human V2 receptor: V1i2 (V1A 152–172 for V2 140–161), V1i3 (V1A 237–303 for V2 225–277). [3H]-AVP (60 Ci/mmol) was from New England Nuclear (Perkin Elmer).
Results
V1A receptor expression in CHO cell line
Previous studies have shown that V1A receptors can activate Gq and Gi linked signalling pathways in response to different agonists (Thibonnier et al., 1993; Abel et al., 2000; Chiu et al., 2002). To examine SP-G-induced biased agonism we stably expressed V1A receptors in CHO-K1 cells. In membrane preparations from these cells, [3H]-AVP bound with high affinity, Kd = 3.3 ± 1.6 nmol·L−1, Bmax = 383 ± 74 fmoles mg protein−1 (n = 4, Fig. 1A). SP-G inhibited [3H]-AVP binding in CHO-V1A cells with a Ki = 3.1 ± 0.6 µmol·L−1 and nH = 1.25 (n = 6). The inhibition of [3H]-AVP binding by SP-G was non-competitive as it significantly decreased saturable binding (Bmax) from 383 ± 74 fmoles mg protein−1 in control cells to 285 ± 25 and 82 ± 12 fmoles mg protein−1 for 1 and 5 µmol·L−1 SP-G (P < 0.05 and P < 0.01, respectively, by Student's t-test), while having no appreciable effect on affinity (Kd = 3.3 ± 1.6, 3.8 ± 1.38 and 4.8 ± 1.6 nmol·L−1 for control, 1 and 5 µmol·L−1 SP-G respectively, P > 0.05, Fig. 1B). Moreover, in kinetic experiments, SP-G at 5 µmol·L−1 significantly accelerated the dissociation of [3H]-AVP from receptor expressing cells (data not shown).
Figure 1.
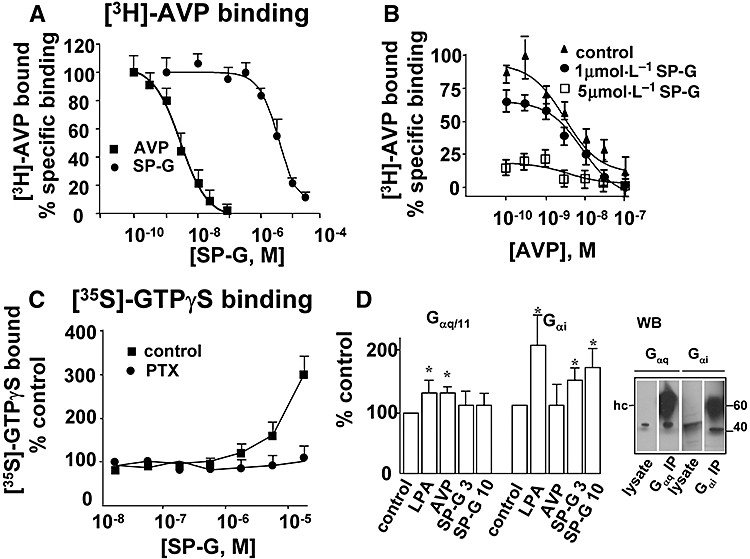
Receptor binding and [35S]-GTPγS binding assay. A. Membranes of CHO-K1 cells stably expressing the V1A receptor were incubated with [3H]-AVP and increasing concentrations of SP-G or unlabelled AVP. Binding isotherms for Kd and Bmax were calculated from AVP competition curves as described in the Methods. B. SP-G inhibition is non-competitive. Membranes were incubated with increasing concentrations of [3H]-AVP in the presence of 1 or 5 µmol·L−1 SP-G as indicated. C. [35S]-GTPγS binding. [35S]-GTPγS binding to the whole membrane fraction was determined as described in the Methods. Membranes were incubated with SP-G in the presence or absence of 100 ng mL−1 PTX for 60 min at 4°C. D. [35S]-GTPγS binding to specific Gα subunits. Membranes were incubated with [35S]-GTPγS in the presence of LPA (1 µmol·L−1), AVP 30 nmol·L−1, or SP-G at 3 or 10 µmol·L−1 as indicated. Solubilised pellets were immunoprecipitated with anti-Gαq/11 or anti-Gαi as described in the Methods. Results represent the mean ± SEM of four independent experiments. (*P < 0.05 vs. untreated control membranes ANOVA with Dunnets's post-test.) Representative Western blots (WB) of whole-cell lysate and immunoprecipitated Gαq/11 or GαI confirm expression of these α subunits in V1A receptor transfected CHO-K1 cells. Immunoprecipitating antibody heavy chain (hc).
We next sought to determine whether SP-G could promote receptor coupling to G-proteins by measuring [35S]-GTPγS binding to V1A receptor transfected cells. This assay measures GTP/GDP exchange on G-proteins and is a measure of Gα activation. As shown in Figure 1C SP-G increased [35S]GTPγS binding to V1A-CHO-K1 membranes. PTX catalyses ADP-ribosylation of the α subunits of Gi-like proteins thus blocking their coupling and activation. Treatment with PTX completely abolished [35S]GTPγS binding induced by SP-G suggesting that SP-G induced coupling to Gi-like proteins. Activation of Gi proteins by SP-G was confirmed by increased [35S]GTPγS binding to Gi proteins immunoprecipitated from solubilised cell membranes. SP-G increased binding to Gαi subunits by 45 ± 10 and 68 ± 9% at 3 and 10 µmol·L−1 respectively (n = 4, Fig. 1D). AVP did not significantly increase activation of Gi suggesting that the natural agonist does not induce V1A receptor coupling to Gi in these cells. Receptor-mediated activation of Gi was confirmed in cells stimulated with lysophosphatidic acid (LPA; 1 µmol·L−1, 102% increase over basal). LPA and AVP but not SP-G stimulated [35S]GTPγS binding to Gq (35 ± 7 and 34 ± 9% increase over basal, respectively, Fig. 1D). These data establish that SP-G selectively activated Gi in V1A receptor transfected cells.
SP-G induced growth inhibition of V1A expressing CHO-K1 cells
We have shown previously that AVP stimulates SCLC cell growth and colony formation (Sethi and Rozengurt, 1991; Sethi et al., 1992). To examine the ability of substance P analogues to inhibit V1A receptor-mediated growth we compared proliferation of V1A receptor transfected CHO-K1 cells with the V1A receptor expressing SCLC cell line H69. As shown in Figure 2A SP-G significantly inhibited growth of H69-SCLC cells in liquid culture (IC50 = 5.1 ± 0.5 µmol·L−1, n = 4) and inhibited colony formation in semi-solid agarose (Fig. 2B, IC50 = 2.5 ± 0.4 µmol·L−1, n = 4). CHO-K1 cells transfected with the human V1A receptor showed a significant increase in proliferation compared with vector transfected cells (Fig. 2C) and formed significantly more colonies in soft agar (Fig. 2D). The expression of the V1A receptor also increased the number of viable cell aggregates formed after 72 h culture in low adhesion culture plates (Fig. 2E). This is in agreement with our previous observations suggesting that V1A receptor expression allows for anchorage- and serum-independent growth consistent with transformation (MacKinnon et al., 2005). Moreover, it suggests that these receptors show some constitutive activity, as wild-type receptor expression alone was sufficient to increase transformation in the absence of exogenously added neuropeptide. This increase in proliferation by V1A receptor expression was significantly blocked by co-incubation with SP-G (IC50 = 5.1 ± 0.9 µmol·L−1, Fig. 2E). Figure 2E shows that PTX (100 ng mL−1) attenuated SP-G-induced inhibition of cell growth in V1A expressing CHO-K1 cells suggesting that SP-G can inhibit growth in part by inducing V1A receptor-mediated Gi activation.
Figure 2.
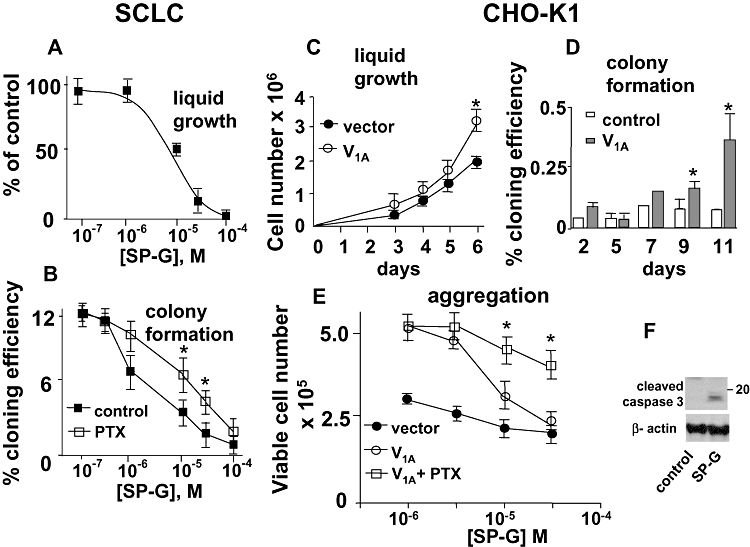
Effect of SP-G on proliferation of SCLC cells and V1A expressing CHO-K1 cells. A, B. H69-SCLC cell growth. The effect of SP-G on liquid growth over 7 days (A) and on colony formation in soft agarose (B) was carried out in the presence or absence of 100 ng mL−1 PTX, as indicated. The results represent the mean ± SEM or four independent experiments. (*P < 0.05, Student's t-test compared with SP-G in non-PTX treated.) C. CHO-K1 cells stably expressing the V1A receptor or empty vector were grown in liquid culture and viable cells counted at the time points indicated. Results represent the mean ± SEM of four experiments. (*P < 0.05 Student's t-test compared with vector transfected cells.) D. Colony formation. Control or V1A receptor expressing CHO-K1 cells were cultured in 0.3% agarose for up to 11 days and colony formation calculated as % cloning efficiency. Results represent the mean ± SEM of four experiments. (*P < 0.05 Student's t-test compared with vector transfected cells.) E. Effect of PTX. CHO-K1 cells were cultured in low adhesion plates in the presence or absence of 100 ng mL−1 PTX for 7 days. Viable cells were counted by propidium iodide exclusion and Coulter counting. Results represent the mean ± SEM of four experiments. (*P < 0.05 Student's t-test compared with untreated V1A receptor expressing cells.) F. SP-G induces cleavage of caspase-3. V1A-CHO-K1 cells were incubated with SP-G for 48 h. Cells were lysed and Western blots probed for cleaved caspase-3.
As has been shown previously, PTX did not affect basal growth of SCLC cells in culture (Codignola et al., 1998). However, PTX attenuated SP-G-induced inhibition of SCLC colony formation (Fig. 2B) suggesting that SP-G-induced growth inhibition is in part mediated via Gi. Our previous work has shown that SP-G induces apoptosis in SCLC cells (MacKinnon et al., 1999; MacKinnon and Sethi, 2003). In Figure 2F we show that SP-G induced cleavage of caspase-3 in V1A receptor expressing CHO-K1 cells suggesting that the anti-proliferative activity, induced by SP-G through the V1A receptor, may be in part due to an induction of apoptosis.
Signalling pathways activated by AVP and SP-G
Arginine vasopressin has been shown to produce a Gq-dependent elevation of intracellular calcium in CHO-K1 cells and other cell types (Laszlo et al., 1991; Thibonnier et al., 1993; Liu and Wess, 1996; Hawtin et al., 2006). In this study, AVP produced an elevation of intracellular calcium in V1A-CHO cells (EC50 = 2.4 ± 0.64 nmol·L−1, Fig. 3A). SP-G had no effect on calcium elevation alone suggesting that SP-G does not stimulate Gq-mediated PLC activation which is in agreement with our previous studies (Waters et al., 2003). SP-G did however inhibit AVP-stimulated increase in intracellular calcium with pA2 = 6.28 ± 0.21 (Fig. 3B). These data suggest that SP-G acts as a competitive antagonist of AVP-mediated intracellular calcium elevation which is mediated by Gq-stimulated PLC activation in V1A-receptor expressing CHO-K1 cells (Thibonnier et al., 1993).
Figure 3.
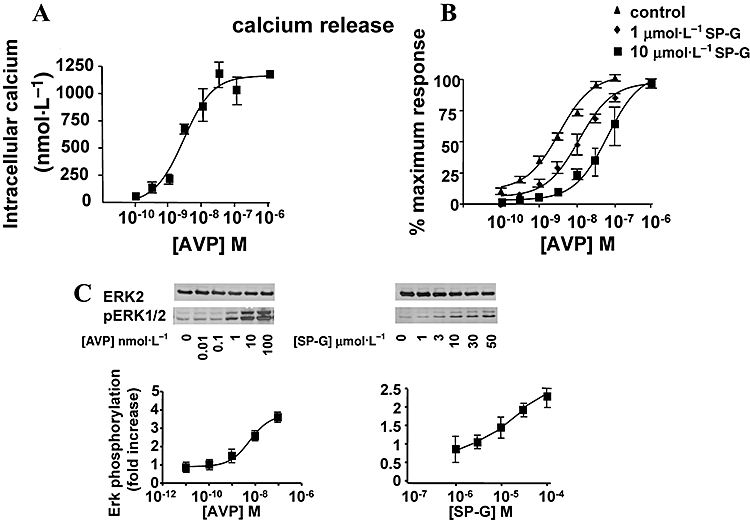
Signalling pathways activated by SP-G. A. Intracellular calcium. Quiescent CHO-K1 cells expressing V1A receptors were incubated with FURA-2-AM for 15 min at 37°C before stimulation with the indicated concentrations of AVP. Ratiometric fluorescence was monitored at 37°C following addition of AVP at the indicated concentrations. Results represent the mean ± SEM of four experiments. B. SP-G inhibits AVP-induced calcium elevation. CHO-K1 cells expressing V1A receptors were incubated with FURA-2AM for 15 min at 37°C and ratiometric fluorescence monitored at 37°C. Concentration response curves to AVP were carried out in control cells and in cells pretreated for 2 min with 1 or 10 µmol·L−1 SP-G. Results are expressed as % maximum control response to AVP and represent the mean ± SEM of four experiments. C. ERK activation. CHO-K1 cells were incubated with AVP (left) or SP-G (right) at the indicated concentrations for 10 min at 37°C. Cells were lysed and Western blots probed for pERK1/2 or total ERK2. Representative blots from four separate experiments are shown, with quantitative summary data below (band densities were quantitated by Image J).
ERK activation
Arginine vasopressin also caused a concentration-dependent stimulation of ERK in cells expressing V1A receptors (Fig. 3C). AVP gave a 3.6-fold stimulation of ERK activity with an EC50 value of 2.8 ± 0.3 nmol·L−1. Although SP-G was an antagonist for calcium elevation, it was also an agonist for ERK stimulation in V1A receptor expressing cells (Fig. 3C) producing a 2.2-fold stimulation at 100 µmol·L−1 and an EC50 of 11.1 ± 1.9 µmol·L−1. SP-G-induced activation of ERK was rapid and sustained. Figure 4 shows that AVP-induced ERK phosphorylation was maximal at 5 min and declined thereafter to baseline levels after 30 min. SP-G-induced ERK phosphorylation was maximal after 10 min and showed sustained activation after 60 min stimulation. We investigated whether ERK activation by SP-G was mediated via Gi and show that SP-G-induced, but not AVP-induced, ERK activation was inhibited by pretreatment with PTX in V1A receptor cells (Fig. 4B). These data confirm that SP-G promotes V1A coupling to Gi proteins which mediate an activation of ERK. Figure 4C shows that the EGF receptor tyrosine kinase inhibitor AG1478 (1 µmol·L−1) inhibited SP-G-induced ERK activation but only partially inhibited AVP-stimulated ERK activation, suggesting different mechanisms of ERK activation by AVP and SP-G in CHO cells.
Figure 4.
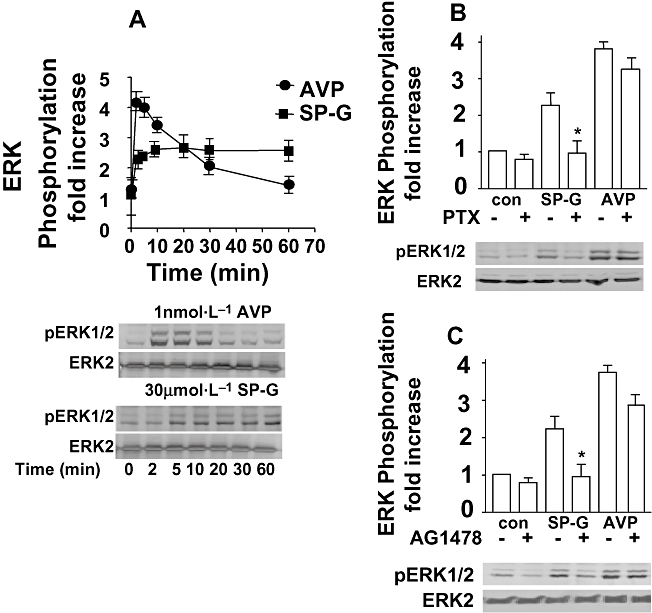
Mechanisms of ERK activation. A. SP-G-induced ERK activation is sustained. CHO-K1 cells expressing V1A receptor were stimulated with 30 µmol·L−1 SP-G or 1 nmol·L−1 AVP for various times prior to lysis. Cells were lysed and immunoblotted for pERK1/2 or total ERK. Representative blots from four separate experiments are shown, below the summary data in the graph [band densities were quantitated by Image J]. B. Effect of PTX. Cells were pretreated for 18 h with 50 ng mL−1 PTX and stimulated with 30 µmol·L−1 SP-G or 1 nmol·L−1 AVP for 5 min. (*P < 0.05 compared with SP-G alone.) C. Effect of AG1478. Cells were incubated with AG1478 (1 µmol·L−1) for 30 min at 37°C prior to stimulation for 5 min with 10 µmol·L−1 SP-G or 1 nmol·L−1 AVP as indicated. Aliquots of cell lysate were resolved by SDS-PAGE and Western blots probed with monoclonal anti-pERK1/2 antibody. Representative Western blots are shown.
V1A receptor chimeras
It has been previously shown that the second intracellular loop of the V1A receptor is essential for V1A receptor coupling to Gq (Liu and Wess, 1996). However, it is not known what region of the receptor may be involved in coupling to Gi. To address this, V1A receptor mutants expressing the second (V1i2) or third (V1i3) intracellular loop of the V2 receptor were transiently transfected into CHO-K1 cells. Table 1 shows that both chimeric receptors bind [3H]-AVP with similar affinity (wild-type Kd 5.71 ± 2.54 nmol·L−1, V1i2Kd 2.64 ± 0.65 nmol·L−1, V1i3Kd 1.14 ± 0.57 nmol·L−1, n = 3); however, there was a marked difference in the ability of the chimeras to activate PLC. AVP at concentrations up to 100 nmol·L−1 was unable to increase generation of total inositol phosphates in V1i2 receptor expressing cells, whereas in V1i3 cells a fourfold increase in inositol phosphates was observed which was comparable to the activation observed in wild-type receptor expressing cells. (EC50 for AVP was 0.89 ± 0.10 and 1.04 ± 0.12 nmol·L−1 in V1A and V1i3 receptor expressing cells respectively, Fig. 5A.) SP-G inhibited AVP-induced inositol phosphate production in V1A and V1i3 cells (Table 1). This suggests that the second intracellular loop of the V1A receptor is essential for V1A receptor activation of PLC.
Table 1.
AVP binding and PLC activation in CHO-K1 cells with V1A receptor chimeras
| V1A | V1i2 | V1i3 | |
|---|---|---|---|
| [3H]-AVP binding | |||
| Kd (nmol·L−1) | 5.7 ± 2.5 | 2.6 ± 0.7 | 1.1 ± 0.6 |
| Bmax (fmoles mg−1) | 151 ± 45 | 103 ± 68 | 226 ± 140 |
| SP-G Ki (µmol·L−1) | 4.2 ± 1.3 | 3.9 ± 1.1 | 1.5 ± 0.6 |
| PLC activation | |||
| SP-G IC50 (µmol·L−1) | 7.1 ± 0.4 | >100 | 1.0 ± 0.1 |
CHO-K1 cells were transiently transfected with V1A receptor chimeras as described in the Methods. The binding parameters Kd and Bmax were calculated from competition binding isotherms with unlabelled AVP. The inhibitory constant Ki was calculated from the IC50 using the equation of Cheng and Prusoff (1973). [3H]-Inositol phosphate formation was measured as described in the Methods. Cells were stimulated with 10 nmol·L−1 AVP for 30 min at 37°C in the presence of various concentrations of SP-G. The results represent the mean ± SEM of three independent experiments.
Figure 5.
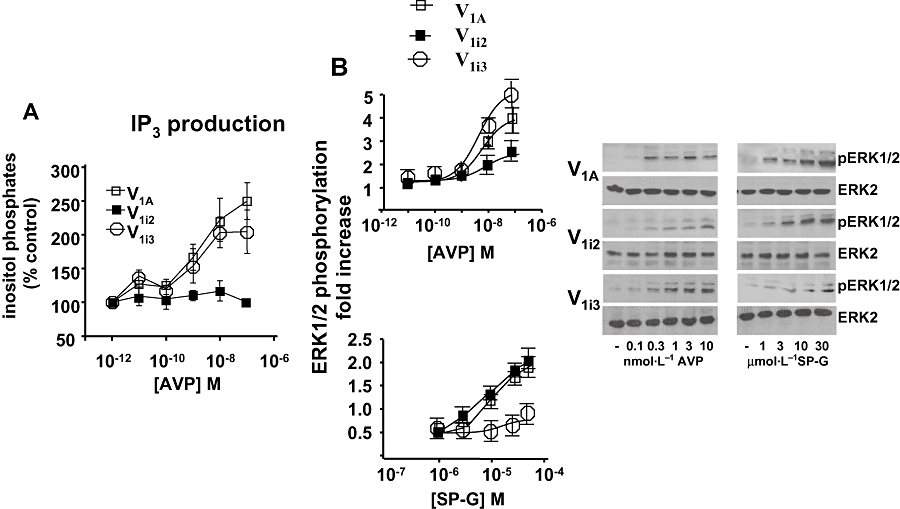
A. PLC activation. [3H]-inositol phosphate formation was measured in cells expressing V1A receptor chimeras and stimulated with the indicated concentrations of AVP for 30 min at 37°C. The results are expressed as % control and represent the mean ± SEM of three independent experiments. B. ERK phosphorylation. Confluent cultures of V1A receptor chimera expressing CHO-K1 cells were rested overnight and stimulated for 5 min with SP-G or AVP, as indicated. Aliquots of cell lysate were resolved by SDS-PAGE and Western blots probed with monoclonal anti-pERK1/2 antibody or anti-ERK2 antibody. Phosphorylation of ERK1/2 from three experiments was quantified by Image J.
Figure 5B shows that the V1i3 chimera potently stimulated ERK in response to AVP, with higher potency than observed in the wild-type receptor. However, SP-G was less able to activate ERK in these cells producing significant stimulation only at 30 µmol·L−1. Conversely, the V1i2 chimera which has the second intracellular loop replaced with that of the V2 receptor, was less able than the wild-type receptor to activate ERK in response to AVP but produced a robust stimulation of ERK in response to SP-G which was similar to its effects in the wild-type cells. It is of note however that the V1i2 chimera was able to activate ERK in response to AVP even though it was unable to activate PLC at these concentrations. These data suggest that SP-G induces a conformational change in the V1A receptor at a region within the third intracellular loop which promotes coupling to Gi and subsequent activation of ERK.
Discussion
The key findings of this study are: (i) SP-G selectively activates Gi and inhibits cell growth of SCLC cells and CHO-K1 cells stably expressing V1A receptors; (ii) SP-G favours coupling to Gi and blocks AVP activation of Gq in V1A CHO-K1 cells thus supporting a biased agonist mechanism; (iii) PTX attenuates SP-G-induced growth inhibition in SCLC cells and V1A CHO-K1 cells suggesting that activation of Gi is an important aspect of its antiproliferative activity; and (iv) Mutant receptors containing the second intracellular loop of the V2 receptor were unable to respond to AVP in terms of PLC activation but were able to activate ERK in response to SP-G suggesting that V1A receptor coupling to Gi involves a region of the receptor outwith the second intracellular loop. Given that a single receptor can couple to more than one G-protein, traditional receptor theory as proposed by Furchgott (1966) would predict that an agonist at the receptor would have similar efficacies for different signalling pathways. However, there are a number of reports that show differential effector activation by agonists that cannot be explained by this mechanism (Berg et al., 1998). For example, α2A receptor agonists show different efficacies for Gi-mediated adenylyl cyclase inhibition and Gs-mediated adenylyl cyclase stimulation in transfected CHO cells (Brink et al., 2000), and the neurotensin receptor-1 agonists EISA-1 and neuromedin B show reverse potency orders for Gq- and Gs-mediated responses (Skrzydelski et al., 2003). This type of activity can be explained by the hypothesis of agonist-dependent trafficking of receptor stimulus (ADTRS) originally described by Kenakin (1995). This hypothesis predicts that when a receptor couples to more than one stimulus, the relative efficacies of a series of agonists may differ depending on their abilities to stabilise different receptor/G-protein activation states. It is therefore theoretically possible to design ‘biased’ agonists which activate a selective subset of responses triggered by the receptor. As yet however there has been little description of such pharmacological agents providing any therapeutic benefit in disease.
In CHO cells expressing V1A receptors, SP-G showed the highest antagonist potency in inhibiting calcium elevation (pA2 = 0.53 µmol·L−1), a Gq-coupled response, but its potency for ERK activation and all other functional responses demonstrated IC50/EC50 values in the low micromolar range. This suggests that stimulation of Gi correlates better with functional effects on growth and this is corroborated by the finding that PTX blocks these responses. Our results illustrate the characteristics of ADTRS, namely that two agents can show reverse efficacies for two pathways and that one (SP-G) directs signalling via a G-protein usually considered to be less efficiently coupled to this receptor. The higher potency of SP-G to antagonise calcium elevation may be reflected by a higher affinity of SP-G for the receptor conformation coupled to Gq. This is adequately explained by the ADTRS hypothesis which states that agonists can differ in their rank order of potency/efficacy between responses mediated by the same receptor. The affinity for SP-G for agonist binding (Ki = 3.1 µmol·L−1) is intermediate between its affinity for inhibition of intracellular calcium and ERK activation. SP-G must therefore interact differently with the receptor to induce a different agonist conformation and would suggest that SP-G must act at a site distinct from the AVP binding site on the V1A receptor. This is corroborated by the finding that SP-G-induced ERK activation was dependent on receptor expression, but was non-competitive in nature as shown by its ability to decrease saturable [3H]-AVP binding and accelerate [3H]-AVP dissociation.
Our data suggest different pathways for ERK activation recruited by AVP and SPG. SP-G-induced ERK activation was abolished by PTX whereas AVP-stimulated ERK activation was largely unaffected. Moreover, the SP-G-induced ERK activation was blocked by the EGFR tyrosine kinase inhibitor AG1478. These data support a multi-track signalling complex leading from the neuropeptide receptor to ERK activation, where one track (stabilised by AVP) is mediated by Gq and most likely involves PKC, whereas another (stabilised by SP-G) feeds into a receptor transactivation pathway (Wetzker and Bohmer, 2003). Although originally described for the EGF receptor activated by thrombin and LPA (Daub et al., 1996), this mechanism occurs in many cell types and via many different GPCRs and RTKs. GPCRs can also induce persistent ERK activation via a G-protein independent recruitment of β-arrestins (see Reiter and Lefkowitz, 2006). Activation of ERK by β-arrestin is slower in onset, persistent and sequestered in the cytosol, whereas G-protein-mediated activation of ERK is normally transient and translocates to the nucleus. The finding that SP-G-induced ERK activation was inhibited by PTX suggests the involvement of Gi and argues against a β-arrestin-mediated pathway.
Persistent ERK activation has been suggested to be consistent with Gi-dependent growth inhibition by the oxytocin ‘biased agonist’ atosiban (Rimoldi et al., 2003; Reversi et al., 2005). Selective activation of Gi has been suggested to inhibit proliferation and induce apoptosis in other endocrine tumours. Specific activation of Gi by GnRH antagonists may induce apoptosis in type 1 GnRH receptor expressing tumour cells (Maudsley et al., 2004). Although activation of ERK is normally associated with cell survival, prolonged ERK activation has been shown to induce apoptosis by RRR-α-tocopheryl succinate (You et al., 2001; Yu et al., 2001) and phenethyl isothiocyanate (Xiao and Singh, 2002), via mechanisms involving p53. In addition, prolonged ERK activation has been shown to have a primary role in the regulation of neuronal cell apoptosis (Cheung and Slack, 2004; Subramaniam et al., 2004), and in particular, sustained ERK is reported to be involved in G1-specific cell cycle arrest of human breast cancer cells (Alblas et al., 1998), NIH 3T3 murine fibroblasts (Sewing et al., 1997) and human myeloblastic leukaemia cells (Yen et al., 1998).
In endocrine cancers such as SCLC, mitogenic neuropeptide receptors such as the V1A receptor and the GRP receptor activate downstream signals which control proliferation and differentiation but also activate death signals leading to JNK activation and apoptosis. An agent that blocks the proliferative effects of Ca2+ mobilising mitogenic neuropeptides, while stimulating receptor-dependent apoptosis, would be highly advantageous in the management of tumour growth. This present study suggests that SP-G acts as a biased agonist promoting an agonist state of the V1A receptor which couples to Gi leading to prolonged activation of ERK but which blocks AVP-induced activation of PLC and subsequent elevation of intracellular Ca2+. Selective activation of Gi coupled with an inhibition of elevation of intracellular Ca2+ may represent a common sequence of events leading to growth arrest and apoptosis in endocrine tumours (Figure 6). As our previous work has shown that SP-G causes Gi-dependent ERK activation in cells expressing GRP receptors, we hypothesise that SP-G may interact with a common binding pocket within these neuropeptide receptors to induce biased agonism.
Figure 6.
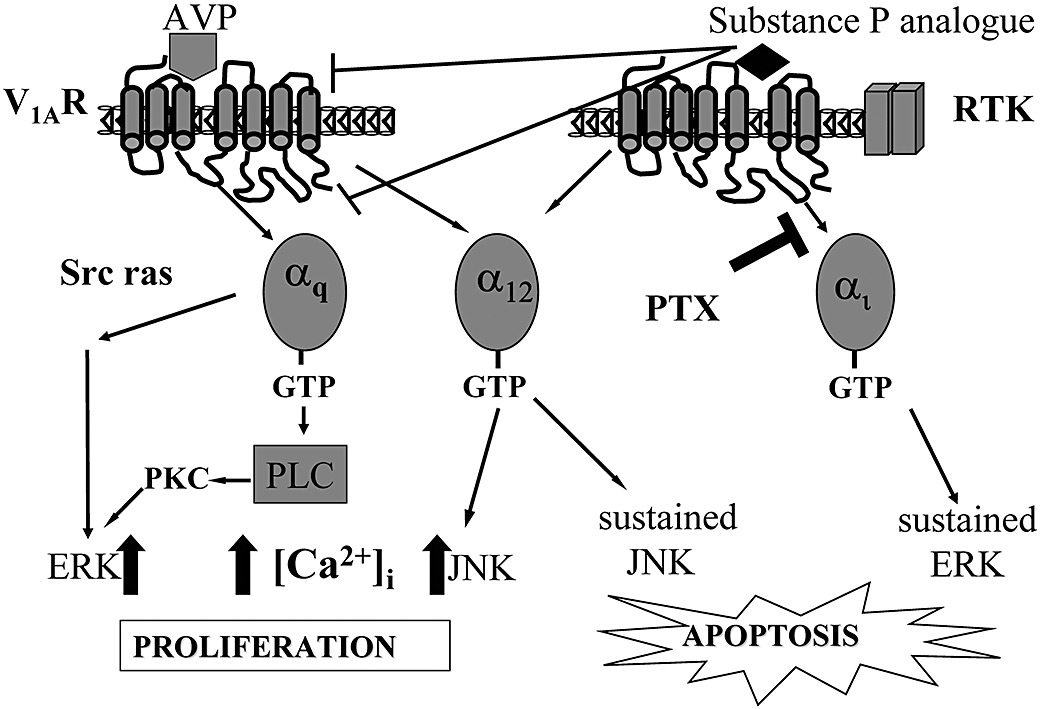
Schematic representation showing activation of V1A-mediated signalling pathways by AVP and substance P analogues. Substance P analogues such as SP-G inhibit calcium mobilisation but induce sustained ERK and JNK activation leading to apoptosis.
Many GPCRs interact with their various G-proteins via their intracellular domains. Previous studies have shown that the second intracellular loop of the V1A receptor is critical to interact with Gq and subsequently activate PLC (Liu and Wess, 1996; Erlenbach and Wess, 1998). Moreover, active peptide mimetics of the second intracellular loop specifically block AVP probably by disrupting intermolecular interactions between the i2 region of endogenous V1A receptors and another core region of the receptor (Demene et al., 2003). The present study shows that replacement of the second intracellular loop with that of the V2 receptor (V1i2) resulted in an abolition of PLC activation without affecting AVP-induced binding of Gq, suggesting that this region is involved in receptor coupling to Gq. However, the V1i2 chimera was able to partially activate ERK in response to AVP suggesting that AVP can activate ERK by additional mechanisms. The V1i2 mutation did not markedly affect the stimulation of ERK by SP-G, although the V1i3 chimera was less able to activate ERK in response to SP-G, than the wild-type receptor. Given that substitution of the second intracellular region of the V1A receptor did not affect SP-G-induced ERK activation coupled with the finding that SP-G increases ERK via Gi suggests that the i2 region may not be involved in V1A receptor coupling to Gi although it is crucial for receptor binding to Gq. This lends further credence to the existence of agonist selective states, as different agonists would be expected to alter receptor conformation to expose these different G-protein interacting sequences leading to selective activation of downstream signalling events.
Our previous results have demonstrated that SCLC tumours which have become resistant to chemotherapy express higher levels of neuropeptide receptors and become more sensitive to substance P analogues (Waters et al., 2003). Through direct activation of anti-proliferative effects, SP-G and its analogues would be more valuable anti-tumour agents than silent antagonists as they would not be dependent on high levels of circulating AVP and may be more beneficial in cancers which have become resistant to conventional chemotherapy.
Acknowledgments
U.T.-H. was supported by a Cancer Research UK PhD studentship. This work was supported in part by the Scottish Hospitals Endowment Research Trust and the British Lung Foundation. T.S. is a recipient of a Wellcome Trust Research Leave Fellowship.
Glossary
Abbreviations
- SP-G
[Arg6, D-Trp7,9, NmePhe8]-substance P (6-11)
- PLC
phospholipase C
- PKC
protein kinase C
- GPCR
G-protein coupled receptor
- ERK
extracellular signal regulated kinase
- JNK
c-jun-N-terminal kinase
- Fura-2-AM
Fura-2-tetraacetoxymethylester
- PBS
phosphate buffered saline
- PMSF
phenyl methyl sulphonyl fluoride
- AVP
arginine vasopressin
- GRP
gastrin releasing peptide
- BSA
bovine serum albumin
- PTX
Pertussis toxin
Conflict of interests
The authors state no conflict of interest.
References
- Abel A, Wittau N, Wieland T, Schultz G, Kalkbrenner F. Cell cycle-dependent coupling of the vasopressin V1a receptor to different G proteins. J Biol Chem. 2000;275:32543–32551. doi: 10.1074/jbc.M002171200. [DOI] [PubMed] [Google Scholar]
- Alblas J, Slager-Davidov R, Steenbergh PH, Sussenbach JS, van der Burg B. The role of MAP kinase in TPA-mediated cell cycle arrest of human breast cancer cells. Oncogene. 1998;16:131–139. doi: 10.1038/sj.onc.1201485. [DOI] [PubMed] [Google Scholar]
- Berg KA, Maayani S, Goldfarb J, Scaramellini C, Leff P, Clarke WP. Effector pathway-dependent relative efficacy at serotonin type 2A and 2C receptors: evidence for agonist-directed trafficking of receptor stimulus. Mol Pharmacol. 1998;54:94–104. [PubMed] [Google Scholar]
- Brink CB, Wade SM, Neubig RR. Agonist-directed trafficking of porcine alpha(2A)-adrenergic receptor signaling in Chinese hamster ovary cells: l-isoproterenol selectively activates G(s) J Pharmacol Exp Ther. 2000;294:539–547. [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Cheung EC, Slack RS. Emerging role for ERK as a key regulator of neuronal apoptosis. Sci STKE. 2004;2004:PE45. doi: 10.1126/stke.2512004pe45. [DOI] [PubMed] [Google Scholar]
- Chiu T, Wu SS, Santiskulvong C, Tangkijvanich P, Yee HF, Jr, Rozengurt E. Vasopressin-mediated mitogenic signaling in intestinal epithelial cells. Am J Physiol Cell Physiol. 2002;282:C434–C450. doi: 10.1152/ajpcell.00240.2001. [DOI] [PubMed] [Google Scholar]
- Clive S, Webb DJ, MacLellan A, Young A, Byrne B, Robson L, et al. Forearm blood flow and local responses to peptide vasodilators: a novel pharmacodynamic measure in the phase I trial of antagonist G, a neuropeptide growth factor antagonist. Clin Cancer Res. 2001;7:3071–3078. [PubMed] [Google Scholar]
- Codignola A, Missale C, Spano P, Clementi F, Carbone E, Sher E. Activation of opioid receptors inhibits neuronal-like calcium channels, distal steps of secretion, and cell proliferation in human small cell lung carcinoma cells. Ann NY Acad Sci. 1998;841:646–650. doi: 10.1111/j.1749-6632.1998.tb10996.x. [DOI] [PubMed] [Google Scholar]
- Coulson JM, Ahmed SI, Quinn JP, Woll PJ. Detection of small cell lung cancer by RT-PCR for neuropeptides, neuropeptide receptors, or a splice variant of the neuron restrictive silencer factor. Methods Mol Med. 2003;75:335–352. doi: 10.1385/1-59259-324-0:335. [DOI] [PubMed] [Google Scholar]
- Cuttitta F, Carney DN, Mulshine J, Moody TW, Fedorko J, Fischler A, et al. Autocrine growth factors in human small cell lung cancer. Cancer Surv. 1985;4:707–727. [PubMed] [Google Scholar]
- Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature. 1996;379:557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- DeBlasi A, O'Reilly K, Motulsky HJ. Calculating receptor number from binding experiments using same compound as radioligand and competitor. Trends Pharmacol Sci. 1989;10:227–229. doi: 10.1016/0165-6147(89)90266-6. [DOI] [PubMed] [Google Scholar]
- Demene H, Granier S, Muller D, Guillon G, Dufour MN, Delsuc MA, et al. Active peptidic mimics of the second intracellular loop of the V(1A) vasopressin receptor are structurally related to the second intracellular rhodopsin loop: a combined 1H NMR and biochemical study. Biochemistry. 2003;42:8204–8213. doi: 10.1021/bi027358n. [DOI] [PubMed] [Google Scholar]
- Erlenbach I, Wess J. Molecular basis of V2 vasopressin receptor/Gs coupling selectivity. J Biol Chem. 1998;273:26549–26558. doi: 10.1074/jbc.273.41.26549. [DOI] [PubMed] [Google Scholar]
- Friedmann AS, Malott KA, Memoli VA, Pai SI, Yu XM, North WG. Products of vasopressin gene expression in small-cell carcinoma of the lung. Br J Cancer. 1994;69:260–263. doi: 10.1038/bjc.1994.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furchgott RF. Metabolic factors that influence contractility of vascular smooth muscle. Bull NY Acad Med. 1966;42:996–1006. [PMC free article] [PubMed] [Google Scholar]
- Ghosh PM, Bedolla R, Thomas CA, Kreisberg JI. Role of protein kinase C in arginine vasopressin-stimulated ERK and p70S6 kinase phosphorylation. J Cell Biochem. 2004;91:1109–1129. doi: 10.1002/jcb.10789. [DOI] [PubMed] [Google Scholar]
- Guha S, Eibl G, Kisfalvi K, Fan RS, Burdick M, Reber H, et al. Broad-spectrum G protein-coupled receptor antagonist, [D-Arg1,D-Trp5,7,9,Leu11]SP: a dual inhibitor of growth and angiogenesis in pancreatic cancer. Cancer Res. 2005;65:2738–2745. doi: 10.1158/0008-5472.CAN-04-3197. [DOI] [PubMed] [Google Scholar]
- Gutkind JS, Crespo P, Xu N, Teramoto H, Coso OA. The pathway connecting m2 receptors to the nucleus involves small GTP-binding proteins acting on divergent MAP kinase cascades. Life Sci. 1997;60:999–1006. doi: 10.1016/s0024-3205(97)00040-4. [DOI] [PubMed] [Google Scholar]
- Hawtin SR, Simms J, Conner M, Lawson Z, Parslow RA, Trim J, et al. Charged extracellular residues, conserved throughout a G-protein-coupled receptor family, are required for ligand binding, receptor activation, and cell-surface expression. J Biol Chem. 2006;281:38478–38488. doi: 10.1074/jbc.M607639200. [DOI] [PubMed] [Google Scholar]
- Jarpe MB, Knall C, Mitchell FM, Buhl AM, Duzic E, Johnson GL. D-Arg1,D-Phe5,D-Trp7,9,Leu11] Substance P acts as a biased agonist toward neuropeptide and chemokine receptors. J Biol Chem. 1998;273:3097–3104. doi: 10.1074/jbc.273.5.3097. [DOI] [PubMed] [Google Scholar]
- Johnson BE, Chute JP, Rushin J, Williams J, Le PT, Venzon D, et al. A prospective study of patients with lung cancer and hyponatremia of malignancy. Am J Respir Crit Care Med. 1997;156:1669–1678. doi: 10.1164/ajrccm.156.5.96-10075. [DOI] [PubMed] [Google Scholar]
- Keegan BP, Memoli VA, North WG. Targeting the neurophysin-related cell surface antigen on small cell lung cancer cells using a monoclonal antibody against the glycopeptide region (MAG-1) of provasopressin. Mol Cancer Ther. 2002;1:1153–1159. [PubMed] [Google Scholar]
- Kenakin T. Agonist-receptor efficacy. II. Agonist trafficking of receptor signals. Trends Pharmacol Sci. 1995;16:232–238. doi: 10.1016/s0165-6147(00)89032-x. [DOI] [PubMed] [Google Scholar]
- Langdon S, Sethi T, Ritchie A, Muir M, Smyth J, Rozengurt E. Broad spectrum neuropeptide antagonists inhibit the growth of small cell lung cancer in vivo. Cancer Res. 1992;52:4554–4557. [PubMed] [Google Scholar]
- Laszlo FA, Laszlo F, Jr, De Wied D. Pharmacology and clinical perspectives of vasopressin antagonists. Pharmacol Rev. 1991;43:73–108. [PubMed] [Google Scholar]
- Liu J, Wess J. Different single receptor domains determine the distinct G protein coupling profiles of members of the vasopressin receptor family. J Biol Chem. 1996;271:8772–8778. doi: 10.1074/jbc.271.15.8772. [DOI] [PubMed] [Google Scholar]
- MacKinnon A, Sethi T. D-Arg6, D-Trp7,9, NmePhe8]-substance P (6-11) activates JNK and induces apoptosis in small cell lung cancer cells via an oxidant-dependent mechanism. Methods Mol Med. 2003;74:299–307. doi: 10.1385/1-59259-323-2:299. [DOI] [PubMed] [Google Scholar]
- MacKinnon AC, Armstrong RA, Waters CM, Cummings J, Smyth JF, Haslett C, et al. Arg6,D-Trp7,9,NmePhe8]-substance P (6-11) activates JNK and induces apoptosis in small cell lung cancer cells via an oxidant-dependent mechanism. Br J Cancer. 1999;80:1026–1034. doi: 10.1038/sj.bjc.6690458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKinnon AC, Waters C, Jodrell D, Haslett C, Sethi T. Bombesin and substance P analogues differentially regulate G-protein coupling to the bombesin receptor. Direct evidence for biased agonism. J Biol Chem. 2001;276:28083–28091. doi: 10.1074/jbc.M009772200. [DOI] [PubMed] [Google Scholar]
- MacKinnon AC, Tufail-Hanif U, Lucas CD, Jodrell D, Haslett C, Sethi T. Expression of V1A and GRP receptors leads to cellular transformation and increased sensitivity to substance-P analogue-induced growth inhibition. Br J Cancer. 2005;92:522–531. doi: 10.1038/sj.bjc.6602366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maudsley S, Davidson L, Pawson AJ, Chan R, de Maturana RL, Millar RP. Gonadotropin-releasing hormone (GnRH) antagonists promote proapoptotic signaling in peripheral reproductive tumor cells by activating a Galphai-coupling state of the type I GnRH receptor. Cancer Res. 2004;64:7533–7544. doi: 10.1158/0008-5472.CAN-04-1360. [DOI] [PubMed] [Google Scholar]
- Moody TW, Chan D, Fahrenkrug J, Jensen RT. Neuropeptides as autocrine growth factors in cancer cells. Curr Pharm Des. 2003;9:495–509. doi: 10.2174/1381612033391621. [DOI] [PubMed] [Google Scholar]
- North WG, Fay MJ, Du J. All three vasopressin receptor sub-types are expressed by small-cell carcinoma. Adv Exp Med Biol. 1998a;449:335–338. doi: 10.1007/978-1-4615-4871-3_42. [DOI] [PubMed] [Google Scholar]
- North WG, Fay MJ, Longo KA, Du J. Expression of all known vasopressin receptor subtypes by small cell tumors implies a multifaceted role for this neuropeptide. Cancer Res. 1998b;58:1866–1871. [PubMed] [Google Scholar]
- Ocejo-Garcia M, Ahmed SI, Coulson JM, Woll PJ. Use of RT-PCR to detect co-expression of neuropeptides and their receptors in lung cancer. Lung Cancer. 2001;33:1–9. doi: 10.1016/s0169-5002(00)00248-8. [DOI] [PubMed] [Google Scholar]
- Reiter E, Lefkowitz RJ. GRKs and beta-arrestins: roles in receptor silencing, trafficking and signaling. Trends Endocrinol Metab. 2006;17:159–165. doi: 10.1016/j.tem.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Reversi A, Rimoldi V, Marrocco T, Cassoni P, Bussolati G, Parenti M, et al. The oxytocin receptor antagonist atosiban inhibits cell growth via a ‘biased agonist’ mechanism. J Biol Chem. 2005;280:16311–16318. doi: 10.1074/jbc.M409945200. [DOI] [PubMed] [Google Scholar]
- Rimoldi V, Reversi A, Taverna E, Rosa P, Francolini M, Cassoni P, et al. Oxytocin receptor elicits different EGFR/MAPK activation patterns depending on its localization in caveolin-1 enriched domains. Oncogene. 2003;22:6054–6060. doi: 10.1038/sj.onc.1206612. [DOI] [PubMed] [Google Scholar]
- Rozengurt E. Signal transduction pathways in the mitogenic response to G protein-coupled neuropeptide receptor agonists. J Cell Physiol. 1998;177:507–517. doi: 10.1002/(SICI)1097-4652(199812)177:4<507::AID-JCP2>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Sethi T, Rozengurt E. Multiple neuropeptides stimulate clonal growth of small cell lung cancer: effects of bradykinin, vasopressin, cholecystokinin, galanin, and neurotensin. Cancer Res. 1991;51:3621–3623. [PubMed] [Google Scholar]
- Sethi T, Langdon S, Smyth J, Rozengurt E. Growth of small cell lung cancer cells: stimulation by multiple neuropeptides and inhibition by broad spectrum antagonists in vitro and in vivo. Cancer Res. 1992;52:2737s–2742s. [PubMed] [Google Scholar]
- Sewing A, Wiseman B, Lloyd AC, Land H. High-intensity Raf signal causes cell cycle arrest mediated by p21Cip1. Mol Cell Biol. 1997;17:5588–5597. doi: 10.1128/mcb.17.9.5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnett-Smith J, Zhukova E, Hsieh N, Jiang X, Rozengurt E. Protein kinase D potentiates DNA synthesis induced by Gq-coupled receptors by increasing the duration of ERK signaling in Swiss 3T3 cells. J Biol Chem. 2004;279:16883–16893. doi: 10.1074/jbc.M313225200. [DOI] [PubMed] [Google Scholar]
- Skrzydelski D, Lhiaubet AM, Lebeau A, Forgez P, Yamada M, Hermans E, et al. Differential involvement of intracellular domains of the rat NTS1 neurotensin receptor in coupling to G proteins: a molecular basis for agonist-directed trafficking of receptor stimulus. Mol Pharmacol. 2003;64:421–429. doi: 10.1124/mol.64.2.421. [DOI] [PubMed] [Google Scholar]
- Subramaniam S, Zirrgiebel U, von Bohlen Und Halbach O, Strelau J, Laliberte C, Kaplan DR, et al. ERK activation promotes neuronal degeneration predominantly through plasma membrane damage and independently of caspase-3. J Cell Biol. 2004;165:357–369. doi: 10.1083/jcb.200403028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibonnier M, Goraya T, Berti-Mattera L. G protein coupling of human platelet V1 vascular vasopressin receptors. Am J Physiol. 1993;264:C1336–C1344. doi: 10.1152/ajpcell.1993.264.5.C1336. [DOI] [PubMed] [Google Scholar]
- Waters CM, MacKinnon AC, Cummings J, Tufail-Hanif U, Jodrell D, Haslett C, et al. Increased gastrin-releasing peptide (GRP) receptor expression in tumour cells confers sensitivity to [Arg6,D-Trp7,9,NmePhe8]-substance P (6-11)-induced growth inhibition. Br J Cancer. 2003;88:1808–1816. doi: 10.1038/sj.bjc.6600957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiland T, Jakobs KH. Measurement of receptor-stimulated guanosine 5′-O-(gamma-thio) triphosphate binding by G proteins. Methods Enzymol. 1994;237:3–13. doi: 10.1016/s0076-6879(94)37048-6. [DOI] [PubMed] [Google Scholar]
- Wetzker R, Bohmer FD. Transactivation joins multiple tracks to the ERK/MAPK cascade. Nat Rev Mol Cell Biol. 2003;4:651–657. doi: 10.1038/nrm1173. [DOI] [PubMed] [Google Scholar]
- Woll PJ, Rozengurt E. D-Arg1,D-Phe5,D-Trp7,9,Leu11]substance P, a potent bombesin antagonist in murine Swiss 3T3 cells, inhibits the growth of human small cell lung cancer cells in vitro. Proc Natl Acad Sci USA. 1988;85:1859–1863. doi: 10.1073/pnas.85.6.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woll PJ, Rozengurt E. Multiple neuropeptides mobilise calcium in small cell lung cancer: effects of vasopressin, bradykinin, cholecystokinin, galanin and neurotensin. Biochem Biophys Res Commun. 1989;164:66–73. doi: 10.1016/0006-291x(89)91683-5. [DOI] [PubMed] [Google Scholar]
- Xiao D, Singh SV. Phenethyl isothiocyanate-induced apoptosis in p53-deficient PC-3 human prostate cancer cell line is mediated by extracellular signal-regulated kinases. Cancer Res. 2002;62:3615–3619. [PubMed] [Google Scholar]
- Yen A, Roberson MS, Varvayanis S, Lee AT. Retinoic acid induced mitogen-activated protein (MAP)/extracellular signal-regulated kinase (ERK) kinase-dependent MAP kinase activation needed to elicit HL-60 cell differentiation and growth arrest. Cancer Res. 1998;58:3163–3172. [PubMed] [Google Scholar]
- You H, Yu W, Sanders BG, Kline K. RRR-alpha-tocopheryl succinate induces MDA-MB-435 and MCF-7 human breast cancer cells to undergo differentiation. Cell Growth Differ. 2001;12:471–480. [PubMed] [Google Scholar]
- Yu W, Liao QY, Hantash FM, Sanders BG, Kline K. Activation of extracellular signal-regulated kinase and c-Jun-NH(2)-terminal kinase but not p38 mitogen-activated protein kinases is required for RRR-alpha-tocopheryl succinate-induced apoptosis of human breast cancer cells. Cancer Res. 2001;61:6569–6576. [PubMed] [Google Scholar]