

Abstract
Background and purpose:
Activation of the persistent sodium current in ischaemic myocardium results in calcium overload which is toxic for the cardiomyocyte. Thus, the activity of 3-(R)-[3-(2-methoxyphenylthio-2-(S)-methylpropyl]amino-3,4-dihydro-2H-1,5 benzoxathiepine bromhydrate (F 15845), a new selective persistent sodium current blocker, in protecting against the effects of cardiac ischaemia was examined, in both in vitro and in vivo models.
Experimental approach:
Electrophysiological studies using patch-clamp and conventional microlelectrode techniques, isolated perfused hearts and models of angina in anaesthetized animals were used to assess the protection afforded by F 15845 against ischaemia-induced changes.
Key results:
F 15845 reduced the persistent sodium current activated by veratridine (IC50 1.58 × 10−6 mol·L−1). F 15845 blocked voltage-gated human cardiac sodium channels in a novel, voltage-dependent manner, selectively affecting steady-state inactivation. F 15845 did not affect action potential shape and basal function of guinea pig isolated perfused hearts but did reduce ischaemia-induced diastolic contracture in this model (IC50 0.64 × 10−6 mol·L−1). In rabbits, F 15845 given i.v. (ED50 0.05 mg·kg−1) or orally (ED50 0.13 mg·kg−1) dose-dependently and powerfully inhibited regional myocardial ischaemia-induced ST segment elevation in the absence of haemodynamic effects, implying direct cardiac activity. In dogs, F 15845 dose-dependently inhibited epicardial ST segment changes (70 ± 8% at 0.63 mg·kg−1) in an experimental angina model of demand ischaemia, again without haemodynamic effects, confirming a direct anti-anginal activity.
Conclusions and implications:
F 15845 is a selective, potent blocker of the persistent sodium current, generated by the human Nav1.5 channel isoforms, and prevents cardiac angina in animal models.
Keywords: F 15845, persistent sodium current, cardiac ischaemia, anti-anginal drugs
Introduction
Angina pectoris is the most common manifestation of coronary artery disease presenting as the initial symptom in almost half of patients (37% of men and 65% of women, Bhatt and Stone, 2006). The therapeutic options in chronic stable angina have expanded in the past few decades, in terms of both interventional and pharmacological means. The therapeutic aims are twofold. The first objective is to improve the prognosis – for example, by using statins, ACE inhibitors and aspirin that target known risk factors. The second objective is to relieve symptoms – for example, through revascularization and the use of anti-anginal drugs. However, an increasing number of patients cannot undergo revascularization because of complicating factors such as age, medical co-morbidities and inadequate coronary anatomy. Many of these patients remain symptomatic despite conventional anti-anginal medications with β-blockers, calcium antagonists and nitrates. So, there is a growing medical need for new anti-anginal treatments and especially for approaches that will cause no or minimal haemodynamic effects. Our work falls precisely within this framework and the present paper describes the properties of 3-(R)-[3-(2-methoxyphenylthio-2-(S)-methylpropyl]amino-3,4-dihydro-2H-1,5 benzoxathiepine bromhydrate (F 15845), a novel agent currently in phase II clinical trials, in protecting against the consequences of cardiac ischaemia.
There is compelling evidence showing that activation of the persistent sodium current results in an accumulation of intracellular sodium and that the concentration of Na+ ([Na+]i) is a function of the duration of ischaemia (Silverman and Stern, 1994; Saint, 2008; Shryock and Belardinelli, 2008). Intracellular sodium is subsequently exchanged for calcium through the Na+/Ca2+ exchanger functioning in the reverse mode (Murphy et al., 1999). This leads in turn to calcium overload, known to be toxic or even lethal for the cell. Overall, it appears that the influx of sodium, utilizing the increased proportion of the population of Na+ channels that have switched into a slow inactivation mode (persistent sodium current), initiates a chain of events responsible for electrical instability such as arrhythmias (Noble and Noble, 2006; Shryock and Belardinelli, 2008), mechanical dysfunction (reduced contractility, increased diastolic tension, Houser, 2005; Saint, 2008; Shryock and Belardinelli, 2008) and mitochondrial dysfunction (energy exhaustion, Saint, 2008). Thus, it is likely that blockade of the persistent sodium current will delay, if not prevent, initiation of that process and therefore attenuate cell damage (Ver Donck et al., 1993). In this study, we describe the first selective blocker of the persistent sodium current, F 15845, and discuss the effects of this compound on the ionic disturbances associated with cardiac ischaemia.
Methods
Animals
All animals were housed and tested in an Association for the Assessment and Accreditation of Laboratory Animal Care-accredited facility in strict compliance with all applicable regulations and the experiments were carried out according to French law and the local ethical committee guidelines for animal research. The ionic channels nomenclature conforms to the BJP's Guide to Receptors and Channels (Alexander et al., 2008).
Ion channel pharmacology
Terminology: in our study, rapid INa represents the normally functioning sodium channels, characterized by a very brief opening and the veratridine-modified functioning sodium channels are called persistent INa and exhibit peak and persistent components.
The sodium channel blocking properties of F 15845 were examined in human embryonic kidney (HEK 293) cells stably transfected with the cloned α subunit of the human sodium channel, Nav1.5, on both rapid and persistent currents by using the patch-clamp technique in the whole cell configuration as described previously (Gellens et al., 1992; Pignier et al., 2007). Similarly, the effects of F 15845 were investigated on the inward rectifier (IK1) and delayed outward potassium currents (IKr and IKs), on T- and L-type calcium current (ICaT and ICaL) recorded in freshly isolated guinea pig ventricular myocytes; on the transient outward potassium current (Ito) in freshly isolated rat ventricular myocytes and on the IKr cloned human ether à gogo in HEK 293 cells by using the patch-clamp technique in the whole cell recording configuration.
The effects of F 15845 on action potential parameters in guinea pig papillary muscle were measured as described using conventional microelectrode techniques (Le Grand et al., 1995).
Isolated perfused guinea pig heart
Female guinea pigs (SPF, Hartley, Charles River, France) were killed with an overdose of sodium pentobarbitone (250 mg·kg−1 i.p.) and the heart (without heparin) was rapidly excised and placed in cold (4°C) modified Krebs medium (see below) until contraction ceased (approximately 15 s). A latex balloon, connected to a pressure transducer (Statham, Spectramed, Oxnard, CA, USA) via a polyethylene cannula, was inserted through the left atrium and mitral valve into the left ventricle for measurements of isovolumic left ventricular pressure. The aorta was cannulated and the heart was mounted on a perfusion apparatus. The heart, surrounded by a thermoregulated jacket, was perfused at 37 ± 1°C at a constant pressure of 800 mm H2O. The perfusate was a modified Krebs buffer that contained (in mmol·L−1) 124.6 NaCl, 4 KCl, 1.1 MgSO4, 0.3 NaH2PO4, 1.8 CaCl2, 24.9 NaHCO3 and 11.1 D(+)-glucose. The solution was continuously gassed with 95% O2–5% CO2, and the pH of the solution was stable at 7.4.
After the hearts began to beat spontaneously, the pressure transducer (Viggo-Spectramed, Oxnard, CA, USA) was connected to a Gould amplifier (Gould Instruments, France). The amplifier was connected to an interface (MP 100, Biopac Systems, Goleta, CA, USA) and the analogue signal was simultaneously digitized and analysed by computer (HP VECTRA VL18, CA, USA) by means of interactive software (Acknowledge III, Biopac Systems). The balloon was inflated with water sufficient to raise the left ventricular end-diastolic pressure (LVEDP) to 10 mmHg. Coronary flow (CF) was measured by a flow probe (Transonic, Ithaca, NY, USA) placed around the aortic cannula. After 45–60 min equilibration, the buffer solution containing vehicle or compound was perfused for 15 min. Global normothermic ischaemia was then induced by clamping perfusate flow for 50 min. Ischaemia caused an immediate and marked decline in LVDP and a rapid arrest of beating. LVEDP increased gradually after approximately 25 min ischaemia reaching relatively stable values (max. variation: 22.2 ± 1.3 mmHg in the presence of the vehicle, n = 15) which determined the maximal diastolic contracture by 35 min ischaemia. The effects of F 15845 were investigated over a concentration range from 0.1 × 10−6 mol·L−1 to 10 × 10−6 mol·L−1 (6 animals per concentration). The percentage of inhibition of contracture was determined by reference to the amplitude of diastolic contracture in the vehicle group versus the amplitude of contracture in the presence of F 15845.
Regional myocardial ischaemia in anaesthetized rabbits
The effects of F 15845 on the consequences of cardiac ischaemia were first examined in a model of supply ischaemia in anaesthetized rabbits. Myocardial ischaemia, induced by a reversible 5 min occlusion of the circumflex coronary artery in pentobarbital-anaesthetized rabbits, produces a large increase in the amplitude of the ST segment determined in lead II of a four-limb electrocardiogram (ECG) (Verscheure et al., 1995). Doses of F 15845 were administered intravenously 5 min prior to occlusion, as a single bolus or orally 2 h prior to occlusion, by gavage.
Intravenous protocol
After the stabilization period, a first 5 min coronary artery occlusion was performed to verify the ST segment elevation. Then the occlusion was fully released for 30 min (reperfusion period). Five min before the end of reperfusion, the compound or its vehicle were administered over 1 min. Subsequently, a second coronary occlusion was performed. Six intravenous doses of F 15845 were evaluated from 0.01 mg·kg−1 to 2.5 mg·kg−1 (six animals per group).
Oral protocol
The effects of the compound or its vehicle on ST segment elevation were evaluated 2 h after gavage. Five oral doses of F 15845 were evaluated from 0.04 mg·kg−1 to 10 mg·kg−1 (6 animals per group).
Maximal amplitudes of the ST segments were taken for calculating the percentage inhibition of ST segment elevation, as follows. The maximal ST segment change occurring during coronary occlusion was measured in control animals and from that a mean group maximal ST change was calculated. The maximal ST segment change was then determined in each F 15845-treated animal and the percent inhibition was calculated, relative to the mean results in the vehicle group.
Regional demand ischaemia in anaesthetized dogs
The potential anti-anginal activity of intravenous F 15845 was evaluated in an anaesthetized dog model of demand ischaemia-evoked epicardial ST segment changes (Sugiyama and Hashimoto, 1999), designed to resemble that which occurs during stable angina. The animals were anaesthetized with propofol (6 mg·kg−1, i.v.) Following placement of a critical stenosis upon the left anterior descending coronary artery of beagle dogs, the left ventricle was driven electrically so as to increase heart rate to 80–90 bpm above baseline for 3 min followed by 30 min. recovery. Successive pacing-recovery cycles were performed at baseline (before F 15845) and 30, 60 and 90 min, after F 15845 administration. The compound was administered as an intravenous bolus after the first period of pacing and infused throughout the subsequent three periods. Four groups of animals were investigated: vehicle (n = 9), F 15845 0.16 mg·kg−1 (n = 6), F 15845 0.63 mg·kg−1 (n = 5) and diltiazem 0.16 mg·kg−1 (n = 9).
Statistical analysis
All values are expressed as means ± SEM. Intragroup statistical analysis of results (drug vs. baseline) was performed by the paired t-test after testing for homogeneity of variance with analysis of variance (anova) with repeated measures. Intergroup statistical analyses of results (drug vs. vehicle) were performed using one-way analysis of variance followed by Dunnett's test when anova was significant. P values less than 0.05 were considered significant (SigmaStat 2.03).
Drugs
F 15845 was synthesized (Le Grand et al., 2008) by the Division of Medicinal Chemistry I, Centre de Recherche Pierre Fabre. F 15845 was dissolved in dimethyl sulphoxide as a 10−2 mol·L−1 stock solution, prepared freshly for each set of in vitro experiments. The highest final concentration of dimethyl sulphoxide was 0.1% (F 15845, 10−5 mol·L−1). Veratridine was purchased from Sigma Chemical (St Louis, MO, USA) and was dissolved in distilled water. F 15845 (salt to base ratio: 1.31) was dissolved in polyethyleneglycol (PEG) 300 for each in vivo experiment. To this solution of F 15845, sterile saline (0.9%) was added to obtain a final solution containing 40% PEG in sterile saline (0.9%).
Results
Effects of F 15845 on human cardiac sodium channel: hNav1.5
In HEK 293 cells transfected with the SCN5A gene which encodes the α-subunit of hNav1.5 (Gellens et al., 1992; Goldin et al., 2000), persistent sodium current induced with veratridine at 4 × 10−5 mol·L−1 was elicited by a depolarizing step from −110 to −30 mV every 5 s. Typical recordings of the effects of F 15845 on persistent sodium current are shown in Figure 1A and its steady-state inactivation in Figure 1B. From 10−6 mol·L−1, F 15845 concentration-dependently reduced the amplitude of the persistent sodium current, measured as the area under the curve, by 22.4 ± 9.8% at 10−6 mol·L−1 (n = 5; P < 0.05) and by 40.0 ± 6.5% (n = 7; P < 0.05) at 10−5 mol·L−1 (Figure 1C) with an IC50 value of 9.34 × 10−6 mol·L−1 with a 95% confidence interval of [ND–56.2]10−6 mol·L−1 and nH of 0.76 (0.23–2.63). The magnitude of the effect, however, depends on the holding potential (see Figure 1C and legends). Furthermore, F 15845 shifted the persistent sodium current inactivation curve towards hyperpolarized potentials (Figure 1B) by 4.5 mV (−80.5 ± 2.3 mV vs. −84.9 ± 2.5 mV, P < 0.05) and 7.3 mV (−81.3 ± 2.2 mV vs. −88.6 ± 2.6, P < 0.05) at 10−6 mol·L−1 and 10−5 mol·L−1 F 15845 respectively. In contrast, tetrodotoxin did not modify these inactivation parameters (data not shown).
Figure 1.
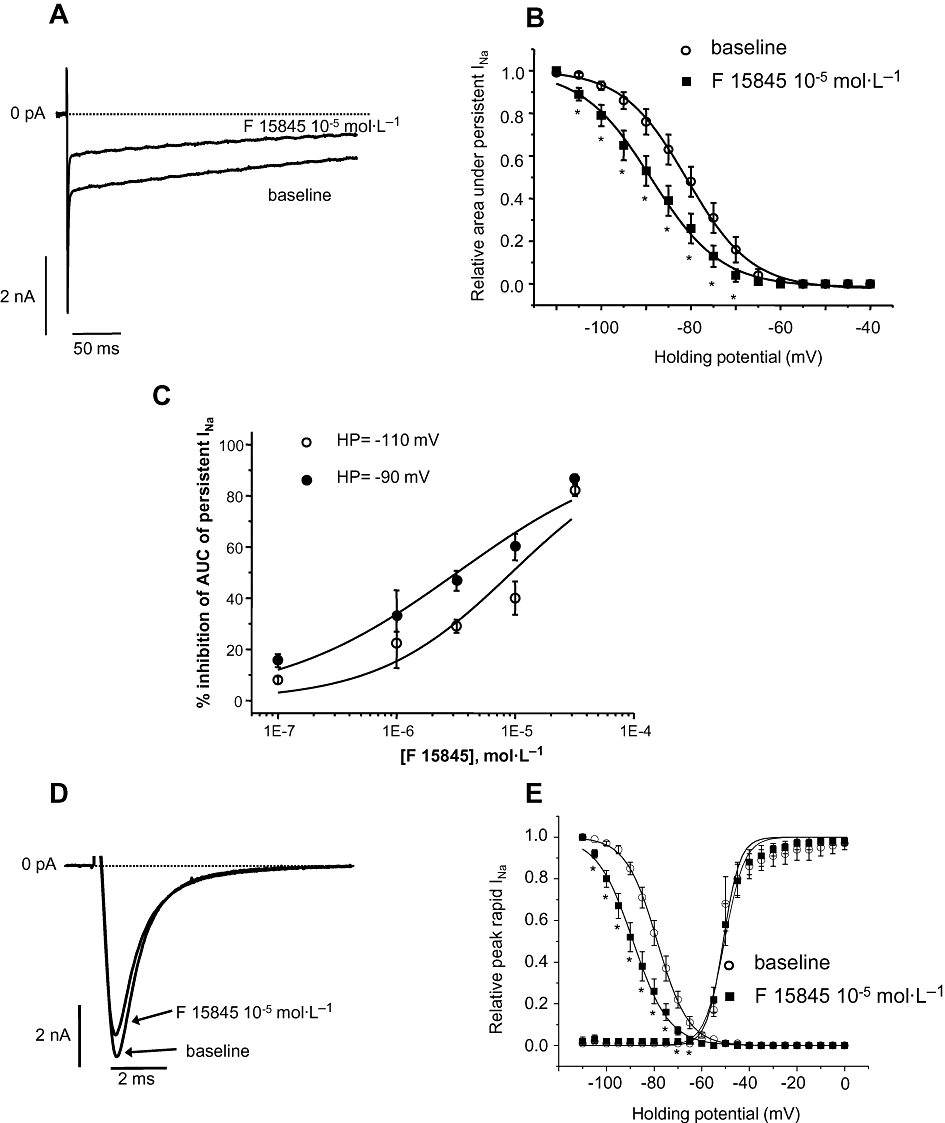
(A) Typical recordings of veratridine-induced persistent sodium current mediated by human embryonic kidney (HEK 293) cells transfected with hNav1.5 (elicited at −30 mV from a holding potential of −110 mV) in presence and absence of 3-(R)-[3-(2-methoxyphenylthio-2-(S)-methylpropyl]amino-3,4-dihydro-2H-1,5 benzoxathiepine bromhydrate (F 15845) (10−5 mol·L−1). (B) Veratridine-induced persistent sodium current steady-state inactivation in absence or presence of F 15845 (10−5 mol·L−1) in HEK 293 cells transfected with hNav1.5. Steady-state inactivation is shifted to hyperpolarized potentials by F 15845 with mean V0.5 values of −80.5 ± 2.3 mV and −84.9 ± 2.5 mV (n = 7, P < 0.05) in vehicle and in presence of F 15845 respectively. Data are means ± SEM. n = 7; *P < 0.05 compared with corresponding vehicle. (C) Voltage-dependent sodium channel blocking effects of F 15845 from 10−7–3.2 × 10−5 mol·L−1 on persistent sodium current. As the holding potential (HP) depolarizes from −110 to −90 mV, the inhibition of persistent sodium current by F 15845 increases significantly. This unique property of sodium channel blockade renders F 15845 selective for depolarized (i.e. ischaemic) cardiac tissue. Data are means ± SEM. n = 5–10. (D) Typical recordings of peak (rapid) sodium current mediated by HEK 293 cells transfected with hNav1.5 in presence and absence of F 15845 (10 µmol). Note a very weak inhibition on peak current elicited at −30 mV from a holding potential of −110 mV. (E) Peak sodium current activation and inactivation curves in absence or presence of F 15845 (10 µmol) in HEK 293 cells transfected with hNav1.5. Note shift of steady-state inactivation to hyperpolarized potentials by F 15845 without significantly affecting peak current activation parameters. Data are expressed as mean ± SEM. n = 4–5.
F 15845 blocked the sodium channel in a voltage-dependent manner. Thus, at a holding potential of −80 mV, F 15845 diminished the amplitude of the persistent sodium current by 52.5 ± 10.4% at 10−6 mol·L−1 (n = 5) and by 70.0 ± 4.9% at 10−5 mol·L−1 (n = 7; Figure 1B). The IC50 value of inhibition of persistent sodium current elicited from a holding potential of −90 mV (Figure 1C) was 3.25 × 10−6 mol·L−1 with a 95% confidence interval of [1.4–7.1]10−6 mol·L−1 and nH of 0.57 (0.31–0.99). This voltage-dependence of F 15845 may render the compound more effective when the cell is depolarized which is precisely the condition of cardiac tissue subjected to an ischaemic situation. On the other hand, when the rapid sodium current was elicited by a depolarizing step (from −110 to −30 mV), F 15845 only weakly reduced the amplitude of this current, 6.7 ± 2.5% at 10−6 mol·L−1 (n = 9, P < 0.05) and 23.4 ± 5.4% at 10−5 mol·L−1 (n = 5, P < 0.05; Figure 1D, phasic blockade). In comparison, tetrodotoxin markedly reduced the rapid sodium current, 28.5 ± 5.2 and 79.8 ± 1.8% at 10−6 and 10−5 mol·L−1 respectively (n = 5, P < 0.05,). The impact of F 15845 on rapid sodium current steady-state activation and inactivation curves are shown in Figure 1E. At 10−5 mol·L−1, F 15845 shifts the steady-state inactivation leftwards without interfering with activation curves (Figure 1E) with mean V0.5 values of activation of −49.7 ± 1.9 mV (vehicle) and −49.9 ± 1.7 mV (F 15845, 10−5 mol·L−1, n = 4) and V0.5 values of steady-state inactivation of −78.7 ± 1.6 mV (vehicle) and −88.7 ± 1.9 (F 15845, 10−5 mol·L−1, n = 5, P < 0.05).
Effects of F 15845 on persistent sodium current in freshly isolated guinea pig cardiomyocytes
Figure 2A shows superimposed typical recordings of veratridine-induced persistent sodium current in absence and presence of F 15845 in freshly isolated guinea pig ventricular cardiomyocytes. As shown in Figure 2A, F 15845 (10−5 mol·L−1) markedly reduced the amplitude of the persistent sodium current obtained with a short depolarizing step (50 ms) from −80 to −10 mV. This effect was only partially reversible following 7–8 min washout with the vehicle. Furthermore, F 15845 (10−7 to 10−4 mol·L−1) concentration dependently reduced the amplitude of the persistent sodium current (Figure 2B) with an IC50 value of 1.58 × 10−6 mol·L−1 with 95% confidence limits of [0.53–3.79] × 10−6 mol·L−1 and nH of 0.45 (0.23–1.01) which is in the same range of potency as that reported above using HEK 293 cells transfected with the SCN5A gene.
Figure 2.
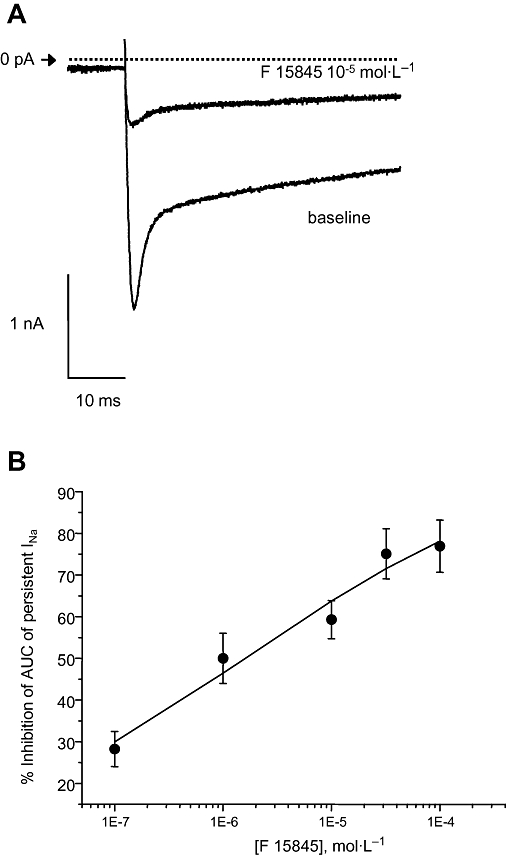
(A) Superimposed typical recordings of veratridine-induced persistent sodium current evoked by a depolarizing step from a holding potential of −80 to −10 mV in the absence and presence of F 15845 (10−5 mol·L−1). (B) Effects of F 15845 (10−7 mol·L−1–10−4 mol·L−1) on veratridine-induced persistent sodium current evoked by a depolarizing step from a holding potential of −80 to −10 mV. Total quantity of charge (area under the curve) which represented the persistent sodium current was obtained at the end of 3 min superfusion and was determined with respect to stabilized baseline values. Data are expressed as mean ± SEM.
Selectivity of F 15845 for the persistent sodium current
F 15845 did not affect the major Ca2+ and K+ channels that take part in the electrical activities of the myocardium (Figure 3A); for instance, at a fixed concentration of 10−5 mol·L−1, F 15845 reduced neuronal rat INav1.2 by 16.0 ± 5.1% (n = 6, P < 0.05). Selectivity of F 15845 towards potassium channels was assessed on HEK 293 cells transfected with human ERG channels with a mean inhibition value of 15.3 ± 7.0% (n = 9, NS), on rat Ito and IK1 currents with mean inhibition of 24 ± 4% (n = 7, P < 0.05) and 11 ± 5% (n = 7, NS) for Ito and IK1 respectively. Similarly to IK1, guinea pig IKr and IKs were not significantly modified by 10−5 mol·L−1 F 15845 with 10 ± 6% (n = 9) and 9 ± 7% (n = 8) inhibition for IKr and IKs respectively. Guinea pig T- and L-type calcium currents were also not significantly altered by 10−5 mol·L−1 F 15845 with mean inhibition values of −6 ± 14% (n = 6) and 16 ± 6% (n = 6) for ICa-T and ICa-L respectively. The selectivity of F 15845 was further assessed in the guinea pig papillary muscle, a particularly sensitive model (Le Grand et al., 1995; Sakmannn et al., 2000; Song et al., 2004) that detects any effect on action potential duration (APD). F 15845 (10−7 to 10−5 mol·L−1) did not interfere with the course of the action potential, either at 50% (APD50) or at 90% (APD90) repolarization. Importantly, other parameters (i.e. resting potential, overshoot, dV/dtmax or action potential amplitude, Figure 3B) were not affected even at the highest concentration studied (10−5 mol·L−1). At low stimulation rates, F 15845 at 10−5 mol·L−1 (Figure 3C) had no effect on APD50 (F 15845, max. variation: 8.5 ± 2.9 ms, n = 6, NS; vehicle, max. variation: 19.2 ± 5.8 ms, n = 6) and APD90 (F 15845, max. variation: 9.1 ± 3.1 ms, n = 6, NS; vehicle, max. variation: 20.3 ± 7.2 ms, n = 6). These data demonstrated that F 15845 was devoid of significant effect on APD in normal, i.e. non-ischaemic, conditions.
Figure 3.
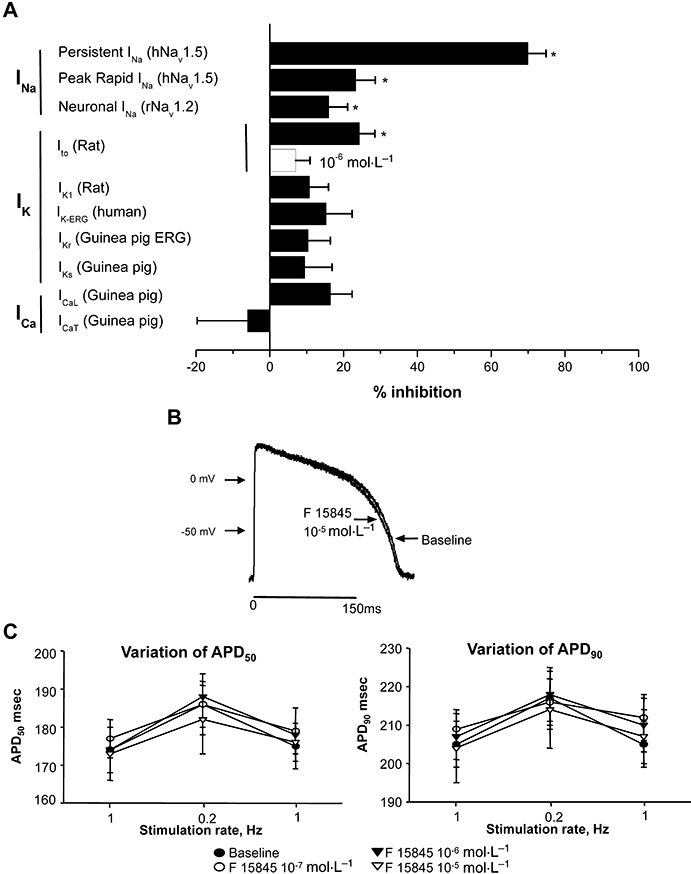
(A) Selectivity of F 15845 on the main cardiac ionic currents. Persistent INa (human Nav1.5 transfected in HEK 293 cells) was induced with veratridine (4 × 10−5 mol·L−1) and elicited at −30 mV from a potential value of −80 mV. Rapid INa was elicited at −30 mV from a holding potential of −80 mV. Neuronal INa (rat Nav1.2 transfected in HEK 293 cells) was elicited at −30 mV from a potential value of −110 mV. Ito was recorded in freshly isolated rat ventricular myocytes and was elicited by a depolarizing step to +60 mV from a holding potential value of −80 mV. Inward rectifier (IK1) and delayed outward potassium currents (IKr and IKs) were recorded in freshly isolated guinea pig ventricular myocytes. IK1 was measured at −120 mV to −10 mV (20 s, 0.01 Hz) from a holding potential value of −80 mV. IKr and IKs were elicited by a depolarizing step (3 s, +60 mV) from a holding potential value of −40 mV. HERG K+ current was investigated in HEK 293 cells transfected with the KCNH2 gene and measured as outward current upon repolarization to −50 mV from a depolarization step to +60 mV. L- and T-type calcium currents were investigated in freshly isolated guinea pig ventricular myocytes. Calcium currents were elicited by incremental depolarizing steps from a holding potential value of −80 to +60 mV every 5 s. Peak T- and L-type calcium currents were measured at −30 and +10 mV respectively. (B) Effects of F 15845 on action potentials recorded in guinea pig papillary muscles. Typical action potential recordings in absence or presence of F 15845 (10−5 mol·L−1). (C) F 15845-induced changes in APD50 (left panel) and in APD90 (right panel) at a low stimulation rate (0.2 Hz) in guinea pig papillary muscles. F 15845 (10−7 mol·L−1–10−5 mol·L−1) and vehicle were tested. APD50 and APD90 values obtained at the end of 15 min superfusion were determined with respect to stabilized baseline values. Data are mean ± SEM. APD, action potential duration; F 15845, 3-(R)-[3-(2-methoxyphenylthio-2-(S)-methylpropyl]amino-3,4-dihydro-2H-1,5 benzoxathiepine bromhydrate; HEK, human embryonic kidney; HERG, human ether à gogo.
On the other hand, a conventional Nav1.5 channel blocker, quinidine, concentration dependently prolonged APD50 (mean max. variation: +14.8 ± 3.7 ms, P < 0.001, n = 5) and APD90 (mean max. variation: +19.5 ± 3.9 ms, P < 0.001, n = 5) from 10−7 to 10−5 mol·L−1. Quinidine over the same concentration range also decreased dV/dt (mean max. variation: −47.9 ± 16.9 V/s, P < 0.01, n = 5). This last result confirmed that F 15845 did not exhibit the pharmacological profile of conventional Nav1.5 blockers (class I antiarrhythmic agents), such as quinidine.
Effects of F 15845 on cardiac basal function in an isolated perfused guinea pig heart
The effects of F 15845 on baseline left ventricular function, heart rate and CF were determined after 10 and 15 min of drug perfusion. At a lower concentration (10−7 and 3.2 × 10−7 mol·L−1), F 15845 failed to statistically significantly affect CF, heart rate and left ventricular function (Figure 4). The highest concentration of F 15845 (10−6 mol·L−1) slightly but significantly decreased LVDP (Figure 4D; max. variation: −10.4 ± 2.6 mmHg; n = 6, P < 0.01) by reduction of the systolic pressure (Figure 4C; max. variation: −9.7 ± 2.3 mmHg; n = 6, NS). Similarly, F 15845 (10−6 mol·L−1) increased CF (Figure 4A; max. variation: +7.1 ± 2.1 ml·min−1·g−1, n = 6, P < 0.01) whereas it was devoid of significant effect on heart rate (Figure 4B; max. variation: −2.2 ± 6.3 bpm, n = 6, NS). Collectively, these results demonstrate that F 15845 (10−7 to 10−6 mol·L−1) did not exert major effects on haemodynamic functions in isolated perfused hearts.
Figure 4.
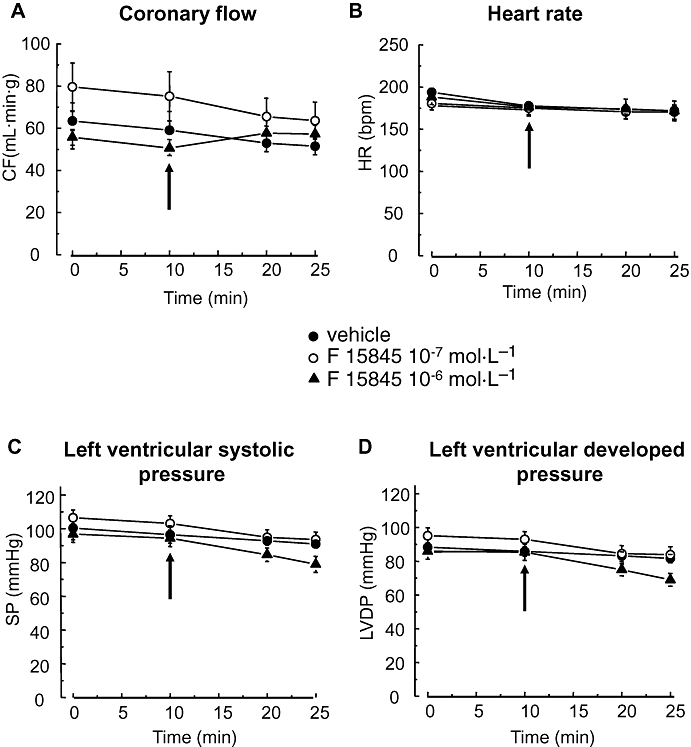
Lack of effects of 3-(R)-[3-(2-methoxyphenylthio-2-(S)-methylpropyl]amino-3,4-dihydro-2H-1,5 benzoxathiepine bromhydrate (F 15845) on basal cardiac function in an isolated perfused guinea pig heart. Coronary flow (CF) (A), heart rate (HR) (B), left ventricular systolic pressure (SP) (C) and left ventricular developed pressure (LVDP) (D) were measured before and after giving F 15845 (10−7 mol·L−1 or 10−7 mol·L−1, n = 6 for each concentration) and vehicle (n = 15). Arrows indicate the time of drug or vehicle administration. Data are mean ± SEM.
Protective effects of F 15845 on ischaemia-induced diastolic contracture
The protective effects of F 15845 were then investigated in a guinea pig isolated and perfused heart preparation which was subjected to a global 50 min ischaemia. Mean changes in LVEDP are shown in Figure 5A. Ischaemia caused an immediate and marked decline in LVDP and a rapid arrest of beating. LVEDP increased gradually after approximately 25 min ischaemia reaching relatively stable values (max. variation: 22.2 ± 1.3 mmHg in the presence of the vehicle, n = 15) which determined the maximal diastolic contracture by 35 min ischaemia. F 15845 (10−7 to 10−5 mol·L−1) concentration-dependently reduced ischaemia-induced diastolic contracture (Figure 5B) with a geometric mean IC50 value of 0.64 × 10−6 mol·L−1 and 95% confidence limits of [0.43–0.94] × 10−6 mol·L−1. Moreover, F 15845 (3.2 × 10−7 mol·L−1) significantly reduced diastolic contracture (inhibition: 27.8 ± 7.4%, n = 6, P < 0.001) without any significant effects on basal cardiac function (Figure 4). Thus, F 15845 is highly effective in preventing ischaemic contracture whereas conventional Nav1.5 channel blockers (class I antiarrhythmic drugs) quinidine (10−7–10−6 mol·L−1), lidocaine (10−6–10−4 mol·L−1) and flecainide (10−7–3.2 10−6 mol·L−1) were devoid of significant protective effects during ischaemia (maximal inhibitions of diastolic contracture: 29.4 ± 7.2%, n = 6, NS with 10−5 mol·L−1 quinidine; 13.4 ± 12.1%, n = 6, NS with 10−4 mol·L−1 lidocaine and 15.8 ± 5.7%, n = 8, NS with 3.2 × 10−6 mol·L−1 flecainide).
Figure 5.
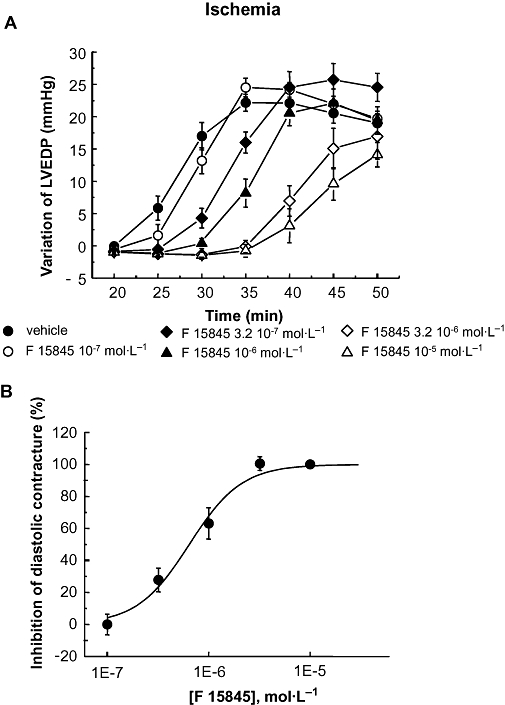
(A) Effects of F 15845 (10−7 mol·L−1–10−5 mol·L−1n = 6 for each concentration) and vehicle (n = 15) upon diastolic contracture during global ischemia. The LVEDP values obtained at various time points during ischemia were determined with respect to stabilized values at the end of 10 min ischemia. (B) Inhibition of diastolic contracture-induced by F 15845 (10−7 mol·L−1–10−5 mol·L−1). The inhibition of LVEDP values obtained at 50 min of ischemia were normalized with respect to LVEDP values obtained at the end of 50 min ischemia in the vehicle group. Data are mean ± SEM. F 15845, 3-(R)-[3-(2-methoxyphenylthio-2-(S)-methylpropyl]amino-3,4-dihydro-2H-1,5 benzoxathiepine bromhydrate; LVEDP, left ventricular end-diastolic pressure.
Protective activity of F 15845 in two in vivo models of cardiac ischaemia
The protective effects of F 15845 were examined in a supply ischaemia model in anaesthetized rabbits subjected to a transient (5 min) coronary occlusion that produces a large increase in the amplitude of the ST segment of the ECG (Verscheure et al., 1995). F 15845 from 0.04 mg·kg−1 (i.v.) upwards, dose-dependently inhibited ischaemia-induced ST segment elevation with an ED50 of 0.05 mg·kg−1 (see also, Le Grand et al., 2008). In contrast to all available anti-ischaemic drugs, the ST segment was normalized without effects on arterial pressure and heart rate (Figure 6B), indicating that F 15845 exerts a potent, direct cardiac protective activity. F 15845, after i.v. injection, reaches maximal protective activity at 0.08 mg·kg−1 and this level of activity was maintained at higher doses (Le Grand et al., 2008). The conventional Nav1.5 blocker, lidocaine (2.5 mg·kg−1) produced only poor protective effects (27 ± 6% inhibition of ST segment elevation, n = 6, NS), indicating limited anti-anginal activities compared with F 15845.
Figure 6.
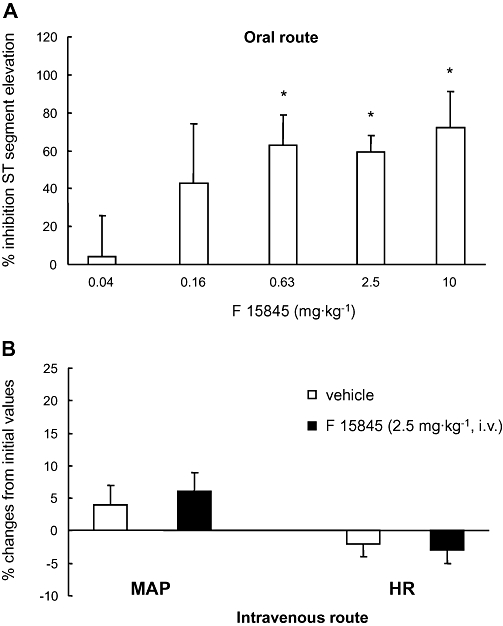
Cardio-protective effects of 3-(R)-[3-(2-methoxyphenylthio-2-(S)-methylpropyl]amino-3,4-dihydro-2H-1,5 benzoxathiepine bromhydrate (F 15845) in anaesthetized rabbits subjected to 5 min coronary artery occlusion, as assessed by inhibition of ischaemia-induced ST segment changes in the four-limb electrocardiogram. (A) Inhibition (%) of ST segment elevation following oral F 15845 (0.04–10 mg·kg−1) administered 2 h prior to coronary artery occlusion. (B) Mean maximal changes in mean arterial pressure (MAP, left side) and heart rate (HR, right side) induced by intravenous F 15845 (2.5 mg·kg−1) administered 5 min prior to coronary occlusion. Data are mean ± SEM. *P < 0.05 versus vehicle.
Likewise, oral administration of F 15845 counteracted ST segment changes from 0.04 mg·kg−1 upwards (ED50 0.13 mg·kg−1; Figure 6A) and this over a wide range of doses (Figure 6A). The narrow oral-to-intravenous ED50 ratio in rabbits suggests that F 15845 is orally bioavailable.
The protective effects of F 15845 were also investigated in a demand ischaemia model. In anaesthetized dogs having a coronary stenosis, and subjected to left atrial pacing for 3 min periods to induced epicardial ST segment changes (Sugiyama and Hashimoto, 1999), F 15845 dose-dependently inhibited ST segment changes at 0.16 mg·kg−1 bolus plus infusion of 0.16 mg·kg−1·h−1 (n = 6; P < 0.05) and at 0.63 mg·kg−1 bolus plus infusion of 0.63 mg·kg−1·h−1 (n = 5; P < 0.05; Figure 7A). Consistent with the data obtained in the rabbit model, there was no haemodynamic effect at protective doses, in contrast to the effects of the L-type calcium channel inhibitor diltiazem at 0.16 mg·kg−1 (Figure 7B,C). In addition, F 15845 was devoid of significant effects on the QT interval (212 ± 12 ms for baseline vs. 216 ± 11 ms after 90 min infusion with 0.63 mg·kg−1 F 15845, n = 5, NS).
Figure 7.
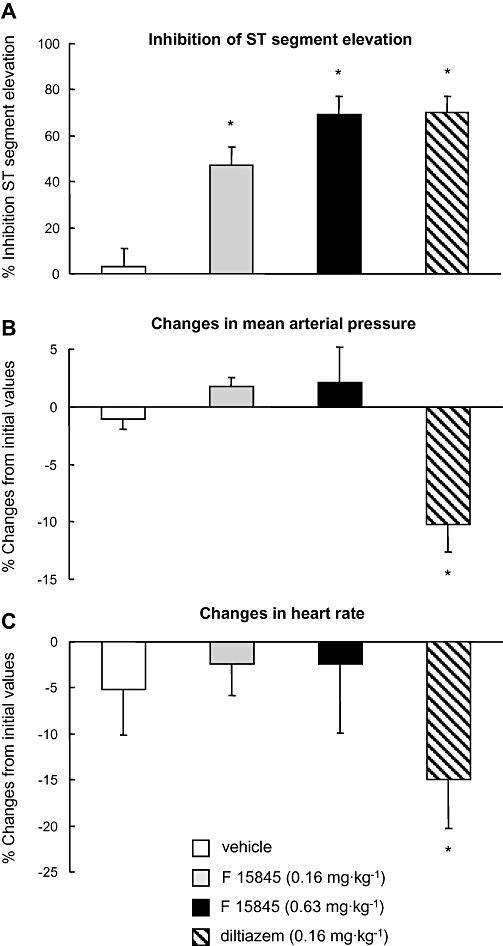
Anti-anginal activity of F 15845 in a canine model of demand-ischaemia-induced epicardial ST segment changes. Following the placement of a critical stenosis on a branch of the LAD coronary artery, heart rate was increased to 80–90 bpm over baseline by left atrial pacing and then allowed to recover for 30 min. Drugs were administered as i.v. bolus after the first period of pacing and infused throughout the subsequent three periods. (A) Maximal inhibition (%) of ST segment elevation obtained by F 15845 (0.16 and 0.63 mg·kg−1 i.v. bolus and infusion) and diltiazem (0.16 mg·kg−1 i.v. bolus and infusion). Note equivalent maximal anti-ischaemic effects of F 15845 and diltiazem. (B–C) Mean maximal changes in mean arterial pressure (B) and heart rate (C) induced by F 15845 and diltiazem. Note absence of effects of F 15845 at either dose, and marked effects of diltiazem. Data are mean ± SEM. *P < 0.05 vs. corresponding vehicle. F 15845 was given as a 0.16 mg·kg−1 i.v. bolus and 0.16 mg·kg−1·h−1 infusionor as a 0.63 mg·kg−1 i.v. bolus and 0.63 mg·kg−1·h−1 infusion. Similarly, diltiazem was given as a 0.16 mg·kg−1 i.v. bolus and 0.16 mg·kg−1·h−1 infusion. F 15845, 3-(R)-[3-(2-methoxyphenylthio-2-(S)-methylpropyl]amino-3,4-dihydro-2H-1,5 benzoxathiepine bromhydrate; LAD, left anterior descending.
Discussion
F 15845 is a selective, potent blocker of the persistent sodium current generated by the Nav1.5 channel isoform. This molecular mechanism underlies the in vitro and in vivo anti-ischaemic activities observed with F 15845.
Voltage-gated sodium channels are transiently activated on depolarization of the cardiac cell membrane. The INa flowing through these channels is responsible for the upstroke (phase 0) of the action potential. Depolarization of the cell membrane leads to a rapid increase of INa that lasts for a few milliseconds before sodium channels inactivate, i.e. change to a non-conducting state. Recovery of each channel from inactivation requires repolarization of the cell membrane and a change of the conformation of the channel from an inactivated to a resting closed state. Although most sodium channels are inactivated within a few milliseconds and remain closed and non-conducting throughout the plateau phase of the cardiac action potential, a small percentage of channels either do not close, or close and then reopen (Liu et al., 1992; Clancy et al., 2003). These channels may continue to open and close spontaneously during the action potential plateau for reasons that are not completely understood. These late channel openings allow a persistent current of sodium ions to enter myocardial cells throughout systole (Saint, 2006). This current has been referred to as late, sustained or persistent to distinguish it from the peak rapid or transient INa. Thus, although the amplitude of persistent INa is small, because it persists for hundreds of milliseconds, the influx of sodium by this mechanism may be substantial (Noble and Noble, 2006). The magnitude of persistent INa in canine mid-myocardial cells and Purkinje fibres is greater than that in either epicardial or endocardial myocytes (Noble and Noble, 2006). The duration of action potentials in mid-myocardial cells and Purkinje fibres is also longer than in other cardiac cells (Noble and Noble, 2006). A role for persistent INa as a contributor to prolongation of APD is suggested by observations showing that lidocaine and tetrodotoxin shorten APDs of Purkinje fibres and ventricular mid-myocardial myocytes (Wasserstrom and Salata, 1988; Le Grand et al., 1995; Le Grand et al., 2008). Therefore, F 15845, as a potent blocker of persistent sodium current, concentration-dependently shortened APD of rabbit Purkinje fibres (data not shown) whereas it was devoid of significant effect on APD from endocardial ventricular myocytes of guinea pig papillary muscle, even at the highest concentration.
In recent years, it has become clear that several experimental and pathological conditions can significantly increase the persistent component of the sodium channel current (Belardinelli et al., 2004b; Valdivia et al., 2005). These conditions include: (i) exposure of myocytes to peptides (e.g. the sea anemone toxin, ATX-II, Belardinelli et al., 2006), chemicals (e.g. veratridine, Pignier et al., 2007) and oxygen-derived free radicals that slow the rate of inactivation of the sodium channel (Saint, 2008); (ii) mutations in the sodium channel gene SCN5A that are associated with the long QT-3 syndrome (Bennett et al., 1995); and (iii) pathological states such as hypoxia, heart failure and post-myocardial infarction. It has been shown that hypoxia increases the amplitude of ‘persistent tetrodotoxin-sensitive sodium current’ (i.e. persistent INa) in rat ventricular myocytes (Saint, 2006). The hypoxia-induced increase of persistent INa was observed experimentally at both the whole cell and the single channel current levels (Saint, 2006). The mechanism of the hypoxia-induced increase of persistent INa has not been fully elucidated. Nevertheless, there is evidence that specific domains in an intracellular loop of the α-subunit of the sodium channel that take part in channel inactivation and are phosphorylated by protein kinase C (Vassilev et al., 1988).
Sodium influx through persistent INa appears then to be a major contributor to the rise of [Na+]i that is observed during ischaemia (Baetz et al., 2003) and hypoxia (Saint, 2006). Exposure of hearts to ischaemia is known to increase lysophosphatidylcholine, palmitoyl-L-carnitine and reactive oxygen species (such as hydrogen peroxide), and these substances are themselves reported to increase persistent INa (Undrovinas et al., 1992; Wu and Corr, 1994; Ward and Giles, 1997; Belardinelli et al., 2004b). The specific reduction of the rapid sodium current (e.g. by tetrodotoxin and lidocaine) has been shown to only partially reduce the rise in [Na+]i in rat ventricular myocytes and isolated hearts during hypoxia and ischaemia respectively (Haigney et al., 1994; Ju et al., 1996; Van Emous et al., 1997). Further, the effects of sodium channel blockers are associated with an improvement of contractile function and with reduction of the hypoxia/ischaemia-induced increase in [Ca2+]i. Although, persistent INa is more sensitive to block by tetrodotoxin than is peak INa, it is also reduced by lidocaine, mexiletine, R-56865, flecainide, amiodarone and ranolazine (Shryock and Belardinelli, 2008).
At pharmacologically active doses, F 15845 does not affect peak Na+ current in normal polarized myocytes. This feature clearly distinguishes F 15845 from conventional Nav1.5 blockers (i.e. class I anti-arrhythmics, local anesthetics and anti-convulsants). All these drugs markedly reduce peak Na+ current leading to negative dromotropic effects. We have shown that F 15845 selectively inhibits the persistent component of the Na+ current and that the activity of F 15845 on that particular component of the current is enhanced under depolarizing conditions, i.e. as exist during ischaemia. In sharp contrast, conventional Nav1.5 blockers are more effective on peak sodium current than on ischaemia-induced persistent sodium current. During ischaemia, the persistent, inward sodium current (mediated by Nav1.5) increases and that Na+ entry triggers a deleterious chain of events ending in cell injury or death. By selectively blocking the persistent Na+ current in depolarized myocytes, F 15845 can prevent or at least, delay Na+ overload and its consequences (Le Grand et al., 1995). Another distinctive feature of F 15845 arises from the nature of its interaction with Nav1.5 channel. The blockade of the persistent sodium current by F 15845 occurs in a defined and limited range of membrane potentials. Experimentally, we have demonstrated that inhibition of the persistent Na+ current is most effective under depolarizing conditions which, therefore, renders the compound ‘ischaemia-selective’ (Figs 5 and 8). In addition, because the persistent sodium current has no role in non-ischaemic cardiomyocytes, F 15845 is devoid of major effect in normal physiological conditions. To our knowledge, such an advantageous profile has not been found previously, even for ranolazine (Belardinelli et al., 2004a; Shryock and Belardinelli, 2008).
Figure 8.
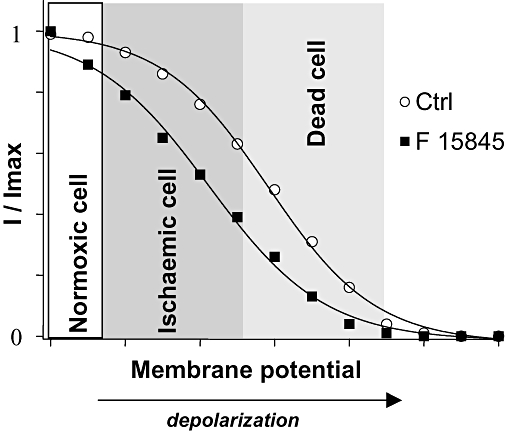
Ischaemia-selectivity of 3-(R)-[3-(2-methoxyphenylthio-2-(S)-methylpropyl]amino-3,4-dihydro-2H-1,5 benzoxathiepine bromhydrate (F 15845). The effect of F 15845 on steady-state inactivation parameters of sodium channels confers ischaemia-selectivity on F 15845, as explained in the cartoon. During ischaemia, resting membrane potential depolarizes as a response to metabolic changes modifying electrophysiological equilibrium. The effects of F 15845 on peak rapid sodium current in normoxic cells may be limited by the normally polarized resting membrane potential (white area) where only few sodium channels are in the inactivated state. In ischaemic cells (gray area), persistent sodium current appears and some sodium channels are in their inactivated state, but the persistent sodium channels are more readily blocked by F 15845, which therefore has a greater effect on sodium current. [Correction added after online publication 8 January 2008: in the above figure, erroneous breaks in the curves were corrected]
These in vitro observations were confirmed in models of myocardial ischaemia in anaesthetized animals. In a model of brief (5 min), reversible coronary artery occlusion-induced regional ischaemia (Verscheure et al., 1995), F 15845 produced potent, dose-dependent inhibition of ischaemia-induced ST segment elevation following acute i.v. (ED50 0.05 mg·kg−1) or oral (ED50 0.13 mg·kg−1) administration. Importantly, these effects occurred in the absence of haemodynamic changes. Interestingly, the human dose of 10 mg, as used in clinical trials, yields a plasma level of 1.1 µg·mL−1 (study currently ongoing) which is close to the active in vitro concentrations described in the present results and with the plasma concentration in the dog model (0.9 µg·mL−1), at the end of perfusion with the highest dose of 0.63 mg·kg−1. In humans as in dogs, the compound is highly bound (>99%) to plasma proteins;. Repeated myocardial ischaemia enhanced the protective activity of F 15845, which can be explained by prominent sodium channel blockade by F 15845 at depolarized (i.e. ischaemic) potentials. Protection by F 15845 against the ischaemia-induced effects lasted for more than 4 h, while non-significant activity was still detected 24 h post administration (data not shown). In comparison, the anti-anginal drugs, atenolol and diltiazem, also produced clear-cut protective activity following either i.v. or oral administration, but the activity was seen over a narrow dose-range, and was associated with haemodynamic changes. In the case of F 15845, the oral to i.v. ED50 ratio is compatible with good oral bioavability.
The protective effects of F 15845 are superior to other (non-antianginal) drugs investigated, as the sodium channel blocker, lidocaine and the Na+/H+ exchanger inhibitor, cariporide, produced little or no inhibition of ischaemia-induced ST segment elevation.
The potential anti-anginal activity of F 15845 was evaluated in a canine model of demand ischaemia-evoked ST segment changes (Sugiyama and Hashimoto, 1999). F 15845 dose-dependently inhibited ischaemia-induced ST segment elevation from 0.16 mg·kg−1 i.v. in the absence of cardiac or systemic haemodynamic effects. Equipotent inhibition of ischaemia-induced ST segment elevation was produced by diltiazem, but this was accompanied with marked cardiac and systemic haemodynamic effects, in accordance with previous findings (Grover and Parham, 1987; Allely and Alps, 1989).
Collectively, the present results demonstrate that F 15845 protected against the effects of cardiac ischaemia and preserved cardiac viability in established models of angina. These effects occurred in the absence of haemodynamic cardiac unloading, which represents a clear advantage of F 15845 over available anti-anginal agents, including the ‘new generation’ anti-anginal drug, ivabradine (Ruzyllo et al., 2007). F 15845 has high selectivity for cardiac voltage-gated sodium channels, versus voltage-gated K+, Ca2+ channels, and it does not interact with Na+–H+ or Na+–Ca2+ exchangers. Finally, it is devoid of pro-arrhythmic activity and has no effect on cardiomyocytes in non-ischaemic conditions.
We strongly believe that F 15845 represents a novel therapeutic opportunity for the oral prophylactic and symptomatic treatment of angina pectoris and for preserving myocardial viability during ischaemic situations.
Acknowledgments
We are indebted to our colleagues in the Division of Cardiovascular Diseases II and Medicinal Chemistry I for performing the experiments described in the present manuscript. A special thanks to the Department of Zootechnie for assistance with large animal husbandry, our colleagues at the Department of General Pharmacology (Campans) for carrying out the canine angina model studies and to A. Newman-Tancredi for English revision. We thank V. Bhaglou for secretarial assistance.
Glossary
Abbreviations:
- APD
action potential duration
- AUC
area under the curve
- CF
coronary flow
- F 15845
3-(R)-[3-(2-methoxyphenylthio-2-(S)-methylpropyl]amino-3,4-dihydro-2H-1,5 benzoxathiepine bromhydrate
- HEK
human embryonic kidney
- LVEDP
left ventricular end-diastolic pressure; ST interval duration between the wave S and T of the four limbs ECG
Conflict of interest
The authors declare that they have no competing financial interests.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl.)(2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allely MC, Alps BJ. A comparison of the effects of a series of anti-anginal agents in a novel canine model of transient myocardial ischaemia. Br J Pharmacol. 1989;96(4):977–985. doi: 10.1111/j.1476-5381.1989.tb11910.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baetz D, Bernard M, Pinet C, Tamareille S, Chattou S, El Banani H, et al. Different pathways for sodium entry in cardiac cells during ischaemia and early reperfusion. Mol Cell Biochem. 2003;242:115–120. [PubMed] [Google Scholar]
- Belardinelli L, Antzelevitch C, Fraser H. Inhibition of late (sustained/persistent) sodium current: a potential drug target to reduce intracellular sodium-dependent calcium overload and its detrimental effects on cardiomyocyte function. Eur Heart J. 2004a;6:13–17. [Google Scholar]
- Belardinelli L, Shryock JC, Fraser H. The mechanism of ranolazine action to reduce ischaemia-induced diastolic dysfunction. Eur Heart J. 2004b;8(Suppl.A):A10–A13. [Google Scholar]
- Belardinelli L, Shryock JC, Fraser H. Inhibition of the late sodium current as potential cardioprotective principle: effects of the late sodium current inhibitor ranolazine. Heart. 2006;92:iv6–iv14. doi: 10.1136/hrt.2005.078790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett PB, Yazawa K, Makita N, George AL. Molecular mechanism for an inherited arrhythmia. Nature. 1995;376:683–685. doi: 10.1038/376683a0. [DOI] [PubMed] [Google Scholar]
- Bhatt AB, Stone PH. Current strategies for the prevention of angina in patients with stable coronary artery disease. Curr Opin Cardiol. 2006;21:492–502. doi: 10.1097/01.hco.0000240588.22086.43. [DOI] [PubMed] [Google Scholar]
- Clancy CE, Tateyama M, Liu H, Wehrens XH, Kass RH. Non-equilibrium gating in cardiac Na+ channels: an original mechanism of arrhythmia. Circulation. 2003;107:2233–2237. doi: 10.1161/01.CIR.0000069273.51375.BD. [DOI] [PubMed] [Google Scholar]
- Gellens M, George A, Chen L, Chahine M, Horn R, Bardin R, et al. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc Natl Acad Sci USA. 1992;89:554–558. doi: 10.1073/pnas.89.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin AL, Barchi RL, Caldwell JH, Hofmann F, Howe JR, Hunter JC, et al. Wood JN, Catterall WA. Nomenclature of voltage-gated sodium channels. Neuron. 2000;28(2):365–368. doi: 10.1016/s0896-6273(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Grover GJ, Parham CS. The effect of diltiazem on ST-segment elevation and myocardial blood flow distribution during pacing-induced ischaemia. Eur J Pharmacol. 1987;143(1):109–117. doi: 10.1016/0014-2999(87)90740-0. [DOI] [PubMed] [Google Scholar]
- Haigney MCP, Lakatta EG, Stern MD, Silverman HS. Sodium channel blockade reduces hypoxic sodium loading and sodium-dependent calcium loading. Circulation. 1994;90:391–399. doi: 10.1161/01.cir.90.1.391. [DOI] [PubMed] [Google Scholar]
- Houser SR. Can novel therapies for arrhythmias caused by spontaneous sarcoplasmic reticulum Ca2+ release be developed by using mouse model? Circ Res. 2005;96:1031–1032. doi: 10.1161/01.RES.0000168921.55931.63. [DOI] [PubMed] [Google Scholar]
- Ju YK, Saint DA, Gage PW. Hypoxia increases persistent sodium current in rat ventricular myocytes. J Physiol. 1996;497:337–347. doi: 10.1113/jphysiol.1996.sp021772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Grand B, Vié B, Talmant JM, Coraboeuf E, John GW. Alleviation of contractile dysfunction in ischaemic hearts by slowly inactivating Na+ current blockers. Am J Physiol. 1995;269:H533–H540. doi: 10.1152/ajpheart.1995.269.2.H533. [DOI] [PubMed] [Google Scholar]
- Le Grand B, Pignier C, Létienne R, Cuisiat F, Rolland F, Mas A, et al. Sodium late current blockers in ischemia reperfusion: is the bullet magic? J Med Chem. 2008;51:3856–3866. doi: 10.1021/jm800100z. [DOI] [PubMed] [Google Scholar]
- Liu Y, DeFelice U, Mazzanti M. Na channels that remain open throughout the cardiac action potential plateau. Biophys J. 1992;63:654–662. doi: 10.1016/S0006-3495(92)81635-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy E, Cross H, Steenbergen C. Sodium regulation during ischaemia versus reperfusion and its role in injury. Circ Res. 1999;84:1401–1406. doi: 10.1161/01.res.84.12.1469. [DOI] [PubMed] [Google Scholar]
- Noble D, Noble PJ. Late sodium current in the pathophysiology of cardiovascular disease: consequences of sodium-calcium overload. Heart. 2006;92:V1–V5. doi: 10.1136/hrt.2005.078782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignier C, Revenaz C, Rauly-Lestienne I, Cussac D, Delhon A, Gardette J, et al. Direct protective effects of poly-unsaturated fatty acids, DHA and EPA, against activation of cardiac late sodium current: a mechanism for ischaemia selectivity. Basic Res Cardiol. 2007;102:553–564. doi: 10.1007/s00395-007-0676-x. [DOI] [PubMed] [Google Scholar]
- Ruzyllo W, Tendera M, Ford I, Fox KM. Antianginal efficacy and safety of ivabradine compared with amlodipine in patients with stable effort angina pectoris: a 3-month randomized, double-blind, multicentre, noninferiority trial. Drugs. 2007;67:393–405. doi: 10.2165/00003495-200767030-00005. [DOI] [PubMed] [Google Scholar]
- Saint DA. The role of the persistent Na+ current during cardiac ischaemia and hypoxia. J Cardiovasc Electrophysiol. 2006;17:S96–S103. doi: 10.1111/j.1540-8167.2006.00390.x. [DOI] [PubMed] [Google Scholar]
- Saint DA. The cardiac persistent sodium current: an appealing therapeutic target? Br J Pharmacol. 2008;153:1133–1142. doi: 10.1038/sj.bjp.0707492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakmannn BFAS, Spindler AJ, Bryant SM, Linz KW, Noble D. Distribution of a persistent sodium current across the ventricular wall in guinea pigs. Circ Res. 2000;87:910–914. doi: 10.1161/01.res.87.10.910. [DOI] [PubMed] [Google Scholar]
- Shryock JC, Belardinelli L. Inhibition of late sodium current to reduce electrical and mechanical dysfunction of ischaemic myocardium. Br J Pharmacol. 2008;153:1128–1132. doi: 10.1038/sj.bjp.0707522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman HS, Stern MD. Ionic basis of ischaemic cardiac injury: insights from cellular studies. Cardiovasc Res. 1994;28:581–567. doi: 10.1093/cvr/28.5.581. [DOI] [PubMed] [Google Scholar]
- Song Y, Shryock JC, Wu L, Belardinelli L. Antagonism by ranolazine of the pro-arrhythmic effects of increasing late INa in guinea pig ventricular myocytes. J Cardiovasc Pharmacol. 2004;44:191–199. doi: 10.1097/00005344-200408000-00008. [DOI] [PubMed] [Google Scholar]
- Sugiyama A, Hashimoto K. Antiischaemic effects of CP-060S, an inhibitor of pathologically modified sodium channels, assessed in the canine experimental model of angina pectoris. J Cardiovasc Pharmacol. 1999;33(1):70–77. doi: 10.1097/00005344-199901000-00011. [DOI] [PubMed] [Google Scholar]
- Undrovinas AI, Fleidervish IA, Makielski JC. Inward sodium current at resting potentials in single cardiac myocytes induced by the ischaemic metabolite lysophosphatidylcholine. Circ Res. 1992;71:1231–1241. doi: 10.1161/01.res.71.5.1231. [DOI] [PubMed] [Google Scholar]
- Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, et al. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. 2005;38:475–483. doi: 10.1016/j.yjmcc.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Van Emous JG, Nederhoff MG, Ruigrok TJ, Van Echteld CJ. The role of the Na+ channel in the accumulation of intracellular Na+ during myocardial ischaemia: consequence for post-ischaemic recovery. J Mol Cell Cardiol. 1997;29:85–96. doi: 10.1006/jmcc.1996.0254. [DOI] [PubMed] [Google Scholar]
- Vassilev PM, Scheuer T, Catterall WA. Identification of an intracellular peptide segment involved in sodium channel inactivation. Science. 1988;241:1658–1661. doi: 10.1126/science.241.4873.1658. [DOI] [PubMed] [Google Scholar]
- Ver Donck L, Borgers M, Verdonck F. Inhibition of sodium and calcium overload pathology in the myocardium: a new cytoprotective principle. Cardiovasc Res. 1993;27:349–357. doi: 10.1093/cvr/27.3.349. [DOI] [PubMed] [Google Scholar]
- Verscheure Y, Pouget G, De Courtois F, Le Grand B, John GW. Attenuation by R 56865, a novel cytoprotective drug of regional myocardial ischaemia- and reperfusion-induced electrocardiographic disturbances in anaesthetized rabbits. J Cardiovasc Pharmacol. 1995;25:126–133. doi: 10.1097/00005344-199501000-00020. [DOI] [PubMed] [Google Scholar]
- Ward CA, Giles WR. Ionic mechanism of the effects of hydrogen peroxide in rat ventricular myocytes. J Physiol. 1997;500:631–642. doi: 10.1113/jphysiol.1997.sp022048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserstrom JA, Salata JJ. Basis for tetrodotoxin and lidocaine effects on action potentials in dog ventricular myocytes. Am J Physiol. 1988;254:H1157–H1166. doi: 10.1152/ajpheart.1988.254.6.H1157. [DOI] [PubMed] [Google Scholar]
- Wu J, Corr PB. Palmitoyl carnitine modifies sodium currents and induces transient inward current in ventricular myocytes. Am J Physiol. 1994;266:H1034–H1046. doi: 10.1152/ajpheart.1994.266.3.H1034. [DOI] [PubMed] [Google Scholar]