

Abstract
Background and purpose:
Levosimendan acts as a vasodilator through the opening of ATP-sensitive K+ channels (KATP) channels. Moreover, the coronary vasodilatation caused by levosimendan in anaesthetized pigs has recently been found to be abolished by the nitric oxide synthase (NOS) inhibitor Nω-nitro-L-arginine methyl ester, indicating that nitric oxide (NO) has a role in the vascular effects of levosimendan. However, the intracellular pathway leading to NO production caused by levosimendan has not yet been investigated. Thus, the purpose of the present study was to examine the effects of levosimendan on NO production and to evaluate the intracellular signalling pathway involved.
Experimental approach:
In porcine coronary endothelial cells (CEC), the release of NO in response to levosimendan was examined in the presence and absence of Nω-nitro-L-arginine methyl ester, an adenylyl cyclase inhibitor, KATP channel agonists and antagonists, and inhibitors of intracellular protein kinases. In addition, the role of Akt, ERK, p38 and eNOS was investigated through Western blot analysis.
Key results:
Levosimendan caused a concentration-dependent and K+-related increase of NO production. This effect was amplified by the mitochondrial KATP channel agonist, but not by the selective plasma membrane KATP channel agonist. The response of CEC to levosimendan was prevented by the KATP channel blockers, the adenylyl cyclase inhibitor and the Akt, ERK, p38 inhibitors. Western blot analysis showed that phosphorylation of the above kinases lead to eNOS activation.
Conclusions and implications:
In CEC levosimendan induced eNOS-dependent NO production through Akt, ERK and p38. This intracellular pathway is associated with the opening of mitochondrial KATP channels and involves cAMP.
Keywords: Levosimendan, coronary endothelial cells, nitric oxide production, KATP channel, p38, ERK, Akt involvement
Introduction
Levosimendan, a pyridazinone-dinitrile derivative, which elicits a positive inotropic effect by a combination of an increase in myofilament Ca2+ sensitivity and an inhibition of phosphodiesterase III (Takahashi and Endoh, 2005), exerts beneficial effects on myocardial performance. Levosimendan also induces arteriolar and venous dilatation. The vasodilator effect of levosimendan has been demonstrated in several vasculatures such as mesenteric arteries and skeletal muscle microvessels in rats (Erdei et al., 2006) and coronary arteries in various animal models (Kaheinen et al., 2001; Grossini et al., 2005). Also in humans, levosimendan administration has been shown to induce vasodilatation in coronary and internal mammary arteries (Michaels et al., 2005; Akar et al., 2007) and in saphenus and portal veins (Pataricza et al., 2000; Höhn et al., 2004). The vasodilatation exerted by levosimendan has been attributed to the inhibition of phosphodiesterase III (Haikala and Linden, 1995), the desensitization of contractile proteins to Ca2+ (Bowman et al., 1999) and to the opening of ATP-sensitive K+ channels (KATP; Yokoshiki et al., 1997; Yildiz, 2007), which are expressed on the surface of cell membranes in vascular smooth muscle cells and cardiomyocytes and in the cardiac mitochondrial inner membrane (Zoltán et al., 2005). It is noteworthy that, recently, a role for nitric oxide (NO) in the mechanism of action of vasodilatation has also been suggested. The coronary vasodilatation caused by levosimendan administration in anaesthetized pigs was, in fact, prevented by the nitric oxide synthase (NOS) blocker Nω-nitro-L-arginine methyl ester (L-NAME) (Grossini et al., 2005). It is notable that NO has been shown to be associated with the KATP channel. Nitric oxide not only operates through the production of cGMP and the activation of PKG, but also through the opening of KATP channels via the phosphorylation of serine-threonine residues (Nelson and Quayle, 1995; Han et al., 2002; Jackson, 2005). Indeed, the NO-cGMP-PKG signalling pathway has been found to induce anoxic preconditioning through activation of KATP in rat and rabbit hearts (Cuong et al., 2002; Das and Sarkar, 2007). Recently, Das and Sarkar (2007) showed that the beneficial effects exerted by levosimendan against ischaemia/reperfusion injuries in anaesthetized rabbits could be attenuated by both the mitochondrial KATP channel blocker 5-hydroxydecanoate (5HD), and the inhibition of NOS. In addition, reports in the literature indicate that the opening of KATP is involved in endothelial NO release. In cerebral vessels, intraluminal application of the KATP channel opener nicorandil (1 µmol·L−1) causes an NO-dependent vasodilatation, which can be blocked by the KATP channel blocker glibenclamide (Janigro et al., 1997).
Protein kinases such as PI3-kinase, p38MAPK and ERK have been shown to have a role in the intracellular signalling involved in both KATP channel opening and NO production, (Oldenburg et al., 2002; Lorenz et al., 2004; Wyatt et al., 2004; Xu et al., 2004; Molinari et al., 2007; Grossini et al., 2008) and it has been proposed that they act as an upstream or downstream trigger linking the surface receptors and the intracellular pathways. It is noteworthy that in guinea-pig hearts levosimendan pre-treatment was very effective at reducing infarct size through an increase of ERK activity (du Toit et al., 2008).
Thus, in the present study we examined the effects of levosimendan on NO production in endothelial cells isolated from porcine coronary arteries so as to evaluate the role of KATP channels in the above effects. Moreover, the involvement of PI3K/Akt, ERK/MAPKs and p38/MAPK pathways leading to eNOS activation was investigated.
We found that levosimendan induces eNOS and NO production through the phosphorylation of PI3K/Akt, ERK/MAPKs and p38/MAPK and the opening of KATP channels.
Methods
Cell preparation
Coronary endothelial cells (CEC) were obtained from anaesthetized prepubescent pigs. The coronary artery was isolated, cut and washed three times in physiological sterile solution and then transferred in a sterile culture dish containing a specific culture medium (EGM-2, endothelial growth media-2) with the addition of hEGF, hydrocortisone, gentamycin-amphotericin-B 1000, 2% foetal bovine serum, vascular endothelial growth factor, human fibroblast growth factor-basic, recombinant analogue insulin-like growth factor 1 human, ascorbic acid, heparin and 1% penicillin-streptomycin-glutamine. The coronary arterial segment was opened longitudinally under sterile conditions and the endothelial cells were obtained by scraping the luminal surface of the coronary artery with a sterile bistoury. The collected cells were plated into 0.1% gelatine-coated 100 mm culture dish with EGM-2 supplemented with 1% penicillin-streptomycin-glutamine and maintained at 37°C with 5% CO2. After 72 h, 10 ml of fresh medium was added and the incubation continued for 48 h. After this time, the cells formed a monolayer and were subcultured. The cells used for the experiments were obtained from passage 3 to passage 5. Before the experiments the CEC were tested for endothelial cell marker 34 to verify their endothelial origin.
After the cells had reached confluence, they were used for NO production measurement (first series of experiments) and protein phosphorylation (second series of experiments). In the first series, 1 × 105 cells were plated in 0.1% gelatine-coated 24-well plates in EGM-2 complete medium for 24 h at 37°C with 5% CO2. At the end of this time, the cells were washed with phosphate-buffered saline 1× (PBS 1×) and maintained for 4–6 h in Dulbecco's modified Eagle's medium supplemented with 1% penicillin-streptomycin-glutamine without foetal calf serum and red phenol (starvation medium). For the measurement of ATP production, 1 × 104 CEC were plated in 0.1% gelatine-coated 96-well plates in EGM-2 complete medium for 8 h at 37°C with 5% CO2. After adhesion, CEC were maintained in the starvation medium for 4 h before the treatment with the same agents and conditions used for the Griess assay.
In the second series of experiments, the cells were plated on 0.1% gelatine-coated dishes with EGM-2 complete medium. The cells at confluence were washed with PBS 1× and then incubated with starvation medium overnight at 37°C with 5% CO2.
NO measurement
NO production was measured in the sample's supernatants in which an equal volume of Griess reagents was added, following the manufacturer's instructions. After 10 min, the absorbance at 570 nm was measured by a spectrophotometer (BS1000 Spectra Count). NO production was quantified by reference to a standard nitrate curve, which was generated in the medium used for the experiments, as previously described (Molinari et al., 2007; Grossini et al., 2008) and expressed as a percentage.
The results obtained with the Griess method have also been validated through the diaminofluorescein fluorophore system (DAF-FM) used as previously described (Grossini et al., 2008; John et al., 2008). Briefly, the cells were plated in 24-well plates coated with 0.1% gelatine and maintained in starvation medium for 4 h before treatment with the same agents and conditions used for the Griess assay. At the end of the stimulation periods, the cells were washed with sterile PBS 1× and incubated with 4-amino-5methylamino-2′,7′-difluorofluorescin diacetate (DAF-FM) diacetate (0.8 µmol·L−1) for 25 min in the dark in an incubator at 37°C with 5% CO2. The cell-bathing medium was taken for measurement of fluorescence by use of a fluorescence spectrophotometer. Fluorescence excitation and emission were 495 and 515 nm respectively. To determine the range of reliable fluorescence readouts, a standard curve for DAF-FM diacetate fluorescence was generated using detanonoate (0.01–200 µmol·L−1; John et al., 2008) incubated with DAF-FM diacetate (0.8 µmol·L−1) for 25 min in the dark in an incubator at 37°C with 5% CO2. The fluorescence of samples was measured by a fluorescence spectrophotometer at λex/em 495/515 nm.
For the ATP assay, after each stimulation the medium was aspirated and the cells were immediately treated with the components of the ATP assay kit (nucleotide releasing buffer, ATP monitoring enzyme, enzyme reconstitution buffer, ATP). Luminescence was measured 1 min after the addition of ATP monitoring enzyme in a Lucy 1 microplate luminometer (Rosys Anthos, Wals, Austria). Some samples were lysed by addition of an ice-cold Ripa buffer (50 mmol·L−1 HEPES, 150 mmol·L−1 NaCl, 0.1% SDS, 1% Triton-X100, 1% sodium deoxycholate, 10% glycerol, 1.5 mmol·L−1 MgCl2, 1 mmol·L−1 EGTA, 1 mmol·L−1 NaF). The total protein in each well was measured using a bicinconinic acid protein (BCA) assay, and luminescence was expressed as µmol of ATP g−1 protein.
Western blot analysis
After stimulation with the same agents used for NO detection, in the second series of experiments, CEC were washed with ice-cold PBS 1×, supplemented with 1:200 sodium orthovanadate and lysed in an iced-Ripa-buffer supplemented with 1:200 sodium orthovanadate and 1:100 protease inhibitor cocktail. The cells were dissolved by incubation for 30 min at 4°C and the proteins extracted were quantified by using BCA. Twenty-five micrograms of cell lysate proteins was dissolved in Laemmly buffer 5×, heated for 5 min at 95°C and resolved on 10% SDS-PAGE. After electrophoresis, proteins were transferred to PVDF membranes, which were incubated overnight at 4°C with specific antibodies: anti-phospho-ERK (Thr202/Tyr204), anti-ERK1/2, anti-phospho-Akt (Ser473), anti-Akt, anti-phospho-p38 (Thr180/Tyr180), anti-p38, anti-phospho-eNOS (Thr495), anti-eNOS. The membranes were washed and then incubated with horseradish peroxidase-coupled goat anti-rabbit IgG and horseradish peroxidase-coupled goat anti-mouse IgG for 45 min, and were developed by use of a non-radioactive method using Western lightning chemiluminescence. Phosphorylated protein expression was normalized through specific total protein expression and verified through β-actin detection, and quantified by measuring the relative optical density of the bands.
Experimental design
NO production
In the first series of experiments, the NO in the culture supernatants of CEC was measured through the Griess method and verified through DAF-FM. The experiments were performed either in the absence of K+ or with 5 mmol·L−1 K+ in the starvation medium of CEC. The effects of levosimendan, dissolved in a solution containing ethanol anhydrous, citrate acid anhydrous and povidone (vehicle), on NO production were examined in the absence or presence of the NO synthase inhibitor L-NAME (10 mmol·L−1), the non-specific KATP channel agonist cromakalim (1 µmol·L−1), the specific plasma membrane KATP channel agonist P1075 (1 µmol·L−1), the specific mitochondrial KATP channel agonist diazoxide (5 µmol·L−1), the non-specific KATP channel antagonist glibenclamide (1 µmol·L−1 for 20 min), the specific mitochondrial KATP channel antagonist 5HD (1 µmol·L−1), the adenylyl cyclase activator and blocker, forskolin (1 µmol·L−1) and 2′5′-dideoxyadenosine, respectively (1 µmol·L−1) and of the PDEIII inhibitor cilostazol (10 µmol·L−1). The role of p38 MAPK, PI3K and MEK1 was tested by performing experiments in the presence of their specific inhibitors SB203580 (1 µmol·L−1), wortmannin (100 nmol·L−1) and UO126 (10 µmol·L−1), respectively, which were dissolved in dimethyl sulphoxide and allowed to act for 20 min before the addition of the other agents. The vehicles of inhibitors and of levosimendan were tested in the basal medium without addition of the agents whereas ACh (10 µmol·L−1) was used as a positive control.
In some experiments, the effects of levosimendan on NO production were examined in the presence of high K+ concentrations (10, 20, 30, 40, 60, 80 mmol·L−1) in the starvation medium.
As the effects of levosimendan on KATP channel could be related to changes in ATP production, some experiments were performed to address this issue. In particular, the effects of levosimendan on ATP production were examined in the absence and presence of cromakalim, P1075 and diazoxide at the same concentration used before.
Western blot analysis
To determine the effects of levosimendan, in the second series of experiments, the CEC were stimulated with the same agents used for NO detection and the effects on phosphorylation of ERK, Akt and p38 were examined as previously described.
Data analysis and statistical procedures
Student's t-test for paired values and anova were used to examine the changes in NO production and protein phosphorylation caused by levosimendan. Data represent means of at least five independent experiments for each experimental protocol and are expressed as means ± SD (range). A P-value < 0.05 was considered statistically significant.
Drugs
Levosimendan was obtained from Orion Pharma (Espoo, Finland); KCl, cromakalin, diazoxide, L-NAME, 2′5′-dideoxyadenosine, glibenclamide, 5HD, detanonoate, wortmannin, cilostazol, dimethyl sulphoxide, acetylcholine, forskolin, sodium orthovanadate, protease inhibitors cocktail, PBS, HEPES, NaCl, SDS, Triton-X100, sodium deoxycholate, glycerol, MgCl2, EGTA, NaF, gelatine, penicillin-streptomycin-glutamine, Dulbecco's modified Eagle's medium were obtained from Sigma-Aldrich; P1075 was obtained from Tocris Bioscience (Ellisville, Missouri, USA); EGM-2, hEGF, hydrocortisone, gentamycin-amphotericin-B 1000, foetal bovine serum, vascular endothelial growth factor, human fibroblast growth factor-basic, recombinant analogue insulin-like growth factor 1 human, ascorbic acid and heparin were purchased from Lonza, Inc. (Basle, Switzerland); Griess reagent system, SB203580 and UO126 were obtained from Promega Corporation (Madison, WI, USA). ATP assay kit was purchased from EMD Biosciences (San Diego, CA, USA); DAF-FM was purchased from Molecular Probes (Eugene, OR, USA); BCA was obtained from Pierce (Rockford, IL, USA); SDS-PAGE, molecular weights and PVDF membranes were purchased from Bio-Rad Laboratories (Hercules, CA, USA). Anti-phospho-ERK, anti-ERK1/2, anti-phospho-AktSer473, anti-Akt, anti-phospho-p38Thr180/Tyr180, anti-p38, anti-phospho-eNOSThr495, anti-eNOS and horseradish peroxidase-coupled goat anti-rabbit IgG were obtained from Cell Signaling Technologies (Beverly, MA, USA). Horseradish peroxidase-coupled goat anti-mouse IgG and β-actin were purchased from Alexis Biochemicals (San Diego, CA, USA). Western Lightning Chemiluminescence was obtained from Perkin Elmer (Boston, MA, USA). All drugs and ion channels nomenclature conforms with the BJP's Guide to Receptors and Channels (Alexander et al., 2008).
Results
First series of experiments
Concentration–response study
The effects of levosimendan on NO release were examined in CEC, isolated and maintained as described in Methods. As illustrated in Figure 1A,B, 1 min stimulation of CEC with levosimendan (0.01–10 µmol·L−1) caused a concentration-dependent NO release, which was detected by both the Griess system and DAF-FM. The NO production caused by 10 µmol·L−1 levosimendan amounted to 45.2 ± 3.5% (P < 0.05). In the presence of 5 mmol·L−1 K+, the effects of levosimendan were significantly amplified (Figure 1A,B; P < 0.05). At 10 µmol·L−1, in fact, the NO production caused by levosimendan amounted to 59.2 ± 4.3% (P < 0.05). This concentration of levosimendan was maintained for all successive experiments.
Figure 1.
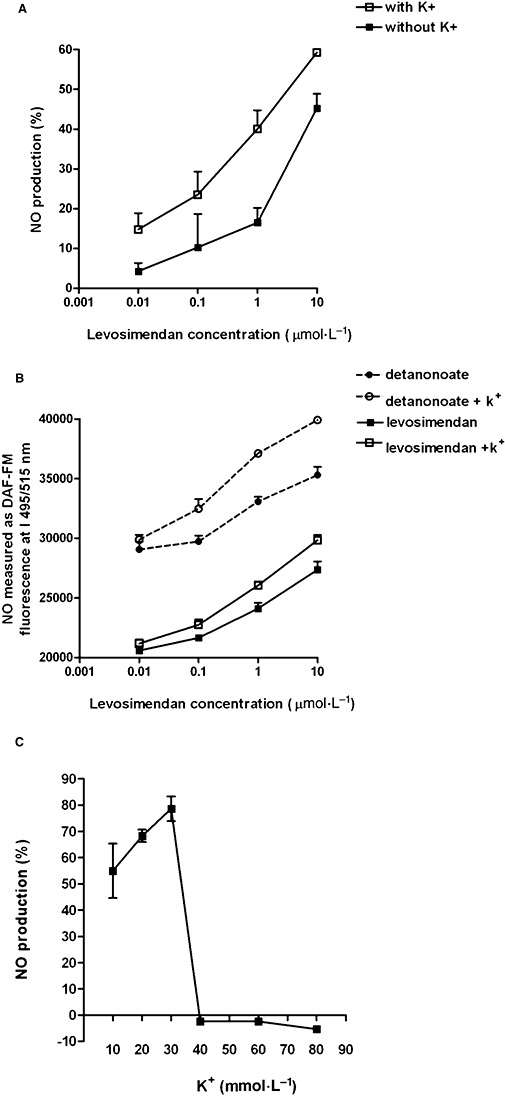
Changes in the levels of NO produced in response to levosimendan. In (A) and (B), changes in the level of NO were determined by the Griess method and the DAF-FM diacetate fluorescence system respectively. The results were obtained with levosimendan (0.01–10 µmol·L−1) in the presence or absence of 5 mmol·L−1 K+. The calibration curve for DAF-FM was obtained with detanonoate (0.01–10 µmol·L−1). In (C), changes in the level of NO, determined by the Griess method, induced by 10 µmol·L−1 levosimendan in the presence of high K+ concentrations (10, 20, 30, 40, 60, 80 mmol·L−1). The data are shown as a percentage change from control (means ± SD). DAF-FM, 4-amino-5methylamino-2′,7′-difluorofluorescin diacetate.
Effects of levosimendan on NO production detected through the Griess method
To verify the intracellular pathway involved in NO production caused by levosimendan and the role of the KATP channel, CEC were treated with various agents in the presence and absence of 5 mmol·L−1 K+ in the medium. ACh, used as positive control, induced the release of similar amounts of NO in the absence and presence of 5 mmol·L−1 K+ (Figure 2A,B; Table 1). The vehicle of levosimendan did not induce any significant changes in NO production at any given concentration (P > 0.05). The effects of various agents alone or together on NO release are presented in Table 1.
Figure 2.
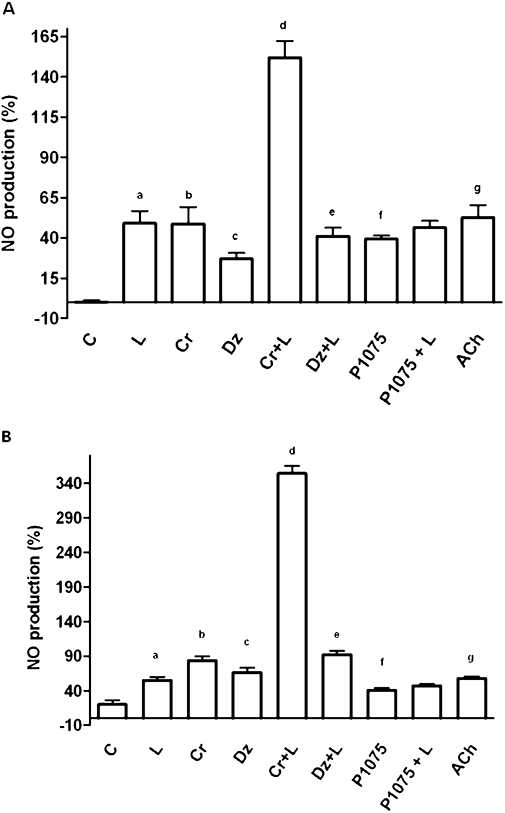
Changes in the levels of NO produced in response to the addition of various agents, as determined by the Griess method. In (A) and (B), changes in the level of NO induced by levosimendan in the absence or presence of K+ respectively. C = control; L = levosimendan (10 µmol·L−1); Cr = cromakalim (1 µmol·L−1); Dz = diazoxide (5 µmol·L−1); P1075 (1 µmol·L−1); Cr + L = co-stimulation; Dz + L = co-stimulation; P1075 + L = co-stimulation; ACh, acetylcholine (10 µmol·L−1). The data are shown as a percentage change from control (means ± SD) a, b, c, f, g P < 0.05 vs control; d P < 0.05 vs b; e P < 0.05 vs c.
Table 1.
Changes in the level of NO production induced by various agents
| Without 5 mmol·L−1 K+ | With 5 mmol·L−1 K+ | |
|---|---|---|
| Vehicle | 1.2 ± 4.8 | 4.2 ± 2.8 |
| Dimethyl sulphoxide | 2 ± 1 | 4 ± 3 |
| Cromakalim | 46.8 ± 10.5 | 83.5 ± 5.8 |
| P1075 | 39.3 ± 2 | 40.6 ± 3.7 |
| Diazoxide | 27 ± 3.6 | 66 ± 7 |
| L-NAME + levosimendan | −1.1 ± 2.1 | −1 ± 2 |
| L-NAME + cromakalim + levosimendan | −1 ± 2.8 | −2.6 ± 1.1 |
| L-NAME + diazoxide + levosimendan | −1.3 ± 0.5 | −1.3 ± 0.5 |
| 2′5′-dideoxyadenosine + levosimendan | −4 ± 3.3 | −0.6 ± 1.5 |
| 2′5′-dideoxyadenosine + cromakalim + levosimendan | −5 ± 4.4 | −4.4 ± 5.7 |
| 2′5′-dideoxyadenosine + diazoxide + levosimendan | −1.6 ± 0.5 | −1 ± 1.7 |
| Forskolin | 51 ± 6.3 | 53 ± 2 |
| 2′5′-dideoxyadenosine + forskolin | −4 ± 2.5 | −1.7 ± 0.5 |
| Glibenclamide + levosimendan | −4.6 ± 2.4 | −1.4 ± 1.8 |
| Glibenclamide + cromakalim + levosimendan | −4.3 ± 3.3 | −1.6 ± 2.5 |
| Glibenclamide + P1075 | −3.3 ± 1.5 | −3 ± 2 |
| Glibenclamide + P1075 + levosimendan | −3.3 ± 2.5 | −3.3 ± 0.6 |
| 5-hydroxydecanoate + levosimendan | −3 ± 3 | −3.2 ± 2.1 |
| 5-hydroxydecanoate + diazoxide + levosimendan | −2 ± 1 | −1 ± 1.7 |
| Inhibitors + levosimendan | −2.5 ± 3 | −13 ± 2 |
| Inhibitors + cromakalim + levosimendan | −0.2 ± 2.2 | −0.2 ± 1 |
| Inhibitors + diazoxide + levosimendan | −1.3 ± 1.3 | −2 ± 1.7 |
| Acetylcholine | 52.6 ± 7.8 | 57.7 ± 7.8 |
The values presented are the percentage changes (means ± SD) in NO production determined by the Griess method. Vehicle of 10 µmol·L−1 levosimendan, ethanol anhydrous, citrate acid anhydrous and povidone; the inhibitors used were wortmannin, SB203580, UO126. All results were replicated in five independent experiments.
L-NAME, Nω-nitro-L-arginine methyl ester.
In the absence of K+, the treatment of CEC with the non-specific KATP channel agonist cromakalim (1 µmol·L−1) or the specific mitochondrial KATP channel agonist diazoxide (5 µmol·L−1) caused an increase of NO production (P < 0.05). In the presence of levosimendan, the above effects were amplified (Figure 2A; P < 0.05). It is notable that although the treatment of CEC with the specific plasma membrane KATP channel agonist P1075 (1 µmol·L−1) increased NO release compared with control (P < 0.05), this effect was not amplified in the presence of levosimendan (P > 0.05; Figure 2A).
In the presence of 5 mmol·L−1 K+, 10 µmol·L−1 levosimendan potentiated, the effects of 1 µmol·L−1 cromakalim and 5 µmol·L−1 diazoxide on NO release by about 353% and 39% respectively. These effects were significantly higher than the ones obtained in the samples stimulated in the absence of 5 mmol·L−1 K+ (P < 0.05; Figure 2B). In contrast, the plasma membrane KATP agonist P1075 failed to potentiate the effects of levosimendan on NO production (P > 0.05; Figure 2B).
The treatment of CEC with 10 mmol·L−1 L-NAME abolished both the effects of cromakalim and diazoxide given alone and in the presence of levosimendan either in the absence or presence of K+ (P > 0.05; Table 1). Interestingly, all the effects of levosimendan on NO production were also abolished in cells pre-treated for 15 min with 1 µmol·L−1 2′5′-dideoxyadenosine; this treatment also prevented the NO produced in response to co-stimulation with levosimendan and cromakalim or levosimendan and diazoxide (P > 0.05; Table 1).
The involvement of the KATP channel in the effects of levosimendan on NO production was also confirmed by experiments performed in the presence of 1 µmol·L−1 glibenclamide and 1 µmol·L−1 5HD. In the samples pre-treated for 15–30 min with either the non-specific or the specific KATP channel antagonist, 10 µmol·L−1 levosimendan failed to induce any effects on NO production irrespective of the presence of K+ in the medium (P > 0.05; Table 1). These results in particular confirm the role of the mitochondrial KATP channel in the mechanisms of action of the cardiovascular effects of levosimendan (Kopustinskiene et al., 2004; Das and Sarkar, 2007).
In addition, the experiments performed to determine whether levosimendan is able to increase the production of ATP showed that levosimendan, either alone or in the presence of cromakalim, P1075 or diazoxide, does not cause any significant changes in ATP content in respect of control values (which amounted to 5.25 ± 1.9 µmol·g−1 protein; P > 0.05; not shown). This finding supports the hypothesis of a direct action of levosimendan on the KATP channel.
Moreover, in CEC pre-treated with the p38 MAPK inhibitor SB203580 (1 µmol·L−1), PI3K inhibitor wortmannin (100 nmol·L−1), or the MEK1 inhibitor UO126 (10 µmol·L−1), any changes in the NO produced in response to 10 µmol·L−1 levosimendan given alone or in association with 1 µmol·L−1 cromakalim or 5 µmol·L−1 diazoxide were abolished (P > 0.05; Table 1).
Finally, cilostazol (10 µmol·L−1) failed to influence the response of CEC to 10 µmol·L−1 levosimendan, (P > 0.05), as shown in Figure 3A. It is noteworthy that the effects of cilostazol on NO production were not influenced by glibenclamide or by the KATP channels agonists (P > 0.05; Figure 3B). This finding shows that levosimendan and cilostazol do not share common pharmacological characteristics.
Figure 3.
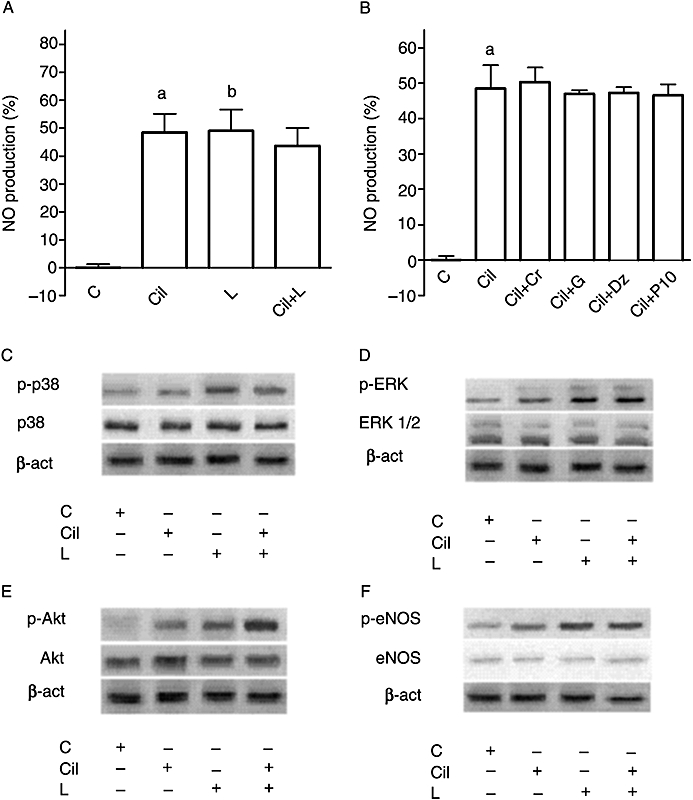
Effects of cilostazol on the response of coronary endothelial cells to levosimendan. In (A), changes in the levels of NO produced, determined by the Griess method, in response to 10 µmol·L−1 levosimendan in the absence or presence of cilostazol (Cil; 10 µmol·L−1). The data are shown as a percentage change from control (means ± SD). a, b P < 0.05 vs control. B. Changes in the levels of NO produced, determined by the Griess method, in response to cilostazol in the presence of KATP channel agonists and antagonists. G, glibenclamide (1 µmol·L−1). Other abbreviations are the same as in Figure 2. Cil + Cr = co-stimulation; Cil + P1075 = co-stimulation; Cil + Dz = co-stimulation; Cil + G = co-stimulation. a P < 0.05 vs control. C–F. Effects of 10 µmol·L−1 levosimendan on p38, ERK, Akt and eNOS phosphorylation in the absence and presence of cilostazol.
Effects of levosimendan on NO production in the presence of high K+ in the medium
As shown in Figure 1C, increasing the K+ in the medium potentiated the effects of levosimendan on NO production. This effect peaked at 30 mmol·L−1 K+ (P < 0.05) and when K+ in the medium was higher than 30 mmol·L−1 levosimendan failed to induce any changes in NO production in CEC (P > 0.05).
Second series of experiments
Western blot analysis
To confirm the involvement of p38, ERK, Akt and eNOS in the signalling activated by levosimendan, the rate of phosphorylation of p38, ERK, Akt and eNOS was examined in the same experimental conditions used previously. As depicted in Figures 4 and 5, immunoblots and densitometric analysis showed that in the absence of K+, levosimendan alone and in the presence of cromakalim or diazoxide activated p38, ERK, Akt and eNOS; this effect was amplified in the presence of 5 mmol·L−1 K+ in the medium. As shown in Figure 3C–F, cilostazol failed to induce changes in the effects of 10 µmol·L−1 levosimendan on p38, ERK, Akt and eNOS phosphorylation, which confirmed the findings observed in the first series of experiments regarding the role of cilostazol. L-NAME, 2′5′-dideoxyadenosine, SB203580, UO126, wortmannin, glibenclamide and 5HD abolished all the above effects (P > 0.05).
Figure 4.
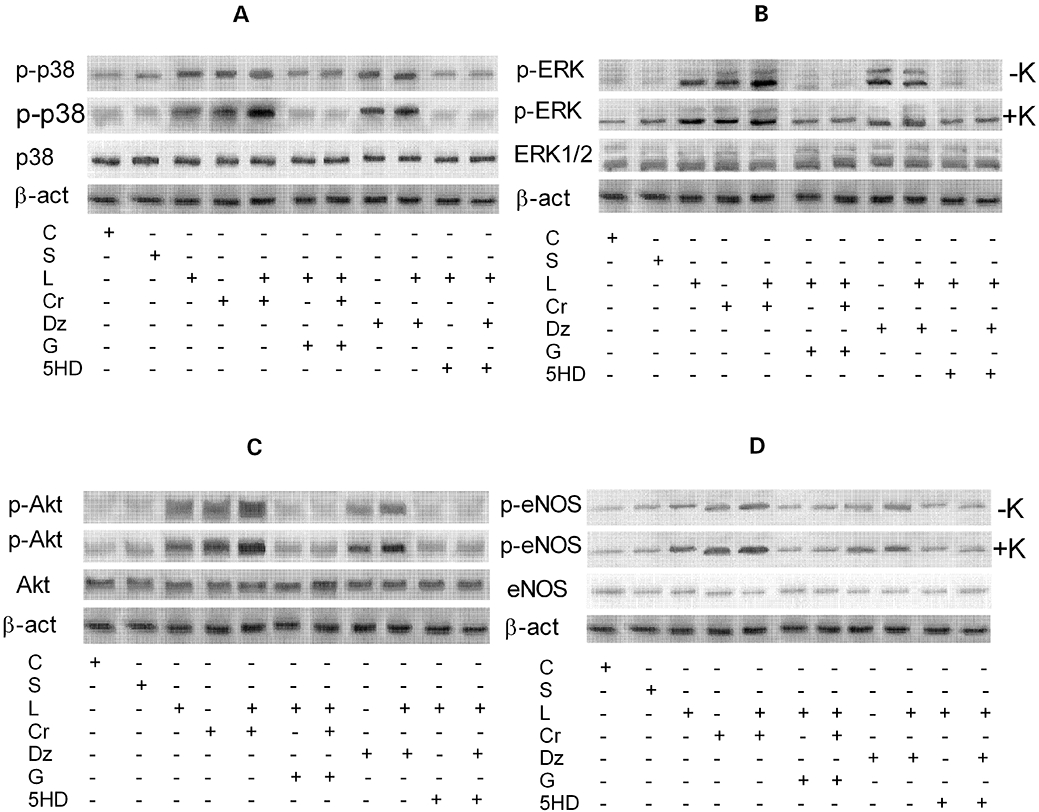
Western blot analysis of the effects of levosimendan on the activation of protein kinases in coronary endothelial cells. A–D. Effects of 10 µmol·L−1 levosimendan on p38, ERK, Akt and eNOS, respectively, in the absence and presence of various agents. S = vehicle of levosimendan; Cr + L = co-stimulation; G + L = co-stimulation; G + Cr + L = co-stimulation; Dz + L = co-stimulation; 5HD + L = co-stimulation; 5HD + Dz + L = co-stimulation. The abbreviations used are the same as in Figures 2 and 3.
Figure 5.
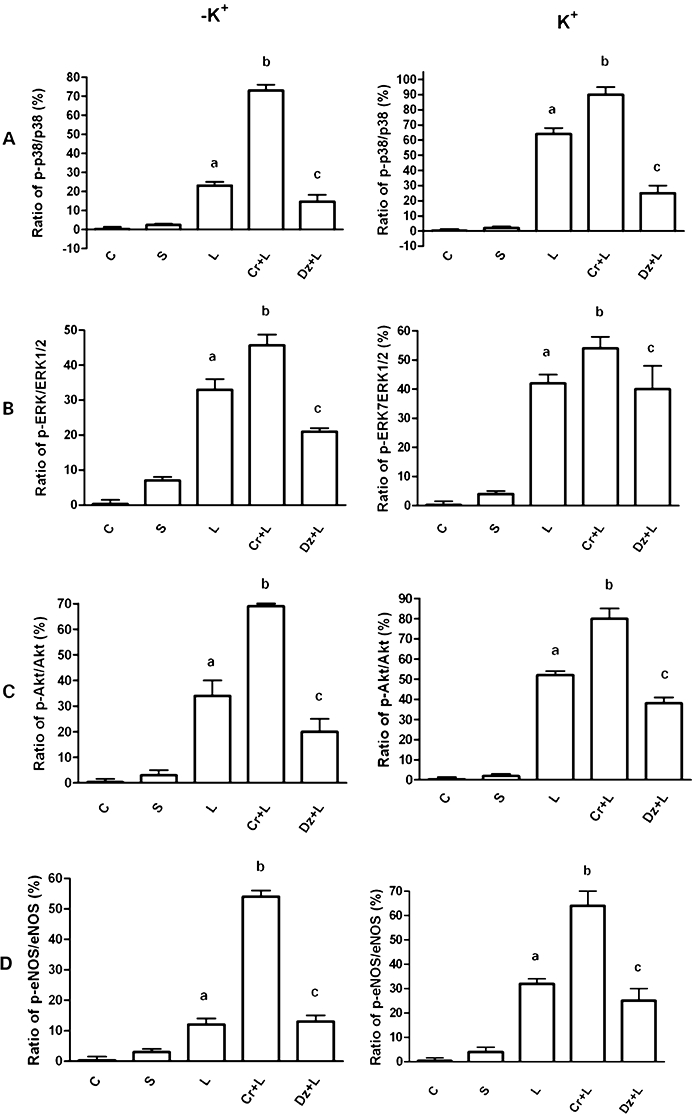
Densitometric analysis of the effects of levosimendan on the activation of protein kinases in coronary endothelial cells. A-D. Effects of 10 µmol·L−1 levosimendan on p38, ERK, Akt and eNOS, respectively, in the absence and presence of various agents. Cr + L = co-stimulation; Dz + L = co-stimulation. The abbreviations are the same as in Figures 2–4. The data are presented as means ± SD. a, c P < 0.05 vs control; b P < 0.05 vs a.
Discussion
The present study demonstrates for the first time that in porcine CECs levosimendan causes NO production through eNOS activation by opening mitochondrial KATP channels. Moreover, the involvement of cAMP and of p38, ERK, Akt pathways has been highlighted.
The results obtained in the present study confirm the ones previously observed in experiments performed in anaesthetized pigs, where the coronary vasodilatation induced by levosimendan was abolished by pre-treatment with the NOS inhibitor L-NAME (Grossini et al., 2005). In porcine CECs levosimendan, given at a concentration similar to the one used in the pigs, was able to induce a concentration-related NO production detected through the Griess method and verified through the NO-sensitive fluorescent dye DAF-FM. Both methods have been previously shown to measure the changes in NO release in porcine aortic endothelial cells induced by various agents (Molinari et al., 2007; Grossini et al., 2008). Moreover, the DAF-FM system has been widely adopted for measuring the intracellular production of NO in endothelial cells (Koyama et al., 2002; Kimura et al., 2004). The effects of levosimendan were blocked by pre-treatment of cells with L-NAME at a concentration able to prevent the effects of genistein in porcine aortic endothelial cells (Grossini et al., 2008).
It is noteworthy that in this study, the effects of levosimendan on NO production were found to be associated with KATP channel opening. Pre-treatment of cells with either the non-specific KATP channel blocker glibenclamide or with 5HD, which is highly selective for the mitochondrial subtype of KATP channel and is widely used in studies regarding cardioprotection (Kopustinskiene et al., 2001; Andrukhiv et al., 2006; Pasdois et al., 2006; 2007), completely prevented all effects of levosimendan on NO production. Indeed, the findings obtained in the presence of 5HD show that the mithocondrial KATP channel is involved in the effects of levosimendan on NO release; this was also confirmed in the experiments performed with diazoxide given as a co-stimulus. In the present study, levosimendan potentiated the NO production induced by the selective mithocondrial KATP channel agonist; with regard to this result, it is noteworthy that the cardioprotection mediated by diazoxide in rats has been found to be associated with the preservation of mitochondrial integrity and function (Simoncíkováet al., 2007).
Also the findings obtained in CEC pre-treated with P1075, which has been reported to be highly selective for the plasma membrane KATP channel (Gross and Fryer, 1999; Baczkóet al., 2005), exclude the involvement of the sarcolemmal type of KATP channel in the effects of levosimendan; in the presence of P1075, the response of CEC to levosimendan was not significantly increased. Moreover, the absence of changes in cellular ATP content in response to levosimendan supports the hypothesis that levosimendan has a direct action on the KATP channel. These findings are also in agreement with results from the literature that show the mitochondrial subtype of KATP channel has a central role in the anti-ischaemic effects of levosimendan. Indeed in isolated heart mitochondria, levosimendan was found to act as a more powerful activator of K+ flux to mitochondrial matrix than either diazoxide or pinacidil (Kopustinskiene et al., 2004). Even though the results obtained in this study suggest the involvement of mithocondrial KATP channels in the effects of levosimendan, the ability of levosimendan to target mitochondrial KATP channels remains to be clearly demonstrated and further experiments need to be performed in order to address this issue. The release of NO induced by KATP channel opening in endothelial cells could represent a further mechanism of action of vasodilatation caused by levosimendan and confirms the relationship between KATP channel activation and NO release. For example, adenosine released from the endothelial cells was found to induce the synthesis of NO through the opening KATP channels (Bryan and Marshall, 1999) and, similarly, in rat aorta and brain microvascular endothelial cells, the KATP channel opener pinacidil was found to modulate the release of endothelial factors (Janigro et al., 1993).
The KATP channel has been proposed as a target for levosimendan in isolated mesenteric artery myocytes (Yokoshiki et al., 1997), small coronary arteries of the dog (Kersten et al., 2000) and human portal and saphenous veins (Pataricza et al., 2000; Höhn et al., 2004). Opening of K+ channel induces hyperpolarization of the membrane, inhibits the inward Ca2+ current by decreasing the opening of the L-type Ca2+ channel, and activates the Na+-Ca2+ exchanger to extrude Ca2+ (Nelson and Quayle, 1995). Both the inhibition of inward Ca2+ current and the activation of the Na+-Ca2+ exchanger act to lower intracellular Ca2+ and thereby promote vasorelaxation. Attenuation of levosimendan-induced dilatation of coronary arteries during concomitant administration of the KATP channel antagonist glibenclamide strengthens the crucial role of KATP channels in this setting (Kersten et al., 2000; Kaheinen et al., 2001). Indeed, it has been suggested that in endothelial cells the membrane potential may play an important role in controlling the extent of agonist-induced Ca2+ influx and, thereby, the release of endothelial NO (Busse et al., 1991). As endothelial cells do not appear to express L-type Ca2+ channels, hyperpolarization caused by the opening of endothelial KATP channels could induce Ca2+ influx and stimulation of endothelium-derived NO (Herrera et al., 1998).
As shown by Nilius and Droogmans (2001), the membrane potential, as well as capacitance and input resistance of vascular endothelial cells, vary considerably between different cell types and conditions of cell isolation and culturing. The resting membrane potential in several endothelial cell types has been shown to have a bimodal distribution, consisting of cells with a resting potential between −70 and −60 mV and cells with a resting potential between −40 and −10 mV. Experiments performed in the latter type of cells showed that they respond to a raised K+ concentration by hyperpolarization and then by depolarization. This model suggests that hyperpolarization and depolarization can occur in response to an increase in K+ and the type of response is dependent on the initial and final K+ concentration.
Thus, we hypothesize that the treatment of CEC with levosimendan would induce an initial strong hyperpolarization, for K+ concentration lower than 30 mmol·L−1, followed by a late depolarization, for K+ concentration higher than 30 mmol·L−1. The hyperpolarization induced by levosimendan increases its direct effect on NO production through either the potentiation of the intracellular signalling leading to eNOS activation or through an increase of Ca2+ influx. Indeed, levosimendan has been shown to hyperpolarize the arterial myocytes through activation of a glibenclamide-sensitive K+ channel (Yokoshiki et al., 1997). On the other hand, the depolarization of the membrane potential caused by a high extracellular K+ concentration could interfere with the hyperpolarizing effects of levosimendan, thus abolishing the response of CEC to this compound. Indeed, the effects of levosimendan on NO production were related to K+ concentration in the medium, being completely abolished in presence of a K+ concentration higher than 30 mmol·L−1. It is noteworthy that Yokoshiki et al. (1997) found that a high K+ concentration (up to 45 mmol·L−1) compromised the efficacy of levosimendan as a vasodilator. In that study, resting membrane potential approached the equilibrium potential of K+, thereby attenuating the hyperpolarizing actions of K+-channel activation, which would agree with the hypothesis derived from the results of the present study.
In addition, Sheng and Braun (2007) recently showed that clamping the membrane potential to 0 mV by a brief exposure to high external KCl was able to block both the agonist-induced membrane hyperpolarization and NO synthesis. Moreover, high external KCl has been shown to reduce the release of endothelial releasing factors from populations of agonist-stimulated endothelial cells (Luckhoff and Busse, 1990). Also in the rat mesenteric artery 80 mmol·L−1 KCl has been found to prevent NO production induced by ACh (Stankevicius et al., 2006).
The activity of eNOS, one of three isoenzymes transforming L-arginine to L-citrulline and NO, is regulated by signal transduction pathways that involve various phosphorylation events. Akt kinase activates eNOS by directly phosphorylating the enzyme at Ser-1179 (Boo et al., 2002). Akt itself is phosphorylated and activated by PI3 kinase, which in turn is activated by various agonists (Fulton et al., 2001). Also MAP kinases, important mediators of signal transduction from the cell surface to the nucleus, have been found to modulate eNOS activation (Ito et al., 2002). Indeed, in porcine aortic endothelial cells p38, ERK, Akt pathways have recently been implicated in the effects of genistein and prolactin on NO production (Molinari et al., 2007; Grossini et al., 2008).
In the present study, the effects of levosimendan on NO production were completely prevented by pre-treatment of CEC with SB203580, wortmannin, UO126 at concentrations known to abolish the in vitro effects of prolactin and genistein (Molinari et al., 2007; Grossini et al., 2008) and similar to those found to block intracellular pathways associated with the effects of ERK, p38 and Akt in endothelial cells (Luo et al., 2007; Padmasekar et al., 2007). Hence, these results show, for the first time, that the above kinases are involved in the eNOS phosphorylation induced by levosimendan, thus leading to NO production. These findings were also confirmed by Western blot analysis; these results showed that levosimendan induced the phosporylation of ERK, p38, Akt and eNOS. It is noteworthy that, recently, levosimendan pre-treatment was shown to protect guinea-pig isolated hearts against ischaemia/reperfusion injury by increasing ERK activity (du Toit et al., 2008).
Finally, in the present study the effects of levosimendan on NO production were completely prevented by the adenylyl cyclase blocker 2′5′-dideoxyadenosine, at a concentration that prevented the effects of forskolin in endothelial cells both in this and in previous studies (Polte and Schröder, 1998; Grossini et al., 2008). Hence, even if the intracellular cAMP content was not determined, the above findings show that cAMP is involved in the effects of levosimendan on NO production in porcine CECs. Indeed, in previous studies the accumulation of cAMP was found to be involved with the vasorelaxation of porcine-isolated coronary arteries and in the inotropic effects of levosimendan in the mammalian heart (Bokník et al., 1997; Gruhn et al., 1998). As there are no data in the literature indicating that levosimendan activates adenylyl cyclase, the generation of cyclic AMP induced by levosimendan could be due to its potent inhibitory effect on PDEIII (Endoh, 2007).
The results obtained in the present study with the PDEIII-specific inhibitor cilostazol seem to confirm the role of levosimendan as a strong PDEIII inhibitor and are in agreement with previous findings (Haikala and Linden, 1995; Gruhn et al., 1998; Endoh, 2007). It is also noteworthy that cilostazol has been shown to induce NO release through a PKA-dependent eNOS phosphorylation/dephosphorylation in CEC that exhibited a different susceptibility to glibenclamide and KATP agonists from those used in the present study (Hashimoto et al., 2006). The finding that levosimendan and cilostazol have different pharmacological characteristics supports the hypothesis that the action of levosimendan may be ascribed to the structure of the compound.
Taken together, the results from this study identify a novel mechanism of action of levosimendan associated with KATP channel opening and NO production in CEC. Levosimendan could initiate cytoplasmic signalling events leading to cAMP production and phosphorylation of factors such as ERK1/2, p38 and Akt, which ultimately could induce activation of eNOS either directly or through the opening of KATP channels. As stated previously, however, the ability of levosimendan to target mitochondrial KATP channels remains to be clearly demonstrated, although the finding that cAMP-dependent signals are involved in the rise of NO induced by levosimendan supports this hypothesis. Hence, activation of protein kinases may lead to membrane hyperpolarization and increase the driving force for L-arginine entry (Sobrevia et al., 1995; Bogle et al., 1996), which could induce activation of eNOS.
Acknowledgments
This research has received generous sponsorship from Università del Piemonte Orientale ‘A. Avogadro’. We thank the Azienda Ospedaliera Maggiore della Carità di Novara and Regione Piemonte for their help.
Glossary
Abbreviations:
- L-NAME
Nω-nitro-L-arginine methyl ester
- 5HD
5-hydroxydecanoate
- CEC
coronary endothelial cells
- EGM2
endothelial growth medium 2
- hEGF
human epidermal growth factor
- VEGF
vascular endothelial growth factor
- hFGF-B
human fibroblast growth factor-basic
- R3-IGF-1
recombinant analogue insulin-like growth factor 1 human
- PBS
phosphate-buffer saline
- DMEM
Dulbecco's modified Eagle's medium FCS, foetal calf serum
- DAF-FM
4-amino-5methylamino-2’,7’-difluorofluorescin diacetate
- SDS
sodium dodecyl sulphate
- NaF
sodium fluoride
- BCA
bicinchoninic acid
- PVDF
polyvinylidene fluoride
- DMSO
dimethyl sulphoxide
- CD34
endothelial cell marker 34
Conflicts of interest
The Authors of this paper have no conflict of interest in connection with the submitted article.
References
- Akar F, Manavbasi Y, Parlar AI, Ulus AT, Katircioglu SF. The gender differences in the relaxation to levosimendan of human internal mammary artery. Cardiovasc Drugs Ther. 2007;21:331–338. doi: 10.1007/s10557-007-6047-x. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition (2008 revision) Br J Pharmacol. 2008;153(Suppl.)(2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrukhiv A, Costa AD, West IC, Garlid KD. Opening mitoKATP increases superoxide generation from complex I of the electron transport chain. Am J Physiol Heart Circ Physiol. 2006;291:H2067–H2074. doi: 10.1152/ajpheart.00272.2006. [DOI] [PubMed] [Google Scholar]
- Baczkó I, Jones L, McGuigan CF, Manning Fox JE, Gandhi M, Giles WR, et al. Plasma membrane KATP channel-mediated cardioprotection involves posthypoxic reductions in calcium overload and contractile dysfunction: mechanistic insights into cardioplegia. FASEB J. 2005;19:980–982. doi: 10.1096/fj.04-3008fje. [DOI] [PubMed] [Google Scholar]
- Bogle RG, Baydoun AR, Pearson JD, Mann GE. Regulation of L-arginine transport and nitric oxide release in superfused porcine aortic endothelial cells. J Physiol. 1996;490:229–241. doi: 10.1113/jphysiol.1996.sp021138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokník P, Neumann J, Kaspareit G, Schmitz W, Scholz H, Vahlensieck U, et al. Mechanisms of the contractile effects of levosimendan in the mammalian heart. J Pharmacol Exp Ther. 1997;280:277–283. [PubMed] [Google Scholar]
- Boo YC, Hwang J, Sykes M. Shear stress stimulates phosphorylation of eNOS at Ser (635) by a protein kinase A-dependent mechanism. Am J Physiol Heart Circ Physiol. 2002;283:H1819–H1828. doi: 10.1152/ajpheart.00214.2002. [DOI] [PubMed] [Google Scholar]
- Bowman P, Haikala H, Paul RJ. Levosimendan, a calcium sensitizer in cardiac muscle, induces relaxation in coronary smooth muscle through calcium desensitization. J Pharmacol Exp Ther. 1999;288:316–325. [PubMed] [Google Scholar]
- Bryan PT, Marshall JM. Cellular mechanisms by which adenosine induces vasodilatation in rat skeletal muscle: significance for systemic hypoxia. J Physiol. 1999;514:163–175. doi: 10.1111/j.1469-7793.1999.163af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busse R, Lückhoff A, Mülsch A. Cellular mechanisms controlling EDRF/NO formation in endothelial cells. Basic Res Cardiol. 1991;86:7–16. doi: 10.1007/978-3-642-72461-9_2. [DOI] [PubMed] [Google Scholar]
- Cuong DV, Kim N, Youm JB, Joo H, Warda M, Lee JW, et al. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature. 2002;416:337–339. doi: 10.1038/416337a. [DOI] [PubMed] [Google Scholar]
- Das B, Sarkar C. Pharmacological preconditioning by levosimendan is mediated by inducible nitric oxide synthase and mitochondrial KATP channel activation in the in vivo anesthetized rabbit heart model. Vascul Pharmacol. 2007;47:248–256. doi: 10.1016/j.vph.2007.06.008. [DOI] [PubMed] [Google Scholar]
- Endoh M. Could Ca2+ sensitizers rescue patients from chronic congestive heart failure? Br J Pharmacol. 2007;150:826–828. doi: 10.1038/sj.bjp.0707163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdei N, Papp Z, Pollesello P, Edes I, Bagi Z. The levosimendan metabolite OR-1896 elicits vasodilation by activating the K(ATP) and BK(Ca) channels in rat isolated arterioles. Br J Pharmacol. 2006;148:696–702. doi: 10.1038/sj.bjp.0706781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulton D, Gratton JP, Sessa WC. Post-translational control of endothelial nitric oxide synthase: why isn't calcium/calmodulin enough? J Pharmacol Exp Ther. 2001;299:818–824. [PubMed] [Google Scholar]
- Gross GJ, Fryer MR. Sarcolemmal versus mitochondrial ATP-sensitive K+ channels and myocardial preconditioning. Circ Res. 1999;84:973–979. doi: 10.1161/01.res.84.9.973. [DOI] [PubMed] [Google Scholar]
- Grossini E, Caimmi PP, Molinari C, Teodori G, Vacca G. Hemodynamic effect of intracoronary administration of levosimendan in the anesthetized pig. J Cardiovasc Pharmacol. 2005;46:333–342. doi: 10.1097/01.fjc.0000175435.41541.6f. [DOI] [PubMed] [Google Scholar]
- Grossini E, Molinari C, Mary DA, Uberti F, Caimmi PP, Surico N, et al. Intracoronary genistein acutely increases coronary blood flow in anesthetized pigs through β adrenergic mediated nitric oxide release and estrogenic receptors. Endocrinology. 2008;149:2678–2687. doi: 10.1210/en.2007-1361. [DOI] [PubMed] [Google Scholar]
- Gruhn N, Nielsen-Kudsk JE, Theilgaard S, Bang L, Olesen SP, Aldershvile J. Coronary vasorelaxant effect of levosimendan, a new inodilator with calcium-sensitizing properties. J Cardiovasc Pharmacol. 1998;31:741–749. doi: 10.1097/00005344-199805000-00013. [DOI] [PubMed] [Google Scholar]
- Haikala H, Linden IB. Mechanisms of action of calcium sensitizing drugs. J Cardiovasc Pharmacol. 1995;26:S10–S19. [PubMed] [Google Scholar]
- Han J, Kim N, Joo H, Kim E, Earm YE. ATP-sensitive K+ channel activation by nitric oxide and protein kinase G in rabbit ventricular myocytes. Am J Physiol Heart Circ Physiol. 2002;283:H1545–H1554. doi: 10.1152/ajpheart.01052.2001. [DOI] [PubMed] [Google Scholar]
- Hashimoto A, Miyakoda G, Hirose Y, Mori T. Activation of endothelial nitric oxide synthase by cilostazol via a cAMP/protein kinase A- and phosphatidylinositol 3-kinase/Akt-dependent mechanism. Atherosclerosis. 2006;189:350–357. doi: 10.1016/j.atherosclerosis.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Herrera GM, Resta TC, Candelaria JJ, Walker BR. Maintained vasodilatory response to cromakalim after inhibition of nitric oxide synthesis. J Cardiovasc Pharmacol. 1998;31:921–929. doi: 10.1097/00005344-199806000-00017. [DOI] [PubMed] [Google Scholar]
- Höhn J, Pataricza J, Petri A, Tóth GK, Balogh A, Varró A, et al. Levosimendan interacts with potassium channel blockers in human saphenous veins. Basic Clin Pharmacol Toxicol. 2004;94:271–273. doi: 10.1111/j.1742-7843.2004.pto940603.x. [DOI] [PubMed] [Google Scholar]
- Ito C, Kusano E, Akimoto T. Cilostazol enhances IL-1beta-induced NO production and apoptosis in rat vascular smooth muscle via PKA-dependent pathway. Cell Signal. 2002;14:625–632. doi: 10.1016/s0898-6568(02)00004-9. [DOI] [PubMed] [Google Scholar]
- Jackson WF. Potassium channels in the peripheral microcirculation. Microcirculation. 2005;12:113–127. doi: 10.1080/10739680590896072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janigro D, West GA, Gordon EL, Winn HR. ATP-sensitive K+ channels in rat aorta and brain microvascular endothelial cells. Am J Physiol. 1993;265:C812–C821. doi: 10.1152/ajpcell.1993.265.3.C812. [DOI] [PubMed] [Google Scholar]
- Janigro D, Nguyen TS, Meno J, West GA, Winn HR. Endothelium-dependent regulation of cerebrovascular tone by extracellular and intracellular ATP. Am J Physiol. 1997;273:H878–H885. doi: 10.1152/ajpheart.1997.273.2.H878. [DOI] [PubMed] [Google Scholar]
- John TA, Ibe BO, Raj JU. Regulation of endothelial nitric oxide synthase: involvement of protein kinase G 1beta, serine 116 phosphorylation and lipid structures. Clin Exp Pharmacol Physiol. 2008;35:148–158. doi: 10.1111/j.1440-1681.2007.04801.x. [DOI] [PubMed] [Google Scholar]
- Kaheinen P, Pollesello P, Levijoki J, Haikala H. Levosimendan increases diastolic coronary flow in isolated guinea-pig heart by opening ATP-sensitive potassium channels. J Cardiovasc Pharmacol. 2001;37:367–374. doi: 10.1097/00005344-200104000-00003. [DOI] [PubMed] [Google Scholar]
- Kersten JR, Montgomery MW, Pagel PS, Warltier DC. Levosimendan, a new positive inotropic drug, decreases myocardial infarct size via activation of K(ATP) channels. Anesth Analg. 2000;90:5–11. doi: 10.1097/00000539-200001000-00003. [DOI] [PubMed] [Google Scholar]
- Kimura C, Oike M, Ohnaka K, Nose Y, Ito Y. Constitutive nitric oxide production in bovine aortic and brain microvascular endothelial cells: a comparative study. J Physiol. 2004;554:721–730. doi: 10.1113/jphysiol.2003.057059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopustinskiene DM, Pollesello P, Saris NE. Levosimendan is a mitochondrial K(ATP) channel opener. Eur J Pharmacol. 2001;428:311–314. doi: 10.1016/s0014-2999(01)01350-4. [DOI] [PubMed] [Google Scholar]
- Kopustinskiene DM, Pollesello P, Saris NE. Potassium-specific effects of levosimendan on heart mitochondria. Biochem Pharmacol. 2004;68:807–812. doi: 10.1016/j.bcp.2004.05.018. [DOI] [PubMed] [Google Scholar]
- Koyama T, Kimura C, Park SJ, Oike M, Ito Y. Functional implications of Ca2+ mobilizing properties for nitric oxide production in aortic endothelium. Life Sci. 2002;72:511–520. doi: 10.1016/s0024-3205(02)02246-4. [DOI] [PubMed] [Google Scholar]
- Lorenz M, Wessler S, Follmann E, Michaelis W, Düsterhöft T, Baumann G, et al. A constituent of green tea, epigallocatechin-3-gallate, activates endothelial nitric oxide synthase by a phosphatidylinositol-3-OH-kinase-, cAMP-dependent protein kinase-, and Akt-dependent pathway and leads to endothelial-dependent vasorelaxation. J Biol Chem. 2004;279:6190–6195. doi: 10.1074/jbc.M309114200. [DOI] [PubMed] [Google Scholar]
- Luckhoff A, Busse R. Calcium influx into endothelial cells and formation of endothelium-derived relaxing factor is controlled by the membrane potential. Pflugers Arch. 1990;416:305–311. doi: 10.1007/BF00392067. [DOI] [PubMed] [Google Scholar]
- Luo SS, Sugimoto K, Fujii S, Takemasa T, Fu SB, Yamashita K. Role of heat shock protein 70 in induction of stress fiber formation in rat arterial endothelial cells in response to stretch stress. Acta Histochem Cytochem. 2007;40:9–17. doi: 10.1267/ahc.06011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaels AD, McKeown B, Kostal M, Vakharia KT, Jordan MV, Gerber IL, et al. Effects of intravenous levosimendan on human coronary vasomotor regulation, left ventricular wall stress, and myocardial oxygen uptake. Circulation. 2005;111:1504–1509. doi: 10.1161/01.CIR.0000159252.82444.22. [DOI] [PubMed] [Google Scholar]
- Molinari C, Grossini E, Mary DA, Uberti F, Ghigo E, Ribichini F, et al. Prolactin induces regional vasoconstriction through the beta2-adrenergic and nitric oxide mechanisms. Endocrinology. 2007;148:4080–4090. doi: 10.1210/en.2006-1577. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol. 1995;268:C799–C822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- Nilius B, Droogmans G. Ion channels and their functional role in vascular endothelium. Physiol Rev. 2001;81:1415–1459. doi: 10.1152/physrev.2001.81.4.1415. [DOI] [PubMed] [Google Scholar]
- Oldenburg O, Qin Q, Sharma A, Cohen M, Downey J, Benoit J. Acetylcholine leads to free radical production dependent on KATP channels, Gi proteins, phosphatidylinositol 3-kinase and tyrosine kinase. Cardiovasc Res. 2002;55:544–552. doi: 10.1016/s0008-6363(02)00332-2. [DOI] [PubMed] [Google Scholar]
- Padmasekar M, Nandigama R, Wartenberg M, Schluter KD, Sauer H. The acute phase protein alpha2-macroglobulin induces rat ventricular cardiomyocyte hypertrophy via ERK1,2 and PI3-kinase/Akt pathways. Cardiovasc Res. 2007;75:118–128. doi: 10.1016/j.cardiores.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Pasdois P, Beauvoit B, Tariosse L, Vinassa B, Bonoron-Adèle S, Santos PD. MitoK(ATP)-dependent changes in mitochondrial volume and in complex II activity during ischemic and pharmacological preconditioning of Langendorff-perfused rat heart. J Bioenerg Biomembr. 2006;38:101–112. doi: 10.1007/s10863-006-9016-3. [DOI] [PubMed] [Google Scholar]
- Pasdois P, Quinlan CL, Rissa A, Tariosse L, Vinassa B, Costa AD, et al. Ouabain protects rat hearts against ischemia-reperfusion injury via pathway involving src kinase, mitoKATP, and ROS. Am J Physiol Heart Circ Physiol. 2007;292:H1470–H1478. doi: 10.1152/ajpheart.00877.2006. [DOI] [PubMed] [Google Scholar]
- Pataricza J, Hõhn J, Petri A, Balogh A, Papp JG. Comparison of the vasorelaxing effect of cromakalim and the new inodilator, levosimendan, in human isolated portal vein. J Pharm Pharmacol. 2000;52:213–217. doi: 10.1211/0022357001773715. [DOI] [PubMed] [Google Scholar]
- Polte T, Schröder H. Cyclic AMP mediates endothelial protection by nitric oxide. Biochem Biophys Res Commun. 1998;20:460–465. doi: 10.1006/bbrc.1998.9486. [DOI] [PubMed] [Google Scholar]
- Sheng JZ, Braun AP. Small- and intermediate-conductance Ca2+-activated K+ channels directly control agonist-evoked nitric oxide synthesis in human vascular endothelial cells. Am J Physiol Cell Physiol. 2007;293:C458–C467. doi: 10.1152/ajpcell.00036.2007. [DOI] [PubMed] [Google Scholar]
- Simoncíková P, Ravingerová T, Andelová E, Tribulová N, Barancík M. Changes in rat myocardium associated with modulation of ischemic tolerance by diazoxide. Gen Physiol Biophys. 2007;26:75–85. [PubMed] [Google Scholar]
- Sobrevia L, Cesare P, Yudilevich DL, Mann GE. Diabetes-induced activation of system y+ and nitric oxide synthase in human endothelial cells: association with membrane hyperpolarization. J Physiol. 1995;489:183–192. doi: 10.1113/jphysiol.1995.sp021040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankevicius E, Lopez-Valverde V, Rivera L, Hughes AD, Mulvany MJ, Simonsen U. Combination of Ca2+ -activated K+ channel blockers inhibits acetylcholine-evoked nitric oxide release in rat superior mesenteric artery. Br J Pharmacol. 2006;149:560–572. doi: 10.1038/sj.bjp.0706886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi R, Endoh M. Dual regulation of myofilament Ca2+ sensitivity by levosimendan in normal and acidotic conditions in aequorin-loaded canine ventricular myocardium. Br J Pharmacol. 2005;145:1143–1152. doi: 10.1038/sj.bjp.0706292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- du Toit EF, Genis A, Opie LH, Pollesello P, Lochner A. A role for the RISK pathway and K(ATP) channels in pre- and post-conditioning induced by levosimendan in the isolated guinea pig heart. Br J Pharmacol. 2008;154:41–50. doi: 10.1038/bjp.2008.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt WA, Steinert JR, Mann GE. Modulaiton of the L-arginine/nitric oxide signalling pathway in vascular endothelial cells. Biochem Soc Symp. 2004;71:143–156. doi: 10.1042/bss0710143. [DOI] [PubMed] [Google Scholar]
- Xu Z, Ji X, Boysen PG. Exogenous nitric oxide generates ROS and induces cardioprotection: involvement of PKG, mitochondrial KATP channels, and ERK. Am J Physiol Heart Circ Physiol. 2004;286:H1433–H1440. doi: 10.1152/ajpheart.00882.2003. [DOI] [PubMed] [Google Scholar]
- Yildiz O. Vasodilating mechanisms of levosimendan: involvement of K+ channels. J Pharmacol Sci. 2007;104:1–5. doi: 10.1254/jphs.cp0060010. [DOI] [PubMed] [Google Scholar]
- Yokoshiki H, Katsube Y, Sunagawa M, Sperelakis N. Levosimendan, a novel Ca++ sensitizer, activates the glibenclamide sensitive K+ channel in rat arterial myocytes. Eur J Pharmacol. 1997;333:249–259. doi: 10.1016/s0014-2999(97)01108-4. [DOI] [PubMed] [Google Scholar]
- Zoltán P, Kálmán C, Pollesello P, Haikala H, Édes I. Pharmacological mechanisms contributing to the clinical efficacy of levosimendan. Cardiovasc Drug Rev. 2005;23:71–98. doi: 10.1111/j.1527-3466.2005.tb00158.x. [DOI] [PubMed] [Google Scholar]