

Abstract
Background and purpose:
Histamine H3 receptor antagonists are currently being evaluated for their potential use in a number of central nervous system disorders including Alzheimer's Disease (AD). To date, little is known about the state of H3 receptors in AD.
Experimental approach:
In the present study we used the radiolabelled H3 receptor antagonist [3H]GSK189254 to investigate H3 receptor binding in the amyloid over-expressing double mutant APPswe × PSI.MI46V (TASTPM) transgenic mouse model of AD and in post-mortem human AD brain samples.
Key results:
No significant differences in specific H3 receptor binding were observed between wild type and TASTPM mice in the cortex, hippocampus or hypothalamus. Specific [3H]GSK189254 binding was detected in sections of human medial frontal cortex from AD brains of varying disease severity (Braak stages I–VI). With more quantitative analysis in a larger cohort, we observed that H3 receptor densities were not significantly different between AD and age-matched control brains in both frontal and temporal cortical regions. However, within the AD group, [3H]GSK189254 binding density in frontal cortex was higher in individuals with more severe dementia prior to death.
Conclusions and implications:
The maintenance of H3 receptor integrity observed in the various stages of AD in this study is important, given the potential use of H3 antagonists as a novel therapeutic approach for the symptomatic treatment of AD.
Keywords: H3 receptor, Alzheimer's Disease, [3H]GSK189254, TASTPM mouse, neocortex
Introduction
Alzheimer's Disease (AD) is characterized by deficits in a number of neurotransmitter systems which are believed to result in cognitive dysfunction as well as neuropsychiatric behaviour. Loss of cholinergic neurons in the basal forebrain is one of the most prominent and consistent events occurring in AD (Whitehouse et al., 1982), and provided the rationale for the development of cholinergic replacement therapies such as acetylcholinesterase inhibitors (Bartus et al., 1982). Prominent cell loss also occurs in glutamatergic pyramidal neurons of the cortex and hippocampus (Greenamyre et al., 1988), while deficits in 5-hydroxytryptaminergic, GABAergic, noradrenergic and dopaminergic pathways have also been described, although the degree to which these correlate with cognitive and/or behavioural changes in AD can vary (Ramirez et al., 2005). The density of certain receptors linked to these neurotransmitter pathways are modulated in AD brain, including 5-HT2A receptors which are decreased in temporal cortex compared with age-matched controls (Lai et al., 2005).
The histaminergic system has also been implicated in AD, although its importance is difficult to assess due to a number of conflicting reports (Fernández-Novoa and Cacabelos, 2001). For example, histamine levels in AD brains have been reported to be increased in areas such as temporal and frontal cortex, basal ganglia and hippocampus (Cacabelos et al., 1989). However, other studies have shown decreases in histamine content in the hypothalamus, hippocampus and temporal cortex of AD brains (Mazurkiewicz-Kwilecki and Nsonwah, 1989; Panula et al., 1998). Histaminergic cell bodies are solely located in the tuberomamillary nucleus (TMN) of the posterior hypothalamus (Brown et al., 2001). Despite some reports showing the occurrence of neurofibrillary tangles in the TMN of AD patients, the distribution and number of histaminergic cell bodies was very similar to that of normal brains (Airaksinen et al., 1991). In contrast, another group showed a significant reduction in large-sized histamine containing neurons in the TMN where numerous neurofibrillary tangles were found, indicative of a central histaminergic dysfunction (Nakamura et al., 1993). High levels of histamine have also been reported in cerebrospinal fluid and serum of AD patients, although this may originate from mast cells as well as the central nervous system (CNS) (Fernández-Novoa and Cacabelos, 2001).
The physiological effects of histamine are mediated through four G-protein-coupled 7-transmembrane receptor subtypes, namely H1, H2, H3 and H4 (Brown et al., 2001; nomenclature follows Alexander et al., 2008). Few studies have investigated histamine receptor subtypes in AD brain. A positron emission tomography study has demonstrated a decrease in frontal and temporal H1 receptors in AD patients (Higuchi et al., 2000), while the number of H2 receptors in temporal cortex and striatum has been reported to be normal in AD post-mortem brains (Perry et al., 1998). We recently showed preliminary evidence for qualitatively normal H3 receptor binding in AD medial temporal cortex (Medhurst et al., 2007), while there are no reports to date describing H4 receptors in AD brain.
Among the histamine receptor subtypes, H3 receptors play an important regulatory role in the CNS. Activation of H3 autoreceptors can inhibit histamine synthesis and release from histaminergic neurons (Arrang et al., 1983), while activation of H3 heteroreceptors can inhibit release of other neurotransmitters such acetylcholine, noradrenaline, dopamine and 5-HT from non-histaminergic neurons (Schlicker et al., 1994; Blandina et al., 1996; Brown et al., 2001). Conversely, blockade of H3 receptors with selective antagonists can increase the release of neurotransmitters involved in cognitive processes (Fox et al., 2005; Medhurst et al., 2007). Selective H3 receptor antagonists have been shown to improve performance in a diverse range of rodent cognition paradigms (Hancock and Fox, 2004; Witkin and Nelson, 2004; Medhurst et al., 2007), and can also increase wakefulness (Brown et al., 2001; Barbier et al., 2004). This has led to the development of H3 receptor antagonists for the potential treatment of several CNS disorders including cognitive dysfunction in AD (Passani et al., 2004; Esbenshade et al., 2008).
GSK189254 is a novel, highly selective H3 receptor antagonist which shows efficacy in a number of cognition paradigms in rats (Medhurst et al., 2007). Given the limited information on H3 receptors in AD, we investigated H3 receptor binding using [3H]GSK189254 in the double mutant APPswe × PSI.MI46V (TASTPM) transgenic mouse model of AD. In these mice, over-expression of both human amyloid precursor protein (hAPP695swe) and presenilin-1 (PS1.M146V) transgenes results in β-amyloid (Aβ) deposition from 3 months and cognitive deficits from 6 to 8 months of age (Howlett et al., 2004). In addition, we carried out a detailed analysis of H3 receptor binding with [3H]GSK189254 in human post-mortem AD neocortex samples using both autoradiography and saturation binding studies. These studies demonstrate maintenance of H3 receptor integrity even in severe AD, an important finding given that H3 receptor antagonists are being pursued as a novel symptomatic treatment for AD.
Methods
TASTPM transgenic mice
All experimental procedures were conducted in compliance with the Home Office Guidance on the operation of the Animals (Scientific Procedures) Act 1986 under the authority granted in personal and project licenses, and procedures were reviewed and approved by the GlaxoSmithKline Procedures Review panel. Adequate measures were taken throughout to minimize pain or discomfort. TASTPM transgenic mice over-expressing human amyloid precursor protein and presenilin-1 cDNAs harbouring the swedish and M146V mutations respectively were generated as previously described (Richardson et al., 2003; Howlett et al., 2004). Thirteen or 16-month-old wild type (WT) and TASTPM mice were used for saturation binding (n = 5 per group) and autoradiography (n = 6–8 per group) studies respectively, ages by which significant cognitive deficit and Aβ load would have been present for over 6 months (Howlett et al., 2004).
Human brain tissues
For autoradiography studies, human medial frontal gyrus tissues of varying disease severity [AD Braak stages I, II, IV, V and VI; Braak and Braak (1991), male or female, ages 72–90 years, non-neurological cause of death] were obtained from the Netherlands Brain Bank following informed patient consent, approval of local ethics and GlaxoSmithKline human tissue committees and compliance with the Human Tissue Act (2006). Twenty µm frozen sections were prepared as previously described (Roberts et al., 2004). AD plaque pathology was confirmed in adjacent sections using monoclonal 1E8 antibody (1 : 1000 dilution) raised against the 13–27 fragment of Aβ as previously described (Howlett et al., 2004).
For saturation binding studies, tissues from a well-characterized cohort of community-based, longitudinally follow-up AD patients (Hope et al., 1997; 1999) were used. Cognitive functioning was assessed 4-monthly from study recruitment to death (mean follow-up 3.5 years) with the Mini-Mental State Examination (MMSE, scores of 0–30, Folstein et al., 1975). At death, informed consent was obtained from next of kin prior to the removal of brain, and tissues from the frontal (orbitofrontal gyrus, Brodmann area 11) and temporal (mid-temporal gyrus, Brodmann area 21) cortices of a maximum of 27 AD patients as well as 12 non-neurological controls were dissected, homogenized and stored at −75°C as previously described (Lai et al., 2003). Tissues from both regions were not available for all subjects, and n values for each assay are specified in Table 1. All samples were from severe AD (Braak V–VI) in this cohort.
Table 1.
Demographic and [3H]GSK189254 saturation binding variables of controls and AD patients
| Controls maximum n = 12 | AD maximum n = 27 | |
|---|---|---|
| Age (years) | 75 ± 3 | 81 ± 2 |
| Sex (M/F) | 6/6 | 11/16 |
| Post-mortem interval (h) | 43 ± 8 | 38 ± 5 |
| [3H]GSK189254 | ||
| Frontal cortex | ||
| KD | 39.6 ± 4 (9) | 73.8 ± 20 (19) |
| Bmax | 14.7 ± 1.0 (9) | 12.3 ± 1.4 (19) |
| Temporal cortex | ||
| KD | 39.6 ± 6 (6) | 46.4 ± 4 (22) |
| Bmax | 10.0 ± 2.0 (6) | 11.1 ± 0.9 (22) |
Data are mean ± SEM. Numbers in parentheses represent the n available for each region.
AD, Alzheimer's Disease; Bmax, binding density (in fmol mg−1 protein); F, female; KD, binding affinity (in pM); M, male.
H3 receptor autoradiography
Autoradiography studies were carried out based on previous methods (Roberts et al., 2004; Medhurst et al., 2007). Twenty µm frozen sections of human medial frontal gyrus, TASTPM or WT mouse brains were thaw-mounted on to gelatin-coated slides and stored at −80°C until time of assay. Sections were incubated in assay buffer [50 mmol·L−1 Tris-HCl, pH 7.7 and 5 mmol·L−1 ethylenediaminetetraacetic acid (EDTA)] containing 1 nmol·L−1[3H]GSK189254 for 60 min at room temperature (22°C). On anatomically adjacent sections, non-specific binding was determined in the presence of 10 µmol·L−1 imetit. Following incubation, all sections were rinsed five times for 3 min at 4°C in Tris-HCl buffer with the addition of 5 mmol·L−1 MgCl2. The sections were then quickly dipped in distilled water at 4°C to remove buffer salts and dried in a stream of cool air. Once dried, the sections were analysed by digital autoradiography using a Beta-Imager 2000 Instrument (Biospace, Paris, France). Adjacent sections were also stained with cresyl fast violet to allow for anatomical orientation. Amyloid plaque pathology was confirmed in adjacent sections using monoclonal 1E8 (1:1000) antibody raised against the 13–27 fragment of Aβ as previously described (Howlett et al., 2004).
In vitro H3 receptor saturation binding
H3 receptor binding in WT and TASTPM mouse brains was determined based on methods previously described using [3H]GSK189254 (Medhurst et al., 2007). For preparation of membranes, tissue from mouse whole brain (∼13-month-old TASTPM and age-matched WT control) was resuspended (1 g of tissue to 10 mL) in 50 mmol·L−1 Tris-HCl, 140 mmol·L−1 NaCl, 1 mmol·L−1 EDTA buffer (pH 7.4 at 4°C) and homogenized using a polytron P10 (2 × 10 s bursts at full speed). The homogenate was centrifuged at approximately 48 000×g in a Sorval Evolution RC centrifuge at 4°C for 20 min (SS34 rotor). The pellet was rinsed with water, resuspended in assay buffer (50 mmol·L−1 Tris-HCl, pH 7.4) and centrifuged as before. The final cell pellet was resuspended in assay buffer and frozen at −80°C until required.
Membranes (corresponding to approximately 20 µg of protein per well) and [3H]GSK189254 (20 nmol·L−1–0.02 nmol·L−1) were incubated in polypropylene tubes in a final volume of 200 µL of 50 mmol·L−1 Tris-HCl, pH 7.7, at 25°C containing 5 mmol·L−1 EDTA. Non-specific binding was determined in the presence of 10 µmol·L−1 Imetit. Reactions were conducted at 30°C for 45 min. The experiments were terminated by rapid filtration through Whatman GF/B filters (Whatman, Maidstone, UK) [presoaked in 0.3% (v/v) polyethyleneimine (PEI)]. The filters were washed with 4 × 2 mL aliquots of ice-cold buffer containing 50 mmol·L−1 Tris-HCl, pH 7.7, at 25°C and 5 mmol·L−1 MgCl2. Filters were dried, and added to vials each containing 4 ml of Ultima Gold MV scintillation fluid (Hewlett Packard, Palo Alto, CA, USA). Radioactivity was determined by liquid scintillation spectrometry using a Packard Tri-Carb 2500TR liquid scintillation counter (PerkinElmer Life and Analytical sciences, Boston, MA, USA). Protein concentrations were determined using the Bradford assay method (Bio-Rad protein assay Kit; Bio-Rad, York, UK) with bovine serum albumin as a standard.
For human AD studies, aliquots of frozen brain homogenates were thawed, diluted in assay buffer (50 mmol·L−1 Tris-HCl, pH 7.4) and added to six concentrations (0.05–5 nmol·L−1) of [3H]GSK189254 in triplicate for 2 h at 25°C. A concurrent series of assay tubes were set up with the addition of 10 µmol·L−1 unlabelled thioperamide maleate to define non-specific binding, which constituted <10% of total binding. An aliquot of the diluted homogenate was used for protein determination using the Coomasie blue method (Pierce Biotech Inc., Rockford, IL, USA). Incubation was terminated by rapid filtration in a cell harvester (Molecular Devices Ltd., Sunnyvale, CA, USA) with ice-cold sodium phosphate buffer through 0.1% PEI-treated GF/B glassfibre filters (Whatman BDS, Maidstone, UK). Filters were then dried and membrane-bound radioactivity was measured by liquid scintillation spectrometry with a Wallac Beta counter.
Data analysis
For autoradiography studies in TASTPM and WT mice, [3H]GSK189254 binding in cortex, hippocampus and hypothalamus was measured from five sections per animal and six measurements per region. This was determined previously to be a statistically validated number of sections. The levels of specific bound radioactivity were determined via the Beta-imager by counting the number of beta particles from delineated areas and the results are expressed as mean specific binding counts per minute per square millimeter (cpm·mm−2; n = 6–8 animals per group), and the data were analysed using a repeated measures anova approach (using Statistica v6.0 StatSoft Inc. software). For human brain, the levels of specific bound radioactivity in the brain areas were determined via the Beta-imager by counting the number of beta particles from delineated whole section areas (delineated area in the range of 86–252 mm2 across all sections measured with a mean delineated section area of 155.60 mm2) and the radioligand binding signal was expressed as counts per minute per section (cpm per section) for each Braak stage.
For saturation binding studies in TASTPM and WT mice, specific binding was analysed to estimate binding parameters Bmax (total number of binding sites) and KD (binding affinity) using GraphPad Prism 3.0 by GraphPad Software Inc. (San Diego, CA, USA). For human AD brain saturation binding studies with [3H]GSK189254, Scatchard transformation of data was performed using EBDA and LIGAND software (McPherson, 1985) to calculate KD and Bmax. Dementia severity (mean of the last five MMSE scores before death, i.e. MMSE5) was correlated with Bmax using Pearson's product moment as previously described (Lai et al., 2003; 2005). In all cases, binding isotherms were best fitted to single sites with Hill coefficients (NH) around 1.
Chemicals
[3H]GSK189254 (6-[(3-cyclobutyl-2,3,4,5-tetrahydro-1H-3-benzazepin-7-yl)oxy]-N-methyl-3-pyridinecarboxamide hydrochloride; specific activity 81 Ci·mmol−1) was prepared through a contract with GE Healthcare, UK. Imetit and thioperamide maleate were obtained from Tocris Cookson Inc. (Bristol, UK). All other chemicals were obtained from Invitrogen (Paisley, UK) or Sigma-Aldrich Co. (St Louis, MO) and were of reagent grade.
Results
TASTPM mice
Autoradiographic analysis of coronal brain sections (Figure 1A) from 16-month-old WT (i) and TASTPM (ii) mice revealed extensive specific [3H]GSK189254 binding (>80%) within the cerebral cortex, hippocampus and hypothalamus, while binding levels were negligible following co-incubation with 10 µmol·L−1 imetit to define non-specific binding (iii). The presence of amyloid plaques observed in TASTPM mice (v) confirmed significant amyloid load in comparison with the lack of plaques seen in WT mice (iv). Quantification of H3 receptor binding revealed no significant differences between WT and TASTPM mice (n = 6–8 per group) in cortex, hippocampus and hypothalamus (Figure 1B).
Figure 1.
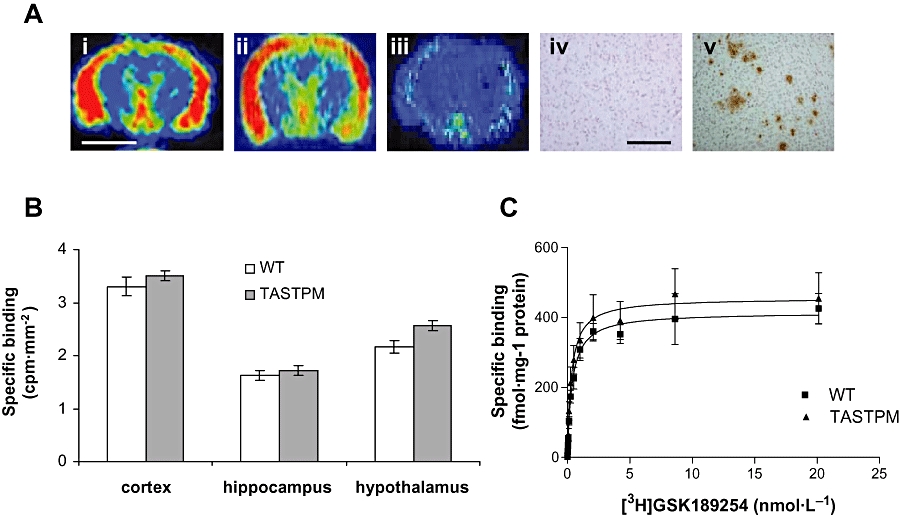
H3 receptor binding in TASTPM and WT mice measured using [3H]GSK189254 and real time autoradiography. (A) Representative pseudo-coloured images of coronal brain sections showing specific [3H]GSK189254 binding in cortex and hypothalamus of WT (i) and TASTPM mouse (ii), but negligible binding was observed in the presence of 10 µmol·L−1 imetit used to determine non-specific binding (iii). Scale bar = 2 mm. Lack of 1E8 staining shown in WT mouse (iv) but significant 1E8 staining of amyloid plaques in cortex of TASTPM mouse (v). Scale bar = 100 µm. (B) Quantitative bar graphs showing specific [3H]GSK189254 binding in cortex, hippocampus and hypothalamus of WT and TASTPM mice (mean ± SEM; n = 4–6 per group). No significant differences were observed between WT and TASTPM mice in any of these brain regions. Non-specific binding was determined in the presence of 10 µmol·L−1 imetit. (C) Saturation binding analysis for [3H]GSK189254 to H3 receptors in whole brain of WT and TASTPM mice (n = 5 pooled brains per group). Mean saturation binding curves (±SEM) are shown. No significant differences were observed in Bmax or KD calculated from individual saturation binding curves. TASTPM, double mutant APPswe × PSI.MI.MI46V transgenic mouse; WT, wild type.
Saturation binding was carried out in whole brain membranes from 13-month-old WT and TASTPM mice using [3H]GSK189254 (Figure 1C). Specific binding represented >90% total binding. Saturation analysis with [3H]GSK189254 in WT and TASTPM mice yielded Bmax values of 428 ± 64 and 455 ± 104 fmol·mg−1 protein respectively and KD values of 0.4 ± 0.02 and 0.33 ± 0.04 nmol·L−1 respectively, with no significant differences being observed between the groups (P > 0.05, Student's t-test).
Human AD brain H3 receptor autoradiography
Specific [3H]GSK189254 binding (>75%) was observed in sections of medial frontal AD cortex from Braak stages 0–1, II, IV, V and VI (Figure 2 lower panels). Plaque pathology was also evident in adjacent sections stained with the 1E8 antibody to total Aβ (Figure 2 upper panels). While quantitative comparisons of H3 receptor binding in AD versus control brains was restricted in this cohort due to limited number of brains available, specific H3 receptor binding was clearly saturable at all disease stages (Figure 3).
Figure 2.
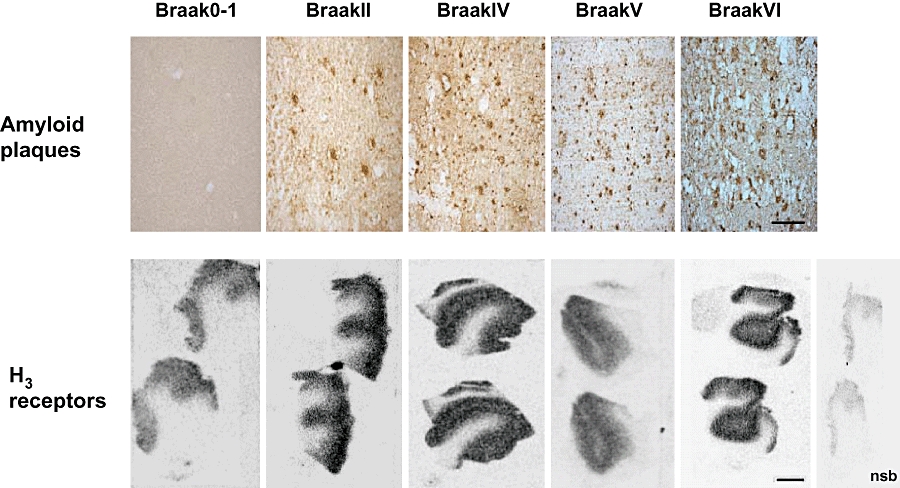
In vitro autoradiography of [3H]GSK189254 binding to human brain H3 receptors in medial frontal cortex from different Braak stages of AD. Upper panels show increasing Aβ plaque pathology across AD Braak stages 0-I, II, IV, V and VI detected using 1E8 antibody to total Aβ (scale bar 25 µm). Lower panels show specific H3 receptor binding maintained across all Braak stages even where severe plaque pathology is present (scale bar 1 mm). Non-specific binding was determined in the presence of 10 µmol·L−1 imetit. Aβ, β-amyloid; AD, Alzheimer's Disease.
Figure 3.
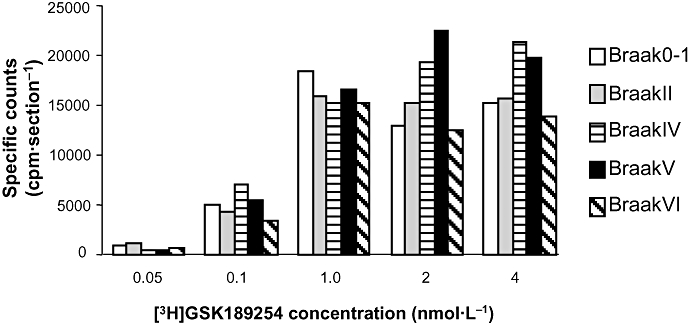
Semi-quantitative analysis of [3H]GSK189254 specific binding to human brain H3 receptors in medial frontal cortex from different Braak stages of AD determined using autoradiography. Comparable binding levels were observed in each Braak stage across the radioligand concentrations tested and binding appeared saturable. Data shown are from each individual expressed as specific binding in cpm per section. Non-specific binding was determined in the presence of 10 µmol·L−1 imetit. AD, Alzheimer's Disease.
Human AD brain H3 receptor saturation binding
We also performed quantitative analysis of H3 receptor binding by [3H]GSK189254 saturation assays in a separate, larger cohort of AD and control brains (see Methods). A representative plot of binding data obtained from AD frontal cortex is shown in Figure 4A. Full saturation of binding was achieved, and [3H]GSK189254 binding was of high specificity (>90% total) at radioligand concentrations around KD. The mean frontal and temporal cortex H3 receptor binding parameters and demographic details from control and AD patients are shown in Table 1. The age and post-mortem interval of controls and AD subjects were matched, and there were no differences in [3H]GSK189254 binding parameters between the two groups (Student's t-test, P > 0.05). Interestingly, within the AD group, frontal cortex [3H]GSK189254 Bmax correlated negatively with the mean of the last five MMSE scores before death (Figure 4B; MMSE5). MMSE5, rather than pre-death MMSE, was used as an indicator of dementia severity to avoid floor effects associated with prolonged terminal states typically seen in AD (Lai et al., 2003). Therefore, in the AD group, [3H]GSK189254 binding density was higher in individuals that had more severe dementia (lower MMSE5) prior to death. However, this association was not observed in the temporal cortex (Figure 4C).
Figure 4.
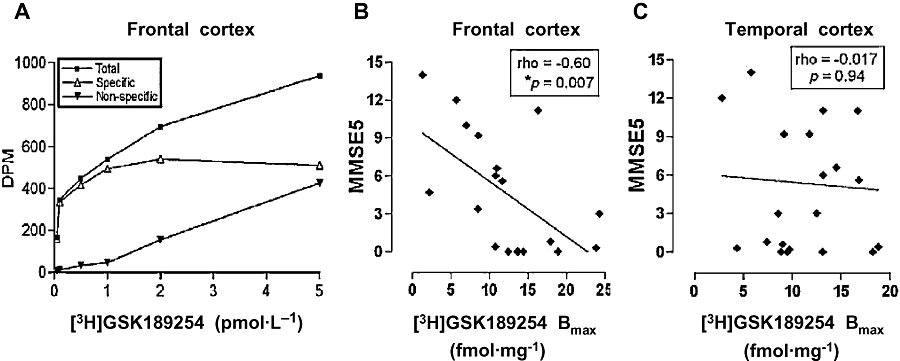
(A) Representative saturation binding plot of [3H]GSK189254 in post-mortem frontal cortex of an AD patient, with KD = 68 pM and Bmax = 14.1 fmol·mg−1 protein. DPM represents disintegrations per minute. (B, C) Correlation between MMSE5 (mean of last five Mini-Mental Examination scores before death) and Bmax (in fmol·mg−1 protein) for frontal (n = 19) and temporal (n = 22) cortex respectively. *Significant Spearman correlation (P < 0.05). AD, Alzheimer's Disease; DPM, disintegrations per min; MMSE, Mini-Mental State Examination.
Discussion
In the present study, we have shown that H3 receptor binding does not significantly alter in amyloid over-expressing TASTPM transgenic mice compared with WT mice, or in human AD brain compared with control brain. These results showing preservation of H3 receptors even in late stage AD are important, given that H3 receptor antagonists are currently being considered as a novel approach for the symptomatic treatment of AD.
Progressive cognitive decline is a key characteristic of AD and related dementias, and enhancing cognitive performance in these diseases represents a complex challenge, given the involvement of numerous neurotransmitter systems and brain regions (Corey-Bloom, 2002). Current therapies such as cholinesterase inhibitors provide only minimal benefit to a subset of patients and for a limited period of time, so a number of alternative symptomatic strategies are being pursued, including the development of selective histamine H3 receptor antagonists (Johnson et al., 2004).
It is well established that H3 receptors can modulate the release of multiple neurotransmitters involved in cognitive processes through a pre-synaptic inhibitory mechanism (Blandina et al., 1996; Fox et al., 2005). By blocking this feedback loop, structurally diverse non-imidazole H3 antagonists including GSK189254, ABT-239 and BF2.649 have been shown to increase the release of a number of neurotransmitters including acetylcholine and dopamine in the cortex, consistent with blockade of H3 heteroreceptors (Fox et al., 2005; Ligneau et al., 2007; Medhurst et al., 2007; Esbenshade et al., 2008). In addition, efficacious effects of multiple H3 receptor antagonists have been described in a huge array of rodent cognition paradigms involving different aspects of learning and memory, and the involvement of different neural substrates. These have been previously reviewed in detail and the majority of studies support pro-cognitive effects of H3 receptor antagonists (Witkin and Nelson, 2004; Esbenshade et al., 2008). For example, GSK189254 shows efficacy in a diverse battery of cognition models in rats (passive avoidance, water maze, attentional set shift and novel object recognition paradigms) when dosed acutely or repeatedly (Medhurst et al., 2007). These effects on multiple neurotransmitters coupled with their pro-cognitive benefit makes H3 receptor antagonists an attractive therapeutic approach for a number of CNS disorders including AD, other dementias and cognitive dysfunction in schizophrenia (Passani et al., 2004).
Given the interest in the potential development of H3 receptor antagonists for AD, we were interested in determining the integrity of H3 receptors in a transgenic mouse model of AD, as well as in human post-mortem AD brains. We have previously shown that [3H]GSK189254 is a suitable radioligand for studying H3 receptor binding in rat as well as human brain, although its affinity for the rat H3 receptor is ∼10-fold lower for the human receptor (Medhurst et al., 2007). Dense specific binding was observed in rat striatal, cortical, thalamic, hippocampal and hypothalamic areas as well as substantia nigra, with minimal binding in white matter areas, consistent with previous studies using other H3 receptor radioligands (Pollard et al., 1993; Barbier et al., 2004). In the current study, we observed a similar binding affinity and distribution pattern in mouse brain compared with the rat, with H3 receptor binding being particularly prominent in cerebral cortex, hippocampus and hypothalamus. Using both autoradiography and saturation binding analysis in homogenates, no differences were observed in H3 receptor density between TASTPM and WT mice, despite a significant amyloid load being present in cortical areas of TASTPM mice at 13–16 months of age. Given that it was previously not possible to distinguish degrees of cognitive deficit in these mice (Howlett et al., 2004), we did not investigate any possible correlation between H3 receptor expression and severity of cognitive deficit as we did with the MMSE5 in human AD brains. However, while significant amyloidosis and cognitive deficits (determined using the object recognition paradigm) are observed in this model, neurodegeneration is not a major pathological component (Howlett et al., 2004; 2008), so this may account for the lack of changes in neuronal H3 receptors observed in the current study. We therefore also investigated H3 receptor binding in human AD brains where neurodegeneration would have clearly occurred.
[3H]GSK189254 has previously been shown to label specific H3 receptor binding sites in human control and AD medial temporal cortex, with localization appearing consistent with a neuronal localization (Medhurst et al., 2007) in agreement with other studies (Martinez-Mir et al., 1990; Anichtchik et al., 2001). In the present study, we extended these preliminary observations to the medial frontal cortex of the same individuals, and into another very well-characterized cohort of AD and control medial and temporal cortex samples that have been described previously (Hope et al., 1997; 1999). Using autoradiography, specific H3 receptor binding was observed in medial frontal cortex samples of individuals with AD pathology of Braak stage I, II, IV, V and VI. These data were similar to our previous observations showing H3 receptor binding in medial temporal cortex of the same individuals, suggesting that H3 receptor expression is prevalent throughout the disease process and even in severe disease within two cortical areas predominantly affected by AD pathology. Unfortunately, a robust quantitative comparison in this cohort of AD brains compared with controls was not possible due to the limited sample number available, although saturable binding in each individual subject appeared comparable across the different Braak stages.
However, we were able to quantify [3H]GSK189254 binding using cortical homogenates from a larger cohort of AD (Braak V–VI) and control brains with more detailed clinical information regarding disease severity in terms of cognitive function. The KD for [3H]GSK189254 in human brain was ∼10-fold lower than that observed in TASTPM and WT mice, consistent with previous species differences in pharmacology observed between human and rat H3 receptors (Medhurst et al., 2007). Both control and AD neocortex exhibited low levels of [3H]GSK189254 binding (Bmax ∼ 10–15 fmol·mg−1) which were not significantly different between the two groups, consistent with the autoradiography studies in the smaller cohort described earlier. This suggests that H3 receptors may not be directly involved in AD, but may become significant as a negative modulator of neurotransmitter systems especially those that are damaged in severe disease, serving to exacerbate the deficits. The majority of cortical H3 receptors are believed to be heteroreceptors on intrinsic neurons mediating cholinergic and monoaminergic function rather than autoreceptors on histaminergic neurons, although these may also be present (Cumming et al., 1991; Pollard et al., 1993; Blandina et al., 1996; Fox et al., 2005). Therefore, it would follow that an H3 receptor antagonist could potentially ameliorate neurotransmitter deficit and improve cognition insofar as these cognitive processes are mediated by cholinergic and monoaminergic neurotransmitter systems.
Interestingly, although there were no differences between AD and control brains, frontal cortex [3H]GSK189254 binding density within the AD group appeared higher in patients with more severe dementia prior to death (based on MMSE5), although this was not the case in temporal cortex. The increase in H3 receptor binding in frontal cortex is difficult to comprehend. Perhaps it is a compensation mechanism to counteract changes elsewhere in the histaminergic system in severe AD such as a decrease in frontal cortex H1 receptors, although a decrease in these receptors is also observed in the temporal cortex (Higuchi et al., 2000). In addition, the functional consequence of increased H3 receptor density could be a further decrease in cognitive neurotransmitters and hence further exacerbation of cognitive deficits, and so would not be a positive compensatory effect. Alternatively, the increase in H3 receptor binding in brains of individuals with more severe dementia could be simply related to loss of cholinergic neurons. The reason for the regional difference observed between frontal and temporal cortex is also not clear, although it may reflect differences in AD pathology in different cortical regions. One could speculate that it reflects a region-specific difference in function of the H3 receptor, or that the functioning of systems being modulated by H3 receptors (e.g. acetylcholine and monoamines) has reached a nadir and are incapable of being activated due to the early selective degeneration of the temporal lobe compared with the frontal cortex (Wilcock and Esiri, 1987; Scahill et al., 2002). However, given the relatively small sample size, further studies are required to explore this potential difference in a larger cohort of AD and control brains.
To our knowledge, this is the first quantitative study assessing H3 receptor binding density in AD brains. In studies with brains from other neurodegenerative conditions such as Parkinson's Disease, H3 receptor binding was either increased or unchanged (Anichtchik et al., 2001), highlighting the importance of studying different cohorts. The fact that we consistently observed H3 binding in severe dementia in both our cohorts strengthens the conclusion that H3 receptors appear relatively spared in AD, and are available for potential therapeutic blockade by H3 antagonists.
While we have demonstrated little difference in H3 receptor binding in AD versus control brain, this does not provide information on whether receptor coupling or other downstream events are affected. For example, given that H3 receptor antagonists can increase the release of various neurotransmitters (Schlicker et al., 1994; Blandina et al., 1996), the effects of this potential therapeutic intervention in AD could be modulated if there were changes in density of other receptors mediating the responses to the increased neurotransmitters released following H3 receptor blockade. Further detailed studies are required to understand whether mechanisms downstream from the H3 receptor are affected in AD, and could result in different responses to H3 receptor antagonists than in non-diseased individuals.
In conclusion, we have shown in the TASTPM mouse model of AD where significant Aβ plaque load is present, that H3 receptor binding was no different from WT mice. In addition, we have demonstrated both qualitatively and quantitatively that H3 receptor binding is preserved throughout different Braak stages of AD, and does not appear significantly different in AD brains compared with age-matched controls. This maintenance of H3 receptor integrity is important, given the potential use of H3 antagonists as a novel therapeutic approach for the symptomatic treatment of AD.
Acknowledgments
The authors would like to thank Professor Margaret Esiri for the provision of post-mortem tissue, and Dr Janet Keene and Professor Tony Hope for providing clinical data. In Singapore, this study was supported by grants from the Department Clinical Research, Singapore General Hospital (DCR/P06/2006) to MKPL, and a Faculty start-up grant to CPLHC.
Glossary
Abbreviations:
- Aβ
β-amyloidy
- AD
Alzheimer' Disease
- MMSE
Mini-Mental State Examination
- PEI
polyethyleneimine
- TMN
tuberomamillary nucleus
- WT
wild type
Conflict of interest
AD Medhurst, JC Roberts, SH Brown and S Roman are employees of GlaxoSmithKline.
References
- Airaksinen MS, Paetau A, Paljarvi L, Reinikainen K, Riekkinen P, Suomalainen R, et al. Histamine neurons in human hypothalamus: anatomy in normal and Alzheimer diseased brains. Neuroscience. 1991;44:465–481. doi: 10.1016/0306-4522(91)90070-5. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (3rd edn) 2008;153(Suppl.)(2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anichtchik OV, Peitsaro N, Rinne JO, Kalimo H, Panula P. Distribution and modulation of histamine H(3) receptors in basal ganglia and frontal cortex of healthy controls and patients with Parkinson's disease. Neurobiol Dis. 2001;8:707–716. doi: 10.1006/nbdi.2001.0413. [DOI] [PubMed] [Google Scholar]
- Arrang JM, Garbarg M, Schwartz JC. Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature. 1983;302:832–837. doi: 10.1038/302832a0. [DOI] [PubMed] [Google Scholar]
- Barbier AJ, Berridge C, Dugovic C, Laposky AD, Wilson SJ, Boggs J, et al. Acute wake-promoting actions of JNJ-5207852, a novel, diamine-based H3 antagonist. Br J Pharmacol. 2004;143:649–661. doi: 10.1038/sj.bjp.0705964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartus RT, Dean RL, III, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217:408–414. doi: 10.1126/science.7046051. [DOI] [PubMed] [Google Scholar]
- Blandina P, Giorgetti M, Bartolini L, Cecchi M, Timmerman H, Leurs R, et al. Inhibition of cortical acetylcholine release and cognitive performance by histamine H3 receptor activation in rats. Br J Pharmacol. 1996;119:1656–1664. doi: 10.1111/j.1476-5381.1996.tb16086.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Brown RE, Stevens DR, Haas HL. The physiology of brain histamine. Prog Neurobiol. 2001;63:637–672. doi: 10.1016/s0301-0082(00)00039-3. [DOI] [PubMed] [Google Scholar]
- Cacabelos R, Yamatodani A, Niigawa H, Hariguchi S, Tada K, Nishimura T, et al. Brain histamine in Alzheimer's disease. Methods Find Exp Clin Pharmacol. 1989;11:353–360. [PubMed] [Google Scholar]
- Corey-Bloom J. The ABC of Alzheimer's disease: cognitive changes and their management in Alzheimer's disease and related dementias. Int Psychogeriatr. 2002;14(Suppl.)(1):51–75. doi: 10.1017/s1041610203008664. [DOI] [PubMed] [Google Scholar]
- Cumming P, Shaw C, Vincent SR. High affinity histamine binding site is the H3 receptor: characterization and autoradiographic localization in rat brain. Synapse. 1991;8:144–151. doi: 10.1002/syn.890080208. [DOI] [PubMed] [Google Scholar]
- Esbenshade TA, Browman KE, Bitner RS, Strakhova M, Cowart MD, Brioni JD. The histamine H3 receptor: an attractive target for the treatment of cognitive disorders. Br J Pharmacol. 2008;154:1166–1181. doi: 10.1038/bjp.2008.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Novoa L, Cacabelos R. Histamine function in brain disorders. Behav Brain Res. 2001;124:213–233. doi: 10.1016/s0166-4328(01)00215-7. [DOI] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR. ‘Mini-mental state’. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- Fox GB, Esbenshade TA, Pan JB, Radek RJ, Krueger KM, Yao BB, et al. Pharmacological properties of ABT-239 [4-(2-{2-[(2R)-2-Methylpyrrolidinyl]ethyl}-benzofuran-5-yl)benzonitrile]: II. Neurophysiological characterization and broad preclinical efficacy in cognition and schizophrenia of a potent and selective histamine H3 receptor antagonist. J Pharmacol Exp Ther. 2005;313:176–190. doi: 10.1124/jpet.104.078402. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT, Maragos WF, Albin RL, Penney JB, Young AB. Glutamate transmission and toxicity in Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry. 1988;12:421–430. doi: 10.1016/0278-5846(88)90102-9. [DOI] [PubMed] [Google Scholar]
- Hancock AA, Fox GB. Perspectives on cognitive domains, H3 receptor ligands and neurological disease. Expert Opin Investig Drugs. 2004;13:1237–1248. doi: 10.1517/13543784.13.10.1237. [DOI] [PubMed] [Google Scholar]
- Higuchi M, Yanai K, Okamura N, Meguro K, Arai H, Itoh M, et al. Histamine H(1) receptors in patients with Alzheimer's disease assessed by positron emission tomography. Neuroscience. 2000;99:721–729. doi: 10.1016/s0306-4522(00)00230-x. [DOI] [PubMed] [Google Scholar]
- Hope T, Keene J, Gedling K, Cooper S, Fairburn C, Jacoby R. Behaviour changes in dementia. 1: point of entry data of a prospective study. Int J Geriatr Psychiatry. 1997;12:1062–1073. doi: 10.1002/(sici)1099-1166(199711)12:11<1062::aid-gps675>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Hope T, Keene J, Fairburn CG, Jacoby R, McShane R. Natural history of behavioural changes and psychiatric symptoms in Alzheimer's disease. A longitudinal study. Br J Psychiatry. 1999;174:39–44. doi: 10.1192/bjp.174.1.39. [DOI] [PubMed] [Google Scholar]
- Howlett DR, Richardson JC, Austin A, Parsons AA, Bate ST, Davies DC, et al. Cognitive correlates of Abeta deposition in male and female mice bearing amyloid precursor protein and presenilin-1 mutant transgenes. Brain Res. 2004;1017:130–136. doi: 10.1016/j.brainres.2004.05.029. [DOI] [PubMed] [Google Scholar]
- Howlett DR, Bowler K, Soden PE, Riddell D, Davis JB, Richardson JC, et al. Abeta deposition and related pathology in an APP × PS1 transgenic mouse model of Alzheimer's disease. Histol Histopathol. 2008;23:67–76. doi: 10.14670/HH-23.67. [DOI] [PubMed] [Google Scholar]
- Johnson CN, Roland A, Upton N. New symptomatic strategies in Alzheimer's disease. Drug Discov Today Ther Strateg. 2004;1:13–19. [Google Scholar]
- Lai MK, Tsang SW, Francis PT, Esiri MM, Keene J, Hope T, et al. Reduced serotonin 5-HT1A receptor binding in the temporal cortex correlates with aggressive behavior in Alzheimer disease. Brain Res. 2003;974:82–87. doi: 10.1016/s0006-8993(03)02554-x. [DOI] [PubMed] [Google Scholar]
- Lai MK, Tsang SW, Alder JT, Keene J, Hope T, Esiri MM, et al. Loss of serotonin 5-HT2A receptors in the postmortem temporal cortex correlates with rate of cognitive decline in Alzheimer's disease. Psychopharmacology (Berl) 2005;179:673–677. doi: 10.1007/s00213-004-2077-2. [DOI] [PubMed] [Google Scholar]
- Ligneau X, Perrin D, Landais L, Camelin JC, Calmels TP, Berrebi-Bertrand I, et al. BF2.649 [1-{3-[3-(4-Chlorophenyl)propoxy]propyl}piperidine, hydrochloride], a nonimidazole inverse agonist/antagonist at the human histamine H3 receptor: Preclinical pharmacology. J Pharmacol Exp Ther. 2007;320:365–375. doi: 10.1124/jpet.106.111039. [DOI] [PubMed] [Google Scholar]
- McPherson GA. Analysis of radioligand binding experiments. A collection of computer programs for the IBM PC. J Pharmacol Methods. 1985;14:213–228. doi: 10.1016/0160-5402(85)90034-8. [DOI] [PubMed] [Google Scholar]
- Martinez-Mir MI, Pollard H, Moreau J, Arrang JM, Ruat M, Traiffort E, et al. Three histamine receptors (H1, H2 and H3) visualized in the brain of human and non-human primates. Brain Res. 1990;526:322–327. doi: 10.1016/0006-8993(90)91240-h. [DOI] [PubMed] [Google Scholar]
- Mazurkiewicz-Kwilecki IM, Nsonwah S. Changes in the regional brain histamine and histidine levels in postmortem brains of Alzheimer patients. Can J Physiol Pharmacol. 1989;67:75–78. doi: 10.1139/y89-013. [DOI] [PubMed] [Google Scholar]
- Medhurst AD, Atkins AR, Beresford IJ, Brackenborough K, Briggs MA, Calver AR, et al. GSK189254, a novel H3 receptor antagonist that binds to histamine H3 receptors in Alzheimer's disease brain and improves cognitive performance in preclinical models19. J Pharmacol Exp Ther. 2007;321:1032–1045. doi: 10.1124/jpet.107.120311. [DOI] [PubMed] [Google Scholar]
- Nakamura S, Takemura M, Ohnishi K, Suenaga T, Nishimura M, Akiguchi I, et al. Loss of large neurons and occurrence of neurofibrillary tangles in the tuberomammillary nucleus of patients with Alzheimer's disease. Neurosci Lett. 1993;151:196–199. doi: 10.1016/0304-3940(93)90019-h. [DOI] [PubMed] [Google Scholar]
- Panula P, Rinne J, Kuokkanen K, Eriksson KS, Sallmen T, Kalimo H, et al. Neuronal histamine deficit in Alzheimer's disease. Neuroscience. 1998;82:993–997. doi: 10.1016/s0306-4522(97)00353-9. [DOI] [PubMed] [Google Scholar]
- Passani MB, Lin JS, Hancock A, Crochet S, Blandina P. The histamine H3 receptor as a novel therapeutic target for cognitive and sleep disorders. Trends Pharmacol Sci. 2004;25:618–625. doi: 10.1016/j.tips.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Perry E, Court J, Goodchild R, Griffiths M, Jaros E, Johnson M, et al. Clinical neurochemistry: developments in dementia research based on brain bank material. J Neural Transm. 1998;105:915–933. doi: 10.1007/s007020050102. [DOI] [PubMed] [Google Scholar]
- Pollard H, Moreau J, Arrang JM, Schwartz JC. A detailed autoradiographic mapping of histamine H3 receptors in rat brain areas. Neuroscience. 1993;52:169–189. doi: 10.1016/0306-4522(93)90191-h. [DOI] [PubMed] [Google Scholar]
- Ramirez MJ, Aisa B, Garcia-Alloza M, Gil-Bea FJ, Marcos B. Involvement of the serotonergic system in cognitive and behavioural symptoms of Alzheimer's disease. Curr Psychiatry Rev. 2005;1:337–343. [Google Scholar]
- Richardson JC, Kendal CE, Anderson R, Priest F, Gower E, Soden P, et al. Ultrastructural and behavioural changes precede amyloid deposition in a transgenic model of Alzheimer's disease. Neuroscience. 2003;122:213–228. doi: 10.1016/s0306-4522(03)00389-0. [DOI] [PubMed] [Google Scholar]
- Roberts JC, Davis JB, Benham CD. 3H]Resiniferatoxin autoradiography in the CNS of wild-type and TRPV1 null mice defines TRPV1 (VR-1) protein distribution145. Brain Res. 2004;995:176–183. doi: 10.1016/j.brainres.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Scahill RI, Schott JM, Stevens JM, Rossor MN, Fox NC. Mapping the evolution of regional atrophy in Alzheimer's disease: unbiased analysis of fluid-registered serial MRI. Proc Natl Acad Sci USA. 2002;99:4703–4707. doi: 10.1073/pnas.052587399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlicker E, Malinowska B, Kathmann M, Gothert M. Modulation of neurotransmitter release via histamine H3 heteroreceptors. Fundam Clin Pharmacol. 1994;8:128–137. doi: 10.1111/j.1472-8206.1994.tb00789.x. [DOI] [PubMed] [Google Scholar]
- Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR. Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215:1237–1239. doi: 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- Wilcock GK, Esiri MM. Asymmetry of pathology in Alzheimer's disease. J Neurol Neurosurg Psychiatry. 1987;50:1384–1386. doi: 10.1136/jnnp.50.10.1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witkin JM, Nelson DL. Selective histamine H3 receptor antagonists for treatment of cognitive deficiencies and other disorders of the central nervous system. Pharmacol Ther. 2004;103:1–20. doi: 10.1016/j.pharmthera.2004.05.001. [DOI] [PubMed] [Google Scholar]