

Abstract
Background and purpose:
Histamine H3 receptor antagonists are currently being evaluated in clinical trials for a number of central nervous system disorders including narcolepsy. These agents can increase wakefulness (W) in cats and rodents following acute administration, but their effects after repeat dosing have not been reported previously.
Experimental approach:
EEG and EMG recordings were used to investigate the effects of acute and repeat administration of the novel H3 antagonist GSK189254 on the sleep–wake cycle in wild-type (Ox+/+) and orexin knockout (Ox−/−) mice, the latter being genetically susceptible to narcoleptic episodes. In addition, we investigated H3 and H1 receptor expression in this model using radioligand binding and autoradiography.
Key results:
In Ox+/+ and Ox−/− mice, acute administration of GSK189254 (3 and 10 mg·kg−1 p.o.) increased W and decreased slow wave and paradoxical sleep to a similar degree to modafinil (64 mg·kg−1), while it reduced narcoleptic episodes in Ox−/− mice. After twice daily dosing for 8 days, the effect of GSK189254 (10 mg·kg−1) on W in both Ox+/+ and Ox−/− mice was significantly reduced, while the effect on narcoleptic episodes in Ox−/− mice was significantly increased. Binding studies revealed no significant differences in H3 or H1 receptor expression between Ox+/+ and Ox−/− mice.
Conclusions and implications:
These studies provide further evidence to support the potential use of H3 antagonists in the treatment of narcolepsy and excessive daytime sleepiness. Moreover, the differential effects observed on W and narcoleptic episodes following repeat dosing could have important implications in clinical studies.
Keywords: Orexin knockout mice, GSK189254, H3 receptor antagonist, narcolepsy, modafinil, sleep–wake cycle
Introduction
Histamine plays a major role in the control of arousal and the sleep–wake cycle (Lin, 2000; Brown et al., 2001; Haas and Panula, 2003). Histaminergic neurons fire in a wake-specific pattern (Vanni-Mercier et al., 2003; Takahashi et al., 2006), and histamine release is subject to circadian variation being higher in periods of wakefulness (W) (Brown et al., 2001). Histamine can increase wake and decrease slow wave sleep (SWS) when administered into cerebral structures important for sleep–wake control in the cat (Lin et al., 1996). In contrast, decreased W occurs following inhibition of histamine synthesis, either pharmacologically using histidine decarboxylase inhibitors or genetically as in histidine decarboxylase knockout mice (Monti et al., 1996; Parmentier et al., 2002).
The wake-promoting effects of histamine are believed to be mediated via histamine H1 receptors in the central nervous system (CNS). It is well documented that brain penetrant H1 antagonists (classical antihistamines) can cause sedation in humans (Roth et al., 1987) and affect psychomotor performance (Richardson et al., 2002). H1 antagonists also show similar sedative activity in a number of preclinical species including rats (Saitou et al., 1999).
Histamine can also exert its effects via other histamine receptors including presynaptic H3 receptors. H3 receptor activation results in the inhibition of histamine synthesis and release from histaminergic neurons via an autoreceptor function (Arrang et al., 1983), and the inhibition of release of other neurotransmitters, such as acetylcholine, dopamine, 5-HT and noradrenaline, from non-histaminergic neurons via a heteroreceptor function (Schlicker et al., 1994; Blandina et el., 1996). Conversely, in vivo blockade of H3 receptors with selective antagonists/inverse agonists can increase the release of histamine (Arrang et al., 1988) and other neurotransmitters (Fox et al., 2005; Medhurst et al., 2007) involved in cognition and arousal.
H3 receptor agonists have been shown to produce sedation in preclinical species while H3 antagonists have the opposite effect and can increase W (Monti et al., 1996; Ligneau et al., 1998; Lin, 2000; Barbier et al., 2004). Transgenic mice devoid of the H3 receptor display characteristics of behavioural inactivation and are unresponsive to the wake-promoting actions of H3 receptor antagonists (Toyota et al., 2002). The increase in wakefulness observed with H3 antagonists can be blocked with H1 antagonists such as mepyramine (Lin et al., 1990), while a recent study in H1 receptor knockout mice confirmed that the W effect of the H3 antagonist ciproxifan is dependent on the presence of H1 receptors (Parmentier et al., 2007). These effects on W have led to the hypothesis that H3 receptor antagonists may be beneficial in diseases where excessive daytime sleepiness occurs, such as in narcolepsy. Moreover, histamine levels have been shown to be decreased in human narcoleptics and in dogs genetically susceptible to narcolepsy (Nishino et al., 2001; Kanbayashi et al., 2004), while H3 antagonists can reverse food-elicited cataplexy in narcoleptic dogs (Bonaventure et al., 2007). In addition, H3 antagonists have recently been shown to increase W in narcoleptic mice (Lin et al., 2008).
A number of H3 antagonists/inverse agonists are currently in clinical trials to evaluate their potential utility in various CNS disorders, such as narcolepsy, Alzheimer's disease, schizophrenia and attention-deficit hyperactivity disorder (Celanire et al., 2005; Esbenshade et al., 2008; Lin et al., 2008). Recently, tiprolisant (BF2.642; Ligneau et al., 2007) has been shown to improve excessive daytime sleepiness in narcoleptic patients in a small phase II clinical trial (Lin et al., 2008), consistent with a potential elevation of histamine in humans following blockade of H3 autoreceptors.
GSK189254 {6-[(3-cyclobutyl-2,3,4,5-tetrahydro-1H-3-benzazepin-7-yl)oxy]-N-methyl-3-pyridinecarboxamide hydrochloride} is a highly potent and selective H3 receptor antagonist/inverse agonist that shows efficacy in a number of cognition paradigms and pain models in rats after acute and repeat administration for up to 8 days (Medhurst et al., 2007; 2008). However, certain effects of other H3 antagonists have been shown to tolerate out after repeat dosing in preclinical species (Pan et al., 2006), although the effect of repeat dosing on the sleep–wake cycle has not been reported previously. In the present study, we therefore investigated the effects of acute and repeat administration of GSK189254 on the sleep–wake cycle in wild-type (Ox+/+) and orexin knockout (Ox−/−) mice that are genetically susceptible to narcoleptic episodes. Ox−/− mice show direct transitions from W to paradoxical sleep (PS)/rapid eye movement (REM) sleep (also known as direct REM sleep, DREMs), a characteristic symptom in human narcolepsy (Mignot, 2005; Billiard et al., 2006). In addition, we investigated whether there were any changes in H3 and H1 receptor levels in Ox−/− mice compared with Ox+/+ mice. These studies reveal interesting differences in the effects of repeat dosing of an H3 antagonist on W and narcoleptic episodes in the mouse model of narcolepsy.
Methods
Animals and surgery
All experiments followed EEC (86/609/EEC) and French National Committee (decree 87/848) directives and every effort was made to minimize any pain and discomfort and the number of animals used. Procedures were also reviewed and approved by the GlaxoSmithKline Procedures Review panel. Orexin knockout (Ox−/−) mice were generated as previously described (Lin et al., 2008). Prepro-orexin KO (Ox−/−) mice were descendants of the mouse strain generated by Chemelli et al. (1999) and kept on C57BL/6J genomic background by five to nine more backcrosses during the present study. To obtain experimental animals, male Ox−/− mice were backcrossed with female wild-type (Ox+/+) mice, and the generated Ox+/− mice were crossed between them resulting in both heterozygotes and homozygotes. Only littermate homozygote Ox+/+ and Ox−/− mice (n= 14 pairs) were used in this study to ensure that any detected phenotype resulted from the deletion of prepro-orexin gene rather than the genetic heterogeneity between individual animals. Mouse genotypes were determined as previously described using tail biopsies performed at 4 weeks of age (Chemelli et al., 1999; Lin et al., 2008).
At the age of 11–13 weeks and with a body weight of 30 ± 2 g, mice used for EEG and sleep–wake studies were chronically implanted, under deep gas anaesthesia using isoflurane (2%, 200 mL·min−1) and a TEM anaesthesia system, with six cortical electrodes (gold-plated tinned copper wire, Ø= 0.4 mm) and three muscle electrodes (fluorocarbon-coated gold-plated stainless steel wire, Ø= 0.03 mm) to record the EEG and EMG, and to monitor the sleep–wake cycle. All electrodes were previously soldered to a multi-channel electrical connector and each was separately insulated with a covering of heat-shrinkable polyolefin/polyester tubing. Cortical electrodes were inserted into the dura through three pairs of holes (Ø= 0.5 mm) made in the skull, located respectively in the frontal (1 mm lateral and anterior to the bregma), parietal (1 mm lateral to the midline at the midpoint between the bregma and lambda) and occipital (2 mm lateral to the midline and 1 mm anterior to the lambda) cortices. Muscle electrodes were inserted into the neck muscles. Finally, the electrode assembly was anchored and fixed to the skull with Super-Bond and dental cement. This implantation allows stable and long-lasting polygraphic recordings (Parmentier et al., 2002; Lin et al., 2008).
Polygraphic recording in the mouse and data acquisition and analysis
After surgery, the animals were housed individually in transparent barrels (Ø= 20 cm, height = 30 cm) placed in an insulated sound-proof recording room maintained at an ambient temperature of 22 ± 1°C and on a 12 h light/dark cycle (lights on at 07 h 00 min), standard food and water being available ad libitum. A video camera with infrared and digital time recording capabilities was set up in the recording room to observe and score, when necessary, the animals' behaviour during the light or dark phase. After a 7 day recovery period, mice were habituated to the recording cable for 7 days before polygraphic recordings were started. Cortical EEG and EMG signals were amplified, digitized with a resolution of 256 and 128 Hz respectively, and computed on a CED 1401 Plus. Using a Spike2 script and with the assistance of spectral analysis using the fast Fourier transform, polygraphic records were visually scored by 30 s epochs for W, SWS and PS/REM sleep according to previously described criteria validated for mice (Valatx, 1971; Valatx and Bugat, 1974; Parmentier et al., 2002). DREMs onset episodes, also called narcoleptic episodes or sleep onset REM periods (Chemelli et al., 1999; Mignot, 2005), were defined as the occurrence of REM sleep directly from W, namely a REM episode that follows directly a W episode lasting more than 60 s without being preceded by any cortical slow activity of more than 5 s during the 60 s.
Drug administration and experimental procedures in the mouse
After recovery from the surgery and habituation to the recording cables, each mouse was subjected to a recording session of two continuous days, beginning at 07 h 00 min. During these periods, the animals were left undisturbed to obtain baseline parameters on the cortical EEG, sleep–wake cycle and circadian rhythm. After baseline recordings, animals were subjected to cortical EEG and sleep–wake recordings following administration of either a vehicle (placebo) or drug (GSK189254 or modafinil). The vehicle consisted of 0.05 mL NaCl at 0.9% containing methylcellulose at 1%. The drug doses were expressed as compound weight. They were dissolved in the vehicle, fresh before each administration, and were administered orally using a mouse gavage probe. The following dosing schedules were investigated:
(1)Effect of acute administration of GSK189254 (3 and 10 mg·kg−1, p.o.) on the sleep–wake cycle in Ox+/+ and Ox−/− mice, dosed at 10 h 00 min (i.e. 3 h after lights on) – mice exhibit maximal sleep between 10 and 16 h (Parmentier et al., 2002; Lin et al., 2008), so this period was appropriate for detecting any awakening effect.
(2)Effects of acute administration of GSK189254 (10 mg·kg−1, p.o.) on narcoleptic episodes, dosed at 18 h 45 min just before lights-off phase at 19 h 00 min – both Ox+/+ and Ox−/− mice were active and awake at this time, and Ox−/− mice display narcoleptic attacks/DREMs almost exclusively during the lights-off phase. This period was therefore appropriate for detecting any anti-narcoleptic effects. Modafinil (64 mg·kg−1, p.o.) was used as a comparator as previously described (Lin et al., 2008), because of its wide use in sleep medicine including narcolepsy.
(3)Effects of repeat oral administration (8 days b.i.d. at 10 h 00 min and 19 h 00 min) of GSK189254 (10 mg·kg−1) either on W or narcoleptic episodes during the lights-off phase. We sought to investigate whether the effects of GSK189254 (10 mg·kg−1) on W or narcolepsy seen during acute dosing were maintained.
Oral doses of GSK189254 (3 and 10 mg·kg−1) were selected to achieve >80% receptor occupancy. The ED50 in CD1 mice had previously been determined to be ∼0.6 mg·kg−1 in ex vivo binding assays 1–2 h post dose (B. Crook, unpubl. obs.). The pharmacokinetic properties of GSK189254 in CD1 mice are comparable to rats, with the plasma half life following oral dosing being around 1.7 h and pharmacokinetics being linear with dose (M. Briggs, unpubl. obs.). Concentrations of GSK189254 determined in CD1 mice were around 1–3 µmol·L−1 following oral dosing of 3 and 10 mg·kg−1 and there was no evidence for drug accumulation. These brain concentrations are well above the Ki required for efficacy as previously reported for a number of other H3 antagonists (Barbier et al., 2004; Medhurst et al., 2007).
The order of drug administration was randomized and each animal received both placebo and drug treatment. Polygraphic recordings were made immediately after any administration and were maintained for 12 h or during the whole lights-off period (12 h). Two acute administrations were separated by a 7 day washout period. In the repeat dose studies, recordings commenced on day 9 after the dose at 10 h 00 min.
Autoradiography
Receptor autoradiography was carried out based on methods previously described (Roberts et al., 2004; Medhurst et al., 2007). Frozen half brains from drug naïve Ox+/+ and Ox−/− mice (n= 4 per group) were sectioned coronally (12 µm), thaw-mounted onto gelatin-coated slides and stored at −80°C until use. For H3 receptor binding, sections were incubated with 1 nmol·L−1[3H]-GSK189254 at room temperature for 60 min (Medhurst et al., 2007). Non-specific binding was determined in the presence of 10 µmol·L−1 imetit on adjacent slides. For H1 receptor binding, sections were incubated with 5 nmol·L−1[3H]-mepyramine for 45 min at room temperature (Palacios et al., 1981). Non-specific binding was determined in the presence of 10 µmol·L−1 chlorpheniramine maleate on adjacent slides. After incubation, all sections were rinsed, dried and analysed by digital autoradiography using a Beta-Imager 2000 instrument as previously described (Medhurst et al., 2007). Ten measurements were made from each brain area (cortex, hippocampus and hypothalamus) for each animal (five sections per animal and two measurements per region).
Saturation binding in brain homogenates
H3 and H1 receptor saturation binding experiments were carried out in cortical membranes from Ox+/+ and Ox−/− mice based on methodology previously described (Tran et al., 1978; Medhurst et al., 2007). Half brains were homogenized using a polytron P10 (2 × 10 s bursts at full speed) in 30 volumes of 50 mmol·L−1 Tris-HCl, pH 7.7, at 25°C containing 5 mmol·L−1 ethylenediamineteraacetic acid buffer. The homogenate was centrifuged in a Sorval Evolution RC centrifuge at 4°C for 20 min (SS34 rotor – approximately 48 000× g). The final pellet was resuspended in 10 mL buffer.
Membranes (corresponding to approximately 20 µg of protein per well) and [3H]-GSK189254 (0.02–20 nmol·L−1) for H3 binding, or [3H]-mepyramine (0.05–50 nmol·L−1) for H1 binding were incubated in polypropylene tubes in a final volume of 200 µL of 50 mmol·L−1 Tris-HCl, pH 7.7, at 25°C containing 5 mmol·L−1 ethylenediamineteraacetic acid. Non-specific binding was determined in the presence of 10 µmol·L−1 imetit for H3 and 10 µmol·L−1 chlorpheniramine for H1. Reactions were conducted at 30°C for 45 min. The experiments were terminated by rapid filtration through Whatman GF/B filters [presoaked in 0.3% (v/v) polyethyleneimine]. The filters were washed with 4 × 2 mL aliquots of ice-cold buffer containing 50 mmol·L−1 Tris-HCl, pH 7.7, at 25°C and 5 mmol·L−1 mgCl2. Filters were dried and added to vials each containing 4 mL of Ultima Gold MV scintillation fluid. Radioactivity was determined by liquid scintillation spectrometry using a Packard Tri-Carb 2500TR liquid scintillation counter. Protein concentrations were determined using the Bradford assay method (Bio-Rad protein assay Kit) with bovine serum albumin as a standard.
Statistical analysis
The sleep–wake amounts obtained after GSK189254 or modafinil dosing were compared with those obtained with vehicle. Individual animals acted as their own controls. Using Excel and/or Statgrafy software, one-way and multiple factorial anova and the post hoc Student's t-test (two-tailed) were applied to compare and evaluate any difference in the effects obtained between: (i) compound and vehicle; (ii) compounds; (iii) doses; (iv) the first and the last administrations of GSK189254; and (v) mouse genotypes.
For autoradiography studies [3H]-GSK189254 and [3H]-mepyramine binding signals in cortex, hippocampus and hypothalamus were measured from five sections per animal and two measurements per region. This was determined previously to be a statistically validated number of sections. The results are expressed as mean specific binding cpm mm−2 (n= 4 animals per group), and the data were analysed using a repeated-measures anova approach. For homogenate binding studies, H3 and H1 specific binding was analysed to estimate binding parameters Bmax (reflective of total number of binding sites) and KD (reflective of the binding affinity) using GraphPad Prism 3.0 by GraphPad Software Inc (San Diego, CA, USA).
Materials
GSK189254 {6-[(3-cyclobutyl-2,3,4,5-tetrahydro-1H-3-benzazepin-7-yl)oxy]-N-methyl-3-pyridinecarboxamide hydrochloride} was synthesized at GlaxoSmithKline (Harlow, UK). [3H]-GSK189254 (specific activity 81 Ci·mmol−1) was prepared through a contract with GE Healthcare. Modafinil was obtained from Laboratoires L. Lafon (Paris), imetit from Tocris Cookson Inc (Bristol UK) and chlorpheniramine from Sigma. [3H]-mepyramine (specific activity 20–30 Ci·mmol−1) was obtained from Amersham Bioscience (UK).
TEM anaesthesia system (Bordeaux, France), cortical electrodes (gold-plated tinned copper wire, Ø= 0.4 mm, Filotex, Draveil, France) and the muscle electrodes (fluorocarbon-coated gold-plated stainless steel wire, Ø= 0.03 mm, Cooner Wire Chatworth, CA, USA); Super-Bond (Sun Medical Co., Shiga, Japan). The CED 1401 Plus was from Cambridge (UK). The mouse gavage probe (20G) was obtained from Phymep (Paris). The Beta-Imager 2000 instrument was obtained from Biospace (Paris, France); Whatman GF/B filters (Whatman, Maidstone, UK); Ultima Gold MV scintillation fluid (Hewlett Packard, Palo Alto, CA); Packard Tri-Carb 2500TR liquid scintillation counter (PerkinElmer Life and Analytical sciences, Boston, MA); Bio-Rad protein assay Kit, Bio-Rad (York, UK); GraphPad Software Inc (San Diego, CA, USA).
Results
Effects of acute administration of GSK189254 on the sleep–wake cycle in Ox+/+ and Ox−/− littermates during a 4 h recording in the lights-on period
In wild-type Ox+/+ mice during the lights-on phase (when the mice slept most of the time at baseline), acute oral administration of GSK189254 (3 and 10 mg·kg−1) caused an increase in W and a corresponding decrease in SWS and PS compared with vehicle-treated mice. Representative examples of hypnograms from Ox+/+ mice are shown in Figure 1A. In EEG spectral analysis, these effects were manifest as a suppression of neocortical slow activity (including δ range 0.8–2.5 Hz) and spindles (8–15 Hz) and an increase in power spectral density of neocortical fast rhythms (β and γ bands, mainly 30–60 Hz), resulting in marked enhancement of cortical activation (i.e. low voltage and fast electrical activity). Very similar effects on the sleep–wake cycle and cortical EEG were observed in Ox−/− littermates following acute oral administration of GSK189254 (3 and 10 mg·kg−1 p.o.), and representative examples of hypnograms from Ox−/− mice are shown in Figure 1B.
Figure 1.
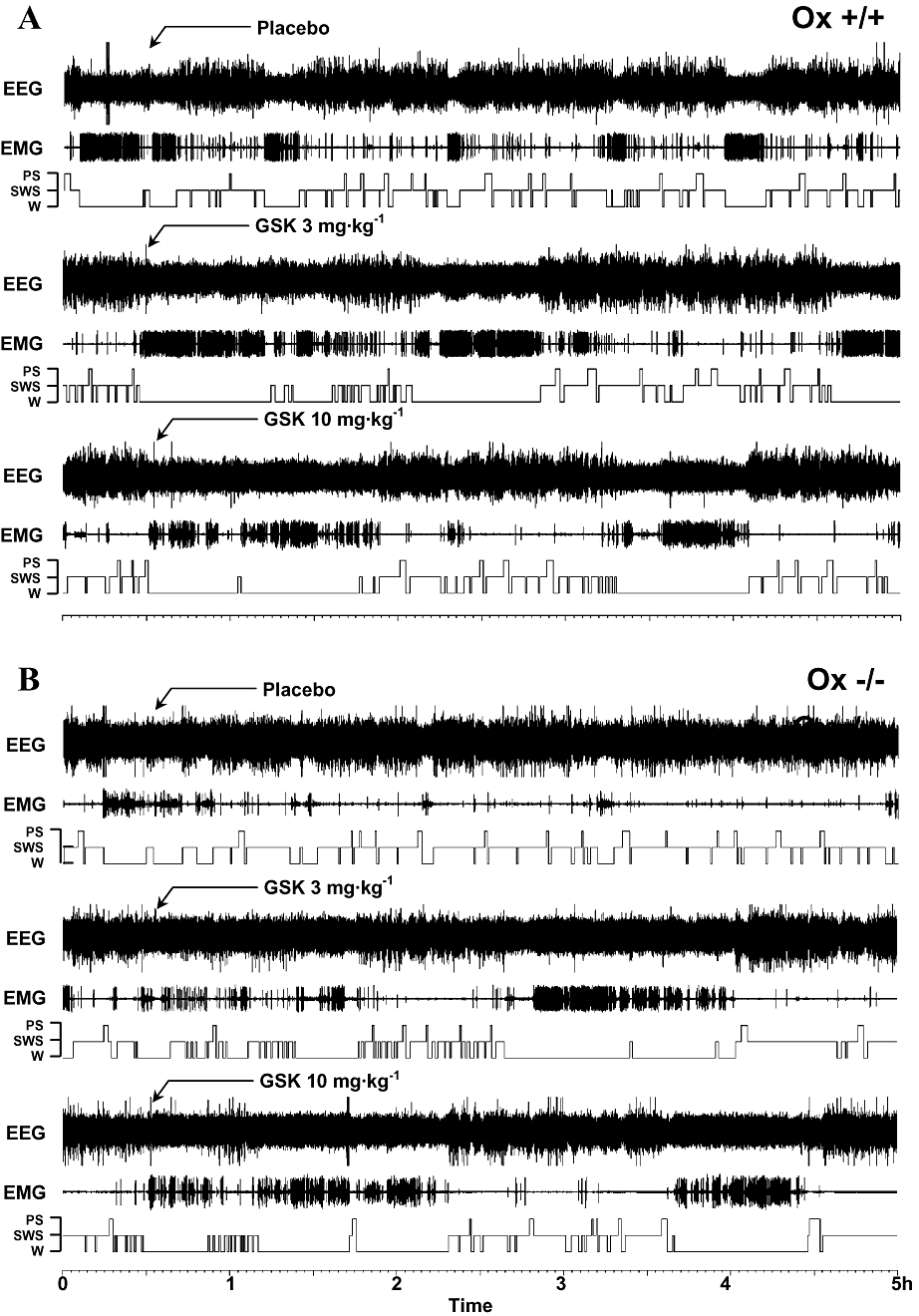
Representative examples of hypnograms showing the sleep–wake cycle in (A) wild-type (Ox+/+) and (B) Ox−/− mice over a 5 h period in the lights-on phase. Arrows show where vehicle (placebo) and GSK189254 (3 and 10 mg·kg−1) were administered. GSK189254 increased wakefulness (W) and decreased slow wave sleep (SWS) and paradoxical sleep (PS) in both Ox+/+ and Ox−/− mice compared with vehicle-treated mice.
As shown in the analysis of sleep–wake states over the 4 h post-dose period (Figure 2A), acute single administration of GSK189254 (3 and 10 mg·kg−1) to wild-type Ox+/+ mice significantly increased (P < 0.05, t-test between groups after significant anova) the duration of W compared with vehicle-treated (placebo control) mice. The increase in W occurred at the expense of SWS and PS, which was reduced significantly during the same period (P < 0.05, t-test between groups after significant anova). Similar effects following acute administration of GSK189254 (3 and 10 mg·kg−1) were observed in Ox−/− mice (Figure 2B), with duration of W significantly increased, and SWS and PS significantly reduced (P < 0.05, t-test between groups after significant anova).
Figure 2.
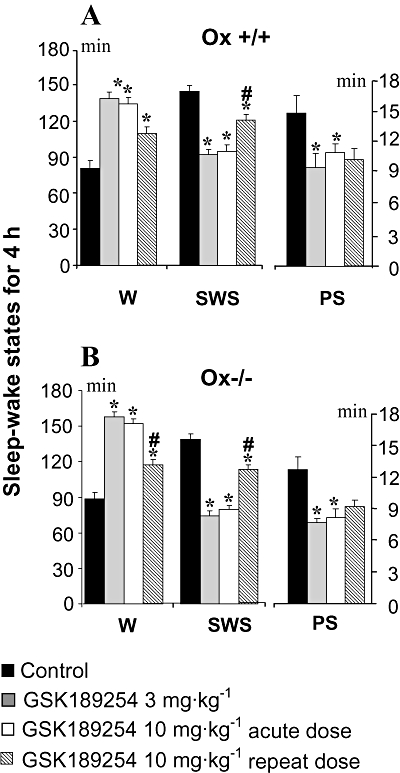
Effects of acute and repeat administration of GSK189254 on mean duration (±SEM) of sleep–wake stages in a 4 h recording within the lights-on period in (A) wild-type (Ox+/+) and (B) Ox−/− mice (n= 14 per group). GSK189254 (3 and 10 mg·kg−1) significantly increased wakefulness (W) and decreased slow wave sleep (SWS) and paradoxical sleep (PS) following acute dosing. Repeat administration of GSK189254 (10 mg·kg−1, twice daily for 8 days) resulted in a reduction in effects on W, SWS and PS compared with acute dosing with 10 mg·kg−1. *P < 0.05, t-test between groups (compared with vehicle-treated mice) after significant anova. #P < 0.05, t-test between groups (compared with acute 10 mg·kg−1 dose group) after significant anova.
Effects of repeat dosing of GSK189254 on the sleep–wake cycle in Ox+/+ and Ox−/− littermates during a 4 h recording in the lights-on period
We were also interested in exploring the effects of repeat dosing with GSK189254 given that this would potentially be required for future therapeutic use in narcolepsy or other sleep disorders. Both Ox+/+ and Ox−/− mice were treated with GSK189254 (3 and 10 mg·kg−1 p.o.) twice a day for 8 days respectively at 10 h 00 min and 19 h 00 min and effects on the sleep–wake cycle were studied on the ninth day when GSK189254 was also administered at 10 h 00 min and 19 h 00 min.
During the lights-on phase when untreated mice were sleepy and inactive at baseline, GSK189254 (10 mg·kg−1, p.o.) was still able to significantly increase W and decrease SWS and PS (P < 0.05, t-test between groups after significant anova) following 8 days of repeat dosing in both Ox+/+ and Ox−/− mice (Figure 2A and B respectively). However, these effects were significantly but not completely reduced compared with those observed following a single acute dose of GSK189254 (10 mg·kg−1, p.o.; P < 0.05, t-test between groups after significant anova). Thus, after repeat dosing, GSK189254 maintained about half its waking potency compared with that seen with acute dosing. For example, during the 4 h recording session, there was an increase by 38% in W in both Ox+/+ and Ox−/− mice following repeat dosing, compared with a 66% (Ox+/+) and 69% (Ox−/−) increase seen with acute dosing. In both Ox+/+ and Ox−/− mice, the decrease in SWS following repeat dosing was also about a half of that seen with acute dosing, whereas the amount of PS was similar between acute and repeat dosing (Figure 2).
Cumulative effects of acute and repeat dosing of GSK189254 on the sleep–wake cycle in Ox+/+ and Ox−/− littermates over 8 h during the lights-on period
We also measured the cumulative effects of GSK189254 on the sleep–wake cycle over an 8 h period to get an indication of whether any sleep rebound occurred after the waking effect. During the recovery periods between the fifth and eighth hours after dosing, total amount of W and SWS in both Ox+/+ and Ox−/− mice remained significantly increased and decreased respectively, indicating that no significant sleep rebound occurred following the effect of GSK189254 on W (Figure 3A and B respectively). It should be noted that there is not a prolonged action of GSK189254 as the data in Figure 3 (and also Figure 5) represent hourly cumulative values of sleep–wake amounts. Hourly analysis showed that the effects of GSK189254 did not last more than 4 h, hence the 4 h sleep–wake amounts presented in Figures 2 and 4. The amount of PS also remained significantly lower than that of control for 8 h in both Ox+/+ and Ox−/− mice (Figure 3A and B respectively). During the lights-on period, the effects on sleep–wake amount elicited by single doses of 3 and 10 mg·kg−1 GSK189254 were similar (Figures 2 and 3), although the activating effect of GSK189254 on the EEG appeared to be greater with 10 mg·kg−1 than with 3 mg·kg−1 (Figure 1). A similar ratio between the effects of acute dosing and those after repeated dosing to that observed during the 4 h measurements (Figure 2) was found for all the sleep–wake states in the 8 h cumulative data set (Figure 3).
Figure 3.
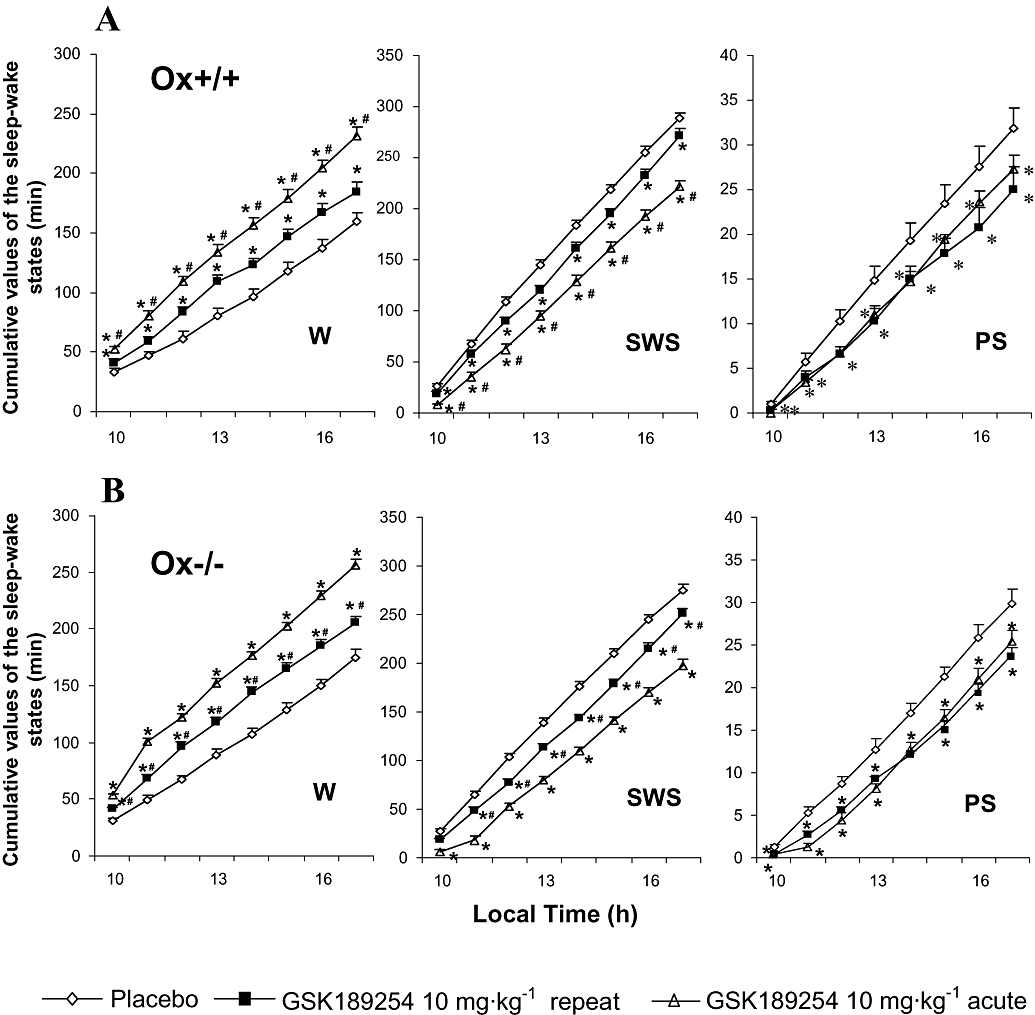
Cumulative effects of acute and repeat administration of GSK189254 on mean duration of sleep–wake stages over 8 h of the lights-on period in (A) wild-type (Ox+/+) and (B) Ox−/− mice (n= 14 per group). GSK189254 (3 and 10 mg·kg−1) significantly increased wakefulness (W) and decreased slow wave sleep (SWS) and paradoxical sleep (PS) following acute dosing. Repeat administration of GSK189254 (10 mg·kg−1, twice daily for 8 days) resulted in a reduction in effects on W, SWS and PS compared with acute dosing with 10 mg·kg−1. *P < 0.05, t-test between groups (compared with vehicle-treated mice) after significant anova. #P < 0.05, t-test between groups (compared with acute 10 mg·kg−1 dose group) after significant anova. There was no evidence of sleep rebound and effects of GSK189254 lasted for the 8 h duration of measurements.
Figure 5.
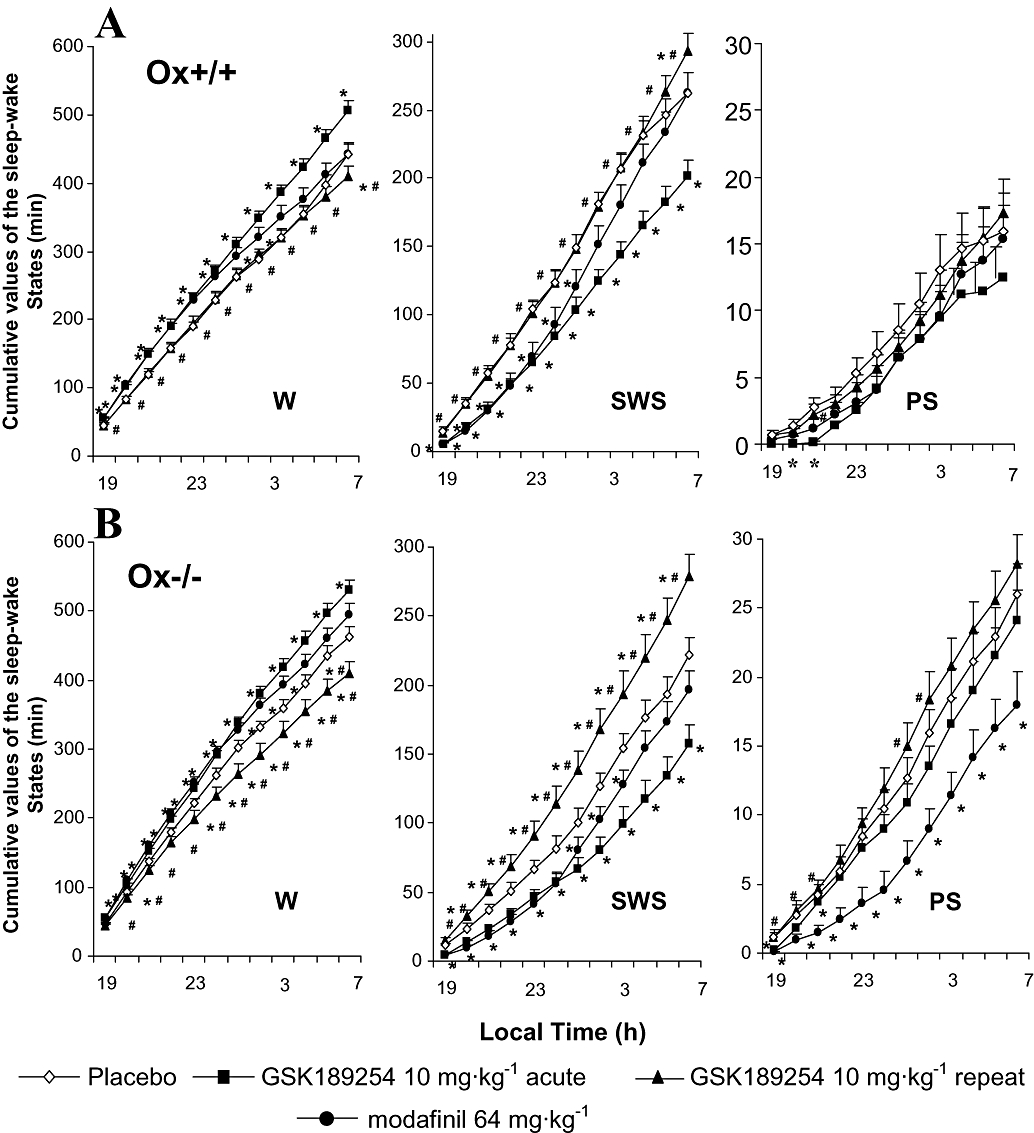
Cumulative effects of acute and repeat administration of GSK189254 and acute administration of modafinil on mean duration of sleep–wake stages over 12 h of the lights-off period in (A) wild-type (Ox+/+) and (B) Ox−/− mice (n= 14 per group). GSK189254 (3 and 10 mg·kg−1) significantly increased wakefulness (W) and decreased slow wave sleep (SWS) and paradoxical sleep (PS) following acute dosing, as did modafinil (64 mg·kg−1). Repeat administration of GSK189254 (10 mg·kg−1, twice daily for 8 days) resulted in a reduction in effects on W, SWS and PS compared with acute dosing with 10 mg·kg−1. *P < 0.05, t-test between groups (compared with vehicle-treated mice) after significant anova. #P < 0.05, t-test between groups (compared with acute 10 mg·kg−1 dose group) after significant anova. There was no evidence of sleep rebound and effects of GSK189254 lasted for the 12 h duration of measurements.
Figure 4.
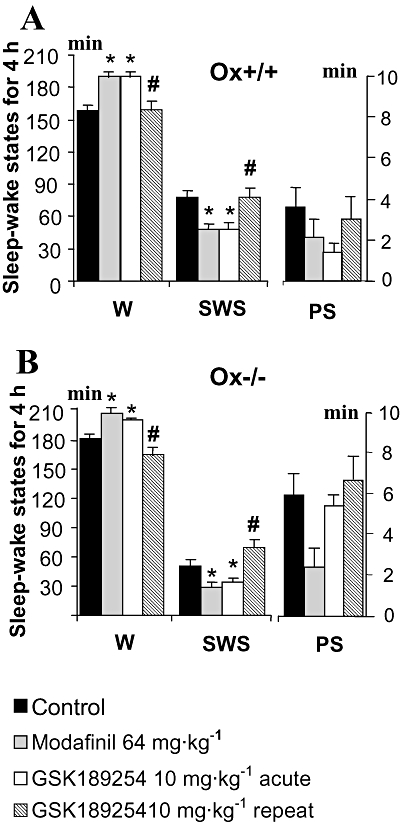
Effects of acute and repeat administration of GSK189254 on mean duration (±SEM) of sleep–wake stages in a 4 h recording within the lights-off period in (A) wild-type (Ox+/+) and (B) Ox−/− mice (n= 14 per group). GSK189254 (3 and 10 mg·kg−1) significantly increased wakefulness (W) and decreased slow wave sleep (SWS) and paradoxical sleep (PS) following acute dosing. Repeat administration of GSK189254 (10 mg·kg−1, twice daily for 8 days) had no effect on W, SWS and PS compared with acute dosing with 10 mg·kg−1 on Ox+/+ mice. In Ox−/− mice, GSK189254 actually reduced W and increased SWS and PS, effects that were the reverse of those seen with acute dosing. *P < 0.05, t-test between groups (compared with vehicle-treated mice) after significant anova. #P < 0.05, t-test between groups (compared with acute 10 mg·kg−1 dose group) after significant anova.
Acute effects of GSK189254 and modafinil on the sleep–wake cycle in Ox+/+ and Ox−/− littermates during a 4 h recording in the lights-off phase
Given the clear arousing effect observed in the lights-on phase, a 10 mg·kg−1 dose was chosen to assess the effects of acute administration of GSK189254 on the sleep–wake cycle of Ox+/+ and Ox−/− mice during the lights-off phase, when the animals were already highly awake and active. For comparison, modafinil, a wake-promoting compound (Lin et al., 1996; Parmentier et al., 2007) now used in sleep medicine for the treatment of narcolepsy and hypersomnia, was also applied to mice at a dose of 64 mg·kg−1. During 4 h post-dose recording, GSK189254 still significantly (P < 0.05, t-test between groups after significant anova) increased the time spent in W, decreased SWS and to a lesser extent decreased PS in both Ox+/+ (Figure 4A) and Ox−/− (Figure 4B) mice when applied just before lights-off, although these effects were less marked than those observed in the lights-on period, as expected given the higher baseline W and general activity. Modafinil also increased W and decreased SWS and PS in both Ox+/+ (Figure 4A) and Ox−/− mice (Figure 4B). During a recording session of 4 h, the effects of GSK189254 (10 mg·kg−1) on sleep–wake states appeared to be similar to those of modafinil in Ox+/+ mice (Figure 4A) but marginally less in Ox−/− mice (Figure 4B).
Effects of repeat dosing of GSK189254 on the sleep–wake cycle in Ox+/+ and Ox−/− littermates during a 4 h recording in the lights-off phase
In Ox+/+ mice during a 4 h recording period in the lights-off phase when untreated mice are highly awake and active, GSK189254 (10 mg·kg−1 p.o.) had no clear effect on W, SWS and PS following 8 days of b.i.d. dosing (Figure 4A) compared with vehicle-treated animals. This was in complete contrast to the effects of a single dose of GSK189254. Interestingly after repeat dosing in Ox−/− mice, GSK189254 (10 mg·kg−1) significantly decreased W and increased SWS (P < 0.05, t-test between groups after significant anova) and to a lesser extent, PS (Figure 4B). These effects in Ox−/− mice were opposite to those observed following a single dose.
Cumulative effects of acute and repeat dosing of GSK189254 on the sleep–wake cycle in Ox+/+ and Ox−/− littermates over 12 h of the lights-off period
As in the lights-on phase, we also measured the cumulative effects of acute and repeat administration of GSK189254 on the sleep–wake cycle over an extended time period (in this case 12 h) to get an indication whether any sleep rebound occurred after the waking effect. In Ox+/+ mice, the cumulative effects of acute administration of GSK189254 (10 mg·kg−1) on W and SWS were somewhat greater than those seen with modafinil, while the effects on PS were comparable (Figure 5A). In Ox−/− mice the effect of GSK189254 at 10 mg·kg−1 on W and SWS appeared slightly greater than that of modafinil during the 12 h recording session whereas the decrease in PS seen with GSK189254 appeared less than that seen with modafinil (Figure 5B). In terms of repeat dosing effects with GSK189254 in the 12 h recording period in Ox+/+ mice, there was no difference in W, SWS or PS compared with vehicle-treated animals, in contrast to the effects of acute dosing (Figure 5A). However, in Ox−/− mice, W duration was significantly reduced, while SWS, and to a lesser extent, PS, were significantly increased compared with vehicle-treated mice, an opposite effect to that observed with acute dosing (Figure 5B) and consistent with observations over the 4 h time frame (Figure 4B).
Acute effects of GSK189254 and modafinil on narcoleptic episodes in Ox−/− mice during lights-off
As shown in previous studies (Chemelli et al., 1999; Lin et al., 2008), Ox−/−mice display, almost exclusively during lights-off phase, a narcoleptic phenotype characterized by DREMs. In spite of their similar effect on W, GSK189254 and modafinil showed a different profile against narcoleptic episodes when applied to Ox−/− mice in the current study.
The waking effect of GSK189254 was accompanied by a significant decrease in the narcoleptic phenotype in Ox−/− mice. During the first 4 h after dosing (Figure 6A), the number of DREMs was significantly reduced (P < 0.05, t-test between groups after significant anova). In addition, the mean cumulative number (Figure 6C) and duration (Figure 6B) of narcoleptic episodes were significantly reduced by GSK189254 (P < 0.05, t-test between groups after significant anova).
Figure 6.
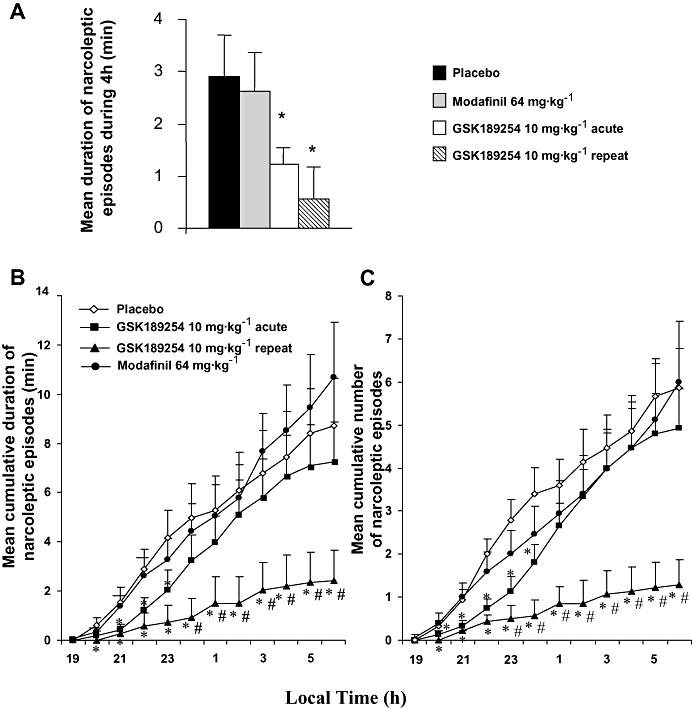
Effects of acute and repeat administration of GSK189254 and effect of acute administration of modafinil on narcoleptic attacks in Ox−/− mice in the lights-off period compared with vehicle-treated mice. (A) Mean (±SEM) number of narcoleptic episodes in the 4 h period after dosing. (B) Mean (±SEM) cumulative duration of narcoleptic attacks and (C) mean (±SEM) cumulative number of narcoleptic attacks over the 12 h lights-off period. *P < 0.05, t-test between groups (compared with vehicle-treated mice) after significant anova. #P < 0.05, t-test between groups (compared with acute 10 mg·kg−1 dose group) after significant anova.
In contrast, despite causing an increase in W and a reduction in SWS and PS, modafinil had no effect on the narcolepsy phenotype in Ox−/− mice. As shown in Figure 6, the total duration and cumulated number of narcoleptic attacks after modafinil dosing were not significantly different to those obtained in vehicle-treated mice throughout the 12 h recording period.
Repeat dose effects of GSK189254 on narcolepsy in Ox−/−mice
To our surprise, both the number and duration of narcoleptic episodes were dramatically reduced following 8 days of repeat dosing of GSK189254 (10 mg·kg−1, b.i.d.). The anti-narcoleptic effect of GSK189254 was significantly greater after repeat dosing than after acute dosing, with narcoleptic attacks being quasi-absent for 6 h and their occurrence remaining very rare during the whole lights-off period; that is, the mean number per night of narcoleptic attacks was reduced from 6 to 1 and the total duration from 8 to 2 min (Figure 6).
Autoradiography
Autoradiographic analysis of coronal half brain sections from Ox+/+ and Ox−/− mice revealed extensive specific [3H]-GSK189254 binding (>90%) to H3 receptors within the cerebral cortex, hippocampus and hypothalamus, while binding levels were negligible following co-incubation with 10 µmol·L−1 imetit to define non-specific binding (Figure 7A, left panel). Quantification of H3 binding revealed no significant differences between Ox+/+ and Ox−/− mice in cortex, hippocampus and hypothalamus (Figure 7B, left panel). Autoradiographic analysis in adjacent coronal half brain sections using [3H]-mepyramine showed H1 receptor expression in similar areas to H3 receptors but at much lower density (Figure 7A and B, right panels). Binding levels were negligible following co-incubation with 10 µmol·L−1 chlorpheniramine to define non-specific binding. No significant differences were observed in H1 receptor density between Ox+/+ and Ox−/− mice in cortex, hippocampus and hypothalamus (P > 0.05; repeated-measures anova).
Figure 7.
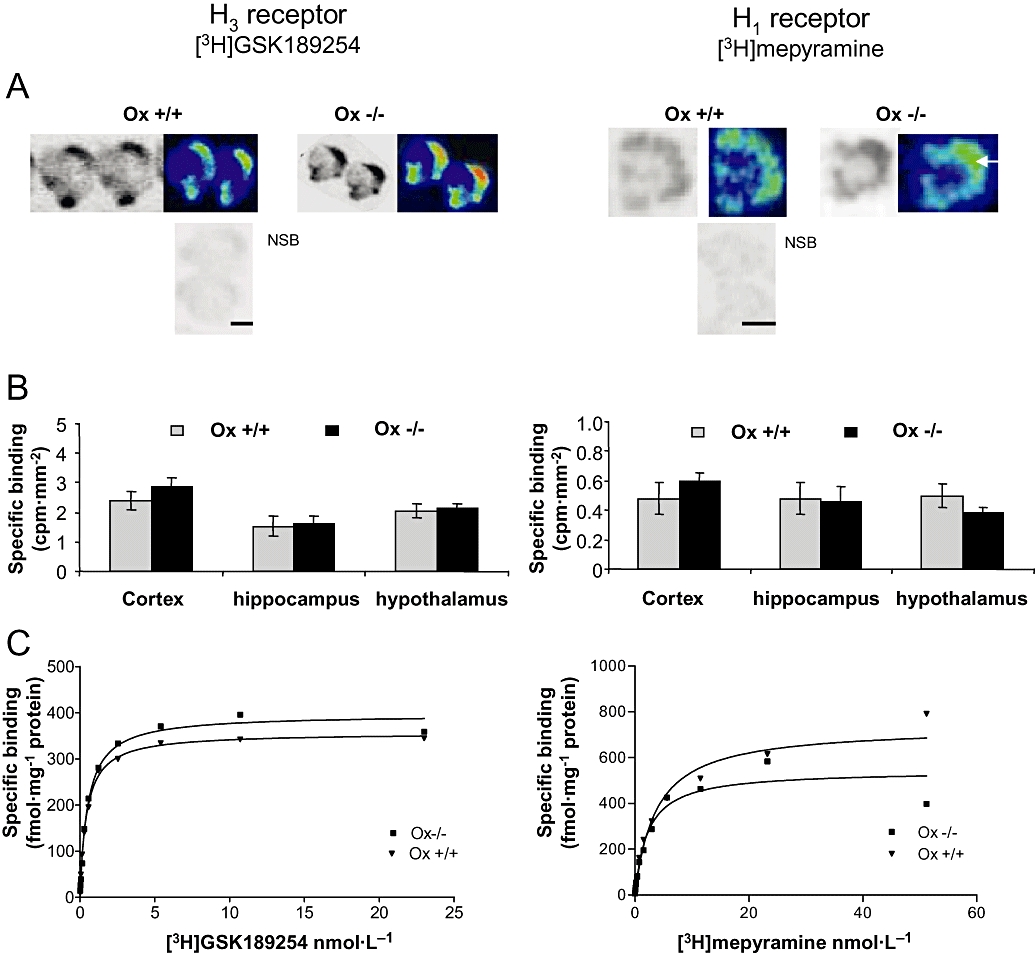
H3 (left panels) and H1 (right panels) receptor binding in Ox+/+ and Ox−/− mice measured using [3H]-GSK189254 and [3H]-mepyramine respectively, with real-time autoradiography. (A) Representative pseudo-coloured images of coronal half brain sections showing specific [3H]-GSK189254 and [3H]-mepyramine binding in cortex and hypothalamus of Ox+/+ and Ox−/− mice. Non-specific binding (NSB) for H3 and H1 was determined in the presence of 10 µmol·L−1 imetit and 10 µmol·L−1 chlorphenimramine respectively. Scale bars = 1 mm. (B) Quantitative histograms showing specific [3H]-GSK189254 and [3H]-mepyramine binding in cortex, hippocampus and hypothalamus of Ox+/+ and Ox−/− mice (mean ± SEM, n= 4 per group). No significant differences were observed between Ox+/+ and Ox−/− mice in any of these brain regions. (C) Saturation binding analysis for [3H]-GSK189254 to H3 receptors and [3H]-mepyramine to H1 receptors in whole brain of Ox+/+ and Ox−/− mice. Representative curves are shown. No significant differences were observed in mean Bmax or KD (n= 3–4 per group) as shown in Results section.
Saturation binding
H3 and H1 receptor saturation binding analysis was carried out in half brain homogenates from Ox+/+ (n= 3) and Ox−/− (n= 4) brains using [3H]-GSK189254 and [3H]-mepyramine respectively (Figure 7C). Specific binding represented >90% of total binding for both ligands, similar to that observed in autoradiography. Saturation analysis for H3 receptors with [3H]-GSK189254 in Ox+/+ and Ox−/− mice yielded Bmax values of 390 ± 21 and 350 ± 17 fmol mg−1 respectively, and KD values of 0.46 ± 0.03 and 0.5 ± 0.04 nmol·L−1 respectively, with no significant differences (P > 0.05, Student's t-test) being observed between the groups. Saturation analysis for H1 receptors with [3H]-mepyramine in Ox+/+ and Ox−/− mice yielded Bmax values of 750 ± 107 and 589 ± 85 fmol mg−1 respectively and KD values of 4 ± 1.2 and 3 ± 0.66 nmol·L−1 respectively, with no significant differences (P > 0.05, Student's t-test) being observed between the groups. This lack of significant difference in Bmax or KD between Ox+/+ and Ox−/− mice for H3 and H1 receptor binding was consistent with the lack of differences in binding observed in autoradiography studies.
Discussion
In the present study, we have identified differential effects of the novel H3 receptor antagonist GSK189254 following acute and repeat oral administration, in both narcoleptic Ox−/− mice and in wild-type littermates (Ox+/+). While the effects of GSK189254 on the sleep–wake cycle decreased following repeat dosing in Ox+/+ and Ox−/− mice, its effects on narcoleptic episodes in Ox−/− mice increased, compared with acute administration. These data are important given that several H3 antagonists/inverse agonists are currently being evaluated in clinical trials for a number of CNS indications including narcolepsy (Celanire et al., 2005; Esbenshade et al., 2008; Lin et al., 2008).
In the present study, acute administration of GSK189254 significantly increased W and decreased SWS and PS in both Ox+/+ and Ox−/− mice consistent with previous data with structurally unrelated H3 antagonists/inverse agonists, such as BF2.649 (tiprolisant), JNJ-5207852, thioperamide and ciproxifan, in several species, including rats, cats and mice, and also Ox−/− mice (Lin et al., 1990; 2008; Monti et al., 1991; Ligneau et al., 1998; 2007; Barbier et al., 2004; Bonaventure et al., 2007; Parmentier et al., 2007). Evidence accumulated from studies with H1 antagonists, as well as H1 and H3 receptor knockout mice suggest that the increase in W observed with H3 antagonists is mediated by an increase in histamine release following blockade of H3 autoreceptors, which then activates postsynaptic H1 receptors resulting in increased cortical arousal (Lin et al., 1990; Toyota et al., 2002; Parmentier et al., 2007). The fact that we observed no difference in the effect of GSK189254 on the sleep–wake cycle between Ox+/+ and Ox−/− would suggest that H3 receptor function is not affected in this model. This is in contrast to another study that suggested that Ox−/− mice were more susceptible to the H3 antagonist JNJ10181457, although this compound is thought to be a neutral antagonist rather than exhibiting inverse agonist properties (Fujiki et al., 2006; Bonaventure et al., 2007).
It is interesting to note that the doses of GSK189254 required to induce W in the present study (i.e. 3 and 10 mg·kg−1) were somewhat higher than those used in cognition studies previously in rats (Medhurst et al., 2007). These doses were known to give ∼80% and 90% occupancy respectively in ex vivo binding studies in CD1 mice 1–2 h after oral administration where brain concentrations of ∼1–3 µmol·L−1 were achieved (M. Briggs, unpubl. obs.). This suggests that higher H3 receptor occupancy (>80%) is probably required to induce effects on W than to improve cognition, consistent with recent findings in rats with a number of H3 antagonists (Le et al., 2008).
The Ox−/− mouse model is not only useful for investigating the effects of drugs on the sleep–wake cycle, but for the assessment of potential anti-narcoleptic agents (Lin et al., 2008). This model displays episodes known as DREMs, which are one of the characteristic symptoms of human narcolepsy (Chemelli et al., 1999; Mignot, 2005; Lin et al., 2008). Previously, it has been reported that modafinil does not alleviate DREMs but increases W in Ox−/− mice (Lin et al., 2008), and we confirmed this in the present study. In contrast to modafinil, acute administration of GSK189254 to Ox−/− mice significantly reduced the number and duration of DREMs, in addition to increasing W. These data are consistent with a previous study with the H3 inverse agonist BF2.649 (tiprolisant; Lin et al., 2008), and suggests that H3 antagonists/inverse agonists may potentially offer a dual approach to alleviating narcoleptic attacks and excessive daytime sleepiness in narcoleptics. Other symptoms observed in narcoleptics can include cataplexy and cognitive deficits (Mignot, 2005; Dauvilliers et al., 2007) and recent preclinical data support the possibility that H3 antagonists might help alleviate these. For example, H3 antagonists can increase cognitive performance in rats (e.g. Fox et al., 2005; Medhurst et al., 2007; Esbenshade et al., 2008) and reduce cataplexy in narcoleptic dogs (Bonaventure et al., 2007). Thus H3 antagonists may offer advantages over current therapies, which tend to help to alleviate some but not all symptoms (Billiard et al., 2006; Siegel and Boehmer, 2006).
Recent studies have demonstrated that certain effects of particular H3 antagonists may be susceptible to tolerance in preclinical species. For example, the effects of ciproxifan on food intake and locomotor activity were decreased following repeat dosing, but the effects of a non-imidazole H3 antagonist A304121 were maintained (Pan et al., 2006). However, in other studies tolerance has not been observed. For example, efficacious effects of ABT-239 in cognition models (Fox et al., 2005) and of GSK189254 in cognition and pain models were observed after repeat dosing for several days (Medhurst et al., 2007; 2008). While there is a wealth of evidence to support the acute effects of H3 antagonists on the sleep–wake cycle, to our knowledge there are no previous reports describing the effects of repeat administration of H3 antagonists. We were therefore keen to investigate the effects of repeat dosing with GSK189254 (twice daily for 8 days) on the sleep–wake cycle in Ox+/+ and Ox−/− mice and DREMs in Ox−/− mice.
To our surprise, the effects of GSK189254 on the sleep–wake cycle (i.e. increased W and decreased SWS and PS) seen after acute administration were significantly reduced following repeat dosing in both Ox+/+ and Ox−/− mice. In addition, in the lights-off phase there was a change in direction of effects of GSK189254 in Ox−/− mice compared with acute dosing. In marked contrast, the beneficial effects of GSK189254 on narcoleptic attacks (i.e. reduction in duration and number of DREMs) in Ox−/− mice were further improved significantly following repeat dosing compared with acute administration. Interestingly, this dramatic effect observed on narcoleptic episodes following repeat dosing with GSK189254 in Ox−/− mice was comparable to the effect observed in the same model with co-administration of BF2.649 and modafinil (Lin et al., 2008).
The divergence of effects observed with GSK189254 on the sleep–wake cycle compared with narcoleptic episodes after repeat dosing is both intriguing and unexpected. It is likely that the W effects are mediated by histaminergic neurons, while narcoleptic episodes may be driven additionally by the noradrenergic system (Nishino et al., 2000; Lin et al., 2008). It could be hypothesized that the W effects of GSK189254 in Ox−/− mice are primarily driven by increased histamine release following autoreceptor blockade, while the anti-narcoleptic effects are driven primarily by the increased monoamine release (most likely noradrenaline) apparent following heteroreceptor blockade. Therefore, it might be possible that heteroreceptors are potentially less susceptible to tolerance than autoreceptors, although complex repeat dose microdialysis studies would be required to investigate this further in these mice.
Further support for this hypothesis is provided by the observations that GSK189254 shows similar efficacy in rat cognition and pain models following single or repeat (up to 8 days b.i.d.) dosing (Medhurst et al., 2007; 2008), in contrast to the reduction in effect on W observed in the present study with repeat dosing compared with acute administration. Efficacy observed with H3 antagonists in these cognition and pain models is more likely to involve heteroreceptor blockade (e.g. facilitation of acetylcholine release), compared with the sleep–wake cycle when autoreceptor blockade (i.e. facilitation of histamine release) is likely to be important (Passani et al., 2004; Esbenshade et al., 2008). Another possibility is that different H3 receptor splice variants are involved (Hancock et al., 2003) and that they have different susceptibility to tolerance. However, it is not known which of the described splice variants are autoreceptors and which are heteroreceptors, as well as how each variant contributes to different behavioural responses.
The mechanism of behavioural tolerance observed in the current sleep–wake studies in Ox+/+ and Ox−/− mice is not clear. Further studies are required with other H3 antagonists to confirm this phenomenon, given that previous reports demonstrating tolerance in other behavioural parameters were compound specific (Pan et al., 2006). It is possible that H3 receptors are up-regulated following repeat dosing of H3 antagonists in Ox−/− mice, as previously described in rats (Pan et al., 2006) and wild-type mice (Morisset et al., 2000), although this up-regulation was dependent on brain region measured and the H3 antagonist investigated. Alternatively, other targets affected by enhanced neurotransmitter release following H3 receptor blockade, such as H1 receptors, acetylcholine or dopamine receptors, could be modulated in some way. However, we were not able to investigate this in the current available cohort of mice.
The current results are potentially important for the future clinical investigation of H3 antagonists not only in narcolepsy, but in other CNS disorders including Alzheimer's disease. While the potential tolerance to the W effects of H3 antagonists could be a downside for the treatment of narcolepsy, it could be beneficial in Alzheimer's disease where long-term W at night would be highly undesirable. In addition, the increase in effect on narcoleptic episodes could be inferred following repeat dosing. Recently, a small clinical study provided the first evidence for a potential beneficial effect of an H3 inverse agonist in human narcoleptics after 1 week of dosing (Lin et al., 2008) and the results of further long-term dosing studies are eagerly awaited.
Finally, as part of the present study we investigated H3 and H1 receptor binding in Ox−/− mice because there was no information on the status of these receptors in narcolepsy, despite histamine levels being shown to be decreased in human narcoleptics and narcoleptic Dobermans, indicative of a deficit in the histaminergic system (Nishino et al., 2000). Using autoradiography and saturation binding in brain homogenates, we did not observe any differences in [3H]-GSK189254 binding to H3 receptors or [3H]-mepyramine binding to H1 receptors between Ox+/+ and Ox−/− mice. This is consistent with a previous report, which eluded to the fact that H3 receptor mRNA levels in hypothalamus, cortex and hippocampus were not significantly different between Ox+/+ and Ox−/− mice (Lin et al., 2008). In addition, no changes in turnover of histamine and other monamines have been observed in Ox−/− mice previously, and tiprolisant had a similar effect on turnover in Ox+/+ and Ox−/− mice (Lin et al., 2008).
In conclusion, the present study provides further supportive data for the potential use of H3 antagonists in narcolepsy. However, following repeat dosing, the effects of the H3 antagonist GSK189254 on W and narcoleptic episodes in Ox−/− mice were differentially affected. Further studies are required to understand the mechanistic basis of these differences and their relevance to human narcolepsy.
Acknowledgments
We kindly thank Dr Masashi Yanagisawa (Howard Hughes Medical Institute, University of Texas Southwestern Medical Center, Dallas, TX 75390-8584, USA) for the original supply of Ox−/− mouse strain. The fellowship of RX Guo and M Zhang was provided by Institut Franco–Chinois (Lyon).
Conflict of interest
A.D.M., J.C.R., S.H.B. and N.U. are employed by GlaxoSmithKline.
References
- Arrang JM, Garbarg M, Schwartz JC. Auto-inhibition of brain histamine release mediated by a novel class (H3) of histamine receptor. Nature. 1983;302:832–837. doi: 10.1038/302832a0. [DOI] [PubMed] [Google Scholar]
- Arrang JM, Garbarg M, Lancelot JC, Lecomte JM, Pollard H, Robba M, et al. Potential interest in powerful and specific ligands for the histamine H3 receptor. Allerg Immunol (Paris) 1988;20:327–329. 331. [PubMed] [Google Scholar]
- Barbier AJ, Berridge C, Dugovic C, Laposky AD, Wilson SJ, Boggs J, et al. Acute wake-promoting actions of JNJ-5207852, a novel, diamine-based H3 antagonist. Br J Pharmacol. 2004;143:649–661. doi: 10.1038/sj.bjp.0705964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billiard M, Bassetti C, Dauvilliers Y, Dolenc-Groselj L, Lammers GJ, Mayer G, et al. EFNS guidelines on management of narcolepsy. Eur J Neurol. 2006;13:1035–1048. doi: 10.1111/j.1468-1331.2006.01473.x. [DOI] [PubMed] [Google Scholar]
- Blandina P, Giorgetti M, Bartolini L, Cecchi M, Timmerman H, Leurs R, et al. Inhibition of cortical acetylcholine release and cognitive performance by histamine H3 receptor activation in rats. Br J Pharmacol. 1996;119:1656–1664. doi: 10.1111/j.1476-5381.1996.tb16086.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaventure P, Letavic M, Dugovic C, Wilson S, Aluisio L, Pudiak C, et al. Histamine H3 receptor antagonists: from target identification to drug leads. Biochem Pharmacol. 2007;73:1084–1096. doi: 10.1016/j.bcp.2006.10.031. [DOI] [PubMed] [Google Scholar]
- Brown RE, Stevens DR, Haas HL. The physiology of brain histamine. Prog Neurobiol. 2001;63:637–672. doi: 10.1016/s0301-0082(00)00039-3. [DOI] [PubMed] [Google Scholar]
- Celanire S, Wijtmans M, Talaga P, Leurs R, De Esch IJ. Keynote review: histamine H3 receptor antagonists reach out for the clinic. Drug Discov Today. 2005;10:1613–1627. doi: 10.1016/S1359-6446(05)03625-1. [DOI] [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, et al. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- Dauvilliers Y, Arnulf I, Mignot E. Narcolepsy with cataplexy. Lancet. 2007;369:499–511. doi: 10.1016/S0140-6736(07)60237-2. [DOI] [PubMed] [Google Scholar]
- Esbenshade TA, Browman KE, Bitner RS, Strakhova M, Cowart MD, Brioni JD. The histamine H3 receptor: an attractive target for the treatment of cognitive disorders. Br J Pharmacol. 2008;154:1166–1181. doi: 10.1038/bjp.2008.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox GB, Esbenshade TA, Pan JB, Radek RJ, Krueger KM, Yao BB, et al. Pharmacological properties of ABT-239 [4-(2-{2-[(2R)-2-Methylpyrrolidinyl]ethyl}-benzofuran-5-yl)benzonitrile]: II. Neurophysiological characterization and broad preclinical efficacy in cognition and schizophrenia of a potent and selective histamine H3 receptor antagonist. J Pharmacol Exp Ther. 2005;313:176–190. doi: 10.1124/jpet.104.078402. [DOI] [PubMed] [Google Scholar]
- Fujiki N, Cheng T, Yoshino F, Nishino S. Specificity of direct transition from wake to REM sleep in orexin/ataxin-3 narcoleptic mice. Sleep. 2006;29:A225. doi: 10.1016/j.expneurol.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock AA, Esbenshade TA, Krueger KM, Yao BB. Genetic and pharmacological aspect of histamine H3 receptor heterogeneity. Life Sci. 2003;73:3043–3072. doi: 10.1016/j.lfs.2003.06.003. [DOI] [PubMed] [Google Scholar]
- Haas H, Panula P. The role of histamine and the tuberomamillary nucleus in the nervous system. Nat Rev Neurosci. 2003;4:121–130. doi: 10.1038/nrn1034. [DOI] [PubMed] [Google Scholar]
- Kanbayashi T, Kodama T, Kondo H, Satoh S, Miyazaki N, Kuroda K, et al. CSF histamine and noradrenaline contents in narcolepsy and other sleep disorders. Sleep. 2004;27:A236. [Google Scholar]
- Le S, Gruner JA, Mathiasen JR, Marino MJ, Schaffhauser H. Correlation between ex vivo receptor occupancy and wake-promoting activity of selective H3 receptor antagonists. J Pharmacol Exp Ther. 2008;325:902–909. doi: 10.1124/jpet.107.135343. [DOI] [PubMed] [Google Scholar]
- Ligneau X, Lin J, Vanni-Mercier G, Jouvet M, Muir JL, Ganellin CR, et al. Neurochemical and behavioral effects of ciproxifan, a potent histamine H3-receptor antagonist. J Pharmacol Exp Ther. 1998;287:658–666. [PubMed] [Google Scholar]
- Ligneau X, Perrin D, Landais L, Camelin JC, Calmels TP, Berrebi-Bertrand I, et al. BF2.649 [1-{3-[3-(4-Chlorophenyl)propoxy]propyl}piperidine, hydrochloride], a nonimidazole inverse agonist/antagonist at the human histamine H3 receptor: preclinical pharmacology. J Pharmacol Exp Ther. 2007;320:365–375. doi: 10.1124/jpet.106.111039. [DOI] [PubMed] [Google Scholar]
- Lin JS. Brain structures and mechanisms involved in the control of cortical activation and wakefulness, with emphasis on the posterior hypothalamus and histaminergic neurons. Sleep Med Rev. 2000;4:471–503. doi: 10.1053/smrv.2000.0116. [DOI] [PubMed] [Google Scholar]
- Lin JS, Sakai K, Vanni-Mercier G, Arrang JM, Garbarg M, Schwartz JC, et al. Involvement of histaminergic neurons in arousal mechanisms demonstrated with H3-receptor ligands in the cat. Brain Res. 1990;523:325–330. doi: 10.1016/0006-8993(90)91508-e. [DOI] [PubMed] [Google Scholar]
- Lin JS, Hou Y, Sakai K, Jouvet M. Histaminergic descending inputs to the mesopontine tegmentum and their role in the control of cortical activation and wakefulness in the cat. J Neurosci. 1996;16:1523–1537. doi: 10.1523/JNEUROSCI.16-04-01523.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JS, Dauvilliers Y, Arnulf I, Bastuji H, Anaclet C, Parmentier R, et al. An inverse agonist of the histamine H(3) receptor improves wakefulness in narcolepsy: studies in orexin−/− mice and patients. Neurobiol Dis. 2008;30:74–83. doi: 10.1016/j.nbd.2007.12.003. [DOI] [PubMed] [Google Scholar]
- Medhurst AD, Atkins AR, Beresford IJ, Brackenborough K, Briggs MA, Calver AR, et al. GSK189254, a novel H3 receptor antagonist that binds to histamine H3 receptors in Alzheimer's disease brain and improves cognitive performance in preclinical models. J Pharmacol Exp Ther. 2007;321:1032–1045. doi: 10.1124/jpet.107.120311. [DOI] [PubMed] [Google Scholar]
- Medhurst SJ, Collins SD, Billinton A, Bingham S, Dalziel RG, Brass A, et al. Novel histamine H3 receptor antagonists GSK189254 and GSK334429 are efficacious in surgically-induced and virally-induced rat models of neuropathic pain. Pain. 2008;138:61–69. doi: 10.1016/j.pain.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Mignot E. Narcolepsy: pharmacology, pathophysiology and genetics. In: Kryger MH, Roth T, Dement WC, editors. Principles and Practice of Sleep Medicine. Philadelphia: Elsevier Saunders; 2005. pp. 761–779. [Google Scholar]
- Monti JM, Jantos H, Boussard M, Altier H, Orellana C, Olivera S. Effects of selective activation or blockade of the histamine H3 receptor on sleep and wakefulness. Eur J Pharmacol. 1991;205:283–287. doi: 10.1016/0014-2999(91)90911-9. [DOI] [PubMed] [Google Scholar]
- Monti JM, Jantos H, Ponzoni A, Monti D. Sleep and waking during acute histamine H3 agonist BP 2.94 or H3 antagonist carboperamide (MR 16155) administration in rats. Neuropsychopharmacology. 1996;15:31–35. doi: 10.1016/0893-133X(95)00151-3. [DOI] [PubMed] [Google Scholar]
- Morisset S, Traiffort E, Arrang JM, Schwartz JC. Changes in histamine H3 receptor responsiveness in mouse brain. J Neurochem. 2000;74:339–346. doi: 10.1046/j.1471-4159.2000.0740339.x. [DOI] [PubMed] [Google Scholar]
- Nishino S, Okura M, Mignot E. Narcolepsy: genetic predisposition and neuropharmacological mechanisms. Sleep Med Rev. 2000;4:57–99. doi: 10.1053/smrv.1999.0069. [DOI] [PubMed] [Google Scholar]
- Nishino S, Fujiki N, Ripley B, Sakurai E, Kato M, Watanabe T, et al. Decreased brain histamine content in hypocretin/orexin receptor-2 mutated narcoleptic dogs. Neurosci Lett. 2001;313:125–128. doi: 10.1016/s0304-3940(01)02270-4. [DOI] [PubMed] [Google Scholar]
- Palacios JM, Wamsley JK, Kuhar MJ. The distribution of histamine H1-receptors in the rat brain: an autoradiographic study. Neuroscience. 1981;6:15–37. doi: 10.1016/0306-4522(81)90240-2. [DOI] [PubMed] [Google Scholar]
- Pan JB, Yao BB, Miller TR, Kroeger PE, Bennani YL, Komater VA, et al. Evidence for tolerance following repeated dosing in rats with ciproxifan, but not with A-304121. Life Sci. 2006;79:1366–1379. doi: 10.1016/j.lfs.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Parmentier R, Ohtsu H, Djebbara-Hannas Z, Valatx JL, Watanabe T, Lin JS. Anatomical, physiological, and pharmacological characteristics of histidine decarboxylase knock-out mice: evidence for the role of brain histamine in behavioral and sleep-wake control. J Neurosci. 2002;22:7695–7711. doi: 10.1523/JNEUROSCI.22-17-07695.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmentier R, Anaclet C, Guhennec C, Brousseau E, Bricout D, Giboulot T, et al. The brain H3-receptor as a novel therapeutic target for vigilance and sleep-wake disorders. Biochem Pharmacol. 2007;73:1157–1171. doi: 10.1016/j.bcp.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Passani MB, Lin JS, Hancock A, Crochet S, Blandina P. The histamine H3 receptor as a novel therapeutic target for cognitive and sleep disorders. Trends Pharmacol Sci. 2004;25:618–625. doi: 10.1016/j.tips.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Richardson GS, Roehrs TA, Rosenthal L, Koshorek G, Roth T. Tolerance to daytime sedative effects of H1 antihistamines. J Clin Psychopharmacol. 2002;22:511–515. doi: 10.1097/00004714-200210000-00012. [DOI] [PubMed] [Google Scholar]
- Roberts JC, Davis JB, Benham CD. 3H]Resiniferatoxin autoradiography in the CNS of wild-type and TRPV1 null mice defines TRPV1 (VR-1) protein distribution. Brain Res. 2004;995:176–183. doi: 10.1016/j.brainres.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Roth T, Roehrs T, Koshorek G, Sicklesteel J, Zorick F. Sedative effects of antihistamines. J Allergy Clin Immunol. 1987;80:94–98. doi: 10.1016/s0091-6749(87)80197-5. [DOI] [PubMed] [Google Scholar]
- Saitou K, Kaneko Y, Sugimoto Y, Chen Z, Kamei C. Slow wave sleep-inducing effects of first generation H1-antagonists. Biol Pharm Bull. 1999;22:1079–1082. doi: 10.1248/bpb.22.1079. [DOI] [PubMed] [Google Scholar]
- Schlicker E, Malinowska B, Kathmann M, Gothert M. Modulation of neurotransmitter release via histamine H3 heteroreceptors. Fundam Clin Pharmacol. 1994;8:128–137. doi: 10.1111/j.1472-8206.1994.tb00789.x. [DOI] [PubMed] [Google Scholar]
- Siegel JM, Boehmer LN. Narcolepsy and the hypocretin system–where motion meets emotion. Nat Clin Pract Neurol. 2006;2:548–556. doi: 10.1038/ncpneuro0300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Lin JS, Sakai K. Neuronal activity of histaminergic tuberomammillary neurons during wake-sleep states in the mouse. J Neurosci. 2006;26:10292–10298. doi: 10.1523/JNEUROSCI.2341-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyota H, Dugovic C, Koehl M, Laposky AD, Weber C, Ngo K, et al. Behavioral characterization of mice lacking histamine H(3) receptors. Mol Pharmacol. 2002;62:389–397. doi: 10.1124/mol.62.2.389. [DOI] [PubMed] [Google Scholar]
- Tran VT, Chang RS, Snyder SH. Histamine H1 receptors identified in mammalian brain membranes with [3H]mepyramine. Proc Natl Acad Sci USA. 1978;75:6290–6294. doi: 10.1073/pnas.75.12.6290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valatx JL. Long-term recording of cerebral, muscular and ocular electric activities in mice. C R Seances Soc Biol Fil. 1971;165:112–115. [PubMed] [Google Scholar]
- Valatx JL, Bugat R. Genetic factors as determinants of the waking-sleep cycle in the mouse. Brain Res. 1974;69:315–330. doi: 10.1016/0006-8993(74)90009-2. [DOI] [PubMed] [Google Scholar]
- Vanni-Mercier G, Gigout S, Debilly G, Lin JS. Waking selective neurons in the posterior hypothalamus and their response to histamine H3-receptor ligands: an electrophysiological study in freely moving cats. Behav Brain Res. 2003;144:227–241. doi: 10.1016/s0166-4328(03)00091-3. [DOI] [PubMed] [Google Scholar]