

Abstract
For a long time the structural and molecular features of mammalian histidine decarboxylase (EC 4.1.1.22), the enzyme that produces histamine, have evaded characterization. We overcome the experimental problems for the study of this enzyme by using a computer-based modelling and simulation approach, and have now the conditions to use histidine decarboxylase as a target in histamine pharmacology. In this review, we present the recent (last 5 years) advances in the structure–function relationship of histidine decarboxylase and the strategy for the discovery of new drugs.
Keywords: histamine, histidine decarboxylase, QM/MM, virtual screening, homology modelling, molecular dynamics
Introduction and historical background
Histamine has many different and important roles in mammalian physiopathology. Among other physiopathological processes, it is involved in allergy and other inflammatory responses, gastric acid secretion, bone loss, the control of sleep and food intake, and schizophrenia (Jorgensen et al., 2007; Haas et al., 2008; Ohtsu, 2008; Schubert and Peura, 2008). Currently, the effects of histamine are controlled by using modulators (mainly antagonists), tailored as specifically as possible to block the histamine binding to one of the four known membrane histamine receptors (H1, H2, H3 and H4). All of these subtypes are homologous G protein-coupled receptors specialized to elicit particular intracellular signals for different physiopathological effects (Kuramasu et al., 2006; Gurevich and Gurevich, 2008; Thurmond et al., 2008). The present strategy used to control unwanted effects of histamine involves finding a specific antagonist that will interfere with a given undesirable effect of the amine on a particular process, with the lowest possible side-effects on other cell types that express different histamine receptor subtypes specific for other physiological effects (see other chapters in this number). However, in practice, this is not an easy task due to the homology among the receptor proteins and the similarities in their methods of binding a common small biomolecule, namely histamine. As such, cross-reaction is one of the major problems of this strategy. In addition, the effective blockage of a given receptor by a specific antagonist may avoid one physiological effect of histamine, but does not prevent the higher synthesis and/or release of histamine that characterizes many histamine-related diseases. An increase in endogenous/newly synthesized histamine can have multiple consequences at both cellular and systemic levels; these effects need further molecular characterization to be controlled (Tanaka and Ichikawa, 2006). How can we control histamine synthesis?
Histamine is produced by α-decarboxylation of histidine and this reaction is catalysed by histidine decarboxylase (HDC). In mammals and other eukaryotic organisms, as well as in Gram-negative bacteria, HDC is a pyridoxal-5'-phosphate (PLP)-dependent enzyme (EC 4.1.1.22) expressed only in a small number of cell types, mainly mast cells, neurons located in the posterior hypothalamus and gastric enterochromaphin-like cells (Medina et al., 2003; 2005). Some important aspects of HDC regulation of histamine synthesis have been elucidated in gastric cells (Chen et al., 2006; Ai et al., 2007), but not much is known of this process in other cell types where HDC expression seems to be associated with poorly-characterized cell differentiation processes and/or subject to alterations in the control of cell proliferation (Fitz et al., 2008; Tachibana et al., 2008). At present, there is no clear strategy for controlling histamine production by interfering with HDC expression.
For a long time, a characterization of the activity of these HDC enzymes has been restricted by their extreme instability. There are no X-rays depicting the structure of any PLP-dependent HDC. In mammals, the problem is even more complex, as the enzyme needs to undergo post-translational maturation to reach its active form. The precursor of the active enzyme seems to be a fusion of a fragment homologous to other PLP-dependent amino acid decarboxylases, and a C-terminal portion (around 160 amino acids), with an unknown role (apart from inhibition of the enzymic activity) and no obvious homology with other functional proteins. This terminal fragment is lost during the activation of the enzyme (Engel et al., 1996; Fleming et al., 2004a), but its intracellular function and the proteolytic mechanisms for both its cleavage and the degradation of the active enzyme (both very rapid and complex processes) have not been completely elucidated. Different proteolytic mechanisms have been proposed: calpains, proteasome and caspase-9 (Viguera et al., 1994; Olmo et al., 1999; 2000; Rodriguez-Agudo et al., 2000; Furuta et al., 2007), but stabilization of the enzyme seems to be neither a useful nor a feasible target for a specific intervention affecting mammalian HDC levels.
In spite of its instability, even in purified preparations (Olmo et al., 2000; Fleming et al., 2004b), putative inhibitors able to bind directly to the protein have been tested, in vitro, using cell-free extracts and recombinant mammalian HDC (Olmo et al., 2002; Rodriguez-Caso et al., 2003a). In fact, several substrate analogues and natural products have been described as direct HDC inhibitors; these include α-fluromethylhistidine (α-FMH), histidine methyl ester (HME) (DeGraw et al., 1977) and a compound derived from green tea, epigallocatechin-3-gallate (EGCG) (Rodriguez-Caso et al., 2003a,b). In the case of the substrate analogues (α-FMH and HME), their mechanisms of action seem to be completely understood. Both analogues react with PLP. HME cannot be decarboxylated and blocks the enzyme in the external aldimine state; α-FMH proceeds to decarboxylation but then forms inactive derivatives of the PLP-product adduct, which are then released slowly from the catalytic site. Nevertheless, these are not specific inhibitors for the mammalian enzyme, since they also act on the homologous PLP-dependent HDC of enterobacteria (Bhattacharjee and Snell, 1990). Thus, their usefulness as therapeutic agents seems to be very limited. Of the many different natural products, EGCG is the one with the greatest inhibitory capacity against mammalian HDC (Nitta et al., 2007), with promising anti-inflammatory effects when assayed in mast cells and monocytes (Melgarejo et al., 2007). It binds to the enzyme and seems to change the PLP conformation inside the catalytic site, so blocking its reaction with the substrate (Rodriguez-Caso et al., 2003b). Nevertheless, the nature of the binding is not yet known. In addition, EGCG is not a specific inhibitor of mammalian HDC, since it is also able to bind and effectively inactivate aromatic L-amino acid (or Dopa) decarboxylase (DDC) (Bertoldi et al., 2001), another important element of our neurological and neuroendocrine system; DDC is the enzyme responsible for the synthesis of the neurotransmitters 5-hydroxytryptamine and dopamine (Haavik et al., 2008). In summary, specificity is the problem in constructing a strategy against the histamine-producing enzyme (Moya-Garcia et al., 2006). The ideal inhibitor should be able to inhibit the mammalian enzyme but have minimal effects on other enzymes present in the human organism and necessary for its homeostasis.
The enterobacterial HDC and both mammalian DDC and HDC are homologous enzymes; all belong to the DDC group II (Sandmeier et al., 1994). Their evolutionary relationships have recently been characterized (Moya-Garcia et al., 2006). The search for specific new inhibitors able to discriminate between these requires a deep structural and functional knowledge, to detect relevant differences among their structures. Then chemical structures, or chemical modifications of previously known structures, that bind preferentially to only one of them (in our case, mammalian HDC) can be designed. Only the structure for pig DDC has been elucidated from the DDC group II; however, its high sequence identity (higher than 50%) with mammalian HDC (active fragment) allowed us to obtain a 3-D model of the latter using comparative modelling techniques (Baker and Sali, 2001). This model was experimentally validated by results obtained with more than 25 direct mutants that were assayed by different biophysical techniques (Fleming et al., 2004b). The initial review of the structure–function relationship of mammalian HDC integrated all the previous information about this enzyme based on its structural characteristics (Moya-Garcia et al., 2005). More recently, the decarboxylation reaction (the rate-limiting step for histamine synthesis) has been analysed by applying a combined strategy of quantum mechanics (QM) and molecular mechanics (MM) simulations on the external aldimine (PLP-histidine) complex located in the catalytic site of the enzyme (Moya-Garcia et al., 2008). Therefore, the exact location of all residues involved in this reaction and their behaviour along the reaction is now known, facilitating the search for new potential inhibitory compounds for this reaction. All this previous information is highly valuable for the construction of in silicio experiments aimed at finding new drugs.
Today, the field of in silicio drug development is very attractive, active and fertile, but is still very new. Genomic and proteomic studies produce vast amounts of information, facilitating the identification of new therapeutically relevant targets, which allows the generation of libraries of compounds with rational chemical combinations. The technique called virtual screening (VS) uses computers to search databases of millions of compounds (already synthesized or not) for those chemical entities able to interact with a given target, thus able to interfere with its activity (Shoichet, 2004). These chemicals can then be tested against the target in order to obtain new candidates for a specific drug. In addition to the essential role played by the advances in experimental and theoretical fields, the incredible progress in computer technology has been decisive in our understanding of biological structures and the processes in which they are involved. Modelling unknown structures from bare sequences, long simulations of enzymes and complex multimeric structures, and large-scale VS experiments are now performed routinely thanks to the availability of fast processors at modest prices. However, the expected revolution in rational drug discovery has not yet arrived, despite all these advances. The main limitations are the availability of reliable structural models for the target (having at hand a 3-D structure of the target in most of the cases is not enough) (Davis et al., 2003) and the inclusion (at accurate levels) of some important effects such as the environment (solvent, ions, metal atoms, and so on) (Morreale et al., 2007), entropic losses (of both conformation and configuration) (Carlsson and Aqvist, 2005) or the flexibility of the target (the most striking and elusive point) (Cozzini et al., 2008).
In this review, we focus on computer-aided drug discovery in cases where the structure of the target has been obtained by means of comparative modelling, refined by simulation of molecular dynamics (MD), and finally, on inhibitors that have been found using rational drug discovery through VS experiments. A brief description of these methods and their application to HDC are presented in the following sections. Figure 1 shows the strategy scheme used in the case of mammalian HDC.
Figure 1.
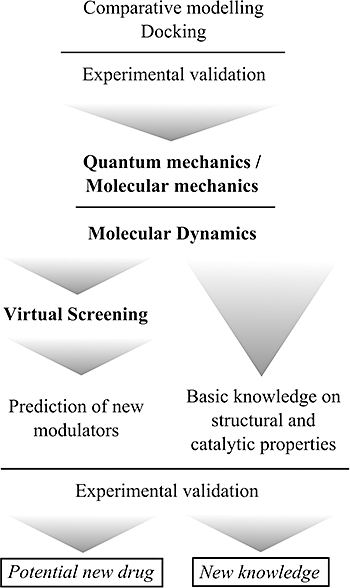
Sequential scheme of combined computer-based and experimental approaches followed in our studies on mammalian histidine decarboxylase over the last 5 years.
Comparative modelling versus experimental structure determination; the example of histidine decarboxylase
Knowing the ligand structure is a linear process, but things are much more complicated for the receptor. Protein structures are usually determined using powerful experimental techniques such as X-ray crystallography, cryoelectron microscopy and nuclear magnetic resonance (NMR). The development of these techniques, together with advances in protein expression and purification, microcrystallization (Abola et al., 2000) and the use of synchrotron light (Sorensen et al., 2006), has led to a separate discipline referred to as Structural Genomics (Chandonia and Brenner, 2006). Because of this development, the growth of the Protein Data Bank (PDB, i.e. the number of known protein structures) (Berman et al., 2007), and the number of potential pharmacological targets, has been exponential.
Nevertheless, there is a growing gap between the number of known structures and sequences; that is, the number of newly discovered protein sequences grows faster. For example, over the last 4 years, the number of sequences in the comprehensive Swiss-Prot/TrEMBL database (Boutet et al., 2007) increased by a factor of 5.44, while the number of protein structures deposited in the PDB increased by only a factor of 1.85. Therefore, the expanding field of Structural Genomics benefits from advances in computer methods for determining protein structure; comparative or homology modelling (Marti-Renom et al., 2000; Xiang, 2006) attempts to bridge this sequence-structure gap.
The technique relies on the observation that, during evolution, protein structure is more stable and changes much more slowly than the underlying sequence, so similar sequences adopt practically identical structures, and distantly related sequences will fold into similar structures (Chothia and Lesk, 1986; Sander and Schneider, 1991). Thus, the unknown structure of a target protein (not to be mistaken with the drug target) can be inferred from the structure of a template protein if there is enough sequence homology between them. In order to obtain a reliable model, the threshold for sequence identity depends on the number of aligned residues, but is usually over 30%. It is important to stress that, in some cases, homology between proteins is not clear from pairwise methods such as sequence alignment. Profile-based methods can be more sensitive in homology detection; since the important factor in obtaining a reliable model is the existence of homology between target and template, the scope of comparative modelling methods can be extended, in some cases, to low sequence identities between target and template (Tramontano and Morea, 2003). Briefly, a homology modelling protocol is carried out in a few sequential steps, as described elsewhere (Marti-Renom et al., 2000; Baker and Sali, 2001; Fiser and Sali, 2003; John and Sali, 2003): finding known structures related to the target sequence (templates), aligning the target sequence with the templates, building the model, and finally assessing and validating the model.
The applications of a protein structure model depend on its accuracy, which tends to decrease as the evolutionary distance between target and templates increases, so the target-template sequence identity is a good indication of the quality of a given model. Fortunately, a protein structure model does not have to be perfect to be helpful in biomedicine or biotechnology; however, the type of problem that can be tackled with a particular model does depend on its quality (Marti-Renom et al., 2000), ranging from prediction of the approximate biochemical function (with models based on less than 30% sequence identity, at the low end of the accuracy spectrum), to predictions of important features in the target protein that do not occur in the template structure (with medium-resolution models). Moreover, the average quality of models at the highest end of the accuracy spectrum, those based on more than 50% sequence identity, is similar to that of low-resolution X-ray structures (Baker and Sali, 2001). The alignments on which these models are based contain almost no errors, so, among other applications, they can be used for structure-based drug discovery, small ligand docking and prediction of detailed ligand–protein interactions.
It is generally assumed that docking to comparative models is more challenging and less successful than docking to crystallographic structures. However, little work has been done to obtain quantitative information about the accuracy of docking to homology models, to determine in detail why the results are inferior to those obtained from experimentally determined structures. In many examples, protein homology models have supported the discovery of the optimum compounds for potency and selectivity (for a detailed review and examples see Hillisch et al., 2004; Jacobson and Sali, 2004).
The PLP-dependent HDCs are good examples of proteins for which characterization by biophysical techniques has so far been impossible. The instability of the protein makes it inconceivable to reach that goal. However, comparative modelling, together with computer simulations (see below), is allowing us to gain insights into this molecular system, with a final aim of revealing new intervention strategies. In 2003, the first model of an active version of mammalian HDC was obtained (Rodriguez-Caso et al., 2003a). The predicted structure was validated experimentally (Fleming et al., 2004b); this allowed us to integrate the previous experimental information on the enzyme, obtaining new insights into the most promising targets at which to interfere with the protein's activity (the catalytic centre and dimerization surface). In fact, the prokaryotic and eukaryotic enzyme are homologous enzymes; however, from the comparative modelling analysis of both types of enzymes, it is clear that they do differ, mostly in residues located in the substrate binding sites and in the N-terminus of their respective monomers (Moya-Garcia et al., 2006). A homology model of the Gram-negative Klebsiella planticola HDC was built, using as a template the structure of human PLP-dependent glutamate decarboxylase (GAD, EC 4.1.1.15), recently determined experimentally (Fenalti et al., 2007). The two enzymes share a sequence identity of 24%. Figure 2 shows a comparison of the monomer of both enzymes. These deduced differences, together with other simulation-based structure-function studies, can help to identify inhibitors more selective for any of the homologous enzymes that can coexist in the human body (mammalian and enterobacterial HDCs and Dopa decarboxylases).
Figure 2.
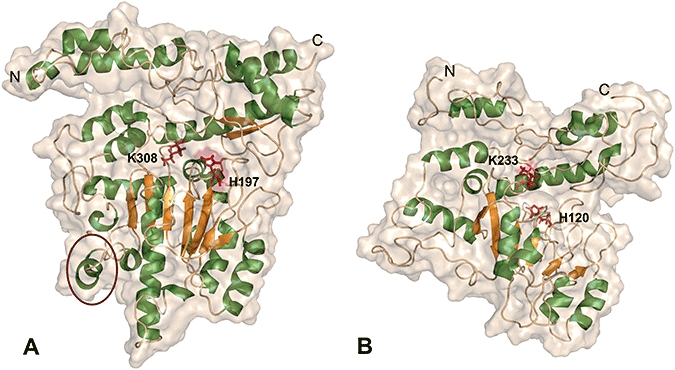
Structural models of both mammalian (A) and bacterial (B) PLP-dependent histidine decarboxylase (HDC) monomers. For orientation, amino(N)- and carboxy (C)-termini are shown. PLP-interacting residues K308 and H197 are depicted as dark red sticks with similar perspective. Flexible loop region of mammalian HDC, predicted to both interact and move during substrate binding, is surrounded by a dark red circle. As can be observed, the corresponding region in bacterial HDC is distorted, in agreement with the suspected differences between the enzymes in both the substrate binding surface and the quaternary structures (for further information see Moya-Garcia et al., 2005; 2006). Images were made using PyMOL (DeLano, 2002).
Molecular dynamics: a more realistic approach to protein function
In structure-based drug discovery, knowing the structure of the drug target is often not enough. Structure determines function, but it is not easy to deduce many of the activities or properties of a protein just from its structure. A protein cannot be merely reduced to its description as a rigid and static structure, as it is a dynamical reality with conformational fluctuations in time and substantial changes in the presence of ligands. Although the global structure is a key element in the function of a protein, its flexibility is an essential factor that modulates the relationship between structure and function. To go further, flexibility, understood as the capacity for conformational change in response to external stimuli, is part of the nature of all proteins and molecular systems. Thus, it is essential to understand how and why proteins change their conformations in order to be able to control and understand their biological functions. This dynamical nature needs to be considered in the study of the ligand–target interactions. Thus, for many applications, such as VS experiments, it is advisable to represent the protein as an ensemble of different conformations that describe the inherent flexibility of the system, although there is not a clear consensus on how the ensemble should be represented. There are examples where many conformations were used (obtained from different X-ray or NMR structures or generated by computerized sampling techniques as Monte Carlo or MD) (Totrov and Abagyan, 2008), or where only one was used that comprised information of the entire ensemble (averaging energies or coordinates of many single conformations) (Osterberg et al., 2002).
This blurry description is not enough to represent the protein flexibility properly, a time-dependent feature of macromolecular systems that is one of the most difficult yet essential to understand. MD is a powerful computer technique used to study flexibility (Hansson et al., 2002; Norberg and Nilsson, 2003). It is valuable for understanding the dynamic behaviour of proteins at different timescales, from the fast internal fluctuations to the slower and more global movements that constitute conformational changes, or, eventually, the folding of a polypeptide into the native structure of a protein (Snow et al., 2005). Furthermore, the explicit effects of solvent molecules and ions on the protein structure and stability can, and should, be taken into account to obtain accurate temporal averages of important structural and thermodynamic properties of the system under study, especially the binding energy, which are of utmost importance in the field of drug discovery.
Molecular dynamics simulates molecular time-dependent events in proteins and other biological macromolecules using the laws of classical mechanics. In particular, Newton's equations of motion are applied for an atomistic representation of a molecular system (balls for atoms and string for bonds) by employing MM force fields based on empirically deduced interaction potentials or derived from more complex quantum calculations. The energy of a molecular system within the force field approximation is a sum of different terms accounting for the distortion of the system as compared with an idealized structure where bonded (bond stretching, angle bending and torsions) and non-bonded (van der Waals and electrostatics) interaction terms are included. The main differences between the most widely used force fields [AMBER (Case et al., 2004), GROMOS (van Gunsteren et al., 1996), CHARMM (Brooks et al., 1983), and so on] are due to parametrical issues and the functional form of the different terms entering in the force field equation. Although atomic charges are explicitly included, they remain constant over the simulation in most of the force fields (new polarized force fields are now emerging to ameliorate this drawback, see Xie et al., 2008), precluding the use of MM force fields in systems undergoing chemical reactions. To model such changes adequately, as in processes such as bond-breaking/-forming, charge-transfer or electronic excitation, it is necessary to rely on the more accurate approximation obtained with QM. As noted above, it is also essential to introduce the effect of the environment. It has been demonstrated that significant changes can occur both in the biological structures and in the reactivity profile due to environmental influences. Therefore, in cases where chemical reactions need to be modelled while also taking into account environmental effects, it is essential to use a method that can account for both. Due to its high computer costs, the application of QM is still limited to relatively small systems consisting of up to tens or several hundreds of atoms, or even smaller systems when using higher levels of theory.
A solution will be a combined method able to treat the main atoms involved in the reaction with QM and the rest with MM. These methods will join the accuracy of the QM description with the low computer costs of MM; these are called hybrid methods (QM/MM) and have become very popular (Warshel, 2003). These methods are being used in the study of reactions of biological interest (Garcia-Viloca et al., 2004; Marti et al., 2004) and it has been demonstrated they can be used to identify key residues in catalysis (Ridder et al., 2003), resolve mechanistic questions and verify the fundamental principles of catalysis (Marti et al., 2004). Potential contributions, obtained from this modelling of enzyme reactions, to drug discovery have recently been reviewed by Raha et al. (2007) and Mulholland (2005). These include the identification of key catalytic residues and the reaction mechanism leading to the identification of transition states and other intermediates, the prediction of drug metabolism and the accurate calculation of the free energy of binding.
Our group has applied simulation techniques and MD techniques, by using the hybrid methodology QM/MM, to unravel the basis of the mammalian HDC catalytic mechanism (Moya-Garcia et al., 2008). In this study, we examined the decarboxylation of the intermediate cofactor-substrate adduct (the external aldimine) in the enzymatic environment (catalysed reaction) and in an aqueous environment. In each case, the reaction environment was explicitly considered and the energy used for each process was calculated. From a comparison of the reactions in the two conditions, we obtained the differentiating elements that explain the catalysis by mammalian HDC. We consider this extensively evaluated computer model of the mammalian HDC structure, in its cofactor-substrate adduct bound state, to be the first step towards performing high-throughput screening in silicio.
Virtual screening techniques: searching for new molecules
The type of strategy employed in VS depends on the structural information that is readily available, and can be performed even if the structure of the target is not known [using pharmacophores (Sun, 2008), similarity techniques applied to known active ligands (Bajorath, 2001), and so on]. The most favourable case is when both of the structures (the target and the ligand) are known. Here, docking-based techniques are very promising, although far from being completely successful (Warren et al., 2006). In docking, the problem is to find out of the many possible ways a ligand can be positioned within a binding site, the appropriate one that triggers/inhibits the biological activity of the target. To discern which position is the best, each of them is scored according to their reaction with the target. This is done by means of a mathematical function, the scoring function, built to capture the essential events that occur when a ligand binds to a target (Warren et al., 2006). Much of the uncertainty (although not all) in docking and scoring protocols has its roots in the definition of these functions. VS is an extension of the docking procedure, actually performed with a small number of molecules, to handle millions of them. In this case, the objective is somehow different than that of docking: success is achieved if we are good at separating the active compounds from the inactive ones, and if, at least, some of the active compounds are found at the top of a list based on the above-mentioned scoring function. The explosion in the use of VS in the last decade can be understood when it is considered that although more money is invested in R&D projects, there are fewer newly discovered drugs reaching the market (Smith, 2002). This fact has fuelled the development of many different docking algorithms and sophisticated scoring functions (Sousa et al., 2006).
A VS protocol is a sequence of filters that increase in complexity to reduce the number of molecules subjected to experimental assays to a tractable amount. It is customary to start with molecules fulfilling Lipinski's rule of five (Lipinski et al., 2001) and possibly imposing some other constraints, such as adequate solubility or certain kind of chemotypes (focalized libraries). Nevertheless, brute force approximation can be also employed, based on the idea that the more molecules you can test, the higher the probability of finding promising candidates. An increasing number of databases are available to start with, for example, ZINC, with over 8 million compounds available (Irwin and Shoichet, 2005). Other databases such as DUD are useful for testing a VS protocol prior to undertaking a search for new molecules, so one can assess the performance of the protocol to decide if it is appropriate for a particular problem (Huang et al., 2006).
To facilitate the choice of a particular protocol, or to compare different protocols, we have developed VSDMIP (Virtual Screening Data Management on an Integrated Platform) (Gil-Redondo et al., 2009), a flexible fully automated computer platform that combines all the steps needed to generate a short list of candidates from a database of 2-D molecular structures. In brief, the VSDMIP protocol consists of (i) a database; (ii) a library of service interfaces and plugins; and (iii) a set of workflows and implementing commands. All of the data and VS results from small molecules (ligands) are stored within VSDMIP. The user controls the platform through different command line utilities and configures it using XML files. VSDMIP currently runs on Linux/x86 platforms and has been successfully implemented in the MareNostrum supercomputer at the Barcelona Supercomputing Center (BSC), making it possible to screen 4 million compounds (the actual size of our molecular database) in less than 1 month.
In general, the steps needed to initiate a VS study can be broadly divided into the preparation of the target and the ligands. (1) For the target, starting from its 3-D structure, we (i) choose only the domains surrounding the active site, (ii) add missing loops and atoms (especially if they are close to the binding site), (iii) add hydrogen atoms, (iv) assign atom types and atomic charges, and finally (v) characterize the active site. This last step entails limiting the active site by a box (where subsequent docking will be performed) and making the space covered by the box discrete with grid points spanning the three-dimensional space. Each grid point contains information on the interaction of an atomic probe atom (representing common atom types in pharmacologically relevant molecules), including the electrostatic interaction and the possibility of forming hydrogen bonds between the ligand and the residues at the binding site. (2) For the ligands, we (i) start with a 2-D topological representation of the structures (SMILES string, see Weininger, 1988) in order to avoid bias related to the conformations, (ii) transform them into 3-D (adding hydrogen atoms and generating tautomers, steroisomers, and different ring conformations if necessary), (iii) assign atomic radii and charges, and (iv) perform conformational analysis with ALFA (Gil-Redondo, 2006).
Each ligand is then docked into the target active site with CDOCK (Perez and Ortiz, 2001), our docking software, which includes a movement/evaluation/refinement strategy: (i) translate and rotate the ligand in each grid point; (ii) evaluate the energy for each configuration generated; and (iii) refine the best configurations generated using a rigid body SIMPLEX optimization program (Nelder and Mead, 1965). Finally, the best configuration of all is taken as the docking result. The scoring function implemented in CDOCK accounts for van der Waals and electrostatic forces, as well as hydrogen–bond interactions. It also includes a solvation correction term based on an implicit model (Morreale et al., 2007). Within VSDMIP, the docking step can be preceded by a docking method such as DOCK (Kuntz et al., 1982) or FRED (OpenEye Scientific Software, Inc.), configurated in a less accurate but faster way, or it can be replaced by Autodock (Morris et al., 1998).
Representing the protein as a grid imposes the rigidity constraint into the docking calculations, which is one of the main drawbacks in computer-aided drug discovery based on structure. To overcome this drawback, the best molecules classified are submitted to a short MD simulation in an explicit solvent. MM-GBSA analysis is performed on selected snapshots to obtain an estimate of the free energy of binding (entropy not included) for each compound (Massova and Kollman, 2000). This is the value employed for the definitive classification. A visual inspection of each of the final best-ranked candidates is always mandatory.
The first attempt of drug discovery based on mammalian HDC structure
As far as we know, there has been no attempt to perform high-throughput screening on mammalian HDC in order to find new inhibitors with a potential pharmacological use, although there is interest in the characterization of this enzyme as a pharmacological target. The HDC inhibitors known to date are substrate analogues and were developed in the 1970s (DeGraw et al., 1977). Recently a new strategy for developing new inhibitors based on the external aldimine, which is the common intermediate of the transformation of all amino acids catalysed by PLP-dependent enzymes, has been reported (Wu et al., 2008). However, the authors do not use computer modelling to guide their inhibitor design rationale. They try to elucidate the structure–activity relationship of their synthesized compounds based on a rough computer model of the active site of human HDC, together with the presumed intracellular form of the compound with the highest inhibition rate, namely pyridoxyl-histidine methyl ester.
Docking of known inhibitors; validation of the model
We were able to build a high-quality structural model for HDC. It shows the relevant structural features and reliably reproduces the behaviour of the enzyme. We were able to reveal key amino acids for the activity and stability of HDC (Rodriguez-Caso et al., 2003a; Fleming et al., 2004b) and we discern particular features of the reaction mechanism, with full atomic details (Moya-Garcia et al., 2008). Nevertheless, we submitted our model to an additional validation test to check whether it can be used to discover new HDC inhibitors with potential pharmacological use. We used the above-mentioned VSDMIP with the natural substrate histidine and the two well-known inhibitors α-FMH and HME. A full standard VS protocol was followed.
Characterization of the receptor was carried out as explained above. The active site pocket was determined by a 5 Å box centred at histidine in its external aldimine conformation and using interaction grids of 0.5 Å spacing. Once the active site was demarcated, the set of test ligands was docked using CDOCK. The best results from the docking process were obtained from the database and visualized in PyMol (DeLano, 2002). The coordinates and topology files of the receptor-ligand complexes were then generated using the program LEaP of the AMBER Molecular Dynamics package (Case et al., 2005). PLP and ligand parameters were obtained with Antechamber module. The systems were solvated, neutralized and submitted to a common protocol of energy minimization and MD simulation using the program sander from AMBER package. A 2 ns production stage at a temperature of 300 K was followed by a cooling process, in which the temperature was decreased gradually from 300 to 292 K.
The minimum energy configuration for each ligand with the HDC active site, according to the scoring function implemented in CDOCK, were obtained and checked visually. All of them showed similar total binding energy values, ranging from −18 kcal·mol−1 (for both HME and histidine) to −11 kcal·mol−1 for FMH. These energy values together with those obtained from the previous VS carried out using a larger number of compounds, showed that we are dealing with a closed active site, which is in agreement with the structural information derived from our HDC homology model and other PLP-dependent decarboxylases structures, whose active sites are frequently buried in the dimerization surface. Our calculations, for the pathway connecting the active site with the outside solvent, show that the substrate needs to pass over a channel of about 40 Å in length inside the enzyme to reach the active site.
The best configurations docked for histidine and the two inhibitors computed show a satisfactory fit to the active site, with the proper orientation to proceed with the transaldimination reaction and form the external aldimine with the cofactor, as can be seen in Figure 3. Both PLP and ligand were able to move freely in the active site during the MD simulation since no local restrictions were imposed, resulting in a slight separation between them. As a result of previous VS studies, where new inhibitors were found and successfully tested, the structures of receptor-inhibitor complexes were then determined by means of X-ray diffraction in order to check the similarity of the configuration predicted after docking and the validation protocol with the one obtained by crystallography (Warren et al., 2006). In only a few cases, VS-derived complexes resembled those structures obtained by experimental means, indicating that the computer-derived approaches are not yet capable of giving us a perfect image of the active site of an enzyme when binding any ligand, but they are reasonably accurate for determining the accommodation and stability of those compounds attracted to the active site pocket.
Figure 3.
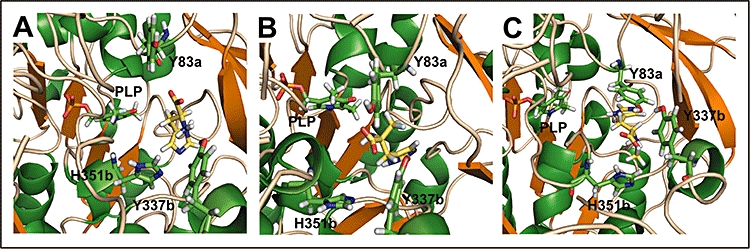
Final configurations of the docked histidine substrate (A), αFMH (B) and HME (C) into the HDC active site (depicted in yellow sticks) after screening with VSDMIP and molecular dynamics simulation. Residues previously described to be located in the proximity of the natural substrate and the cofactor pyridoxal-5'-phosphate (PLP) are shown as green sticks. Suffixes ‘a’ and ‘b’ indicate the residue-containing monomers, monomer ‘a’ being the one that binds PLP.
Preliminary results of the docking process in our VS study over the ZINC 7 compound database (Irwin and Shoichet, 2005), which comprises 4 million molecules, showed the method tended to fail when trying to fit compounds with a large number of atoms. This would significantly reduce the number of suitable candidates as potential inhibitors of HDC activity. These results are in agreement with those observed by Wu et al. (2008).
On the other hand, those compounds identified with suitable configurations after docking are arranged in the active site as they can make interactions with key residues involved in stabilization of the substrate (Moya-Garcia et al., 2008). Y83 and Y337, which have been determined as important residues in favouring the reception of the ligand into the active site (Rodriguez-Caso et al., 2003a; Fleming et al., 2004b), as well as H351 and H197, are located in the proximity of our best-docked compounds. Further refinement of these preliminary results by means of MD simulations will consolidate these interactions or even reveal new ones that were not obvious just after the docking process.
Discussion and conclusions
Due to the pleiotropic effects of histamine, and on the basis of results obtained with HDC knockout mice (Jorgensen et al., 2007; Haas et al., 2008; Ohtsu, 2008; Schubert and Peura, 2008), it is possible that selective, direct inhibition of HDC could have many different secondary effects. To be able to control the production of histamine by this method, either locally (for instance, topical use) or at the systemic level, rather than just interfering with the reception of the amine on target cells, could have important therapeutic consequences in physiopathological situations where either the local or circulating levels of histamine are excessive due to abnormal histamine production. Also, given the important roles of histamine in the central nervous system (Wijtmans et al., 2008; Zhao et al., 2008), special attention should be paid to the ability of any HDC inhibitor (or its derivatives) to cross the blood-brain barrier, and this should be evaluated by experimental and/or in silicio approaches (Kortagere et al., 2008; Malakoutikhah et al., 2008). In addition, therapies combining both HDC inhibitors and histamine receptor agonists/antagonists should not be ruled out.
This review presents an example of not only a potentially interesting protein for pharmacology, but also a drug target that has been very difficult to characterize by experimental approaches and, consequently, to use efficiently for drug discovery. By changing the strategy, that is by combining in silicio and experimental techniques, the structural and catalytic properties of HDC are now known and this knowledge can be used to discover potential, new antihistamine drugs. In addition, this strategy can be applied to many other proteins related to amine metabolism, immunology and drug discovery in general, to solve other pending problems in biomedicine, biotechnology and pharmacology. From an economical point of view, it is obvious that this strategy would also be convenient for the pharmacological industry, since the in silicio approach can save significant investment in experimental protein chemistry techniques and high-throughput screening protocols.
Acknowledgments
The CIBER de Enfermedades Raras is an initiative of the ISCIII. This work was supported by Grant SAF2008-02522, Ministerio de Ciencia e Innovación Work at the CBM-SO was partially supported by a grant from ‘Comunidad de Madrid’ thorough BIPEDD project (SBIO-0214–2006). We also acknowledge the generous allocation of computer time at the BSC.
Glossary
Abbreviations:
- α-FMH
α-fluromethylhistidine
- DDC
L-amino acid decarboxylase
- EGCG
epigallocatechin-3-gallate
- GAD
glutamate decarboxylase
- GBSA
generalized born surface area
- HDC
histidine decarboxylase
- HME
histidine methyl esther
- MD
molecular dynamics
- MM
molecular mechanics
- NMR
nuclear magnetic resonance
- PLP
pyridoxal-5'-phosphate
- QM
quantum mechanics
- VS
virtual screening
Conflict of interest
The authors state no conflict of interest.
References
- Abola E, Kuhn P, Earnest T, Stevens RC. Automation of X-ray crystallography. Nat Struct Biol. 2000;7(Suppl.):973–977. doi: 10.1038/80754. [DOI] [PubMed] [Google Scholar]
- Ai W, Zheng H, Yang X, Liu Y, Wang TC. Tip60 functions as a potential corepressor of KLF4 in regulation of HDC promoter activity. Nucleic Acids Res. 2007;35:6137–6149. doi: 10.1093/nar/gkm656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajorath J. Selected concepts and investigations in compound classification, molecular descriptor analysis, and virtual screening. J Chem Inf Comput Sci. 2001;41:233–245. doi: 10.1021/ci0001482. [DOI] [PubMed] [Google Scholar]
- Baker D, Sali A. Protein structure prediction and structural genomics. Science. 2001;294:93–96. doi: 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
- Berman H, Henrick K, Nakamura H, Markley JL. The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2007;35:D301–D303. doi: 10.1093/nar/gkl971. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertoldi M, Gonsalvi M, Voltattorni CB. Green tea polyphenols: novel irreversible inhibitors of dopa decarboxylase. Biochem Biophys Res Commun. 2001;284:90–93. doi: 10.1006/bbrc.2001.4945. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee MK, Snell EE. Pyridoxal 5'-phosphate-dependent histidine decarboxylase. Mechanism of inactivation by alpha-fluoromethylhistidine. J Biol Chem. 1990;265:6664–6668. [PubMed] [Google Scholar]
- Boutet E, Lieberherr D, Tognolli M, Schneider M, Bairoch A. UniProtKB/Swiss-Prot: the manually annotated section of the UniProt KnowledgeBase. Methods Mol Biol. 2007;406:89–112. [Google Scholar]
- Brooks BR, Bruccoleri RE, Olafson DJ, States DJ, Swaminathan S, Karplus M. CHARMM: a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- Carlsson J, Aqvist J. Absolute and relative entropies from computer simulation with applications to ligand binding. J Phys Chem. 2005;109:6448–6456. doi: 10.1021/jp046022f. [DOI] [PubMed] [Google Scholar]
- Case DA, Darden TA, Cheatham TE, Simmerling CL, Wang J, Duke RE, et al. AMBER. Vol. 8. San Francisco: University of California; 2004. [Google Scholar]
- Case DA, Cheatham TE, III, Darden T, Gohlke H, Luo R, Merz KM, Jr, et al. The Amber biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandonia JM, Brenner SE. The impact of structural genomics: expectations and outcomes. Science. 2006;311:347–351. doi: 10.1126/science.1121018. [DOI] [PubMed] [Google Scholar]
- Chen D, Aihara T, Zhao CM, Hakanson R, Okabe S. Differentiation of the gastric mucosa. I. Role of histamine in control of function and integrity of oxyntic mucosa: understanding gastric physiology through disruption of targeted genes. Am J Physiol Gastrointest Liver Physiol. 2006;291:G539–G544. doi: 10.1152/ajpgi.00178.2006. [DOI] [PubMed] [Google Scholar]
- Chothia C, Lesk AM. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986;5:823–826. doi: 10.1002/j.1460-2075.1986.tb04288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzini P, Kellogg GE, Spyrakis F, Abraham DJ, Costantino G, Emerson A, et al. Target flexibility: an emerging consideration in drug discovery and design. J Med Chem. 2008;51:6237–6255. doi: 10.1021/jm800562d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AM, Teague SJ, Kleywegt GJ. Application and limitations of X-ray crystallographic data in structure-based ligand and drug design. Angew Chem Int Ed Engl. 2003;42:2718–2736. doi: 10.1002/anie.200200539. [DOI] [PubMed] [Google Scholar]
- DeGraw JI, Engstrom J, Ellis M, Johnson HL. Potential histidine decarboxylase inhibitors. 1. α- and β-Substituted histidine analogs. J Med Chem. 1977;20:1671–1674. doi: 10.1021/jm00222a027. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
- Engel N, Olmo MT, Coleman CS, Medina MA, Pegg AE, Sanchez-Jimenez F. Experimental evidence for structure-activity features in common between mammalian histidine decarboxylase and ornithine decarboxylase. Biochem J. 1996;320:365–368. doi: 10.1042/bj3200365. Pt 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenalti G, Law RH, Buckle AM, Langendorf C, Tuck K, Rosado CJ, et al. GABA production by glutamic acid decarboxylase is regulated by a dynamic catalytic loop. Nat Struct Mol Biol. 2007;14:280–286. doi: 10.1038/nsmb1228. [DOI] [PubMed] [Google Scholar]
- Fiser A, Sali A. Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol. 2003;374:461–491. doi: 10.1016/S0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
- Fitz LJ, Brennan A, Wood CR, Goldman SJ, Kasaian MT. Activation-induced cellular accumulation of histamine in immature but not mature murine mast cells. Cell Mol Life Sci. 2008;65:1585–1595. doi: 10.1007/s00018-008-8106-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming JV, Fajardo I, Langlois MR, Sanchez-Jimenez F, Wang TC. The C-terminus of rat L-histidine decarboxylase specifically inhibits enzymic activity and disrupts pyridoxal phosphate-dependent interactions with L-histidine substrate analogues. Biochem J. 2004a;381:769–778. doi: 10.1042/BJ20031553. Pt 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming JV, Sanchez-Jimenez F, Moya-Garcia AA, Langlois MR, Wang TC. Mapping of catalytically important residues in the rat L-histidine decarboxylase enzyme using bioinformatic and site-directed mutagenesis approaches. Biochem J. 2004b;379:253–261. doi: 10.1042/BJ20031525. Pt 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuta K, Nakayama K, Sugimoto Y, Ichikawa A, Tanaka S. Activation of histidine decarboxylase through post-translational cleavage by caspase-9 in a mouse mastocytoma P-815. J Biol Chem. 2007;282:13438–13446. doi: 10.1074/jbc.M609943200. [DOI] [PubMed] [Google Scholar]
- Garcia-Viloca M, Gao J, Karplus M, Truhlar DG. How enzymes work: analysis by modern rate theory and computer simulations. Science. 2004;303:186–195. doi: 10.1126/science.1088172. [DOI] [PubMed] [Google Scholar]
- Gil-Redondo R. Master Thesis: Implementación de una plataforma para el cribado virtual de quimiotecas. Madrid: UNED edn; 2006. [Google Scholar]
- Gil-Redondo R, Estrada J, Morreale A, Herranz F, Sancho J, Ortiz AR. VSDMIP: virtual screening data management on an integrated platform. J Comput Aided Mol Des. 2009;23:171–184. doi: 10.1007/s10822-008-9249-9. [DOI] [PubMed] [Google Scholar]
- van Gunsteren WF, Billeter SR, Eising AA, Hünenberger PH, Krüger P, Mark AE, et al. Biomolecular Simulation: The GROMOS96 Manual and User Guide. Zürich: Vdf Hochschulverlag; 1996. [Google Scholar]
- Gurevich VV, Gurevich EV. Rich tapestry of G protein-coupled receptor signaling and regulatory mechanisms. Mol Pharmacol. 2008;74:312–316. doi: 10.1124/mol.108.049015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas HL, Sergeeva OA, Selbach O. Histamine in the nervous system. Physiol Rev. 2008;88:1183–1241. doi: 10.1152/physrev.00043.2007. [DOI] [PubMed] [Google Scholar]
- Haavik J, Blau N, Thony B. Mutations in human monoamine-related neurotransmitter pathway genes. Hum Mutat. 2008;29:891–902. doi: 10.1002/humu.20700. [DOI] [PubMed] [Google Scholar]
- Hansson T, Oostenbrink C, van Gunsteren W. Molecular dynamics simulations. Curr Opin Struct Biol. 2002;12:190–196. doi: 10.1016/s0959-440x(02)00308-1. [DOI] [PubMed] [Google Scholar]
- Hillisch A, Pineda LF, Hilgenfeld R. Utility of homology models in the drug discovery process. Drug Discov Today. 2004;9:659–669. doi: 10.1016/S1359-6446(04)03196-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N, Shoichet BK, Irwin JJ. Benchmarking sets for molecular docking. J Med Chem. 2006;49:6789–6801. doi: 10.1021/jm0608356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin JJ, Shoichet BK. ZINC – a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson M, Sali A. Comparative protein structure modeling and its applications to drug discovery. Annu Rep Med Chem. 2004;39:259–276. [Google Scholar]
- John B, Sali A. Comparative protein structure modeling by iterative alignment, model building and model assessment. Nucleic Acids Res. 2003;31:3982–3992. doi: 10.1093/nar/gkg460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen EA, Knigge U, Warberg J, Kjaer A. Histamine and the regulation of body weight. Neuroendocrinology. 2007;86:210–214. doi: 10.1159/000108341. [DOI] [PubMed] [Google Scholar]
- Kortagere S, Chekmarev D, Welsh WJ, Ekins S. New predictive models for blood-brain barrier permeability of drug-like molecules. Pharm Res. 2008;25:1836–1845. doi: 10.1007/s11095-008-9584-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntz ID, Blaney JM, Oatley SJ, Landridge R, Ferrin TE. A geometric approach to macromolecule – ligand interactions. J Mol Biol. 1982;161:269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- Kuramasu A, Sukegawa J, Yanagisawa T, Yanai K. Recent advances in molecular pharmacology of the histamine systems: roles of C-terminal tails of histamine receptors. J Pharmacol Sci. 2006;101:7–11. doi: 10.1254/jphs.fmj06001x3. [DOI] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Malakoutikhah M, Teixido M, Giralt E. Toward an optimal blood-brain barrier shuttle by synthesis and evaluation of peptide libraries. J Med Chem. 2008;51:4881–4889. doi: 10.1021/jm800156z. [DOI] [PubMed] [Google Scholar]
- Marti S, Roca M, Andres J, Moliner V, Silla E, Tunon I, et al. Theoretical insights in enzyme catalysis. Chem Soc Rev. 2004;33:98–107. doi: 10.1039/b301875j. [DOI] [PubMed] [Google Scholar]
- Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- Massova I, Kollman PA. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect Drug Discov Des. 2000;200:113–135. [Google Scholar]
- Medina MA, Urdiales JL, Rodriguez-Caso C, Ramirez FJ, Sanchez-Jimenez F. Biogenic amines and polyamines: similar biochemistry for different physiological missions and biomedical applications. Crit Rev Biochem Mol Biol. 2003;38:23–59. doi: 10.1080/713609209. [DOI] [PubMed] [Google Scholar]
- Medina MA, Correa-Fiz F, Rodriguez-Caso C, Sanchez-Jimenez F. A comprehensive view of polyamine and histamine metabolism to the light of new technologies. J Cell Mol Med. 2005;9:854–864. doi: 10.1111/j.1582-4934.2005.tb00384.x. [DOI] [PubMed] [Google Scholar]
- Melgarejo E, Medina MA, Sanchez-Jimenez F, Botana LM, Dominguez M, Escribano L, et al. Epigallocatechin-3-gallate interferes with mast cell adhesiveness, migration and its potential to recruit monocytes. Cell Mol Life Sci. 2007;64:2690–2701. doi: 10.1007/s00018-007-7331-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morreale A, Gil-Redondo R, Ortiz AR. A new implicit solvent model for protein-ligand docking. Proteins. 2007;67:606–616. doi: 10.1002/prot.21269. [DOI] [PubMed] [Google Scholar]
- Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- Moya-Garcia AA, Medina MA, Sanchez-Jimenez F. Mammalian histidine decarboxylase: from structure to function. Bioessays. 2005;27:57–63. doi: 10.1002/bies.20174. [DOI] [PubMed] [Google Scholar]
- Moya-Garcia AA, Pino-Angeles A, Sanchez-Jimenez F. New structural insights to help in the search for selective inhibitors of mammalian pyridoxal 5'-phosphate-dependent histidine decarboxylase. 4. Synthesis, metabolism and release of histamine. Inflamm Res. 2006;55(Suppl.)(1):S55–S56. doi: 10.1007/s00011-005-0040-2. [DOI] [PubMed] [Google Scholar]
- Moya-Garcia AA, Ruiz-Pernia J, Marti S, Sanchez-Jimenez F, Tunon I. Analysis of the decarboxylation step in mammalian histidine decarboxylase. A computational study. J Biol Chem. 2008;283:12393–12401. doi: 10.1074/jbc.M707434200. [DOI] [PubMed] [Google Scholar]
- Mulholland AJ. Modelling enzyme reaction mechanisms, specificity and catalysis. Drug Discov Today. 2005;10:1393–1402. doi: 10.1016/S1359-6446(05)03611-1. [DOI] [PubMed] [Google Scholar]
- Nelder JA, Mead R. A simplex method for function minimization. Comput J. 1965;7:308–313. [Google Scholar]
- Nitta Y, Kikuzaki H, Ueno H. Food components inhibiting recombinant human histidine decarboxylase activity. J Agric Food Chem. 2007;55:299–304. doi: 10.1021/jf062392k. [DOI] [PubMed] [Google Scholar]
- Norberg J, Nilsson L. Advances in biomolecular simulations: methodology and recent applications. Q Rev Biophys. 2003;36:257–306. doi: 10.1017/s0033583503003895. [DOI] [PubMed] [Google Scholar]
- Ohtsu H. Progress in allergy signal research on mast cells: the role of histamine in immunological and cardiovascular disease and the transporting system of histamine in the cell. J Pharmacol Sci. 2008;106:347–353. doi: 10.1254/jphs.fm0070294. [DOI] [PubMed] [Google Scholar]
- Olmo MT, Rodriguez-Agudo D, Medina MA, Sanchez-Jimenez F. The pest regions containing C-termini of mammalian ornithine decarboxylase and histidine decarboxylase play different roles in protein degradation. Biochem Biophys Res Commun. 1999;257:269–272. doi: 10.1006/bbrc.1999.0456. [DOI] [PubMed] [Google Scholar]
- Olmo MT, Urdiales JL, Pegg AE, Medina MA, Sanchez-Jimenez F. In vitro study of proteolytic degradation of rat histidine decarboxylase. Eur J Biochem. 2000;267:1527–1531. doi: 10.1046/j.1432-1327.2000.01153.x. [DOI] [PubMed] [Google Scholar]
- Olmo MT, Sanchez-Jimenez F, Medina MA, Hayashi H. Spectroscopic analysis of recombinant rat histidine decarboxylase. J Biochem. 2002;132:433–439. doi: 10.1093/oxfordjournals.jbchem.a003240. [DOI] [PubMed] [Google Scholar]
- Osterberg F, Morris GM, Sanner MF, Olson AJ, Goodsell DS. Automated docking to multiple target structures: incorporation of protein mobility and structural water heterogeneity in AutoDock. Proteins. 2002;46:34–40. doi: 10.1002/prot.10028. [DOI] [PubMed] [Google Scholar]
- Perez C, Ortiz AR. Evaluation of docking functions for protein-ligand docking. J Med Chem. 2001;44:3768–3785. doi: 10.1021/jm010141r. [DOI] [PubMed] [Google Scholar]
- Raha K, Peters MB, Wang B, Yu N, Wollacott AM, Westerhoff LM, et al. The role of quantum mechanics in structure-based drug design. Drug Discov Today. 2007;12:725–731. doi: 10.1016/j.drudis.2007.07.006. [DOI] [PubMed] [Google Scholar]
- Ridder L, Harvey JN, Rietjens IMCM, Vervoort J, Mulholland AJ. Ab initio qm/mm modeling of the hydroxylation step in p-hydroxybenzoate hydroxylase. J Phys Chem B. 2003;107:2118–2126. [Google Scholar]
- Rodriguez-Agudo D, Olmo MT, Sanchez-Jimenez F, Medina MA. Rat histidine decarboxylase is a substrate for m-calpain in vitro. Biochem Biophys Res Commun. 2000;271:777–781. doi: 10.1006/bbrc.2000.2715. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Caso C, Rodriguez-Agudo D, Moya-Garcia AA, Fajardo I, Medina MA, Subramaniam V, et al. Local changes in the catalytic site of mammalian histidine decarboxylase can affect its global conformation and stability. Eur J Biochem. 2003a;270:4376–4387. doi: 10.1046/j.1432-1033.2003.03834.x. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Caso C, Rodriguez-Agudo D, Sanchez-Jimenez F, Medina MA. Green tea epigallocatechin-3-gallate is an inhibitor of mammalian histidine decarboxylase. Cell Mol Life Sci. 2003b;60:1760–1763. doi: 10.1007/s00018-003-3135-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander C, Schneider R. Database of homology-derived protein structures and the structural meaning of sequence alignment. Proteins. 1991;9:56–68. doi: 10.1002/prot.340090107. [DOI] [PubMed] [Google Scholar]
- Sandmeier E, Hale TI, Christen P. Multiple evolutionary origin of pyridoxal-5'-phosphate-dependent amino acid decarboxylases. Eur J Biochem. 1994;221:997–1002. doi: 10.1111/j.1432-1033.1994.tb18816.x. [DOI] [PubMed] [Google Scholar]
- Schubert ML, Peura DA. Control of gastric acid secretion in health and disease. Gastroenterology. 2008;134:1842–1860. doi: 10.1053/j.gastro.2008.05.021. [DOI] [PubMed] [Google Scholar]
- Shoichet BK. Virtual screening of chemical libraries. Nature. 2004;432:862–865. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. Screening for drug discovery: the leading question. Nature. 2002;418:453–459. doi: 10.1038/418453a. [DOI] [PubMed] [Google Scholar]
- Snow CD, Sorin EJ, Rhee YM, Pande VS. How well can simulation predict protein folding kinetics and thermodynamics? Annu Rev Biophys Biomol Struct. 2005;34:43–69. doi: 10.1146/annurev.biophys.34.040204.144447. [DOI] [PubMed] [Google Scholar]
- Sorensen TL, McAuley KE, Flaig R, Duke EM. New light for science: synchrotron radiation in structural medicine. Trends Biotechnol. 2006;24:500–508. doi: 10.1016/j.tibtech.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Sousa SF, Fernandes PA, Ramos MJ. Protein-ligand docking: current status and future challenges. Proteins. 2006;65:15–26. doi: 10.1002/prot.21082. [DOI] [PubMed] [Google Scholar]
- Sun H. Pharmacophore-based virtual screening. Curr Med Chem. 2008;15:1018–1024. doi: 10.2174/092986708784049630. [DOI] [PubMed] [Google Scholar]
- Tachibana M, Wada K, Katayama K, Kamisaki Y, Maeyama K, Kadowaki T, et al. Activation of peroxisome proliferator-activated receptor gamma suppresses mast cell maturation involved in allergic diseases. Allergy. 2008;63:1136–1147. doi: 10.1111/j.1398-9995.2008.01677.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Ichikawa A. Recent advances in molecular pharmacology of the histamine systems: immune regulatory roles of histamine produced by leukocytes. J Pharmacol Sci. 2006;101:19–23. doi: 10.1254/jphs.fmj06001x5. [DOI] [PubMed] [Google Scholar]
- Thurmond RL, Gelfand EW, Dunford PJ. The role of histamine H1 and H4 receptors in allergic inflammation: the search for new antihistamines. Nat Rev Drug Discov. 2008;7:41–53. doi: 10.1038/nrd2465. [DOI] [PubMed] [Google Scholar]
- Totrov M, Abagyan R. Flexible ligand docking to multiple receptor conformations: a practical alternative. Curr Opin Struct Biol. 2008;18:178–184. doi: 10.1016/j.sbi.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramontano A, Morea V. Exploiting evolutionary relationships for predicting protein structures. Biotechnol Bioeng. 2003;84:756–762. doi: 10.1002/bit.10850. [DOI] [PubMed] [Google Scholar]
- Viguera E, Trelles O, Urdiales JL, Matés JM, Sánchez-Jiménez F. Mammalian L-amino acid decarboxylases producing 1,4-diamines: analogies among differences. Trends Biochem Sci. 1994;19:318–319. doi: 10.1016/0968-0004(94)90069-8. [DOI] [PubMed] [Google Scholar]
- Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH, et al. A critical assessment of docking programs and scoring functions. J Med Chem. 2006;49:5912–5931. doi: 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- Warshel A. Computer simulations of enzyme catalysis: methods, progress, and insights. Annu Rev Biophys Biomol Struct. 2003;32:425–443. doi: 10.1146/annurev.biophys.32.110601.141807. [DOI] [PubMed] [Google Scholar]
- Weininger D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J Chem Inf Model. 1988;28:31–36. [Google Scholar]
- Wijtmans M, Celanire S, Snip E, Gillard MR, Gelens E, Collart PP, et al. 4-benzyl-1H-imidazoles with oxazoline termini as histamine H3 receptor agonists. J Med Chem. 2008;51:2944–2953. doi: 10.1021/jm7014149. [DOI] [PubMed] [Google Scholar]
- Wu F, Yu J, Gehring H. Inhibitory and structural studies of novel coenzyme-substrate analogs of human histidine decarboxylase. FASEB J. 2008;22:890–897. doi: 10.1096/fj.07-9566com. [DOI] [PubMed] [Google Scholar]
- Xiang Z. Advances in homology protein structure modeling. Curr Protein Pept Sci. 2006;7:217–227. doi: 10.2174/138920306777452312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Song L, Truhlar DG, Gao J. The variational explicit polarization potential and analytical first derivative of energy: towards a next generation force field. J Chem Phys. 2008;128:234108. doi: 10.1063/1.2936122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Sun M, Bennani YL, Gopalakrishnan SM, Witte DG, Miller TR, et al. The alkaloid conessine and analogues as potent histamine H3 receptor antagonists. J Med Chem. 2008;51:5423–5430. doi: 10.1021/jm8003625. [DOI] [PubMed] [Google Scholar]