

Abstract
Background and purpose:
Prostanoid EP4 receptor antagonists may have therapeutic utility in the treatment of migraine since EP4 receptors have been shown to be involved in prostaglandin (PG)E2-induced cerebral vascular dilatation, which may be an important contributor to migraine pain. This study reports the pharmacological characterization of BGC20-1531, a novel EP4 receptor antagonist.
Experimental approach:
BGC20-1531 was characterized in radioligand binding and in vitro functional assays employing recombinant and native EP4 receptors. Changes in canine carotid haemodynamics were used to assess the pharmacodynamic profile of BGC20-1531 in vivo.
Key results:
BGC20-1531 exhibited high affinity at recombinant human EP4 receptors expressed in cell lines (pKB 7.6) and native EP4 receptors in human cerebral and meningeal artery (pKB 7.6–7.8) but showed no appreciable affinity at a wide range of other receptors (including other prostanoid receptors), channels, transporters and enzymes (pKi < 5). BGC20-1531 competitively antagonized PGE2-induced vasodilatation of human middle cerebral (pKB 7.8) and meningeal (pKB 7.6) arteries in vitro, but had no effect on responses induced by PGE2 on coronary, pulmonary or renal arteries in vitro. BGC20-1531 (1–10 mg·kg−1 i.v.) caused a dose-dependent antagonism of the PGE2-induced increase in canine carotid blood flow in vivo.
Conclusions and implications:
BGC20-1531 is a potent and selective antagonist at EP4 receptors in vitro and in vivo, with the potential to alleviate the symptoms of migraine that result from cerebral vasodilatation. BGC20-1531 is currently in clinical development for the treatment of migraine headache.
Keywords: EP4 receptor, PGE2, migraine, pain
Introduction
Migraine is characterized by intermittent neurovascular headache with certain associated features, such as sensitivity to light, sound or movement, and often with nausea or vomiting (Goadsby and Olesen, 1996). Migraine affects 10–20% of the world population and is listed in the top 20 causes of disabling conditions, and in the top four neurological disabling conditions by the World Health Organization. This condition accounts for an estimated 30 million days of lost productivity, at a cost of up to $17 billion per annum in the USA alone (Hu et al., 1999).
Migraine is a complex disorder and the mechanism underlying migraine attacks remains to be fully elucidated. While cerebrovascular dilatation has long been known to be associated with the aetiology of the disorder, its contribution to the occurrence of head pain and associated aura remains to be established. Furthermore, extensive research in the field has led to additional theories as to the mechanistic basis of migraine, including the neurological (neuronal sensitization, particularly of the trigeminal nerve), and the neurogenic (dural inflammation resulting in the release of inflammatory neuropeptides) (Goadsby, 2007). There is now good reason to believe that cortical spreading depression is involved at least in the aura phase in migraine, although there is less evidence for its involvement in head pain (Richter and Lehmenkühler, 2008).
At present, the most commonly used therapies in the treatment of acute migraine are paracetamol, non-steroidal anti-inflammatory drugs (NSAIDs) and triptans (5-HT1B/1D receptor agonists) (Adelman and Adelman, 2001; Goadsby et al., 2002). A recent survey assessing the level of satisfaction of patients with their current migraine therapy has revealed that a significant proportion of migraineurs do not experience sufficient relief of symptoms or are otherwise dissatisfied with current treatment (Bigal et al., 2007). Thus, while triptans and NSAIDs are effective in some migraineurs, there remains a significant need for novel agents that can act via alternative mechanisms to (i) treat acute migraine attacks in patients for whom triptans and NSAIDs are unsuitable due to their adverse effect profile; (ii) treat acute migraine attacks in patients for whom triptans and NSAIDs are insufficiently effective; and (iii) complement current pharmacological therapy in patients who only experience partial relief with triptans and NSAIDs.
Prostaglandin E2(PGE2) has long been recognized as an important mediator of pain and inflammation, and considerable clinical and experimental evidence implicates it in the pathogenesis of migraine. Elevated levels of PGE2 occur in the jugular venous blood (Sarchielli et al., 2000), plasma (Nattero et al., 1989) and saliva (Vardi et al., 1983; Tuca et al., 1989) of migraineurs undergoing an attack. In peripheral blood monocytes isolated from migraineurs, nitric oxide, a potential migraine trigger, elevates PGE2release twofold over that observed in non-migraineurs (Stirparo et al., 2000). Furthermore, intravenous infusion of prostaglandins can trigger migraine-like symptoms in migraineurs (Carlson et al., 1968; Peatfield et al., 1981). As previously indicated, the pain of migraine is associated with aberrant relaxation of cerebral and cranial blood vessels, and PGE2has been shown to be a potent and direct vasodilator of human cerebral and meningeal arteries (Davis et al., 2004). In addition, in vivo and in vitro chemical stimulation of dura mater (Zimmermann et al., 2002), trigeminal neurones (Jenkins et al., 2001), afferent nerves (Hua et al., 1994) and sensory afferents (Friese et al., 1997; Boku et al., 2001) cause PGE2release.
The actions of PGE2 are mediated primarily via prostanoid EP receptors, of which there are four subtypes, EP1, EP2, EP3 and EP4, each of which exhibits nanomolar affinity for the endogenous prostanoid, PGE2 (Coleman et al., 1994a). While EP1 and EP3 receptors mediate smooth muscle contraction, EP2 and EP4 receptors have previously been shown to exert relaxant effects on vascular smooth muscle (Negishi et al., 1995).
Rank orders of agonist potencies pointed to a role for EP4 receptors in the cerebral vasodilatory response to PGE2 in human middle cerebral and meningeal arteries (Davis et al., 2004). Pre-treatment with EP4 receptor antagonists (AH23848, EP4A) caused concentration-related rightward, surmountable displacement of PGE2 concentration-effect curves (Davis et al., 2004). The antagonist potencies in blocking the effects of PGE2 were consistent with their affinities for the human recombinant EP4 receptor (Coleman et al., 1994b).
In addition, although sensory neurons are capable of expressing multiple EP receptor subtypes, the use of reverse transcription polymerase chain reaction and antisense oligonucleotides of EP4 mRNA, have shown that PGE2-induced augmentation of peptide release and sensitization of sensory neurons are mediated by EP4 receptors (Southall and Vasko, 2001). As PGE2-induced cerebrovascular dilatation and neuronal sensitization may contribute to migraine pain, these data suggest that antagonists at EP4 receptors may have therapeutic utility in the treatment of migraine.
Based on the hypothesis that EP4 receptor blockade will prove useful in alleviating migraine pain, whether through its inhibition of cerebrovascular dilatation or its direct anti-hyperalgesic activity, a lead identification and optimization programme was initiated which resulted in the generation of a novel small molecule antagonist at EP4 receptors, BGC20-1531 (N-(4-[4-(5-methoxypyridin-2-yl)phenoxymethyl]-5-methylfuran-2-carbonyl)-2-methylbenzenesulphonamide sodium salt) (Figure 1). Here we describe, for the first time, the pharmacological characteristics of this molecule in vitro and in vivo.
Figure 1.
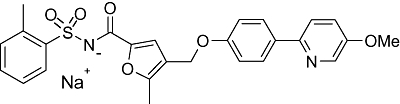
The chemical structure of BGC20-1531 (N-(4-[4-(5-methoxypyridin-2-yl)phenoxymethyl]-5-methylfuran-2-carbonyl)-2-methylbenzenesulphonamide sodium salt).
Methods
Selectivity and specificity
Broad spectrum specificity screening of BGC20-1531 was carried out by Cerep SA (http://www.cerep.com/) against a range of seven transmembrane (7TM) receptors, transmitter-gated channels, ion channels, nuclear receptors, transporters and enzymes (nomenclature utilized conforms to the BJP's Guide to Receptors and Channels; Alexander et al., 2008). The IC50values (concentration causing a half-maximal inhibition of control values), EC50 values (concentration causing a half-maximal stimulation of control values) and Hill coefficients (nH) were determined by non-linear regression analysis of the concentration-response curves using Hill equation curve fitting. In each experiment, the respective reference compound was tested concurrently with BGC20-1531 in order to assess the assay suitability and the data were compared with historical values determined at Cerep SA.
Human recombinant EP4 receptors – radioligand displacement binding
Membranes were prepared from cells stably transfected with human EP receptor cDNA. In brief, human embryonic kidney-293 Epstein Barr nuclear antigen (HEK-293 EBNA) cells were cultured to confluency, scraped from culture flasks, and centrifuged (800× g, 8 min, 4°C). Cells were twice washed in ice-cold homogenization buffer containing (mmol·L−1) 10 Tris-HCl, 1 ethylenediaminetetraacetic acid, 2 Na (EDTA), 250 sucrose, 1 phenylmethylsulphonyl fluoride, 0.3 indomethacin, pH 7.4, then homogenized and re-centrifuged as before. The supernatant was stored on ice and pellets re-homogenized and re-spun. Supernatants were pooled and centrifuged at 40 000× g for 10 min at 4°C. Resultant membrane pellets were stored at −80°C until use.
Membranes expressing human EP4, EP3 or EP2 receptors were incubated in Millipore (MHVBN45) plates containing assay buffer [10 mmol·L−1 2-(N-morpholino)ethanesulphonic acid (MES) pH 6.0; 10 mmol·L−1 MgCl2; 1 mmol·L−1 EDTA, 3 µmol·L−1 indomethacin], radiolabelled [3H] PGE2 and 0.1–10 000 nmol·L−1 concentrations of compounds. Incubations were performed at 30°C for 60 min to allow equilibrium to be reached. Non-specific binding was determined in the presence of 10 µmol·L−1 PGE2. Bound and free radiolabel was separated by vacuum manifold filtration using appropriate wash buffers (10 mmol·L−1 MES pH 6.0; 10 mmol·L−1 MgCl2), and bound radiolabel was determined by scintillation counting. The affinity or pKi of each compound for each receptor was calculated from the concentration causing 50% radioligand displacement (IC50) using the Cheng-Prusoff equation: Ki = IC50/(1 + ([[3H] PGE2]/KD).
Saturation analysis was also performed using 3 nmol·L−1 PGE2 or 10 nmol·L−1 BGC20-1531 in the presence of increasing concentrations of radiolabelled [3H] PGE2. Non-specific binding was determined in the presence of 10 µmol·L−1 PGE2. Saturation data were transformed in the form of a Scatchard plot.
Human recombinant EP4 receptors – functional studies
The ability of BGC20-1531 to antagonize PGE2-mediated functional responses mediated via the human EP4 receptor was determined by measuring concentration-dependent PGE2-stimulated cAMP accumulation.
Transfected HEK-293 cells were cultured at 37°C in a 5% CO2 incubator, in 96-well poly-L-lysine coated plates at a density of 50 000 cells per well. Culture medium was Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% foetal bovine serum, 100 U·mL−1 penicillin, 100 ng·mL−1 streptomycin, 2.5 µg·mL−1 fungizone, 2 mmol·L−1 glutamine, together with an appropriate selection antibiotic, Zeocin (200 µg·mL−1), hygromycin (200 µg·mL−1) and geneticin (G418, 250 µg·mL−1).
Cells were cultured to confluency (3–4 days) prior to use. Medium was removed and replaced with DMEM, containing 1 mmol·L−1 3-isobutyl-1-methylxanthine and BGC20-1531 (concentration range 0.1, 1.0 and 10 µmol·L−1) or vehicle (0.1% DMSO) for 60 min, 37°C. PGE2 (concentration range 0.0001 to 10 µmol·L−1) was subsequently added for 15 min, 37°C, before the reaction was terminated by the addition of 1 mol·L−1 HCl. Plates were stored at −80°C until assay, then rapidly thawed and samples neutralized by the addition of 1 mol·L−1 NaOH. Plates were centrifuged (1000× g, 5 min) before the supernatant was transferred to a fresh 96 well plate, containing assay buffer (in mmol·L−1) 50 Tris, 10 EDTA, 0.1% bovine serum albumin, pH 7; 8 µg cAMP binding protein and ≈2 nmol·L−1[3H]cAMP, then incubated on ice for 2 h. Bound and free radiolabel were separated by rapid vacuum filtration and scintillation counting was used to determine bound radioligand. The amount of cAMP generated was determined from a standard curve. Comparison of the rightward displacement of the PGE2-stimulated cAMP accumulation by BGC20-1531 was used to estimate the antagonist affinity (pKB). The mean concentration ratio (CR) was plotted as log (CR-1) against log antagonist molar concentration, according to the method of Arunlakshana and Schild (1959). If the slope of the plot did not differ significantly from unity, it was constrained to unity to calculate an apparent pKBvalue. Where only one concentration of BGC20-1531 was tested, a slope of unity was assumed and pA2 estimated using the Gaddum-Schild equation (where pA2 = log[CR − 1] − log[antagonist]).
EP4 receptors in human cerebral and meningeal arteries in vitro
All samples of human tissue were obtained through medically qualified intermediaries at the Netherlands Brain Bank with the informed consent of the donor or donor's next of kin, and with approval of the Local Research Ethics Committee (LREC). Tissues were rapidly removed at autopsy, transported on wet ice in phosphate-buffered saline (PBS), and stored in Krebs' buffer at 4°C until the experiment, which was carried out within 72 h of tissue removal from the patient. Viable cerebral arteries were obtained from 19 donors (11 male, 8 female) with an age range of 59–88 years.
Sections of middle cerebral artery were carefully removed from samples of human cerebral vasculature containing an intact circle of Willis using sharp dissection scissors. Regions of tissue which appeared to be atherosclerotic on visual inspection were not used. Intact rings of middle cerebral arteries, 2–3 mm in length and 1–2 mm internal diameter, were suspended between stainless steel hooks in 10 mL organ baths containing oxygenated (95% O2/5% CO2) Krebs' buffer containing (in mmol·L−1): NaCl 118.2, KCl 4.69, MgSO4.7H2O 1.18, K2HPO4 1.19, glucose 11.1, NaHCO3 25.0, CaCl2.6H2O 2.5, indomethacin 0.003, pH 7.4 at 37°C. Tissues were placed under a tension equivalent to 5 mN and left to equilibrate for a period of at least 60 min. Responses were recorded using isometric transducers coupled to an Apple Macintosh computer via a MacLab interface. Following the equilibration period, a cumulative contractile concentration-response curve to phenylephrine (minimum of 5 min contact time of each concentration of agonist) was constructed in all tissues. After washout, an approximate EC50 concentration of phenylephrine (1 µmol·L−1) was added to obtain a stable contraction. Tissues were subsequently exposed to increasing concentrations of PGE2, in the absence or presence of BGC20-1531 (incubated for 60 min prior to exposure to PGE2). Untreated tissues were run in parallel to agonist-treated tissue, to serve as time-matched controls. Experiments on the relaxant effects of prostanoids were performed in the presence of the thromboxane (TP) receptor antagonist [1R-[1(Z),2,3,5]]-(+)-7-[5-([1,1′-biphenyl]-4-ylmethoxy)-3-hydroxy-2-(1-piperidinyl)cyclopentyl]-4-heptanoic acid (GR32191) (1 µmol·L−1) (Lumley et al., 1989); as in the absence of GR32191, PGE2 caused vasodilatation at low concentrations but vasoconstriction at concentrations greater than 0.1 µmol·L−1 (n = 3, data not shown). The maximal relaxation of tissues was determined at the end of each experiment by the addition of prostacyclin (1 µmol·L−1).
The ability of BGC20-1531 (1 µmol·L−1) to reverse an established PGE2-mediated relaxation in cerebral arteries was also determined. Tissues were pre-contracted with 1 µmol·L−1 phenylephrine before relaxation was induced by the addition of 100 nmol·L−1 PGE2. BGC20-1531 (1 µmol·L−1) was subsequently added to observe possible reversal of the PGE2– induced vasorelaxation.
Meningeal arteries were dissected from the dura mater and intact rings (internal diameter <1 mm) were mounted as previously described. Following the equilibration period, a cumulative concentration-response curve to phenylephrine (0.01–100 µmol·L−1) was generated in all tissues. After washout, tissues were pre-incubated (60 min) with either BGC20-1531 (0.03, 0.1 or 1 µmol·L−1) or vehicle (0.1% DMSO) and then pre-contracted with an ≈EC70 concentration of phenylephrine (10 µmol·L−1). Once a stable phenylephrine contraction was obtained, tissues were exposed to increasing concentrations of PGE2 (0.001–100 µmol·L−1, log units). Prostacyclin (1 µmol·L−1) was added at the end of the PGE2 concentration-effect curve and the responses to PGE2 were normalized relative to the prostacyclin response.
Human coronary, pulmonary and renal arteries in vitro
All samples of human tissue were obtained through medically qualified intermediaries with the informed consent of the donor or the donor's next of kin, and with approval of the LREC. Tissues were transported on wet ice in PBS and stored in Krebs' buffer at 4°C until the experiment could be performed within 25 h of tissue removal from the patient. Coronary artery was obtained from five donors (four male, one female; age range 32–59), pulmonary artery was from four donors (two male, two female; age range 23–66) and renal arteries were obtained from four donors (four male; age range 37–63).
Sections of right proximal, distal and left coronary artery, pulmonary and renal artery were dissected from whole heart, lungs or kidneys, respectively, which were obtained from multi-organ donors. Sections of all arteries were cut into intact rings of approximately 2–3 mm in length (internal diameter 3–5 mm) and suspended between stainless steel hooks in 10 mL organ baths containing oxygenated (95% O2/5% CO2) Krebs' buffer, containing (in mmol·L−1): 118.2 NaCl, 4.69 KCl, 1.18 MgSO4·7H2O, 1.19 K2HPO4, 11.1 glucose, 25.0 NaHCO3, 2.5 CaCl2·6H2O, 0.003 indomethacin, pH 7.4 at 37°C. Responses were recorded using isometric transducers (Pioden, UK) coupled to a computer via a MacLab interface.
Tissues were placed under a tension equivalent to 10 mN and left to equilibrate for a period of at least 60 min. Tissue viability was subsequently assessed by challenge with 120 mmol·L−1 KCl and the generation of a cumulative contractile concentration-response curve to phenylephrine (0.001–100 µmol·L−1). In coronary artery experiments, isoprenaline (1 µmol·L−1) was also added at the start of each equilibration to reduce spontaneous activity. Tissues were subsequently washed with Krebs' solution and pre-treated for 30 min with a TP receptor antagonist (either SQ29548 or GR32191; 1 µmol·L−1). Tissues were either pre-contracted with a sub-maximal concentration of phenylephrine (1 µmol·L−1) or left untreated prior to incubation with BGC20-1531 (1 µmol·L−1) or DMSO (0.1%) for 30 min. A PGE2 concentration-response curve (0.001–10 µmol·L−1, log units) was subsequently constructed, followed by the addition of 1 µmol·L−1 PGI2 (prostacyclin) and 120 mmol·L−1 KCl, to allow normalization of the response.
Canine native EP4 receptors in vitro
All animal studies were conducted in accordance with Animals (Scientific Procedures) Act 1986, with UK Home Office Guidance on the implementation of the Act and with all applicable Codes of Practice for the care and housing of laboratory animals.
Organs were obtained from three male and three female beagle dogs (mean ± SEM ages 17.7 ± 0.6, 16.5 ± 0.9 months respectively). Brains and dura were transported in PBS and stored in Krebs' buffer at 4°C until the experiment could be performed, within 72 h of tissue removal.
Meningeal arteries were dissected from the dura mater and intact rings (outer diameter ≈300–500 µm) were mounted on 40 µm tungsten wires in Mulvany-Halpern wire myographs at a tension equivalent to that generated at 0.9 times the diameter of the vessel at 100 mm Hg. Arterial rings were maintained in a static 5 mL bath at 37°C in oxygenated (95% O2/5% CO2) Krebs' buffer containing (in mmol·L−1): 118.2 NaCl, 4.69 KCl, 1.18 MgSO4·7H2O, 1.19 K2HPO4, 11.1 glucose, 25.0 NaHCO3, 2.5 CaCl2·6H2O, 0.003 indomethacin, pH 7.4. Following several washes and an equilibration period, tissues were incubated with GR32191 (1 µmol·L−1), as in its absence PGE2 caused vasoconstriction at high concentrations. Viability was assessed by challenge with a single concentration of phenylephrine (10 µmol·L−1). Following washout, a cumulative concentration-response curve to phenylephrine (0.01–100 µmol·L−1, log units) was generated in all tissues. After a further washout and equilibration stage, tissues were pre-incubated for 60 min with either BGC20-1531 (0.03, 0.1 or 1 µmol·L−1) or its vehicle (0.1% DMSO) and then pre-contracted with an ≈EC50–70 concentration of phenylephrine (1–3 µmol·L−1). Once a stable phenylephrine contraction was maintained, tissues were exposed to increasing concentrations of PGE2 (0.01–10 µmol·L−1). Prostacyclin (1 µmol·L−1) was added at the end of the concentration-response curve. Responses to PGE2 were subsequently normalized relative to the response to prostacyclin. A pA2 estimate for BGC20-1531 was calculated using the Gaddum-Schild equation, where pA2 = log [CR − 1] − log[antagonist].
Canine native EP4 receptors in vivo – effects on PGE2-mediated carotid vasodilator responses
Four beagle dogs (Marshall BioResources, North Rose, NY; 6.5 kg) were anaesthetized, intubated and artificially ventilated. For induction of anaesthesia, inhalant anaesthetic (sevoflurane) was delivered through a facemask. An i.v. catheter was placed and the animals were anaesthetized with a bolus dose of pentobarbital (20 mg·kg−1 i.v.) followed by an intravenous infusion at a rate between 2 and 20 mg·kg−1·h−1 (adjusted to maintain a proper plane of anaesthesia). Total volume flow was approximately 5 mL·kg−1·h−1. A midline incision was made on the ventral neck. The left common carotid artery exposed and a flow probe (Transonic) placed around the vessel to measure carotid artery blood flow. A side branch of the vessel (e.g. cranial thyroid) was isolated and a needle or cannula inserted for the injection of PGE2 into the carotid artery. An incision was made to expose the femoral artery which was cannulated for measurement of arterial pressure and heart rate (HR). The femoral vein was cannulated for injection of vehicle and BGC20-1531. The animal was then allowed to stabilize for at least 30 min after instrumentation.
Blood pressure [systolic, diastolic and mean arterial pressure (MAP)], HR, carotid blood flow (CBF) and carotid vascular resistance were continuously recorded on a computerized data acquisition system.
After stabilization, PGE2 was administered by bolus dosing at 0.02 µg·kg−1 directly into the carotid artery via the needle or catheter previously placed in a side branch. This dose was increased to 0.04 µg·kg−1 to achieve a measurable and reproducible response. Changes in MAP, HR and CBF response were recorded for 15 min. This process was repeated three times to establish the baseline response of each animal to intra-arterial PGE2 injections. After the baseline response was established, the dose-response profile to BGC20-1531 or vehicle was determined. For each dose level, the dose of test drug was administered intravenously (femoral vein catheter) as a slow bolus over approximately 1 min. Within 10 min after dosing, a 3 mL blood sample was obtained. Following the removal of the blood sample, and 15 min after the previous PGE2 challenge, the PGE2 challenge was repeated. After each dosing episode and PGE2 challenge, haemodynamics were allowed to stabilize and the process was repeated for each of the five dosing levels (vehicle and 1, 3, 10 and 10 mg·kg−1 i.v.). Following the final dosing episode, additional plasma samples were obtained at 25 and 35 min after the last dose was administered. The PGE2 challenge was repeated immediately after obtaining the plasma samples.
Data analysis
All values shown in the text and figures are mean ± SEM. Differences between groups were tested by one-way anova and unpaired two-tailed Student's t-test. P values less than 0.05 at 95% confidence level were considered significant.
Materials
PGE2, prostacyclin, PGD2, PGE1-OH, 11-deoxy PGE1 and PGF2 were obtained from Alexis Corp, UK and GR32191 was a kind gift from GlaxoSmithKline, UK.
Results
Affinity, selectivity and specificity for human EP4 prostanoid receptors
In membrane binding studies, BGC20-1531 displaced [3H]-PGE2 from human recombinant EP4 prostanoid receptors (stably expressed in HEK-293 EBNA cells) with a pKi of 7.9 ± 0.1 (11.7 nmol·L−1; n = 3, Figure 2A). BGC20-1531 exhibited negligible affinity for both EP2and EP3 receptors, displacing 17.4 ± 6.5% and 2.7 ± 1.9% of [3H]-PGE2 at concentrations of 10 µmol·L−1 respectively (Figure 2A). Furthermore, BGC20-1531 displayed negligible affinity for other members of the prostanoid receptor family (mean pKi values derived from radioligand binding studies using human recombinant receptors as follows: EP1 < 5, EP2 < 5, EP3 < 5, EP4 7.9, TP < 5, DP 5.6 and IP < 5). Scatchard analysis (Figure 2B) of the saturation binding of BGC20-1531 reveals a profile consistent with simple competitive binding with [3H]-PGE2 at the human recombinant EP4 receptor.
Figure 2.
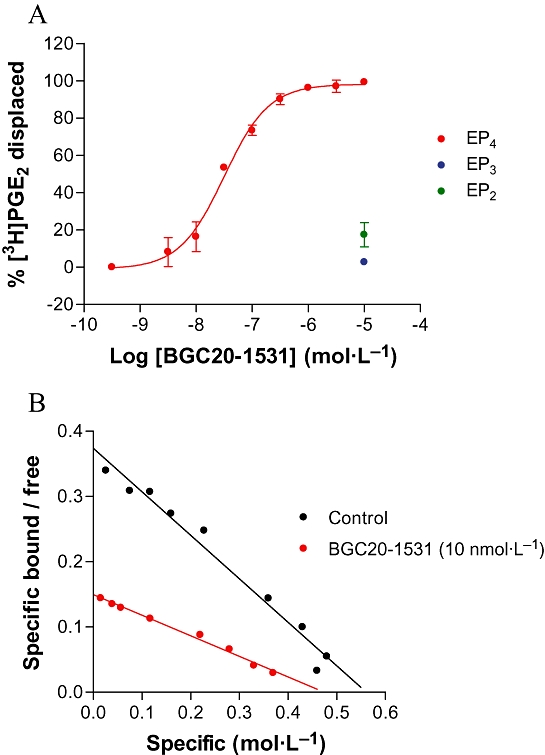
Radioligand displacement binding by BGC20-1531 at recombinant human EP4, EP3 and EP2 receptors stably expressed in HEK-293 EBNA cells (A). Data are representative of mean ± SEM derived from three independent experiments (EP2, EP3, EP4). Scatchard plot generated from [3H]-PGE2 saturation binding data, in the presence and absence of BGC20-1531 (10 nmol·L−1; n = 2) (B).
In addition to the above human recombinant EP receptors, the affinity of BGC20-1531 for a range of 42 7TM receptors, nine transmitter-gated channels, eight ion channels, four nuclear receptors, five cell-surface transporters and 16 enzymes has also been tested.
Binding assays comprised those for the 7TM receptors adenosine A1 (h), A2A (h) and A3 (h), adrenoceptor α1 (non-selective), α2 (non-selective), β1 (h) and β2 (h), imidazoline I1 and I2, angiotensin AT1 (h) and AT2 (h), bradykinin B1 (h) and B2 (h), cannabinoid CB1 (h) and CB2 (h), cholecystokinin CCK1 and CCK2, corticotrophin-releasing factor CRF1, dopamine D1 (h), D2 (h), D3 (h) and D4 (h), endothelin ETA (h) and ETB (h), histamine H1 (h), H2 (h) and H3, leukotriene CysLT1 (h), melanocortin MC4 (h), acetylcholine muscarinic M (non-selective), tachykinin NK1 (h), NK2 (h) and NK3 (h), neuropeptide Y (non-selective), opioid (non-selective) and N/OFQ (h), prostanoid TP and IP, P2Y, 5-HT (non-selective), thyrotrophin-releasing hormone TRH, and vasopressin V1a (h) and V2 (h); the transmitter-gated channels, acetylcholine nicotinic α4, benzodiazepine, GABAA, glutamate (ionotropic) AMPA, kainate, NMDA and PCP, P2X; the ion channels, calcium Cav1.1, Cav1.2 and Cav1 potassium KIR, KV and KCa, sodium NaV1.2 and chloride CIC; the nuclear receptors, glucocorticoid GR (h), oestrogen ER (h), progesterone PR (h) and androgen AR (h); and the cell-surface transporters, monoamine NA (h), DA (h), and SERT (h), GABA GAT, and choline CHT.
Enzyme assays comprised those for the enzymes 3′,5′-cyclic nucleotide phosphodiesterases 1 (h), 2 (h), 3 (h), 4 (h) and 5 (h), adenylyl cyclase, guanylyl cyclase, protein kinase C, L-tyrosine hydroxylase, acetylcholinesterase (h), catechol-O-methyl transferase, GABA transaminase, monoamine oxidase(MAO)-A (h), MAO-B (h), phenylethanolamine-N-methyl transferase, and ATPase (Na+/K+).
Based on screening data at a concentration of 10 µmol·L−1, BGC20-1531 caused less than 50% inhibition of binding (corresponding to a pKi < 5) at all targets described above (data not shown).
Antagonism of responses mediated by human recombinant EP4 receptors in cell-based assays
In adherent HEK-293 EBNA cells that stably express the human EP4 receptor, increasing concentrations of BGC20-1531 caused surmountable, competitive antagonism of PGE2-mediated cAMP accumulation (Figure 3A and 3B). Schild analysis (slope of 0.95) resulted in an estimated mean pKB for BGC20-1531 of 7.6 (n = 3 at each concentration; Figure 3C). PGE2was also shown not to stimulate cAMP accumulation in non-transfected HEK-293 cells, demonstrating that PGE2-induced cAMP accumulation is mediated via EP4 receptor stimulation (n = 3, data not shown). Furthermore, at concentrations of 1 and 10 µmol·L−1, BGC20-1531 had no effect on forskolin-stimulated cAMP accumulation in these cells, suggesting that BGC20-1531 acts as a specific antagonist of PGE2 (n = 3, data not shown).
Figure 3.
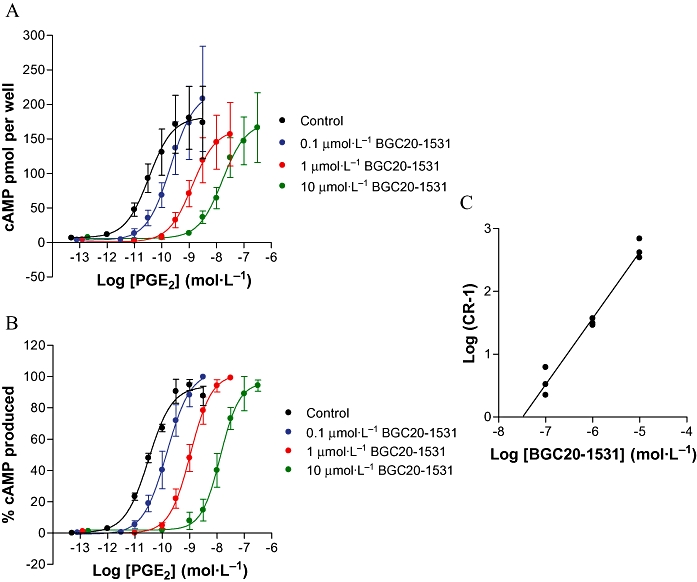
PGE2-stimulated cAMP accumulation in HEK-293 cells stably expressing the EP4 prostanoid receptor in the absence or presence of BGC20-1531. BGC20-1531 was pre-incubated with cells for 1 h prior to 15 min stimulation with PGE2. Data are representative of mean ± SEM derived from three independent experiments at each concentration and are expressed as cAMP pmol per well (A) and percentage normalized to that obtained in response to maximal PGE2 stimulation (B). Both these sets of data show a surmountable competitive antagonism of cAMP accumulation. In (C) is shown a Schild analysis of the antagonism of PGE2-stimulated cAMP accumulation in transfected HEK-293 cells by increasing concentrations of BGC20-1531 (C). A best fit linear regression of unity is shown.
Antagonism of the effects of PGE2 in human cerebral and meningeal arteries in vitro
Addition of phenylephrine (10 µmol·L−1) caused reproducible contractions of isolated preparations of the human middle cerebral artery. Contractions observed at all phenylephrine concentrations were well maintained. Application of increasing concentrations of PGE2 to cerebral artery rings pre-contracted with phenylephrine produced concentration-dependent relaxations to basal levels (Figure 4A).
Figure 4.
PGE2-stimulated vasodilatation in human cerebral artery rings in the absence or presence of BGC20-1531. In (A), cerebral artery rings were pre-incubated with BGC20-1531 30 min prior to stimulation with PGE2, and demonstrated surmountable competitive antagonism. Data are representative of mean ± SEM derived from 4–5 independent experiments at each concentration, and are normalized to the maximal relaxation caused by PGI2. In (B), Schild analysis of the antagonism of EP4 receptor mediated PGE2-stimulated vasodilatation in human middle cerebral artery by increasing concentrations of BGC20-1531 is shown. Data are from 4–5 independent experiments at each concentration and a linear regression slope of unity is shown.
In human middle cerebral arterial rings pre-contracted with phenylephrine (10 µmol·L−1), BGC20-1531 antagonized PGE2-mediated vasorelaxation with a pKB of 7.8 (n = 4–5 at each concentration). The antagonism appeared to be competitive (Figure 4A), and Schild analysis of the antagonism showed a slope not significantly different from unity (Figure 4B).
In human middle meningeal artery rings, again pre-contracted with phenylephrine (10 µmol·L−1), BGC20-1531 antagonized PGE2-mediated vasorelaxation with a pKB of 7.6 ± 0.1 (n = 3–5 at each concentration; Figure 5A). The antagonism was apparently competitive in nature, and Schild analysis of the antagonism showed a slope not significantly different from unity (Figure 5B).
Figure 5.
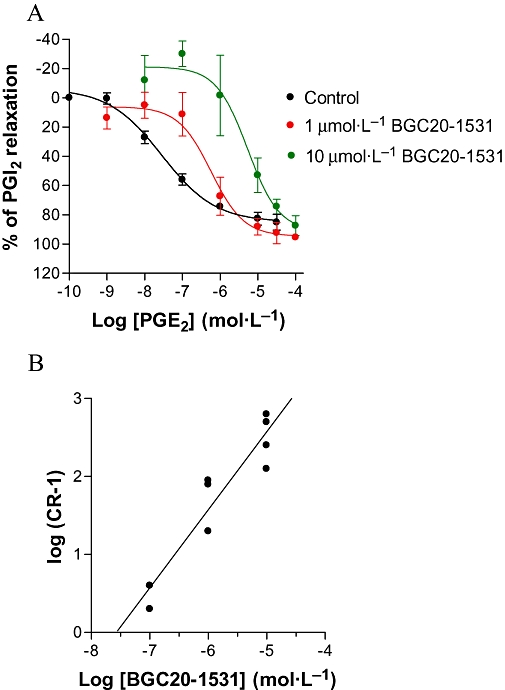
Effects of BGC20-1531 on PGE2-stimulated relaxation of human middle meningeal artery rings. In (A), BGC20-1531 (1 or 10 µmol·L−1) was pre-incubated with tissues 30 min prior to PGE2 stimulation, and demonstrated surmountable competitive antagonism. Data obtained in the presence of 0.1 µmol·L−1 BGC20-1531 have been omitted for ease of graphical interpretation. Data are representative of mean ± SEM derived from 3–5 independent experiments at each concentration, and are normalized to the maximal relaxation to PGI2. In (B), a Schild analysis of the antagonism of EP4 receptor mediated PGE2-stimulated vasodilatation in human middle meningeal artery by increasing concentrations of BGC20-1531 is shown. Data are from 3–5 independent experiments at each concentration and a linear regression slope of unity is shown.
For BGC20-1531 to be useful in providing acute pain relief during an episode of migraine, it would be given after the point at which PGE2has begun to evoke vasodilatation of pain-sensitive cerebral vessels. Attempts were made to model this situation in vitro under conditions where PGE2-induced relaxation of cerebral vessels was already established (more relevant to migraine). Under these conditions and as illustrated in Figure 6, BGC20-1531 (1 µmol·L−1) demonstrated a rapid and complete reversal of an established vasorelaxant response to PGE2(100 nmol·L−1) in human middle cerebral arterial rings (by 169 ± 34%, n = 3). A similar rapid reversal of an established PGE2 response (100 nmol·L−1) was observed with BGC20-1531 (1 µmol·L−1) in human middle meningeal artery (by 95 ± 21%, n = 3). These data suggest that BGC20-1531 will offer pain relief after a migraine attack has commenced, if the pain is caused by PGE2- induced cerebral vasodilatation.
Figure 6.
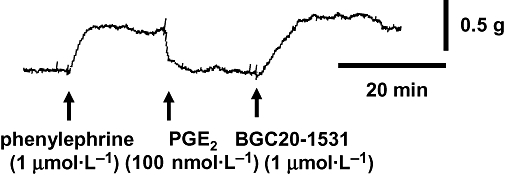
Reversal of an established PGE2-mediated relaxation in rings of human middle cerebral artery, by BGC20-1531 (1 µmol·L−1). The tissue was pre-contracted with phenylephrine (1 µmol·L−1), before vasodilatation was induced by the addition of PGE2 (100 nmol·L−1). BGC20-1531 induced a rapid and complete reversal of the PGE2 response (by 133%). The response to BGC20-1531 shown here is representative of data from three independent experiments.
In summary, BGC20-1531 is a potent antagonist of the human prostanoid EP4 receptor. The affinity and potency of the antagonist demonstrated in cell lines expressing recombinant human EP4 receptors (pKi = 7.9 and pKB = 7.6) are in line with the antagonist potency estimates at native human EP4 receptors expressed in middle cerebral and meningeal arteries (pKB 7.6–7.8).
Potential for cardiovascular, pulmonary or renal side effects of BGC20-1531
In human coronary, pulmonary and renal arteries in vitro, PGE2 was found to cause concentration-dependent vasoconstriction [pEC50 (mol·L−1) 7.2 ± 0.1, 7.8 ± 0.2, 6.4 ± 0.2 respectively], and not vasodilatation as observed in the cerebral and meningeal arteries. The magnitude of the response mediated by PGE2 was greater in coronary than in pulmonary than in renal arteries. In all three vessel types, BGC20-1531 alone was shown to have no effect on basal or PGE2-induced elevated tone (data not shown, n = 3–4 each preparation). BGC20-1531 also failed to exhibit any significant antagonism of PGE2-induced vasoconstriction in coronary, pulmonary or renal arteries respectively (Figure 7).
Figure 7.
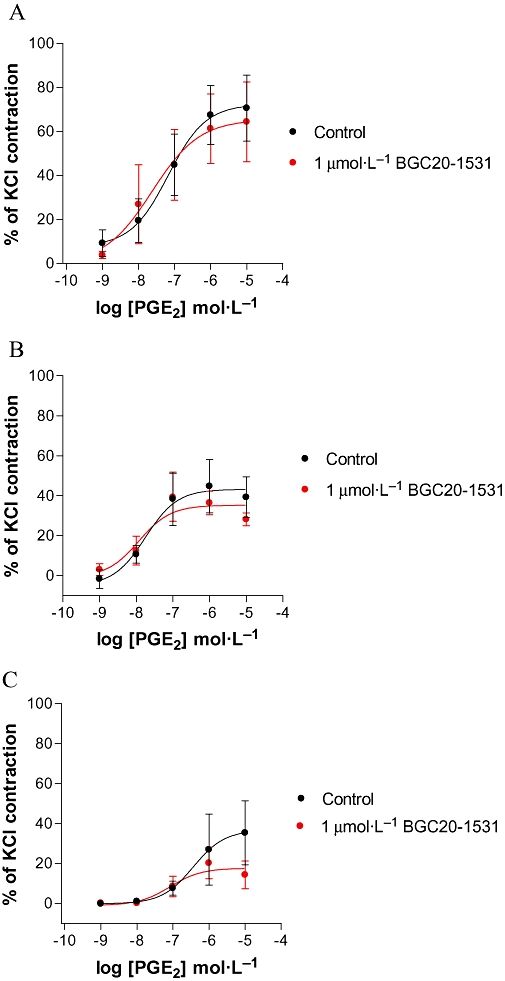
PGE2-stimulated contraction of rings from human (A) coronary, (B) pulmonary and (C) renal arteries in the absence or presence of BGC20-1531 (1 µmol·L−1). Either vehicle (DMSO 0.1%) or BGC20-1531 were incubated with tissues 30 min prior to PGE2 stimulation. BGC20-1531 had no effect on the PGE2-mediated contraction of human coronary, pulmonary or renal arteries. Data are representative of mean ± SEM derived from four independent experiments and are normalized to the maximal contractile response observed to KCl (120 mmol·L−1).
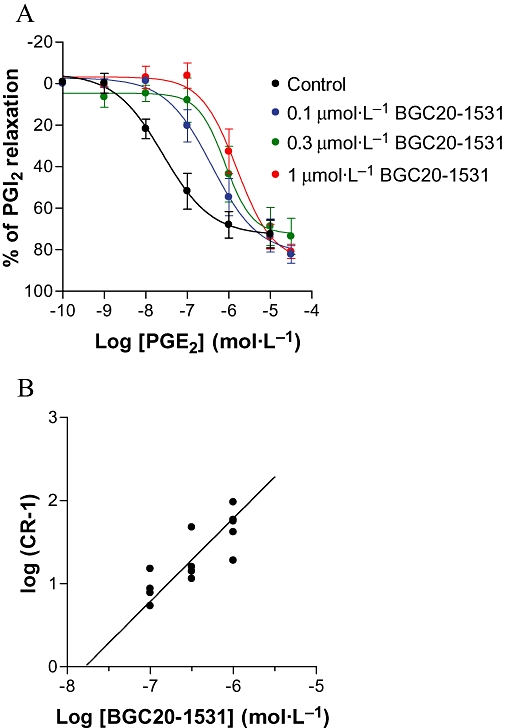
Antagonism of the effects of PGE2 in canine middle meningeal arteries in vitro
In preparations from each of four animals, PGE2 caused concentration-dependent relaxation of phenylephrine pre-contracted (1–3 µmol·L−1) canine middle meningeal arterial rings, exhibiting a mean pEC50 of 7.6 ± 0.2 (EC50 = 25 nmol·L−1, Figure 8A,B). BGC20-1531 (0.1, 0.3 or 1 µmol·L−1, 60 min incubation) showed competitive antagonism of PGE2-induced relaxation of canine meningeal artery (Figure 8B), exhibiting a mean pA2of 7.7 (n = 8 tissues from four animals at three BGC20-1531 concentrations). Hence, the functional effects of BGC20-1531 at canine EP4 receptors expressed in the middle meningeal artery are comparable to those observed in human vessels (pKB 7.6–7.8).
Figure 8.
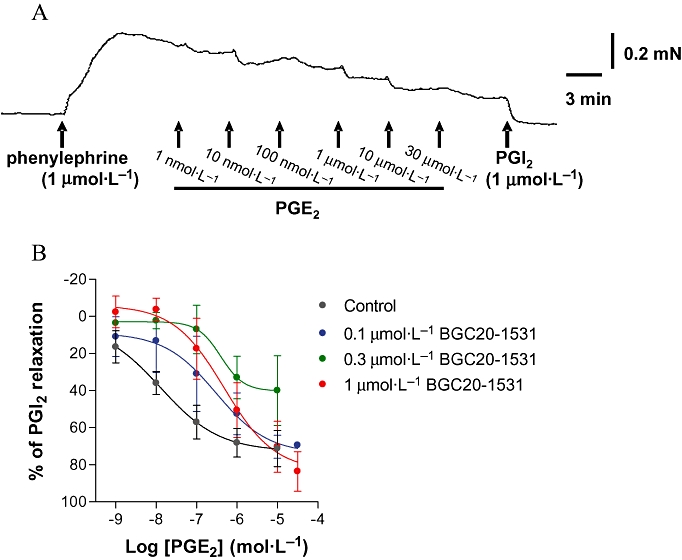
PGE2 induced relaxation of rings of canine meningeal artery, pre-contracted with phenylephrine and antagonism by the EP4 receptor antagonist, BGC20-1531. (A) Typical concentration-dependent relaxant responses to PGE2 (1 nmol·L−1 to 30 µmol·L−1) in phenylephrine pre-contracted canine middle meningeal artery rings in vitro. Representative data are shown from one of four independent experiments. (B) Concentration-dependent rightward shift of relaxant concentration-effect curves to PGE2. Rings of meningeal artery were pre-incubated for 60 min with vehicle, 0.1 µmol·L−1, 0.3 µmol·L−1 or 1 µmol·L−1 BGC20-1531, then cumulatively concentration-dependently relaxed with PGE2. Data are expressed as percentage of the relaxation produced by prostacyclin (1 µmol·L−1) and are expressed as mean ± SEM derived from four dogs.
Antagonism of the effects of PGE2 on canine carotid vascular bed in vivo
Inhibition of PGE2-induced haemodynamic responses in the canine carotid vascular bed was investigated as a suitable pharmacodynamic model and to establish an in vivo dose–response relationship of this novel EP4 receptor antagonist.
Prostaglandin E2 (0.04 µg·kg−1, dose volume 0.5 mL·kg−1) injected into the carotid artery caused an increase in CBF, with a corresponding decrease in carotid resistance (CR). Reproducible responses to repeated PGE2 challenges were seen with mean reductions in CBF of 52%, 49% and 41% seen on the first, second and third applications, respectively; corresponding mean decreases in CR were measured to be 34, 31 and 28% respectively. These doses of PGE2 had no effect on MAP, indicating that the dose and route of administration used in the study induced effects restricted to the carotid vascular bed.
There was no effect of BGC20-1531 on either MAP or HR. However, BGC20-1531 (1–10 mg·kg−1 i.v.) caused a dose-dependent antagonism of the PGE2-induced increase in CBF and a corresponding antagonism of the PGE2-mediated decrease in CR, with an ID50 ≈ 5 mg·kg−1 (Figure 9). In contrast, there was no effect of the vehicle on either CBF or CR. Blood samples were taken at regular intervals throughout the study, each blood sample being taken prior to PGE2 injection, and analysed for BGC20-1531 concentration by LC-MS. There was an apparent correlation between the plasma concentration of BGC20-1531 and the degree of antagonism of PGE2-induced carotid vasodilatation. Therefore, BGC20-1531 displayed dose-dependent efficacy in this pharmacodynamic model.
Figure 9.
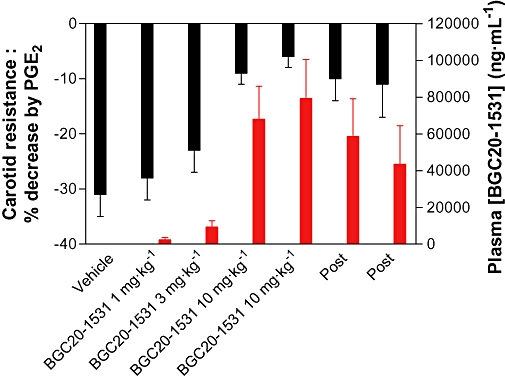
Effects of BGC20-1531 on PGE2-induced changes in common carotid resistance in anaesthetized dogs. Resistance in the vascular bed served by the common carotid artery was decreased by close intra-arterial injection of PGE2 (0.04 µg·kg−1, 15 min apart), as shown on the left hand axis. Ten minutes after a dose of vehicle or BGC20-1531 (1, 3, 10 and 10 mg·kg−1 i.v.) and immediately before a test dose of PGE2, blood samples were taken for measurement of BGC20-1531 levels in plasma (right hand axis). The effects of PGE2 (decreased carotid resistance) were markedly reduced by 10 mg·kg−1 of BGC20-1531. As the plasma levels of BGC20-1531 fell (values marked Post), the effects of the test doses of PGE2 returned. Carotid resistance is presented as % change from baseline and the plasma level of BGC20-1531 as ng·mL−1; data are mean ± SEM (n = 4).
Discussion and conclusions
The novel small molecule BGC20-1531 was identified through a lead identification and medicinal chemistry optimization programme. Emphasis was placed on the use of human recombinant receptors and disease relevant, ethically donated human tissues to investigate the pharmacological profile of BGC20-1531. Thus, radioligand binding studies confirmed nanomolar affinity for the human EP4 receptor. Furthermore, at the recombinant human EP4 receptor, BGC20-1531 was devoid of agonist activity and competitively antagonised PGE2-mediated cAMP accumulation.
Importantly, in vitro, BGC20-1531 was also shown to be a potent antagonist of the vasodilator effects of PGE2 on the vessels implicated in migraine pain. Thus, BGC20-1531 acts as a competitive antagonist of PGE2 on middle cerebral and meningeal arteries. Moreover, BGC20-1531 rapidly reversed a pre-existing PGE2-induced dilator response in such vessels, and hence is likely to provide pain relief during an episode of migraine that has already commenced, that is, where the PGE2-induced cerebral dilatation and associated pain are already established. The possibility that antagonists at EP4 receptors may have therapeutic utility in the treatment of migraine is strengthened by reports that both administration of an EP4 receptor antagonist (AH23848) and EP4 receptor knockdown in mutant mice result in attenuated inflammation-induced thermal and mechanical behavioural hypersensitivity (Lin et al., 2006). More recently, another EP4 receptor antagonist (CJ-023,423) has been shown to display anti-hyperalgesic effects in animal models of acute (mechanical hyperalgesia in the carrageenan model) and chronic inflammatory pain, induced by complete Freund's adjuvant (Nakao et al., 2007). These observations suggest that blockade of EP4 receptors may prove a valid target for the pharmacological treatment of inflammatory pain.
Inhibition of haemodynamic responses in the carotid vascular bed was utilized to investigate the pharmacodynamic profile of BGC20-1531. Previous work (Lumley et al., 1982) indicated that PGE2 induced increases in CBF in dogs, possibly reflecting dilatation within the cranial vasculature. The validity of this model to predict responses in human cranial vasculature was achieved through demonstrations both that PGE2 relaxed canine cranial vessels, and that BGC20-1531 inhibited this relaxant effect in the same way as in human vessels. Meningeal arteries only were used in this study, as middle cerebral arteries from the dog proved too small to prepare in the same way as their human counterpart. Studies were then conducted in anaesthetized dogs to evaluate the pharmacodynamic properties of BGC20-1531, where external CBF was measured following intra-arterial administration of PGE2 in the absence and presence of BGC20-1531. This study revealed that BGC20-1531 also behaves as a selective EP4 receptor antagonist in vivo, causing dose- and plasma drug concentration-dependent antagonism of the increase in carotid artery blood flow seen in response to intra-arterial administration of PGE2.
While no current experimental animal model of migraine completely reflects the pathophysiology observed in man, various models have been developed which reflect distinct facets of this clinically heterogeneous disorder (Eikermann-Haerter and Moskowitz, 2008). While these animal models have significant shortcomings, their use in order to profile existing treatments has contributed to promising new drugs that are now being developed and brought to the clinic. One such model is the measurement of drug-induced changes in carotid haemodynamics (as described above), which has previously been used to characterize drugs believed to act in part via a vascular mechanism (Connor et al., 1997; Kapoor et al., 2003). It is encouraging therefore that BGC20-1531 has been shown to be effective in this model.
Triptans are generally recognized as the gold standard for pharmacological intervention in acute migraine, but while they remain a therapeutic and commercial success story, they are not without limitations. First, while they are relatively selective constrictors of the cerebral vasculature, which may contribute at least in part to their anti-migraine activity, they can also constrict coronary and pulmonary vasculature (Dodick, 2004), which may be responsible for the incidence in some patients of chest symptoms similar to angina. Indeed, in rare instances, triptans have been associated with myocardial infarction. For this reason, triptans are contraindicated in patients with ischaemic heart disease, uncontrolled hypertension and cerebrovascular disease (Papademetriou, 2004). The fact that BGC20-1531 did not cause constriction of human coronary, pulmonary and renal arteries, and that it had no effect on vasoconstriction mediated by PGE2suggests that BGC20-1531 will not have any effects on the vascular tone of coronary, pulmonary or renal arteries. These data are consistent with the view that, unlike the triptans, BGC20-1531 would also be acceptable for the treatment of migraine in patients with co-existing cardiovascular disease.
In migraineurs who are suitable for treatment with a triptan, efficacy is initially reasonably good, with approximately 60% of patients showing an improvement in headache at 2 h post-dose, and approximately 20% patients being pain free at the same time point (Ferrari et al., 2001). However, studies measuring the effectiveness of the triptans in providing sustained pain relief have revealed relatively low results, ranging from 20.9% to 42.6% of the cases (Diener et al., 2005; Loder et al., 2005; Winner et al., 2005; Cady et al., 2006). It is interesting that a number of independent, double-blind, placebo-controlled clinical studies have demonstrated that NSAIDs (inhibitors of prostanoid synthesis) are as effective as triptans in alleviating migraine and tension headache (Diener, 1999; Kellstein et al., 2000; Codispoti et al., 2001; Dib et al., 2002; Prior et al., 2002). This not only suggests that prostanoids play a significant role in the aetiology of the migraine syndrome, but it also raises the possibility that EP4receptor antagonists which selectively block the action of prostanoids involved in the migraine cascade should exhibit efficacy as anti-migraine agents at least equal to that of triptans and NSAIDs.
Non-steroidal anti-inflammatory drugs act through inhibiting PG synthesis by inhibiting the activities of cyclooxygenase (COX) enzymes. Two isoforms of COX have been identified, COX-1 and COX-2. In the main, COX-2 is induced in the presence of inflammation, whereas COX-1 is generally constitutively expressed, and is thought to play an essential role for example in normal gastrointestinal and renal function. NSAIDs inhibit both isoforms; inhibition of COX-2 is primarily associated with the therapeutic effects of NSAIDs, while inhibition of COX-1 is thought to cause the adverse gastrointestinal effects such as gastric erosion, ulceration and haemorrhage which limit their use (Cryer, 2000). BGC20-1531 is a highly selective antagonist at prostanoid EP4 receptors, displaying negligible affinity for other prostanoid receptors (EP1, EP2, EP3, TP, IP and DP), or a broad range of other receptors, ion channels, enzymes or transporters. This suggests that BGC20-1531 is likely to block the EP4-mediated vasodilatation of cerebral vessels, as well as EP4-mediated hyperalgesia, but is not likely to influence functions mediated by other prostanoid receptor subtypes. More specifically, BGC20-1531 is unlikely to antagonize the beneficial effects of PGs in the gastrointestinal tract, inhibition of which is known to result in gastric erosion, ulceration and haemorrhage (Araki et al., 2000). Furthermore, there is a wealth of evidence for the anti-inflammatory effects of PGE2 acting via EP2 in a number of systems (Meja et al., 1997; Aronoff et al., 2004; Burelout et al., 2004; Ahmad et al., 2006), which would not be affected by BGC20-1531. Hence, BGC20-1531 is expected to show an improved adverse effect and efficacy profile compared with NSAIDs. Consistent with this hypothesis, an EP4receptor antagonist (CJ-42794), given orally, did not produce any damage in the rat gastrointestinal mucosa, while an NSAID (indomethacin) caused gross lesions (Takeuchi et al., 2007).
One of the most promising additional approaches to migraine therapy is the development of calcitonin gene related peptide (CGRP) receptor antagonists. It is well established that there is a close relationship between CGRP and PGE2. The release of CGRP from sensory nerve fibres results in the subsequent appearance of PGE2 from the dura mater (Rich et al., 1996; Ebersberger et al., 1999). Conversely, PGE2can itself cause the release of CGRP, not only from cultured trigeminal neurones in vitro (Jenkins et al., 2001), but also from sensory afferents in vivo (Friese et al., 1997; Boku et al., 2001). It is very interesting, therefore, that positive results in clinical trials with the CGRP receptor antagonists BIBN4096 (olcegepant) (Rudolf et al., 2005; Doods et al., 2007) and, more recently, MK-0974 (Ho et al., 2008), have provided proof of a role for CGRP in migraine. The development of CGRP receptor antagonists for migraine is still in its infancy, but these early clinical results are encouraging.
In conclusion, BGC20-1531 is a potent and selective EP4receptor antagonist that has the potential to alleviate the symptoms of migraine caused by dilatation of cerebral blood vessels, with an improved side-effect profile in comparison with existing therapies. These data suggest that BGC20-1531 is suitable for clinical development as a ‘first in class’ approach for the treatment of migraine headache.
Acknowledgments
R J Davis was study director of the BGC20-1531 programme at Pharmagene Laboratories Ltd. He and the other authors would like to acknowledge the support of RL Sheldrick, S Williams, C Murdoch, J A Wade and I Cousins who were involved in the pharmacological characterisation at Pharmagene Laboratories Ltd (now Asterand UK Ltd) and J Sutton, C Newton, D Daley and N Harris who contributed to the lead identification and medicinal chemistry programme at Argenta Discovery Ltd. We would also like to thank A Woodrooffe (Asterand UK Ltd) for her assistance and GlaxoSmithKline UK for their kind provision of GR32191.
Glossary
Abbreviations:
- BGC20-1531
N-(4-[4-(5-Methoxypyridin-2-yl)phenoxymethyl]-5-methylfuran-2-carbonyl)-2-methylbenzenesulphonamide sodium salt
- CBF
carotid blood flow
- CGRP
calcitonin gene related peptide
- COX
cyclooxygenase
- CR
carotid resistance
- CSD
cortical spreading depression
- DMEM
Dulbecco's modified Eagle's medium
- DMSO
dimethylsulphoxide
- EBNA
Epstein-Barr nuclear antigen
- EDTA
ethylenediaminetetraacetic acid
- HEK
human embryonic kidney
- HR
heart rate
- LREC
Local Research Ethics Committee
- MAP
mean arterial pressure
- MES
2-(N-morpholino)ethanesulphonic acid
- NSAID
non-steroidal anti-inflammatory drug
- PBS
phosphate-buffered saline
- PGE2
prostaglandin E2
- PGI2
prostacylin
- RT-PCR
reverse transcription polymerase chain reaction
- 7TM receptor
the family C seven transmembrane receptor
- TP receptor
thromboxane receptor
- WHO
World Health Organisation
References
- Adelman JU, Adelman RD. Current options for the prevention and treatment of migraine. Clin Ther. 2001;23:772–788. doi: 10.1016/s0149-2918(01)80069-2. [DOI] [PubMed] [Google Scholar]
- Ahmad AS, Zhuang H, Echeverria V, Dore SJ. Stimulation of prostaglandin EP2 receptors prevents NMDA-induced excitotoxicity. J Neurotrauma. 2006;23(12):1895–1903. doi: 10.1089/neu.2006.23.1895. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition (2008 revision) Br J Pharmacol. 2008;153(Suppl.)(2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki H, Ukawa H, Sugawa Y, Yagi K, Suzuki K, Takeuchi K. The roles of prostaglandin E receptor subtypes in the cytoprotective action of prostaglandin E2 in rat stomach. Aliment Pharmacol Ther. 2000;14:116–124. doi: 10.1046/j.1365-2036.2000.014s1116.x. [DOI] [PubMed] [Google Scholar]
- Aronoff DM, Canetti C, Peters-Golden M. Prostaglandin E2 inhibits alveolar macrophage phagocytosis through an E-prostanoid 2 receptor-mediated increase in intracellular cyclic AMP. J Immunol. 2004;173:559–565. doi: 10.4049/jimmunol.173.1.559. [DOI] [PubMed] [Google Scholar]
- Arunlakshana O, Schild HO. Some quantitative use of drug antagonists. Br J Pharmacol Chemother. 1959;257:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigal M, Rapoport A, Aurora S, Sheftell F, Tepper S, Dahlof C. Satisfaction with current migraine therapy: experience from 3 centers in US and Sweden. Headache. 2007;47:475–479. doi: 10.1111/j.1526-4610.2007.00752.x. [DOI] [PubMed] [Google Scholar]
- Boku K, Ohno T, Saeki T, Hayashi H, Hayashi I, Katori M, et al. Adaptive cytoprotection mediated by prostaglandin I2 is attributed to sensitisation of CGRP-containing sensory nerves. Gastroenterology. 2001;120:134–143. doi: 10.1053/gast.2001.20916. [DOI] [PubMed] [Google Scholar]
- Burelout C, Thibault N, Levasseur S, Simard S, Naccache PH, Bourgoin SG. Prostaglandin E2 inhibits the phospholipase D pathway stimulated by formyl-methionyl-leucyl-phenylalanine in human neutrophils. Involvement of EP2 receptors and phosphatidylinositol 3-kinase gamma. Mol Pharmacol. 2004;66:293–301. doi: 10.1124/mol.66.2.293. [DOI] [PubMed] [Google Scholar]
- Cady R, Martin V, Mauskop A, Rodgers A, Hustad CM, Ramsay KE, et al. Efficacy of Rizatriptan 10 mg administered early in a migraine attack. Headache. 2006;46:914–924. doi: 10.1111/j.1526-4610.2006.00466.x. [DOI] [PubMed] [Google Scholar]
- Carlson LA, Ekelund LG, Orö L. Clinical and metabolic effects of different doses of prostaglandin E1 in man. Prostaglandin and related factors. Acta Med Scand. 1968;83:423–430. doi: 10.1111/j.0954-6820.1968.tb10502.x. [DOI] [PubMed] [Google Scholar]
- Codispoti JR, Prior MJ, Fu M, Harte CM, Nelson EB. Efficacy of non-prescription doses of ibuprofen for treating migraine headache. A randomised controlled trial. Headache. 2001;41:665–679. doi: 10.1046/j.1526-4610.2001.041007665.x. [DOI] [PubMed] [Google Scholar]
- Coleman RA, Smith WL, Narumiya S. VIII. Internation Union of Pharmacology Classification of prostanoid receptors: properties, distribution, and structure of the receptors and their subtypes. Pharmacol Rev. 1994a;46:205–229. [PubMed] [Google Scholar]
- Coleman RA, Grix SP, Head SA, Louttit JB, Mallet A, Sheldrick RL. A novel inhibitory prostanoid receptor in piglet saphenous vein. Prostaglandins. 1994b;47:151–168. doi: 10.1016/0090-6980(94)90084-1. [DOI] [PubMed] [Google Scholar]
- Connor HE, Feniuk W, Beattie DT, North PC, Oxford AW, Saynor DA, et al. Naratriptan: biological profile in animal models relevant to migraine. Cephalalgia. 1997;17:145–152. doi: 10.1046/j.1468-2982.1997.1703145.x. [DOI] [PubMed] [Google Scholar]
- Cryer B. NSAID gastrointestinal toxicity. Curr Opin Gastroenterol. 2000;16(6):495–502. doi: 10.1097/00001574-200011000-00006. [DOI] [PubMed] [Google Scholar]
- Davis RJ, Murdoch CM, Ali M, Purbrick S, Ravid R, Baxter GS, et al. EP4 prostanoid receptor mediated vasodilatation of human middle cerebral arteries. Br J Pharmacol. 2004;141:580–585. doi: 10.1038/sj.bjp.0705645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib M, Massiou H, Weber M, Henry P, Garcia-Acosta S, Bousser MG. Efficacy of oral ketoprofen in acute migraine: a double-blind randomised clinical trial. Neurology. 2002;58:1660–1665. doi: 10.1212/wnl.58.11.1660. [DOI] [PubMed] [Google Scholar]
- Diener HC. Efficacy and safety of intravenous acetylsalicylic acid lysinate compared to subcutaneous sumatriptan and parenteral placebo in the acute treatment of migraine. A double-blind, double-dummy, randomised, multicenter, parallel group study. Cephalalgia. 1999;19:581–588. doi: 10.1046/j.1468-2982.1999.019006581.x. [DOI] [PubMed] [Google Scholar]
- Diener HC, Gendolla A, Gebert I, Beneke M. Almotriptan in migraine patients who respond poorly to oral sumatriptan: a double-blind, randomized trial. Headache. 2005;45:874–882. doi: 10.1111/j.1526-4610.2005.05151.x. [DOI] [PubMed] [Google Scholar]
- Dodick DW. Triptans and chest symptoms: the role of pulmonary vasoconstriction. Cephalalgia. 2004;24:298–304. doi: 10.1111/j.1468-2982.2004.00675.x. [DOI] [PubMed] [Google Scholar]
- Doods H, Arndt K, Rudolf K, Just S. CGRP antagonists: unravelling the role of CGRP in migraine. Trends Pharmacol Sci. 2007;28:580–587. doi: 10.1016/j.tips.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Ebersberger A, Averbeck B, Messlinger K, Reeh PW. Release of substance P, calcitonin gene-related peptide and prostaglandin E2 from rat dura mater encephali following electrical and chemical stimulation in vitro. Neuroscience. 1999;89:901–907. doi: 10.1016/s0306-4522(98)00366-2. [DOI] [PubMed] [Google Scholar]
- Eikermann-Haerter K, Moskowitz MA. Animal models of migraine headache and aura. Curr Opin Neurol. 2008;21:294–300. doi: 10.1097/WCO.0b013e3282fc25de. [DOI] [PubMed] [Google Scholar]
- Ferrari MD, Roon KI, Lipton RB, Goadsby PJ. Oral triptans (serotonin 5-HT(1B/1D) agonists) in acute migraine treatment: a meta-analysis of 53 trials. Lancet. 2001;358:1668–1675. doi: 10.1016/S0140-6736(01)06711-3. [DOI] [PubMed] [Google Scholar]
- Friese N, Diop L, Chevalier E, Angel F, Riviere PJ, Dahl SG. Involvement of prostaglandins and CGRP-dependent sensory afferents in peritoneal irritation-induced visceral pain. Regul Pept. 1997;70:1–7. doi: 10.1016/s0167-0115(97)02141-1. [DOI] [PubMed] [Google Scholar]
- Goadsby PJ. Recent advances in understanding migraine mechanisms, molecules and therapeutics. Trends Mol Med. 2007;13:39–44. doi: 10.1016/j.molmed.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Goadsby PJ, Olesen J. Migraine: diagnosis and treatment in the 1990's. BMJ. 1996;312:1279–1282. doi: 10.1136/bmj.312.7041.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goadsby PJ, Lipton RB, Ferrari MD. Migraine – current understanding and treatment. N Engl J Med. 2002;346:257–270. doi: 10.1056/NEJMra010917. [DOI] [PubMed] [Google Scholar]
- Ho TW, Mannix LK, Fan X, Assaid C, Furtek C, Jones CJ, et al. Randomized control trial of an oral CGRP antagonist, MK-0974, in acute treatment of migraine. Neurology. 2008;70:1304–1312. doi: 10.1212/01.WNL.0000286940.29755.61. [DOI] [PubMed] [Google Scholar]
- Hu XH, Markson LE, Lipton RB, Stewart WF, Berger ML. Burden of migraine in the United States. Arch Intern Med. 1999;159:813–818. doi: 10.1001/archinte.159.8.813. [DOI] [PubMed] [Google Scholar]
- Hua XY, Jinno S, Back SM, Tam EK, Yaksh TL. Multiple mechanisms for the effects of capsaicin, bradykinin and nicotine on CGRP release from tracheal afferent nerves: role of prostaglandins, sympathetic nerves and mast cells. Neuropharmacology. 1994;33:1147–1154. doi: 10.1016/s0028-3908(05)80004-8. [DOI] [PubMed] [Google Scholar]
- Jenkins DW, Feniuk W, Humphrey PPA. Characterisation of the prostanoid receptor types involved in mediating calcitonin gene-related peptide release from cultured rat trigeminal neurones. Br J Pharmacol. 2001;134:1296–1302. doi: 10.1038/sj.bjp.0704357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor K, Arulmani U, Heiligers JP, Willems EW, Doods H, Villalon CM, et al. Effects of BIBN4096BS on cardiac output distribution and on CGRP-induced carotid heamodynamic responses in the pig. Eur J Pharmacol. 2003;475:69–77. doi: 10.1016/s0014-2999(03)02082-x. [DOI] [PubMed] [Google Scholar]
- Kellstein DE, Lipton RB, Geetha R, Koronkiewicz K, Evans FT, Stewart WF, et al. Evaluation of a novel solubilised formulation of ibuprofen in the treatment of migraine headache: a randomised, double-blind, placebo-controlled, dose-ranging study. Cephalalgia. 2000;20:223–243. doi: 10.1046/j.1468-2982.2000.00055.x. [DOI] [PubMed] [Google Scholar]
- Lin C-R, Amaya F, Barrett L, Wang H, Takada J, Samad TA, et al. Prostaglandin E2 receptor EP4 contributes to inflammatory pain hypersensitivity. J Pharmacol Exp Ther. 2006;319:1096–1103. doi: 10.1124/jpet.106.105569. [DOI] [PubMed] [Google Scholar]
- Loder E, Goldstein R, Biondi D. Placebo effects in oral triptan trials: the scientific and ethical rationale for continued use of placebo controls. Cephalalgia. 2005;25:124–131. doi: 10.1111/j.1468-2982.2004.00817.x. [DOI] [PubMed] [Google Scholar]
- Lumley P, Humphrey PP, Kennedy I, Coleman RA. Comparison of the potencies of some prostaglandins as vasodilators in three vascular beds of the anaesthetised dog. Eur J Pharmacol. 1982;16:421–430. doi: 10.1016/0014-2999(82)90107-8. [DOI] [PubMed] [Google Scholar]
- Lumley P, White BP, Humphrey PP. GR32191, a highly potent and specific thromboxane A2 receptor blocking drug on platelets and vascular and airways smooth muscle, in vitro. Br J Pharmacol. 1989;97:783–794. doi: 10.1111/j.1476-5381.1989.tb12017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meja KK, Barnes PJ, Giembycz MA. Characterization of the prostanoid receptor(s) on human blood monocytes at which prostaglandin E2 inhibits lipopolysaccharide-induced tumour necrosis factor-alpha generation. Br J Pharmacol. 1997;122:149–157. doi: 10.1038/sj.bjp.0701360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao K, Murase A, Ohshiro H, Okumura T, Taniguchi K, Murata Y, et al. CJ-023,423, a novel, potent and selective prostaglandin EP4 receptor antagonist with antihyperalgesic properties. J Pharmacol Exp Ther. 2007;322:686–694. doi: 10.1124/jpet.107.122010. [DOI] [PubMed] [Google Scholar]
- Nattero G, Allais G, De Lorenzo C, Benedetto C, Zonca M, Melzi E, et al. Relevance of prostaglandins in true menstrual migraine. Headache. 1989;29:232–237. doi: 10.1111/j.1526-4610.1989.hed22904233.x. [DOI] [PubMed] [Google Scholar]
- Negishi M, Sugimoto Y, Ichikawa A. Molecular mechanisms of diverse actions of prostanoid receptors. Biochim Biophys Acta. 1995;1259:109–120. doi: 10.1016/0005-2760(95)00146-4. [DOI] [PubMed] [Google Scholar]
- Papademetriou V. Cardiovascular risk assessment and triptans. Headache. 2004;44:S31–9. doi: 10.1111/j.1526-4610.2004.04106.x. [DOI] [PubMed] [Google Scholar]
- Peatfield RC, Gavel MJ, Rose FC. The effect of infused prostacyclin in migraine and cluster headache. Headache. 1981;21:190–195. doi: 10.1111/j.1526-4610.1981.hed2105190.x. [DOI] [PubMed] [Google Scholar]
- Prior MJ, Cooper KM, May LG, Bowen DL. Efficacy and safety of acetaminophen and naproxen in the treatment of tension-type headache. A randomised, double blind, placebo-controlled trial. Cephalalgia. 2002;22:232–240. doi: 10.1046/j.1468-2982.2002.00419.x. [DOI] [PubMed] [Google Scholar]
- Rich G, Yober EJ, Prokuski L, Moore SA. Prostaglanclin production in cultured cerebral microvascular smooth muscle is serum dependent. Am J Physiol. 1996;270:C1379–C1387. doi: 10.1152/ajpcell.1996.270.5.C1379. [DOI] [PubMed] [Google Scholar]
- Richter F, Lehmenkühler A. Cortical Spreading depression (CSD): a neurophysiological correlate of migraine aura. Schmerz. 2008;22:544–6. 548–550. doi: 10.1007/s00482-008-0653-9. [DOI] [PubMed] [Google Scholar]
- Rudolf K, Eberlein W, Engel W, Pieper H, Entzeroth M, Hallermayer G, et al. Development of human calcitonin gene-related peptide (CGRP) receptor antagonists. 1. Potent and selective small molecule CGRP antagonists. 1-[N2-[3,5-dibromo-N-[[4-(3,4-dihydro-2(1H)-oxoquinazolin-3-yl)-1-piperidinyl]carbonyl]-D-tyrosyl]-l-lysyl]-4-(4-pyridinyl)piperazine: the first CGRP antagonist for clinical trials in acute migraine. J Med Chem. 2005;48:5921–5931. doi: 10.1021/jm0490641. [DOI] [PubMed] [Google Scholar]
- Sarchielli P, Alberti A, Codini M, Floridi A, Gallai V. Nitric oxide metabolites, prostaglandin and trigeminal vasoactive peptides in internal jugular vein blood during spontaneous migraine attacks. Cephalalgia. 2000;20:907–918. doi: 10.1046/j.1468-2982.2000.00146.x. [DOI] [PubMed] [Google Scholar]
- Southall MD, Vasko MR. Prostaglandin receptor subtypes, EP3C and EP4, mediate the prostaglandin E2-induced cAMP production and sensitization of sensory neurons. J Biol Chem. 2001;276:16083–16091. doi: 10.1074/jbc.M011408200. [DOI] [PubMed] [Google Scholar]
- Stirparo G, Zicari A, Favilla M, Lipari M, Martelleti P. Linked activation of nitric oxide synthase and cyclooxygenase in peripheral monocytes of asymptomatic migraine without aura patients. Cephalalgia. 2000;20:100–106. doi: 10.1046/j.1468-2982.2000.00025.x. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, Tanaka A, Kato S, Aihara E, Amagase K. Effect of (S)-4-(1-(5-chloro-2-(4-fluorophenyoxy)benzamido)ethyl) benzoic acid (CJ-42794), a selective antagonist of prostaglandin E receptor subtype 4, on ulcerogenic and healing responses in rat gastrointestinal mucosa. J Pharmacol Exp Ther. 2007;322:903–912. doi: 10.1124/jpet.107.122978. [DOI] [PubMed] [Google Scholar]
- Tuca JO, Planas JM, Parellada PP. Increase in PGE2 and TXA2 in the saliva of common migraine patients. Actions of calcium channel blockers. Headache. 1989;29:498–501. doi: 10.1111/j.1526-4610.1989.hed2908498.x. [DOI] [PubMed] [Google Scholar]
- Vardi J, Flechter S, Alguati A, Regev I, Ayalon D. PGE2 levels in the saliva of common migraineous women. Headache. 1983;23:59–61. doi: 10.1111/j.1526-4610.1983.hed2302059.x. [DOI] [PubMed] [Google Scholar]
- Winner P, Landy S, Richardson M, Ames M. Early intervention in migraine with sumatriptan tablets 50 mg versus 100 mg: a pooled analysis of data from six clinical trials. Clin Ther. 2005;27:1785–1794. doi: 10.1016/j.clinthera.2005.11.009. [DOI] [PubMed] [Google Scholar]
- Zimmermann K, Reeh PW, Averbeck B. ATP can enhance the proton-induced CGRP release through P2Y receptors and secondary PGE2 release in isolated rat dura. Pain. 2002;97:259–265. doi: 10.1016/S0304-3959(02)00027-1. [DOI] [PubMed] [Google Scholar]