

Abstract
Background and purpose:
Functional interactions between the G protein-coupled dopamine D1 and histamine H3 receptors have been described in the brain. In the present study we investigated the existence of D1–H3 receptor heteromers and their biochemical characteristics.
Experimental approach:
D1–H3 receptor heteromerization was studied in mammalian transfected cells with Bioluminescence Resonance Energy Transfer and binding assays. Furthermore, signalling through mitogen-activated protein kinase (MAPK) and adenylyl cyclase pathways was studied in co-transfected cells and compared with cells transfected with either D1 or H3 receptors.
Key results:
Bioluminescence Resonance Energy Transfer and binding assays confirmed that D1 and H3 receptors can heteromerize. Activation of histamine H3 receptors did not lead to signalling towards the MAPK pathway unless dopamine D1 receptors were co-expressed. Also, dopamine D1 receptors, usually coupled to Gs proteins and leading to increases in cAMP, did not couple to Gs but to Gi in co-transfected cells. Furthermore, signalling via each receptor was blocked not only by a selective antagonist but also by an antagonist of the partner receptor.
Conclusions and implications:
D1–H3 receptor heteromers constitute unique devices that can direct dopaminergic and histaminergic signalling towards the MAPK pathway in a Gs-independent and Gi-dependent manner. An antagonist of one of the receptor units in the D1–H3 receptor heteromer can induce conformational changes in the other receptor unit and block specific signals originating in the heteromer. This gives rise to unsuspected therapeutic potentials for G protein-coupled receptor antagonists.
Keywords: dopaminergic transmission, histaminergic transmission, receptor heteromers, signal transduction, dopamine D1 receptor, histamine H3 receptor, MAPK pathway, bioluminescent resonance energy transfer
Introduction
Although with some initial resistance from the scientific community, the existence of neurotransmitter receptor heteromers is becoming accepted. Neurotransmitter receptors cannot only be considered as single functional units, but as forming part of multimolecular aggregates localized in the plane of the plasma membrane, which can contain other interacting proteins, including receptors for the same or other neurotransmitters (Agnati et al., 2003; 2005; Franco et al., 2003; Bockaert et al., 2004). The functional significance of receptor heteromers is however just beginning to be understood. It is becoming clear that heteromerization of neurotransmitter receptors leads to functional entities that possess different biochemical characteristics with respect to the individual components of the heteromer. Thus, the quantitative or qualitative aspects of the signalling generated by stimulation of either receptor unit in the heteromer are different from those obtained during co-activation (Ferréet al., 2007; 2009; Franco et al., 2007; Rashid et al., 2007).
The striatum is the main input structure of the basal ganglia, which are subcortical structures involved in the processing of information related with the performance and learning of complex motor acts. GABAergic striatal efferent neurons constitute more than 95% of the striatal neuronal population (Gerfen, 2004). There are two subtypes of GABAergic striatal efferent neurons: GABAergic dynorphinergic neurons, which express the peptide dynorphin and dopamine D1 receptors, and GABAergic enkephalinergic neurons, which express the peptide enkephalin and dopamine D2 receptors (Gerfen, 2004). Histamine is an important neuromodulator of striatal function, and the striatum contains one of the highest densities of histamine H3 receptors in the brain (Pollard et al., 1993; Anichtchik et al., 2001; Brown et al., 2001). Both D1 receptors and H3 receptors are co-expressed in striatal GABAergic dynorphinergic neurons (Ryu et al., 1994; Pillot et al., 2002), where they have been reported to establish functional interactions (Arias-Montano et al., 2001; Sanchez-Lemus and Arias-Montano, 2004). In the present study we show that heteromerization of dopamine D1 receptors and histamine H3 receptors, produces dramatic changes in G protein coupling and signalling in human cell lines. Furthermore, both D1 receptor and H3 receptor antagonists could block the heteromer-mediated signalling, a fact that highlights new possibilities for G protein-coupled receptor (GPCR) pharmacology.
Methods
Expression vectors
A plasmid encoding the cDNA of the human H3 receptor was provided by Johnson & Johnson Pharmaceutical Research & Development, L.L.C. (San Diego, CA, USA). The H3 receptor cDNA without its stop codon was amplified by using sense and antisense primers harbouring a unique EcoRI site. The fragment was then subcloned to be in-frame with enhanced yellow variant of green fluorescent protein (EYFP) into the EcoRI site of pEYFP-N1 (Clontech, Heidelberg, Germany) to provide the plasmid H3 receptor–YFP, which expresses EYFP on the C-terminal ends of the receptor. The human cDNAs for cannabinoid CB1 receptors, 5HT2B receptors or D1 receptors cloned in pcDNA3.1 were amplified without their stop codons using sense and antisense primers harbouring unique BamHI and EcoRI to clone D1 receptors and CB1 receptors in EYFP vector or to clone 5HT2B receptors or D1 receptors in a Renilla luciferase-expressing vector (pcDNA3.1-RLuc). A pcDEF3 plasmid encoding the human cDNA of the H4 receptor fused to EYFP was also used as negative control. The cDNA for the human D1 receptor was also subcloned into BamHI and ApaI restriction sites of the pcDNA3.1/Hygro (Invitrogen, Grand Island, NY, USA) for the cell line stably expressing D1 receptors and H3 receptors. All constructs were verified by nucleotide sequencing. Nomenclature for receptors conforms to the BJP's Guide to Receptors and Channels (Alexander et al., 2008)
Cell culture and transfection
Human embryonic kidney (HEK)-293 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS), 100 units·mL−1 penicillin, 100 µg·mL−1 streptomycin, 2 mmol·L−1 L-glutamine and 100 µg·mL−1 sodium pyruvate (all from Invitrogen), at 37°C in a humidified atmosphere of 5% CO2 For Bioluminescence Resonance Energy Transfer (BRET) experiments cells were seeded in 35 mm diameter wells of 6-well plates, and transient transfection with the corresponding fusion protein cDNAs was performed the following day by using the calcium phosphate precipitation method (Jordan et al., 1996). Cells were harvested for 48 h after transfection and used for BRET experiments. The empty vector pcDNA3.1 was used to equilibrate the total amount of transfected DNA. For extracellular signal-regulated kinase (ERK) experiments, HEK-293 cells were grown to 80% confluence and transfected by using linear polyethylenimine, MW 25 000 (PEI, Polysciences, Eppelheim, Germany) with 5 µg of cDNA corresponding to human H3 receptors or human D1 receptors or both cDNAs at the same time. The empty vector pcDNA3.1 was used to equilibrate the total amount of transfected DNA. Briefly, the plasmid DNA was diluted in 50 µL of medium containing no additives (serum, antibiotics or other protein), and PEI was added (ratio µg DNA : µg PEI, 1:7.5) and incubated for 8 min at room temperature. Medium with 10% FBS was added to the DNA/PEI complex, and the mixture was applied to the cultures. After 2 h incubation, the mixture was replaced for grown medium.
SK-N-MC cells were grown in Eagle's minimal essential medium, supplemented with 10% FBS, 50 units·mL−1 penicillin, 50 µg·mL−1 streptomycin, non-essential amino acids, 2 mmol·L−1 L-glutamine and 50 µg·mL−1 sodium pyruvate at 37°C in a humidified atmosphere of 5% CO2 to 80% confluence. Cells were transiently transfected with 5 µg of cDNA corresponding to human D1 receptors (SK-N-MC/D1) using Lipofectamine™ 2000 (Invitrogen), according to the manufacturer's protocol. To obtain the SK-N-MC cells stably expressing human H3 receptors and human D1 receptors (SK-N-MC/D1H3), the SK-N-MC cells stably expressing the human H3R (SK-N-MC/H3) (provided by Johnson & Johnson Pharmaceutical Research & Development, L.L.C.) were grown to 30–40% confluence in 60 cm2 dishes in presence of 600 µg·mL−1 G418 (Invitrogen) and transfected with the cDNA corresponding to human D1 receptors using Lipofectamine™ 2000. SK-N-MC/D1H3 receptor cells were allowed to recover for 24 h before the addition of G418 and 300 µg·mL−1 hygromycin B (Invitrogen), and the colonies that survived selection were grown and tested by binding experiments and Western blotting.
Immunostaining
For immunocytochemistry, HEK-293 cells were grown on glass coverslips and transiently transfected with 0.1 µg of D1 receptor–RLuc and 0.1 µg H3 receptor–YFP constructs. After 48 h the cells were fixed in 4% paraformaldehyde for 15 min and washed with phosphate-buffered saline containing 20 mmol·L−1 glycine (buffer A) to quench the aldehyde groups. Then, after permeabilization with buffer A containing 0.05% Triton X-100 for 15 min, cells were treated with phosphate-buffered saline containing 1% bovine serum albumin. After 1 h at room temperature, cells expressing D1 receptor–RLuc were labelled with the primary rat monoclonal anti-D1 receptor antibody (1:200, Sigma, St. Louis, MO, USA) for 1 h, washed and stained with the secondary antibody Alexa Fluor®350 Goat anti-rat (1:1000, Invitrogen). The H3 receptor–YFP construct was detected by its fluorescence properties. Samples were rinsed and observed in a Leica SP5 confocal microscope (Leica Microsystems, Mannheim, Germany).
Bioluminescence Resonance Energy Transfer (BRET)
HEK-293 cells were transfected with 250 ng·well−1 of the cDNA construct coding for D1 receptor–RLuc, acting as BRET donor, and increasing amounts (0.5–9 µg·well−1) of the cDNA construct coding for the BRET acceptor H3 receptor–YFP or the negative control H4 receptor–YFP. After 48 h of transfection cells were washed twice with Hanks' balanced salt solution HBSS (137 mmol·L−1 NaCl, 5 mmol·L−1 KCl, 0.34 mmol·L−1 Na2HPO4.12H2O, 0.44 mmol·L−1 KH2PO4, 1.26 mmol·L−1 CaCl2.2H2O, 0.4 mmol·L−1 MgSO4.7H2O, 0.5 mmol·L−1 MgCl2, 10 mmol·L−1 HEPES, pH 7.4) supplemented with 0.1% glucose (w·v−1), detached by gently pipetting and resuspended in the same buffer. Sample protein concentration was determined to control cell number, using a Bradford assay kit (Bio-Rad, Munich, Germany) using bovine serum albumin dilutions as standards. Cell suspension (20 µg of protein) was dispensed in duplicates into 96-well black microplates with a transparent bottom (Porvair, King's Lynn, UK), and the fluorescence was measured using a Mithras LB940 fluorescence-luminiscence detector (Berthold, Bad Wildbad, Germany) with an excitation filter of 485 nm and an emission filter of 535 nm. For BRET measurement, 20 µg of cell suspension were distributed in duplicates into 96-well white opaque microplates (Porvair), and coelenterazine H (Molecular Probes Europe, Leiden, The Netherlands) was added at a final concentration of 5 µmol·L−1. After 1 min the readings were collected by using sequential integration of signals detected at 440–500 nm and 510–590 nm. The same samples were incubated for 10 min, and the luminescence was measured. Cells expressing BRET donors alone were used to determine background. The BRET ratio is defined as [(emission at 510–590)–(emission at 440–500)]–Cf where Cf corresponds to (emission at 510–590)–(emission at 440–500) for the D1 receptor–RLuc construct expressed alone in the same experiment. Curves were fitted by using a non-linear regression equation, assuming a single phase with GraphPad Prism software (San Diego, CA, USA).
Membrane preparation and protein determination
SK-N-MC/D1H3 receptor or transfected HEK-293 cells were harvested by centrifugation at 1500×g for 5 min. Cell pellet was washed twice with phosphate-buffered saline and resuspended in 10 volumes of 50 mmol·L−1 Tris-HCl buffer, pH 7.4. Cell suspensions were disrupted with a Polytron homogenizer (PTA 20 TS rotor, setting 3; Kinematica, Basel, Switzerland) for three 5 s periods, and membranes were obtained by centrifugation at 105 000×g (40 min, 4°C). The pellet was resuspended and centrifuged under the same conditions, stored at −80°C until use. Membranes were washed once more as described above and resuspended in 50 mmol·L−1 Tris-HCl buffer for immediate use. Protein was quantified by the bicinchoninic acid method (Pierce Chemical Co., Rockford, IL, USA) using bovine serum albumin dilutions as standard.
Radioligand binding experiments
Membrane suspensions (0.3 mg of protein per millilitre) were incubated for 1 h at 25°C in 50 mmol·L−1 Tris-HCl buffer, pH 7.4, containing 10 mmol·L−1 MgCl2 with the indicated radioligand in the presence or absence of competing ligands. To obtain competition curves, membranes were incubated with 2.2 nmol·L−1 of the D1 receptor antagonist [3H]SCH 23390 (NEN Perkin Elmer, Wellesley, MA, USA) or with 2.0 nmol·L−1 of the H3 receptor agonist [3H]R-α-methyl histamine ([3H]RAMH, Amersham, Buckinghamshire, UK) and increasing concentrations of the D1 receptor agonist SKF 38393 (Tocris, Ellisville, MO, USA) or H3 receptor agonist R-α-methyl histamine (RAMH) (triplicates of 13 different competitor concentrations from 0.1 nmol·L−1 to 10 µmol·L−1) in the absence or the presence of 10 nmol·L−1 of the H3 receptor agonist RAMH or 100 nmol·L−1 of the D1 receptor agonist SKF 38393 respectively. In all cases, non-specific binding was determined in the presence of an excess of unlabeled ligand [10 µmol·L−1 SCH 23390 (Sigma) for [3H]SCH 23390 binding or 10 µmol·L−1 RAMH for [3H]RAMH binding], and in competition experiments it was confirmed that the value was the same as calculated by extrapolation of the competition curves. Free and membrane-bound ligand were separated by rapid filtration of 500 µL aliquots in a cell harvester (Brandel, Gaithersburg, MD, USA) through Whatman GF/C filters (Brandel) soaked in 0.3% PEI, which were subsequently washed for 5 s with 5 mL of ice-cold Tris-HCl buffer. The filters were incubated with 10 mL of Ecoscint H scintillation cocktail (National Diagnostics, Atlanta, GA, USA) overnight at room temperature, and radioactivity counts were determined by using a Tri-Carb 1600 scintillation counter (PerkinElmer, Boston, MA, USA) with an efficiency of 62%.
Binding data analysis
Due to the homodimeric nature of D1 receptors (O'Dowd et al., 2005; Kong et al., 2006) and H3 receptors (Bakker et al., 2006), binding data from competition experiments were analysed by non-linear regression using the commercial Grafit curve-fitting software (Erithacus Software, Surrey, UK), by fitting data to the two-state dimer receptor model (Franco et al., 2005; 2006; Casadóet al., 2007) and not to the classical two-independent-site model for monomeric receptors that considers two binding sites (high and low affinity binding sites). To calculate the macroscopic equilibrium dissociation constants involved in the binding of the agonist SKF 38393 or RAMH to the D1 receptor or H3 receptor dimer respectively, the following equation for a competition binding experiment (Casadóet al., 2007) was considered:
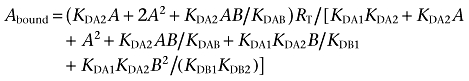 |
(1) |
where A represents the radioligand (the D1 receptor antagonist [3H]SCH 23390 or the H3 receptor agonist [3H]RAMH) concentration, RT is the total amount of receptor dimers and KDA1 and KDA2 are the macroscopic dissociation constants describing the binding of the first and the second radioligand molecule (A) to the dimeric receptor; B represents the assayed competing compound (the D1 receptor agonist SKF 38393 or the H3 receptor agonist RAMH) concentration, and KDB1 and KDB2 are, respectively, the equilibrium dissociation constants of the first and second binding of B; KDAB can be described as a hybrid equilibrium dissociation constant, which is the dissociation constant of B binding to a receptor dimer semi-occupied by A.
Because the radioligand A ([3H]RAMH or [3H]SCH 23390) showed non-cooperative behaviour (Franco et al., 2006); (Casadóet al., 2007), Eqn 1 was simplified to Eqn 2 due to the fact that KDA2= 4KDA1 (see Casadóet al., 2007):
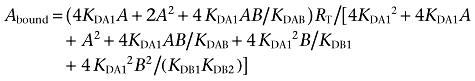 |
(2) |
The dimer homotropic cooperativity (DC) index for the competing ligand B (the agonist SKF 38393) was calculated (see Casadóet al., 2007; Gracia et al., 2008) according to the following expression:
Goodness of fit was tested according to reduced χ2 value given by the non-linear regression programme. The test of significance for two different model population variances was based upon the F-distribution (see Casadóet al., 1990, for details). Using this F-test, a probability greater than 95% (P < 0.05) was considered the criterion to select a more complex model (cooperativity) over the simplest one (non-cooperativity). In all cases, a probability of less than 70% (P > 0.30) resulted when one model was not significantly better than the other.
cAMP determination
The SK-N-MC or transfected HEK-293 cells were grown in 25 cm2 flasks to 80% confluence and incubated in serum-free medium for 16 h before the experiment. The day of experiment the cells were pre-incubated with 50 µmol·L−1 zardaverine (a phosphodiesterase inhibitor; Tocris) for 10 min at 37°C and treated for 10 min with 100 nmol·L−1 RAMH or 1 µmol·L−1 SKF 81297 (Tocris) in the presence or the absence of 10 µmol·L−1 forskolin (Sigma). When indicated, the H3 receptor antagonist thioperamide (Sigma) or the D1 receptor antagonist SCH 23390 (Tocris) were added at 10 µmol·L−1 final concentration and pre-incubated for 5 min before agonist addition. To stop the reaction cells were placed on ice and washed with ice-cold phosphate-buffered saline. The cells were incubated with 200 µL of HClO4 (4%) for 30 min, 1.5 mol·L−1 KOH was added to reach neutral pH, and samples were centrifuged. The supernatant was frozen at −20°C. The accumulation of cAMP was measured with cyclic AMP (3H) assay system (Amersham Biosciences, Uppsala, Sweden) as described in the manual from the manufacturer.
ERK phosphorylation assay
Cells were grown in 25 cm2 flasks to 80% confluence and cultured in serum-free medium for 16 h before the addition of any agent. Cells were treated or not with 10 µmol·L−1 SCH 23390 or 10 µmol·L−1 thioperamide for 30 min before the addition of the agonists 1 µmol·L−1 RAMH or 1 µmol·L−1 SKF 81297 for 2 min. In experiments evaluating Pertussis toxin (PTX), cells were pretreated with the toxin (100 ng·mL−1) for 16 h before ligand addition and in experiments evaluating cholera toxin (CTX), cells were pretreated with the toxin (1 µg·mL−1) for 30 min before ligand addition. At the end of the incubation periods, cells were rinsed with ice-cold phosphate-buffered saline and lysed by the addition of 500 µL of ice-cold lysis buffer (50 mmol·L−1 Tris-HCl pH 7.4, 50 mmol·L−1 NaF, 150 mmol·L−1 NaCl, 45 mmol·L−1β-glycerophosphate, 1% Triton X-100, 20 µmol·L−1 phenyl-arsine oxide, 0.4 mmol·L−1 NaVO4 and protease inhibitor cocktail). The cellular debris was removed by centrifugation at 13 000×g for 5 min at 4°C, and the protein was quantified by the bicinchoninic acid method by using bovine serum albumin dilutions as standard. To determine the level of ERK1/2 phosphorylation, equivalent amounts of protein (10 µg) were separated by electrophoresis on a denaturing 7.5% SDS-polyacrylamide gel and transferred onto PVDF membranes. The membranes were then probed with a mouse anti-phospho-ERK1/2 antibody (Sigma, 1:5000). In order to rule out that the differences observed were due to the application of unequal amounts of lysates, PVDF blots were stripped and probed with a rabbit anti-ERK1/2 antibody that recognizes both, phosphorylated and non-phosphorylated ERK1/2 (Sigma, 1:40 000). Bands were visualized by the addition of anti-mouse HRP conjugated (Dako, Glostrup, Denmark) or anti-rabbit HRP conjugated (Sigma) secondary antibodies, respectively, and SuperSignal West Pico Chemiluminescent Substrate (Pierce). Bands densities were quantified with a LAS-3000 (Fujifilm, Madrid, Spain), and the level of phosphorylated ERK1/2 isoforms was normalized for differences in loading using the total ERK protein band intensities. Quantitative analysis of detected bands was performed by Image Gauge V4.0 software.
Data analysis
Results are given as mean ± SEM. Differences between group means have been tested for significance (P < 0.05) by using Student's t-test for unpaired samples.
Results
Dopamine D1–histamine H3 receptor heteromerization
The BRET approach was used to demonstrate the ability of H3 receptors to heteromerize with D1 receptors. BRET measurements were performed in transiently co-transfected HEK-293 cells by using a constant amount of D1 receptor–RLuc and increasing amounts of H3 receptor–YFP. The subcellular localization of fusion proteins was investigated and the D1 receptor–RLuc and H3 receptor–YFP membrane expression and co-localization is shown in Figure 1A. Fusion of RLuc and YFP to D1 receptors or to H3 receptors did not modify receptor binding parameters (results not shown) or receptor function as determined by cAMP assays (Figure 1B). The correlation between properly folded receptors, determined by ligand binding, and fluorescence or luminescence is shown in Figure 1C. The expression level of the fusion proteins was in the range of 0.05 pmol·mg−1 protein for D1 receptor–RLuc and between 0.3 and 4.5 pmol·mg−1 protein for the different amounts of the transfected cDNA corresponding to H3 receptor–YFP. These data demonstrate that the fusion proteins are not strongly over-expressed at BRET50. A positive and saturable BRET signal was found for the pair D1 receptor–RLuc and H3 receptor–YFP (Figure 1D). From the saturation curve, a BRETmax of 0.034 ± 0.005 units and a BRET50 of 10 ± 4 were calculated. As the human histamine H4 receptor is closely related to the human H3 receptor [31% sequence identity at the protein level, which increases to 54% in the transmembrane region; de Esch et al. (2005)], the pair D1 receptor–RLuc and H4 receptor–YFP was used as a negative control. Also as negative controls the BRET pairs D1 receptor–RLuc and cannabinoid CB1 receptor–YFP or 5HT2B receptor–RLuc and H3 receptor–YFP were used. As shown in Figure 1D the negative controls gave a linear non-specific BRET signal, thus confirming the specificity of the interaction between D1 receptor–RLuc and H3 receptor–YFP in HEK-293 cells.
Figure 1.
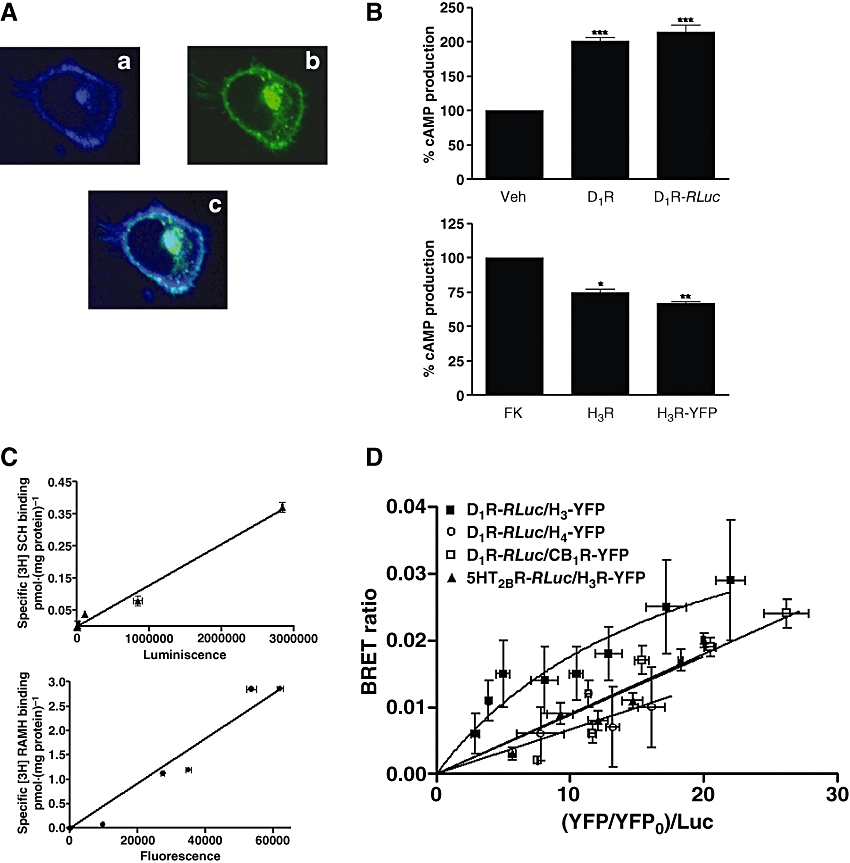
Heteromerization of functional D1 and H3 receptors. (A) Confocal microscopy images of HEK-293 cells expressing D1 receptor–RLuc (0.1 µg plasmid) and H3 receptor–YFP (0.1 µg plasmid). Proteins were identified by fluorescence or by immunocytochemistry. D1 receptor–RLuc immunoreactivity in shown in blue (a), H3 receptor–YFP fluorescence in shown in green (b) and co-localization of D1 receptor–RLuc and H3 receptor–YFP is shown in light blue (c). (B) Functionality of D1 receptor–RLuc (D1R–RLuc) and H3 receptor–YFP (H3R–YFP) constructs. HEK-293 cells transfected with 5 µg of cDNA corresponding to D1 receptors or D1 receptor–RLuc were stimulated with the D1 receptor agonist SKF 81297 (10 µmol·L−1), and HEK-293 cells transfected with 5 µg of cDNA corresponding to H3 receptors or H3 receptor–YFP were treated with 10 µmol·L−1 forskolin plus the H3 receptor agonist RAMH (0.1 µmol·L−1). Results (mean ± SEM; n= 2–4) are expressed as percentage over basal (upper panel) or over forskolin (FK) alone (lower panel); significantly different compared with the basal for D1 receptors and D1 receptor–RLuc or compared with forskolin alone for H3 receptors or H3 receptor–YFP, (non-paired Student's t-test: *P < 0.05, **P < 0.01 and ***P < 0.001). (C) Correlation between 2.1 nmol·L−1[3H]SCH 23390 binding and luminiscence expression (upper panel) or 1.9 nmol·L−1[3H]RAMH binding and fluorescence expression (lower panel) in HEK-293 cell transfected with increasing amounts of cDNA for D1 receptor–RLuc (upper panel) or H3 receptor–YFP (lower panel) (D) D1–H3 receptor heteromerization in HEK-293 cells. BRET experiments were performed with HEK-293 cells co-expressing D1 receptor–RLuc and H3 receptor–YFP, D1 receptor–RLuc and CB1 receptor–YFP, 5HT2B receptor–RLuc and H3 receptor–YFP or D1 receptor–RLuc and H4 receptor–YFP constructs. Co-transfections were performed with increasing amounts of plasmid–YFP (0.5–9 µg cDNA) whereas the plasmid–RLuc construct was maintained constant (250 ng cDNA). Both fluorescence and luminiscence of each sample were measured before every experiment to confirm similar donor expressions (about 250 000 luminescent units) while monitoring the increase acceptor expression (5000–80 000 fluorescent units). The relative amount of BRET is given as the ratio between the fluorescence of the acceptor and the luciferase activity of the donor. YFP0 corresponds to the fluorescence value of cells expressing the donor alone. BRET data are expressed as means ± SD of 3–13 different experiments grouped as a function of the amount of BRET acceptor. [3H]RAMH, [3H]R-α-methyl histamine; BRET, Bioluminescence Resonance Energy Transfer; HEK, human embryonic kidney; RLuc, Renilla luciferase; Veh, vehicle.
Intracellular crosstalk between histamine H3 and dopamine D1 receptors in HEK-293 cells
To investigate potential functional consequences of D1–H3 receptor heteromerization, HEK-293 cells expressing the human D1 receptor and/or the human H3 receptor at amounts giving approximately maximum BRET (Figure 1D) were treated with dopamine or histamine receptor agonists, and signalling was assayed by ERK1/2 phosphorylation. When cells expressing H3 receptors were treated with the selective H3 receptor agonist RAMH, no phosphorylation of ERKs was found (Figure 2A). On the other hand, when cells expressing D1 receptors were activated with the selective D1 receptor agonist SKF 81297, we observed a significant level of ERK1/2 phosphorylation, which was antagonized by the selective D1 receptor antagonist SCH 23390 (Figure 2A). When HEK-293 cells were transfected simultaneously with D1 receptors and H3 receptors, the D1 receptor agonist also activated the mitogen-activated protein kinase (MAPK) pathway, and this effect was blocked by SCH 23390 (Figure 2B). Interestingly, the H3 receptor agonist was also able to induce a significant ERK1/2 phosphorylation in co-transfected cells expressing D1–H3 receptor heteromers (Figure 2B). The specificity of the effect was proven by the blockade of the RAMH-induced effect by the H3 receptor antagonist, thioperamide (Figure 2B). These results indicate that the H3 receptor is able to couple to the MAPK-signalling pathway only in HEK-293 cells expressing D1 receptors and H3 receptors.
Figure 2.
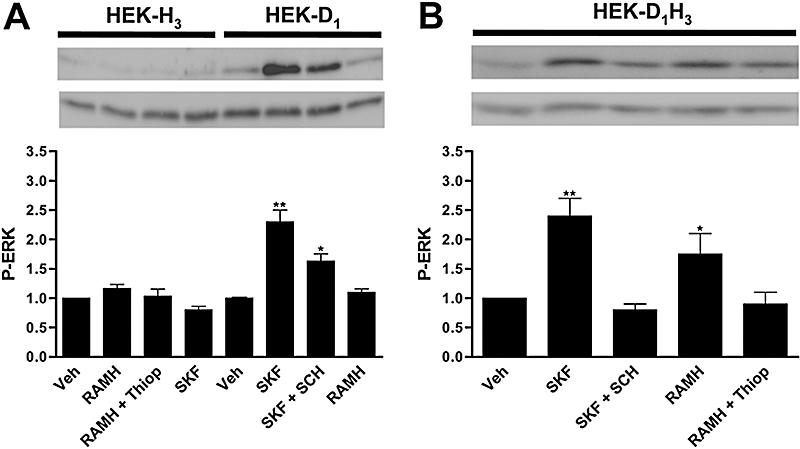
Crosstalk between H3 receptors and D1 receptors in HEK-293 cells. HEK-293 cells transiently expressing H3 receptors (HEK-H3) or D1 receptors (HEK-D1) (A) or both (HEK-D1H3) (B) were treated for 2 min with the H3 receptor agonist RAMH (1 µmol·L−1) or with the D1 receptor agonist SKF 81297 (1 µmol·L−1, SKF), in the presence or in the absence of the H3 receptor antagonist thioperamide (10 µmol·L−1, Thiop) or the D1 receptor antagonist SCH 23390 (10 µmol·L−1, SCH), and ERK1/2 phosphorylation (P-ERK) was determined as indicated in Methods. A representative Western blot is shown in each panel. The immunoreactive bands from three independent experiments were quantified, and values represent the mean ± SEM of fold increase of phosphorylation over the basal levels found in untreated cells. Significant differences with respect to the treatment with vehicle, were calculated by Student's t-test for unpaired samples (*P < 0.05 and **P < 0.01). ERK, extracellular signal-regulated kinase; HEK, human embryonic kidney; RAMH, R-α-methyl histamine; Veh, vehicle.
D1–H3 receptor heteromers in human neuroblastoma cells
For some receptor pairs it is possible to detect the heteromer receptor fingerprint (Ferréet al., 2007; Franco et al., 2007). This fingerprint often consists of intramembrane receptor–receptor interactions, in which changes in ligand binding characteristics of one receptor are obtained when the partner receptor is activated by using membrane preparations in which no intracellular crosstalk occurs (Agnati et al., 2003; El-Asmar et al., 2005; Ferréet al., 2007; Springael et al., 2007; Vilardaga et al., 2008). We investigated the possible existence of D1–H3 receptor intramembrane receptor interactions in SK-N-MC cells as a neuronal cell model. SK-N-MC cells have been used as a good model to transfect H3 receptors (Bongers et al., 2007); nevertheless, some authors have described the presence of D1 receptors in SK-N-MC cells (Sidhu et al., 1999; Chen et al., 2003; Moussa et al., 2006; Robinson et al., 2008) and some controversy exists about the functionality of these receptors, whether they couple to different G proteins (Kimura et al., 1995) and whether they signal (Chen et al., 2004) or not (Chan et al., 2005) towards the MAPK cascade. Our SK-N-MC cell clone expresses less than 0.030 pmol·(mg protein)−1 of D1 receptors [0.009 ± 0.004 pmol·(mg protein)−1 in the parental cell clone and 0.026 ± 0.005 pmol·(mg protein)−1 in the SK-N-MCMC/H3 cell clone], determined as [3H]SCH 23390 maximum binding, that is, at near saturating (>90%) concentrations of the radioligand. It should be noted that the D1 receptor agonist did not induce ERK1/2 phosphorylation neither in SK-N-MCMC/H3 (Figure 3A) or in parental cells (results not shown). Therefore it seems that different SK-N-MC cell clones may give different results.
Figure 3.
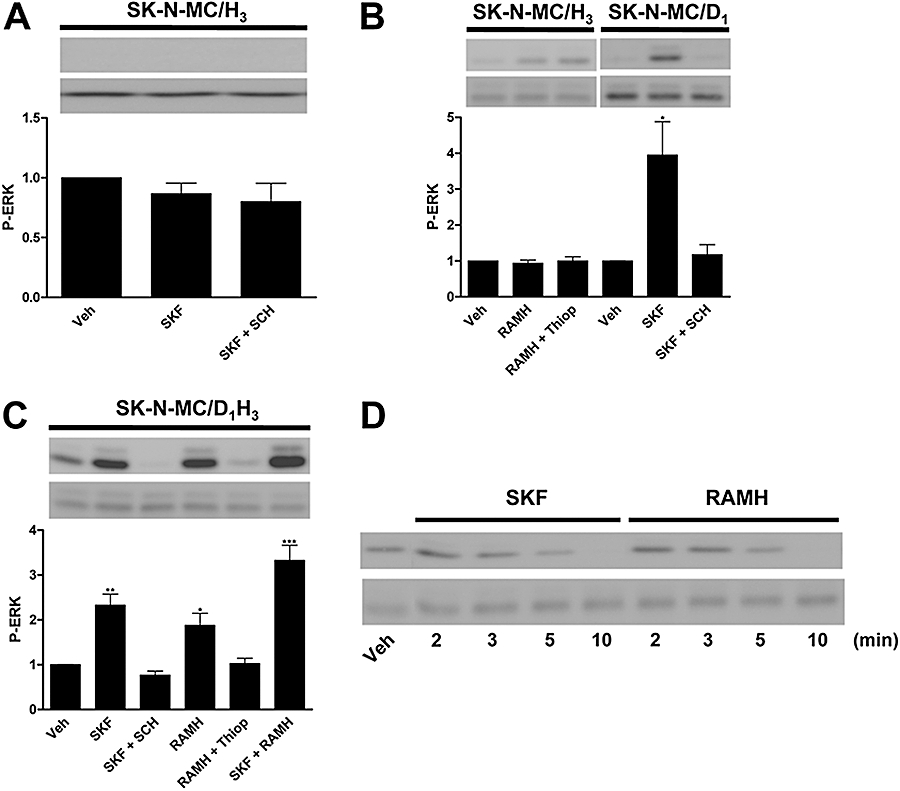
ERK1/2 phosphorylation (P-ERK) via the D1–H3 receptor heteromer in human neuroblastoma cells. SK-N-MC cells expressing H3 receptors (SK-N-MC/H3) or D1 receptors (SK-N-MC/D1) or both (SK-N-MC/D1H3) were treated with the H3 receptor agonist, RAMH (1 µmol·L−1), or with the D1 receptor agonist, SKF 81297 (1 µmol·L−1, SKF) alone or in combination, in the presence or in the absence of the H3 receptor antagonist, thioperamide (10 µmol·L−1, Thiop) or the D1 receptor antagonist, SCH 23390 (10 µmol·L−1, SCH). ERK1/2 phosphorylation was determined as indicated in Methods after 2 min of agonist treatment (A, B and C). In (D) a time–course response of ERK1/2 phosphorylation induced by 1 µmol·L−1 SKF 81297 or 1 µmol·L−1 RAMH in SK-N-MC/D1H3 cells is shown. A representative Western blot is shown in each panel. The immunoreactive bands from three to four experiments were quantified, and values represent the mean ± SEM of fold increase of phosphorylation over the basal levels found in untreated cells. Significant differences were calculated by Student's t-test for unpaired samples (*P < 0.05, **P < 0.01, ***P < 0.001). ERK, extracellular signal-regulated kinase; RAMH, R-α-methyl histamine; Veh, vehicle.
Membranes prepared from SK-N-MC human neuroblastoma cells stably expressing human versions of H3 receptors and D1 receptors (SK-N-MC/D1H3 cells) were used in binding competition experiments with [3H]SCH 23390 (2.2 nmol·L−1) as radioligand and increasing concentrations of SKF 38393 as competitor in the presence and in the absence of RAMH (10 nmol·L−1). Binding data were fitted to the two-state dimer receptor model (Franco et al., 2005; 2006; Casadóet al., 2007), to calculate the macroscopic equilibrium dissociation constants and the cooperativity index. The competition curve was biphasic in the absence of RAMH (significantly better than monophasic; F-test: P < 0.05), showing cooperativity in the D1 receptor agonist binding, but monophasic in the presence of RAMH. Variations in binding parameter values are shown in Table 1. These results indicate that an intramembrane crosstalk occurs between these receptors by which H3 receptor activation induces a shift from a cooperative to a non-cooperative binding and an overall decrease of affinity for the D1 receptor agonist binding. In contrast, D1 receptor stimulation did not influence the agonist binding to H3 receptor. In fact, competition experiments of 2 nmol·L−1[3H]RAMH binding versus increasing RAMH concentrations, performed as indicated in Methods, gave similar RT and KDB1 values for the non-cooperative RAMH binding both in the absence [0.46 ± 0.05 pmol·(mg protein)−1 and 2.9 ± 0.3 nmol·L−1] or presence [0.42 ± 0.04 pmol·(mg protein)−1 and 3.0 ± 0.3 nmol·L−1] of 100 nmol·L−1 SKF 38393.
Table 1.
Parameter values from competition experiments of [3H]SCH 23390 versus SKF 38393 in the presence and in the absence of RAMH (two-state dimer model)
| Agonists | Parameters | |||
|---|---|---|---|---|
| RT[pmol·(mg protein)−1] | KDB1 (nmol·L−1) | KDB2 (µmol·L−1) | DCB | |
| SKF 38393 | 0.436 ± 0.011 | 41 ± 3 | 1.3 ± 0.1 | −0.85 |
| SKF 38393 + RAMH | 0.404 ± 0.007 | 95 ± 9* | − | 0 |
Data are mean ± SEM values of three experiments; DCB, dimer cooperativity index for the binding of SKF 38393; KDB1 and KDB2, equilibrium dissociation constants for the first and second bindings of SKF 38393; RAMH, R-α-methyl histamine; RT, total amount of receptor dimers.
Significantly different compared with the KDB1 value of SKF 38393 alone, P < 0.05.
Signal transduction via D1–H3 receptor heteromers in human neuroblastoma cells
As described above, in HEK-293 cells, H3 receptors were able to mediate activation of the MAPK signalling pathway only through D1–H3 receptor heteromerization, demonstrated by BRET. This characteristic of the heteromer can also be used as a signalling fingerprint to identify the D1–H3 receptor heteromers. Thus, similar biochemical experiments were performed in SK-N-MC/D1H3 cells and cells transfected with only one receptor. As shown in Figure 3B, cells expressing D1 receptors are able to induce ERK1/2 phosphorylation in response to the treatment with the D1 receptor agonist SKF 81297, an effect that was blocked by SCH 23390. In SK-N-MC/H3 cells, RAMH had no effect on ERK1/2 phosphorylation (Figure 3B). However, in SK-N-MC/D1H3 cells both RAMH and SKF 81297 were able to activate the MAPK pathway, and co-activation of the two receptors did not result in synergism (Figure 3C). As shown in Figure 3D, there was no change in the time course of ERK1/2 phosphorylation when the agonists for D1 receptors or H3 receptors were used individually in SK-N-MC/D1H3 cells; the maximum phosphorylation was reached at 2 min and disappeared after 10 min stimulation. Overall the results were qualitatively identical to those obtained by using transiently transfected HEK-293 cells, demonstrating D1–H3 receptor heteromerization in neuroblastoma cells. These results also indicate that H3 receptors are able to couple to the MAPK pathway only in neuroblastoma cells expressing D1–H3 receptor heteromers. Similar experiments were performed by using a mutant version of H3 receptors (R3.50A; Arg 132 substituted by Ala) that is neither able to bind full agonists nor to signal (Appendix S1; Figure S1). The D1 receptor agonist was not able to provide any ERK1/2 phosphorylation signal when D1 receptors were co-expressed with the H3 R3.50A receptors (data not shown). This indicates that the D1 receptor signals towards MAPK via H3 receptors in cells co-expressing both receptors. Interestingly, in SK-N-MC/D1H3 cells, SKF 81297-induced ERK1/2 phosphorylation was reversed not only by SCH 23390, the specific D1 receptor antagonist, but also by thioperamide, the H3 receptor antagonist. Furthermore, RAMH-induced ERK1/2 phosphorylation in these cells was not only antagonized by thioperamide but also by SCH 23390 (Figure 4). It should be noted that both SKF 81297 and SCH 23390 are specific ligands for D1 receptors and do not appreciably interact with H3 receptors, as in SK-N-MC/H3 cells they were not able to reduce the 1.9 nmol·L−1[3H]RAMH binding [0.61 ± 0.02 vs. 0.57 ± 0.02 and 0.54 ± 0.04 pmol·(mg protein)−1 in the presence of 10 µmol·L−1 SKF 81297 or 10 µmol·L−1 SCH 23390, respectively]. Analogously, thioperamide and RAMH are specific H3 receptor ligands, as they were not able to reduce the 1.9 nmol·L−1[3H]SCH 23390 binding to SK-N-MC/D1H3 cells [0.72 ± 0.03 vs. 0.71 ± 0.02 and 0.73 ± 0.01 pmol·(mg protein)−1 in the presence of 10 µmol·L−1 thioperamide or 10 µmol·L−1 RAMH, respectively].
Figure 4.
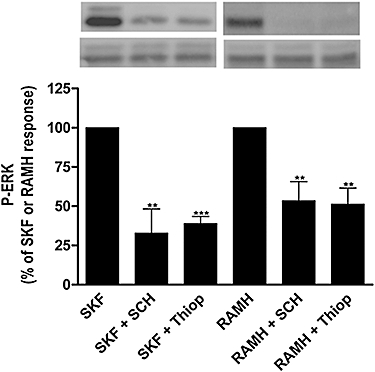
Effect of receptor antagonists on ERK1/2 phosphorylation (P-ERK) via the D1–H3 receptor heteromer in human neuroblastoma cells. SK-N-MC cells expressing H3 receptors and D1 receptors (SK-N-MC/D1H3) were treated with the H3 receptor agonist, RAMH (1 µmol·L−1), or the D1 receptor agonist, SKF 81297 (1 µmol·L−1. SKF), in the presence or in the absence of the H3 receptor antagonist, thioperamide (10 µmol·L−1, Thiop) or the D1 receptor antagonist, SCH 23390 (10 µmol·L−1, SCH). ERK1/2 phosphorylation was determined as indicated in Methods after 2 min of agonist treatment. A representative Western blot is shown. The immunoreactive bands from four experiments were quantified, and values represent the mean ± SEM of percentage of phosphorylation of agonist-treated cells. Significant differences were calculated by Student's t-test for unpaired samples (**P < 0.01, ***P < 0.001). ERK, extracellular signal-regulated kinase; RAMH, R-α-methyl histamine.
As expected from the known coupling of H3 receptor to heterotrimeric Gi proteins (Lovenberg et al., 1999; Drutel et al., 2001; Leurs et al., 2005), RAMH markedly inhibited the 10 µmol·L−1 forskolin-stimulated production of cAMP in SK-N-MC/H3 cells, and this effect was effectively blocked by thioperamide (Figure 5A), showing that in these neuroblastoma cells the H3 receptors are functional. Consistent with the very low D1 receptor expression in parental SK-N-MC cells and with the reported coupling of D1 receptors to Gs proteins (Neve et al., 2004), SKF 81297 was not able to increase cAMP in our SK-N-MC cell clone but was able to increase the intracellular levels of cAMP in SK-N-MC/D1 cells, an effect that was fully blocked by SCH 23390 (Figure 5B). Interestingly, in SK-N-MC/D1H3 cells, RAMH was still able to inhibit the cAMP accumulation induced by forskolin, and thioperamide blocked this action. In the same cell clone, which co-expresses the two receptors, SKF 81297 did not have any significant effect on cAMP production but reduced the forskolin-induced cAMP levels (Figure 5C). This suggests that D1 receptors are signalling in the D1–H3 receptor heteromer by coupling to an inhibitory G protein.
Figure 5.
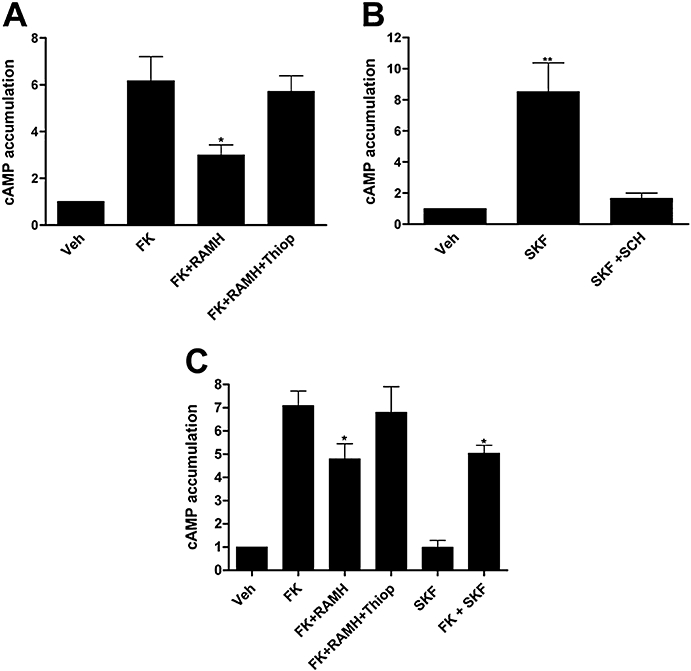
cAMP production by D1–H3 receptor heteromer in human neuroblastoma cells. SK-N-MC cells expressing (A) H3 receptors (SK-N-MC/H3) or (B) D1 receptors (SK-N-MC/D1) or (C) both (SK-N-MC/D1H3) were treated or not with 10 µmol·L−1 forskolin (FK) and the H3 receptor agonist, RAMH (0.1 µmol·L−1), and/or the D1 receptor agonist, SKF 81297 (1 µmol·L−1, SKF). The effect of the H3 receptor antagonist, thioperamide (10 µmol·L−1, Thiop) or the D1 receptor antagonist, SCH 23390 (10 µmol·L−1, SCH) was also assayed. cAMP levels were determined as indicated in Methods. Results are expressed as fold increase over basal levels obtained in untreated cells (mean ± SEM of three to five experiments). Significant differences were calculated by Student's t-test for unpaired samples (*P < 0.05, **P < 0.01). RAMH, R-α-methyl histamine; Veh, vehicle.
Based on the data described above, it is likely that a single heterotrimeric G protein, probably of the Gi/o type, is transducing the signal generated by either dopamine or histamine receptor agonists through the H3–D1 receptor heteromer. To check for this possibility SK-N-MC/D1H3 cells were pretreated with PTX, which specifically inactivates Gi/Go-mediated signalling pathways, or with CTX, which activates adenylyl cyclase by catalysing ADP-ribosylation of the stimulatory Gαs protein. After pretreatment with these toxins, H3 receptors and D1 receptors were activated by using respectively RAMH or SKF 81297. Whereas PTX inhibited the phosphorylation of ERK1/2 induced by RAMH and SKF 81297, CTX had no significant effect on the activation induced by any of the agonists (Figure 6). These results suggest that the activation of MAPK pathway through any of the two receptors in the D1–H3 receptor heteromer depends on Gi coupling.
Figure 6.
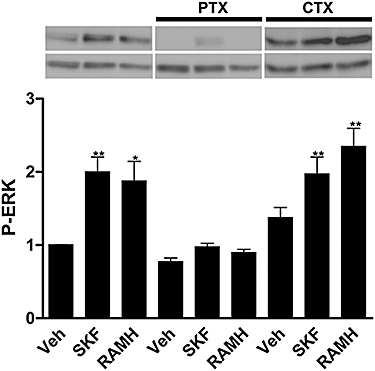
Effect of PTX and CTX on SKF- or RAMH-induced ERK1/2 phosphorylation (P-ERK). SK-N-MC cells expressing H3 receptors and D1 receptors (SK-N-MC/D1H3) were treated with PTX (100 ng·mL−1) for 16 h or with CTX (1 µg·mL−1) for 30 min prior to the addition of the H3 receptor agonist, RAMH (1 µmol·L−1), or the D1 receptor agonist, SKF 81297 (1 µmol·L−1, SKF). ERK1/2 phosphorylation was determined as indicated in Methods. A representative Western blot is shown. The immunoreactive bands from four experiments were quantified, and values represent the mean ± SEM of fold increase of phosphorylation over basal levels found in untreated cells. Significant differences were calculated by Student's t-test for unpaired samples (*P < 0.05 and **P < 0.01). CTX, cholera toxin; ERK, extracellular signal-regulated kinase; RAMH, R-α-methyl histamine; PTX, Pertussis toxin; Veh, vehicle.
Discussion
It seems that most, if not all, members of the GPCR superfamily can exist as homodimers (Bouvier, 2001; Devi, 2001; Marshall, 2001; Rios et al., 2001; George et al., 2002; Franco et al., 2003; Terrillon and Bouvier, 2004; Prinster et al., 2005; Milligan, 2006). The first indication of the existence of GPCR heteromers was obtained with radioligand binding experiments, which showed the existence of biochemical interactions between different GPCRs in brain membrane preparations (Agnati et al., 2003). In this kind of interactions, initially known as ‘intramembrane receptor–receptor interactions’, stimulation of one receptor changes the binding characteristics of another receptor for endogenous or exogenous ligands in crude membrane preparations (Agnati et al., 2003). This implied the lack of involvement of intracellular signalling and suggested some kind of allosteric interaction between adjacent receptors. Thus, at the beginning of the 90s, it was hypothesized that this intramembrane interaction could result from an intermolecular crosstalk, implying receptor heteromerization (Zoli et al., 1993). This is now considered as a biochemical fingerprint of a receptor heteromer (Ferréet al., 2007; Franco et al., 2007). Here we show that D1 receptors and H3 receptors are able to form D1–H3 receptor heteromers by BRET, in transiently transfected human embryonic cells, and by radioligand experiments in SK-N-MC/D1H3 cells, in which a specific H3 receptor agonist led to the disappearance of the cooperative D1 receptor agonist binding and to a significant change in the affinity of the D1 receptor for the agonist.
The crosstalk occurring via receptor heteromers has different components. One of them is the above discussed change in binding characteristics of one receptor upon activation of the partner receptor. Another is the crosstalk at the level of second messengers. For heteromers in which one of the constituent receptors is coupled to Gi/o whereas the other is coupled to Gs proteins, co-activation of the receptors would result not in a functional antagonism but in contradictory messages for the cell. Recent reports are providing clues to solve this conundrum. Significant advances in the case of heteromers for the same neurotransmitter have been achieved (Jordan and Devi, 1999; George et al., 2000; Fan et al., 2005; Ciruela et al., 2006; Rashid et al., 2007). Recent data indicate that in neurons co-expressing D1 receptors, a Gs-coupled receptor, and D2 receptors, a Gi-coupled receptor, D1–D2 receptor heteromers are formed that couple to a Gq protein (Rashid et al., 2007). This makes possible that a single neurotransmitter may increase cAMP levels, decrease cAMP levels or modify intracellular calcium levels depending on whether a given neuron (or microdomain in a neuron) expresses, respectively, the D1 receptor, the D2 receptor or the D1–D2 receptor heteromer. Two different neurotransmitters, dopamine and histamine, can interact with D1–H3 receptor heteromers. In neuroblastoma cells co-expressing D1 receptors and H3 receptors there is a change in the D1 receptor coupling from the Gs to the Gi protein, to which H3 receptors are already coupled. In fact, in the presence of the H3 receptor, D1 receptors were no longer coupled to Gs, and could not activate adenylyl cyclase, but were coupled to Gi, which transduced the signal towards the MAPK pathway. On the other hand, H3 receptors in cells co-expressing the two receptors could signal through both adenylyl cyclase (inhibiting enzyme activity) and MAPK (increasing ERK1/2 phosphorylation). These results indicate that D1–H3 receptor heteromers constitute unique devices to direct dopaminergic and histaminergic signalling towards the MAPK pathway in a Gs-independent and Gi-dependent manner. In the SK-N-MC cell clone stably expressing the human H3 receptors near to physiological receptor densities [0.1–1 pmol·(mg protein)−1], the H3 receptor agonist did not promote ERK1/2 phosphorylation unless the D1 receptor was co-expressed. It has been described that agonist binding to H3 receptors expressed at high densities in Chinese hamster ovary or in COS-7 cells can phosphorylate ERK1/2 (Drutel et al., 2001; Gbahou et al., 2003). In contrast to the cAMP response, the H3 receptor did not exhibit constitutive activation of the MAPK pathway (Gbahou et al., 2003). Whether ERK1/2 phosphorylation in these cells is solely due to the action of Gβγ subunits or to crosstalk with another receptor in these cells remains to be elucidated. In vivo, the first evidence of a positive correlation between ERK phosphorylation and memory improvement was given by Giovannini et al. (2003), who demonstrated an improvement in fear memory by H3 receptor-elicited ERK2 phosphorylation in hippocampal CA3 neurons in which the D1 receptor is co-expressed (Pantazopoulos et al., 2004)
Our results would be in agreement with the recently suggested 1:2 stoichiometry for the G protein: receptor interaction (Herrick-Davis et al., 2005). The results obtained by co-expressing D1 receptors and the mutant version of H3 receptors unable to activate MAPK indicate that GPCR activation results from a dynamic intersubunit interplay as shown in dimeric metabotropic glutamate receptors (Brock et al., 2007). The possibility that better explains the overall results is that D1 receptors are able to signal to the MAP kinase in the absence of the H3 receptor, but that in the presence of this receptor the signalling to ERK is mediated by the H3 receptor and not via the D1 receptor. Then, in the presence of non-functional H3 receptors, D1 receptor agonists are unable to produce ERK phosphorylation. Interestingly, not only the antagonist of their respective receptors but also the antagonist of the partner receptor counteracted the effect of D1 receptor or H3 receptor activation. Thus, an antagonist of one of the receptor units in the D1–H3 receptor heteromer can induce conformational changes in the other receptor unit and block specific signals originating in the heteromer. This fact broadens the therapeutic potential for GPCR antagonists.
Acknowledgments
This work was supported by Grants from Spanish Ministerio de Ciencia y Tecnología (SAF2005-00170 to E.I.C. SAF2006-05481 to R.F), Grant 060110 from Fundació La Marató de TV3 (RF) and by the intramural funds of the National Institute on Drug Abuse (SF). We acknowledge Dr Timothy A Esbenshade (Abbott Laboratories) for the kind gift of [3H]-A-349821. Sergi Ferré is employed by NIDA-NIH. No claim to US government work.
Glossary
Abbreviations:
- [3H]RAMH
[3H]R-α-methyl histamine
- BRET
Bioluminescence Resonance Energy Transfer
- CTX
cholera toxin
- EYFP
enhanced yellow variant of green fluorescent protein
- GPCR
G protein-coupled receptor
- PEI
polyethylenimine
- PTX
Pertussis toxin
- RAMH
R-α-methyl histamine
- RLuc
Renilla luciferase
Conflict of interest
The authors state no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Appendix S1
Figure S1 Binding and signalling of wild type or mutant (R3.50A) H3R in transfected HEK-293 cells. (A, B) HEK- 293 cells co-transfected with pTATAlucNEO/CRE121-3 (pTLNC121-3) CRE-luciferase reporter gene, and either the wild type or the mutant version of human H3R (Arg 132 substituted by Ala; see Methods) were treated with a full (R-αmethyl histamine) or an inverse (A-349821) agonist and the activity of the reporter gene was recorded (see Methods). (C, D) Binding to membranes from cells transfected with either the wild type or the mutant version of human H3R were performed by using (see Methods) either radiolabelled full (NAMH) or inverse (A-349821) agonists.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Agnati LF, Ferré S, Lluis C, Franco R, Fuxe K. Molecular mechanisms and therapeutical implications of intramembrane receptor/receptor interactions among heptahelical receptors with examples from the striatopallidal GABA neurons. Pharmacol Rev. 2003;55:509–550. doi: 10.1124/pr.55.3.2. [DOI] [PubMed] [Google Scholar]
- Agnati LF, Fuxe K, Ferré S. How receptor mosaics decode transmitter signals. Possible relevance of cooperativity. Trends Biochem Sci. 2005;30:188–193. doi: 10.1016/j.tibs.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (3rd) 2008;153(Suppl.)(2):S1–S209. doi: 10.1038/sj.bjp.0707746. edn (2008 revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anichtchik OV, Peitsaro N, Rinne JO, Kalimo H, Panula P. Distribution and modulation of histamine H(3) receptors in basal ganglia and frontal cortex of healthy controls and patients with Parkinson. s disease. Neurobiol Dis. 2001;8:707–716. doi: 10.1006/nbdi.2001.0413. [DOI] [PubMed] [Google Scholar]
- Arias-Montano JA, Floran B, Garcia M, Aceves J, Young JM. Histamine H(3) receptor-mediated inhibition of depolarization-induced, dopamine D(1) receptor-dependent release of [(3)H]-gamma-aminobutryic acid from rat striatal slices. Br J Pharmacol. 2001;33:165–171. doi: 10.1038/sj.bjp.0704053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker RA, Lozada AF, van Marle A, Shenton FC, Drutel G, Karlstedt K, et al. Discovery of naturally occurring splice variants of the rat histamine H3 receptor that act as dominant-negative isoforms. Mol Pharmacol. 2006;69:1194–1206. doi: 10.1124/mol.105.019299. [DOI] [PubMed] [Google Scholar]
- Bockaert J, Fagni L, Dumuis A, Marin P. GPCR interacting proteins (GIP) Pharmacol Ther. 2004;103:203–221. doi: 10.1016/j.pharmthera.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Bongers G, Sallmen T, Passani MB, Mariottini C, Wendelin D, Lozada A, et al. The Akt/GSK-3beta axis as a new signaling pathway of the histamine H(3) receptor. J Neurochem. 2007;103:248–258. doi: 10.1111/j.1471-4159.2007.04752.x. [DOI] [PubMed] [Google Scholar]
- Bouvier M. Oligomerization of G-protein-coupled transmitter receptors. Nat Rev Neurosci. 2001;2:274–286. doi: 10.1038/35067575. [DOI] [PubMed] [Google Scholar]
- Brock C, Oueslati N, Soler S, Boudier L, Rondard P, Pin JP. Activation of a dimeric metabotropic glutamate receptor by intersubunit rearrangement. J Biol Chem. 2007;282:33000–33008. doi: 10.1074/jbc.M702542200. [DOI] [PubMed] [Google Scholar]
- Brown RE, Stevens DR, Haas HL. The physiology of brain histamine. Prog Neurobiol. 2001;63:637–672. doi: 10.1016/s0301-0082(00)00039-3. [DOI] [PubMed] [Google Scholar]
- Casadó V, Cantí C, Mallol J, Canela EI, Lluís C, Franco R. Solubilization of A1 adenosine receptor from pig brain. Characterization and evidence of the role of the cell membrane on the coexistence of the high and low-affinity states. J Neurosci Res. 1990;26:461–473. doi: 10.1002/jnr.490260409. [DOI] [PubMed] [Google Scholar]
- Casadó V, Cortés A, Ciruela F, Mallol J, Ferré S, Lluis C, et al. Old and new ways to calculate the affinity of agonists and antagonists interacting with G-protein-coupled monomeric and dimeric receptors: the receptor-dimer cooperativity index. Pharmacol Ther. 2007;116:343–354. doi: 10.1016/j.pharmthera.2007.05.010. [DOI] [PubMed] [Google Scholar]
- Chan AS, Yeung WW, Wong YH. Integration of G protein signals by extracellular signal-regulated protein kinases in SK-N-MC neuroepithelioma cells. J Neurochem. 2005;94:1457–1470. doi: 10.1111/j.1471-4159.2005.03304.x. [DOI] [PubMed] [Google Scholar]
- Chen J, Wersinger C, Sidhu A. Chronic stimulation of D1 dopamine receptors in human SK-N-MC neuroblastoma cells induces nitric-oxide synthase activation and cytotoxicity. J Biol Chem. 2003;278:28089–28100. doi: 10.1074/jbc.M303094200. [DOI] [PubMed] [Google Scholar]
- Chen J, Rusnak M, Luedtke RR, Sidhu A. D1 dopamine receptor mediates dopamine-induced cytotoxicity via the ERK signal cascade. J Biol Chem. 2004;279:39317–33930. doi: 10.1074/jbc.M403891200. [DOI] [PubMed] [Google Scholar]
- Ciruela F, Casadó V, Rodrigues RJ, Lujan R, Burgueno J, Canals M, et al. Presynaptic control of striatal glutamatergic neurotransmission by adenosine A1-A2A receptor heteromers. J Neurosci. 2006;26:2080–2087. doi: 10.1523/JNEUROSCI.3574-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi LA. Heterodimerization of G-protein-coupled receptors: pharmacology, signaling and trafficking. Trends Pharmacol Sci. 2001;22:532–537. doi: 10.1016/s0165-6147(00)01799-5. [DOI] [PubMed] [Google Scholar]
- Drutel G, Peitsaro N, Karlstedt K, Wieland K, Smit MJ, Timmerman H, et al. Identification of rat H3 receptor isoforms with different brain expression and signaling properties. Mol Pharmacol. 2001;59:1–8. [PubMed] [Google Scholar]
- El-Asmar L, Springael JY, Ballet S, Andrieu EU, Vassart G, Parmentier M. Evidence for negative binding cooperativity within CCR5-CCR2b heterodimers. Mol Pharmacol. 2005;67:460–469. doi: 10.1124/mol.104.003624. [DOI] [PubMed] [Google Scholar]
- de Esch IJ, Thurmond RL, Jongejan A, Leurs R. The histamine H4 receptor as a new therapeutic target for inflammation. Trends Pharmacol Sci. 2005;26:462–469. doi: 10.1016/j.tips.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Fan T, Varghese G, Nguyen T, Tse R, O'Dowd BF, George SR. A role for the distal carboxyl tails in generating the novel pharmacology and G protein activation profile of mu and delta opioid receptor hetero-oligomers. J Biol Chem. 2005;280:38478–38488. doi: 10.1074/jbc.M505644200. [DOI] [PubMed] [Google Scholar]
- Ferré S, Ciruela F, Woods AS, Lluis C, Franco R. Functional relevance of neurotransmitter receptor heteromers in the central nervous system. Trends Neurosci. 2007;30:440–446. doi: 10.1016/j.tins.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Ferré S, Baler R, Bouvier M, Caron MG, Devi LA, Durroux T, et al. Building a new conceptual framework for receptor heteromers. Nat Chem Biol. 2009;5:131–134. doi: 10.1038/nchembio0309-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R, Canals M, Marcellino D, Ferré S, Agnati L, Mallol J, et al. Regulation of heptaspanning-membrane-receptor function by dimerization and clustering. Trends Biochem Sci. 2003;28:238–243. doi: 10.1016/S0968-0004(03)00065-3. [DOI] [PubMed] [Google Scholar]
- Franco R, Casadó V, Mallol J, Ferré S, Fuxe K, Cortés A, et al. Dimer-based model for heptaspanning membrane receptors. Trends Biochem Sci. 2005;30:360–366. doi: 10.1016/j.tibs.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Franco R, Casadó V, Mallol J, Ferrada C, Ferré S, Fuxe K, et al. The two-state dimer receptor model: a general model for receptor dimers. Mol Pharmacol. 2006;69:1905–1912. doi: 10.1124/mol.105.020685. [DOI] [PubMed] [Google Scholar]
- Franco R, Casadó V, Cortés A, Ferrada C, Mallol J, Woods A, et al. Basic concepts in G-protein-coupled receptor homo- and heterodimerization. Sci World J. 2007;7:48–57. doi: 10.1100/tsw.2007.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gbahou F, Rouleau A, Morisset S, Parmentier R, Crochet S, Lin JS, et al. Protean agonism at histamine H3 receptors in vitro and in vivo. Proc Natl Acad Sci USA. 2003;100:11086–11091. doi: 10.1073/pnas.1932276100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George SR, Fan T, Xie Z, Tse R, Tam V, Varghese G, et al. Oligomerization of mu- and delta-opioid receptors. Generation of novel functional properties. J Biol Chem. 2000;275:26128–26135. doi: 10.1074/jbc.M000345200. [DOI] [PubMed] [Google Scholar]
- George SR, O'Dowd BF, Lee SP. G-protein-coupled receptor oligomerization and its potential for drug discovery. Nat Rev Drug Discov. 2002;1:808–820. doi: 10.1038/nrd913. [DOI] [PubMed] [Google Scholar]
- Gerfen CR. Basal ganglia. In: Paxinos G, editor. The Rat Nervous System. Amsterdam: Elsevier Academis Press; 2004. pp. 445–508. [Google Scholar]
- Giovannini MG, Efoudebe M, Passani MB, Baldi E, Bucherelli C, Giachi F, et al. Improvement in fear memory by histamine-elicited ERK2 activation in hippocampal CA3 cells. J Neurosci. 2003;23:9016–9023. doi: 10.1523/JNEUROSCI.23-27-09016.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracia E, Cortés A, Meana JJ, García-Sevilla J, Herhsfield MS, Canela EI, et al. Human adenosine deaminase as an allosteric modulator of human A adenosine receptor: abolishment of negative cooperativity for [H](R)-PIA binding to the caudate nucleus. J Neurochem. 2008;107:161–170. doi: 10.1111/j.1471-4159.2008.05602.x. [DOI] [PubMed] [Google Scholar]
- Herrick-Davis K, Grinde E, Harrigan TJ, Mazurkiewicz JE. Inhibition of serotonin 5-hydroxytryptamine 2C receptor function through heterodimerization: receptor dimers bind two molecules of ligand and one G-protein. J Biol Chem. 2005;280:40144–40151. doi: 10.1074/jbc.M507396200. [DOI] [PubMed] [Google Scholar]
- Jordan M, Schallhorn A, Wurm FM. Transfecting mammalian cells: optimization of critical parameters affecting calcium-phosphate precipitate formation. Nucleic Acids Res. 1996;24:596–601. doi: 10.1093/nar/24.4.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan BA, Devi LA. G-protein-coupled receptor heterodimerization modulates receptor function. Nature. 1999;399:697–700. doi: 10.1038/21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K, White BH, Sidhu A. Coupling of human D-1 dopamine receptors to different guanine nucleotide binding proteins. Evidence that D-1 dopamine receptors can couple to both Gs and G(o) J Biol Chem. 1995;270:14672–14678. doi: 10.1074/jbc.270.24.14672. [DOI] [PubMed] [Google Scholar]
- Kong MM, Fan T, Varghese G, O'Dowd BF, George SR. Agonist-induced cell surface trafficking of an intracellularly sequestered D1 dopamine receptor homo-oligomer. Mol Pharmacol. 2006;70:78–89. doi: 10.1124/mol.105.021246. [DOI] [PubMed] [Google Scholar]
- Leurs R, Bakker RA, Timmerman H, de Esch IJ. The histamine H3 receptor: from gene cloning to H3 receptor drugs. Nat Rev Drug Discov. 2005;4:107–120. doi: 10.1038/nrd1631. [DOI] [PubMed] [Google Scholar]
- Lovenberg TW, Roland BL, Wilson SJ, Jiang X, Pyati J, Huvar A, et al. Cloning and functional expression of the human histamine H3 receptor. Mol Pharmacol. 1999;55:1101–1107. [PubMed] [Google Scholar]
- Marshall FH. Heterodimerization of G-protein-coupled receptors in the CNS. Curr Opin Pharmacol. 2001;1:40–44. doi: 10.1016/s1471-4892(01)00001-7. [DOI] [PubMed] [Google Scholar]
- Milligan G. G-protein-coupled receptor heterodimers: pharmacology, function and relevance to drug discovery. Drug Discov Today. 2006;11:541–549. doi: 10.1016/j.drudis.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Moussa CE, Tomita Y, Sidhu A. Dopamine D1 receptor-mediated toxicity in human SK-N-MC neuroblastoma cells. Neurochem Int. 2006;48:226–234. doi: 10.1016/j.neuint.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Neve KA, Seamans JK, Trantham-Davidson H. Dopamine receptor signaling. J Recept Signal Transduct Res. 2004;24:165–205. doi: 10.1081/rrs-200029981. [DOI] [PubMed] [Google Scholar]
- O'Dowd BF, Ji X, Alijaniaram M, Rajaram KD, Kong MM, Rashid A, et al. Dopamine receptor oligomerization visualized in living cells. J Biol Chem. 2005;280:37225–33735. doi: 10.1074/jbc.M504562200. [DOI] [PubMed] [Google Scholar]
- Pillot C, Heron A, Cochois V, Tardivel-Lacombe J, Ligneau X, Schwartz JC, et al. A detailed mapping of the histamine H(3) receptor and its gene transcripts in rat brain. Neuroscience. 2002;114:173–193. doi: 10.1016/s0306-4522(02)00135-5. [DOI] [PubMed] [Google Scholar]
- Pantazopoulos H, Stone D, Walsh J, Benes FM. Differences in the cellular distribution of D1 receptor mRNA in the hippocampus of bipolars and schizophrenics. Synapse. 2004;54:147–155. doi: 10.1002/syn.20076. [DOI] [PubMed] [Google Scholar]
- Pollard H, Moreau J, Arrang JM, Schwartz JC. A detailed autoradiographic mapping of histamine H3 receptors in rat brain areas. Neuroscience. 1993;52:169–189. doi: 10.1016/0306-4522(93)90191-h. [DOI] [PubMed] [Google Scholar]
- Prinster SC, Hague C, Hall RA. Heterodimerization of G protein-coupled receptors: specificity and functional significance. Pharmacol Rev. 2005;57:289–298. doi: 10.1124/pr.57.3.1. [DOI] [PubMed] [Google Scholar]
- Rashid AJ, So CH, Kong MMC, Furtak T, El-Ghundi M, Cheng R, et al. D1–D2 dopamine receptor heterooligomers with unique pharmacology are coupled to rapid activation of Gq/11 in. the striatum. Proc Natl Acad Sci USA. 2007;104:654–659. doi: 10.1073/pnas.0604049104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios CD, Jordan BA, Gomes I, Devi LA. G-protein-coupled receptor dimerization: modulation of receptor function. Pharmacol Ther. 2001;92:71–87. doi: 10.1016/s0163-7258(01)00160-7. [DOI] [PubMed] [Google Scholar]
- Robinson P, Lebel M, Cyr M. Dopamine D1 receptor-mediated aggregation of N-terminal fragments of mutant huntingtin and cell death in a neuroblastoma cell line. Neuroscience. 2008;153:762–772. doi: 10.1016/j.neuroscience.2008.02.052. [DOI] [PubMed] [Google Scholar]
- Ryu JH, Yanai K, Iwata R, Ido T, Watanabe T. Heterogeneous distributions of histamine H3, dopamine D1 and D2 receptors in rat brain. Neuroreport. 1994;5:621–624. doi: 10.1097/00001756-199401000-00022. [DOI] [PubMed] [Google Scholar]
- Sanchez-Lemus E, Arias-Montano JA. Histamine H3 receptor activation inhibits dopamine D1 receptor-induced cAMP accumulation in rat striatal slices. Neurosci Lett. 2004;364:179–184. doi: 10.1016/j.neulet.2004.04.045. [DOI] [PubMed] [Google Scholar]
- Sidhu A, Olde B, Humblot N, Kimura K, Gardner N. Regulation of human D1 dopamine receptor function and gene expression in SK-N-MC neuroblastoma cells. Neuroscience. 1999;9:537–547. doi: 10.1016/s0306-4522(98)00555-7. [DOI] [PubMed] [Google Scholar]
- Springael JY, Urizar E, Costagliola S, Vassart G, Parmentier M. Allosteric properties of G protein-coupled receptor oligomers. Pharmacol Ther. 2007;115:410–418. doi: 10.1016/j.pharmthera.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Terrillon S, Bouvier M. Roles of G-protein-coupled receptor dimerization. EMBO Rep. 2004;5:30–34. doi: 10.1038/sj.embor.7400052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilardaga JP, Nikolaev VO, Lorenz K, Ferrandon S, Zhuang Z, Lohse MJ. Conformational cross-talk between alpha2A-adrenergic and mu-opioid receptors controls cell signaling. Nat Chem Biol. 2008;4:126–131. doi: 10.1038/nchembio.64. [DOI] [PubMed] [Google Scholar]
- Zoli M, Agnati LF, Hedlund PB, Li XM, Ferré S, Fuxe K. Receptor-receptor interactions as an integrative mechanism in nerve cells. Mol Neurobiol. 1993;7:293–334. doi: 10.1007/BF02769180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.