

Abstract
Background and purpose
Benzylidene-anabaseines (BAs) are partial agonists of the α7 nicotinic acetylcholine receptor (nAChR) but their mechanism(s) of action are unknown. Our study explores several possibilities, including direct interactions of BAs with the nAChR channel.
Experimental approach
Functional and radioligand-binding assays were used to examine the interaction of two BA analogues, 3-(2,4-dimethoxybenzylidene)-anabaseine (DMXBA) and its primary metabolite 3-(4-hydroxy-2-methoxybenzylidene)-anabaseine (4OH-DMXBA) with both agonist and non-competitive antagonist (NCA)-binding sites on muscle-type nAChRs.
Key results
Both BAs non-competitively inhibited ACh activation of human fetal muscle nAChRs and sterically inhibited the specific binding of the NCAs [piperidyl-3,4-3H(N)]-(N-(1-(2-thienyl)cyclohexyl)-3,4-piperidine ([3H]TCP) and [3H]dizocilpine to Torpedo nAChRs in the desensitized state. These compounds modulated [3H]tetracaine, [14C]amobarbital and [3H]TCP binding to resting nAChRs by allosteric mechanisms. Both BAs enhanced [3H]TCP binding when the nAChR was initially in the resting but activatable state, suggesting that both compounds desensitized the Torpedo nAChR. Although DMXBA failed to activate human fetal muscle nAChRs, 4OH-DMXBA was found to be a partial agonist. [3H]Nicotine competition-binding experiments confirmed that 4OH-DMXBA has higher affinity than DMXBA for the agonist sites, and that DMXBA is also a competitive antagonist.
Conclusions and implications
3-(4-hydroxy-2-methoxybenzylidene)-anabaseine is a partial agonist for human fetal muscle nAChRs, whereas DMXBA only has competitive and NCA activities. The NCA-binding site for BAs overlaps both the phencyclidine-and dizocilpine-binding sites in the desensitized Torpedo nAChR ion channel. The desensitizing property of BAs suggests another possible mode of non-competitive inhibition in addition to direct channel-blocking mechanisms.
Keywords: Nicotinic acetylcholine receptor, benzylidine-anabaseine, partial agonist, non-competitive antagonist, DMXBA, GTS-21, 4OH-DMXBA, 4OH-GTS-21, PCP
Introduction
Nicotinic acetylcholine receptors (nAChRs) are members of a superfamily of genetically linked ligand-gated ion channels that includes GABAA, GABAC, glycine and 5-HT3 receptors (nomenclature follows Alexander et al., 2008). A variety of nAChRs have been found in mammals (Arias, 2006; Kalamida et al., 2007). In the peripheral nervous system, the two most abundant nAChRs have traditionally been classified as muscle-type (α1β1δγ/ε; embryonic/adult) and ganglionic nAChRs (e.g. primarily α3β4 subunit-containing receptors). The embryonic nAChR is similar to the Torpedo nAChR. In the central nervous system, nAChRs can be classified into two major subclasses: receptors that bind the competitive antagonist α-bungarotoxin (α-BTx) with high affinity but the agonist nicotine with low affinity, such as the primarily α7-containing receptors, and nAChRs that bind nicotine with high affinity but α-BTx with low affinity, primarily those containing β2 subunits expressed with α4 and/or α6 subunits. Over the past two decades, numerous studies have provided a much more detailed knowledge of the mechanisms by which nAChRs are activated or inhibited by various ligands, and there is currently much interest in the possible therapeutic utility of some of these compounds.
Anabaseine is a marine toxin that, like nicotine, potently stimulates a wide variety of nicotinic receptors (Kem et al., 1997 2006). In contrast to anabaseine, some 3-(benzylidene)-anabaseines (BAs), such as 3-(2,4-dimethoxybenzylidene)-anabaseine (DMXBA) and its hydroxy metabolites including 3-(4-hydroxy-2-methoxybenzylidene)-anabaseine (4OH-DMXBA), are partial agonists only at α7 nAChRs (de Fiebre et al., 1995; Meyer et al., 1997; Meyer et al., 1998; Papke et al., 2000; Kem et al., 2004). Phase I studies to evaluate DMXBA as a drug candidate for the treatment of cognitive dysfunction have been completed with promising results (Kitagawa et al., 2003; Olincy et al., 2006; Freedman et al., 2008). As DMXBA and its primary phase I metabolite, 4OH-DMXBA, are lead compounds for the further development of therapies for several diseases in which α7 nAChR expression or function is abnormal, it is important to understand the mechanisms by which they affect this and other nAChRs. For example, the molecular basis of partial agonism by BAs at α7 nAChRs is not well understood. Partial agonism may be due to one or more mechanisms: (i) once bound to the nAChR agonist sites, BAs may not maximally increase the probability of channel opening; (ii) BAs may effectively induce the opening of the ion channel, but at the same time they may block the ion channel by binding to one or more non-competitive antagonist (NCA) site(s), the so-called agonist self-inhibitory mechanism (reviewed in Arias, 1996); and (iii) BAs may increase the desensitization process, through interactions either at the ACh-binding sites and/or by binding to NCA sites within the ion channel. Previous electrophysiological studies (de Fiebre et al., 1995; Meyer et al., 1998; Uteshev et al., 2002) of BA action have provided some support for mechanism (ii), but non-competitive inhibition was not demonstrated.
In this study our primary goals were to: (i) demonstrate the NCA action of the two BAs upon muscle-type nAChRs (both human fetal and Torpedo nAChRs); (ii) characterize and locate the putative NCA-binding sites for the BAs on the Torpedo nAChR; and (iii) determine the influence of nAChR conformational state upon interaction of BAs with the NCA sites. We selected the Torpedo nAChR for this initial analysis of BA interaction because the α7 nAChR has not yet been expressed at levels that would allow us to measure binding to known NCA sites that have been well characterized for the Torpedo nAChR (Arias et al., 2002a 2006b; Arias and Bhumireddy, 2005).
Here, we initially demonstrate, using a cultured cell line (TE671) expressing human fetal muscle nAChRs, that 4OH-DMXBA is a partial agonist, whereas DMXBA only has competitive and non-competitive antagonistic activities. We compare the binding properties of BAs with the agonist sites by means of [3H]nicotine competition experiments. We then characterize the binding of the BAs to Torpedo nAChR NCA-binding sites by radioligand competition experiments using well-known NCAs such as [piperidyl-3,4-3H(N)]-(N-(1-(2-thienyl)cyclohexyl)-3,4-piperidine ([3H]TCP), a structural and functional analogue of phencyclidine (PCP) (Arias et al., 2003 2006a), [3H]dizocilpine (Arias et al., 2001a), [3H]tetracaine (Middleton et al., 1999; Gallagher and Cohen, 1999) and [14C]amobarbital (Arias et al., 2001b).
Methods
Functional analysis of nAChR inhibition using cultured cells
TE671 cells naturally expressing human fetal muscle nAChRs obtained from the ATCC were cultured and used in 96-well FlexStation experiments, as previously described (Lee et al., 2006). A membrane potential-sensing Molecular Devices proprietary dye (blue dye kit) was used to measure the kinetics of membrane depolarization resulting from sudden addition of 11 different concentrations of ACh alone or in the presence of one of six concentrations of DMXBA or 4OH-DMXBA. TE671 cells were maintained in a culture medium consisting of Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 U·mL−1 penicillin and 100 µg·mL−1 streptomycin, and grown in 75 cm2 culture flasks housed in a humidified incubator at 37°C in an atmosphere of 5% CO2. They were seeded at a density of roughly 3–6 × 104 cells per well into 96-well flat-bottom black-walled culture plates that had been coated with 50 µL per well of 50 µg·mL−1 poly-D-lysine (70–150 kDa) and grown to near confluence overnight in 75 µL of culture medium. The fluorescent dye solution was prepared by dissolving one vial of the dye kit into 36 mL Hanks' balanced salt solution (HBSS) supplemented with 20 mmol·L−1 HEPES buffer (pH 7.4). The 96-well cell culture plates were equilibrated to room temperature for 10 min, and then the cells were washed once with 100 µL HBSS-HEPES before adding 30 µL of the membrane potential dye solution to each well. The cells were incubated with the dye for 45 min before agonist addition and recording. Serial dilutions of a compound for concentration-response analysis were prepared in 96-well V-bottom plates by addition of the required volume of a methanolic stock solution. After evaporation of the methanol, the compound in each well was redissolved in HBSS-HEPES (pH 7.4).
Changes in membrane potential resulting from sudden addition of ACh or KCl were measured in a FlexStation fluorimeter (Molecular Devices). Excitation and emission wavelengths were set to 530 nm and 565 nm respectively; the cut-off wavelength was 550 nm. The lipophilic anionic bis-oxonol dye crosses the membrane when it is depolarized, resulting in a significant enhancement of its fluorescence. A reading was taken every 1.44 s for 200 s, a total of 139 readings per well. The first 16 s were used as a basal reading. At 17 s, 30 µL of a test compound was added to assess agonist activity. A 30 µL pulse of concentrated KCl (to attain a final concentration of 40 mmol·L−1 KCl) in saline was added to identical wells of cells not exposed to ACh or either BA to serve as a depolarizing calibrant and to correct for differences in dye loading and cell counts between experiments. Peak fluorescent (depolarizing) response (F) was initially normalized to the maximal calibrant (KCl, final concentration 40 mmol·L−1) response: F = (FMax,Compound−FBasal)/(FMax,Calibrant−FBasal). These F values were then expressed relative to the maximum response generated by ACh alone. Normalized F values were then fitted to a four-parameter logistic equation with variable slope using the Prism software (GraphPad Software, San Diego, CA, USA), for fitting sigmoidal concentration-response curves to estimate the BA concentration that produces 50% inhibition of agonist activity (IC50) and maximal degree of activation for each agonist curve.
Preparation of Torpedo membranes for radioligand-binding experiments
Receptor rich membranes were prepared from frozen Torpedo californica electric organs obtained from Aquatic Research Consultants (San Pedro, CA, USA) by differential and sucrose density gradient centrifugation, as described previously (Pedersen et al., 1986). Total Torpedo nAChR membrane protein was determined using the bicinchoninic acid protein assay (Thermo Fisher Scientific, Rockford, IL, USA) with bovine serum albumin as standard. Specific activities of these membrane preparations were determined by the decrease in dansyltrimethylamine (6.6 µmol·L−1) fluorescence produced by the titration of suberyldicholine into receptor suspensions (0.3 mg·mL−1) in the presence of 100 µmol·L−1 PCP and ranged from 1.0 to 1.2 nmol of suberyldicholine-binding sites (mg total protein)−1 (0.5–0.6 nmol nAChR mg protein−1). The nAChR membrane preparations were stored at −80°C in 12% sucrose.
Benzylidine-anabaseine-induced inhibition of agonist and NCA binding to nAChRs
In order to determine the binding affinity of DMXBA and 4OH-DMXBA for the agonist sites, [3H]nicotine-binding competition experiments were performed. To determine the NCA-binding site location for BAs, we also studied the effect of these BAs on the binding of several NCAs including [3H]dizocilpine, [3H]TCP, [3H]tetracaine and [14C]amobarbital to nAChRs in different conformational states. In this regard, Torpedo membranes were suspended in binding saline buffer containing 50 mmol·L−1 Tris–HCl, 120 mmol·L−1 NaCl, 5 mmol·L−1 KCl, 2 mmol·L−1 CaCl2, 1 mmol·L−1 MgCl2, pH 7.4, with 6.9 nmol·L−1[3H]nicotine (in the absence of any other ligand), or alternatively with 10 nmol·L−1[3H]dizocilpine or 5 nmol·L−1[3H]TCP [in presence of 1 mmol·L−1 carbamylcholine (CCh) to stabilize the desensitized state], or 6 nmol·L−1[3H]tetracaine, 13 µmol·L−1[14C]amobarbital, or 5 nmol·L−1[3H]TCP (either in the absence of any ligand, or in the presence of 1 µmol·L−1α-BTx to stabilize the resting state), and preincubated for 1 h. α-BTx is a competitive antagonist that maintains the nAChR in the resting state (Moore and McCarthy, 1995). Non-specific binding was determined in the presence of 1 mmol·L−1 CCh ([3H]nicotine experiments), 100 µmol·L−1 PCP ([3H]TCP experiments), 500 µmol·L−1 dizocilpine ([3H]dizocilpine experiments), 100 µmol·L−1 tetracaine ([3H]tetracaine experiments) or 400 µmol·L−1 amobarbital ([14C]amobarbital experiments). The membrane suspension was aliquoted equally to tubes and increasing concentrations of DMXBA or 4OH-DMXBA (i.e. 0.01 nmol·L−1–500 µmol·L−1; depending on the experiment type) were then added. After incubating 2 h, nAChR-bound radioligand was then separated from free radioligand by a filtration assay using a 48-sample harvester system with GF/B Whatman filters (Brandel Inc., Gaithersburg, MD, USA), previously soaked with 0.5% polyethylenimine for 30 min. The membrane-containing filters were transferred to scintillation vials with 3 mL of Bio-Safe II (Research Product International Corp, Mount Prospect, IL, USA), and the radioactivity was determined using a Beckman LS6500 scintillation counter (Beckman Coulter, Inc., Fullerton, CA, USA).
The concentration–response data were curve-fitted by non-linear least squares analysis using the Prism software, where the corresponding IC50 values were calculated using the following equation
| (1) |
where θ is the fractional amount of the radioligand bound in the presence of inhibitor at a concentration [L] compared with the amount of the radioligand bound in the absence of inhibitor (total binding). IC50 is the inhibitor concentration at which θ= 0.5 (50% bound), and nH is the Hill coefficient. The observed IC50 values from the competition experiments described above were transformed into inhibition constant (Ki) values using the Cheng–Prusoff relationship (Cheng and Prusoff, 1973)
| (2) |
where [ligand] and Kdligand are the initial concentration and the dissociation constant (Kd) for [3H]nicotine (Middleton and Cohen, 1991). The calculated Ki and nH values for DMXBA and 4OH-DMXBA binding to the agonist sites are summarized in Table 1.
Table 1.
Binding affinity of benzylidine-anabaseine analogues for the Torpedo nAChR agonist-binding sites
| Benzylidine-anabaseine analogue | Kia(µmol·L−1) | nHb |
|---|---|---|
| DMXBA | 10.6 ± 0.8 | 1.27 ± 0.11 |
| 4OH-DMXBA | 2.7 ± 0.2 | 1.07 ± 0.06 |
Data shown are the means ± SD, from two to three experiments each performed in triplicate.
Inhibition constants were calculated from Figure 3 using eq. 2.
Hill coefficients.
4OH-DMXBA, 3-(4-hydroxy-2-methoxybenzylidene)-anabaseine; DMXBA (or GTS-21), 3-(2,4-dimethoxybenzylidene)-anabaseine; nAChR, nicotinic acetylcholine receptor.
Taking into account that the nAChR presents one binding site for [3H]dizocilpine (Arias et al., 2001a) and [3H]TCP (Arias et al., 2003) in the desensitized state, the observed IC50 values were transformed into Ki values using eq. 2 (Cheng and Prusoff, 1973). In this case, [ligand] is the initial concentration of [3H]dizocilpine or [3H]TCP, and Kdligand is the Kd for [3H]dizocilpine (4.8 µmol·L−1; Arias et al., 2001a) or [3H]TCP (0.25 µmol·L−1; Pagán et al., 2001) in the desensitized state. The calculated Ki and nH values for DMXBA and 4OH-DMXBA binding to the NCA sites are summarized in Table 2.
Table 2.
Effect of benzylidine-anabaseines on [3H]dizocilpine, [3H]TCP, [3H]tetracaine and [14C]amobarbital binding to the nAChR in different conformational states
| Radioligand | Conformational state | DMXBA | 4OH-DMXBA | ||
|---|---|---|---|---|---|
| Kiaor IC50b(µmol·L−1) | nHc | Kiaor IC50b(µmol·L−1) | nHc | ||
| [3H]TCP | Desensitized | 13 ± 1a | 0.91 ± 0.06 | 48 ± 5a | 0.90 ± 0.08 |
| [3H]Dizocilpine | Desensitized | 39 ± 8a | 0.92 ± 0.16 | 84 ± 11a | 0.70 ± 0.07 |
| [3H]Tetracaine | Resting | 262 ± 10b | 2.1 ± 0.2 | 0.79 ± 0.50d | ∼1.3 |
| [14C]Amobarbital | Resting | 95 ± 9b | >3 | 46 ± 32b | ∼0.4 |
Data shown are the means ± SD, from 2–3 experiments each performed in triplicate.
Inhibition constants were calculated from Figures 4A ([3H]TCP), 4B ([3H] dizocilpine), 5A ([3H]tetracaine) and 5B ([14C]amobarbital), using eq. 2.
Drug concentration to produce 50% binding inhibition. These values were determined by non-linear least-squares fit according to eq. 1.
Hill coefficients.
This apparent EC50 value is obtained from the 4OH-DMXBA-induced enhancement of [3H]tetracaine binding to the resting AChR.
[3H]TCP, [piperidyl-3,4-3H(N)]-(N-(1-(2-thienyl)cyclohexyl)-3,4-piperidine; 4OH-DMXBA, 3-(4-hydroxy-2-methoxybenzylidene)-anabaseine; DMXBA (or GTS-21), 3-(2,4-dimethoxybenzylidene)-anabaseine; nAChR, nicotinic acetylcholine receptor.
Schild-type plots for DMXBA-induced inhibition of [3H]TCP binding
In order to have a better indication whether BAs inhibit [3H]TCP binding to the nAChR by a steric or allosteric mechanism, DMXBA-induced inhibition of [3H]TCP-binding experiments was performed at increasing initial concentrations of unlabelled PCP (i.e. 3.1, 6.2 and 9.3 µmol·L−1). The rationale of this experiment is based on the expectation that, for a higher initial concentration of the radioligand, a higher concentration of the competitor will be necessary to produce a total inhibition of radioligand binding. This is consistent with Schild-type analysis (Schild, 1949). From these competition curves, the apparent IC50 values were obtained according to eq. 1. Then, we plot the ratio between the IC50 values for DMXBA determined at different initial concentrations of unlabelled PCP under the IC50 control values (no PCP added) versus the initial PCP concentration (IC50PCP/IC50control vs. [PCP]initial) (for more details, see the study by Arias et al., 2002b). A linear relationship from this modified Schild-type plot would indicate a competitive interaction, whereas a non-linear relationship would suggest an allosteric mechanism of inhibition (Schild, 1949; Arias et al., 2002b).
Drugs, chemical reagents and other materials
[Piperidyl-3,4-3H(N)]-N-(1-(2-thienyl)cyclohexyl)-3,4-piperidine ([3H]TCP) (42–45 Ci·mmol−1), (+)-[3-3H]dizocilpine (21.7 Ci·mmol−1) and [3H]nicotine (77 Ci·mmol−1) were obtained from PerkinElmer Life Sciences Products, Inc. (Boston, MA, USA). [14C]Amobarbital (50 Ci·mol−1) was obtained from American Radiolabeled Chemicals, Inc. (St. Louis, MO, USA). [3H]Tetracaine (36 Ci·mmol−1) was a gift from Dr Jonathan Cohen (Harvard Medical School, Boston, MA, USA). All radioligands were stored in ethanol at −20°C. CCh chloride, acetylcholine chloride (ACh), suberyldicholine dichloride and polyethylenimine were purchased from Sigma Chemical Co. (St. Louis, MO, USA). [1-(Dimethylamino) naphthalene-5-sulphonamido]ethyltrimethylammonium perchlorate (dansyltrimethylamine) was purchased from Pierce Chemical Co. (Rockford, IL, USA). α-BTx was obtained from Molecular Probes (Eugene, OR, USA). (+)-Dizocilpine maleate was purchased from Tocris (Ellisville, MO, USA). DMXBA dihydrochloride (DMXBA.2HCl) and 4OH-DMXBA (4OH-DMXBA.2HCl) were synthesized as described elsewhere (Kem et al., 2004). Amobarbital hydrochloride and PCP hydrochloride were obtained through the National Institute on Drug Abuse (NIDA) (NIH, Bethesda, MD, USA). Fetal bovine serum and penicillin/streptomycin were obtained from Media Tech, Inc. (Herndon, VA, USA), Dulbecco's modified Eagle's medium was purchased from the American Type Culture Collection (Manassas, VA, USA) and the fluorescence dye kit was purchased from Molecular Devices (Sunnyvale, CA, USA). Salts were of analytical grade.
Results
Inhibition of ACh-mediated depolarization of TE671 cells
3-(2,4-dimethoxybenzylidene)-anabaseine and 4OH-DMXBA inhibited the TE671 cell response to ACh in a non-competitive manner (Figure 1). The entire ACh concentration-response curve was depressed by each BA concentration tested. The effects of DMXBA or 4OH-DMXBA on the ACh EC50 values were concentration independent, as would be expected for non-competitive inhibition. The IC50 values for BA inhibition of ACh response obtained from several experiments were 6.6 ± 1.2 µmol·L−1 for DMXBA and 10.0 ± 2.8 µmol·L−1 for 4OH-DMXBA (Figure 1C).
Figure 1.
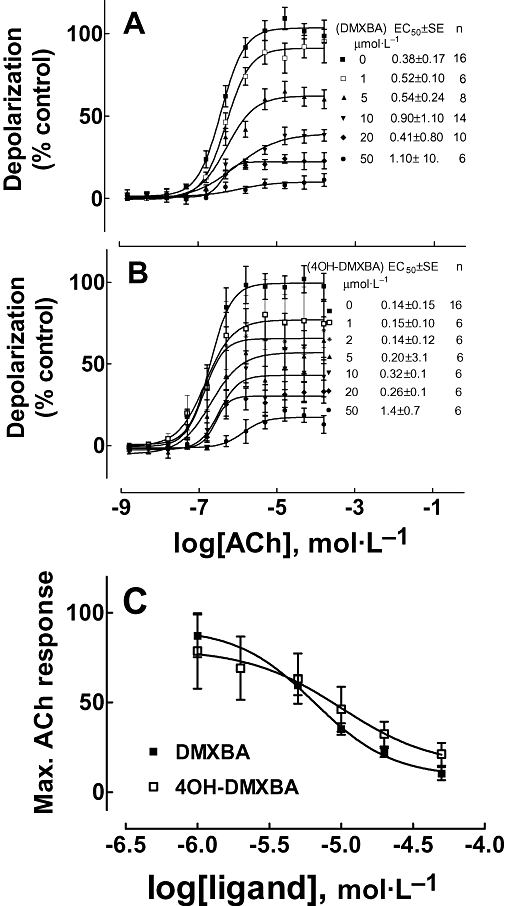
Non-competitive inhibition of ACh-induced membrane depolarization in TE671 cells expressing the human fetal muscle nAChR by DMXBA (A) and 4OH-DMXBA (B). Each curve was fitted using Prism software as described in Methods section. The drug was manually added to the cells in programmed wells at least 10 min before ACh was added by the FlexStation program (n, number of wells tested at each ACh concentration). The ACh response at high concentrations in the control curve is less than maximal due to the desensitizing effect being greater at high concentrations. (C) Maximal ACh response for varying concentrations of DMXBA [taken from (A)] and 4OH-DMXBA [taken from (B)] were plotted to estimate the IC50 values for DMXBA (6.6 ± 1.2 µmol·L−1) and 4OH-DMXBA (10.0 ± 2.8 µmol·L−1). Data shown are means ± SEM from the number of assays shown. 4OH-DMXBA, 3-(4-hydroxy-2-methoxybenzylidene)-anabaseine; DMXBA (or GTS-21), 3-(2,4-dimethoxybenzylidene)-anabaseine; nAChR, nicotinic acetylcholine receptor.
Although DMXBA failed to show any depolarizing action on the TE671 cells when tested over a wide range of concentrations (2 nmol·L−1–200 µmol·L−1, data not shown), HPLC-pure 4OH-DMXBA consistently activated TE671 cell nAChRs with an EC50∼2 µmol·L−1 and a maximal effect that was 40–50% of the ACh response (Figure 2A). This indicates that although DMXBA binds to the agonist sites (see Figure 3), it does not present agonist properties. The depolarizing action elicited by 4OH-DMXBA was blocked by 10 nmol·L−1α-BTx in an insurmountable manner, as this toxin binds in a quasi-irreversible manner to skeletal muscle nAChRs and thus prevents competition by ACh during the short period of ACh exposure (Figure 2B). The stimulatory effect of 4OH-DMXBA was unexpected, as a 100 µmol·L−1 concentration was previously reported to lack agonist activity at the adult mouse skeletal muscle nAChR expressed in the Xenopus oocyte (Meyer et al., 1998). However, we subsequently learned that the fetal-type nAChR, but not the adult type, is activated by d-tubocurarine and some other competitive antagonists of the adult, ε-subunit expressing muscle receptor (Ziskind and Dennis, 1978; Kopta and Steinbach, 1994; Fletcher and Steinbach, 1996).
Figure 2.
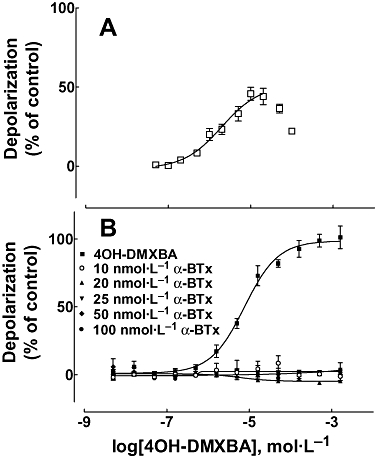
(A) Intrinsic agonist effect of 4OH-DMXBA on human fetal muscle nAChR-containing TE671 cells (n = 14; where the error bars are SD). A depression of the response was observed at the highest concentrations and is likely due to desensitization and/or channel block by 4OH-DMXBA. The curve shown was calculated with Prism where the points obtained with the two highest concentrations were omitted from the analysis. The obtained EC50 value was 1.98 ± 0.15 µmol·L−1, and nH= 0.94 ± 0.23. (B) The agonist activity of 4OH-DMXBA was inhibited by preincubation with α-BTx. Here, 4OH-DMXBA effects are normalized with respect to its maximal effect, rather than to the maximal ACh effect as in (A). Data shown are means ± SD from four separate assays. 4OH-DMXBA, 3-(4-hydroxy-2-methoxybenzylidene)-anabaseine; α-BTx, α-bungarotoxin; DMXBA (or GTS-21), 3-(2,4-dimethoxybenzylidene)-anabaseine; nAChR, nicotinic acetylcholine receptor; nH, Hill coefficient.
Figure 3.
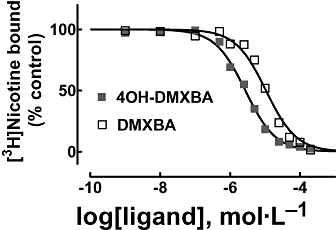
Inhibition of [3H]nicotine binding to Torpedo nAChRs induced by BA analogues. nAChR-rich membranes (0.3 µmol·L−1 nAChR) were equilibrated (2 h) with 6.9 nmol·L−1[3H]nicotine, and increasing concentrations (i.e. 1 nmol·L−1–200 µmol·L−1) of DMXBA or 4OH-DMXBA respectively. Non-specific binding was determined in the presence of 1 mmol·L−1 CCh. The IC50 and nH values were obtained by non-linear least-squares fit according to eq. 1. Subsequently, the Ki values were calculated using eq. 2. The calculated Ki and nH values are summarized in Table 1. Values shown are the averages of two different experiments. 4OH-DMXBA, 3-(4-hydroxy-2-methoxybenzylidene)-anabaseine; BA, 3-(benzylidene)-anabaseine; CCh, carbamylcholine; DMXBA (or GTS-21), 3-(2,4-dimethoxybenzylidene)-anabaseine; nAChR, nicotinic acetylcholine receptor; nH, Hill coefficient.
Benzylidene-anabaseine inhibition of [3H]nicotine binding to Torpedo nAChRs
To determine the binding affinity of each analogue for the Torpedo nAChR agonist sites, [3H]nicotine-binding competition experiments were performed (see Figure 3). The results indicate that 4OH-DMXBA has higher affinity than DMXBA for the agonist-binding sites (Table 1). The fact that the calculated nH values for DMXBA and 4OH-DMXBA are close to one (Table 1) indicates that both analogues inhibit [3H]nicotine binding in a non-cooperative manner. This suggests that BAs do not discriminate between the high-and low-affinity agonist-binding sites and thus, BAs bind to both agonist sites in the same concentration range. In addition, our data indicate that the two BAs possess much lower binding affinities for the Torpedo nAChR relative to the rat brain α7 nAChR, where the DMXBA and 4OH-DMXBA Ki values determined by [125I]α-BTx displacement experiments were ∼0.13 and ∼0.24 µmol·L−1 respectively (Kem et al., 2004).
Effect of benzylidene-anabaseines on [3H]dizocilpine, [3H]TCP, [3H]tetracaine and [14C]amobarbital binding to the Torpedo nAChR
The binding sites for [3H]dizocilpine (Arias et al., 2001a) and [3H]TCP (Arias et al., 2002b 2003 2006a) in the desensitized state, and for [3H]tetracaine (Middleton et al., 1999), [14C]amobarbital (Arias et al., 2001b) and [3H]TCP (Arias et al., 2002b 2003 2006a) in the resting state, have been previously characterized. Thus, we sought to determine the BA-binding site location relative to these NCA-binding loci. To this end, we determined the effect of DMXBA and 4OH-DMXBA on either [3H]TCP, [3H]dizocilpine, [3H]tetracaine or [14C]amobarbital maximal binding to Torpedo nAChRs in different conformational states. Figure 4 shows the displacing effects of the two BAs on the binding of [3H]TCP (Figure 4A) and [3H]dizocilpine (Figure 4B), respectively, to the desensitized nAChR. The data obtained from these plots indicate that DMXBA has higher affinity than 4OH-DMXBA for both the TCP-and dizocilpine-binding sites. The fact that the calculated nH values are close to unity (Table 2) suggests that BAs inhibit both [3H]dizocilpine and [3H]TCP binding in a non-cooperative manner. This further suggests that BAs interact with just a single binding site. Thus, we suggest that the BA-binding site overlaps both PCP and dizocilpine loci.
Figure 4.
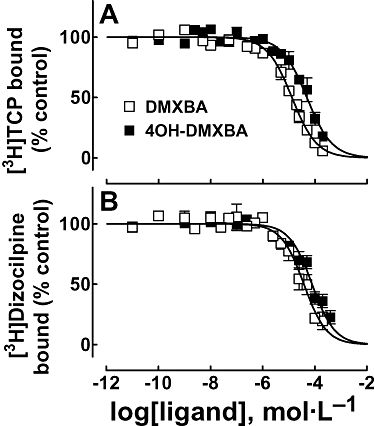
Effect of BAs on [3H]TCP (A) and [3H]dizocilpine (B) binding to Torpedo nAChRs in the desensitized state. Inhibition of [3H]TCP (A) or [3H]dizocilpine (B) binding to the desensitized nAChR elicited by DMXBA or 4OH-DMXBA respectively. nAChR-rich membranes (0.3 µmol·L−1 nAChR) were equilibrated (2 h) with 5 nmol·L−1[3H]TCP or 10 nmol·L−1[3H]dizocilpine, in the presence of 1 mmol·L−1 CCh (nAChRs are mainly in the desensitized state), and increasing concentrations of the BA (e.g. 0.01 nmol·L−1–500 µmol·L−1; depending on the experiment type). Non-specific binding was determined in the presence of 100 µmol·L−1 PCP or 500 µmol·L−1 dizocilpine respectively. The IC50 and nH values were obtained by non-linear least-squares fit according to eq. 1. Subsequently, the Ki values were calculated using eq. 2. The calculated Ki and nH values are summarized in Table 2. Values shown are the means ± SD from two to three separate experiments, each one performed in triplicate. [3H]TCP, [piperidyl-3,4-3H(N)]-(N-(1-(2-thienyl)cyclohexyl)-3,4-piperidine; 4OH-DMXBA, 3-(4-hydroxy-2-methoxybenzylidene)-anabaseine; BA, 3-(benzylidene)-anabaseine; DMXBA (or GTS-21), 3-(2,4-dimethoxybenzylidene)-anabaseine; Ki, inhibition constant; nH, Hill coefficient.
Figure 5 shows the effect of DMXBA and 4OH-DMXBA on [3H]tetracaine (Figure 5A) and [14C]amobarbital (Figure 5B) binding to the nAChR when the resting state predominates (in the presence of α-BTx). Considering that tetracaine and amobarbital bind preferably to the resting nAChR (Middleton et al., 1999; Arias et al., 2001b), we did not study the interaction of BAs to these binding sites in the desensitized state. DMXBA displaces 100% of [3H]tetracaine specific binding to the resting nAChR (see Figure 5A). The calculated IC50 value (262 ± 10 µmol·L−1; Table 2) for DMXBA is in the same concentration range as that obtained by DMXBA-induced inhibition of [3H]TCP-binding experiments in the resting state (172 ± 12 µmol·L−1; Table 3). However, the observed nH value was higher than unity, indicating that DMXBA inhibits [3H]tetracaine binding in a cooperative manner. This suggests that DMXBA interacts with more than one binding site and that DMXBA probably inhibits [3H]tetracaine binding through an allosteric mechanism. On the contrary, 4OH-DMXBA enhances [3H]tetracaine binding with apparent EC50∼0.8 µmol·L−1 (Table 2). This result argues against the idea of any steric competition between these two molecules, and rules out the possibility that the 4OH-DMXBA locus overlaps the tetracaine-binding site in the resting ion channel. Moreover, this finding supports the hypothesis that the binding of 4OH-DMXBA to its locus provokes a local conformational change that allows the tetracaine molecule to fit more readily within its binding domain.
Figure 5.
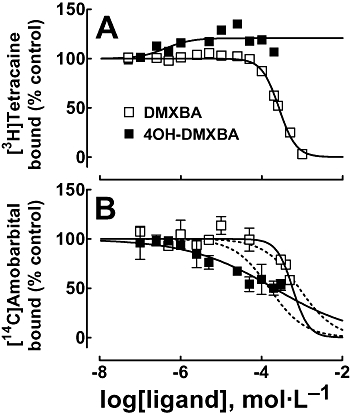
Effect of BAs on [3H]tetracaine (A) and [14C]amobarbital (B) binding to Torpedo nAChRs in the resting state. Inhibition of [3H]tetracaine (A) or [14C]amobarbital (B) binding to the resting nAChR elicited by DMXBA or 4OH-DMXBA respectively. nAChR-rich membranes (0.3 µmol·L−1 AChR) were equilibrated (2 h) with 6 nmol·L−1[3H]tetracaine or 13 µmol·L−1[14C]amobarbital, in the presence of 1 µmol·L−1α-BTx (nAChRs are mainly in the resting state), and increasing concentrations of the BA (e.g. 0.1–1000 µmol·L−1; depending on the experiment type). Non-specific binding was determined in the presence of 100 µmol·L−1 tetracaine or 400 µmol·L−1 amobarbital respectively. The apparent IC50 values for DMXBA and 4OH-DMXBA, the EC50 value for 4OH-DMXBA (A), and the respective nH values were obtained by non-linear least-squares fit according to eq. 1. In the case of (B), a variable (−) or constant (nH= 1) nH value (---) was considered for the sake of comparison. The IC50, EC50 and nH values are summarized in Table 2. Values shown are the result of one out of two experiments (A) or the mean ± SD from two experiments performed in triplicate (B). 4OH-DMXBA, 3-(4-hydroxy-2-methoxybenzylidene)-anabaseine; α-BTx, α-bungarotoxin; BA, 3-(benzylidene)-anabaseine; DMXBA (or GTS-21), 3-(2,4-dimethoxybenzylidene)-anabaseine; nAChR, nicotinic acetylcholine receptor; nH, Hill coefficient.
Table 3.
Modulation of [3H]TCP binding to Torpedo nAChRs elicited by benzylidine-anabaseine analogues
| Benzylidine-anabaseine analogue | Apparent EC50a(µmol·L−1) | nHc | IC50b(µmol·L−1) | nHc |
|---|---|---|---|---|
| DMXBA | 3.9 ± 1.2 | 0.79 ± 0.03 | 172 ± 12 | 2.04 ± 0.25 |
| 4OH-DMXBA | 0.21 ± 0.02 | 1.37 ± 0.17 | 611 ± 65 | 1.91 ± 1.10 |
Data shown are the means ± SD, from two to three experiments each performed in triplicate.
BA concentration required to produce 50% binding enhancement as a measure of AChR desensitization. These values were obtained using nAChRs in the absence of any ligand (mostly in the resting but activatable state), from the portion of the plot where drugs enhance radioligand binding (Figure 7).
Drug concentration to produce 50% binding inhibition. These values were obtained using nAChRs in the resting state (in the presence of α-BTx), from the portion of the plot where drugs inhibit radioligand binding (Figure 7).
Hill coefficients.
[3H]TCP, [piperidyl-3,4-3H(N)]-(N-(1-(2-thienyl)cyclohexyl)-3,4-piperidine; 4OH-DMXBA, 3-(4-hydroxy-2-methoxybenzylidene)-anabaseine; α-BTx, α-bungarotoxin; BA, 3-(benzylidene)-anabaseine; DMXBA (or GTS-21), 3-(2,4-dimethoxybenzylidene)-anabaseine; nAChR, nicotinic acetylcholine receptor.
Regarding the [14C]amobarbital-binding experiments (Figure 5B), both DMXBA and 4OH-DMXBA displace only ∼50% [14C]amobarbital binding at maximal concentrations, with apparent nH values different from unity. Additional sigmoid curves fitting the experimental data with nH= 1 are included for the sake of comparison (Figure 5B). In this regard, an unmistakable difference can be observed between both fitted curves. These results suggest that DMXBA and 4OH-DMXBA do not bind to the barbiturate locus in the resting ion channel. The possibility of two allosterically linked binding domains, the PCP and the barbiturate loci bridged by the tetracaine domain, in the resting ion channel has been previously proposed (Gallagher et al., 2001; Arias et al., 2002b 2003 2006a). Thus, this result is in agreement with the previous experimental conclusions indicating that the binding site for DMXBA and 4OH-DMXBA does not overlap the tetracaine locus (Figure 5A).
Schild-type plots for DMXBA-induced inhibition of [3H]TCP binding
Although competition-binding results suggest that BAs inhibit [3H]TCP (Figure 4A) binding to the desensitized nAChR in a steric fashion, we cannot rule out that this competition is produced by a potent allosteric mechanism as well. In this regard, we determined whether BAs inhibit [3H]TCP binding to the desensitized nAChR in a steric or allosteric manner by Schild-type analyses (Schild, 1949). Figure 6A shows the DMXBA-induced inhibition of [3H]TCP binding at different initial concentrations of unlabelled PCP. At increased initial PCP concentrations, the plots were shifted to the right, indicating that higher DMXBA amounts are required to inhibit the binding of [3H]TCP at increased PCP concentrations (see Figure 6A). By means of a modified Schild-type plot (Figure 6B), we found a linear relationship between the initial concentration of PCP and the extent of radioligand inhibition with goodness of fit values (r2) of 0.98 (Figure 6B). This suggests that DMXBA inhibits [3H]TCP by a steric mechanism. Thus, we may infer that the DMXBA locus overlaps the PCP-binding site in the desensitized state.
Figure 6.
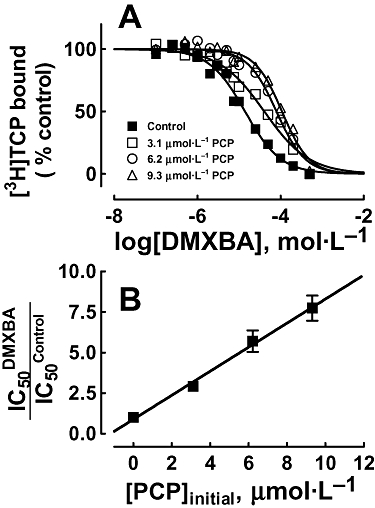
Schild-plot for DMXBA-induced inhibition of [3H]TCP binding to the desensitized Torpedo nAChR. (A) nAChR-rich membranes (0.3 µmol·L−1 nAChR) were equilibrated with [3H]TCP, in the presence of 1 mmol·L−1 CCh (desensitized state), at initial PCP concentrations of 0 (control), 3.1, 6.2 and 9.3 µmol·L−1 respectively. The apparent IC50 values were calculated by non-linear least-squares fit according to eq. 1. Values shown are the averages of experiments performed in triplicate. (B) Modified Schild-plot for DMXBA-induced inhibition of [3H]TCP binding. The plot shows a linear relationship with a r2 value of 0.98, suggesting that DMXBA inhibits [3H]TCP binding by a steric mechanism. Values shown are the means ± SD from one to three experiments performed in triplicate. [3H]TCP, [piperidyl-3,4-3H(N)]-(N-(1-(2-thienyl)cyclohexyl)-3,4-piperidine; CCh, carbamylcholine; DMXBA (or GTS-21), 3-(2,4-dimethoxybenzylidene)-anabaseine; nAChR, nicotinic acetylcholine receptor; PCP, phencyclidine.
Modulation of [3H]TCP binding to Torpedo nAChRs elicited by benzylidine-anabaseines
In order to determine the mechanisms of inhibition elicited by BAs, we studied the effect of DMXBA and 4OH-DMXBA on the binding of the NCA [3H]TCP to Torpedo nAChRs in distinct conformational states. Figure 7 shows that in the absence of any ligand, where the nAChR is mainly in the resting (but activatable) state, both DMXBA and 4OH-DMXBA enhance [3H]TCP binding in the concentration range between 0.01 nmol·L−1 and ∼10 µmol·L−1. Taking into account that [3H]TCP binds with ∼3-fold higher affinity to the desensitized (Pagán et al., 2001) than to the resting state (Arias et al., 2003), we can conclude that BAs enhance [3H]TCP binding by inducing nAChR desensitization. To quantify this [3H]TCP-binding enhancement, we calculated the concentration to produce 50% binding enhancement (apparent EC50) as a measure of the apparent desensitizing properties of the drug. The apparent EC50 values for DMXBA and 4OH-DMXBA are 3.9 ± 1.2 and 0.21 ± 0.02 µmol·L−1 (Table 3), respectively, indicating that 4OH-DMXBA is a more potent desensitizing agent than DMXBA. The fact that 4OH-DMXBA increases [3H]TCP binding by ∼180%, whereas DMXBA increases [3H]TCP binding by ∼120%, also indicates that 4OH-DMXBA is a more efficacious desensitizing agent than DMXBA. Moreover, both drugs start inhibiting [3H]TCP binding at concentrations higher than ∼10 µmol·L−1.
Figure 7.
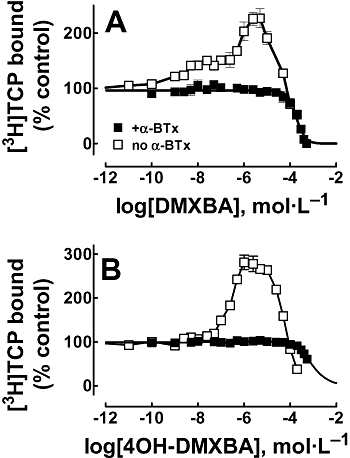
BA-induced modulation of [3H]TCP binding to Torpedo nAChRs in different conformational states. Modulation of [3H]TCP binding elicited by DMXBA (A) or 4OH-DMXBA (B) in the activatable (no α-BTx) and in the resting (+α-BTx) states respectively. nAChR native membranes (0.3 µmol·L−1 nAChR) were equilibrated (1 h) with 5 nmol·L−1[3H]TCP, in the absence (the nAChR is in the resting but activatable state) or in the presence of 1 µmol·L−1α-BTx (the nAChR is maintained resting state), and increasing concentrations of the BA (i.e. 0.01 nmol·L−1–500 µmol·L−1). The IC50 and nH values were obtained by non-linear least-squares fit according to eq. 1, whereas the apparent EC50 values were obtained using an equation similar to eq. 1, where apparent EC50 instead of IC50 was used. The calculated apparent EC50, IC50 and nH values are summarized in Table 3. Shown is the mean ± SD from one experiment performed in triplicate. [3H]TCP, [piperidyl-3,4-3H(N)]-(N-(1-(2-thienyl)cyclohexyl)-3,4-piperidine; 4OH-DMXBA, 3-(4-hydroxy-2-methoxybenzylidene)-anabaseine; α-BTx, α-bungarotoxin; BA, 3-(benzylidene)-anabaseine; DMXBA (or GTS-21), 3-(2,4-dimethoxybenzylidene)-anabaseine; nAChR, nicotinic acetylcholine receptor.
Figure 7 also shows that neither DMXBA nor 4OH-DMXBA enhances [3H]TCP binding when the nAChR is stabilized in the resting state with a high concentration (1 µmol·L−1) of α-BTx. This result indicates that BAs do not induce any additional conformational change when the nAChR is arrested in the resting state (in the presence of α-BTx), and supports the above conclusion that BAs induce the desensitization process. Moreover, DMXBA inhibits ∼100% [3H]TCP binding, whereas 4OH-DMXBA inhibits [3H]TCP binding only partially (<50%), even at very high concentrations. The observed IC50 values are very high (∼200–600 µmol·L−1; see Table 3), suggesting that if BAs bind to the [3H]TCP-binding site in the resting state, they do so with very low affinity. The fact that the nH values are higher than unity (see Table 3) indicates a cooperative interaction, supporting the idea that the BA-binding site in fact does not overlap the PCP locus in the resting state. Considering that the PCP locus partially overlaps the tetracaine-binding domain in the resting ion channel (Arias et al., 2002b 2003 2006a); reviewed in (Arias et al., 2002a 2006b; Arias and Bhumireddy, 2005), this latter conclusion also fits very well with the previous results indicating that the BA-binding site does not overlap the tetracaine locus (see Figure 5A).
Discussion and conclusions
The TE671 cell FlexStation data (Figure 1) of DMXBA and 4OH-DMXBA inhibition of the depolarizing effect of ACh indicates a non-competitive antagonism of nAChR function under the conditions of BA preincubation. Inhibition by DMXBA when administered simultaneously with ACh yielded essentially the same results (not shown). The micromolar concentrations at which inhibition by DMXBA was observed (IC50s ∼7 µmol·L−1) were much higher (>20-fold) than the plasma concentrations attained by DMXBA in the preclinical animal tests (Mahnir et al., 1998; Azuma et al., 1999; Kem et al., 2004) and various clinical tests (Kitagawa et al., 2003; Olincy et al., 2006; Freedman et al., 2008). Furthermore, it is known that about 96% of DMXBA is bound in plasma (Kem et al., 2004). Thus, the extent to which the DMXBA might inhibit muscle-type, α7-type and possibly other nAChRs in vivo is uncertain. A significant fraction of α7 nAChRs, the presumed clinical targets for DMXBA central actions, are known to be tonically active at concentrations significantly below those required for synchronous activation of all α7 receptors (Papke and Thinschmidt, 1998; Papke et al., 2000; Uteshev et al., 2002).
The significant partial agonist activity of 4OH-DMXBA on the human fetal muscle nAChR resembles the observed agonist activity of d-tubocurarine on fetal mouse muscle nAChRs (Ziskind and Dennis, 1978; Kopta and Steinbach, 1994; Fletcher and Steinbach, 1996). Indeed, both molecules contain hydroxyl groups which could conceivably form important H-bonds with amino acid side chains within the ACh-binding site. d-Tubocurarine does not stimulate adult muscle-type nAChRs which contain the ε subunit instead of the γ subunit (Ziskind and Dennis, 1978; Kopta and Steinbach, 1994). A clinical implication of our finding is that pregnant women should not be given either of these two BAs until their possible contracting effect on fetal muscle is further investigated.
Our radioligand competition experiments indicate that BAs effectively inhibit the binding of [3H]dizocilpine and [3H]TCP (the structural and functional analogue of the dissociative general anesthetic PCP) to desensitized nAChRs (Table 2). Interestingly, DMXBA has ∼2–4 times higher affinity for either NCA-binding site than 4OH-DMXBA. Practically, the same ratio was observed in our results from the non-competitive inhibition of ACh-induced nAChRs by BAs (see Figure 1). However, the opposite is true for the binding to the agonist sites, where 4OH-DMXBA has ∼4-fold higher affinity than DMXBA (see Table 1). This result might explain why the 4OH metabolite has ∼4-fold higher potency and ∼2-fold higher efficacy to activate the human α7 nAChR than that for DMXBA, if a similar binding affinity situation holds for the α7 receptor. Considering that drug efficacy and potency properties are influenced by several mechanisms including binding, activation, gating and self-inhibition, it may be that 4OH-DMXBA has a lesser propensity to self-inhibition than DMXBA at the α7 nAChR ion channel and that this may be at least one reason for its higher efficacy at this receptor. While 4OH-DMXBA displays more favourable agonist properties than DMXBA at the α7 nAChR, its ability to activate human muscle-type nAChRs might lead to adverse effects, especially in the fetus.
Schild-type analyses indicate that DMXBA displaces [3H]TCP binding from its high-affinity site in the desensitized nAChR by a steric (competitive) mechanism (Figure 6). These data suggest that the BA-binding site overlaps the high-affinity PCP locus in the desensitized state. Previous studies aimed at characterizing the high-affinity PCP locus have only been partially successful (reviewed in Arias et al., 2002a 2006b; Arias and Bhumireddy, 2005). For instance, photoaffinity labelling studies using [3H]ethidium diazide, which binds with high affinity to the PCP locus, helped to determine the structural components of this site in the desensitized state (Pratt et al., 2000). The results indicated that residues Leu251 at position 9′ (i.e. the leucine ring) and Ser252 at position 10′ from the α1-M2 transmembrane segment as well as other unknown amino acids located in the M1 and M2 transmembrane segments from the δ subunit are structurally involved in this particular site. On the other hand, the dizocilpine locus was suggested to be in a domain different from that for the fluorescent NCAs, crystal violet and ethidium, based on Schild-plot analyses as well as [125I]dizocilpine photoaffinity labelling studies (Arias et al., 2001a). More recently, however, Schild-type analyses of dizocilpine-induced inhibition of [3H]TCP binding indicate that dizocilpine and PCP bind to overlapping sites in the desensitized nAChR (Arias, unpublished experiments). Consequently, our data are in accord with a model where the desensitized Torpedo ion channel bears overlapping binding sites for BAs, PCP and dizocilpine. The fact that different mutations in the ion channel increase the self-inhibitory effect of DMXBA on nAChRs (Papke, 2002), supports the idea that certain amino acids in the ion channel are possible sites for the antagonist action of DMXBA.
Regarding the location of the BA-binding site in the resting state, our results indicate that DMXBA inhibits [3H]tetracaine binding in an allosteric fashion, whereas 4OH-DMXBA enhances [3H]tetracaine binding. These results rule out the possibility that the BA locus overlaps the tetracaine-binding site. Other experimental data (Papke, 2002) indicating that tetracaine does not protect the receptor from the inhibition produced by DMXBA is in accord with our results. In addition, both BAs partially inhibit [14C]amobarbital and inhibit [3H]TCP binding to nAChRs arrested in the resting state (in the presence of α-BTx) in an allosteric manner. These data indicate that BAs bind neither to the barbiturate nor to the PCP site in the resting state. The fact that BAs enhance [3H]TCP binding when the nAChR is in the resting but activatable state (in the absence of α-BTx) (see Figure 7) also supports the same conclusion.
Based on photoaffinity labelling results using Torpedo nAChRs in the resting state, the tetracaine-binding site has been located in a long portion of the M2 transmembrane domain that involves residues from position 5′ to 13′ (i.e. the valine ring) (Gallagher and Cohen, 1999; Middleton et al., 1999) and probably extending to the outer ring at position 20′ (Arias et al., 2002a). The tetracaine-binding domain partially overlaps the barbiturate locus in approximately the middle of the ion channel (between position 9′ and 13′) (Arias et al., 2001b) as well as the PCP site, which is located after the valine ring (i.e. position 13′) and close to the outer ring (i.e. position 20′) (Arias et al., 2001b 2002b 2003 2006a; Gallagher et al., 2001). Considering our data and the results from different laboratories, we can envision a model where the NCA-binding site for BAs in the resting Torpedo nAChR is located in a domain different from the tetracaine site (i.e. between position 5′ and 20′). A possibility is that relatively lipophilic BAs allosterically modulate [3H]tetracaine, [14C]amobarbital and [3H]TCP binding by interacting with hydrophobic sites at the lipid-protein interface, as different local anesthetics do (Horváth et al., 1990; Mantipragada et al., 2003).
Previous electrophysiological studies indicated that BAs, in addition to activating α7 nAChRs with differing potency, inhibit muscle-(e.g. α12β1γδ) and neuronal-type nAChRs (e.g. α4β2 and α3β4) (Meyer et al., 1998; Papke, 2002; Papke et al., 2000 2004; Uteshev et al., 2002 2003). One possible mechanism of BA nAChR inhibition, besides low affinity for the open state upon binding to the agonist-binding site and/or direct ion channel block mechanisms, would be enhancement of desensitization. In fact, BAs may modulate the function of the α7 nAChR in tuberomammillary histamine neurons from the posterior hypothalamus by a combination of activation, blocking and desensitization processes (Papke et al., 2000; Uteshev et al., 2002 2003). In this regard, the BA-induced desensitization observed as BA-enhanced [3H]TCP (see Figure 7) binding to the Torpedo nAChR in the resting but activatable state (in the absence of α-BTx) supports these previous experiments. Our results using Torpedo nAChRs also indicate that 4OH-DMXBA is a more potent and efficacious desensitizing agent than DMXBA (see Table 3). Our present observation that 4OH-DMXBA is a rather potent partial agonist for the human fetal muscle nAChR, whereas DMXBA lacks agonist effects on this receptor subtype (see Figure 2), predicts that 4OH-DMXBA would be a more potent desensitizer of this nAChR, and maybe of the α7 nAChR. Direct interaction of BAs with the nAChR ion channel may also favour nAChR desensitization.
Another way of considering the inhibitory activity of DMXBA at the nAChR would be to compare its affinity for the different conformational states of the receptor. As the nAChR subtype in Torpedo electric organs and in the human TE671 cell line is the same as fetal muscle nAChRs (α12β1γδ), we can assume that the structural and functional properties of these nAChRs are basically the same and thus, we can infer that DMXBA interacts with the muscle receptor ion channel with the following relative affinities: open (IC50∼7 µmol·L−1; see Figure 1) > desensitized (Ki= 13 ± 1 µmol·L−1; see Table 2) >> resting (Ki= 172 ± 12 µmol·L−1; see Table 3). Considering these relative affinities, we can assume that under in vivo conditions, DMXBA would interact initially with the open ion channel, after the receptor is activated by ACh. Desensitization of the receptor could then occur, leading to a more profound functional inhibition. Further experiments are needed to determine which of the various possible inhibitory mechanisms discussed above operate to limit nAChR activation by DMXBA and other BAs. A better understanding of these non-competitive inhibitory phenomena should facilitate the design of more efficacious BA-type α7 nAChR agonists with lower activity on muscle-type nAChRs.
Acknowledgments
This research was supported by research grants from the Science Foundation Arizona and Stardust Foundation, and from the Office of Research and Sponsored Programs and College of Pharmacy, Midwestern University (H.R.A.), by intramural grants from Texas Tech University Health Sciences Center (School of Medicine Seed Grant; Roger Alan Valkenaar Estate; the South Plains Foundation, M.P.B.), by NIH Grant MH-61412 (R. Freedman, Univ. Colorado Heath Sciences Center, P.I.; W.R.K., Co-Invest.) and Comentis Inc. (W.R.K., P.I.). We thank Dr R. Lukas (Barrow Neurological Institute, Phoenix, AZ, USA) for comments on the manuscript, and Jorgelina Castillo Arias and Paulina Iacoban for technical support. The authors also thank NIDA (NIH, Bethesda, MD, USA) for its gift of amobarbital and PCP.
Glossary
Abbreviations
- [3H]TCP
[piperidyl-3,4-3H(N)]-(N-(1-(2-thienyl)cyclohexyl)-3,4-piperidine
- 4OH-DMXBA
3-(4-hydroxy-2-methoxybenzylidene)-anabaseine
- α-BTx
α-bungarotoxin
- BA
3-(benzylidene)-anabaseine
- CCh
carbamylcholine
- DMXBA (or GTS-21)
3-(2,4-dimethoxybenzylidene)-anabaseine
- Kd
dissociation constant
- Ki
inhibition constant
- nAChR
nicotinic acetylcholine receptor
- NCA
non-competitive antagonist
- nH
Hill coefficient
- PCP
phencyclidine
Conflicts of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (3rd edn.) 2008;153(Suppl.)(2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias HR. Agonist self-inhibitory binding site of the nicotinic acetylcholine receptor. J Neurosci Res. 1996;44:97–105. doi: 10.1002/(SICI)1097-4547(19960415)44:2<97::AID-JNR1>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Arias HR. Ligand-gated ion channel receptor superfamilies. In: Arias HR, editor. Biological and Biophysical Aspects of Ligand-Gated Ion Channel Receptor Superfamilies. Kerala, India: Research Signpost; 2006. pp. 1–25. [Google Scholar]
- Arias HR, Bhumireddy P. Anesthetics as chemical tools to study the structure and function of nicotinic acetylcholine receptors. Curr Protein Pept Sci. 2005;6:451–472. doi: 10.2174/138920305774329331. [DOI] [PubMed] [Google Scholar]
- Arias HR, McCardy EA, Blanton MP. Characterization of the dizocilpine binding site on the nicotinic acetylcholine receptor. Mol Pharmacol. 2001a;59:1051–1060. doi: 10.1124/mol.59.5.1051. [DOI] [PubMed] [Google Scholar]
- Arias HR, McCardy EA, Gallagher MJ, Blanton MB. Interaction of barbiturate analogs with the Torpedo californica nicotinic acetylcholine receptor ion channel. Mol Pharmacol. 2001b;60:497–506. [PubMed] [Google Scholar]
- Arias HR, Kem WR, Trudell JR, Blanton MP. Unique general anesthetic binding sites within distinct conformational states of the nicotinic acetylcholine receptor. Int Rev Neurobiol. 2002a;54:1–50. doi: 10.1016/s0074-7742(03)54002-8. [DOI] [PubMed] [Google Scholar]
- Arias HR, McCardy EA, Bayer EZ, Gallagher MJ, Blanton MB. Allosterically linked noncompetitive antagonist binding sites in the resting nicotinic acetylcholine receptor ion channel. Arch Biochem Biophys. 2002b;403:121–131. doi: 10.1016/S0003-9861(02)00214-X. [DOI] [PubMed] [Google Scholar]
- Arias HR, Trudell JR, Bayer EZ, Hester B, McCardy EA, Blanton MB. Noncompetitive antagonist binding sites in the Torpedo nicotinic acetylcholine receptor ion channel. Structure-activity relationship studies using adamantane derivatives. Biochemistry. 2003;42:7358–7370. doi: 10.1021/bi034052n. [DOI] [PubMed] [Google Scholar]
- Arias HR, Bhumireddy P, Spitzmaul G, Trudell JR, Bouzat C. Molecular mechanisms and binding site location for the noncompetitive antagonist crystal violet on nicotinic acetylcholine receptors. Biochemistry. 2006a;45:2014–2026. doi: 10.1021/bi051752e. [DOI] [PubMed] [Google Scholar]
- Arias HR, Bhumireddy P, Bouzat C. Molecular mechanisms and binding site locations for noncompetitive antagonists of nicotinic acetylcholine receptors. Int J Biochem Cell Biol. 2006b;38:1254–1276. doi: 10.1016/j.biocel.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Azuma R, Komuro H, Korsch BH, Andre JC, Onnagawa O, Black SR, et al. Metabolism and disposition of GTS-21, a novel drug for Alzheimer's disease. Xenobiot. 1999;29:747–762. doi: 10.1080/004982599238362. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- de Fiebre CM, Meyer EM, Henry J, Muraskin SI, Kem W, Papke R. Characterization of a series of anabaseine-derived compounds reveals that the 3-(4)-dimethylaminocinnamylidine derivative is a selective agonist at neuronal nicotinic α7/125I-α-bungarotoxin receptor subtypes. Mol Pharmacol. 1995;47:164–171. [PubMed] [Google Scholar]
- Fletcher GH, Steinbach JH. Ability of nondepolarizing neuromuscular blocking drugs to act as partial agonists at fetal and adult mouse muscle nicotinic receptors. Mol Pharmacol. 1996;49:938–947. [PubMed] [Google Scholar]
- Freedman R, Olincy A, Buchanan RW, Harris JG, Gold JM, Kem WR, et al. Initial phase 2 trial of a nicotinic agonist in schizophrenia. J Gen Psychiat. 2008;165:1040–1047. doi: 10.1176/appi.ajp.2008.07071135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher MJ, Cohen JB. Identification of amino acids of the Torpedo nicotinic acetylcholine receptor contributing to the binding site for the noncompetitive antagonist [3H]tetracaine. Mol Pharmacol. 1999;56:300–307. doi: 10.1124/mol.56.2.300. [DOI] [PubMed] [Google Scholar]
- Gallagher MJ, Chiara DC, Cohen JB. Interactions between 3-(trifluoromethyl)-3-(m-[(125)I]iodophenyl)diazirine and tetracaine, phencyclidine, or histrionicotoxin in the Torpedo species nicotinic acetylcholine receptor ion channel. Mol Pharmacol. 2001;59:1514–1522. doi: 10.1124/mol.59.6.1514. [DOI] [PubMed] [Google Scholar]
- Horváth LI, Arias HR, Hankovszky HO, Hideg K, Barrantes FJ, Marsh D. Association of spin-labeled local anesthetics at the hydrophobic surface of acetylcholine receptor in native membranes from Torpedo marmorata. Biochemistry. 1990;29:8707–8713. doi: 10.1021/bi00489a029. [DOI] [PubMed] [Google Scholar]
- Kalamida D, Poulas K, Avramopoulou V, Fostieri E, Lagoumintzis G, Lazaridis K, et al. Muscle and neuronal nicotinic acetylcholine receptors: structure, function and pathogenicity. FEBS J. 2007;274:3799–3845. doi: 10.1111/j.1742-4658.2007.05935.x. [DOI] [PubMed] [Google Scholar]
- Kem WR, Mahnir VM, Papke R, Lingle C. Anabaseine is a potent agonist upon muscle and neuronal alpha-bungarotoxin sensitive nicotinic receptors. J Pharmacol Exp Ther. 1997;283:979–992. [PubMed] [Google Scholar]
- Kem WR, Mahnir VM, Prokai L, Papke RL, Cao X, LeFrancois S, et al. Hydroxy metabolites of the Alzheimer's drug candidate 3-[(2,4-dimethoxy)benzylidene]-anabaseine dihydrochloride (GTS-21): Their molecular properties, interactions with brain nicotinic receptors, and brain penetration. Mol Pharmacol. 2004;65:56–67. doi: 10.1124/mol.65.1.56. [DOI] [PubMed] [Google Scholar]
- Kem WR, Soti F, LeFrancois S, Wildeboer K, MacDougall K, Wei D, et al. The nemertine toxin anabaseine and its derivative DMXBA (GTS-21): chemical and pharmacological properties. Marine Drugs (Marine Toxins and Ion Channels) 2006;4:255–273. [Google Scholar]
- Kitagawa H, Takenouchi T, Azuma R, Wesnes KA, Kramer WG, Clody DE, et al. Safety, pharmacokinetics, and effects on cognitive function of multiple doses of GTS-21 in healthy, male volunteers. Neuropsychopharmacology. 2003;28:542–551. doi: 10.1038/sj.npp.1300028. [DOI] [PubMed] [Google Scholar]
- Kopta C, Steinbach JH. Comparison of mammalian adult and fetal nicotinic acetylcholine receptors stably expressed in fibroblasts. J Neurosci. 1994;14:3922–3933. doi: 10.1523/JNEUROSCI.14-06-03922.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ST, Wildeboer K, Panter KE, Kem WR, Gardner DR, Molyneux RJ, et al. Relative toxicities and neuromuscular nicotinic receptor agonistic potencies of anabasine enantiomers and anabaseine. Neurotoxicol Teratol. 2006;28:220–228. doi: 10.1016/j.ntt.2005.12.010. [DOI] [PubMed] [Google Scholar]
- Mahnir VM, Lin B, Prokai-Tatrai K, Kem WR. Pharmacokinetics and urinary excretion of DMXBA (GTS-21), a compound enhancing cognition. Biopharm Drug Disp. 1998;19:147–151. doi: 10.1002/(sici)1099-081x(199804)19:3<147::aid-bdd77>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Mantipragada SB, Horváth LI, Arias HR, Schwarzmann G, Sandhoff K, Barrantes FJ, Marsh D. Lipid-protein interactions and the effect of local anesthetics in acetylcholine receptor-rich membranes from Torpedo marmorata electric organ. Biochemistry. 2003;42:9167–9175. doi: 10.1021/bi034485q. [DOI] [PubMed] [Google Scholar]
- Meyer EM, Tay ET, Papke RL, Meyers C, Huang GL, de Fiebre CM. 3-[2,4-Dimethoxybenzylidene]anabaseine (DMXB) selectively activates rat α7 receptors and improves memory-related behaviors in a mecamylamine-sensitive manner. Brain Res. 1997;768:49–56. doi: 10.1016/s0006-8993(97)00536-2. [DOI] [PubMed] [Google Scholar]
- Meyer EM, Kuryatov A, Gerzanich V, Lindstrom J, Papke RL. Analysis of 3-(4-hydroxy, 2-methoxybenzylidene)anabaseine selectivity and activity at human and rat α7 nicotinic receptors. J Pharm Exp Ther. 1998;287:918–925. [PubMed] [Google Scholar]
- Middleton RE, Cohen JB. Mapping of the acetylcholine binding site of the nicotinic acetylcholine receptor: [3H]nicotine as an agonist photoaffinity label. Biochemistry. 1991;30:6987–6997. doi: 10.1021/bi00242a026. [DOI] [PubMed] [Google Scholar]
- Middleton RE, Strand NP, Cohen JB. Photoaffinity labeling the Torpedo nicotinic acetylcholine receptor with [3H]tetracaine, a nondesensitizing noncompetitive antagonist. Mol Pharmacol. 1999;56:290–299. doi: 10.1124/mol.56.2.290. [DOI] [PubMed] [Google Scholar]
- Moore MA, McCarthy MP. Snake venom toxins, unlike smaller antagonists, appear to stabilize a resting state conformation of the nicotinic acetylcholine receptor. Biochim Biophys Acta. 1995;1235:336–342. doi: 10.1016/0005-2736(95)80022-8. [DOI] [PubMed] [Google Scholar]
- Olincy A, Harris JG, Johnson LL, Pender V, Kongs S, Allensworth D, et al. Proof-of-concept trial of an α7-nicotinic agonist in schizophrenia. Arch Gen Psychiatry. 2006;63:630–638. doi: 10.1001/archpsyc.63.6.630. [DOI] [PubMed] [Google Scholar]
- Pagán OR, Eterovic VA, García M, Vergne D, Basilio CM, Rodriguez AD, et al. Cembranoid and long-chain alkanol sites on the nicotinic acetylcholine receptor and their allosteric interaction. Biochemistry. 2001;40:11121–11130. doi: 10.1021/bi0112255. [DOI] [PubMed] [Google Scholar]
- Papke RL. Enhanced inhibition of a mutant neuronal nicotinic acetylcholine receptor by agonists: protection of function by (E)-N-methyl-4-(3-pyridinyl)-3-butene-1-amine (TC-2403) J Pharm Exp Ther. 2002;301:765–773. doi: 10.1124/jpet.301.2.765. [DOI] [PubMed] [Google Scholar]
- Papke RL, Thinschmidt JS. The correction of α7 nicotinic acetylcholine receptor concentration-response relationships in Xenopus oocytes. Neurosci Lett. 1998;256:163–166. doi: 10.1016/s0304-3940(98)00786-1. [DOI] [PubMed] [Google Scholar]
- Papke RL, Meyer E, Nutter T, Uteshev VV. α7 Receptor-selective agonists and modes of α7 receptor activation. Eur J Pharmacol. 2000;393:179–195. doi: 10.1016/s0014-2999(00)00009-1. [DOI] [PubMed] [Google Scholar]
- Papke RL, Meyer EM, Lavieri S, Bollampally SR, Papke TA, Horenstein NA, et al. Effects at a distance in α7 nAChR selective agonists: benzylidene substitutions that regulate potency and efficacy. Neuropharmacology. 2004;46:1023–1038. doi: 10.1016/j.neuropharm.2004.01.005. [DOI] [PubMed] [Google Scholar]
- Pratt MB, Pedersen SE, Cohen JB. Identification of the sites of incorporation of [3H]ethidium diazide within the Torpedo nicotinic acetylcholine receptor ion channel. Biochemistry. 2000;39:11452–11462. doi: 10.1021/bi0011680. [DOI] [PubMed] [Google Scholar]
- Pedersen SE, Dreyer EB, Cohen JB. Location of ligand-binding sites on the nicotinic acetylcholine receptor α-subunit. J Biol Chem. 1986;261:13735–13743. [PubMed] [Google Scholar]
- Schild HO. pAx and competitive drug antagonism. Br J Pharmacol. 1949;4:277–280. doi: 10.1111/j.1476-5381.1949.tb00548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uteshev VV, Meyer EM, Papke RL. Activation and inhibition of native neuronal α-bungarotoxin-sensitive nicotinic ACh receptors. Brain Res. 2002;948:33–46. doi: 10.1016/s0006-8993(02)02946-3. [DOI] [PubMed] [Google Scholar]
- Uteshev VV, Meyer EM, Papke RL. Regulation of neuronal function by choline and 4OH-GTS-21 through α7 nicotinic receptors. J Neurophysiol. 2003;89:1797–1806. doi: 10.1152/jn.00943.2002. [DOI] [PubMed] [Google Scholar]
- Ziskind L, Dennis MJ. Depolarizing effect of curare on embryonic rat muscles. Nature. 1978;276:622–623. doi: 10.1038/276622a0. [DOI] [PubMed] [Google Scholar]