

Abstract
Background and purpose:
Activation of post-synaptic 5-HT1A receptors may provide enhanced therapy against depression. We describe the signal transduction profile of F15599, a novel 5-HT1A receptor agonist.
Experimental approach:
F15599 was compared with a chemical congener, F13714, and with (+)8-OH-DPAT in models of signal transduction in vitro and ex vivo.
Key results:
F15599 was highly selective for 5-HT1A receptors in binding experiments and in [35S]-GTPγS autoradiography of rat brain, where F15599 increased labelling in regions expressing 5-HT1A receptors. In cell lines expressing h5-HT1A receptors, F15599 more potently stimulated extracellular signal-regulated kinase (ERK1/2) phosphorylation, compared with G-protein activation, internalization of h5-HT1A receptors or inhibition of cAMP accumulation. F13714, (+)8-OH-DPAT and 5-HT displayed a different rank order of potency for these responses. F15599 stimulated [35S]-GTPγS binding more potently in frontal cortex than raphe. F15599, unlike 5-HT, more potently and efficaciously stimulated Gαi than Gαo activation. In rat prefrontal cortex (a region expressing post-synaptic 5-HT1A receptors), F15599 potently activated ERK1/2 phosphorylation and strongly induced c-fos mRNA expression. In contrast, in raphe regions (expressing pre-synaptic 5-HT1A receptors) F15599 only weakly or did not induce c-fos mRNA expression. Finally, despite its more modest affinity in vitro, F15599 bound to 5-HT1A receptors in vivo almost as potently as F13714.
Conclusions and implications:
F15599 showed a distinctive activation profiles for 5-HT1A receptor-mediated signalling pathways, unlike those of reference agonists and consistent with functional selectivity at 5-HT1A receptors. In rat, F15599 potently activated signalling in prefrontal cortex, a feature likely to underlie its beneficial effects in models of depression and cognition.
Keywords: antidepressant, serotonin, 5-HT1A, G-protein, ERK1/2 phosphorylation, autoradiography, c-fos, agonist-directed trafficking of receptor signalling
Introduction
5-HT1A receptors play a key role in 5-hydroxytryptaminergic transmission due to their localization both as pre-synaptic receptors located on 5-HT cell bodies in the raphe nuclei and as post-synaptic receptors in brain areas associated with mood, cognition and pain (Lucki, 1998; Colpaert et al., 2002; Blier and Ward, 2003; Colpaert, 2006; Millan, 2006). In particular, 5-HT1A receptors are profoundly involved in the mechanism of action of antidepressants (Blier and Ward, 2003; Celada et al., 2004). Thus, 5-HT1A receptor agonists are potently active in rodent models of depression such as the forced swim test, completely reversing upon acute administration the immobility that is taken as a measure of ‘helplessness’ (De Vry and Schreiber, 1993; De Vry, 1995; Koek et al., 1998; 2001). Conversely, the antidepressant-like activities in rats of the selective 5-HT (serotonin) reuptake inhibitor (SSRI), fluoxetine, the selective noradrenaline reuptake inhibitor, desipramine and the non-selective (5-HT and noradrenaline) re-uptake inhibitor, venlafaxine are inhibited by 5-HT1A receptor antagonists (Detke et al., 1995; De Vry et al., 2004; Berrocoso and Mico, 2008). Further, the influence of fluoxetine in a strain of mice selected for their ‘depressive’ behaviour is accompanied by changes in 5-HT1A receptor expression levels (Naudon et al., 2002; El Yacoubi et al., 2003). In electrophysiological studies in rats, a variety of antidepressant treatments, as well as 5-HT1A agonists, share a common influence: desensitization of pre-synaptic 5-HT1A receptors in the raphe nuclei and increased response of post-synaptic 5-HT1A receptors in limbic regions (e.g. hippocampus; Haddjeri et al., 1998; 1999). When 5-HT neurons are lesioned, eliminating pre-synaptic 5-HT1A receptors, antidepressant-like actions of 5-HT1A agonists in mice are maintained, indicating that reduction of depressive-like behaviour is mediated by post-synaptic 5-HT1A receptors (Matsuda et al., 1995). Further, post-synaptic 5-HT1A receptor control of hippocampal pyramidal neuron firing is amplified by preferential antagonism of pre-synaptic 5-HT1A receptors with pindolol (Romero et al., 1996; Béïque et al., 2000). The involvement of 5-HT1A receptors in depressive states is supported by the decrease in both forebrain and dorsal raphe 5-HT1A receptor binding potential in positron emission tomography scans of depressed humans and primates (Bhagwagar et al., 2004; Shively et al., 2006). Further, antidepressant treatment normalizes 5-HT1A receptor binding potential in depressed subjects (Parsey et al., 2005). Several weeks of treatment with SSRIs are necessary for pre-synaptic 5-HT1A receptors to be desensitized (Invernizzi et al., 1994; Rutter et al., 1994) allowing antidepressant efficacy to manifest itself. Accordingly, combined treatment of depressed patients with SSRIs and pindolol accelerates the onset of therapeutic efficacy of antidepressants (see Artigas et al., 2006). 5-HT1A partial agonists (such as the anxiolytics, buspirone and gepirone) are not effective as antidepressants in monotherapy, probably due to insufficient efficacy at post-synaptic 5-HT1A receptors and/or metabolic instability. However, when feedback inhibition of pre-synaptic 5-HT1A receptors was blocked with pindolol, buspirone did exert clinical antidepressant influence, presumably through activation of post-synaptic 5-HT1A receptors (Blier et al., 1997).
All these data strongly suggest that direct targeting of post-synaptic 5-HT1A receptors with a high-efficacy agonist may provide accelerated and more effective antidepressant therapy. Indeed, although only a single 5-HT1A receptor gene sequence has been identified, post-synaptic 5-HT1A receptors exhibit distinct control of downstream signal transduction events. Thus, chronic administration of fluoxetine or imipramine desensitizes 5-HT1A receptor-mediated G-protein activation in the raphe, hypothalamus and lateral septum but not in frontal cortex or hippocampus (Hensler, 2002; Pejchal et al., 2002; Shen et al., 2002; Castro et al., 2003; see Hensler, 2003 for review). Go and Gi2 G-proteins are down-regulated, but Gs up-regulated, in the prefrontal cortex of suicide victims (Dwivedi et al., 2002). Indeed, Gs localization in brain of depressed suicides is preferentially shifted to lipid raft domains, with decreased coupling to adenylyl cyclase (Donati et al., 2008). Pre-synaptic 5-HT1A receptors in raphe are preferentially coupled to Gi3 G-proteins whereas post-synaptic 5-HT1A receptors in hippocampus and cortex preferentially couple to Go and Gi3 proteins (Mannoury et al., 2006). Further, post-synaptic 5-HT1A receptors in hypothalamus also couple to other G-protein subtypes (Gi1 and Gz; Raap et al., 1999; Serres et al., 2000). Pre-synaptic 5-HT1A receptors in raphe inhibit inositol phosphate but not adenylyl cyclase formation in rat tissue (Johnson et al., 1997; Marazziti et al., 2002). In contrast, post-synaptic 5-HT1A receptors in hippocampus and cortex inhibit adenylyl cyclase but not inositol phosphate formation (Clarke et al., 1996; Johnson et al., 1997). In ex vivo experiments, 5-HT1A receptor activation stimulates extracellular signal-regulated kinase (ERK1/2) phosphorylation in the raphe and the paraventricular nucleus (Sullivan et al., 2005; Crane et al., 2007) but inhibits it in the hippocampus (Chen et al., 2002; Crane et al., 2007), indicating brain region specificity in a ‘downstream’ signalling response. A single-nucleotide polymorphism (C-1019G substitution; Lesch and Gutknecht, 2004) impairs repression of the 5-HT1A promoter in raphe cells, leading to excessive expression of pre-synaptic 5-HT1A receptors (Lemonde et al., 2003; Parsey et al., 2006a). In patients receiving antidepressant treatments, the C-1019G polymorphism is associated with higher levels of remission failure (Lemonde et al., 2004; Parsey et al., 2006b), probably due to overactive pre-synaptic 5-HT1A receptors. In contrast, in cortical neurons, the C-1019G polymorphism leads to 5-HT1A receptor hypofunction (Czesak et al., 2006), suggesting that increased stimulation of receptors in cortex is necessary to alleviate depressive symptoms.
Thus, drugs exhibiting 5-HT1A receptor functional selectivity (i.e. agonist-directed trafficking of receptor signalling; Kenakin, 1995; Newman-Tancredi et al., 2003) would be expected to exert brain region-specific activity on 5-hydroxytryptaminergic neurotransmission. Recently, we identified a novel, highly selective and efficacious 5-HT1A receptor agonist, F15599. Neurochemical, electrophysiological and behavioural data indicate that this compound preferentially activates post-synaptic 5-HT1A receptors. Thus, F15599 influences frontal cortex pyramidal neuron electrical activity at doses that are an order of magnitude lower than those that inhibit raphe neuron electrical activity (Celada et al., 2007). Further, F15599 increases dopamine release in frontal cortex at low doses (reflecting post-synaptic 5-HT1A receptor activation) whereas higher doses are necessary to inhibit 5-HT release in hippocampus (a pre-synaptic 5-HT1A receptor response; Assiéet al., 2006a). The preferential activation of post-synaptic 5-HT1A receptors by F15599 is accompanied by a remarkable capacity to reverse memory/cognition deficits (Auclair et al., 2007) and potent activity in the forced swim test in rats (Assiéet al., 2006a).
The present study describes the receptor interaction and signal transduction profile of F15599 both in vitro and ex vivo in comparison with a chemical congener, F13714 and the more active (+) enantiomer of the prototypical 5-HT1A receptor agonist, 8-OH-DPAT. These ligands displayed distinct signalling profiles, suggesting that functional selectivity of F15599 for specific receptor activation responses is likely to underlie its favourable profile in models of cognitive and mood deficits.
Methods
The activity of F15599 and reference agonists at 5-HT1A receptors was characterized using a series of methods designed to investigate different levels of drug action. These included: (i) ligand binding, to determine affinity directly at 5-HT1A receptors in vitro and in vivo receptor occupancy in mice; (ii) in vitro signal transduction measures representing a cascade of responses to 5-HT1A agonism. These included ‘total’ G-protein activation (i.e. without distinguishing the subtypes of G-protein involved), Gαi and Gαo G-protein subtype targeting, adenylyl cyclase inhibition, ERK1/2 phosphorylation and receptor internalization. The purpose was to determine whether the pattern of activation of these different signalling responses was agonist-specific; and (iii) Ex vivo measures of ERK1/2 phosphorylation and c-fos gene expression in various brain regions in rat. The object was to determine whether F15599 influenced these responses in a brain region-specific manner. Details of the methods employed are described below. The molecular target nomenclature (receptors, ion channels, etc.) is expressed according to the BJP Guide to Receptors and Channels (Alexander et al., 2008).
Animals
Animals were housed and tested in an Association for the Assessment and Accreditation of Laboratory Animal Care International-accredited facility in strict compliance with all applicable regulations and the protocols were carried out in compliance with French regulations and with local ethical committee guidelines for animal research.
Competition binding methods
Competition binding experiments were carried out using the radioligands, buffer and incubation conditions outlined in Table 1 and described by Newman-Tancredi et al. (2007). Experiments at native rat receptors employed brains of male Sprague–Dawley rats [Ico: OFA SD (S.P.F. Caw); Iffa Credo, France], weighing 180–200 g. Animals were decapitated and brains were rapidly removed and stored at −80°C before use in binding assays. For native 5-HT2C receptor binding assays (Pazos et al., 1985a,b), pig cortex was obtained from the local slaughter house.
Table 1.
In vitro competition binding methods for determination of affinity at various monoamine receptors
| Receptor | Tissue | [3H]Radioligand (nmol·L−1) | Kd (nmol·L−1) | Non-specific (µmol·L−1) | Incubation (h) temperature (°C) | Buffer |
|---|---|---|---|---|---|---|
| h 5-HT1A | CHO cells | 8-OH-DPAT (1) | 0.68 | 5-HT (10) | 2, 23 | A |
| r 5-HT1A | Rat hippocampus | 8-OH-DPAT (1) | 0.75 | 5-HT (10) | 2, 23 | B |
| r 5-HT1B | Rat cortex | GR125,743 (1) | 0.36 | 5-HT (10) | 2, 23 | C |
| r 5-HT2A | Rat cortex | Ketanserin (0.2) | 3.1 | Methysergide (10) | 0.5, 23 | C |
| p 5-HT2C | Pig cortex | Mesulergine (1) | 4.8 | Mianserin (10) | 1, 23 | C |
| r α1 | Rat cortex | Prazosin (0.2) | 0.07 | Phentolamine (5) | 2, 23 | B |
| r α2 | Rat cortex | RX 821002 (0.5) | 0.49 | Phentolamine (5) | 2, 23 | B |
| r D1 | Rat striatum | SCH 23390 (1) | 0.45 | SKF 38393 (10) | 2, 37 | D |
| r D2 | Rat striatum | Nemonapride (0.2) | 0.05 | (+)Butaclamol (1) | 2, 23 | D |
Further methodological details are given in Newman-Tancredi et al. (2007).
D1, D2, dopamine receptors; h, human; p, porcine; r, rat; α1, α2, α-adrenoceptors; buffer A, HEPES 20 mmol·L−1 pH 7.4; buffer B, Tris HCl 50 mmol·L−1 pH 7.4; buffer C, Tris HCl 50 mmol·L−1 pH 7.4, pargyline 10 µmol·L−1, CaCl2 4 mmol·L−1, ascorbic acid 0.1%; buffer D, Tris HCl 50 mmol·L−1 pH 7.4, NaCl 120 mmol·L−1, KCl 5 mmol·L−1.
Binding experiments at human 5-HT1A receptors were carried out using membranes from stably transfected CHO cells as described previously (see references in Table 2). Experiments were terminated by rapid filtration through Whatman GF-B filters. Radioactivity retained on the filters was measured by liquid scintillation spectroscopy. Data from native tissue receptors were analysed using the non-linear curve fitting program KELL RADLIG version 6 (Biosoft, Cambridge, UK; McPherson, 1985) and pKi values are from at least three experiments, each comprising six to seven concentrations differing by one log unit interval. Data from human cloned receptor binding experiments were analysed using GraphPad Prism (GraphPad Software Inc., San Diego, CA), and pKi values are expressed as mean ± SEM of at least three experiments each comprising 7–10 concentrations differing by 0.5 or 1 log unit interval.
Table 2.
Summary of methods for determination of functional responses at recombinant human and native rat 5-HT1A receptors in vitro
| Functional measure | Tissue/cell line | Incubation buffer | Incubation time (min) and temperature (°C) | Literature reference |
|---|---|---|---|---|
| ‘Total’ G-protein activationa | C6-h5-HT1A glial cell memb | Buffer A | 30, 23 | Koek et al. (2001) |
| ‘Total’ G-protein activation | HeLa-h5-HT1A cell memb | Buffer B | 60, 30 | Newman-Tancredi et al. (2005) |
| Gαi activation | HeLa-h5-HT1A cell memb | Buffer C | 60, 23 | Newman-Tancredi et al. (2002) |
| cAMP formation | HeLa-h5-HT1A cell memb | Buffer D | 10, 23 | Newman-Tancredi et al. (2005) |
| ERK1/2 phosphorylation | CHO-h5-HT1A whole cells | RPMI serum-free | 5, 37 | Bruins Slot et al. (2006) |
| 5-HT1A internalization | HEK293-h5-HT1A whole cells | DMEM serum-free | 120, 37 | Heusler et al. (2008) |
| ‘Total’ G-protein activation | Rat hippocampal memb | Buffer E | 60, 37 | Newman-Tancredi et al. (2005) |
| Gαo activation | Rat hippocampal memb | Buffer F | 60, 23 | Martel et al. (2006) |
G-protein activation experiments were carried out by [35S]-GTPγS binding to cell membrane (memb) preparations. Other measures (cAMP formation and ERK1/2 phosphorylation and receptor internalization) were carried out on whole cells.
‘Total’ G-protein activation refers to [35S]-GTPγS binding that does not distinguish between the G-protein subtypes involved.
Buffer A, 20 mmol·L−1 HEPES, pH 7.4, 100 mmol·L−1 NaCl, 3 mmol·L−1 MgCl2, 30 µmol·L−1 GDP, 0.2 mmol·L−1 ascorbic acid; buffer B, 20 mmol·L−1 HEPES, pH 7.4, 100 mmol·L−1 NaCl, 10 mmol·L−1 MgCl2, 30 µmol·L−1 GDP, 10 µmol·L−1 pargyline; buffer C, 20 mmol·L−1 HEPES, pH 7.4, 100 mmol·L−1 NaCl, 3 mmol·L−1 MgCl2, 3 µmol·L−1 GDP; buffer D, DMEM + 10 mmol·L−1 HEPES, pH 7.4, 100 µmol·L−1 forskolin, 100 µmol·L−1 isobutylmethylxanthine; buffer E, 50 mmol·L−1 HEPES, pH 7.4, 150 mmol·L−1 NaCl, 5 mmol·L−1 MgCl2, 100 µmol·L−1 GDP, 0.2 mmol·L−1 EDTA, 0.2 mmol·L−1 DTT; buffer F, 20 mmol·L−1 HEPES, pH 7.4, 100 mmol·L−1 NaCl, 5 mmol·L−1 MgCl2, 50 µmol·L−1 GDP, 0.2 mmol·L−1 EDTA, 0.2 mmol·L−1 DTT.
Functional responses to F15599 at recombinant human 5-HT1A receptors
The agonist properties of F15599 at h5-HT1A receptors expressed in transfected cell lines were determined in vitro for measures of signal transduction representing different levels of intracellular responses: activation of G-proteins, inhibition of adenylyl cyclase activity, phosphorylation of ERK1/2 and induction of (HA-tagged) h5-HT1A receptor internalization. The methodologies employed were as described previously in studies of h5-HT1A receptor signalling (see literature references and methodological summary shown in Table 2). In the case of G-protein activation experiments, two test conditions were used: [35S]-GTPγS binding to C6-h5-HT1A glial cell membranes and to HeLa-h5-HT1A cell membranes. Membranes from these cell lines express similar levels of h5-HT1A receptors (1110 and 1280 fmol receptor per mg protein respectively). C6-h5-HT1A glial cells have been previously shown to be suitable to discriminate agonists with high efficacy (Pauwels et al., 1997). Thus, lower Emax values are observed under these conditions, relative to 5-HT. HeLa-h5-HT1A cells are more sensitive and partial agonists are more easily detected therein (Cosi and Koek, 2000). These cells were used in order to provide a comparison with experiments on cAMP inhibition and Gαi3 activation, which were carried out in the same cell line.
Concentration-response isotherms were analysed by non-linear regression, using GraphPad Prism. The value of the minimum and maximum asymptotes was not fixed. The latter (Emax) is expressed as a percentage of the effect observed with 5-HT (10 µmol·L−1). KB values of antagonists for inhibition of agonist action were calculated according to Lazareno and Birdsall (1993):
where IC50 = inhibitory concentration50 of antagonist, agonist = concentration of agonist in the test and EC50 = effective concentration50 of agonist.
G-protein activation by F15599 at native rat 5-HT1A receptors in rat brain regions
Following death and removal of brain from the skull, brains were rapidly cooled over liquid nitrogen before dissection. Large brain structures (frontal cortex and hippocampus) were dissected macroscopically while other structures (lateral septum, entorhinal cortex and raphe nuclei) were micro-dissected on a cold plate as described by Palkovits and Brownstein (1987). Briefly, micro-dissections were performed with a 1 mm circular punch on 300-µm-thick coronal sections in the range from 2400 to 600 µm anterior to bregma for lateral septum, from 4000 to 8400 µm posterior to bregma for entorhinal cortex, and from 6900 to 8400 µm posterior to bregma for dorsal and median raphe. Tissues and punches were collected in microtubes, frozen on dry ice and stored at −80°C until used.
On the day of experiment, tissues were suspended in 20 volumes (v/w) of ice cold 50 mmol·L−1 HEPES containing 150 mmol·L−1 NaCl, 0.2 mmol·L−1 EDTA, 10 µmol·L−1 pargyline (pH 7.4 at room temperature), 1 mmol·L−1 GTP, homogenized and incubated at 37°C for 10 min in order to dissociate the endogenous neurotransmitters from the receptors. The homogenate was centrifuged at 20 000× g for 15 min at 4°C. The pellet was washed twice by two successive runs of homogenization/centrifugation in the same buffer containing successively 1 mmol·L−1 and 100 µmol·L−1 GDP instead of GTP. The final pellet was resuspended in 600 volumes (v/w from original weight) of the same buffer containing 100 µmol·L−1 GDP and 5 mmol·L−1 MgCl2. Membranes (30–40 µg protein per tube) were incubated in the presence of different concentrations of drugs ranging from 10−10 to 10−5 mol·L−1, together with 0.1 nmol·L−1 [35S]-GTPγS for 1 h at 37°C. Non-specific binding was determined with 10 µmol·L−1 unlabelled GTPγS. Other experimental controls included basal and 5-HT (10 µmol·L−1)-stimulated [35S]-GTPγS binding.
G-protein subtype targeting experiments using scintillation proximity assay
Rat hippocampi were homogenized in cold HEPES-dithiothreitol (DTT) buffer (20 mmol·L−1 HEPES pH 7.0, 0.2 mmol·L−1 EDTA and 0.2 mmol·L−1 DTT) with 1 mmol·L−1 GTP using a Polytron homogenizer: GTP was added to induce dissociation of endogeneous ligand. Homogenates were incubated at 35°C for 15 min to favour endogenous ligand dissociation, and washed by two cycles of centrifugation at 20 000× g for 15 min at 4°C and resuspension in cold HEPES-DTT with 1 mmol·L−1 GDP. Final pellets were resuspended in 300 volumes (based on original tissue weight) of HEPES-DTT buffer containing 100 mmol·L−1 NaCl, 5 mmol·L−1 MgCl2 and 50 µmol·L−1 GDP (rat hippocampus assay buffer) to give a final protein concentration of 8 µg per well. Membranes from HeLa-h5-HT1A cells were diluted in 20 mmol·L−1 HEPES pH 7.0 containing 100 mmol·L−1 NaCl, 3 mmol·L−1 MgCl2 and 3 µmol·L−1 GDP (HeLa-h5-HT1A assay buffer) to give a final protein concentration of 50 µg per well. All reactions were performed at room temperature using 96-well plates, in a final volume of 250 µL of the appropriate assay buffer. Membranes were incubated at room temperature with drugs (10−11 to 10−5 mol·L−1), buffer (to define basal) or 10 µmol·L−1 GTPγS (to define non-specific) and 0.4 nmol·L−1 (rat hippocampus) or 0.2 nmol·L−1 (HeLa-h5-HT1A) [35S]-GTPγS for 60 min. Incubations were stopped by adding Nonidet NP-40 and the plate was agitated for another 30 min before addition of 0.2 µg anti-Gα-selective antibodies to each well. The antibodies used were mouse monoclonal anti-Gαi1 and anti-Gαo from Biomol (Plymouth Meeting, PA). Antibody specificity was assessed by Western blot as described (Cussac et al., 2002; 2004). Anti-Gαo is highly selective (Martel et al., 2007) while some cross-reactivity was observed for anti-Gαi1 antibody with Gαi3 (Cussac et al, 2002). As HeLa-h5-HT1A cells express undetectable levels of Gαi1 (D. Cussac, unpubl. obs.), the response detected in this cell line with anti-Gαi1 antibodies is more likely to correspond to a stimulation of Gαi3 proteins. The primary antibodies were left to react for 60 min under agitation before adding 50 µL of the secondary antibodies [anti-mouse coupled to scintillation proximity assay (SPA) beads, GE Healthcare, Amersham, UK], diluted according to manufacturer's recommendations. The secondary antibodies were left to react for another 60 min and the plate was centrifuged at 1000× g for 15 min at room temperature and radioactivity was immediately measured on a Top-Count scintillation counter (Perkin-Elmer Life Sciences, Courtaboeuf, France). Raw data [disintegrations per minute (DPM)] were analysed by subtracting non-specific binding and expressed as percentage of basal [35S]-GTPγS binding. All pharmacological parameters were derived from sigmoid non-linear regression using Graphpad Prism.
Functional autoradiography: specific activation of 5-HT1A receptors by F15599
[35S]-GTPγS autoradiography was carried out essentially as described by Newman-Tancredi et al. (2003). Frozen rat brains were cut horizontally in 20 µm serial sections using a cryostat at −20°C, fixed on microscope slides and were kept frozen until assayed. The assay was performed by pre-incubating the slides at room temperature for 15 min in buffer A (50 mmol·L−1 HEPES buffer containing 150 mmol·L−1 NaCl, 0.2 mmol·L−1 EGTA and 0.2 mmol·L−1 DTT) plus 2.5 mmol·L−1 GTP, 15 min in buffer A plus 2.5 mmol·L−1 GDP and 15 min in buffer A with 2.5 mmol·L−1 GDP, 10 mmol·L−1 MgCl2 and 100 mU·mL−1 adenosine deaminase (= buffer B) plus antagonist. Sections were then incubated for 60 min at 37°C in buffer B containing 0.05 nmol·L−1 [35S]-GTPγS and drugs. Basal [35S]-GTPγS binding was defined as that observed in the absence of drugs, and non-specific binding was defined as the binding in the presence of 100 µmol·L−1 unlabelled GTPγS. At the end of the incubation period, sections were rapidly washed twice for 2 min in cold buffer B (4°C), and rapidly dried under a flow of cold air. Dried sections together with [14C] radioactivity standards were placed in X-ray cassettes, apposed to Biomax films and exposed for 4.5 days. Films were developed and radioactivity on sections was quantified using an image analysis system (AIS system, InterFocus Ltd, Linton, UK). Grey levels were converted to nCi·g−1 equivalents using [14C] radioactivity standards, and radioactivity was measured on each structures/sections.
In vivo 5-HT1A receptor binding of F15599 in mice
Male NMRI mice [Ico: NMRI (S.P.F.) Han; Iffa Credo, France], weighing 20–22 g upon arrival, were group-housed (12 mice per cage) in the animal facility, under controlled conditions (12/12 h light/dark cycle: lights on at 7.00 a.m.; ambient temperature 21 ± 1°C; humidity 55 ± 5%), with rodent food (AO4, UAR, France) and filtered (0.2 µm pore diameter) tap water available ad libitum. At least 5 days were allowed for adaptation before the mice were used in the experiments. The doses of compounds were expressed as the weight of the base. The compounds were dissolved in distilled water and the administration volume was 10 mL·kg−1.
On the day of the experiment, mice were housed individually with free access to food and water. Each animal was treated with the drug or saline and returned to its cage. Thirty minutes later, the mouse was gently introduced and maintained in a Plexiglas cylinder, with the tail protruding, and 4 µCi in 0.2 mL (48 pmol) of a [3H]WAY100635 solution was slowly injected into the caudal vein and the mouse was returned to its cage. One hour after administration of the test compound, the animal was decapitated, the frontal cortex and hippocampus dissected out, weighed and kept frozen on dry ice. At the end of the experiment, the tissue was thawed and homogenized (10 s with an Ultra-Turrax) in 3 mL distilled water. The radioactivity in 0.5 mL samples was counted using liquid scintillation spectroscopy (Packard, TriCarb 2500). Data were expressed as mean ± SEM dpm·(mg wet tissue weight)−1 (n = 5 to 8 animals per group). Data were analysed using GraphPad Prism software to determine the ID50. For each compound and each brain region, the maximum value of the curve was constrained at the value of control animals. Dose–response data were analysed by one-way anova, followed by Dunnett's test.
Modulation of ERK1/2 phosphorylation by F15599 in rat brain
Adult male Sprague–Dawley rats were injected intraperitoneally (i.p.) with F15599 (0.04–2.5 mg·kg−1) or saline (n = 6 per group). Thirty minutes after injection, rats were decapitated. In 5-HT1A receptor antagonist studies, WAY100635 (0.63 mg·kg−1) or saline was injected subcutaneously 30 min prior to the i.p. injection of F15599 (0.63 mg·kg−1 i.p.) or saline, and rats were decapitated 30 min after the i.p. injection. The 0.63 mg·kg−1 dose of WAY100635 was chosen from previous data showing this dose to antagonize the effects of the other selective high-efficacy 5-HT1A receptor agonists, F13640 (Bardin et al., 2003; Buritova et al., 2003) and F13714 (Assiéet al., 2006b) in the rat.
Following decapitation, rat brains were cooled in vaporized liquid nitrogen for 1 min and the brain regions of interest (entire hippocampus, prefrontal cortex and entire hypothalamus) then immediately dissected. Each dissected brain region was put into Lysing Matrix D tube (MP, Illkirch, France) filled with 700 µL of cell extraction buffer at 4°C (Biosource, Nivelles, Belgium) supplemented with protease inhibitors, phosphatase inhibitors and urea (7 mol·L−1). Tissues were homogenized, centrifuged and supernatant was stored frozen until pERK1/2 quantification. The latter was carried out by quantitative ELISA assay (Bruins Slot et al., 2006), using kits from Biosource (ref. KHO0091) according to the manufacturer's instructions. Samples of homogenized rat brain tissues were assayed for content of pERK1/2 and total ERK1/2 (n = 6 and 3 rats per group respectively). For each brain region studied, effects of the treatments were compared by anova with the Fisher test). The pair-wise comparisons of treatment groups were carried out by the contrast method (using the Student's t distribution). The significance level was fixed at 5%.
Induction of c-fos mRNA expression by F15599 in rat brain
For quantitation of c-fos mRNA, rats were treated as above for ERK1/2 phosphorylation and tissue specimens from eight different brain regions (see Figure 7 later) were dissected. These were mechanically homogenized and total RNAs were extracted using a commercially available kit (Rneasy Mini Kit; Qiagen, France) and including a DNAse treatment step (Qiagen, France) following the manufacturer's protocol. The quality and quantity of the RNA samples were determined by analysis using an Agilent 2001 Bioanalyser (France). 100 ng RNA (for raphe nuclei) and 1 µg RNA (for others structures) were reverse-transcribed in a 15 µL final volume following the Iscript cDNA synthesis protocol from BioRad (France). Cycle settings were as follows: 5 min at room temperature to allow primer alignment, then 45 min at 42°C and 5 min at 85°C.
Figure 7.
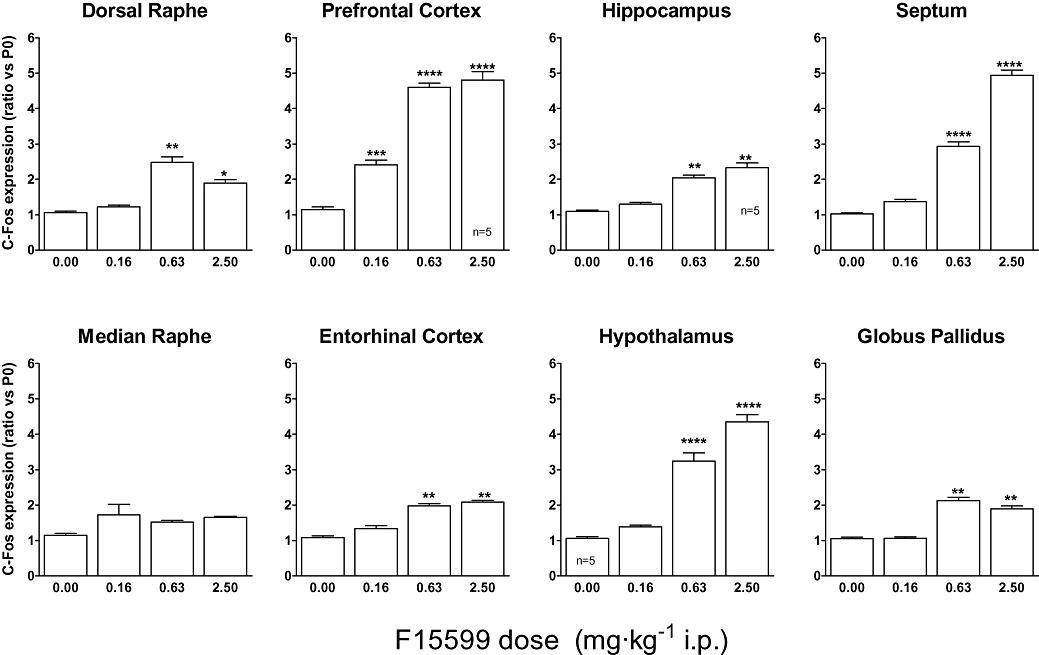
Influence of F15599 on c-fos mRNA induction by F15599 in different rat brain regions. The mean value of the control group (saline-treated animals) was set at 1 (basal value) using Arbp (i.e. P0) as the reference gene for normalization. All data were expressed as fold induction relative to basal value. Except where indicated otherwise, bars represent the mean ± SEM of values from six animals. anova with Tukey's test was used to evaluate differences in c-fos expression between treatment groups. The significance level is fixed at 5%.
Quantitative polymerase chain reaction measures were performed in an IQ thermocycler in 96-well microtitre plates using a 25 µL final volume with a mix composed of 400 nmol·L−1 of specific primers, 7.3 µL of water, 12.5 µL of the iQ SYBR Green Supermix from Biorad (France) and 5 µL of diluted cDNA. Thermal cycling parameters were 2 min at 95°C, followed by 40 cycles at 94°C for 30 s, 30 s at 60°Cj and 30 s at 72°C. To avoid amplification of contaminating genomic DNA, c-fos primers were placed in two different exons and one of the housekeeping gene Arbp primers (acidic ribosomal phosphoprotein P0) was placed at a junction between two exons. Primers were designed from sequences in the Genbank and Ensembl database using Primer 3 software and I.D.T. (Integrated DNA technologies) software in order to check the absence of dimer or short hairpin sequences. Each primer pair was then validated to verify that their amplification efficacy was ≥95%, and their amplicons were specific (as verified by a melting curve). Primer alignment was then validated by blast on NCBI. Primer sequences were as follows: c-fos forward (NM_022197) AAGGGAAAGGAATAAGATGG and c-fos reverse TCTTCAAGTTGATCTGTCTCC (94 bp product); Arbp forward (NM_022402) GGTACCATTGAAATCCTGAG and Arbp reverse GTTCAACATGTTCAGCAGTG (90 bp product). Results, calculated using the comparative 2ΔΔCt method, are shown as a ratio of c-fos expression relative to that of the housekeeping Arbp (i.e. P0) gene. Arbp was previously chosen among six housekeeping genes as being the most stable between different treatments. Histograms (see Figure 7 later) show mean ± SEM from n = 6 rats per group. Statistical analysis using sas software was conducted by anova, followed by Tukey's test, to evaluate differences in c-fos expression between treatment groups. The significance level was fixed at 5%. The mean value of the control group (saline-treated animals) was set at 1 (basal value) and all data were normalized versus the saline-treated group.
Drugs
F15599 (3-Chloro-4-fluorophenyl-(4-fluoro-4-{[(5-methylpyrimidin-2-ylmethyl)-amino]-methyl}-piperidin-1-yl)-methanone) and F13714 (3-chloro-4-fluorophenyl-(4-fluoro-4-{[(5-methyl-6-methylaminopyridin-2-ylmethyl)-amino]-methyl}-piperidin-1-yl-methanone) were synthesized by Jean-Louis Maurel, Medicinal Chemistry Division 1, Centre de Recherche Pierre Fabre (Castres, France), as described earlier (Maurel et al., 2007; Figure 1). 5-HT creatinine sulphate, (+)8-hydroxy-dipropylaminotryptamine [(+)8-OH-DPAT] bromohydrate, were purchased from Sigma RBI (St. Quentin Fallavier, France). Drugs were dissolved in distilled water or 10% DMSO at 10−3 mol·L−1, and subsequent dilutions were prepared in the appropriate assay buffer.
Figure 1.
[3H]8-OH-DPAT (TRK.850: 160–240 Ci·mmol−1), [3H]GR125,743 (TRK.1046: 50–86 Ci·mmol−1), [3H]mesulergine (TRK.845: 70–85 Ci·mmol−1), [3H]SCH23390 (TRK.876: 60–90 Ci·mmol−1), [3H]RX821002 (TRK.914: 40–70 Ci·mmol−1), [3H]citalopram (TRK.1068: 60–86 Ci·mmol−1), [3H]prazosin (TRK.843: 65–85 Ci·mmol−1), [3H]WAY100635 (TRK.1034: 60–86 Ci·mmol−1) and [35S]-GTPγS (1000–1200 Ci·mmol−1) were purchased from GE Healthcare. [3H]ketanserin (NET-791: 60–90 Ci·mmol−1) and [3H]YM-09151-2 (i.e. [3H]nemonapride; NET-1004: 70–87 Ci·mmol−1) were purchased from Perkin-Elmer Life Sciences.
Results
Receptor binding profile of F15599 at native and recombinant human receptors
In competition binding experiments, F15599 had affinity for both native rat and cloned human 5-HT1A receptors, with pKi values of about 8.5 (Table 3). The affinity of F15599 is an order of magnitude less than that of 8-OH-DPAT and 35- to 70-fold less than that of F13714. F15599 has no measurable affinity (up to 10 µmol·L−1) for other native receptor binding sites examined (Table 3). In a receptor screen carried out by Cerep (La Celle Levescault, France; data on file), F15599 did not interact significantly with a panoply of other sites including h5-HT2A, h5-HT2B, h5-HT2C, h5-HT3, 5-HT4, h5-HT5A, h5-HT6, h5-HT7, adrenergic, dopaminergic, muscarinic, histaminergic, adenosine, opiate, cannabinoid, benzodiazepine, GABA, sigma (less than 50% inhibition at 10 µmol·L−1). F15599 also did not significantly interact with calcium (verapamil site), potassium, sodium or chloride channels and did not inhibit MAO-A or MAO-B enzyme activities. F15599 did not interact with 5-HT, dopamine or noradrenaline transporters. Thus, F15599 was 1000-fold selective for 5-HT1A receptors versus all other receptors and binding sites examined.
Table 3.
Comparison of affinities of F15599, F13714 and (+)8-OH-DPAT for various native receptors determined by competition binding in vitro
| Receptor | Tissue | F15599 | F13714 | (+)8-OH-DPAT |
|---|---|---|---|---|
| h 5-HT1A | CHO cells | 8.57 ± 0.05 | 10.40 ± 0.09 (68) | 9.50 ± 0.03 (9) |
| r 5-HT1A | Rat hippocampus | 8.47 ± 0.08 | 10.01 ± 0.01 (35) | 9.15 ± 0.11 (5) |
| r 5-HT1B | Rat cortex | <5 | <5 | 5.28 ± 0.18 |
| r 5-HT2A | Rat cortex | <5 | <5 | <5 |
| p 5-HT2C | Pig cortex | <5 | <5 | 5.12 ± 0.05 |
| r α1 | Rat cortex | <5 | 7.17 ± 0.05 | 5.82 ± 0.12 |
| r α2 | Rat cortex | <5 | 5.58 ± 0.03 | 6.52 ± 0.04 |
| r D1 | Rat striatum | <5 | 5.59 ± 0.03 | <5 |
| r D2 | Rat striatum | <5 | 5.89 ± 0.11 | 6.08 ± 0.03 |
Affinity is expressed as pKi ± SEM Numbers in brackets indicate the affinity ratio versus F15599.
D1, D2, dopamine receptors; h, human; p, porcine; r, rat; α1, α2, α-adrenoceptors.
Activation by F15599 of recombinant human 5-HT1A receptors
In a rigorous in vitro system that discriminates high-efficacy agonists ([35S]-GTPγS binding in membranes from C6 glial-h5-HT1A cells), F15599 increased G-protein activation with a pEC50 of 6.41 ± 0.06 and a maximal response of 70 ± 1% relative to that induced by 10 µmol·L−1 5-HT (mean ± SEM, n = 3). The efficacy of F15599 was higher, but its potency lower, than that of F13714 (pEC50 8.31 ± 0.18, Emax 61 ± 5), (+)8-OH-DPAT (pEC50 7.16 ± 0.09, Emax 55 ± 2) and 5-HT (pEC50 6.64 ± 0.14, Emax 94 ± 7). In HeLa-h5-HT1A cell membranes (Figure 2; Table 4), F15599 maximally increased G-protein activation (Emax 102%) relative to 5-HT. Maximal efficacy was also observed for inhibition of cyclic AMP formation in the same cell line. However, in all cases, the potency of F15599 was one to two orders of magnitude less than that of F13714 and (+)8-OH-DPAT. In CHO-h5-HT1A cells, F15599 efficaciously stimulated ERK1/2 phosphorylation (pERK) and, in HEK293-h5-HT1A cells, F15599 stimulated HA-tagged h5-HT1A receptor internalization.
Figure 2.
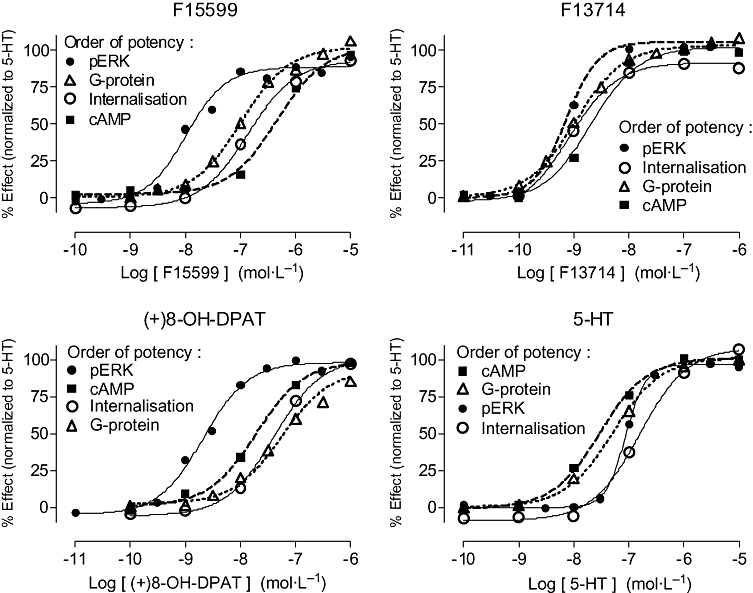
Influence of F15599 and reference agonists on: (i) stimulation of G-protein activation using [35S]-GTPγS binding at h5-HT1A receptors in HeLa cell membranes; (ii) inhibition of cAMP accumulation in HeLa-h5-HT1A cells; (iii) stimulation of ERK1/2 phosphorylation at h5-HT1A receptors expressed in CHO cells; and (iv) induction of HA-tagged h5-HT1A receptor internalization in HEK-293 cells. Data are expressed as percentage of the effect induced by a maximally effective concentration of 5-HT (10 µmol·L−1). Values are mean ± SEM from at least three experiments performed in triplicate or in duplicate. The order of potency of the responses for the different transduction tests is agonist-dependent (see Table 4).
Table 4.
Efficacy and potency of F15599 and comparators for stimulation of signal transduction responses in cell lines stably expressing h5-HT1A receptors
| Efficacy model |
F15599 |
F13714 |
(+)8-OH-DPAT |
5-HT |
||||
|---|---|---|---|---|---|---|---|---|
| Emax (%) | pEC50 | Emax (%) | pEC50 | Emax (%) | pEC50 | Emax (%) | pEC50 | |
| Total G-proteina | 102 ± 3 | 6.97 ± 0.08 | 73 ± 14 | 9.02 ± 0.12 | 107 ± 3 | 7.17 ± 0.05 | 101 ± 9 | 7.32 ± 0.05 |
| cAMP inhibition | 95 ± 1 | 6.46 ± 0.10 | 100 ± 1 | 8.67 ± 0.02 | 99 ± 1 | 7.77 ± 0.38 | 100 ± 1 | 7.45 ± 0.18 |
| ERK1/2 phosphorylation | 102 ± 9 | 7.81 ± 0.13 | 108 ± 13 | 9.07 ± 0.11 | 100 ± 3b | 8.74 ± 0.06b | 95 ± 8b | 6.86 ± 0.09b |
| Receptor internalization | 93 ± 7 | 6.80 ± 0.04 | 92 ± 7 | 9.06 ± 0.16 | 101 ± 3 | 7.38 ± 0.02 | 108 ± 6c | 6.80 ± 0.12c |
Efficacy (Emax) values are expressed as % of the stimulation induced by saturating concentrations (10 µmol·L−1) of 5-HT.
‘Total’ G-protein activation refers to [35S]-GTPγS binding that does not distinguish between the G-protein subtypes involved: compare with data in Table 6 on Gαi and Gαo activation.
F15599 exhibited a distinctive order of potency for these responses: the pEC50 for ERK1/2 phosphorylation (7.81; Table 4) was greater than that for G-protein activation (6.97), which was similar to that for internalization (6.80), which was greater than that for inhibition of cAMP formation (6.46). (+)8-OH-DPAT also showed the highest potency for ERK1/2 phosphorylation but lowest potency for G-protein activation. In contrast, F13714 showed similar potency for ERK1/2 phosphorylation, internalization and G-protein activation. 5-HT showed its highest potency for G-protein activation and inhibition of cAMP formation, with lower potency for ERK1/2 phosphorylation and receptor internalization. Hence, the order of potencies for the different signalling pathways was different for each of the agonists tested (Table 4).
Activation by F15599 of native rat 5-HT1A receptors in membranes from different brain regions
The influence of F15599 on G-protein activation ([35S]-GTPγS binding) in different brain regions was diverse (Table 5). Agonist efficacy, relative to 5-HT (10 µmol·L−1), varied from 54% in prefrontal cortex to 78% in hippocampal membranes. However, the stimulation by 5-HT is partially due to receptors other than 5-HT1A (Newman-Tancredi et al., 2003) such that the relative efficacy of a 5-HT1A receptor ‘full agonist’ is necessarily less than 100%. As regards agonist potency, compounds differed between brain regions. Thus, in prefrontal cortex, F15599 was 2.2-fold more potent than in hippocampus, and sixfold more potent than in dorsal raphe (based on EC50 ratios). In comparison, in prefrontal cortex, F13714 was 1.5-fold more potent than in hippocampus and 2.9-fold more potent than in dorsal raphe. In contrast, (+)8-OH-DPAT did not differ in potency between prefrontal cortex and hippocampus. In addition, a low slope value was observed for concentration–response curves in raphe membranes and for (+)8-OH-DPAT in prefrontal cortex (see Table 5, legend), suggesting the presence of heterogeneous populations of G-proteins.
Table 5.
Efficacy and potency of F15599 and comparators for stimulation of [35S]-GTPγS binding to membranes prepared from various brain regions
| Tissue/cell line |
F15599 |
F13714 |
(+)8-OH-DPAT |
5-HT |
||||
|---|---|---|---|---|---|---|---|---|
| Emax (%) | pEC50 | Emax (%) | pEC50 | Emax (%) | pEC50 | Emax (%) | pEC50 | |
| Prefrontal cortex | 54 ± 3 | 7.20 ± 0.04 | 54 ± 6 | 9.19 ± 0.08 | 65 ± 2 | 7.62 ± 0.14 | 92 ± 5 | 7.25 ± 0.05 |
| Entorhinal cortex | 63 ± 7 | 7.00 ± 0.14 | 65 ± 12 | 9.14 ± 0.01 | 75 ± 6 | 7.41 ± 0.05 | 101 ± 7 | 7.07 ± 0.08 |
| Hippocampus | 78 ± 2 | 6.85 ± 0.03 | 77 ± 2 | 9.02 ± 0.04 | 77 ± 3 | 7.59 ± 0.04 | 101 ± 1 | 6.99 ± 0.03 |
| Dorsal raphe | 68 ± 8 | 6.42 ± 0.12 | 64 ± 4 | 8.73 ± 0.08 | n.d. | n.d. | ||
| Median raphe | 57 ± 2 | 6.62 ± 0.06 | 57 ± 2 | 8.82 ± 0.07 | n.d. | n.d. | ||
Efficacy (Emax) values are expressed as % of the stimulation induced by saturating concentrations (10 µmol·L−1) of 5-HT. Slope values of the [35S]-GTPγS binding isotherms did not significantly differ from unity except in: (i) dorsal raphe F15599 0.62 ± 0.07, F13714 0.76 ± 0.03; (ii) median raphe F13714 0.69 ± 0.03; and (iii) prefrontal cortex (+)8-OH-DPAT 0.59 ± 0.06.
n.d., not determined.
In separate experiments, activation of G-proteins induced by F15599 (10 µmol·L−1) (assayed as [35S]-GTPγS binding) in rat hippocampal membranes was completely antagonized by WAY100635, indicating that it is specifically mediated by 5-HT1A receptors (pKB = 9.27 ± 0.03, n = 3).
Preferential activation by F15599 of Gαi over Gαo G-proteins
In a G-protein subtype targeting procedure (antibody capture coupled to detection by SPA), F15599 activated Gαi subunits in HeLa-h5-HT1A cell membranes with modest potency (pEC50 6.96) but remarkably high efficacy (Emax 122%), exceeding that of F13714, (+)8-OH-DPAT and even 5-HT (Figure 3; Table 6).
Figure 3.
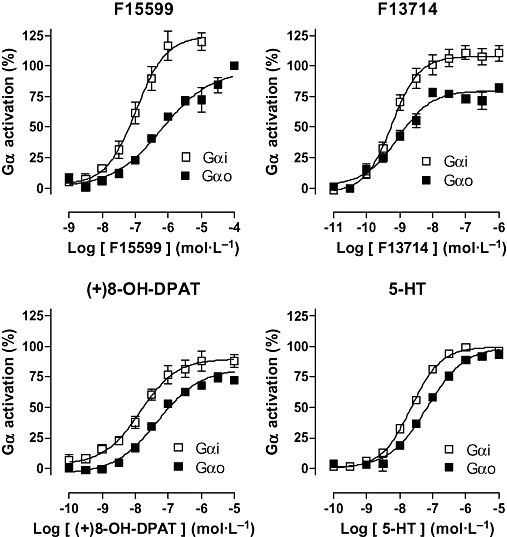
Influence of F15599 and reference agonists on G-protein subtype activation. Gαi subunits in HeLa-h5-HT1A cell membranes and Gαo subunits in rat hippocampal membranes were targeted by [35S]-GTPγS binding followed by antibody capture coupled to detection by scintillation proximity assay. Data are expressed as percentage of the effect induced by a maximally effective concentration of 5-HT (10 µmol·L−1). Values are mean ± SEM from at least three experiments performed in triplicate or in duplicate.
Table 6.
Efficacy and potency of F15599 and comparators for stimulation of [35S]-GTPγS binding to Gαi and Gαo subunits
| Tissue/cell line |
F15599 |
F13714 |
(+)8-OH-DPAT |
5-HT |
||||
|---|---|---|---|---|---|---|---|---|
| Emax (%) | pEC50 | Emax (%) | pEC50 | Emax (%) | pEC50 | Emax (%) | pEC50 | |
| Gαi (HeLa-h5-HT1A membranes) | 122 ± 8 | 6.96 ± 0.08 | 111 ± 6 | 9.15 ± 0.08 | 90 ± 8 | 7.79 ± 0.13 | 100 ± 1 | 7.63 ± 0.04 |
| Gαo (hippocampus) | 103 ± 8 | 6.07 ± 0.06 | 83 ± 6 | 9.11 ± 0.16 | 82 ± 4 | 7.37 ± 0.06 | 100 ± 1 | 7.18 ± 0.07 |
Efficacy (Emax) values are expressed as % of the stimulation induced by saturating concentrations (10 µmol·L−1) of 5-HT.
F15599 also activated Gαo subunits in rat hippocampal membranes with high efficacy, similar to that of 5-HT, and exceeding that of F13714 and (+)8-OH-DPAT. However, the potency of F15599 (pEC50 6.07) was eightfold less than for Gαi activation and markedly less than that for activation of ‘total’ hippocampal G-proteins (i.e. without distinguishing the subtypes; pEC50 6.85; Table 5). In contrast, F13714 and (+)8-OH-DPAT exhibited potency for Gαo that was similar to that for Gαi activation. Furthermore, they were more potent for Gαo activation in hippocampal membranes than for total G-protein activation in the same preparation (compare data in Tables 5 and 6).
Agonism by F15599 at native rat 5-HT1A receptors: functional autoradiography
The influence of a saturating concentration (100 µmol·L−1) of F15599 was examined on [35S]-GTPγS binding to horizontal rat brain sections. Incubation with F15599 increased [35S]-GTPγS binding to hippocampus, lateral septum and limbic cortex, areas that express high levels of 5-HT1A receptors (Figure 4). The effect of F15599 was blocked by co-incubation with the selective 5-HT1A receptor antagonist, WAY100635 (10 µmol·L−1). A low concentration (0.1 µmol·L−1) of F13714 exhibited a similar pattern of G-protein activation that was also abolished by WAY100635.
Figure 4.
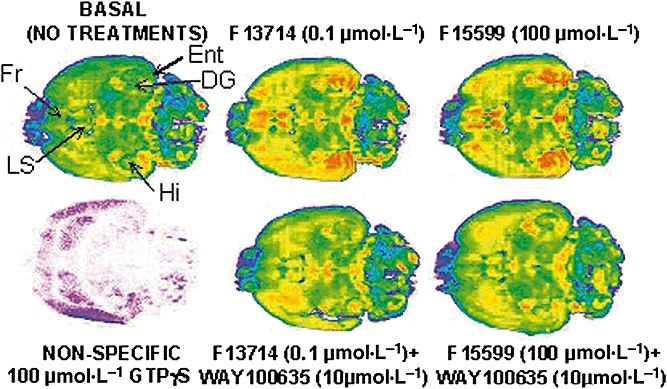
Influence of F15599 and F13714 on G-protein activation in horizontal brain sections. Pseudo-colour images of sections incubated with [35S]-GTPγS and drug treatments: blue/pink indicates low levels of activation while red represents high activation. Images are from a representative experiment. Measurements were performed on sets of serial sections from six animals, each set representing all treatments presented. DG, dentate gyrus; Ent, entorhinal cortex; Fr, frontal cortex; Hi, hippocampus; LS, lateral septum.
In vivo occupancy of 5-HT1A receptors by F15599
The 5-HT1A receptors in mouse brain were labelled in vivo with [3H]WAY100635. Control (saline-treated) animals exhibited radiolabelling of 517 ± 22 dpm·(mg wet weight)−1 in the frontal cortex and 507 ± 22 dpm·(mg wet weight)−1 in the hippocampus. F15599 and F13714 [0.16–640 mg·kg−1 per os (p.o.)] dose-dependently inhibited [3H]WAY100635 binding in these brain structures. The ID50 values were similar in both brain regions (Figure 5). F15599: 2.5 mg·kg−1 [95% confidence interval (CI): 1.0 to 6.4] in frontal cortex and 2.2 mg·kg−1 (95% CI: 0.57 to 8.7) in hippocampus. F13714: 0.95 mg·kg−1 (95% CI: 0.69 to 1.3) in frontal cortex and 0.95 mg·kg−1 (95% CI: 0.63 to 1.23) in hippocampus. The maximal inhibition of labelling by F15599 was similar to that produced by F13714 (26 ± 6% binding remaining, compared with 22 ± 3%, respectively, in cortex). However, the inhibition curve of F15599 was noticeably shallower than that of F13714, possibly indicating the presence of more than one population of 5-HT1A binding sites.
Figure 5.
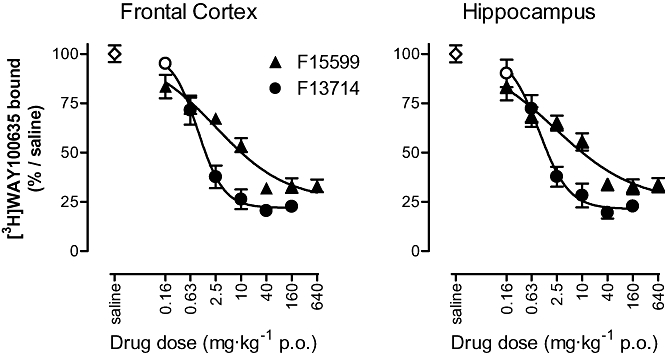
Inhibition of [3H]WAY100635 binding in vivo in mouse hippocampus and frontal cortex. Compounds were administered per os (p.o.) 30 min before intravenous injection of the radiotracer. Animals were then killed 30 min later. Each point is the mean ± SEM of five animals. Filled symbols indicate significant difference (P < 0.05) from control group (one-way anova followed by Dunnett's test).
Modulation of ERK1/2 phosphorylation in rat brain
In preliminary experiments, the induction of ERK1/2 phosphorylation by F15599 was time-dependent: anova indicated a significant effect of time in the hippocampus (F4,25 = 8.53, P < 0.001), prefrontal cortex (F4,25 = 7.09, P < 0.001) and hypothalamus (F4,25 = 4.14, P < 0.05; data not shown). A decrease in pERK1/2 levels is observed in hippocampus, while an increase was observed in prefrontal cortex and hypothalamus. A treatment interval of 30 min (used in all subsequent experiments) was determined, permitting analysis of the effects in the three brain regions.
Dose–response experiments showed a significant effect of treatment in hippocampus (F4,25 = 8.42, P < 0.001), prefrontal cortex (F4,25 = 20.15, P < 0.0001) and hypothalamus (F4,25 = 9.97, P < 0.0001). The 0.63 mg·kg−1 dose of F15599 was the first to reduce hippocampal pERK1/2 levels (−47% relative to basal values, P < 0.0001) and to increase pERK1/2 levels in both the prefrontal cortex and hypothalamus (+53% and +74% relative to basal values, P < 0.001 for both; Figure 6, left-hand graphs).
Figure 6.
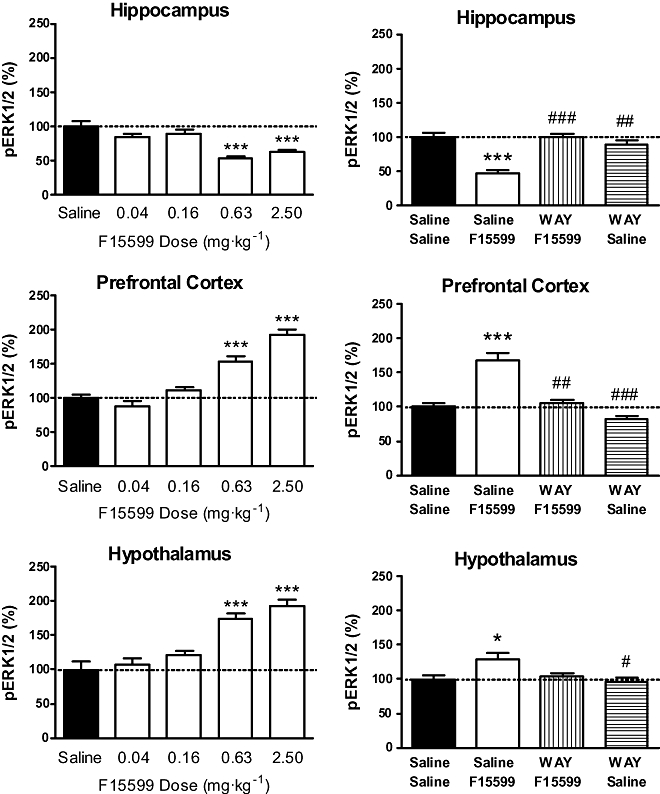
Influence of F15599 on phospho-ERK1/2 (pERK1/2) levels in rat hippocampus, prefrontal cortex and hypothalamus at 30 min post injection. F15599 was tested either alone (left-hand graphs) or following pre-treatment with WAY100635 (0.63 mg·kg−1 subcutaneously; right-hand graphs). The mean value of the control group (saline-treated animals) was set at 100 (basal value) and all data were expressed as percentages relative to basal value (%). Data represent the mean ± SEM (n = 12, two measures for each of six rats per group). anova analyses were performed on raw data (see Methods): *P < 0.05, **P < 0.01 and ***P < 0.001 as compared with saline/saline + saline group; #P < 0.05, ##P < 0.01 and ###P < 0.001 as compared with saline + F15599 group.
Pre-treatment with WAY100635 (0.63 mg·kg−1 subcutaneously) did not modify pERK1/2 levels in saline-injected animals but abolished the F15599 (0.63 mg·kg−1 i.p.)-induced decrease in hippocampal pERK1/2 levels (P < 0.001 as compared with saline-saline group) and increase in pERK1/2 levels in prefrontal cortex (P < 0.001 as compared with saline-saline group; Figure 6, right-hand graphs). Although in this interaction study with WAY100635, F15599 did not significantly modify the pERK1/2 levels in hypothalamus, the slight increase observed with F15599 was reversed by WAY100635 (Figure 6, right-hand graphs).
In control experiments, quantitative ELISA assays were carried out to determine total ERK1/2 levels, showing that these were not significantly modified by treatment with various doses of F15599 (n = 3 rats per group, hippocampus F4,10 = 0.24, P = 0.907; prefrontal cortex F4,10 = 1.13, P = 0.395; hypothalamus F4,10 = 0.71, P = 0.602 respectively; data not shown).
Induction of c-fos expression in rat brain
The action of F15599 on c-fos mRNA was determined using quantitative polymerase chain reaction with Arbp as the housekeeping gene, in different rat brain regions 30 min after i.p. injection of different doses of compound. The most potent action of F15599 was observed in prefrontal cortex where increased c-fos expression was observed at the lowest dose tested (0.16 mg·kg−1). In other brain regions expressing post-synaptic 5-HT1A receptors (hippocampus, septum, entorhinal cortex, hypothalamus), F15599 induced c-fos expression at doses of 0.63 mg·kg−1 (Figure 7). In brain regions expressing pre-synaptic 5-HT1A receptors, F15599 induced some c-fos mRNA expression in the dorsal raphe nucleus but was totally inactive in median raphe. The magnitude of induction of c-fos mRNA expression by F15599 was highly diverse according to the brain area examined: a strong induction of c-fos was observed in prefrontal cortex, septum and hypothalamus (four- to five-fold induction relative to vehicle-treated animals) whereas a modest induction of c-fos was observed in dorsal raphe, entorhinal cortex, hippocampus and globus pallidus (2- to 2.5-fold, Figure 7).
Discussion
The main finding of this study is that F15599 is a highly selective, high-efficacy 5-HT1A receptor agonist that exhibits distinctive signal transduction properties. F15599 is substantially less potent in vitro than a closely related analogue, F13714, or the prototypical agonist, (+)8-OH-DPAT, but potently occupies 5-HT1A receptors in vivo and activates ERK1/2 phosphorylation ex vivo. Notably, whereas F15599 exhibits little activation of c-fos expression in dorsal or median raphe, it potently stimulates c-fos in prefrontal cortex, hypothalamus and septum, suggesting preferential activation of 5-HT1A receptors located in brain regions where those receptors are post-synaptic.
Affinity and selectivity of F15599 at 5-HT1A receptors
F15599 exhibited exceptional selectivity for 5-HT1A receptors. This was demonstrated in binding experiments where no interaction was detected with a large number of other receptors, binding sites and enzymes. The selectivity of F15599 was greater than 1000-fold for all sites examined. The selectivity of F15599 for 5-HT1A was also greater than that of its congener, F13714 and of (+)8-OH-DPAT (Table 3). Notably, the latter has some affinity for 5-HT7 receptors (Sprouse et al., 2004), whereas F15599 did not interact with this site. The selectivity of F15599 was supported by the observation that it stimulated G-protein activation in functional ([35S]-GTPγS) autoradiography experiments in those brain regions that express 5-HT1A receptors. Further, the selective 5-HT1A receptor antagonist, WAY100635, abolished the effects of F15599 (Figure 4). These data are consistent with the observation that the neurochemical and behavioural effects of F15599 are all also abolished by 5-HT1A antagonism, reflecting its remarkable selectivity (see Introduction and Assié et al., 2006a; Auclair et al., 2007; Celada et al., 2007). It is interesting that the affinity of F15599 at h5-HT1A receptors was 9- and 68-fold less than that of (+)8-OH-DPAT or F13714 (Table 3). This observation corresponds to in vitro measures of potency for signal transduction. However, such differences in potency are not observed for in vivo binding at mouse brain 5-HT1A receptors. Thus, the ID50 values determined for inhibition of the binding of the radiotracer (the selective 5-HT1A receptor antagonist, [3H]WAY100635) were less than threefold higher for F15599 than for F13714 (Figure 5). The unexpectedly potent in vivo affinity of F15599 cannot be readily attributed to a potential active metabolite arising from pharmacokinetic properties (in rat, F15599 possesses plasma half-life greater than 24 h, F. Sautel, unpubl. obs.), although other differences, such as lipophilicity, permeability and/or blood-brain barrier penetration, may play a role. Nevertheless, taken together with the frontal cortex preference of F15599 seen in other models (see Introduction), the data suggest that the potent 5-HT1A receptor occupancy observed for F15599 arises from its agonist–receptor interaction, possibly involving high affinity for receptor populations (not subtypes) coupled to specific G-protein subtypes. This conclusion is supported by the observation that the dose–response curve for F15599 was substantially shallower than that for F13714, suggesting that different conformations or affinity states of 5-HT1A receptors may be distinguished by F15599 but not F13714.
Agonist efficacy of F15599 at human and rat 5-HT1A receptors in vitro
In all the tests of signal transduction, F15599 displayed high agonist efficacy (see Tables 4–6) at both cloned human and native rat 5-HT1A receptors. Indeed, the response to F15599 in all the systems tested is comparable with that observed for F13714 or (+)8-OH-DPAT [note: (+)OH-DPAT possesses approximately twofold greater efficacy than (−)8-OH-DPAT; Lejeune et al., 1997)]. F15599 is also much more efficacious than buspirone (Newman-Tancredi et al., 2005), a clinically used anxiolytic. In contrast to its efficacy, the potency of F15599 in functional tests is generally modest, with pEC50 values one to two orders of magnitude lower than for (+)8-OH-DPAT and F13714, consistent with the lower binding affinity of F15599 (see above). Interestingly, F15599 exhibits a distinctive pattern of stimulation of signalling responses. Thus, F15599 was more potent for stimulation of ERK1/2 phosphorylation compared with G-protein activation, receptor internalization and adenylyl cyclase inhibition (Table 4). Indeed, each of the four agonists displayed a distinct order of potency for these signalling responses (Figure 2) and the more ‘downstream’ responses were not always more potently activated than activation of G-proteins or adenylyl cyclase inhibition. This indicates that there is not a systematic ‘signal amplification’ between receptor activation and intracellular signalling cascades and suggests that the agonists are preferentially directing responses to specific signalling cascades. Although the underlying mechanisms that produce the differential profiles of the 5-HT1A agonists require further elucidation, the data suggest that F15599 and the other agonists exhibit functional selectivity (Kenakin, 2007). Given that all the second messenger responses are G-protein-mediated, the differences in signalling preference may result from activation of distinct G-protein subtypes. We therefore performed [35S]-GTPγS binding experiments on membranes prepared from different brain regions (Table 5). The data showed that the potency (pEC50 values) of the agonists vary according to brain region, with F15599 exhibiting greater potency in membranes from frontal cortex than from raphe. In addition, the slope values for the raphe were shallow (see legend of Table 5), an observation that is suggestive of G-protein heterogeneity. Thus, F15599 and the other agonists may differentially influence G-protein coupling in the different brain regions.
This hypothesis is supported by the observation that F15599 displayed enhanced activation of specific G-protein subtypes: its pEC50 for Gαi activation (6.96) was nearly an order of magnitude greater than that for Gαo activation (6.07; Table 6). In contrast, 5-HT displayed similar potencies for both Gα subtypes while F13714 and (+)8-OH-DPAT had intermediate profiles (Figure 3). Taken together, these data suggest that F15599 may display signalling pathway specificity at 5-HT1A receptors due to preferential activation of Gαi subunits. However, it should be noted that Gαi activation was determined in HeLa-h5-HT1A cells because it is difficult to measure in brain tissue using the present antibody capture/SPA methodology, possibly because of low native Gαi expression levels. Further, we detected Gαo G-protein activation but did not differentiate between Gαo subtypes. Therefore, the latter's contribution to the actions of 5-HT1A agonists remains to be evaluated. In addition, other parameters may influence G-protein activation, including RGS proteins, and other intracellular cofactors. Nevertheless, despite these caveats, the concept of G-protein-based ‘functional selectivity’ at 5-HT1A receptors has been suggested previously: ipsapirone, but not rauwolscine, preferentially directs 5-HT1A coupling to Gαi3 rather than Gαi2 (Gettys et al., 1994) and pindolol preferentially activated Gαi3 versus other G-proteins (Newman-Tancredi et al., 2002). Functional selectivity at 5-HT1A receptors is closely associated with the possibility of preferential activity in specific brain regions. Thus, although only a single molecular entity of 5-HT1A receptors has been identified, the affinity of a series of 5-HT receptor agonists was two- to fivefold lower at pre-synaptic 5-HT1A receptors in the raphe than at post-synaptic 5-HT1A receptors in the hippocampus (Johnson et al., 1997), suggesting heterogeneity of 5-HT1A receptor populations and potential for differential drug targeting.
Taken together, these data suggest that F15599 displays preferential activation of Gαi and that this is associated with a specific order of potency on intracellular signalling cascades. Such mechanisms would underlie the post-synaptic cortical preference of F15599 observed in neurotransmitter release and electrophysiological measures (Assié et al., 2006a; Celada et al., 2007).
Influence of F15599 on ERK1/2 phosphorylation in different brain regions ex vivo
In view of the potent in vitro stimulation of ERK1/2 phosphorylation by F15599 (Table 4), it was interesting to examine this response ex vivo in rat. Indeed, ERK1/2 is implicated in both long-term processes, such as neuronal plasticity/differentiation, and short-term influence on spatial memory and mood. 5-HT1A receptors influence ERK1/2 phosphorylation both in vitro (Bruins Slot et al., 2006) and in vivo (Chen et al., 2002; Sullivan et al., 2005; Crane et al., 2007). Stimulation of ERK1/2 phosphorylation by F15599 was time, brain region and dose-dependent (Figure 6). In frontal cortex, ERK1/2 phosphorylation is controlled by activation of post-synaptic 5-HT1A receptors. In contrast, inhibition of ERK1/2 phosphorylation in hippocampus by 5-HT1A receptor agonists is likely to involve inhibition of 5-HT release consequent to activation of raphe-localized 5-HT1A autoreceptors (Chen et al., 2002). Thus, in ex vivo studies, F15599 (0.63 mg·kg−1 i.p.) stimulated ERK1/2 phosphorylation in frontal cortex and reduced it in hippocampus, both of these responses being mediated by 5-HT1A receptors, as shown by their reversal with WAY100635 (Figure 6). In comparison, F13714, but not (+)8-OH-DPAT, stimulated ERK1/2 phosphorylation in frontal cortex at 0.16 mg·kg−1 but reduced ERK1/2 phosphorylation in hippocampus from 0.04 mg·kg−1 i.p. (Buritova et al., 2007), suggesting preferential activation of pre-synaptic 5-HT1A receptors. Analysis of ERK1/2 in hypothalamus, another region expressing post-synaptic receptors, yielded similar tendency to that seen in frontal cortex but with reduced stimulation. Taken together, these data provide additional evidence for potent 5-HT1A receptor activation by F15599 in rodents and support the notion that selective 5-HT1A agonists can differentially regulate signal transduction in different brain regions. It is interesting that h5-HT1A receptor-mediated ERK1/2 phosphorylation in vitro is mediated by Gαi subtypes (Lin et al., 2002). Given the capacity of F15599 to preferentially activate Gαi versus Gαo (see above), this provides a mechanistic underpinning for its potent induction of ERK1/2 phosphorylation in rat, although a specifically designed study would be necessary to identify the precise G-protein subtypes involved.
Influence of F15599 on c-fos mRNA expression
Further evidence for a brain region-selective influence of F15599 was generated from c-fos mRNA expression studies (Figure 7). These showed that induction of c-fos did not directly correlate with the known density of 5-HT1A receptor throughout the rat brain. Thus, F15599 efficaciously stimulated expression of this immediate early gene in the frontal cortex, septum and hypothalamus (regions expressing post-synaptic 5-HT1A receptors) but very little in median or dorsal raphe (the locus of 5-HT1A auto-receptors) or in entorhinal cortex and hippocampus (which express high levels of post-synaptic 5-HT1A receptors) or globus pallidus. In comparison, F13714 potently stimulated c-fos expression in both frontal cortex and also median and dorsal raphe whereas (+)8-OH-DPAT exhibited an intermediate profile (Buritova et al., 2008). These data reinforce the conclusion that selective and efficacious 5-HT1A receptor agonists can induce activation of 5-HT1A receptor signalling in a brain region-dependent manner. In the case of F15599, its activity appears to be preferentially expressed at sub-populations of 5-HT1A receptors found in brain regions with post-synaptic locations. In particular, the potent activation by F15599 of 5-HT1A receptors in the frontal cortex (from 0.16 mg·kg−1) is consistent with electrophysiological data showing that F15599 potently increases cortical pyramidal neuron cell firing in anaesthetized rats at doses 20-fold lower than those that inhibit dorsal raphe neuron firing (Celada et al., 2007). In addition, F15599 also increases dopamine release in the frontal cortex (a post-synaptic 5-HT1A receptor-mediated response) at doses about eightfold lower than those that inhibit 5-HT release in hippocampus (a pre-synaptic 5-HT1A receptor-mediated response; Assié et al., 2006a).
In summary, F15599 is a novel, highly selective and efficacious agonist at 5-HT1A receptors. In a series of in vitro signal transduction assays, it induced a distinctive pattern of responses, unlike that of reference agonists. Although the agonist potency of F15599 in vitro was more modest than that of other agonists (Tables 4 and 5), its in vivo binding affinity was almost as great as that of its potent congener, F13714. F15599 was also remarkably potent for ex vivo stimulation of ERK1/2 phosphorylation, being active at low doses, comparable with those determined for 8-OH-DPAT. F15599, unlike F13714, was more potent for activation of Gαi G-protein subunits than Gαo, suggesting that it exhibited functional selectivity for 5-HT1A receptor signalling. F15599 may, thus, preferentially activate some brain region-specific signal transduction pathways, a conclusion reinforced by the observation that it potently induces c-fos mRNA expression in frontal cortex, a region where 5-HT1A receptors are post-synaptically located, but hardly at all in dorsal or median raphe, regions that express pre-synaptic receptors. Indeed, 5-HT1A receptor signalling in these brain regions is mechanistically distinct, possibly involving different populations of G-protein subtypes. The present data provide a mechanistic basis for interpreting the profile of action of F15599 seen in microdialysis experiments and electrophysiological studies, where it shows marked preference for activation of cortical 5-HT1A receptors and only little activity at pre-synaptic sites (see above). The preferential agonist properties at post-synaptic 5-HT1A receptors displayed by F15599 are likely to underlie its favourable activity in rodent models of depression and cognitive deficits and would be of considerable promise for the treatment of depressive states and other psychiatric disorders.
Acknowledgments
All the authors are employees of Centre de Recherche Pierre Fabre. Nathalie Leduc, Nathalie Danty, Valérie Faucillon, Nathalie Consul-Denjean, Anne-Marie Ormière, Nathalie Delmas, Stéphanie Tardif, Marie-Christine Ailhaud, Géraldine Berrichon, Sabine Callewaert and Yann Leroy are thanked for technical assistance. Sophie Bréand, Jerôme Besse and Audrey Dos Santos are thanked for assistance with statistical analysis.
Glossary
Abbreviations:
- DTT
dithiothreitol
- ERK1/2
extra-cellular signal regulated kinase
- SPA
scintillation proximity assay
- SNRI
selective noradrenaline reuptake inhibitor
- SSRI
selective serotonin reuptake inhibitor
Conflicts of interest
All the authors are employees of Centre de Recherche Pierre Fabre.
References
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (3rd edition) 2008;153(Suppl)(2):S1–209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artigas F, Adell A, Celada P. Pindolol augmentation of antidepressant response. Curr Drug Targets. 2006;7:139–147. doi: 10.2174/138945006775515446. [DOI] [PubMed] [Google Scholar]
- Assié M-B, Bardin L, Depoortère R, Carilla-Durand E, Newman-Tancredi A. F15599, a highly selective serotonin 5-HT1A receptor agonist: in vivo profile in neurochemical and behavioural models of serotonergic activity. Eur Neuropsychopharmacol. 2006a;16:S233. doi: 10.1017/S1461145709991222. [DOI] [PubMed] [Google Scholar]
- Assié M-B, Lomenech H, Ravailhe V, Faucillon V, Newman-Tancredi A. Rapid desensitization of somatodendritic 5-HT1A receptors by chronic administration of the high-efficacy 5-HT1A agonist, F13714: a microdialysis study in the rat. Br J Pharmacol. 2006b;149:170–178. doi: 10.1038/sj.bjp.0706859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auclair A, Bardin L, Depoortère R, Newman-Tancredi A. F15599, a 5-HT1A agonist that preferentially targets post-synaptic receptors in the frontal cortex. III) activity in radent models of cognition. 2007 Neuroscience Meeting Planner. San Diego, CA: Society for Neuroscience, 2007. Online. 170.24.
- Bardin L, Tarayre JP, Malfetes N, Koek W, Colpaert FC. Profound, non-opioid analgesia produced by the high-efficacy 5-HT1A agonist F 13640 in the formalin model of tonic nociceptive pain. Pharmacology. 2003;67:182–194. doi: 10.1159/000068404. [DOI] [PubMed] [Google Scholar]
- Béïque JC, Blier P, De Montigny C, Debonnel G. Potentiation by (−)pindolol of the activation of postsynaptic 5-HT1A receptors induced by venlafaxine. Neuropsychopharmacology. 2000;23:294–306. doi: 10.1016/S0893-133X(00)00112-3. [DOI] [PubMed] [Google Scholar]
- Berrocoso E, Mico JA. Role of serotonin 5-HT1A receptors in the antidepressant-like effect and the antinociceptive effect of venlafaxine in mice. Int J Neuropsychopharmacol. 2008;14:1–11. doi: 10.1017/S1461145708008766. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Bhagwagar Z, Rabiner EA, Sargent PA, Grasby PM, Cowen PJ. Persistent reduction in brain serotonin1A receptor binding in recovered depressed men measured by positron emission tomography with [11C]WAY-100635. Mol Psychiatry. 2004;9:386–392. doi: 10.1038/sj.mp.4001401. [DOI] [PubMed] [Google Scholar]
- Blier P, Ward NM. Is there a role for 5-HT1A agonists in the treatment of depression? Biol Psychiatry. 2003;53:193–203. doi: 10.1016/s0006-3223(02)01643-8. [DOI] [PubMed] [Google Scholar]
- Blier P, Bergeron R, De Montigny C. Selective activation of postsynaptic 5-HT1A receptors induces rapid antidepressant response. Neuropsychopharmacology. 1997;16:333–338. doi: 10.1016/S0893-133X(96)00242-4. [DOI] [PubMed] [Google Scholar]
- Bruins Slot LA, De Vries L, Newman-Tancredi A, Cussac D. Differential profile of antipsychotics at serotonin 5-HT1A and dopamine D2S receptors coupled to extracellular signal-regulated kinase. Eur J Pharmacol. 2006;534:63–70. doi: 10.1016/j.ejphar.2006.01.027. [DOI] [PubMed] [Google Scholar]
- Buritova J, Newman-Tancredi A, Buquet C, Berrichon G, Cussac D. Region specific effects of F15599, a selective serotonin 5-HT1A receptor agonist, on the phosphorylation of MAP-kinase (ERK1/2) in the rat brain: a quantitative ELISA study. Proceedings Société des Neurosciences. 2007;8eme Colloque:F12 p.118. [Google Scholar]
- Buritova J, Tarayre JP, Besson JM, Colpaert FC. The novel analgesic and high-efficacy 5-HT1A receptor agonist, F 13640 induces c-Fos protein expression in spinal cord dorsal horn neurons. Brain Res. 2003;974:212–221. doi: 10.1016/s0006-8993(03)02582-4. [DOI] [PubMed] [Google Scholar]
- Buritova J, Berrichon G, Cathala C, Colpaert F, Cussac D. Region-specific changes in 5-HT1A agonist-induced Extracellular signal-Regulated Kinases 1/2 phosphorylation in rat brain: A quantitative ELISA study. Neuropharmacology. 2008 Sep 17. [Epub ahead of print. [DOI] [PubMed]
- Castro ME, Diaz A, Del Olmo E, Pazos A. Chronic fluoxetine induces changes in G protein coupling at pre and postsynaptic 5-HT1A receptors in rat brain. Neuropharmacology. 2003;44:93–101. doi: 10.1016/s0028-3908(02)00340-4. [DOI] [PubMed] [Google Scholar]
- Celada P, Puig M, Amargós-Bosch M, Adell A, Artigas F. The therapeutic role of 5-HT1A and 5-HT2A receptors in depression. J Psychiatry Neurosci. 2004;29:252–265. [PMC free article] [PubMed] [Google Scholar]
- Celada P, Llado L, Assié M-B, Newman-Tancredi A, Artigas F. F15599, a 5-HT1A agonist that preferentially targets post-synaptic receptors in the frontal cortex: IV) Influence on rat raphe and pyramidal neuronal activity and cortical dopamine release. 2007 Neuroscience Meeting Planner. San Diego, CA: Society for Neuroscience, 2007. Online. 170.27.
- Chen J, Shen C, Meller E. 5-HT1A receptor-mediated regulation of mitogen-activated protein kinase phosphorylation in rat brain. Eur J Pharmacol. 2002;452:155–162. doi: 10.1016/s0014-2999(02)02297-5. [DOI] [PubMed] [Google Scholar]
- Clarke WP, Yocca FD, Maayani S. Lack of 5-hydroxytryptamine1A-mediated inhibition of adenylyl cyclase in dorsal raphe of male and female rats. J Pharmacol Exp Ther. 1996;277:1259–1266. [PubMed] [Google Scholar]
- Colpaert FC. 5-HT1A receptor activation: new molecular and neuroadaptive mechanisms of pain relief. Curr Opin Investig Drugs. 2006;7:40–47. [PubMed] [Google Scholar]
- Colpaert FC, Tarayre JP, Koek W, Pauwels PJ, Bardin L, Xu XJ, et al. Large-amplitude 5-HT1A receptor activation: a new mechanism of profound, central analgesia. Neuropharmacology. 2002;43:945–958. doi: 10.1016/s0028-3908(02)00119-3. [DOI] [PubMed] [Google Scholar]
- Cosi C, Koek W. The putative ‘silent’ 5-HT1A receptor antagonist, WAY 100635, has inverse agonist properties at cloned human 5-HT1A receptors. Eur J Pharmacol. 2000;401:9–15. doi: 10.1016/s0014-2999(00)00410-6. [DOI] [PubMed] [Google Scholar]
- Crane JW, Shimizu K, Carrasco GA, Garcia F, Jia C, Sullivan NR, et al. 5-HT1A receptors mediate (+)8-OH-DPAT-stimulation of extracellular signal-regulated kinase (MAP kinase) in vivo in rat hypothalamus: time dependence and regional differences. Brain Res. 2007;1183:51–59. doi: 10.1016/j.brainres.2007.07.101. [DOI] [PubMed] [Google Scholar]
- Cussac D, Newman-Tancredi A, Duqueyroix D, Pasteau V, Millan MJ. Differential activation of Gq/11 and Gi(3) proteins at 5-hydroxytryptamine (2C) receptors revealed by antibody capture assays: influence of receptor reserve and relationship to agonist-directed trafficking. Mol Pharmacol. 2002;62:578–589. doi: 10.1124/mol.62.3.578. [DOI] [PubMed] [Google Scholar]
- Cussac D, Pasteau V, Millan MJ. Characterization of Gs activation by dopamine D1 receptors using an antibody capture assay: antagonist properties of clozapine. Eur J Pharmacol. 2004;485:111–117. doi: 10.1016/j.ejphar.2003.11.077. [DOI] [PubMed] [Google Scholar]
- Czesak M, Lemonde S, Peterson EA, Rogaeva A, Albert PR. Cell-specific repressor or enhancer activities of Deaf-1 at a serotonin 1A receptor gene polymorphism. J Neurosci. 2006;26:1864–1871. doi: 10.1523/JNEUROSCI.2643-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vry J. 5-HT1A receptor agonists: recent developments and controversial issues. Psychopharmacology. 1995;121:1–26. doi: 10.1007/BF02245588. [DOI] [PubMed] [Google Scholar]
- De Vry J, Schreiber R. Comparison of acute and repeated treatment with the 5-HT(1A) receptor ligands 8-OH-DPAT and ipsapirone in animal models of anxiety and depression. Drug Dev Res. 1993;30:91–103. [Google Scholar]
- De Vry J, Schreiber R, Melon C, Dalmus M, Jentzsch KR. 5-HT1A receptors are differentially involved in the anxiolytic- and antidepressant-like effects of 8-OH-DPAT and fluoxetine in the rat. Eur Neuropsychopharmacol. 2004;14:487–495. doi: 10.1016/j.euroneuro.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Detke MJ, Wieland S, Lucki I. Blockade of the antidepressant-like effects of 8-OH-DPAT, buspirone and desipramine in the rat forced swim test by 5HT1A receptor antagonists. Psychopharmacology. 1995;119:47–54. doi: 10.1007/BF02246053. [DOI] [PubMed] [Google Scholar]
- Donati RJ, Dwivedi Y, Roberts RC, Conley RR, Pandey GN, Rasenick MM. Postmortem brain tissue of depressed suicides reveals increased Gs alpha localization in lipid raft domains where it is less likely to activate adenylyl cyclase. J Neurosci. 2008;28:3042–3050. doi: 10.1523/JNEUROSCI.5713-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi Y, Rizavi HS, Conley RR, Roberts RC, Tamminga CA, Pandey GN. mRNA and protein expression of selective alpha subunits of G proteins are abnormal in prefrontal cortex of suicide victims. Neuropsychopharmacology. 2002;27:499–517. doi: 10.1016/S0893-133X(02)00335-4. [DOI] [PubMed] [Google Scholar]
- El Yacoubi M, Bouali S, Popa D, Naudon L, Leroux-Nicollet I, Hamon M, et al. Behavioral, neurochemical, and electrophysiological characterization of a genetic mouse model of depression. Proc Natl Acad Sci USA. 2003;100:6227–6232. doi: 10.1073/pnas.1034823100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gettys TW, Fields TA, Raymond JR. Selective activation of inhibitory G-protein α-subunits by partial agonists of the human 5-HT1A receptor. Biochemistry. 1994;33:4283–4290. doi: 10.1021/bi00180a024. [DOI] [PubMed] [Google Scholar]
- Haddjeri N, Blier P, De Montigny C. Long-term antidepressant treatments result in a tonic activation of forebrain 5-HT1A receptors. J Neurosci. 1998;18:10150–10156. doi: 10.1523/JNEUROSCI.18-23-10150.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddjeri N, Ortemann C, De Montigny C, Blier P. Effect of sustained administration of the 5-HT1A receptor agonist flesinoxan on rat 5-HT neurotransmission. Eur Neuropsychopharmacol. 1999;9:427–440. doi: 10.1016/s0924-977x(99)00020-6. [DOI] [PubMed] [Google Scholar]
- Hensler JG. Differential regulation of 5-HT1A receptor-G protein interactions in brain following chronic antidepressant administration. Neuropsychopharmacology. 2002;26:565–573. doi: 10.1016/S0893-133X(01)00395-5. [DOI] [PubMed] [Google Scholar]
- Hensler JG. Regulation of 5-HT1A receptor function in brain following agonist or antidepressant administration. Life Sci. 2003;72:1665–1682. doi: 10.1016/s0024-3205(02)02482-7. [DOI] [PubMed] [Google Scholar]
- Heusler P, Newman-Tancredi A, Loock T, Cussac D. Antipsychotics differ in their ability to internalise human dopamine D2S and human serotonin 5-HT1A receptors in HEK293 cells. Eur J Pharmacol. 2008;581:37–46. doi: 10.1016/j.ejphar.2007.11.046. [DOI] [PubMed] [Google Scholar]
- Invernizzi R, Bramante M, Samanin R. Chronic treatment with citalopram facilitates the effect of a challenge dose on cortical serotonin output: role of presynaptic 5-HT1A receptors. Eur J Pharmacol. 1994;260:243–246. doi: 10.1016/0014-2999(94)90344-1. [DOI] [PubMed] [Google Scholar]
- Johnson RG, Fiorella D, Winter JC, Rabin RA. 3H]8-OH-DPAT labels a 5-HT site coupled to inhibition of phosphoinositide hydrolysis in the dorsal raphe. Eur J Pharmacol. 1997;329:99–106. doi: 10.1016/s0014-2999(97)10113-3. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Agonist-receptor efficacy. II. Agonist trafficking of receptor signals. Trends Pharmacol Sci. 1995;16:232–238. doi: 10.1016/s0165-6147(00)89032-x. [DOI] [PubMed] [Google Scholar]
- Kenakin T. Functional selectivity through protean and biased agonism: who steers the ship? Mol Pharmacol. 2007;72:1393–1401. doi: 10.1124/mol.107.040352. [DOI] [PubMed] [Google Scholar]
- Koek W, Patoiseau JF, Assié M-B, Cosi C, Kleven MS, Dupont-Passelaigue E, et al. F 11440, a potent, selective, high efficacy 5-HT1A receptor agonist with marked anxiolytic and antidepressant potential. J Pharmacol Exp Ther. 1998;287:266–283. [PubMed] [Google Scholar]
- Koek W, Vacher B, Cosi C, Assié M-B, Patoiseau JF, Pauwels PJ, et al. 5-HT1A receptor activation and antidepressant-like effects: F 13714 has high efficacy and marked antidepressant potential. Eur J Pharmacol. 2001;420:103–112. doi: 10.1016/s0014-2999(01)01011-1. [DOI] [PubMed] [Google Scholar]
- Lazareno S, Birdsall NJ. Estimation of antagonist Kb from inhibition curves in functional experiments: alternatives to the Cheng-Prusoff equation. Trends Pharmacol Sci. 1993;14:237–239. doi: 10.1016/0165-6147(93)90018-f. [DOI] [PubMed] [Google Scholar]
- Lejeune F, Newman-Tancredi A, Audinot V, Millan MJ. Interactions of (+)- and. )-8- and 7-hydroxy-2-(Di-n- propylamino) tetralin at human (h)D3, hD2 and h serotonin1A receptors and their modulation of the activity of serotoninergic and dopaminergic neurones in rats. J Pharmacol Exp Ther. 1997;280:1241–1249. [PubMed] [Google Scholar]
- Lemonde S, Turecki G, Bakish D, Du LS, Hrdina PD, Bown CD, et al. Impaired repression at a 5-hydroxytryptamine 1A receptor gene polymorphism associated with major depression and suicide. J Neurosci. 2003;23:8788–8799. doi: 10.1523/JNEUROSCI.23-25-08788.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemonde S, Du L, Bakish D, Hrdina P, Albert PR. Association of the C(-1019)G 5-HT1A functional promoter polymorphism with antidepressant response. Int J Neuropsychopharmacol. 2004;7:501–506. doi: 10.1017/S1461145704004699. [DOI] [PubMed] [Google Scholar]
- Lesch KP, Gutknecht L. Focus on The 5-HT1A receptor: emerging role of a gene regulatory variant in psychopathology and pharmacogenetics. Int J Neuropsychopharmacol. 2004;7:381–385. doi: 10.1017/S1461145704004845. [DOI] [PubMed] [Google Scholar]
- Lin SL, Setya S, Johnson-Farley NN, Cowen DS. Differential coupling of 5-HT1 receptors to G proteins of the Gi family. Br J Pharmacol. 2002;136:1072–1078. doi: 10.1038/sj.bjp.0704809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucki I. The spectrum of behaviors influenced by serotonin. Biol Psychiatry. 1998;44:151–162. doi: 10.1016/s0006-3223(98)00139-5. [DOI] [PubMed] [Google Scholar]
- McPherson GA. Analysis of radioligand binding experiments. A collection of computer programs for IBM PC. J Pharmacol Methods. 1985;14:213–228. doi: 10.1016/0160-5402(85)90034-8. [DOI] [PubMed] [Google Scholar]
- Mannoury LC, El Mestikawy S, Hanoun N, Hamon M, Lanfumey L. Regional differences in the coupling of 5-hydroxytryptamine-1A receptors to G proteins in the rat brain. Mol Pharmacol. 2006;70:1013–1021. doi: 10.1124/mol.106.022756. [DOI] [PubMed] [Google Scholar]
- Marazziti D, Palego L, Giromella A, Mazzoni MR, Borsini F, Mayer N, et al. Region-dependent effects of flibanserin and buspirone on adenylyl cyclase activity in the human brain. Int J Neuropsychopharmacol. 2002;5:131–140. doi: 10.1017/S1461145702002869. [DOI] [PubMed] [Google Scholar]
- Martel JC, Cussac D, Assié M-B, Rauly-Lestienne I, Newman-Tancredi A. Antagonism by atypical benzamide antipsychotics of serotonin 5-HT2B receptors: comparison with 5-HT2A, 5-HT2C and dopamine D2 receptors. Neuroscience Meeting Planner. Atlanta, GA: Society for Neuroscience 2006. Online. 93.8.
- Martel JC, Ormiere AM, Leduc N, Assie MB, Cussac D, Newman-Tancredi A. Native rat hippocampal 5-HT1A receptors show constitutive activity. Mol Pharmacol. 2007;71:638–643. doi: 10.1124/mol.106.029769. [DOI] [PubMed] [Google Scholar]
- Matsuda T, Somboonthum P, Suzuki M, Asano S, Baba A. Antidepressant-like effect by postsynaptic 5-HT1A receptor activation in mice. Eur J Pharmacol. 1995;280:235–238. doi: 10.1016/0014-2999(95)00254-i. [DOI] [PubMed] [Google Scholar]
- Maurel JL, Autin JM, Funes P, Newman-Tancredi A, Colpaert F, Vacher B. High-efficacy 5-HT1A agonists for antidepressant treatment: a renewed opportunity. J Med Chem. 2007;50:5024–5033. doi: 10.1021/jm070714l. [DOI] [PubMed] [Google Scholar]
- Millan MJ. Multi-target strategies for the improved treatment of depressive states: conceptual foundations and neuronal substrates, drug discovery and therapeutic application. Pharmacol Ther. 2006;110:135–370. doi: 10.1016/j.pharmthera.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Naudon L, El Yacoubi M, Vaugeois JM, Leroux-Nicollet I, Costentin J. A chronic treatment with fluoxetine decreases 5-HT1A receptors labeling in mice selected as a genetic model of helplessness. Brain Res. 2002;936:68–75. doi: 10.1016/s0006-8993(02)02548-9. [DOI] [PubMed] [Google Scholar]
- Newman-Tancredi A, Cussac D, Marini L, Millan MJ. Antibody capture assay reveals bell-shaped concentration-response isotherms for h5-HT1A receptor-mediated Gαi3 activation: conformational selection by high-efficacy agonists, and relationship to trafficking of receptor signaling. Mol Pharmacol. 2002;62:590–601. doi: 10.1124/mol.62.3.590. [DOI] [PubMed] [Google Scholar]
- Newman-Tancredi A, Rivet JM, Cussac D, Touzard M, Chaput C, Marini L, et al. Comparison of hippocampal G protein activation by 5-HT1A receptor agonists and the atypical antipsychotics clozapine and S16924. Naunyn Schmiedebergs Arch Pharmacol. 2003;368:188–199. doi: 10.1007/s00210-003-0788-2. [DOI] [PubMed] [Google Scholar]
- Newman-Tancredi A, Assié M-B, Leduc N, Ormière A-M, Danty N, Cosi C. Novel antipsychotics activate recombinant human and native rat serotonin 5-HT1A receptors: affinity, efficacy and potential implication for treatment of schizophrenia. Int J Neuropsychopharmacol. 2005;8:1–16. doi: 10.1017/S1461145704005000. [DOI] [PubMed] [Google Scholar]
- Newman-Tancredi A, Assié M-B, Martel JC, Cosi C, Bruins Slot LA, Palmier C, et al. F15063, a potential antipsychotic with D2/D3 antagonist, 5-HT1A agonist and D4 partial agonist properties: I) in vitro, receptor affinity and efficacy profile. Br J Pharmacol. 2007;151:237–252. doi: 10.1038/sj.bjp.0707158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palkovits M, Brownstein MJ. Maps and Guide to Microdissection of the Rat Brain. London: Elsevier; 1987. [Google Scholar]
- Parsey RV, Arango V, Olvet DM, Oquendo MA, Van Heertum RL, Mann JJ. Regional heterogeneity of 5-HT1A receptors in human cerebellum as assessed by positron emission tomography. J Cereb Blood Flow Metab. 2005;25:785–793. doi: 10.1038/sj.jcbfm.9600072. [DOI] [PubMed] [Google Scholar]
- Parsey RV, Olvet DM, Oquendo MA, Huang YY, Ogden RT, Mann JJ. Higher 5-HT1A receptor binding potential during a major depressive episode predicts poor treatment response: preliminary data from a naturalistic study. Neuropsychopharmacology. 2006a;31:1745–1749. doi: 10.1038/sj.npp.1300992. [DOI] [PubMed] [Google Scholar]
- Parsey RV, Oquendo MA, Ogden RT, Olvet DM, Simpson N, Huang YY, et al. Altered serotonin 1A binding in major depression: a [carbonyl-C-11]WAY100635 positron emission tomography study. Biol Psychiatry. 2006b;59:106–113. doi: 10.1016/j.biopsych.2005.06.016. [DOI] [PubMed] [Google Scholar]
- Pauwels PJ, Tardif S, Wurch T, Colpaert FC. Stimulated [35S]-GTPgammaS binding by 5-HT1A receptor agonists in recombinant cell lines – modulation of apparent efficacy by G-protein activation state. Naunyn Schmiedebergs Arch Pharmacol. 1997;356:551–561. doi: 10.1007/pl00005090. [DOI] [PubMed] [Google Scholar]
- Pazos A, Hoyer D, Palacios JM. Mesulergine, a selective serotonin-2 ligand in the rat cortex, does not label these receptors in porcine and human cortex: evidence for species differences in brain serotonin-2 receptors. Eur J Pharmacol. 1985a;106:531–538. doi: 10.1016/0014-2999(84)90056-6. [DOI] [PubMed] [Google Scholar]
- Pazos A, Hoyer D, Palacios JM. The binding of serotonergic ligands to the porcine choroid plexus: characterization of a new type of serotonin recognition site. Eur J Pharmacol. 1985b;106:539–546. doi: 10.1016/0014-2999(84)90057-8. [DOI] [PubMed] [Google Scholar]
- Pejchal T, Foley MA, Kosofsky BE, Waeber C. Chronic fluoxetine treatment selectively uncouples raphe 5-HT1A receptors as measured by [35S]-GTPgammaS autoradiography. Br J Pharmacol. 2002;135:1115–1122. doi: 10.1038/sj.bjp.0704555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raap DK, Evans S, Garcia F, Li Q, Muma NA, Wolf WA, et al. Daily injections of fluoxetine induce dose-dependent desensitization of hypothalamic 5-HT1A receptors: reductions in neuroendocrine responses to 8-OH-DPAT and in levels of Gz and Gi proteins. J Pharmacol Exp Ther. 1999;288:98–106. [PubMed] [Google Scholar]
- Romero L, Hervás I, Artigas F. The 5-HT1A antagonist WAY-100635 selectively potentiates the presynaptic effects of serotonergic antidepressants in rat brain. Neurosci Lett. 1996;219:123–126. doi: 10.1016/s0304-3940(96)13199-2. [DOI] [PubMed] [Google Scholar]
- Rutter JJ, Gundlah C, Auerbach SB. Increase in extracellular serotonin produced by uptake inhibitors is enhanced after chronic treatment with fluoxetine. Neurosci Lett. 1994;171:183–186. doi: 10.1016/0304-3940(94)90635-1. [DOI] [PubMed] [Google Scholar]
- Serres F, Li Q, Garcia F, Raap DK, Battaglia G, Muma NA, et al. Evidence that Gz-proteins couple to hypothalamic 5-HT1A receptors in vivo. J Neurosci. 2000;20:3095–3103. doi: 10.1523/JNEUROSCI.20-09-03095.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C, Li H, Meller E. Repeated treatment with antidepressants differentially alters 5-HT1A agonist-stimulated [35S]-GTPgammaS binding in rat brain regions. Neuropharmacology. 2002;42:1031–1038. doi: 10.1016/s0028-3908(02)00064-3. [DOI] [PubMed] [Google Scholar]
- Shively CA, Friedman DP, Gage HD, Bounds MC, Brown-Proctor C, Blair JB, et al. Behavioral depression and positron emission tomography-determined serotonin 1A receptor binding potential in cynomolgus monkeys. Arch Gen Psychiatry. 2006;63:396–403. doi: 10.1001/archpsyc.63.4.396. [DOI] [PubMed] [Google Scholar]
- Sprouse J, Reynolds L, Li X, Braselton J, Schmidt A. 8-OH-DPAT as a 5-HT7 agonist: phase shifts of the circadian biological clock through increases in cAMP production. Neuropharmacology. 2004;46:52–62. doi: 10.1016/j.neuropharm.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Sullivan NR, Crane JW, Damjanoska KJ, Carrasco GA, D'Souza DN, Garcia F, et al. Tandospirone activates neuroendocrine and ERK (MAP kinase) signaling pathways specifically through 5-HT1A receptor mechanisms in vivo. Naunyn Schmiedebergs Arch Pharmacol. 2005;371:18–26. doi: 10.1007/s00210-004-1005-7. [DOI] [PubMed] [Google Scholar]