

Abstract
With more than 20 molecules in clinical use, monoclonal antibodies have finally come of age as therapeutics, generating a market value of $11 billion in 2004, expected to reach $26 billion by 2010. While delivering interesting results in the treatment of several major diseases including autoimmune, cardiovascular and infectious diseases, cancer and inflammation, clinical trials and research are generating a wealth of useful information, for instance about associations of clinical responses with Fc receptor polymorphisms and the infiltration and recruitment of effector cells into targeted tissues. Some functional limitations of therapeutic antibodies have come to light such as inadequate pharmacokinetics and tissue accessibility as well as impaired interactions with the immune system, and these deficiencies point to areas where additional research is needed. This review aims at giving an overview of the current state of the art and describes the most promising avenues that are being followed to create the next generation of antibody-based therapeutic agents.
This article is part of a themed section on Vector Design and Drug Delivery. For a list of all articles in this section see the end of this paper, or visit: http://www3.interscience.wiley.com/journal/121548564/issueyear?year=2009
Keywords: immunotherapy, monoclonal antibodies, pharmacokinetics, cancer, antibody engineering, glycosylation, Fc receptors, therapeutic antibodies, intrabodies
Therapeutic antibodies: coming of age
First hopes and disappointments
Since 1975, when Kohler and Milstein developed a procedure to efficiently produce monoclonal antibodies (mAbs) (Kohler and Milstein, 1975), it has been widely believed that these molecules would be ideal reagents for imaging and therapy, similar to the magic bullets imagined by Paul Ehrlich at the beginning of the 20th century. Antibodies have been selected during evolution for their ability to bind with high specificity and affinity to a wide variety of molecules and, as they are stable molecules, they seemed to be ideal molecules to be used as targeting reagents. However, the early excitement was rapidly replaced by disappointment when it became clear that these molecules were facing serious problems when used as therapeutics. The first mAbs were murine molecules and were recognized as foreign when injected into patients, leading to their elimination by the patient's immune system. Moreover, in order to be effective, antibodies often need to interact with certain elements of the immune system such as receptors displayed on effector cells or the complement cascade. Because of their murine nature, these early antibodies did not interact properly with components of the human immune system and their biological efficacy was severely restricted.
Antibody engineering
Developments in molecular biology made it possible in the early 90s to clone the genes of IgG molecules (Winter and Milstein, 1991) and, as a result, the genes of mAbs of interest could be cloned in eukaryotic expression vectors. In this way, recombinant versions of any mAb could be obtained from diverse cell lines in a reproducible fashion, and this solved production issues caused by the instability of many hybridoma lines (Chames and Baty, 2000). Cloning antibody genes was the first step towards the modification of antibodies by subcloning, random or directed mutagenesis and molecular evolution procedures, which made it possible to optimize recombinant antibodies at will and ushered the age of antibody engineering (Hoogenboom and Chames, 2000).
Chimeric, humanized and human antibodies
A major application of antibody engineering was the possibility to create chimeric antibodies (Figure 1). The binding activity of IgG molecules is generated by the variable domains of the heavy and light chains. As antibodies are well conserved through evolution, it was possible to create chimeras by fusing murine variable domains, responsible for the binding activity, with human constant domains (Neuberger et al., 1985) leading to the development of a new generation of therapeutic candidates (Reichert et al., 2005) (Figure 1). These chimeric antibodies are 70% human and possess a fully human Fc portion, which makes them considerably less immunogenic in humans and allows them to interact with human effector cells and the complement cascade. With the development of antibody engineering techniques, it became possible to decrease even further the murine part of mAbs by replacing the hypervariable loops of a fully human antibody with the hypervariable loops of the murine antibody of interest, using an approach called complementarity-determining region grafting (Jones et al., 1986). These antibodies, called ‘humanized’, are 85–90% human and are even less immunogenic than chimeric antibodies (Figure 1). However, complementarity-determining region grafting is more technically demanding than a mere fusion, and directed mutagenesis approaches are often needed to restore the affinity present in the murine parental antibody. Most of the approved mAbs in current use are either chimeric or humanized (Table 1).
Figure 1.
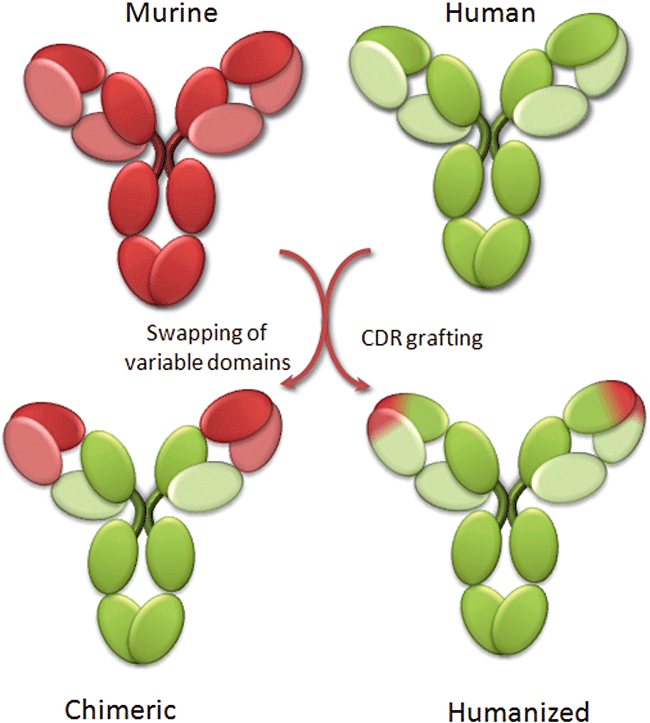
Chimeric and humanized antibodies. Murine sequences are depicted in red and human sequences in green, using light colours for light chain and dark colours for heavy chains.
Table 1.
Monoclonal antibodies approved for therapeutic use
| Generic name | Trade name | Antibody format | Antigen | Approved indication | 13FDA approval | 13EMEA approval |
|---|---|---|---|---|---|---|
| Muromomab | Orthoclone | Murine, IgG2a | CD3 | Allograft rejection in allogeneic renal transplantation | 86/06/19 | NA |
| Abciximab1 | ReoPro | Chimeric, IgG1 | GPIIb/IIIa r | Maintenance of coronary patency | 94/12/22 | NA |
| Rituximab2 | Mabthera | Chimeric, IgG1 | CD20 | CD20-positive B-cell non-Hodgkin's lymphoma | 97/11/26 | 98/06/02 |
| Daclizumab | Zenapax | Humanized, IgG1 | CD25 (Il2r) | Allograft rejection | 97/12/10 | 99/02/26 |
| Basiliximab | Simulect | Chimeric, IgG1 | CD25 (Il2r) | Allograft rejection | 98/05/12 | 98/10/09 |
| Palivizumab | Synagis | Humanized, IgG1 | Protein F | Respiratory syncytial virus (RSV inhibitor) in children | 98/06/19 | 99/08/13 |
| Infliximab | Remicade | Chimeric, IgG1 | TNFα | Crohn's disease and rheumatoid arthritis | 98/08/24 | 99/08/13 |
| Trastuzumab | Herceptin | Humanized, IgG1 | HER2/Neu | Metastatic breast cancer | 98/09/25 | 00/08/28 |
| Etanercept3 | Enbrel | huFcγ1/TNFr | TNFα and β | Autoimmune diseases such as ankylosing spondylitis | 98/11/02 | 00/02/03 |
| Gemtuzumab4 | Mylotarg | Humanized, IgG4 | CD33 | CD33-positive acute myeloid leukemia | 00/05/17 | NA |
| Alemtuzumab5 | Mabcampath | Humanized, IgG1 | CD52 | B-cell chronic lymphocytic leukemia | 01/05/07 | 01/07/06 |
| Ibritomomab6 | Zevalin 90Y | Mouse, IgG1 | CD20 | B-cell non-Hodgkin's lymphoma | 02/02/19 | 04/01/16 |
| Adalimumab7 | Trudexa | Human, IgG1 (PD) | TNFα | Crohn's disease and rheumatoid arthritis | 02/12/31 | 03/09/01 |
| Alefacept3 | Amevive | huFcγ1/LFA-3 | CD2 | Chronic plaque psoriasis | 03/01/30 | NA |
| Omalizumab | Xolair | Humanized, IgG1 | IgE | Treatment of asthma | 03/06/20 | 05/10/25 |
| Tositumomab68 | Bexxar 131I | Murine, IgG2a | CD20 | CD20-positive B-cell non-Hodgkin's lymphoma | 03/06/27 | NA |
| Efalizumab | Raptiva | Humanized, IgG1 | CD11a | Moderate to severe plaque psoriasis | 03/10/27 | 04/09/20 |
| Cetuximab | Erbitux | Chimeric, IgG1 | EGFR | Metastatic colorectal and head and neck carcinoma | 04/02/12 | 04/06/29 |
| Bevacizumab | Avastin | Humanized, IgG1 | VEGF-A | Metastatic colorectal and non-small cell lung carcinoma | 04/02/26 | 05/01/12 |
| Natalizumab9 | Tysabri | Humanized, IgG4 | Integrin-α4 | Multiple sclerosis | 04/11/23 | 06/06/27 |
| Ranibizumab | Lucentis | Humanized, IgG1 | VEGF-A | Wet-type age-related macular degeneration | 06/06/30 | 07/01/22 |
| Panitumumab10 | Vectibis | Human, IgG2 | EGFR | Metastatic colorectal carcinoma | 06/09/27 | 07/12/19 |
| Eculizumab11 | Soliris | Humanized, IgG2/4 | C5 | Paroxysmal nocturnal haemoglobinuria | 07/03/16 | 07/06/20 |
| Certolizumab12 | Cimzia | Humanized, IgG1 | TNFα | Crohn's disease | 08/04/18 | NA |
Abciximab is a Fab fragment.
Rituximab is commercialized under the trade name Rituxan in USA.
These molecules are fusions between the IgG1 Fc portion and a receptor. On 2 May 2008, the FDA placed a black box warning on Etanercept due to a number of serious infections associated with the drug.
Gemtuzumab ‘ozogamicine’ is coupled to calicheamicin, an anti-tumoural antibiotic.
Alemtuzumab is commercialized under the trade name Campath in USA.
Ibritomomab ‘tiuxetan’ and Tositumomab are coupled to radioisotopes.
Adalimumab is commercialized under the trade name Humira in USA.
All approved antibodies have a κ light chain except Tositumomab that has a γ light chain.
Natalizumab was voluntarily withdrawn from the market in February 2005. On 5 June 2006, FDA approved a special restricted distribution programme.
Panitumumab is the first human antibody obtained from humanized mice.
Eculizumab contains a CH1 domain and hinge from IgG2, and CH2–CH3 domains from IgG4.
Certolizumab pegol is a PEGylated humanized Fab fragment.
Year/month/day; NA, not approved in Europe.
EGFR, epidermal growth factor receptor; EMEA, European Medicines Agency; FDA, US Food and Drug Administration; HER, human epidermal growth factor receptor; TNF, tumour necrosis factor; VEGF, vascular endothelial growth factor.
Another major improvement came with the development of in vitro selection methods, the most successful one being phage display. With the ever increasing power of antibody engineering, it became possible to clone entire repertoires of antibody fragment genes, from immunized or non-immunized animals, including humans. A powerful selection method was therefore needed to select from this large number of potential ligands, those able to bind the antigen of choice. The first technique, and still by far the most common one was mainly developed in the laboratory of Greg Winter (McCafferty et al., 1990), and was inspired by earlier work by George Smith (Smith, 1985). Like all in vitro selection methods, this technique relies on the ability to establish a physical link between a protein and the gene encoding this protein, in this case between a protein fused to a filamentous phage capsid protein (p3 or p8) displayed at the surface of phage M13 and its corresponding gene contained in the encapsidated DNA. If the molecule is immunopurified by binding to the antigen of interest, its gene is immediately available, allowing sequencing and further multiplication of the specific clone. Because of these in vitro selection methods, it is now possible to rapidly and efficiently select fully human antibody fragments against virtually any antigen by using ‘universal’, large, non-immunized libraries (Hoogenboom and Chames, 2000). Moreover, the same approach can be used to maximize the affinity of a valuable antibody by creating a secondary library consisting of mutants of the first candidate and performing stringent in vitro selection against the antigen of choice. Phage display and more recently ribosome display have been used to obtain ligands with sub-picomolar affinities for the relevant antigen, outperforming the affinities of most conventional mAbs (Luginbuhl et al., 2006).
During the same decade, a complementary approach was developed to create fully human antibodies. Transgenic ‘humanized’ mice were created by replacing the entire mouse IgG repertoire with a human repertoire (Lonberg, 2008). Upon immunization, these humanized mice produce human IgGs and conventional hybridoma techniques can be used to clone human antibodies with the required properties. This approach has the advantage of yielding in vivo matured antibodies, circumventing the need for additional affinity maturation. Moreover, they directly lead to full-length IgG, which is often the preferred format for therapy. However, humanized mice cannot be used effectively when the immunogen is toxic or when the targeted antigen shares a high degree of homology with its murine ortholog. This latter problem represents a real limitation, as it could sometimes be highly convenient to use a murine model for preclinical characterization and the murine orthologue of a therapeutic target.
Current limitations
The creation of chimeric, humanized or fully human antibodies was a major breakthrough and led to a wave of US Food and Drug Administration (FDA)-approved antibodies. Currently, 22 antibodies are commercialized as therapeutics, mainly for cancer and immune disorders (Table 1). Impressive results have been achieved in cancer therapy, as exemplified by the success met by Rituximab in the treatment of several cancer types. However, mAb-based treatments are nevertheless facing several limitations, which limit their widespread use as therapeutics.
Production costs
Monoclonal antibodies are large (150 kDa) multimeric proteins containing numerous disulphide bonds and post-translational modifications such as glycosylation. They need a sophisticated eukaryotic machinery to be produced in active form. Moreover, most studies have shown that these molecules have to be injected in large amounts to achieve clinical efficacy (e.g. 8–16 doses of 375 mg·m−2, that is, a total amount of 6–12 g per patient for Rituximab; see http://www.rituxan.com). Consequently, the production of therapeutic antibodies necessitates the use of very large cultures of mammalian cells followed by extensive purification steps, under Good Manufacturing Practice conditions, leading to extremely high production costs and limiting the wide use of these drugs. Several alternative production systems in microorganisms and plants are being evaluated at the moment, which might lead to significant progress in the near future (Giritch et al., 2006; Graumann and Premstaller, 2006).
Pharmacokinetics versus tissue penetration
In murine xenograft models, mAbs directed against tumour-specific antigens largely remain in the blood and no more than 20% of the administered dose typically interacts with the tumour (Beckman et al., 2007). This represents probably one of the major limitations faced by mAbs. Antibody uptake by the tumour depends on a subtle balance between favourable pharmacokinetics and efficient penetration and retention in the targeted tissue, and various characteristics of mAbs, such as molecular size, shape, affinity and valency control these properties. mAbs are large molecules that are characterized by very long serum half-lives (Table 2). They far exceed the renal clearance threshold (∼70 kDa), preventing them from being eliminated through the kidney glomeruli. Moreover, the Fc portion of IgG molecules can interact with various receptors expressed at the surface of several cell types, which increase their retention in the circulation. Most importantly, the Fc portion can interact with the neonatal Fc receptor (FcRn) expressed at the surface of several cell types, including vascular endothelium cells, monocytes and macrophages as well as with barrier sites such as the blood–brain interface, the glomerular filter in the kidneys and the intestinal epithelium (Roopenian and Akilesh, 2007). Beside its role in the transport of antenatal maternal antibody to the fetus, across the placenta, FcRn plays an important role in IgG homeostasis. Indeed it has been shown that FcRn extends the serum half-life of IgG and albumin (i.e. ∼90% of serum protein content) from 1 day (the typical half-life of serum protein not freely filtered by the kidneys) to up to several weeks. Most serum proteins are pinocytosed and undergo gradual acidification in endosomes, followed by fusion with lysosomes and hydrolysis. However, IgGs bind to FcRn at low pH, and the complex is carried back to the cell surface, whereupon it dissociates at neutral pH. FcRn therefore serves as a protective carrier that shuttles IgG molecules away from the lysosome and back into the serum, conferring on them an even longer serum half-life.
Table 2.
Properties of antibody isotypes and antibody fragments1
| Isotype | Serum level (mean adult; mg·mL−1) | Biological half-life (days) | Biological Functions | Molecular weight (kDa) |
|---|---|---|---|---|
| IgA1 | 6 | 6 | Pathogen neutralization in mucosal secretions | 160 |
| IgA2 | 6 | 6 | Pathogen neutralization in mucosal secretions | 160 |
| IgD | 3 | 3 | Membrane B-cell receptor | 184 |
| IgE | 2 | 2 | Mast cell histamine release | 188 |
| IgG1 | 21 | 21 | Pathogen neutralization in tissues | 146 |
| Classical complement activation | ||||
| Opsonization | ||||
| NK cell ADCC | ||||
| Transplacental transfer | ||||
| IgG2 | 20 | 20 | Pathogen neutralization in tissues | 146 |
| IgG3 | 7 | 7 | Pathogen neutralization in tissues | 165 |
| Classical complement activation | ||||
| Opsonization | ||||
| NK cell ADCC | ||||
| Transplacental transfer | ||||
| IgG4 | 21 | 21 | Pathogen neutralization in tissues | 146 |
| Transplacental transfer | ||||
| IgM | 10 | 10 | Classical complement activation | 970 |
| Membrane B-cell receptor (monomer) | ||||
| Fab | 0.3–0.8 | 50 | ||
| scFv | 0.1 | 27 | ||
| sdAb | <0.05 | 13 |
Adapted from Janeway CA, Travers P, Walport M and Shlomchick. Immunobiology. Garland publishing, NY, 2001.
ADCC, antibody-dependent, cell-mediated cytotoxicity; NK, natural killer; sdAbs, single-domain antibodies.
While the large size of mAbs and the presence of the Fc region can be advantageous in terms of pharmacokinetics, they can also be a serious handicap. Except in the case of haematological malignancies and diseases, therapeutic antibodies need to penetrate tissues and the extracellular matrix to reach their target cells. Tissue penetration, especially in the case of solid tumours, is a crucial parameter, most of the time severely limiting the overall efficiency of the treatment. Tumours are characterized by heterogeneous and tortuous vasculature, high interstitial fluid pressure and high viscosity of the tumour blood supply. Consequently, mAbs must diffuse against this pressure gradient to penetrate tumours (Beckman et al., 2007). A major determinant of speed of diffusion through tumours is molecular size. The rate of diffusion is approximately inversely proportional to the cube root of molecular weight. Consequently, large macromolecules such as mAbs diffuse poorly explaining why larger tumour masses may be more difficult to treat by mAb therapy. Strikingly, among the nine mAbs approved for cancer therapy, only two (trastuzumab, cetuximab) are targeting solid tumours, whereas over 85% of human cancers are solid tumours, clearly reflecting the current limitation of mAb treatment.
Moreover, another factor, the ‘binding site barrier effect’, can further decrease the penetration of tumours by mAbs (Fujimori et al., 1990). Whereas one might presume that tighter binding is always better, several studies have shown that very high affinities can be suboptimal for therapeutic antibodies that target solid tumours (Adams et al., 2001). Because high affinity antibodies tightly bind their antigen upon the first encounter, that is, at the periphery of the tumour, they do not penetrate deeper inside the tumour until all antigen molecules are saturated in the periphery. By contrast, moderate binders are released from these first encountered antigens and penetrate deeper into the tumour, ultimately leading to uniform intratumoural distribution and higher tumour uptake. The right balance in terms of affinity will lead to an efficient tumour targeting and tumour retention and will also allow some diffusion inside the tumour. Because these properties depend on several factors, including antigen density, internalization, association and dissociation rates, the optimal affinity is not always easy to engineer.
Mode of action and associated limitations
Monoclonal antibodies can have various modes of actions in vitro, and the actual mode of action once injected in patients is not always clear (Borrebaeck and Carlsson, 2001). The simplest mode of action is mere binding of the antibody to its antigen, thereby interfering with its activity and interaction with binding partners. The antigen can be a soluble ligand, and examples of such antibodies include infliximab, adalimumab and certolizumab (anti-TNFα) or bevacizumab (anti-vascular endothelial growth factor). On the other hand, the antibody may target a receptor displayed at the cell surface, block its interaction with a ligand, interfere with a multimerization process or trigger internalization of receptors or apoptosis of targeted cells. Examples of such antibodies include cetuximab and panitumumab [anti-EGFR (epidermal growth factor receptor) or HER1 (human epidermal growth factor receptor)] and trastuzumab (anti-HER2) (Table 1).
In addition, once bound to their antigen, antibodies can also interact in several ways with the immune system of the recipient through their Fc portion. They can recruit the complement cascade through interaction with C1q, ultimately leading to the formation of pores in the targeted cell membrane, or they can recruit effector cells through interaction with the C4b/C2b/C3b complex bound to the target cell surface and the receptor CR1 (complement receptor). However, although most therapeutic antibodies can trigger complement-dependent cytotoxicity in vitro, this has not been demonstrated in vivo so far.
One of the most important mechanisms by which IgG antibodies engage the cellular immune system is via interaction of the Fc domain with Fcγ receptors (FcγRs) (Nimmerjahn and Ravetch, 2007). The human FcγR family contains six known members in three subgroups, including FcγRI (CD64), FcγRIIa,b,c (CD32a,b,c) and FcγRIIIa,b (CD16a,b), expressed by various effector cells of the immune system, including macrophages, neutrophils, dendritic cells and natural killer (NK) cells. The latter cell type is the main agent of antibody-dependent, cell-mediated cytotoxicity (ADCC). These cells can be recruited and activated through the interaction between FcγRIIIa and the Fc region, leading to the formation of an immunological synapse, the release of perforin/granzyme and the establishment of the Fas/FasL interaction, both leading to apoptosis of the target cells. The other cell types mainly lead to the phagocytosis of target cells.
Several pieces of evidence demonstrate that ADCC plays a major role in the in vivo efficacy of mAbs. Several investigators have examined the relationship between FcγR polymorphisms and clinical responses to antibody therapy (Cartron et al., 2002; Weng and Levy, 2003). Around 20% of the White population possess a valine in position 158 of FcγRIIIa, while the rest of the population has a phenylalanine. In vitro, a fivefold higher affinity between FcγRIIIa-V158 and IgG1 Fc compared with FcγRIII-F158 has been reported, leading to a more efficient in vivo ADCC by using peripheral blood mononuclear cells (PBMCs) or purified NKs. Interestingly, several studies demonstrated that Rituximab-treated follicular lymphoma patients who were homozygous for the higher affinity form of FcγRIIIa (V/V158) had significantly prolonged periods of progression-free survival relative to patients heterozygous or homozygous for the lower affinity F158 form. This finding is probably the strongest available demonstration of a major role of ADCC in patients.
Unfortunately the triggering of ADCC by therapeutic antibodies faces several limitations. First of all, as described above, the affinity between the Fc and its receptors plays a crucial role, and the fact that 80% of the population expresses a low affinity variant of the receptor is a major issue. Second, IgG1 molecules are glycosylated in the CH2 domain (Asn 297) of the Fc region. This modification is extremely important as it modulates the affinity of the Fc for FcγRIIIa, thereby modifying the in vivo efficacy of antibodies. More specifically, the presence of fucose residues in the carbohydrate has been shown to decrease ADCC efficiency (Shinkawa et al., 2003). The nature of the carbohydrate moiety is dependent on which enzymes are expressed by the cell line used for antibody production (Siberil et al., 2006). For example, an anti-CD20 chimeric IgG1 produced by the rat hybridoma YB2/0 cell line showed more than 50-fold higher ADCC using purified human PBMCs as effector than the same antibody produced by the Chinese hamster ovary (CHO) cell line, the cell line traditionally used for the production of therapeutic proteins. This difference was attributed to the elevated expression of FUT8, the gene coding for α1,6-fucosyltransferase, in the CHO line (Shinkawa et al., 2003).
The affinity between the antibody and its antigen is not the only issue. A third limitation lies in the fact that therapeutic antibodies have to compete with a high concentration of patient's IgGs for binding to FcγRIIIa. Indeed, the serum concentration of IgG is 8–17 mg·mL−1, and around 66% of those molecules are IgG1 capable of interacting with FcγRIIIa. Strikingly, most antibodies, in order to show a therapeutic effect, have to be injected at high doses to reach a serum concentration between 10 and 100 µg·mL−1, whereas these antibodies lead to a saturating ADCC at 10 ng·mL−1in vitro, that is, in the absence of competing IgGs. Competition with patients' IgGs has been proposed to account for this huge concentration difference (Preithner et al., 2006).
Finally, a fourth limitation of the use of therapeutic antibodies may be their affinity for inhibitory Fc receptors such as FcγRIIb, expressed by B-cells, macrophages, dendritic cells and neutrophils (Nimmerjahn and Ravetch, 2007). Unlike activating receptors possessing a cytoplasmic immunoreceptor tyrosine-based activation motif, which is either encoded directly (FcγRIIa,c) or gained by association with a common immunoreceptor tyrosine-based activation motif γ-chain (FcγRI and FcγRIIIa), FcγRIIb possesses an inhibitory motif in its cytoplasmic domain, and signalling through this receptor negatively regulates effector functions. This signalling leads to a balance between activating and inhibitory signals, which is used by the immune system to control the immune reaction. Like other IgG1, therapeutic antibodies do interact with this receptor, which decreases their overall efficiency.
New avenues
Antibody engineering has played a major role in the development of the first generation of therapeutic antibodies. It is now being used in several ways to obtain a new generation of optimized antibodies with a modified Fc region capable of circumventing some of the limitations described above. However, the potential offered by antibody engineering can go further than optimization and is a way to create entirely new Ig domain-based molecules, not found in nature, which can be tailored to match desired characteristics.
Fc engineering and glycoengineering of therapeutic mAbs
Because mAbs depend on their Fc region for eliciting certain immune reactions, a way to improve their action is to engineer this portion of the antibody. For instance, for most therapeutic applications, a long serum half-life is desirable as it would decrease the need for repetitive injections of the molecule to achieve a therapeutically relevant serum concentration. Accordingly, several groups are attempting to use mutagenesis to increase the affinity of the IgG Fc portion for FcRn at acidic pH (in the endosome) without raising the affinity of the interaction at physiological pH (to allow an efficient release of the antibody in the circulation). On the other hand, some applications such as imaging necessitate very high contrasts, which depend on rapid clearance of the excess of unbound molecules. Some studies have demonstrated the possibility to engineer the Fc portion in order to decrease its affinity for FcRn, leading to shorter serum half-lives and thus better contrast (Vaccaro et al., 2005). Worthy of note, the IgG3 isotype is not efficiently bound by FcRn and consequently, its serum half-life is naturally short (1 week instead of 3 for IgG1) (Table 2).
More importantly, the affinity of the Fc region for various Fc receptors also plays a major role in the effectiveness of therapies. A way to increase the efficiency of therapeutic antibodies would be to increase their binding to activating receptors, namely FcγRI, FcγRIIa and more importantly FcγRIIIa, and decrease their interaction with inhibitory FcγRIIb receptors (Shields et al., 2001; Lazar et al., 2006; Desjarlais et al., 2007).
As NK cells are known to be responsible for most if not all tumour cell lysis observed when PBMCs are used as effector cells and as this cell type typically expresses a unique activating receptor, FcγRIIIa, most mutagenesis studies have tried to increase the affinity of the Fc region for this receptor. Several approaches have been used to reach this goal, including alanine scanning, site-directed mutagenesis, computational structure-based design and selection-based methods. Impressive results have been achieved, with variants possessing up to 100-fold greater affinity for FcγRIIIa, resulting in 100-fold enhanced in vitro ADCC (Siberil et al., 2006; Desjarlais et al., 2007).
The second receptor of importance for therapy is FcγRIIa. This receptor is important for the function of neutrophils, monocytes, macrophages and dendritic cells, although macrophages and dendritic cells also express FcγRIIIa. Macrophages and monocytes can phagocytose opsonized target cells through engagement of FcγRs. They can also induce apoptosis of target cells through the release of reactive nitrogen and oxygen intermediates, or lyse them through the release of cytolytic granules. Perhaps more importantly, macrophages and dendritic cells, being professional antigen presenting cells, facilitate a potentially more robust anti-tumour effect known as cross-priming, by which these cells process and present tumour-derived antigens on their surface class I MHC molecules, thus acquiring the ability to activate T-cells. Cross-priming can activate cytotoxic T lymphocytes that recognize MHC/tumour antigen complexes, ultimately leading to T-cell attack on the tumour cells (Dhodapkar et al., 2002). Remarkably, this process has the potential to lead to a long-lasting adaptive immune response that could protect patients from relapses. Increasing the affinity of human Fc for FcγRIIa is therefore of considerable interest. Unfortunately, the same cells also express the inhibitory FcγRIIb receptor, whose extracellular domain shares 93% of homology with that of FcγRIIa, and the task of selectively increasing the Fc affinity for the activating receptor remains a challenge that has not yet been solved (Armour et al., 2003).
It must be stressed that the clinical relevance of FcγRIIa activation is still not clear, the most compelling piece of evidence being the correlation between the FcγRIIa (R/R131) genotype and progression-free survival in an anti-GD2 antibody and granulocyte-macrophage colony-stimulating factor co-therapy of neuroblastoma using a murine IgG3 (Cheung et al., 2006). Most clinical trials use human IgG1 molecules, which bind the two alleles (H131 and R131) with a similar affinity and thus cannot shed any light on the clinical relevance of FcγRIIa polymorphism. By contrast human IgG2 binds with a 10-fold higher affinity to the H131 allele. Polymorphism studies with panitumumab, an IgG2 anti-EGFR antibody for treatment of cancer of the colon, would therefore be of considerable interest.
The high affinity of the third main receptor, FcγRI, for monomeric IgG (KD = 10−9 mol·L−1) probably precludes the capacity to distinguish between unbound antibody and immune complexes, suggesting that this receptor is not a key player in anti-tumour antibody activity; no engineering studies have been done on this receptor.
Mutagenesis of the Fc region is not the only way to improve its affinity for Fc receptors. As mentioned earlier, the nature of carbohydrates linked to Asn297 of the CH2 domain has a major influence on the affinity of the Fc for FcγRs. Several studies demonstrated that the presence of fucose residues can lead to severely reduced ADCC efficiency. Several academic groups and pharmaceutical companies are presently focusing on the development of new cell lines capable of producing defucosylated mAbs, such as CHO cell lines deleted of the FUT8 gene coding for the enzyme α-1,6-fucosyltransferase, or over-expressing a recombinant β-1,4-N-acetylglucosaminyltransferase III leading to antibodies enriched in bisected and non-fucosylated oligosaccharides (Yamane-Ohnuki et al., 2004). The same kind of studies have also been conducted on non-mammalian expression systems such as yeast, plant and moss (Cox et al., 2006; Li et al., 2006; Nechansky et al., 2007).
Although the effects of such modifications on FcγR interaction need further characterization, it is assumed that defucosylated antibodies have increased affinity for FcγRIIIa/b and the same affinity for other receptors, including FcγRIIb. The clinical impact of these modifications remains to be seen.
Antibody fragments
Antibody engineering has been used to chimerize or humanize mAbs, and more recently to optimize the Fc portion of mAbs. However, many studies have demonstrated that it is possible to produce various antibody fragments that retain the binding activity of the full-length molecule (Figure 2) and to use these new formats in certain specific applications (Holliger and Hudson, 2005). As mentioned earlier, the large size of antibodies limits tumour penetration, and their long serum half-life is not suitable for applications such as radioimmunotherapy or imaging as it would lead to irradiation of healthy tissues and high background respectively. Antibody fragments such as Fab fragments, although they lack an effector function, can be an attractive alternative as they are monovalent and are rapidly eliminated by renal clearance. To compensate for these shortcomings, several groups have proposed new engineered antibody formats (Figure 2).
Figure 2.
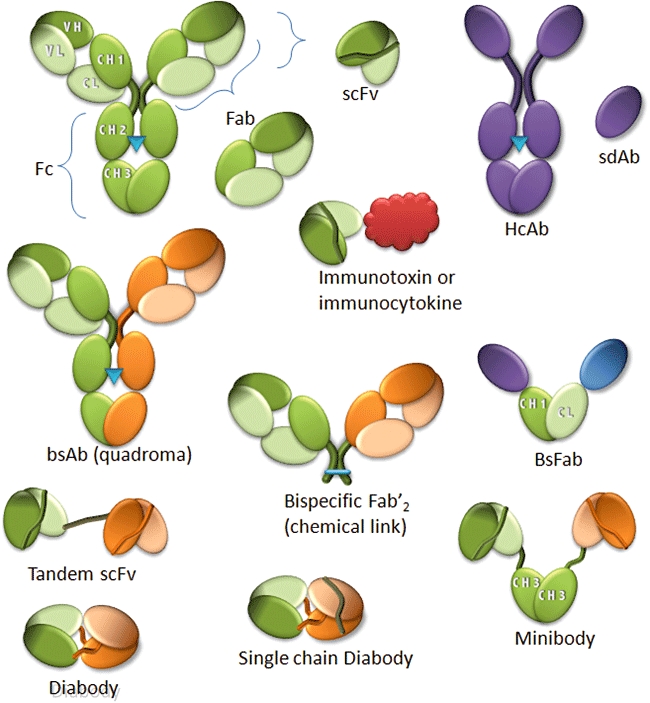
Antibody fragments with therapeutic potential. A conventional antibody is depicted in green (light for light chain, dark for heavy chain, blue triangle indicate the glycosylation site) and the derived fragments (shaded areas represent the binding sites). The orange colour symbolizes a different specificity. HcAb from camelids and their fragments (sdAb for single-domain antibodies) are depicted in mauve or blue. The red molecule represents a cytokine or a toxin. bsAb, bispecific antibodies; bsFab, bispecific Fab fragment; HcAb, heavy chain only antibodies.
scFv fragments (variable domains of the heavy and light chains linked by a flexible linker) were described very early as small fragments capable of retaining the binding activity of the full IgG molecule, albeit in a monovalent fashion (Bird et al., 1988). Because of its very short half-life in serum (∼2 h), this fragment cannot be used as such, although it has been extensively used as a binding moiety when incorporated in more complex molecules (see next section). However, the team of Greg Winter discovered that decreasing the length of the linker between the two domains induced the formation of a dimer, named diabody (Holliger et al., 1993) (Figure 2). Diabodies are compact, medium-size (60 kDa) molecules and can be an interesting choice for imaging purposes or radioimmunotherapy. Besides increasing the molecular weight, the dimerization provides bivalency, which leads to a higher avidity and higher tumour retention. Thus, diabodies provide rapid tissue penetration, high target retention and rapid blood clearance. Because they are rapidly eliminated through the kidneys, they limit the exposure to the bone marrow, which is most often the dose-limiting organ with intact radiolabelled mAbs. Diabodies possess an excellent combination of rapid tumour uptake and clearance for in vivo imaging when labelled with 123I or 111In, or for rapid xenograft visualization by PET when labelled with positron emitters such as 64Cu or 124I (Robinson et al., 2008). Diabodies therefore have a high potential for radiotherapy. Indeed, a single intravenous dose of 90Y-labelled diabody inhibited growth rates of established HER2 tumour xenografts in athymic nude mice (Adams et al., 2004), and even more promising results have been reported with an anti-HER2 diabody coupled to the α-emitting radioisotope 211At, with 60% tumour-free animals 1 year after a single injection of the conjugate in immunodeficient nude mice bearing established HER2/neu-positive tumours (Robinson et al., 2008).
More recently, several teams have shown that it is possible to obtain high affinities using a single variable domain. In 1989, the group of Greg Winter demonstrated that mouse variable domains could be used as binding units (Ward et al., 1989), but this was not further developed because the vast majority of these domains aggregate spontaneously. However, it was later found that camelids and sharks express a type of antibodies devoid of light chains (Figure 2), called heavy chain antibodies (HcAbs) (Hamers-Casterman et al., 1993) and new antigen receptor antibodies (IgNAR) (Dooley et al., 2003) respectively. These antibodies have a single variable domain (called VHH for camelids and V-NAR for sharks), which generates high affinities towards a large spectrum of antigens. These small domains (13 kDa) can be easily produced in bacteria or yeast and are then called domain antibodies (dAbs), single-domain antibodies (sdAbs) or nanobodies. Interestingly, these sdAbs are naturally endowed with very useful characteristics (Holliger and Hudson, 2005), for instance a very high stability, even when the intradomain disulphide bond characteristic of Ig domains is not present, the possibility of binding strongly to epitopes not accessible to conventional antibodies such as enzyme-active sites and a high sequence homology with the human VHIII gene family in the case of VHH. Since then, several studies have demonstrated the possibility to develop human variable domains into stable dAbs (Holliger and Hudson, 2005). These fragments combine the high affinity and specificity of antibodies with the stability and ease of production of small molecules and have the potential to be administered by means other than injection. Several dAbs are being tested in phase I and phase II clinical trials at the moment, including two anti-TNFα against rheumatoid arthritis and Crohn's disease, and an anti-von Willebrand factor used as an anti-thrombotic. Furthermore, dAbs can be very efficiently engineered as targeting moiety of more complex molecular constructions (see next section).
Because of their reduced size, antibody fragments usually penetrate tumours much more rapidly and efficiently than full IgG, but this benefit is counterbalanced by a very short serum half-life that decreases the overall tumour uptake of these small molecules (Table 2). Several academic groups and companies are investigating alternative approaches to increase the serum half-life of antibody fragments, the most promising one being the chemical addition of polyethylene glycol (PEG) residues, which considerably increase the size of the fragments. An example is the certolizumab pegol (Blick and Curran, 2007), a recently approved anti-TNFα PEGylated Fab fragment that has a 14 day serum half-life. PEG linkage (PEGylation) is very efficient for increasing the half-life and scFv stability, conferring improved anti-tumour activity and apparently also reducing immunogenicity (Holliger and Hudson, 2005). Improved circulation time and accumulation in tumours has been demonstrated with PEGylated scFv fragments, tandem scFv (two scFvs linked with a flexible linker) and diabodies. However, these chemical modifications sometimes lead to partial inactivation or decreased affinity of the fragment, and several alternatives are currently being explored. For example, fusion of recombinant antibody fragments to human serum albumin (HSA) can be used to increase the serum half-life without affecting the binding and activity of the fragments, unlike PEGylation. HSA is not eliminated by the kidneys and is actively recycled by its interaction with neonatal receptor FcRn. Fusion proteins containing HSA have been obtained with scFv, tandem scFv and diabodies and showed a large increase in circulation time (Muller et al., 2007).
Interestingly, similar results were obtained by fusing antibody fragments to HSA-binding peptides or proteins. The half-life of an anti-HER2 Fab fragment (derived from trastuzumab) could be increased 10–15-fold by fusing it to short HSA binding peptides selected by phage display. The resulting Fab fragments outperformed the parental IgG in vivo, in terms of efficacy, targeting, tumour accumulation and retention and tumour to blood ratio (Nguyen et al., 2006). Similarly, the group of Ian Tomlinson isolated human dAbs of 12 kDa directed against albumin (albudAbs) (Holt et al., 2008). The serum half-life of these fragments was extended from 45 min to the half-life of their targets (53 h for rats, 35 h in mice), and this extended half-life was conferred to a fused partner such as IL-1. The group of Roland Kontermann improved the circulation time of a single-chain diabody by a factor of 6 by fusing it to an albumin-binding domain from streptococcal protein G (Stork et al., 2007). The same laboratory also fused a tag displaying several N-glycosylation motifs to a single-chain diabody and produced glycosylated diabodies after expression in transfected HEK293 cells (Stork et al., 2008). The half-life of these molecules was increased by a factor of 2–3, albeit at the cost of reduced activity of the fragment in a target cell-dependent IL-2 release assay.
More generally, multimerization has been proposed in order to obtain a good compromise between serum half-life, tumour penetration and multivalency. In addition to diabodies, triabodies or tetrabodies have also been produced by multimerization of scFvs harbouring a short or no linker, leading to high molecular weight and multivalent fragments with increased serum half-lives (Holliger and Hudson, 2005).
Fusions with effector proteins
As they are small and easy to produce in E. coli or in yeast, antibody fragments have also been used as the binding moiety in newly created molecules endowed with new functions. Various proteins have been fused to antibody fragments (Figure 2). For instance, interesting results have been obtained with immunotoxins, consisting of a toxin, such as a fragment of Pseudomonas exotoxin (PE38) without its cell-binding domain, diphtheria toxin or the A chain of ricin, fused to a scFv (Pastan et al., 2007). However, such molecules are immunogenic and are rapidly neutralized by the immune system of recipients. More potent immunotoxins have been created by fusing a human RNase to a human scFv targeting a tumour antigen such as HER2 receptor, leading to a dramatic reduction in tumour volume in animal models (De Lorenzo et al., 2004). Several immunotoxins are being tested in clinical trials at the moment (Pastan et al., 2007).
Antibody-directed enzyme prodrug therapy uses a fusion between a scFv and an enzyme to convert a relatively non-toxic prodrug into a potent cytotoxic drug in the vicinity of the targeted cells, thereby avoiding the effects of the drug on healthy cells. Examples of such molecules include a fusion between a scFv with specificity for TAG-72, a carbohydrate epitope that is over-expressed and exposed on the cell surface in a large fraction of solid malignancies and α-lactamase. This fusion can convert C-Mel, a nitrogen mustard prodrug, into melphalan, a very potent drug (Alderson et al., 2006).
Antibody fragments were also fused to cytokines (immunocytokines). These molecules have the potential to activate the immune system of patients in the vicinity of the tumour, thereby avoiding important side effects traditionally associated with systemic administration of activating cytokines. Tumour-specific scFvs have been fused to various cytokines, including IL-2, IL-15, granulocyte-macrophage colony-stimulating factor, interferon-γ, leading to impressive results (Ebbinghaus et al., 2005; Gillies et al., 2005; Kaspar et al., 2007), and several of these constructs are now tested in the clinic.
Finally, scFv can also be fused to membrane proteins, to create chimeric receptors. ‘T-bodies’ are genetically retargeted T-cells armed with chimeric receptors whose extracellular recognition unit is an antibody fragment such as a scFv and whose intracellular region is derived from lymphocyte stimulating moieties, including CD3ζ and FcεRI-γ (Willemsen et al., 2003). These chimeric receptor constructs are introduced ex vivo into T-cells from peripheral lymphocytes of a given patient by using retroviral vectors. The retargeted T-cells are then infused back into the patient. This approach has been used with success against several targets, including tumours, HIV-infected cells and autoimmune effector cells (Eshhar, 2008). Interestingly, this approach is not HLA-restricted and can thus be used for a wide spectrum of patients and tumours, and thus holds promise for future immunotherapies.
Bispecific antibodies (bsAb)
The idea to create antibodies capable of strong and specific binding with two different antigens is as old as mAbs, and the potential of such molecules has been clearly demonstrated. Developing such molecules was difficult before the development of antibody engineering and cloning of antibody fragments. Since then, several recombinant bsAb fragments have been developed (Figure 2). The small size of these fragments allows efficient tumour penetration, at the expense of a short serum half-life, and several of these molecules are currently being tested in clinical trials (Fischer and Leger, 2007).
The most studied recombinant formats include tandem scFv, minibodies (using ‘knob into hole’ CH3 domains engineered to decrease homodimerization) or bispecific diabodies (Figure 2). The very compact structure of diabodies gives them attractive properties such as good tumour penetration, expression and solubility as well as enhanced stability. Although several preclinical studies have shown the efficiency of these molecules, no clinical trials have yet been reported (Fischer and Leger, 2007). Another format is the single-chain diabody where the two chains of the fragment are linked via an additional linker, thereby combining all domains in a single polypeptide (Figure 2). Several preclinical studies have demonstrated the potency of this format (Muller and Kontermann, 2007). All the described recombinant bsAbs rely on the use of flexible peptide linkers and although these linkers have obvious advantages in terms of antibody engineering, they also have some disadvantages due to their foreign nature, which leads to an unwanted immune response. Furthermore, their flexible nature makes linkers prone to proteolytic cleavage in serum, potentially leading to production issues, poor antibody stability, aggregation and increased immunogenicity (Fischer and Leger, 2007). dAbs can overcome these limitations as they do not require the use of two separate variable domains. Our laboratory has recently developed a format obtained by the direct fusion of two different llama dAbs to human CH1 and Cκ constant domains (Figure 2). The resulting linker-free molecule closely resembles a human Fab fragment but is capable of simultaneously binding to two different antigens, such as a tumour marker [e.g. carcinoembryonic antigen, (Baty et al., 2006)] and an activating receptor [e.g. CD16a (Behar et al., 2008)]. This new format and a bivalent bispecific derivative are currently being evaluated as a NK cell retargeting agent (see below).
Bispecific antibodies have been used in various ways, the most exciting application being the possibility to simultaneously retarget effector cells of the immune system and stimulate them through the interaction with an activating receptor in order to achieve an efficient lysis of tumour cells. Effector cells include T-cells, activated via CD3, or NK cells, neutrophils and macrophages activated via FcγRIIIa,b and FcγRIIa (Fischer and Leger, 2007; Muller and Kontermann, 2007). T-cells have several advantages as they represent the majority of lymphocytes. They are also known to be very motile, and infiltration of T-cells inside tumours has been frequently described. The major drawback of T-cell-based therapy is the requirement for co-signalling through interaction between CD28 and ligands such as B7 for full activation. Consequently, most studies are using either pre-activated T-cells in ex vivo settings, or a combination therapy using a bsAb together with an anti-CD28 mAb. The only exception to this rule is the bispecific T-cell engager format from Micromet AG (Munich, Germany) (Wolf et al., 2005). These molecules are made by the fusion of an anti-CD3 scFv with a tumour-specific scFv such as an anti-CD19 via a short peptide linker (five amino acids). They are capable of retargeting patients T-cells and lead to potent lysis in the absence of co-stimulation. Although not clearly demonstrated, the short distance between the two binding sites of these bispecific molecules might lead to a very efficient establishment of lytic synapses, bypassing the need for CD28 activation (Wolf et al., 2005). One of these molecules, MT103 is giving very encouraging results in clinical trials, including complete remissions, for the treatment of non-Hodgkin's lymphoma. Another bispecific T-cell engager format, MT110 (anti-CD3 x anti-EpCAM) is also being tested at the moment against colorectal cancers (Amann et al., 2008).
Unlike T-cells, NK cells do not need pre-activation and constitutively exhibit cytolytic functions. Although activation of NK cells normally relies on equilibrium between activating and inhibitory signals, the stimulation of FcγRIIIa by itself is sufficient to trigger NK cell activation and cytolysis of the target cells. Moreover, NK cells lead to a potent cytolysis of tumour cells down-regulating their MHC class I molecules. However, these cells represent less than 10% of lymphocytes, and very small numbers of NK cells are found in direct contact with tumour cells, suggesting that NK cells poorly infiltrate tumours (Muller and Kontermann, 2007). bsAb-dependent stimulation of NK cells via FcγRIIIa mimics the natural ADCC process but does not suffer from the various shortcomings described earlier. Indeed, by choosing an anti-FcγRIIIa antibody that does not bind to the epitope used for Fc recognition, polymorphism issues and endogenous IgG competition are avoided. It is also possible to select FcγRIIIa ligands that do not bind to inhibitory Fc receptors (Behar et al., 2008). Furthermore, bsAbs do not require glycosylation. Thus, this approach has considerable potential and is being pursued by several groups (Muller and Kontermann, 2007). To our knowledge, none of these molecules are tested in clinical trials yet.
Finally, and rather unexpectedly, one of the first approaches used to create bsAbs has been applied successfully to create molecules that are performing extremely well in clinical trials. TriomAbs are produced by a quadroma cell line prepared by the fusion of two hybridoma cell lines (mouse IgG2a and rat IgG2b) (Figure 2). This specific combination confers to these antibodies properties that circumvent most of the problems that have hindered the development of this approach (Lindhofer et al., 1995). Indeed, a preferential species-restricted pairing of heavy and light chain allows the correct association of the heavy and light chain of each specificity without production of inactive heteromolecules. Moreover, the resulting chimeric Fc obtained by the interaction of one mouse and one rat heavy chain allows a one step separation of monospecific and bispecific molecules by a pH gradient during the elution step in protein A affinity purification (Lindhofer et al., 1995). Surprisingly, this chimeric Fc is capable of mediating activation of human accessory cells and NK cells and can induce the direct phagocytosis of target cells (Zeidler et al., 1999). TriomAbs can therefore be characterized as trifunctional as they combine binding activities towards target cells via one Fab, to T-cells via the second Fab and to accessory cells such as neutrophils, macrophages and NK cells via the chimeric Fc portion, leading to a very efficient T-cell-dependent lysis. Most importantly, preclinical data demonstrated the induction of long-lasting tumour immunity, considered as the Holy Grail of antibody therapy, probably due to the induction of crosstalk between the various recruited effector cells (Ruf and Lindhofer, 2001; Morecki et al., 2008). The only severe limitation faced by these molecules is their non-human nature, limiting the doses and the number of injections that can be given to patients. However, the most advanced of these molecules, Catumaxomab, an anti-EpCAM x CD3, has recently demonstrated efficacy and safety in a phase II/III clinical trial against malignant ascites, demonstrating the high potential of such molecules for cancer therapy (Shen and Zhu, 2008).
Intrabodies
An intrabody is an antibody that has been designed to be expressed intracellularly by the in-frame incorporation of intracellular, peptidic trafficking signals. This allows the antibody to enter a cellular compartment, which it would normally not enter. Intrabodies have been developed against different target antigens present in various subcellular locations such as the cytosol, nucleus, endoplasmic reticulum (ER), mitochondria, peroxisomes and the plasma membrane (Lo et al., 2008). Intrabodies interact specifically with their target antigens, and this offers the possibility of blocking or modifying specific molecular interactions leading to changes in the biological activity of the target protein. Intrabodies have been divided into two categories that differ in their pathway of biosynthesis and site of action (Stocks, 2006). One category known as retained intrabodies is engineered to be targeted to the ER by conjugating them to the KDEL retention peptide signal, which binds to a KDEL receptor and keeps the intrabodies within the lumen of the ER. As the intrabody binds to its target within the ER lumen this prevents the target molecule from leaving the ER and being expressed on the cell surface. Such ER-targeted intrabodies have been used to decrease the cell surface expression of the oncogenic vascular endothelial growth factor receptor, leading to tumour cell growth inhibition in vitro and in vivo (Boldicke, 2007). The redox conditions in the lumen of the ER favour the formation of disulphide bridges, which stabilize the structure of retained intrabodies. This is in contrast to the second category of intrabodies that are antibodies from which the leader immunoglobulin sequence has been removed and that therefore are expressed on cytoplasmic polysomes and are released into the cytoplasm of the cell. These intrabodies have to fold in the reducing environment of the cytoplasm and in the absence of chaperones and other factors that favour the formation of disulphide bridges and the native antibody structure. This means that this second category of intrabodies must be inherently self-folding and must be more stable than the average antibody framework (Stocks, 2006). Although such intrinsically stable antibody frameworks do exist within the germline repertoire, they are rare and must be specially screened for or engineered by point mutations (Visintin et al., 2002). Such intrabodies are thus more difficult to obtain as an intracellular screen is required to select for molecules that possess intracellular binding activity. However, a general approach based on the use of the two hybrid systems has recently been adapted to the intracellular selection of scFvs capable of binding their epitope in the yeast cytoplasm (Visintin et al., 2004). This technique has the advantage of selecting antibodies based on their specificity as well as their aptitude to work as intrabodies. Very recently, a derivative of this approach has been proposed to directly select intrabodies capable of interfering with a specific interaction, opening the way towards a systematic isolation of intrabodies against most interactions of a given protein network (Visintin et al., 2008).
Although most intrabodies are recombinant scFvs (Manikandan et al., 2007), sdAbs such as human VH and VL and camelid frameworks (Tanaka et al., 2003; Aires da Silva et al., 2004; Paz et al., 2005; Rothbauer et al., 2006; Tanaka et al., 2007) are being increasingly explored as intrabodies. dAbs do not require pairing of two variable domains or the use of a flexible linker, and their intrinsic stability allows them to be efficiently produced even without disulphide bond formation, which makes them excellent candidates as intrabodies. For example, an sdAb-selected against NEF from HIV is capable of blocking the CD4 down-regulation and infectivity enhancement activity of its target when expressed in the cytoplasm of the target cells (D. Baty, M. Chartier, P. Chames, S. Benichou, S. Basmaciogullari, J. Bouchet, unpubl. data).
As intrabodies act intracellularly, they have the potential of interfering with biosynthetic pathways by targeting molecules not previously accessible to antibodies. Most research on intrabodies is driven by their potential therapeutic applications in cancer, viral diseases and neurological disorders. Many preclinical studies have shown that intrabodies, directed to over-expressed cancer-related intracellular receptor domains, signal transduction molecules such as Ras or molecules such as caspases involved in apoptosis, are able to knock out the neoplastic phenotype and inhibit tumour growth (Williams and Zhu, 2006; Lo et al., 2008). Many studies have shown that intrabodies are able to down-regulate viral envelope proteins or viral non-structural proteins and may be useful for instance in the therapy of human immunodeficiency virus, hepatitis and papillomaviruses (Doorbar and Griffin, 2007). In the field of neurological disorders intrabodies have shown promising anti-aggregation and neuroprotective effects against misfolded huntingtin protein in Huntington's disease (Colby et al., 2004), against β-synuclein in Parkinson's disease (Lynch et al., 2008) and against α-amyloid precursor protein in Alzheimer's disease (Paganetti et al., 2005).
Undoubtedly, the major obstacle to obtaining intrabodies of therapeutic value remains the absence of efficient in vivo methods to deliver the genetic material encoding the intrabody to live target cells. All laboratory studies so far have used in vitro model systems where cells in culture are transfected by using expression vectors, whereas clinical applications will require efficient gene therapy protocols that are not yet available (Stocks, 2006). Currently, attempts are made to deliver intrabody genes by using recombinant adenovirus and vaccinia virus vectors or immunoliposomes (Williams and Zhu, 2006). Other approaches involve the use of peptide-based transduction protocols or cationic lipids for mediating the delivery of intrabody protein material to target cells (Matsushita et al., 2005; Courtete et al., 2007). Only if the intrabody delivery bottleneck is solved will this promising technology be able to show its full therapeutic potential.
Conclusion
The second generation of recombinant antibodies has already led to the approval of more than 20 mAbs used for therapy (Table 1), leading to valuable clinical data. Clinical results are now being used to guide antibody engineers to new approaches for making these antibodies even more efficient. At present, a third generation of antibody-derived molecules, potentially much more efficient than conventional mAbs, are being evaluated in early clinical trials. This is an exciting time that should see the emergence of ever more efficient immunotherapeutic molecules, vindicating earlier expectations that antibody engineering will deliver considerable medical benefits.
 |
Glossary
Abbreviations
- CR
complement receptor
- EGFR
epidermal growth factor receptor
- EMEA
European Medicines Agency
- FDA
US Food and Drug Administration
- HER
human epidermal growth factor receptor
- mAb
monoclonal antibody
- PBMC
peripheral blood mononuclear cell
- TNF
tumour necrosis factor
- VEGF
vascular endothelial growth factor
References
- Adams GP, Schier R, McCall AM, Simmons HH, Horak EM, Alpaugh RK, et al. High affinity restricts the localization and tumor penetration of single-chain fv antibody molecules. Cancer Res. 2001;61:4750–4755. [PubMed] [Google Scholar]
- Adams GP, Shaller CC, Dadachova E, Simmons HH, Horak EM, Tesfaye A, et al. A single treatment of yttrium-90-labeled CHX-A″-C6.5 diabody inhibits the growth of established human tumor xenografts in immunodeficient mice. Cancer Res. 2004;64:6200–6206. doi: 10.1158/0008-5472.CAN-03-2382. [DOI] [PubMed] [Google Scholar]
- Aires da Silva F, Santa-Marta M, Freitas-Vieira A, Mascarenhas P, Barahona I, Moniz-Pereira J, et al. Camelized rabbit-derived VH single-domain intrabodies against Vif strongly neutralize HIV-1 infectivity. J Mol Biol. 2004;340:525–542. doi: 10.1016/j.jmb.2004.04.062. [DOI] [PubMed] [Google Scholar]
- Alderson RF, Toki BE, Roberge M, Geng W, Basler J, Chin R, et al. Characterization of a CC49-based single-chain fragment-beta-lactamase fusion protein for antibody-directed enzyme prodrug therapy (ADEPT) Bioconjug Chem. 2006;17:410–418. doi: 10.1021/bc0503521. [DOI] [PubMed] [Google Scholar]
- Amann M, Brischwein K, Lutterbuese P, Parr L, Petersen L, Lorenczewski G, et al. Therapeutic window of MuS110, a single-chain antibody construct bispecific for murine EpCAM and murine CD3. Cancer Res. 2008;68:143–151. doi: 10.1158/0008-5472.CAN-07-2182. [DOI] [PubMed] [Google Scholar]
- Armour KL, van de Winkel JG, Williamson LM, Clark MR. Differential binding to human FcgammaRIIa and FcgammaRIIb receptors by human IgG wildtype and mutant antibodies. Mol Immunol. 2003;40:585–593. doi: 10.1016/j.molimm.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Baty D, Behar G, Chartier M, Pelegrin A, Teillaud JL, Teulon I. Production of antibody formats and immunological applications of said formats. In WO/2006/064136.
- Beckman RA, Weiner LM, Davis HM. Antibody constructs in cancer therapy: protein engineering strategies to improve exposure in solid tumors. Cancer. 2007;109:170–179. doi: 10.1002/cncr.22402. [DOI] [PubMed] [Google Scholar]
- Behar G, Siberil S, Groulet A, Chames P, Pugniere M, Boix C, et al. Isolation and characterization of anti-Fc{gamma}RIII (CD16) llama single-domain antibodies that activate natural killer cells. Protein Eng Des Sel. 2008;21:1–10. doi: 10.1093/protein/gzm064. [DOI] [PubMed] [Google Scholar]
- Bird RE, Hardman KD, Jacobson JW, Johnson S, Kaufman BM, Lee SM, et al. Single-chain antigen-binding proteins [published erratum appears in Science 1989 Apr 28; 244 (4903): 409. Science. 1988;242:423–426. doi: 10.1126/science.3140379. [DOI] [PubMed] [Google Scholar]
- Blick SK, Curran MP. Certolizumab pegol: in Crohn's disease. BioDrugs. 2007;21:195–201. doi: 10.2165/00063030-200721030-00006. discussion 202–3. [DOI] [PubMed] [Google Scholar]
- Boldicke T. Blocking translocation of cell surface molecules from the ER to the cell surface by intracellular antibodies targeted to the ER. J Cell Mol Med. 2007;11:54–70. doi: 10.1111/j.1582-4934.2007.00002.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrebaeck CA, Carlsson R. Human therapeutic antibodies. Curr Opin Pharmacol. 2001;1:404–408. doi: 10.1016/s1471-4892(01)00070-4. [DOI] [PubMed] [Google Scholar]
- Cartron G, Dacheux L, Salles G, Solal-Celigny P, Bardos P, Colombat P, et al. Therapeutic activity of humanized anti-CD20 monoclonal antibody and polymorphism in IgG Fc receptor FcgammaRIIIa gene. Blood. 2002;99:754–758. doi: 10.1182/blood.v99.3.754. [DOI] [PubMed] [Google Scholar]
- Chames P, Baty D. Antibody engineering and its applications in tumor targeting and intracellular immunization. FEMS Microbiol Lett. 2000;189:1–8. doi: 10.1111/j.1574-6968.2000.tb09197.x. [DOI] [PubMed] [Google Scholar]
- Cheung NK, Sowers R, Vickers AJ, Cheung IY, Kushner BH, Gorlick R. FCGR2A polymorphism is correlated with clinical outcome after immunotherapy of neuroblastoma with anti-GD2 antibody and granulocyte macrophage colony-stimulating factor. J Clin Oncol. 2006;24:2885–2890. doi: 10.1200/JCO.2005.04.6011. [DOI] [PubMed] [Google Scholar]
- Colby DW, Chu Y, Cassady JP, Duennwald M, Zazulak H, Webster JM, et al. Potent inhibition of huntingtin aggregation and cytotoxicity by a disulfide bond-free single-domain intracellular antibody. Proc Natl Acad Sci USA. 2004;101:17616–17621. doi: 10.1073/pnas.0408134101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtete J, Sibler AP, Zeder-Lutz G, Dalkara D, Oulad-Abdelghani M, Zuber G, et al. Suppression of cervical carcinoma cell growth by intracytoplasmic codelivery of anti-oncoprotein E6 antibody and small interfering RNA. Mol Cancer Ther. 2007;6:1728–1735. doi: 10.1158/1535-7163.MCT-06-0808. [DOI] [PubMed] [Google Scholar]
- Cox KM, Sterling JD, Regan JT, Gasdaska JR, Frantz KK, Peele CG, et al. Glycan optimization of a human monoclonal antibody in the aquatic plant Lemna minor. Nat Biotechnol. 2006;24:1591–1597. doi: 10.1038/nbt1260. [DOI] [PubMed] [Google Scholar]
- De Lorenzo C, Arciello A, Cozzolino R, Palmer DB, Laccetti P, Piccoli R, et al. A fully human antitumor immunoRNase selective for ErbB-2-positive carcinomas. Cancer Res. 2004;64:4870–4874. doi: 10.1158/0008-5472.CAN-03-3717. [DOI] [PubMed] [Google Scholar]
- Desjarlais JR, Lazar GA, Zhukovsky EA, Chu SY. Optimizing engagement of the immune system by anti-tumor antibodies: an engineer's perspective. Drug Discov Today. 2007;12:898–910. doi: 10.1016/j.drudis.2007.08.009. [DOI] [PubMed] [Google Scholar]
- Dhodapkar KM, Krasovsky J, Williamson B, Dhodapkar MV. Antitumor monoclonal antibodies enhance cross-presentation ofcCellular antigens and the generation of myeloma-specific killer T cells by dendritic cells. J Exp Med. 2002;195:125–133. doi: 10.1084/jem.20011097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley H, Flajnik MF, Porter AJ. Selection and characterization of naturally occurring single-domain (IgNAR) antibody fragments from immunized sharks by phage display. Mol Immunol. 2003;40:25–33. doi: 10.1016/s0161-5890(03)00084-1. [DOI] [PubMed] [Google Scholar]
- Doorbar J, Griffin H. Intrabody strategies for the treatment of human papillomavirus-associated disease. Expert Opin Biol Ther. 2007;7:677–689. doi: 10.1517/14712598.7.5.677. [DOI] [PubMed] [Google Scholar]
- Ebbinghaus C, Ronca R, Kaspar M, Grabulovski D, Berndt A, Kosmehl H, et al. Engineered vascular-targeting antibody-interferon-gamma fusion protein for cancer therapy. Int J Cancer. 2005;116:304–313. doi: 10.1002/ijc.20952. [DOI] [PubMed] [Google Scholar]
- Eshhar Z. The T-body approach: redirecting T cells with antibody specificity. Handb Exp Pharmacol. 2008:329–342. doi: 10.1007/978-3-540-73259-4_14. [DOI] [PubMed] [Google Scholar]
- Fischer N, Leger O. Bispecific antibodies: molecules that enable novel therapeutic strategies. Pathobiology. 2007;74:3–14. doi: 10.1159/000101046. [DOI] [PubMed] [Google Scholar]
- Fujimori K, Covell DG, Fletcher JE, Weinstein JN. A modeling analysis of monoclonal antibody percolation through tumors: a binding-site barrier. J Nucl Med. 1990;31:1191–1198. [PubMed] [Google Scholar]
- Gillies SD, Lan Y, Williams S, Carr F, Forman S, Raubitschek A, et al. An anti-CD20-IL-2 immunocytokine is highly efficacious in a SCID mouse model of established human B lymphoma. Blood. 2005;105:3972–3978. doi: 10.1182/blood-2004-09-3533. [DOI] [PubMed] [Google Scholar]
- Giritch A, Marillonnet S, Engler C, van Eldik G, Botterman J, Klimyuk V, et al. Rapid high-yield expression of full-size IgG antibodies in plants coinfected with noncompeting viral vectors. Proc Natl Acad Sci USA. 2006;103:14701–14706. doi: 10.1073/pnas.0606631103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graumann K, Premstaller A. Manufacturing of recombinant therapeutic proteins in microbial systems. Biotechnol J. 2006;1:164–186. doi: 10.1002/biot.200500051. [DOI] [PubMed] [Google Scholar]
- Hamers-Casterman C, Atarhouch T, Muyldermans S, Robinson G, Hamers C, Songa EB, et al. Naturally occurring antibodies devoid of light chains. Nature. 1993;363:446–448. doi: 10.1038/363446a0. [DOI] [PubMed] [Google Scholar]
- Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol. 2005;23:1126–1136. doi: 10.1038/nbt1142. [DOI] [PubMed] [Google Scholar]
- Holliger P, Prospero T, Winter G. Diabodies’: small bivalent and bispecific antibody fragments. Proc Natl Acad Sci USA. 1993;90:6444–6448. doi: 10.1073/pnas.90.14.6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt LJ, Basran A, Jones K, Chorlton J, Jespers LS, Brewis ND, et al. Anti-serum albumin domain antibodies for extending the half-lives of short lived drugs. Protein Eng Des Sel. 2008;21:283–288. doi: 10.1093/protein/gzm067. [DOI] [PubMed] [Google Scholar]
- Hoogenboom HR, Chames P. Natural and designer binding sites made by phage display technology. Immunol Today. 2000;21:371–378. doi: 10.1016/s0167-5699(00)01667-4. [DOI] [PubMed] [Google Scholar]
- Jones PT, Dear PH, Foote J, Neuberger MS, Winter G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature. 1986;321:522–525. doi: 10.1038/321522a0. [DOI] [PubMed] [Google Scholar]
- Kaspar M, Trachsel E, Neri D. The antibody-mediated targeted delivery of interleukin-15 and GM-CSF to the tumor neovasculature inhibits tumor growth and metastasis. Cancer Res. 2007;67:4940–4948. doi: 10.1158/0008-5472.CAN-07-0283. [DOI] [PubMed] [Google Scholar]
- Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci USA. 2006;103:4005–4010. doi: 10.1073/pnas.0508123103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Sethuraman N, Stadheim TA, Zha D, Prinz B, Ballew N, et al. Optimization of humanized IgGs in glycoengineered Pichia pastoris. Nat Biotechnol. 2006;24:210–215. doi: 10.1038/nbt1178. [DOI] [PubMed] [Google Scholar]
- Lindhofer H, Mocikat R, Steipe B, Thierfelder S. Preferential species-restricted heavy/light chain pairing in rat/mouse quadromas. Implications for a single-step purification of bispecific antibodies. J Immunol. 1995;155:219–225. [PubMed] [Google Scholar]
- Lo AS, Zhu Q, Marasco WA. Intracellular antibodies (intrabodies) and their therapeutic potential. In: Chernajovsky Y, Nissim A, editors. Therapeutic Antibodies. Handbook of Experimental Pharmacology, Volume 181. Berlin Heidelberg: Springer-Verlag; 2008. pp. 343–373. Eds. [DOI] [PubMed] [Google Scholar]
- Lonberg N. Human monoclonal antibodies from transgenic mice. In: Chernajovsky Y, Nissim A, editors. Therapeutic Antibodies. Handbook of Experimental Pharmacology, Volume 181. Berlin Heidelberg: Springer-Verlag; 2008. pp. 69–97. Eds. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luginbuhl B, Kanyo Z, Jones RM, Fletterick RJ, Prusiner SB, Cohen FE, et al. Directed evolution of an anti-prion protein scFv fragment to an affinity of 1 pM and its structural interpretation. J Mol Biol. 2006;363:75–97. doi: 10.1016/j.jmb.2006.07.027. [DOI] [PubMed] [Google Scholar]
- Lynch SM, Zhou C, Messer A. An scFv intrabody against the nonamyloid component of alpha-synuclein reduces intracellular aggregation and toxicity. J Mol Biol. 2008;377:136–147. doi: 10.1016/j.jmb.2007.11.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCafferty J, Griffiths AD, Winter G, Chiswell DJ. Phage antibodies: filamentous phage displaying antibody variable domains. Nature. 1990;348:552–554. doi: 10.1038/348552a0. [DOI] [PubMed] [Google Scholar]
- Manikandan J, Pushparaj PN, Melendez AJ. Protein i: interference at protein level by intrabodies. Front Biosci. 2007;12:1344–1352. doi: 10.2741/2152. [DOI] [PubMed] [Google Scholar]
- Matsushita M, Matsui H. Protein transduction technology. J Mol Med. 2005;83:324–328. doi: 10.1007/s00109-004-0633-1. [DOI] [PubMed] [Google Scholar]
- Morecki S, Lindhofer H, Yacovlev E, Gelfand Y, Ruf P, Slavin S. Induction of long-lasting antitumor immunity by concomitant cell therapy with allogeneic lymphocytes and trifunctional bispecific antibody. Exp Hematol. 2008;36:997–1003. doi: 10.1016/j.exphem.2008.03.005. [DOI] [PubMed] [Google Scholar]
- Muller D, Kontermann RE. Recombinant bispecific antibodies for cellular cancer immunotherapy. Curr Opin Mol Ther. 2007;9:319–326. [PubMed] [Google Scholar]
- Muller D, Karle A, Meissburger B, Hofig I, Stork R, Kontermann RE. Improved pharmacokinetics of recombinant bispecific antibody molecules by fusion to human serum albumin. J Biol Chem. 2007;282:12650–12660. doi: 10.1074/jbc.M700820200. [DOI] [PubMed] [Google Scholar]
- Nechansky A, Schuster M, Jost W, Siegl P, Wiederkum S, Gorr G, et al. Compensation of endogenous IgG mediated inhibition of antibody-dependent cellular cytotoxicity by glyco-engineering of therapeutic antibodies. Mol Immunol. 2007;44:1815–1817. doi: 10.1016/j.molimm.2006.08.013. [DOI] [PubMed] [Google Scholar]
- Neuberger MS, Williams GT, Mitchell EB, Jouhal SS, Flanagan JG, Rabbitts TH. A hapten-specific chimaeric IgE antibody with human physiological effector function. Nature. 1985;314:268–270. doi: 10.1038/314268a0. [DOI] [PubMed] [Google Scholar]
- Nguyen A, Reyes AE, 2nd, Zhang M, McDonald P, Wong WL, Damico LA, et al. The pharmacokinetics of an albumin-binding Fab (AB.Fab) can be modulated as a function of affinity for albumin. Protein Eng Des Sel. 2006;19:291–297. doi: 10.1093/protein/gzl011. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn F, Ravetch JV. Antibodies, Fc receptors and cancer. Curr Opin Immunol. 2007;19:239–245. doi: 10.1016/j.coi.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Paganetti P, Calanca V, Galli C, Stefani M, Molinari M. beta-site specific intrabodies to decrease and prevent generation of Alzheimer's Abeta peptide. J Cell Biol. 2005;168:863–868. doi: 10.1083/jcb.200410047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastan I, Hassan R, FitzGerald DJ, Kreitman RJ. Immunotoxin treatment of cancer. Annu Rev Med. 2007;58:221–237. doi: 10.1146/annurev.med.58.070605.115320. [DOI] [PubMed] [Google Scholar]
- Paz K, Brennan LA, Iacolina M, Doody J, Hadari YR, Zhu Z. Human single-domain neutralizing intrabodies directed against Etk kinase: a novel approach to impair cellular transformation. Mol Cancer Ther. 2005;4:1801–1809. doi: 10.1158/1535-7163.MCT-05-0174. [DOI] [PubMed] [Google Scholar]
- Preithner S, Elm S, Lippold S, Locher M, Wolf A, da Silva AJ, et al. High concentrations of therapeutic IgG1 antibodies are needed to compensate for inhibition of antibody-dependent cellular cytotoxicity by excess endogenous immunoglobulin G. Mol Immunol. 2006;43:1183–1193. doi: 10.1016/j.molimm.2005.07.010. [DOI] [PubMed] [Google Scholar]
- Reichert JM, Rosensweig CJ, Faden LB, Dewitz MC. Monoclonal antibody successes in the clinic. Nat Biotechnol. 2005;23:1073–1078. doi: 10.1038/nbt0905-1073. [DOI] [PubMed] [Google Scholar]
- Robinson MK, Shaller C, Garmestani K, Plascjak PS, Hodge KM, Yuan QA, et al. Effective treatment of established human breast tumor xenografts in immunodeficient mice with a single dose of the alpha-emitting radioisotope astatine-211 conjugated to anti-HER2/neu diabodies. Clin Cancer Res. 2008;14:875–882. doi: 10.1158/1078-0432.CCR-07-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7:715–725. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- Rothbauer U, Zolghadr K, Tillib S, Nowak D, Schermelleh L, Gahl A, et al. Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat Methods. 2006;3:887–889. doi: 10.1038/nmeth953. [DOI] [PubMed] [Google Scholar]
- Ruf P, Lindhofer H. Induction of a long-lasting antitumor immunity by a trifunctional bispecific antibody. Blood. 2001;98:2526–2534. doi: 10.1182/blood.v98.8.2526. [DOI] [PubMed] [Google Scholar]
- Shen J, Zhu Z. Catumaxomab, a rat/murine hybrid trifunctional bispecific monoclonal antibody for the treatment of cancer. Curr Opin Mol Ther. 2008;10:273–284. [PubMed] [Google Scholar]
- Shields RL, Namenuk AK, Hong K, Meng YG, Rae J, Briggs J, et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem. 2001;276:6591–6604. doi: 10.1074/jbc.M009483200. [DOI] [PubMed] [Google Scholar]
- Shinkawa T, Nakamura K, Yamane N, Shoji-Hosaka E, Kanda Y, Sakurada M, et al. The absence of fucose but not the presence of galactose or bisecting N-acetylglucosamine of human IgG1 complex-type oligosaccharides shows the critical role of enhancing antibody-dependent cellular cytotoxicity. J Biol Chem. 2003;278:3466–3473. doi: 10.1074/jbc.M210665200. [DOI] [PubMed] [Google Scholar]
- Siberil S, Dutertre CA, Boix C, Bonnin E, Menez R, Stura E, et al. Molecular aspects of human FcgammaR interactions with IgG: functional and therapeutic consequences. Immunol Lett. 2006;106:111–118. doi: 10.1016/j.imlet.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- Stocks M. Intracellular antibodies: a revolution waiting to happen? Curr Opin Mol Ther. 2006;8:17–23. [PubMed] [Google Scholar]
- Stork R, Muller D, Kontermann RE. A novel tri-functional antibody fusion protein with improved pharmacokinetic properties generated by fusing a bispecific single-chain diabody with an albumin-binding domain from streptococcal protein G. Protein Eng Des Sel. 2007;20:569–576. doi: 10.1093/protein/gzm061. [DOI] [PubMed] [Google Scholar]
- Stork R, Zettlitz KA, Muller D, Rether M, Hanisch FG, Kontermann RE. N-glycosylation as novel strategy to improve pharmacokinetic properties of bispecific single-chain diabodies. J Biol Chem. 2008;283:7804–7812. doi: 10.1074/jbc.M709179200. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Lobato MN, Rabbitts TH. Single domain intracellular antibodies: a minimal fragment for direct in vivo selection of antigen-specific intrabodies. J Mol Biol. 2003;331:1109–1120. doi: 10.1016/s0022-2836(03)00836-2. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Williams RL, Rabbitts TH. Tumour prevention by a single antibody domain targeting the interaction of signal transduction proteins with RAS. Embo J. 2007;26:3250–3259. doi: 10.1038/sj.emboj.7601744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccaro C, Zhou J, Ober RJ, Ward ES. Engineering the Fc region of immunoglobulin G to modulate in vivo antibody levels. Nat Biotechnol. 2005;23:1283–1288. doi: 10.1038/nbt1143. [DOI] [PubMed] [Google Scholar]
- Visintin M, Settanni G, Maritan A, Graziosi S, Marks JD, Cattaneo A. The intracellular antibody capture technology (IACT): towards a consensus sequence for intracellular antibodies. J Mol Biol. 2002;317:73–83. doi: 10.1006/jmbi.2002.5392. [DOI] [PubMed] [Google Scholar]
- Visintin M, Quondam M, Cattaneo A. The intracellular antibody capture technology: towards the high-throughput selection of functional intracellular antibodies for target validation. Methods. 2004;34:200–214. doi: 10.1016/j.ymeth.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Visintin M, Melchionna T, Cannistraci I, Cattaneo A. In vivo selection of intrabodies specifically targeting protein-protein interactions: a general platform for an ‘undruggable’ class of disease targets. J Biotechnol. 2008;135:1–15. doi: 10.1016/j.jbiotec.2008.02.012. [DOI] [PubMed] [Google Scholar]
- Ward ES, Gussow D, Griffiths AD, Jones PT, Winter G. Binding activities of a repertoire of single immunoglobulin variable domains secreted from Escherichia coli[see comments. Nature. 1989;341:544–546. doi: 10.1038/341544a0. [DOI] [PubMed] [Google Scholar]
- Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. 2003;21:3940–3947. doi: 10.1200/JCO.2003.05.013. [DOI] [PubMed] [Google Scholar]
- Willemsen RA, Debets R, Chames P, Bolhuis RL. Genetic engineering of T cell specificity for immunotherapy of cancer. Hum Immunol. 2003;64:56–68. doi: 10.1016/s0198-8859(02)00730-9. [DOI] [PubMed] [Google Scholar]
- Williams BR, Zhu Z. Intrabody-based approaches to cancer therapy: status and prospects. Curr Med Chem. 2006;13:1473–1480. doi: 10.2174/092986706776872899. [DOI] [PubMed] [Google Scholar]
- Winter G, Milstein C. Man-made antibodies. Nature. 1991;349:293–299. doi: 10.1038/349293a0. [DOI] [PubMed] [Google Scholar]
- Wolf E, Hofmeister R, Kufer P, Schlereth B, Baeuerle PA. BiTEs: bispecific antibody constructs with unique anti-tumor activity. Drug Discov Today. 2005;10:1237–1244. doi: 10.1016/S1359-6446(05)03554-3. [DOI] [PubMed] [Google Scholar]
- Yamane-Ohnuki N, Kinoshita S, Inoue-Urakubo M, Kusunoki M, Iida S, Nakano R, et al. Establishment of FUT8 knockout Chinese hamster ovary cells: an ideal host cell line for producing completely defucosylated antibodies with enhanced antibody-dependent cellular cytotoxicity. Biotechnol Bioeng. 2004;87:614–622. doi: 10.1002/bit.20151. [DOI] [PubMed] [Google Scholar]
- Zeidler R, Reisbach G, Wollenberg B, Lang S, Chaubal S, Schmitt B, et al. Simultaneous activation of T cells and accessory cells by a new class of intact bispecific antibody results in efficient tumor cell killing. J Immunol. 1999;163:1246–1252. [PubMed] [Google Scholar]