

Abstract
Background and purpose
Monocytes-macrophages play a key role in the initiation and persistence of inflammatory reactions. Consequently, these cells represent an attractive therapeutic target for switching off overwhelming inflammatory responses. Non-steroidal anti-inflammatory drugs (NSAIDs) are among the most common drugs for the symptomatic treatment of rheumatic diseases. Their effects have been explained on the basis of cyclooxygenase (COX) inhibition. However, some of the actions of these drugs are not related to inhibition of prostaglandin synthesis.
Experimental approach
We examined the effect of oxaprozin on apoptosis of immune complex-activated monocytes in comparison with drugs of the same class, and the signalling pathway that leads activated monocytes exposed to oxaprozin to apoptosis. In particular, we studied the activity of caspase-3, the involvement of IκB kinase (IKK)-nuclear factor κB (NF-κB) system and the activity of X-linked mammalian inhibitor of apoptosis protein (XIAP), Akt and mitogen-activated protein kinase (MAPK) in activated monocytes in the presence of oxaprozin.
Key results
Immune complexes caused the inhibition of monocyte apoptosis. Oxaprozin reversed in a dose-dependent manner immune complex-induced survival of monocytes, without affecting the apoptosis of resting cells. Other NSAIDs are ineffective. The activity of oxaprozin was related to inhibition of Akt activation that, in turn, prevented p38 MAPK, IKK and NF-κB activation. Consistently, the inhibition of NF-κB activation reduced the production of the anti-apoptotic molecule XIAP, leading to uncontrolled activity of caspase 3.
Conclusions and implications
These results suggest that oxaprozin exerts its anti-inflammatory activity also through COX-independent pathways. It is likely that oxaprozin-mediated inhibition of the Akt/IKK/NF-κB pathway contributes to its anti-inflammatory properties.
Keywords: apoptosis, Akt, IKK, NF-κB, NSAIDs, oxaprozin, monocytes, immune complexes
Introduction
The inflammation that characterizes rheumatic diseases is usually accompanied by swelling, stiffness and considerable pain which can limit the ability to perform daily activities, reduce overall joint functions and negatively interfere with health-related quality of life (Pollard et al., 2005; Scott et al., 2005). Understanding the molecular mechanisms that underpin the processes of inflammation and pain is the topic of considerable research, with the aim of developing more effective and selective strategies (Firestein, 2003 2006; Cook and Visvanathan, 2004). Albeit complex, there is a temporal and functional hierarchy of biological events in the inflammation process that involves cytokines and other mediators (Arend and Gabay, 2004). The cytokine cascade is initiated by tumour necrosis factor-α (TNF-α) released by local cells, which promotes further release of other pro-inflammatory molecules, such as interleukin (IL)-1, IL-6 and IL-8 (Feldmann and Maini, 2003). Depending on the type of injurious stimulus and tissue involved, the array of mediators may differ, but generally eicosanoids, and in particular prostaglandins, are involved in the process (Funk, 2001).
Monocytes and macrophages are major regulators of many aspects of the inflammatory reaction. Their activation is largely responsible for the generation of a local pro-inflammatory micro-environment (Solbach et al., 1991; Gordon, 1999; Liew and McInnes, 2002). The involvement of these cells in the pathogenesis of inflammatory diseases such as rheumatoid arthritis is suggested by much experimental evidence. For instance, macrophages are the main sources of cytokines in the rheumatoid joint (Firestein et al., 1990). Furthermore, the administration of an antagonist of monocyte chemotactic protein 1 (MCP-1), a major determinant of the influx of circulating monocytes within tissues (Lu et al., 1998), prevents the development of inflammation in models of arthritis (Gong et al., 1997). Finally, the quantity of monocytes and macrophages, but not of other cells, recruited in inflamed rheumatoid joints correlates with the subsequent development of inflammation-related local tissue destruction (Mulherin et al., 1996). At sites of inflammation, these cells undergo activation of genes coding for various pro-inflammatory mediators, including cytokines, such as TNF, IL-1, IL-6 and chemokines, such as MCP-1 and IL-8, tissue-damaging metalloproteinases and the prostaglandin-producing cyclooxygenase (COX)-2 enzyme (Gordon, 1999). Among multiple activators of monocytes and macrophages, immune complexes (IC) are of particular interest. In fact, IC have been identified in various pathogenic mechanisms involved in different inflammatory diseases including rheumatoid arthritis (Firestein, 2003). On the other hand, activation of monocytes mediated by the fragment crystallizable receptor (FcR) (Marsh et al., 1999; Ottonello et al., 2005) results in their prolonged survival as apoptosis of the cells is inhibited. Consequently, IC-activated monocytes and macrophages appear to be an attractive target for therapeutic approaches proposed to sustain the resolution of inflammatory responses in chronic inflammatory diseases such as rheumatoid arthritis.
Non-steroidal anti-inflammatory drugs (NSAIDs) are among the most widely used therapeutic drugs for the symptomatic treatment of chronic inflammatory diseases and are of particular interest for the treatment of rheumatic disease (Abramson and Weaver, 2005). Among them, oxaprozin, an achiral oxazole-propionic acid derivative (4,5-diphenyl-2-oxazolepropionic acid) (Davies, 1998), is characterized by a relatively high rate of accumulation in inflamed synovium, compared with that detectable in plasma and synovial fluids (Kurowski and Thabe, 1989; Rainsford et al., 2002). In particular, following oral administration of 1200 mg of oxaprozin for 2.5 days to patients with rheumatoid arthritis, the drug concentrations in synovial tissue averaged 25 µg·mL−1 (≅85.2 µmol·L−1), whereas the concentrations in plasma and in synovial fluids were, respectively, 4.9–7.6 µg·mL−1 (16.7–25.9 µmol·L−1) and 10–17 µg·mL−1 (34–58 µmol·L−1) (Todd and Brogden, 1986). When the direct action of oxaprozin on COX was examined, it was found to inhibit COX activities with an IC50 for human platelet COX-1 of 2.2 µmol·L−1 and an IC50 for IL-1-stimulated human synovial cell COX-2 of 36 µmol·L−1 (Kawai et al., 1998). In other words, oxaprozin-mediated inhibition of COX-2 is observed at drug concentrations easily achievable in inflamed joints (Kurowski and Thabe, 1989; Rainsford et al., 2002). Thus, as the biological effects of NSAIDs are generally explained on the basis of their inhibitory action on COXs (Vane and Botting, 1998), a similar inhibitory action of oxaprozin on COX-2 inhibition would be expected to contribute substantially to its anti-inflammatory effects. Nevertheless, some of the actions of NSAIDs seem to be unrelated to their ability to inhibit prostaglandin synthesis (Chiabrando et al., 1989). In keeping with this hypothesis, oxaprozin has been recently demonstrated to be capable of inducing apoptosis of rheumatoid fibroblast-like synovial cells, albeit at supra-physiological concentrations (Yamazaki et al., 2002). Interestingly, aspirin is capable of efficiently inhibiting the activation of nuclear factor κB (NF-κB), a major factor controlling the activation of genes coding for anti-apoptotic molecules in inflammatory cells, by interfering with the activity of IκB kinase (IKK)-β (Yin et al., 1998). In the present study, we showed that oxaprozin, at concentration achievable in inflamed tissues, inhibits the IC-mediated activation of Akt in human monocytes. This in turn prevents the IKK-mediated activation of NF-κB. Consistently, the inhibition of NF-κB activation by oxaprozin reduced the production of the anti-apoptotic molecule X-linked mammalian inhibitor of apoptosis protein (XIAP) in monocytes, thereby leading to cell apoptosis.
Methods
Purification of monocytes
Human monocytes were isolated from fresh leukocyte buffy coats obtained from healthy volunteers, as previously described (Ottonello et al., 2005). Informed consent was obtained from all donors and all procedures were approved by local institutional ethical committees. Mononuclear cells, purified by centrifugation on a Ficoll density gradient, were collected from the interface and washed twice in PBS. Then, cells were separated into monocytes and lymphocytes on a hypotonic Percoll density gradient (1.129 g·mL−1). Subsequently, the upper interface containing monocytes was collected and the resulting cell population was washed twice with PBS and purified again on a second hypotonic Percoll density gradient (1.129 g·mL−1). The upper interface was then collected and washed twice in PBS. Purified monocytes were resuspended in RPMI 1640 medium supplemented with 1% (v/v) heat-inactivated foetal calf serum. Viability of monocytes was more than 98% as determined by ethidium bromide-fluoresceine diacetate assay (Dankberg and Persidsky, 1976) and purity was at least 90% as assessed by flow cytometric analysis (CD14 staining) and non-specific esterase staining. Monocytes were cultured at 1 × 106 per mL in culture medium containing 500 ng·mL−1 polymyxin B.
Preparation of insoluble IC
Immune complexes were prepared by incubating human albumin and rabbit anti-human albumin IgG at equivalence, which was determined, on the basis of quantitative precipitin curves, at the molar ratio 1:5, as previously described (Ottonello et al., 2002). Briefly, albumin and rabbit anti-human albumin IgG were incubated for 2 h at 37°C and thereafter overnight at 4°C. After then, IC were washed three times (500×g per minute, 10 min), resuspended in cold PBS and stored at 4°C. Total protein in the precipitates was determined by bicinchoninic acid (BCA) protein assay.
Acridine orange assay
The percentage of apoptotic cells was measured under the fluorescence microscope by staining the cells with acridine orange and ethidium bromide, as previously described (Ottonello et al., 2005). Acridine orange (100 µg·mL−1) was mixed with 100 µg·mL−1 ethidium bromide in HBBS. Staining solution (1 µL) was mixed with 25 µL of cell suspension (5 × 105). Live cells were determined by the uptake of acridine orange (green fluorescence) and exclusion of ethidium bromide (red fluorescence) stain. Slides were read blindly by two independent observers. Live and dead apoptotic cells were identified by the perinuclear condensation of chromatin stained by acridine orange or ethidium bromide, respectively, and by the formation of apoptotic bodies. Necrotic cells were identified by uniform labelling of the cells with ethidium bromide.
Immunofluorescence flow cytometry of Annexin V-FITC cell binding
Immunofluorescence analysis of Annexin-V binding was performed following the manufacturer's instruction with minor changes, as previously described (Ottonello et al., 2002). Briefly, cells were washed and resuspended in 100 µL isotonic binding buffer. Then, annexin V-FITC (3 µL) was added and, after incubation (15 min), cells were washed and resuspended in ice-cold PBS supplemented with 3% FCS and 0.1% sodium azide. Flow cytometry analysis was performed on an EPICS XL flow cytometer. Living monocytes were gated on the basis of physical properties (forward vs. side light scatter) and at least 2000 living cells were analysed for each sample.
Flow cytometric assessment of monocyte DNA content
Flow cytometric analysis of apoptotic nuclei was carried out as previously described (Ottonello et al., 2002). Briefly, cells were washed and resuspended in 0.5 mL PBS, the cell suspension was added dropwise to 4.5 mL ice-cold 80% ethanol while being vortexed, and kept at −20°C for 24 h. Afterwards, cells were washed twice, propidium iodide was added to a final concentration of 10 µg·mL−1, and the sample was analysed by flow cytometry after an overnight incubation. Flow cytometric analysis was performed on an EPICS XL flow cytometer. As described above, living monocytes were gated on the basis of physical properties (forward vs. side light scatter) and at least 2000 living cells were analysed for each sample.
Electrophoretic mobility shift assay
Monocytes were washed twice with PBS and resuspended in 400 µL of buffer (10 mmol·L−1 HEPES, pH 7.9, 5 mmol·L−1 MgCl2, 10 mmol·L−1 KCl, 1 mmol·L−1 ZnCl2, 0.2 mmol·L−1 EGTA, 1 mmol·L−1 Na3VO4, 10 mmol·L−1 NaF, 0.5 mmol·L−1 DTT, 0.5 mmol·L−1 PMSF, 1 µg·mL−1 leupeptin, 1 µg·mL−1 aprotinin and 1 µg·mL−1 pepstatin A). After the cells had been incubated on ice for 10 min and then lysed by the addition of 50 µL of 10% Nonidet P-40 (1.1% final concentration), the nuclei were harvested by centrifugation. The nuclear pellets were resuspended in 60 µL of extraction buffer (10 mmol·L−1 HEPES, pH 7.9, 5 mmol·L−1 MgCl2, 300 mmol·L−1 NaCl, 1 mmol·L−1 ZnCl2, 0.2 mmol·L−1 EGTA, 25% glycerol, 1 mmol·L−1 Na3VO4, 10 mmol·L−1 NaF, 0.5 mmol·L−1 DTT, 0.5 mmol·L−1 PMSF, 1 µg·mL−1 leupeptin, 1 µg·mL−1 aprotinin and 1 µg·mL−1 pepstatin A) and incubated for 15 min on ice. Nuclear debris was removed by centrifugation (15 700×g for 10 min), and the nuclear protein extract was used for gel-shift analysis. Protein concentration was determined by BCA protein assay. Gel-shift analysis of nuclear extracts was performed using oligonucleotides containing the consensus sequence for NF-κB (5′-AGT TGA GGG GAC TTT CCC AGG-3′) end-labelled with [γ-32P]-ATP using T4 polynucleotide kinase. Typical binding reactions consisted of 10 µg of nuclear extract, 1 ng DNA probe, 2 µg·mL−1 poly[d(I-C)] in a buffer containing 20 mmol·L−1 HEPES, pH 7.9, 50 mmol·L−1 NaCl, 1 mmol·L−1 DTT, 1 mmol·L−1 EDTA, and 5% glycerol and were incubated at 30°C for 20 min. Binding reactions were separated on 6% polyacrylamide gels in a 0.5 TBE buffer system. The gels were transferred to Whatman paper, dried and subjected to autoradiography (Ottonello et al., 2005).
Kinase assay
The IKK activation was measured by a modification of a published radioactive assay (Tang et al., 2003). Cells were lysed in NP40 Buffer (20 mmol·L−1 Tris-HCl pH 7.5, 150 mmol·L−1 NaCl, 10 mmol·L−1 NaF, 1% NP40, 10 µg·mL−1 glycerol, 10 µg·mL−1 aprotinin, 10 µg·mL−1 leupeptin, 1 mmol·L−1 PMSF, 0.5 mmol·L−1 Na3VO4) for 15 min at 4°C. Lysates were precleared with rabbit serum and protein A-agarose (3 h) and immunoprecipitated with 2 µg of anti-IKKγ Ab and protein A-agarose on a rotating wheel (overnight, 4°C). After that, the immunoprecipitates were washed twice with lysis buffer and twice with kinase buffer (20 mmol·L−1 HEPES, pH 7.4, 20 mmol·L−1 MnCl2, 25 mmol·L−1β-glycerophosphate and 2 mmol·L−1 dithiothreitol). Precipitates were incubated in 20 µL kinase buffer containing 40 µmol·L−1 adenosine 5′-triphosphate, 5 µCi γ32P ATP and 2 µg of GST-IkBα substrate (20 min, 30°C). The reaction was stopped by the addition of 5X sodium dodecyl sulphate (SDS) loading buffer and boiled for 5 min. After separation by SDS polyacrylamide gel electrophoresis, the gel was dried and analysed by autoradiography.
Western blot
X-linked mammalian inhibitor of apoptosis protein expression and IκBα, extracellular signal-regulated kinase (ERK) 1/2, Jun N-terminal kinase (JNK) 1/2, and p38 mitogen-activated protein kinase (MAPK) phosphorylation were investigated by Western blot analysis as previously described (Ottonello et al., 2002). Monocytes were incubated for 30 min on ice in lysis buffer containing 20 mmol·L−1 HEPES, pH 7.9, 0.15 mol·L−1 NaCl, 1 mmol·L−1 EDTA, 10% glycerol, 0.5% Nonidet P-40, 2.5 mmol·L−1 DTT, 1 mmol·L−1 PMSF, 10 µg·mL−1 leupeptin, 10 µg·mL−1 aprotinin, 1 µg·mL−1 pepstatin A, 1 mmol·L−1 Na3VO4. Cell lysates were then freeze-thawed twice in liquid nitrogen. Insoluble material was removed by centrifugation at 12 000×g for 15 min at 4°C. Protein content was determined by the BCA protein assay using bovine serum albumin as a standard. Equal amounts of protein (20 µg) were loaded on 12% SDS polyacrylamide gel and boiled for 3 min before being used. Gels were transferred electrophoretically at 4°C onto Hybond-C nitrocellulose membrane overnight at 10 V. Blots were blocked with 5% non-fat dry milk in PBS, followed by incubation with the appropriate Ab. After three washes in 0.5% Tween 20 in PBS, blots were incubated for 1 h with goat anti-mouse horseradish Ig conjugated with horseradish peroxidase. Detection was performed by the enhanced chemiluminescence detection system according to the manufacturer's instructions.
Caspase-3 assay
The assay was performed as previously described (Ottonello et al., 2002). After the appropriate incubation time, monocytes (106) were washed in cold PBS and resuspended in 50 µL of 50 mmol·L−1 NaCl, 2 mmol·L−1 MgCl2, 5 mmol·L−1 EGTA, 2 µg·mL−1 leupeptin, 2 µg·mL−1 aprotin, 10 mmol·L−1 Hepes pH 7.4. After 20 min incubation on ice, cells were lysed by freezing and thawing in liquid nitrogen. The cell lysate was spun (14 000×g, 4°C, 15 min), and the supernatant was removed and diluted to 200 µL in the assay buffer consisting of 25 µmol·L−1 HEPES pH 7.4, 0.1 % CHAPS, 10% glycerol, 1 mmol·L−1 EDTA, 5 mmol·L−1 dithiothreitol, supplemented with 50 µmol·L−1 of the caspase-3 substrate Ac-DEVD-pNa. Then, the enzymatic activity was determined spectrophotometrically by use of a Titertek TwinReader Plus for 60 min at 405 nm assuming an extinction coefficient of 8.8 × 103 mol·L−1·cm–1.
Medium, reagents and materials
RPMI 1640 with 25 mmol·L−1 HEPES (Irvine Scientific, Santa Ana, CA) supplemented with 1% heat-inactivated FCS (ICN Biomedicals s.r.l., Milano, Italy) was used as incubation medium. Dulbecco's PBS and HBSS were from Irvine Scientific. Ficoll-Hypaque was purchased from Seromed (Berlin, Germany). Fluorescein diacetate, ethidium bromide, propidium iodide, acridine orange, human albumin, rabbit anti-human albumin IgG, polymyxin B, percoll, ibuprofen, naproxen and indomethacin were from Sigma-Aldrich S.r.l. (Milano, Italy). Oxaprozin was from Helsinn SA (Pambio-Noranco, Switzerland). IKK inhibitor BMS-345541 (4(2′-aminoethyl)amino-1,8-dimethylimidazo(1,2-a)quinoxaline) and 1L-6-hydroxymethyl-chiro-inositol 2 [(R)-2-O-methyl-3O-octadecylcaromate (AKTI, Akt inhibitor) were from Calbiochem Merck (Darmstadt, Germany). Annexin V-FITC Kit was purchased from Boeringher Ingelheim (Heidelberg, Germany). Ac-Asp-Glu-Val-Asp-p-nitroanilide (Ac-DEVD-pNa) was from Bachem AG (Bubendorf, Switzerland). PD098059 (2′-amino-3′methoxyflavone; MEK inhibitor) was from Biomol Research Laboratories, Inc. (Plymouth Meeting, PA, USA). Mouse anti-human CD14 monoclonal antibody (mAb) was from Becton Dickinson Italia S.p.A (Buccinasco, Italy). Mouse anti-XIAP mAb was from MBL International (Woburn, MA, USA). Rabbit anti-human phospho-IκBα and rabbit anti-human p-IKKα/IKKβ polyclonal Abs were from Cell Signaling Technology (Danvers, MA, USA). Rabbit anti-human IKKγ polyclonal Ab and GST-IkBα protein were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Mouse anti-human tubulin mAb and ATP were from Sigma-Aldrich S.r.l. (Milano, Italy). The anti-phosphorylated ERK AF1018 Ab was from R&D System (Minneapolis, MN, USA), the anti-phosphorylated Akt 1/2/3 sc-16646-R anti-phosphorylated p-JNK sc-6254, anti-ERK1, anti-JNK and anti-Akt polyclonal Abs were from Santa Cruz Biotechnology. Other reagent-grade compounds used were obtained from commercial suppliers. All culture reagents used had an endotoxin level lower than 0.01 ng·mL−1 LPS.
The EPICS XL flow cytometer (Coulter, Hialeah, FL); BCA protein assay (Pierce); oligonucleotides containing the consensus sequence for NF-κB, Santa Cruz Biotechnology; T4 polynucleotide kinase (Promega); Whatman paper, Tewksbury (MA, USA); γ32P ATP, ICN Biomed; Hybond-C nitrocellulose membrane and the enhanced chemiluminescence detection system, Amersham Pharmacia Biotech Italia (Cologno Monzese, Italy); Titertek TwinReader Plus, Flow Lab, Ltd (Irvine, Scotland).
Densitometric and statistical analyses
Images of immunoblots were analysed by scanning densitometry quantified with the Scion Image Analysis program (release beta 2; Scion Corp., Frederick, MD, USA). Data are expressed as mean ± s.d. One-way anova with Bonferroni's post-test was performed using GraphPad InStat version 3.05 for Windows 95 (GraphPad Software, San Diego, CA, USA). Differences were accepted as significant when P < 0.05.
Results
Oxaprozin stimulates apoptosis in human monocytes exposed to IC
Freshly isolated human monocytes were placed in culture and cell apoptosis was evaluated after a 48-h incubation by light microscopic evaluation of acridine orange stained cells. At the end of the incubation period, the mean percentage of apoptotic monocytes was 36.3 ± 8.0% (mean ± 1 s.d., n = 65), without detection of cell necrosis, as evaluated by the ethidium bromide assay. In accord with our previous data (Ottonello et al., 2005), when monocytes were cultured with different doses of IC, a dose-dependent inhibition of apoptosis was observed (% apoptosis in presence of 0 µg·mL−1 IC: 38.0 ± 6.0; 6.25 µg·mL−1 IC: 34.3 ± 5.5; 12.5 µg·mL−1 IC: 20.7.0 ± 6.0; 25 µg·mL−1 IC: 14.3 ± 3.0; 50 µg·mL−1 IC: 9.3 ± 4.9; 100 µg·mL−1 IC: 10.3 ± 2.5, mean ± 1 s.d., n = 3). The concentration of 25 µg·mL−1 IC, capable of significantly inhibiting apoptosis, was chosen for subsequent experiments. As shown in Figure 1A, when added to monocytes in the presence of 25 µg·mL−1 IC, oxaprozin induced apoptosis in a dose-dependent manner. In comparison, the drug did not accelerate apoptosis of resting monocytes, that is, cells incubated in the absence of IC (Figure 1A). The concentration of 50 µmol·L−1 oxaprozin was chosen for subsequent experiments. The ability of the drug to induce apoptosis in monocytes exposed to IC was confirmed by two independent assays. First, the subdiploid DNA content of propidium iodide stained cells was assessed by flow cytometry. As shown in Figure 1B, in absence of oxaprozin, an ipodiploid peak was detectable after incubation of the cells for 48 h, whereas a low amount of subdiploid DNA was evident after cell stimulation with 25 µg·mL−1 IC. On the other hand, when monocytes were co-incubated with 50 µmol·L−1 oxaprozin the inhibitory effect of the IC was almost completely reversed. Second, flow cytometric analysis of the annexin V binding was performed. As shown in Figure 1B, monocytes cultured for 48 h bind annexin V. When stimulated with 25 µg·mL−1 IC, annexin V binding was reduced but it was observed again in the presence of 50 µmol·L−1 oxaprozin. It is of note that oxaprozin did not induce cell necrosis, assessed by testing the cell membrane integrity using both ethidium bromide and propidium iodide assays, and by evaluating the release of LDH during cell cultures (data not shown).
Figure 1.
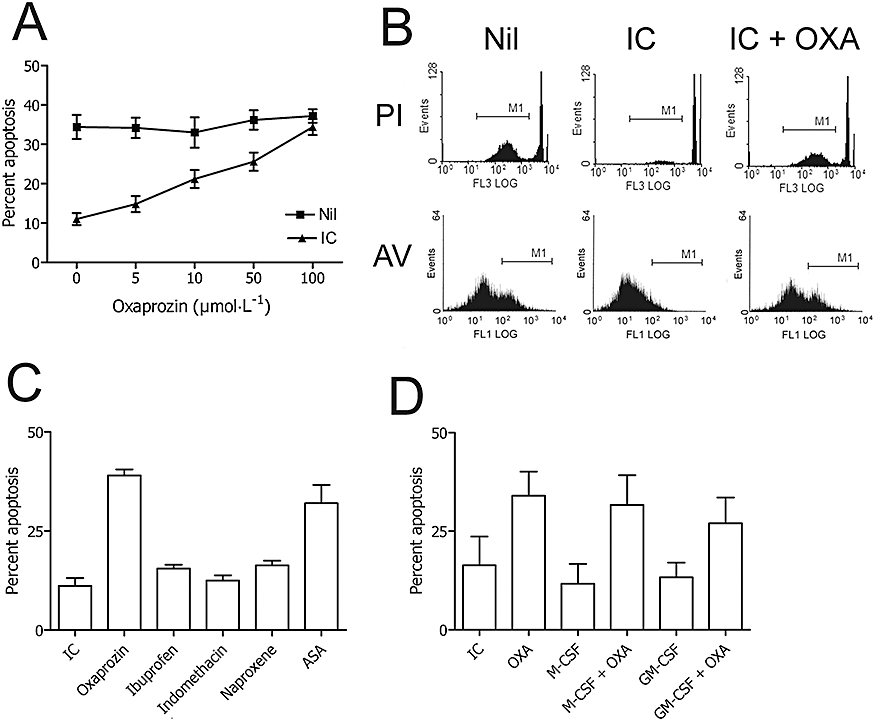
Effects of NSAIDs on cell apoptosis of IC-stimulated monocytes. (A) Microscopic analysis of the apoptosis of human monocytes after acridine orange staining. Monocytes cultured in the absence (Nil) and presence (IC) of IC were exposed to different concentrations of oxaprozin (OXA). Data are expressed as mean ± 1 s.d., n = 3. (B) Flow cytometric analysis of the apoptosis of human monocytes after propidium iodide (PI) and annexin V (AV) staining. (Nil): apoptosis of monocytes cultured in medium. (IC): apoptosis of monocytes cultured in the presence of IC. (IC + OXA): apoptosis of monocytes cultured in the presence of IC plus OXA. Results represent one of three experiments that yielded similar results. (C) Microscopic analysis of the apoptosis of human monocytes after acridine orange staining. Monocytes cultured with IC in the absence (IC) and presence of various NSAIDs used at the following concentrations chosen on the basis of their COX-inhibiting activity: OXA: 50 µmol·L−1; ibuprofen: 68.2 µmol·L−1; indomethacin: 0.29 µmol·L−1; naproxene: 216 µmol·L−1; ASA: 73 µmol·L−1. Data are expressed as mean ± 1 s.d., n = 3. The % spontaneous apoptosis in the absence or presence of OXA (50 µmol·L−1): 33 ± 4 and 36 ± 4, mean ± 1 s.d., n = 3. IC versus OXA: P < 0.001; IC versus ASA: P < 0.001; IC versus ibuprofen, indomethacin or naproxene: ns. (D) Microscopic analysis of human monocytes apoptosis after acridine orange staining. Monocytes cultured with IC (IC) were exposed to OXA, to M-CSF (M-CSF), to OXA after cell treatment with M-CSF (M-CSF + OXA), to GM-CSF (GM-CSF) or to OXA after cell treatment with GM-CSF (GM-CSF + OXA). Data are expressed as mean ± 1 s.d., n = 3. The % spontaneous apoptosis in the absence or presence of OXA (50 µmol·L−1): 38 ± 3 and 37 ± 4, mean ± 1 s.d., n = 3. ASA, acetylsalicylic acid; IC, immune complex; M-CSF, macrophage-colony stimulating factor; NSAIDs, non-steroidal anti-inflammatory drugs.
The activity of oxaprozin was studied in comparison with other NSAIDs, that is, ibuprofen, indomethacin, naproxen and acetylsalicylic acid (ASA), used at an equivalent concentration as COX inhibitors. As shown in Figure 1C, only oxaprozin and ASA were effective inducers of apoptosis of human monocytes cultured in the presence of IC. This suggests that the activity of oxaprozin is not directly related to its COX inhibitory properties. Finally, we evaluated the pro-apoptotic activity of oxaprozin towards monocytes triggered by IC in the presence of macrophage-colony stimulating factor (M-CSF) and GM-CSF. Indeed, inflamed joints are a rich source of myeloid growth factors, that is, M-CSF and GM-CSF (Firestein et al., 1988; Xu et al., 1989), which also contribute to the prolonged survival of inflammatory cells, including monocytes (Bratton et al., 1995; Marsh et al., 1999). As shown in Figure 1D, oxaprozin reversed the anti-apoptotic of IC in presence of either M-CSF or GM-CSF, which is in in vitro conditions resembling the inflammatory microenvironment of an inflamed joint.
Oxaprozin modulates apoptosis of IC-triggered monocytes by a caspase-3-dependent mechanism
Caspase-3 activation is a major executioner in the apoptosis programme of monocytes (Fahy et al., 1999). Thus, we hypothesized that oxaproxin may modulate the apoptosis of monocytes, induced to survive by IC triggering, by a caspase-3-dependent mechanism. Caspase-3 activity was studied in monocytes cultured in conditions identical to those used in the apoptosis assays. As shown in Figure 2A, caspase-3 activity was significantly lower after stimulation by IC than in resting conditions. Furthermore, oxaprozin increased caspase-3 activity in the activated but not in the resting condition (Figure 2A). Consistent with these data, oxaprozin-induced apoptosis of IC-stimulated monocytes was almost completely inhibited by the specific caspase-3 inhibitor Ac-DEVD-CHO (Figure 2B).
Figure 2.
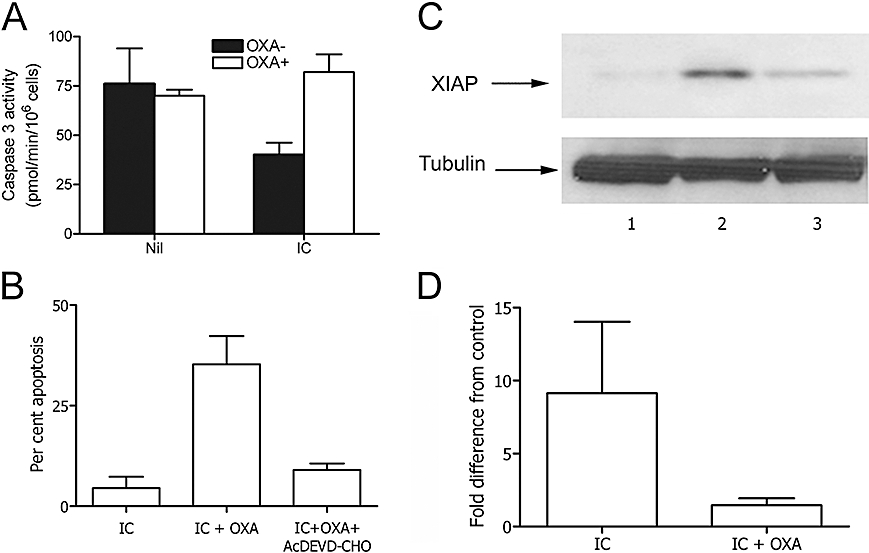
Oxaprozin (OXA)-induced monocyte apoptosis was dependent on caspase-3 activity and XIAP expression. (A) Human monocytes cultured in the absence (Nil) or presence (IC) of IC were exposed to medium (solid columns), or OXA (open columns). Then, caspase-3 activity was determined spectrophotometrically on whole-cell lysates. Results are expressed as the mean ± 1 s.d., n = 4. Without IC: OXA− versus OXA+: ns. With IC: OXA− versus OXA+: P < 0.01. (B) Microscopic analysis of the apoptosis of human monocytes after acridine orange staining. Monocytes were cultured with IC alone (IC), IC plus OXA (IC + OXA) and IC plus OXA in presence of the caspase-3 inhibitor Ac-DEVD-CHO (IC + OXA + Ac-DEVD-CHO). Data are expressed as mean ± 1 s.d., n = 3. The % spontaneous apoptosis in the absence or presence of OXA (50 µmol·L−1): 38 ± 6 and 36 ± 6, mean ± 1 s.d., n = 3. IC versus. IC + OXA: P < 0.01, IC + OXA versus IC + OXA + Ac-DEVD-CHO: P < 0.01. (C) Western blot: monocyte lysates after incubation of the cells in medium (lane 1), IC (lane 2), IC plus OXA (lane 3). (D) Densitometric analysis of Western blot. Data are expressed as the mean ± 1 s.d., n = 3, and presented as fold difference from control (medium). Fold difference from control of OXA alone: 0.17 ± 0.65. Medium versus IC: P < 0.05; IC versus IC + OXA: P < 0.05. IC, immune complex; XIAP, X-linked mammalian inhibitor of apoptosis protein.
It has been demonstrated previously that the activity of monocyte caspase-3 is regulated by the presence of XIAP, one of the most potent components of the family of IAP anti-apoptotic proteins that regulate programmed cell death in different types of cells (Riedl et al., 2001). Therefore, it is conceivable that oxaprozin protects caspase-3 activity by acting on XIAP synthesis. In fact, as shown in Figure 2, monocyte IC triggering caused nearly 10 times more phosphorylation of XIAP in comparison with resting cells (Figure 2C and D), whereas oxaprozin prevented this phenomenon (Figure 2C and D).
Role of the IKK-NF-κB system in oxaprozin-induced apoptosis of IC triggered monocytes
Owing to its capacity to promote the production of IAP proteins including XIAP, the NF-κB system is one of the major intracellular pathways involved in the regulation of apoptosis in several cell types (Lee and Collins, 2001; Li and Verma, 2002). Thus, a detailed analysis of this pathway was performed in unstimulated and IC-triggered monocytes. With this aim, monocytes were cultured with 25 µg·mL−1 IC for 30 min under conditions identical to those used for the apoptotic assays. Then, nuclear and cytoplasmic extracts were obtained from cultured cells and examined in an electrophoretic gel mobility shift assay (NF-κB activity). As shown in Figure 3A, resting, that is, unstimulated, monocytes display a low level of NF-κB activation. Exposure of monocytes to IC induced a sharp increase in NF-κB activation (Figure 3A and B) with a magnitude of binding comparable with that induced by TNF-α, a well-known strong activator of NF-κB (Chaturvedi et al., 1994). The specificity of NF-κB activation was confirmed by the inhibition of IC-dependent DNA binding in the presence of a specific anti-p65 mAb (Figure 3A and B). Finally, NF-κB activation was inhibited by 50 µmol·L−1 oxaprozin (Figure 3A and B).
Figure 3.
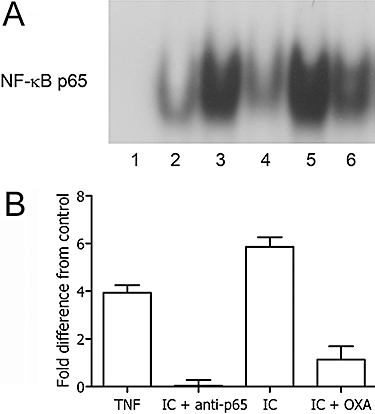
Oxaprozin (OXA) inhibited NF-κB activation in IC-triggered monocytes. (A) EMSA was performed with monocyte nuclear extracts after incubation of the cells in medium (lane 2), TNF-α (positive control) (lane 3), IC in presence of anti-p65 mAb (lane 4), IC (lane 5), IC plus OXA (lane 6). Lane 1: EMSA performed in the presence of a 100-fold excess of unlabelled NF-κB probe. (B) Densitometric analysis of EMSA. Data are expressed as the mean ± 1 s.d., n = 4, and presented as fold difference from control (medium). Fold difference from control of OXA alone: 0.13 ± 0.73. Medium versus TNF-α: P < 0.01; medium versus IC: P < 0.001; IC versus IC + OXA: P < 0.001. EMSA, electrophoretic mobility shift assay; IC, immune complex; NF-κB, nuclear factor κB; TNF-α, tumour necrosis factor-α.
In different immune cells, the anti-apoptotic activity of the NF-κB pathway requires the degradation of its natural inhibitor IκBα, which follows IκBα phosphorylation through IKK (Li and Verma, 2002). Therefore, the effects of oxaprozin on NF-κB might reflect its ability to inhibit cell activation of the IKK system. We explored this hypothesis by studying the effect of oxaprozin on IKK-mediated phosphorylation of IκBα. As shown in Figure 4A and B, the amount of phosphorylated IκB from activated monocytes was dose-dependently inhibited by oxaprozin. Finally, oxaprozin inhibited activation of the IKK system induced by the reagent IκBα (Figure 4C and D).
Figure 4.
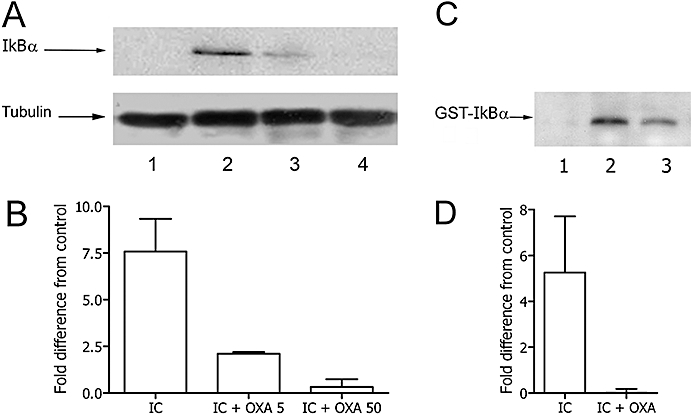
Oxaprozin (OXA) inhibited IκBα phosphorylation in IC-triggered monocytes. (A) Western blot: monocyte lysates after incubation of the cells in medium (lane 1), IC (lane 2), IC plus OXA 5 µmol·L−1 (lane 3), IC plus OXA 50 µmol·L−1 (lane 4). (B) Densitometric analysis of Western blot. Data are expressed as the mean ± 1 s.d., n = 3, and presented as fold difference from control (medium). Fold difference from control of OXA alone: 0.29 ± 0.31. Medium versus IC: P < 0.01; IC versus IC + OXA 50: P < 0.05. (C) Endogenous IKK activity of immunoprecipitates from monocytes incubated in medium (lane 1), with IC (lane 2), with IC plus OXA (lane 3) determined using GST-IκBα as substrate. D. Densitometric analysis of kinase assay. Data are expressed as the mean ± 1 s.d., n = 3, and presented as fold difference from control (medium). Fold difference from control of OXA alone: 0.11 ± 0.35. Medium versus IC: P < 0.05; IC versus IC + OXA: P < 0.05. IC, immune complex; IKK, IκB kinase.
Role of Akt and MAPK on oxaprozin-induced apoptosis of IC triggered monocytes
We have recently demonstrated that IC-mediated inhibition of apoptosis involves the PI3K/Akt and MAPK pathways (Bianchi et al., 2007). These molecules, by regulating the NF-κB activity, are involved in the survival signalling cascade of many cell types. Thus, we next explored the possibility that oxaprozin exerts its inhibitory effect on NF-κB by affecting the activity of the PI3K/Akt and/or MAPK pathways. As shown in Figure 5A and B, the exposure of monocytes to IC resulted in the activation of Akt only in the absence but not in the presence of oxaprozin. Similarly, oxaprozin strongly inhibited IC-induced activation of p38 MAPK and had the same magnitude of activity as the Akt inhibitor AKTI (Figure 5C and D). In contrast, ERK 1/2 activation by IC was unaffected by co-incubation of monocytes with oxaprozin or AKTI (Figure 5E and F). Finally, no activation of JNK1/2 by IC was observed in the absence and presence of oxaprozin or AKTI in comparison with TNF-α, a well-known activator of JNK in monocytes (Figure 5G and H). These data suggest that oxaprozin inhibits IC-induced NF-κB activation by a pathway dependent on Akt and p38 MAPK. Consistent with these data, AKTI inhibited IC-induced IKK activation (Figure 6A and B), IkBα phosphorylation (Figure 6C and D), NF-κB activation (Figure 6E and F) and XIAP phosphorylation (Figure 6G and H) similarly to oxaprozin.
Figure 5.
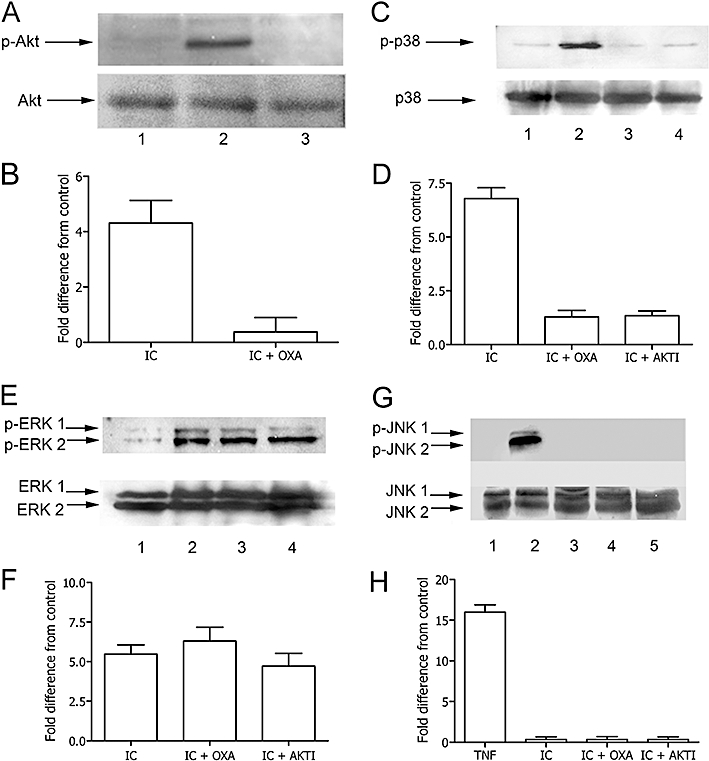
Effect of oxaprozin (OXA) on Akt and MAPK activation in IC-triggered monocytes. (A) Western blot analysis of lysates incubated with specific anti-p-Akt or anti-total Akt: monocyte lysates after incubation of the cells in medium (lane 1), IC (lane 2), IC plus OXA (lane 3). (B) Densitometric analysis of Western blot. Data are expressed as the mean ± 1 s.d., n = 3, and presented as fold difference from control (medium). Fold difference from control of OXA alone: 0.41 ± 0.49. Medium versus IC: P < 0.01; IC versus IC + OXA: P < 0.01. (C) Western blot analysis of lysates incubated with specific anti-p-p38 MAPK or anti-total p38 MAPK: monocyte lysates after incubation of the cells in medium (lane 1), IC (lane 2), IC plus OXA (lane 3), IC plus Akt inhibitor AKTI (lane 4). (D) Densitometric analysis of Western blot. Data are expressed as the mean ± 1 s.d., n = 3, and presented as fold difference from control (medium). Fold difference from control of OXA alone: 0.18 ± 0.17. Medium versus IC: P < 0.001; IC versus IC + OXA: P < 0.01.; IC versus IC + AKTI: P < 0.01. (E) Western blot analysis of lysates incubated with specific anti-p-ERK 1/2 or anti-total ERK 1/2: monocyte lysates after incubation of the cells in medium (lane 1), IC (lane 2), IC plus OXA (lane 3). (F) Densitometric analysis of Western blot. Data are expressed as the mean ± 1 s.d., n = 3, and presented as fold difference from control (medium). Fold difference from control of OXA alone: 0.15 ± 0.33. Medium versus IC, Medium versus IC + OXA, Medium versus IC + AKTI: P < 0.001. (G) Western blot analysis of lysates incubated with specific anti-p-JNK 1/2 or anti-total JNK 1/2: monocyte lysates after incubation of the cells in medium (lane 1), TNF-α (lane 2), IC (lane 3), IC plus OXA (lane 4), IC plus Akt inhibitor AKTI (lane 5). (H) Densitometric analysis of Western blot. Data are expressed as the mean ± 1 s.d., n = 3, and presented as fold difference from control (medium). Fold difference from control of OXA alone: 0.10 ± 0.40. Medium versus TNF-α: P < 0.001. ERK, extracellular signal-regulated kinase; IC, immune complex; JNK, Jun N-terminal kinase; MAPK, mitogen-activated protein kinase; TNF-α, tumour necrosis factor-α.
Figure 6.
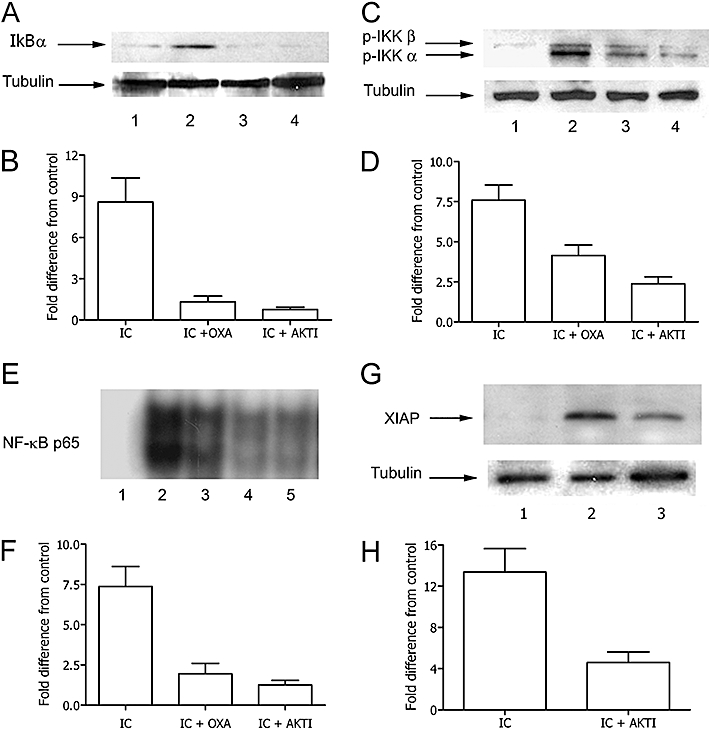
Role of Akt on IKK and NF-κB activation and XIAP expression in IC-triggered monocytes. (A) Western blot: monocyte lysates incubated with specific anti-p-IκBα after incubation of the cells in medium (lane 1), IC (lane 2), IC plus oxaprozin (OXA) (lane 3), IC plus AKTI (lane 4). (B) Densitometric analysis of Western blot. Data are expressed as the mean ± 1 s.d., n = 3, and presented as fold difference from control (medium). Fold difference from control of OXA alone: 0.10 ± 0.10. Medium versus IC: P < 0.01; IC versus IC + OXA: P < 0.05, IC versus IC + AKTI: P < 0.05. C. (C) Western blot analysis of lysates incubated with specific anti-p-IKK or anti-total IKK: monocyte lysates after incubation of the cells in medium (lane 1), IC (lane 2), IC plus OXA (lane 3), IC plus Akt inhibitor AKTI (lane 4). (D) Densitometric analysis of Western blot. Data are expressed as the mean ± 1 s.d., n = 3, and presented as fold difference from control (medium). Fold difference from control of OXA alone: 0.22 ± 0.18. Medium versus IC: P < 0.001. ED: IC versus IC + OXA: P < 0.05, IC versus IC + AKTI: P < 0.01. (E) EMSA was performed with monocyte nuclear extracts after incubation of the cells in medium (lane 5), IC (lane 2), IC plus OXA (lane 3), IC plus AKTI (lane 4). Lane 1: EMSA performed in presence of a 100-fold excess of unlabelled NF-κB probe. (F) Densitometric analysis of EMSA. Data are expressed as the mean ± 1 s.d., n = 3, and presented as fold difference from control (medium). Fold difference from control of OXA alone: 0.38 ± 0.52. Medium versus IC: P < 0.001; IC versus IC + OXA: P < 0.01; IC versus IC + AKTI: P < 0.01. (G) Western blot: monocyte lysates incubated with anti-XIAP after incubation of the cells in medium (lane 1), IC (lane 2), IC plus AKTI (lane 3). (H) Densitometric analysis of Western blot. Data are expressed as the mean ± 1 s.d., n = 3, and presented as fold difference from control (medium). Fold difference from control of OXA alone: 0.24 ± 0.28. Medium versus IC: P < 0.05; IC versus IC + AKTI: P < 0.05. EMSA, electrophoretic mobility shift assay; IC, immune complex; IKK, IκB kinase; NF-κB, nuclear factor κB; XIAP, X-linked mammalian inhibitor of apoptosis protein.
Discussion
In the present study, we showed that IC-activated monocytes are effectively induced to die through activation of an apoptotic pathway when cultured in the presence of the propionic NSAID derivative oxaprozin. In contrast, the drug did not exert any pro-apoptotic activity towards unstimulated resting monocytes. It is noteworthy that the pro-apoptotic action of oxaprozin on activated monocytes cannot be considered a common feature of NSAIDs, as of four other NSAIDs, at an equivalent concentration to the COX inhibitor, only aspirin, but not ibuprofen, indomethacin and naproxen, is capable of affecting the survival of IC-activated monocytes.
Monocytes are relatively inert circulating precursors of tissue macrophages endowed with poor phagocytic capacity (Gordon, 1999). In response to stimulation by chemoattractants, monocytes trans-migrate through endothelium using selectins and integrins and, after leaving the blood stream, they differentiate into macrophages, that is, cells capable of phagocytosis (Gordon, 1999). In fact, binding of IC as well as phagocytosis of opsonized particles triggers their activation through Fc receptor signalling (Gordon, 1999). Consequently, IC-activated macrophages secrete a number of mediators potentially capable of modulating the activity of other cells including lymphocytes, fibroblasts, endothelial cells and neutrophils, as well as an array of histotoxic mediators (Gordon, 1999). This pathway of activation, known as ‘classical pathway’ (Gordon, 1999) is, thinking in a teleological fashion, responsible for the crucial role of monocytes/macrophages in the innate and adaptive immune response involved in host defense. Nevertheless, when uncontrolled, classical activation may result in the tissue damage responsible for organ dysfunction in different pathological conditions including rheumatoid arthritis (Kinne et al., 2000). The unchecked histotoxic activity of IC-activated monocyte/macrophages is favoured by the prolongation of cell survival due to FcR-mediated inhibition of the apoptosis (Marsh et al., 1999; Ottonello et al., 2005). Thus, the induction of monocyte/macrophage apoptosis has been suggested as a possible pharmacological target for the resolution of persistent/chronic inflammation (Kinne et al., 2000; Liu and Pope, 2003). Nevertheless, under specific conditions, monocyte/macrophages may have per se a positive role in the resolution of the inflammatory process (Duffield, 2003). Indeed, tissue injury during inflammation is characterized by an increased burden of apoptotic cells, including infiltrating inflammatory cells, such as neutrophils as well as resident stromal and parenchimal cells. On the other hand, monocyte/macrophages have a crucial role in the disposal of dying cells, in that they are professional scavengers, devoted to the phagocytosis of apoptotic cells (Fadok et al., 1998). This phenomenon has at least two favourable effects: (i) avoidance of secondary necrosis of dying neutrophils and subsequent release into the environment of histotoxic proteases; and (ii) production of anti-inflammatory cytokines by phagocytosing macrophages induces the withdrawal of survival signals for inflammatory cells and normalization of chemokine gradients that allow infiltrating cells to undergo apoptosis or leave through draining lymphatics (Savill et al., 2002). Thus, under these conditions, the non-selective removal of monocytes/macrophages may be detrimental for the resolution of inflammation. Taking into account these observations, the capacity of oxaprozin to act on activated macrophages while sparing resting monocytes, demonstrated in the present study, may represent a useful model for the rationale approach to the pharmacological induction of monocyte/macrophage apoptosis aimed to curb an undesired overwhelming inflammatory process. Consistent with this view, in a mouse model of arthritis, in vivo administration of an immunotoxin capable of inducing apoptosis in activated macrophages, while leaving resting cells unaffected (van Roon et al., 2003), causes the effective elimination of activated macrophages and a decrease in the inflammation and bone erosion resulting in significant inhibition of disease activity (van Vuuren et al., 2006).
The NF-κB system is generally considered a key player in controlling acute and chronic inflammatory reactions and, at least in part, immune responses (Li and Verma, 2002). A coordinated activation of NF-κB occurs in various cell populations involved in the inflammatory response, including monocytes and macrophages, neutrophils, lymphocytes, endothelial cells, fibroblasts and synoviocytes. The activation of this system results in the production of diverse inflammatory and immune response mediators. More than 150 genes susceptible to activation by NF-κB have been identified. They include chemokines, cytokines, adhesion molecules, enzymes, such as for instance COX-2, and also some anti-apoptotic molecules such as XIAP, which in turn favours the survival of monocytes at sites of inflammation (Makarov, 2001; Tak and Firestein, 2001; Pope, 2002). In 1994, Kopp and co-workers demonstrated that high doses of ASA and sodium salicylate are capable of inhibit NF-κB activation (Kopp and Ghosh, 1994). Since then, several observations have been reported regarding the capacity of NSAIDs to inhibit NF-κB activity. However, the bulk of these observations concerns NF-κB-dependent anti-proliferative activity of NSAIDs towards tumour cells (Palayoor et al., 1999; Yamamoto et al., 1999; Takada et al., 2004; Cho et al., 2005). With regard to the anti-NF-κB properties of NSAIDs towards non-proliferating immune cells, data presently available are scant and somewhat contradictory. In particular, ibuprofen inhibits NF-κB activation in T cells at concentrations comparable with those used in the present study (Scheuren et al., 1998). On the contrary, ibuprofen inhibits NF-κB activation in human monocytes only at very high concentrations not achievable in vivo after oral administration (Stuhlmeier et al., 1999). In fact, we found that ibuprofen used at a concentration achievable in vivo is incapable of inhibiting not only monocyte apoptosis but also NF-κB activation and XIAP expression in IC-activated monocytes (data not shown). Furthermore, the lack of anti-apoptotic activity of indomethacin and naproxen observed in the present study is consistent with previous observations demonstrating that these NSAIDs are devoid of anti-NF-κB activity (Kazmi et al., 1995; Lavagno et al., 2004). On the other hand, our results showing that aspirin is capable of inducing apoptosis of monocytes exposed to IC and that NF-kB is required for the protection of activated monocytes from apoptosis support and extend previous findings showing that aspirin and its derivative sodium salicylate, but not ibuprofen are capable of inhibiting NF-κB–mediated adhesion to endothelium of monocytes exposed to LDL (Eisele et al., 2004).
The PI3K/Akt system is a well-identified target for the pharmacological manipulation of cell survival of cancer cells (Cheng et al., 2005). Furthermore, PI3K/Akt plays a crucial role in the recruitment and function of several immune cells. In fact, the inhibition of this system is effective in ameliorating different inflammatory diseases in animal models (Marone et al., 2008). Various mechanisms for the regulation of cell survival by Akt have been reported, including the modulation of transcriptional factors responsible for anti-apoptotic genes. In particular, Akt regulates NF-κB by activating IKK (Viatour et al., 2005). In accord with these observation, in the present study we showed that the IKK-dependent inhibition of IC-activated human monocytes induced by oxaprozin is dependent on the ability of this drug to inhibit Akt activation. In turn, by restraining the activity of Akt, oxaprozin strongly inhibited p38 MAPK phosphorylation. On the contrary, in our experimental conditions, ERK 1/2, although crucial for the survival of monocytes induced by IC triggering (Bianchi et al., 2007), is not involved in oxaprozin-mediated pro-apoptotic activity as oxaprozin did not block the activation of ERK 1/2. Finally, our results do not suggest any role for JNK 1/2 in IC-dependent monocyte survival. Thus, unlike aspirin and sodium salicylate, which inhibit NF-κB by directly acting on IκBα activity (Yin et al., 1998), our data suggest that oxaprozin exerts its NF-κB-inhibiting effect at a proximal level by inhibiting Akt.
Immune complexes activate monocytes interacting with FcRs, and consequently inducing generation of oxidants and various proinflammatory mediators, such as cytokines (TNF, IL-1 and M-CSF), chemokines (MCP-1 and IL-8), tissue-damaging metalloproteinases and the prostaglandin producing COX enzyme (Fernandez et al., 2002). Some of these products are involved in monocyte survival. In fact, protection from apoptosis of IC-activated monocytes appears to be mediated by M-CSF via activation of Akt (Kelley et al., 1999). Also oxidants directly generated by FcR-dependent NADPH oxidase activation or indirectly induced by M-CSF-dependent PI3K activation mediate the inhibition of monocyte apoptosis through activation of ERK 1/2 (Bhatt et al., 2002). In other words, M-CSF generated by FcR-triggering augments monocyte survival through activation of AkT and ERK 1/2. Does oxaprozin promote monocyte apoptosis through inhibition of M-CSF and/or oxidant generation? Taking into account the crucial role of ERK 1/2, an MAPK that is not involved in oxaprozin-mediated activity, this is unlikely. Although data regarding PGE2 and monocyte apoptosis are lacking, PGE2 has been shown to stimulate Akt (Xu et al., 2008) and thus could possibly exert an anti-apoptotic effect on monocytes. Nevertheless, a putative role for this COX-derived product as target for the pro-apoptotic action of oxaprozin shown in the present study is unlikely as under our experimental conditions three different COX inhibitors failed to induce monocyte apoptosis. Thus, the mechanism by which oxaprozin selectively interferes with IC-induced Akt inhibition is still not clear, and should be a matter of future investigation. Nevertheless, this is not an uncommon feature, at least as far as monocytes are concerned. For example, the production of IL-12 from LPS-or IFN-γ-stimulated monocytes can be inhibited by ligands for G(i)-protein-coupled Rs, such as C5a, in a manner strictly dependent on PI3K/Akt and JNK, but not ERK, although the latter MAPK is activated by C5a triggering (la Sala et al., 2005).
In summary, as shown in Figure 7, in our study oxaprozin interferes with the survival pathway of IC-stimulated monocytes by inhibiting Akt (1). This results in the inhibition of IKK activity (2) and consequently in the suppression NF-κB translocation to the nucleus (3) and NF-κB-dependent XIAP production (4). In turn, the death effector caspase-3 can freely exert its proteolytic activity with the subsequent shift from cell survival to cell apoptosis (5).
Figure 7.
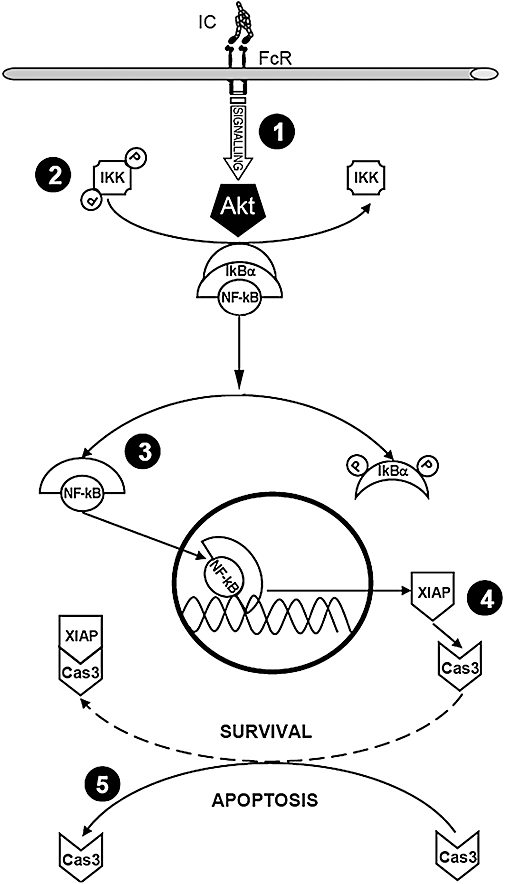
Key points of the survival/apoptosis pathways in immune-complex-stimulated human monocytes. FcR, fragment crystallizable receptor; IC, immune complex; IKK, IκB kinase; NF-κB, nuclear factor κB; XIAP, X-linked mammalian inhibitor of apoptosis protein.
As oxaprozin was shown to inhibit this Akt-dependent pathway at concentrations which are within the therapeutic range of this drug, this novel finding raises new opportunities to develop strategies to control inflammation in rheumatic conditions by acting on systems that are completely different from the well-known and documented COX-dependent pathways.
Glossary
Abbreviations
- COX
cyclooxygenase
- ERK
extracellular signal-regulated kinase
- FcR
fragment crystallizable receptor
- IC
immune complex
- IKK
IκB kinase
- IL
interleukin
- JNK
Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- MCP-1
monocyte chemotactic protein 1
- M-CSF
macrophage-colony stimulating factor
- NF-κB
nuclear factor κB
- NSAIDs
non-steroidal anti-inflammatory drugs
- TNF-α
tumour necrosis factor-α
- XIAP
X-linked mammalian inhibitor of apoptosis protein
Conflict of interest
FD received research funding from HELSINN HEALTHCARE SA, Pambio-Noranco, Switzerland, as a part of contribution to the present work.
References
- Abramson SB, Weaver AL. Current state of therapy for pain and inflammation. Arthritis Res Ther. 2005;7(Suppl. 4):S1–S6. doi: 10.1186/ar1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arend WP, Gabay C. Cytokines in the rheumatic diseases. Rheum Dis Clin North Am. 2004;30(1):41–67. v-vi. doi: 10.1016/S0889-857X(03)00115-7. [DOI] [PubMed] [Google Scholar]
- Bhatt NY, Kelley TW, Khramtsov VV, Wang Y, Lam GK, Clanton TL, et al. Macrophage-colony-stimulating factor-induced activation of extracellular-regulated kinase involves phosphatidylinositol 3-kinase and reactive oxygen species in human monocytes. J Immunol. 2002;169(11):6427–6434. doi: 10.4049/jimmunol.169.11.6427. [DOI] [PubMed] [Google Scholar]
- Bianchi G, Montecucco F, Bertolotto M, Dallegri F, Ottonello L. Immune complexes induce monocyte survival through defined intracellular pathways. Ann N Y Acad Sci. 2007;1095:209–219. doi: 10.1196/annals.1397.025. [DOI] [PubMed] [Google Scholar]
- Bratton DL, Hamid Q, Boguniewicz M, Doherty DE, Kailey JM, Leung DY. Granulocyte macrophage colony-stimulating factor contributes to enhanced monocyte survival in chronic atopic dermatitis. J Clin Invest. 1995;95(1):211–218. doi: 10.1172/JCI117642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi MM, LaPushin R, Aggarwal BB. Tumor necrosis factor and lymphotoxin. Qualitative and quantitative differences in the mediation of early and late cellular response. J Biol Chem. 1994;269(20):14575–14583. [PubMed] [Google Scholar]
- Cheng JQ, Lindsley CW, Cheng GZ, Yang H, Nicosia SV. The Akt/PKB pathway: molecular target for cancer drug discovery. Oncogene. 2005;24(50):7482–7492. doi: 10.1038/sj.onc.1209088. [DOI] [PubMed] [Google Scholar]
- Chiabrando C, Castelli MG, Cozzi E, Fanelli R, Campoleoni A, Balotta C, et al. Antiinflammatory action of salicylates: aspirin is not a prodrug for salicylate against rat carrageenin pleurisy. Eur J Pharmacol. 1989;159(3):257–264. doi: 10.1016/0014-2999(89)90156-8. [DOI] [PubMed] [Google Scholar]
- Cho M, Gwak J, Park S, Won J, Kim DE, Yea SS, et al. Diclofenac attenuates Wnt/beta-catenin signaling in colon cancer cells by activation of NF-kappaB. FEBS Lett. 2005;579(20):4213–4218. doi: 10.1016/j.febslet.2005.06.049. [DOI] [PubMed] [Google Scholar]
- Cook AD, Visvanathan K. Molecular targets in immune-mediated diseases: focus on rheumatoid arthritis. Expert Opin Ther Targets. 2004;8(5):375–390. doi: 10.1517/14728222.8.5.375. [DOI] [PubMed] [Google Scholar]
- Dankberg F, Persidsky MD. A test of granulocyte membrane integrity and phagocytic function. Cryobiology. 1976;13(4):430–432. doi: 10.1016/0011-2240(76)90098-5. [DOI] [PubMed] [Google Scholar]
- Davies N. Clinical pharmacokinetics of oxaprozin. Clin Pharmacokinet. 1998;35:425–436. doi: 10.2165/00003088-199835060-00002. [DOI] [PubMed] [Google Scholar]
- Duffield JS. The inflammatory macrophage: a story of Jekyll and Hyde. Clin Sci (Lond) 2003;104(1):27–38. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- Eisele G, Schwedhelm E, Schieffer B, Tsikas D, Boger RH. Acetylsalicylic acid inhibits monocyte adhesion to endothelial cells by an antioxidative mechanism. J Cardiovasc Pharmacol. 2004;43(4):514–521. doi: 10.1097/00005344-200404000-00006. [DOI] [PubMed] [Google Scholar]
- Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101(4):890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahy RJ, Doseff AI, Wewers MD. Spontaneous human monocyte apoptosis utilizes a caspase-3-dependent pathway that is blocked by endotoxin and is independent of caspase-1. J Immunol. 1999;163(4):1755–1762. [PubMed] [Google Scholar]
- Feldmann M, Maini RN. Lasker Clinical Medical Research Award. TNF defined as a therapeutic target for rheumatoid arthritis and other autoimmune diseases. Nat Med. 2003;9(10):1245–1250. doi: 10.1038/nm939. [DOI] [PubMed] [Google Scholar]
- Fernandez N, Renedo M, Garcia-Rodriguez C, Sanchez Crespo M. Activation of monocytic cells through Fc gamma receptors induces the expression of macrophage-inflammatory protein (MIP)-1 alpha, MIP-1 beta, and RANTES. J Immunol. 2002;169(6):3321–3328. doi: 10.4049/jimmunol.169.6.3321. [DOI] [PubMed] [Google Scholar]
- Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423(6937):356–361. doi: 10.1038/nature01661. [DOI] [PubMed] [Google Scholar]
- Firestein GS. Inhibiting inflammation in rheumatoid arthritis. N Engl J Med. 2006;354(1):80–82. doi: 10.1056/NEJMcibr054344. [DOI] [PubMed] [Google Scholar]
- Firestein GS, Alvaro-Gracia JM, Maki R. Quantitative analysis of cytokine gene expression in rheumatoid arthritis. J Immunol. 1990;144(9):3347–3353. [PubMed] [Google Scholar]
- Firestein GS, Xu WD, Townsend K, Broide D, Alvaro-Gracia J, Glasebrook A, et al. Cytokines in chronic inflammatory arthritis. I. Failure to detect T cell lymphokines (interleukin 2 and interleukin 3) and presence of macrophage colony-stimulating factor (CSF-1) and a novel mast cell growth factor in rheumatoid synovitis. J Exp Med. 1988;168(5):1573–1586. doi: 10.1084/jem.168.5.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294(5548):1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- Gong JH, Ratkay LG, Waterfield JD, Clark-Lewis I. An antagonist of monocyte chemoattractant protein 1 (MCP-1) inhibits arthritis in the MRL-lpr mouse model. J Exp Med. 1997;186(1):131–137. doi: 10.1084/jem.186.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S. Development and distribution of mononuclear phagocytes. In: Gallin J, Snyderman R, editors. Inflammation: Principles and Clincal Correlates. 3rd edn. Philadelphia: Lippincott Williams & Wilkins; 1999. pp. 35–48. [Google Scholar]
- Kawai S, Nishida S, Kato M, Furumaya Y, Okamoto R, Koshino T, et al. Comparison of cyclooxygenase-1 and-2 inhibitory activities of various nonsteroidal anti-inflammatory drugs using human platelets and synovial cells. Eur J Pharmacol. 1998;347(1):87–94. doi: 10.1016/s0014-2999(98)00078-8. [DOI] [PubMed] [Google Scholar]
- Kazmi SM, Plante RK, Visconti V, Taylor GR, Zhou L, Lau CY. Suppression of NF kappa B activation and NF kappa B-dependent gene expression by tepoxalin, a dual inhibitor of cyclooxygenase and 5-lipoxygenase. J Cell Biochem. 1995;57(2):299–310. doi: 10.1002/jcb.240570214. [DOI] [PubMed] [Google Scholar]
- Kelley TW, Graham MM, Doseff AI, Pomerantz RW, Lau SM, Ostrowski MC, et al. Macrophage colony-stimulating factor promotes cell survival through Akt/protein kinase B. J Biol Chem. 1999;274(37):26393–26398. doi: 10.1074/jbc.274.37.26393. [DOI] [PubMed] [Google Scholar]
- Kinne RW, Brauer R, Stuhlmuller B, Palombo-Kinne E, Burmester GR. Macrophages in rheumatoid arthritis. Arthritis Res. 2000;2(3):189–202. doi: 10.1186/ar86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp E, Ghosh S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science. 1994;265(5174):956–959. doi: 10.1126/science.8052854. [DOI] [PubMed] [Google Scholar]
- Kurowski M, Thabe H. The transynovial distribution of oxaprozin. Agents Actions. 1989;27:458–460. doi: 10.1007/BF01972852. [DOI] [PubMed] [Google Scholar]
- la Sala A, Gadina M, Kelsall BL. G(i)-protein-dependent inhibition of IL-12 production is mediated by activation of the phosphatidylinositol 3-kinase-protein 3 kinase B/Akt pathway and JNK. J Immunol. 2005;175(5):2994–2999. doi: 10.4049/jimmunol.175.5.2994. [DOI] [PubMed] [Google Scholar]
- Lavagno L, Gunella G, Bardelli C, Spina S, Fresu LG, Viano I, et al. Anti-inflammatory drugs and tumor necrosis factor-alpha production from monocytes: role of transcription factor NF-kappa B and implication for rheumatoid arthritis therapy. Eur J Pharmacol. 2004;501(1–3):199–208. doi: 10.1016/j.ejphar.2004.07.101. [DOI] [PubMed] [Google Scholar]
- Lee R, Collins T. Nuclear factor-kappaB and cell survival: IAPs call for support. Circ Res. 2001;88(3):262–264. doi: 10.1161/01.res.88.3.262. [DOI] [PubMed] [Google Scholar]
- Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2(10):725–734. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- Liew FY, McInnes IB. The role of innate mediators in inflammatory response. Mol Immunol. 2002;38(12–13):887–890. doi: 10.1016/s0161-5890(02)00014-7. [DOI] [PubMed] [Google Scholar]
- Liu H, Pope RM. The role of apoptosis in rheumatoid arthritis. Curr Opin Pharmacol. 2003;3(3):317–322. doi: 10.1016/s1471-4892(03)00037-7. [DOI] [PubMed] [Google Scholar]
- Lu B, Rutledge BJ, Gu L, Fiorillo J, Lukacs NW, Kunkel SL, et al. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J Exp Med. 1998;187(4):601–608. doi: 10.1084/jem.187.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov SS. NF-kappa B in rheumatoid arthritis: a pivotal regulator of inflammation, hyperplasia, and tissue destruction. Arthritis Res. 2001;3(4):200–206. doi: 10.1186/ar300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marone R, Cmiljanovic V, Giese B, Wymann MP. Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta. 2008;1784(1):159–185. doi: 10.1016/j.bbapap.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Marsh CB, Pomerantz RP, Parker JM, Winnard AV, Mazzaferri EL, Jr, Kelley TM, et al. Regulation of monocyte survival in vitro by deposited IgG: role of macrophage colony-stimulating factor. J Immunol. 1999;162(10):6217–6225. [PubMed] [Google Scholar]
- Mulherin D, Fitzgerald O, Bresnihan B. Synovial tissue macrophage populations and articular damage in rheumatoid arthritis. Arthritis Rheum. 1996;39(1):115–124. doi: 10.1002/art.1780390116. [DOI] [PubMed] [Google Scholar]
- Ottonello L, Bertolotto M, Montecucco F, Dapino P, Dallegri F. Dexamethasone-induced apoptosis of human monocytes exposed to immune complexes. Intervention of CD95-and XIAP-dependent pathways. Int J Immunopathol Pharmacol. 2005;18(3):403–415. doi: 10.1177/039463200501800302. [DOI] [PubMed] [Google Scholar]
- Ottonello L, Frumento G, Arduino N, Bertolotto M, Dapino P, Mancini M, et al. Differential regulation of spontaneous and immune complex-induced neutrophil apoptosis by proinflammatory cytokines. Role of oxidants, Bax and caspase-3. J Leukoc Biol. 2002;72(1):125–132. [PubMed] [Google Scholar]
- Palayoor ST, Youmell MY, Calderwood SK, Coleman CN, Price BD. Constitutive activation of IkappaB kinase alpha and NF-kappaB in prostate cancer cells is inhibited by ibuprofen. Oncogene. 1999;18(51):7389–7394. doi: 10.1038/sj.onc.1203160. [DOI] [PubMed] [Google Scholar]
- Pollard L, Choy EH, Scott DL. The consequences of rheumatoid arthritis: quality of life measures in the individual patient. Clin Exp Rheumatol. 2005;23(5) Suppl. 39:S43–S52. [PubMed] [Google Scholar]
- Pope RM. Apoptosis as a therapeutic tool in rheumatoid arthritis. Nat Rev Immunol. 2002;2(7):527–535. doi: 10.1038/nri846. [DOI] [PubMed] [Google Scholar]
- Rainsford K, Omar H, Ashraf A, Hewson A, Bunning R, Rishiraj R, et al. Recent pharmacodynamic and pharmacokinetic findings on oxaprozin. Inflammopharmacology. 2002;10:185–239. [Google Scholar]
- Riedl SJ, Renatus M, Schwarzenbacher R, Zhou Q, Sun C, Fesik SW, et al. Structural basis for the inhibition of caspase-3 by XIAP. Cell. 2001;104(5):791–800. doi: 10.1016/s0092-8674(01)00274-4. [DOI] [PubMed] [Google Scholar]
- van Roon JA, van Vuuren AJ, Wijngaarden S, Jacobs KM, Bijlsma JW, Lafeber FP, et al. Selective elimination of synovial inflammatory macrophages in rheumatoid arthritis by an Fcgamma receptor I-directed immunotoxin. Arthritis Rheum. 2003;48(5):1229–1238. doi: 10.1002/art.10940. [DOI] [PubMed] [Google Scholar]
- Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2(12):965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- Scheuren N, Bang H, Munster T, Brune K, Pahl A. Modulation of transcription factor NF-kappaB by enantiomers of the nonsteroidal drug ibuprofen. Br J Pharmacol. 1998;123(4):645–652. doi: 10.1038/sj.bjp.0701652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DL, Smith C, Kingsley G. What are the consequences of early rheumatoid arthritis for the individual? Best Pract Res Clin Rheumatol. 2005;19(1):117–136. doi: 10.1016/j.berh.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Solbach W, Moll H, Rollinghoff M. Lymphocytes play the music but the macrophage calls the tune. Immunol Today. 1991;12(1):4–6. doi: 10.1016/0167-5699(91)90103-Z. [DOI] [PubMed] [Google Scholar]
- Stuhlmeier KM, Li H, Kao JJ. Ibuprofen: new explanation for an old phenomenon. Biochem Pharmacol. 1999;57(3):313–320. doi: 10.1016/s0006-2952(98)00301-3. [DOI] [PubMed] [Google Scholar]
- Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J Clin Invest. 2001;107(1):7–11. doi: 10.1172/JCI11830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada Y, Bhardwaj A, Potdar P, Aggarwal BB. Nonsteroidal anti-inflammatory agents differ in their ability to suppress NF-kappaB activation, inhibition of expression of cyclooxygenase-2 and cyclin D1, and abrogation of tumor cell proliferation. Oncogene. 2004;23(57):9247–9258. doi: 10.1038/sj.onc.1208169. [DOI] [PubMed] [Google Scholar]
- Tang ED, Inohara N, Wang CY, Nunez G, Guan KL. Roles for homotypic interactions and transautophosphorylation in IkappaB kinase beta IKKbeta) activation [corrected. J Biol Chem. 2003;278(40):38566–38570. doi: 10.1074/jbc.M304374200. [DOI] [PubMed] [Google Scholar]
- Todd P, Brogden R. Oxaprozin: a preliminary review of its pharmacodynamic and phamacokinetic properties and therapeutic effects. Drugs. 1986;32:291–312. doi: 10.2165/00003495-198632040-00001. [DOI] [PubMed] [Google Scholar]
- Vane JR, Botting RM. Mechanism of action of nonsteroidal anti-inflammatory drugs. Am J Med. 1998;104(3A):2S–8S. doi: 10.1016/s0002-9343(97)00203-9. discussion 21S–22S. [DOI] [PubMed] [Google Scholar]
- Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005;30(1):43–52. doi: 10.1016/j.tibs.2004.11.009. [DOI] [PubMed] [Google Scholar]
- van Vuuren AJ, van Roon JA, Walraven V, Stuij I, Harmsen MC, McLaughlin PM, et al. CD64-directed immunotoxin inhibits arthritis in a novel CD64 transgenic rat model. J Immunol. 2006;176(10):5833–5838. doi: 10.4049/jimmunol.176.10.5833. [DOI] [PubMed] [Google Scholar]
- Xu WD, Firestein GS, Taetle R, Kaushansky K, Zvaifler NJ. Cytokines in chronic inflammatory arthritis. II. Granulocyte-macrophage colony-stimulating factor in rheumatoid synovial effusions. J Clin Invest. 1989;83(3):876–882. doi: 10.1172/JCI113971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XJ, Reichner JS, Mastrofrancesco B, Henry WL, Jr, Albina JE. Prostaglandin E2 suppresses lipopolysaccharide-stimulated IFN-beta production. J Immunol. 2008;180(4):2125–2131. doi: 10.4049/jimmunol.180.4.2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y, Yin MJ, Lin KM, Gaynor RB. Sulindac inhibits activation of the NF-kappaB pathway. J Biol Chem. 1999;274(38):27307–27314. doi: 10.1074/jbc.274.38.27307. [DOI] [PubMed] [Google Scholar]
- Yamazaki R, Kusunoki N, Matsuzaki T, Hashimoto S, Kawai S. Nonsteroidal anti-inflammatory drugs induce apoptosis in association with activation of peroxisome proliferator-activated receptor gamma in rheumatoid synovial cells. J Pharmacol Exp Ther. 2002;302(1):18–25. doi: 10.1124/jpet.302.1.18. [DOI] [PubMed] [Google Scholar]
- Yin MJ, Yamamoto Y, Gaynor RB. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature. 1998;396(6706):77–80. doi: 10.1038/23948. [DOI] [PubMed] [Google Scholar]