

Abstract
Dysregulated innate immune responses to commensal bacteria contribute to the development of inflammatory bowel disease (IBD). TLR4 is overexpressed in the intestinal mucosa of IBD patients and may contribute to uncontrolled inflammation. However, TLR4 is also an important mediator of intestinal repair. The aim of this study is to examine the effect of a TLR4 antagonist on inflammation and intestinal repair in two murine models of IBD. Colitis was induced in C57BL/6J mice with dextran sodium sulfate (DSS) or by transferring CD45Rbhi T cells into RAG1−/− mice. An antibody (Ab) against the TLR4/MD-2 complex or isotype control Ab was administered intraperitoneally during DSS treatment, recovery from DSS colitis, or induction of colitis in RAG1−/− mice. Colitis severity was assessed by disease activity index (DAI) and histology. The effect of the Ab on the inflammatory infiltrate was determined by cell isolation and immunohistochemistry. Mucosal expression of inflammatory mediators was analyzed by real-time PCR and ELISA. Blocking TLR4 at the beginning of DSS administration delayed the development of colitis with significantly lower DAI scores. Anti-TLR4 Ab treatment decreased macrophage and dendritic cell infiltrate and reduced mucosal expression of CCL2, CCL20, TNF-α, and IL-6. Anti-TLR4 Ab treatment during recovery from DSS colitis resulted in defective mucosal healing with lower expression of COX-2, PGE2, and amphiregulin. In contrast, TLR4 blockade had minimal efficacy in ameliorating inflammation in the adoptive transfer model of chronic colitis. Our findings suggest that anti-TLR4 therapy may decrease inflammation in IBD but may also interfere with colonic mucosal healing.
Keywords: innate immunity, inflammatory bowel disease, antigen presenting cells
inflammatory bowel disease (IBD) is characterized by recurrent or chronic inflammation of the intestinal tract in the absence of a specific inciting pathogen (55, 79). Most people do not develop IBD despite the myriad microorganisms that populate the gastrointestinal tract (18, 47). Indeed, intestinal homeostasis is dependent on the ability of the mucosal immune system to tolerate the normal commensal flora (7, 9, 40). However, patients with IBD appear to lose this tolerance and mount an immune response against resident intestinal bacteria (17, 43). Experimental mouse models of IBD have demonstrated that resident luminal bacteria are necessary for the induction and perpetuation of intestinal inflammation since genetically susceptible mice are protected from colitis when raised in germ-free conditions (19). The contribution of bacteria to the pathogenesis of IBD has also been demonstrated clinically since antibiotics have some efficacy in treating Crohn's colitis as well as pouchitis suggesting that decreased bacterial load may ameliorate inflammation (54).
The importance of dysregulated immune responses to bacteria is further supported by the fact that many of the genes associated with IBD encode molecules that play a role in innate immunity, most notably the nucleotide-binding oligomerization domain 2 (NOD-2) gene (7, 79). In addition, the Toll-like receptor 4 (TLR4) gene region and specific TLR4 polymorphisms, Asp299gly and Thr399Ile, have been associated with susceptibility to IBD although the functional significance of these polymorphisms has not been clearly established (21, 50, 72). This evidence suggests that abnormal innate immune responses to commensal bacteria are critical in the initiation and maintenance of the uncontrolled inflammation seen in IBD.
TLRs play a central role in mucosal innate immune regulation and may play a key role in the pathogenesis of IBD. TLRs are evolutionarily conserved transmembrane receptors that recognize pathogen-associated molecular patterns and are key initiators of the immune response. Their activation leads to the production of cytokines, chemokines, and antimicrobial molecules important in the initial innate immune response and priming of antigen specific adaptive immunity. TLR4 recognizes lipopolysaccharide (LPS), which is present in the cell wall of gram-negative bacteria. Interaction of LPS with TLR4 and its coreceptor MD-2 triggers signaling cascades mediated by MyD88-dependent and -independent pathways that lead to the translocation of the transcription factor nuclear factor kappa B (NF-κB) and the production of proinflammatory cytokines and proteins (45, 52).
TLR4 signaling is important in the recruitment of inflammatory cells and the production of inflammatory cytokines in the intestine (29, 41). Our previous studies with TLR4-deficient mice (TLR4−/−) demonstrated that in the dextran sodium sulfate (DSS) model of colitis, TLR4−/− mice have a decreased inflammatory infiltrate in the lamina propria (24, 26). However, TLR4−/− mice also demonstrate defects in mucosal repair in response to DSS-induced injury with decreased epithelial proliferation and increased rectal bleeding, suggesting that TLR4 signaling serves a dual role in the gut as a mediator of both inflammation and mucosal repair (24, 58). TLR4 is normally expressed throughout the intestine at low levels both in the epithelium and in lamina propria mononuclear cells but is increased in IBD (36, 69). Studies have demonstrated that TLR4 is present on epithelial cells in the human colon and that its expression is largely confined to cells in the crypt and is not present on cells lining the lumen (9, 28). TLR4 is also weakly expressed on intestinal dendritic cells and macrophages in noninflamed tissue (35, 64). We have also shown that TLR4 is expressed on CD4+ T cells (23). TLR4 may contribute to the development of colitis in mouse models since its expression is increased in the colon of DSS-treated mice (48, 51). These findings suggest that increased TLR4 expression may contribute to the initiation or perpetuation of intestinal inflammation and may be a potential therapeutic target in the treatment of IBD.
Increased understanding of the pathogenesis of IBD has identified potential targets for immunotherapy. Biologics, such as infliximab, target specific inflammatory mediators but have many side effects. Aside from antibiotics, no therapeutic interventions have focused on the dysregulated interactions between the host and commensal bacteria in IBD patients. In the present study, we sought to investigate the clinical effect of TLR4 blockade in two murine models of colitis. DSS-induced colitis is a useful model for investigating the contributions of TLR4 signaling to innate immune responses since it is not dependent on T or B cells for the induction of inflammation and it is ameliorated by antibiotics (15, 27, 34). Although we and others have previously used TLR4−/− mice in the DSS model, there always exists a question about the developmental effects on the immune system of a complete knockout vs. temporarily dampening of TLR4 signaling. Although TLR4 is thought chiefly to contribute to innate immune responses, we and others have previously demonstrated that T cells express TLRs, including TLR4, and that TLR signaling in T cells may be an important contributor to intestinal inflammation (10, 23, 37, 80). We therefore also investigated the effect of TLR4 blockade in the adoptive transfer model of colitis, which results in chronic colitis and wasting disease mediated by pathological T cell activation.
Since TLR4 serves a dual role as a mediator of both intestinal inflammation and repair, our study had two aims. The first aim was to examine how TLR4 blockade during DSS-induced colitis affects inflammation and clinical colitis severity. The second aim investigated how TLR4 inhibition might affect mucosal healing by administering the antibody during recovery from DSS colitis. Our findings indicate that TLR4 blockade during DSS treatment ameliorates colitis severity by decreasing the recruitment of antigen presenting cells (APCs) to the lamina propria with reduced expression of monocyte-attracting chemokines and proinflammatory cytokines. In contrast, TLR4 blockade during recovery from DSS colitis impairs mucosal healing and epithelial proliferation with decreased colonic expression of COX-2, PGE2, and amphiregulin. Additionally, TLR4 blockade appears to have minimal efficacy in ameliorating inflammation in the transfer model of T cell-mediated chronic colitis, suggesting that TLR4 is more important in modulating acute inflammation than in maintaining chronic colitis. Our findings suggest that anti-TLR4 therapy may decrease inflammation in IBD but may also interfere with mucosal healing.
MATERIALS AND METHODS
Mice and interventions.
C57BL/6J mice were obtained from Jackson Laboratories (Bar Harbor, ME) and kept in specific pathogen-free conditions and fed by free access to a standard diet and water. Eight- to 12-wk-old sex-matched mice were given 2.5% DSS (molecular weight, 36–50 kDa, ICN, Aurora, OH) in their drinking water and were euthanized at the end of 7 days of DSS treatment. For recovery studies, DSS was administered for the first 7 days as indicated. DSS was then removed from the drinking water and mice were euthanized 7 days after cessation of DSS treatment.
T cell adoptive transfer colitis was created by injecting CD4+CD62L+ cells to RAG1−/− mice obtained from Jackson Laboratories. Spleen and mesenteric lymph nodes (MLNs) were taken from C57BL/6J mice. Single cell suspensions from the spleen and MLNs were prepared to isolate CD4+CD62L+ T cells by using the MACS magnetic separation system (Miltenyi Biotec, Auburn, CA). Over 98% of the purified cells were CD45Rbhigh populations, confirmed by flow cytometry (data not shown). Isolated cells were resuspended in sterile PBS, and 5 × 105 CD4+CD62L+ cells were injected intraperitoneally (ip) into each RAG1−/− mouse (3, 56). Mice were euthanized 5 wk after T cell transfer. All experiments were performed according to the Mount Sinai School of Medicine Animal Experimental Ethics Committee guidelines and were approved by the Mount Sinai Institutional Animal Care and Use Committee.
A rat IgG2b monoclonal antibody toward the mouse TLR4/MD-2 complex called 1A6 or an isotype-matched control was injected ip at a dose of 20 mg/kg at the given time points (Fig. 1A, Fig. 5A, and Fig. 7A). Monoclonal antibody, 1A6, targets the TLR4/MD-2 complex and has previously been described as being protective in a mouse model of sepsis (67). To generate 1A6, male Wistar rats were immunized three times every second week by subcutaneous application of 1 × 106 CHO cells overexpressing TLR4 and MD-2 suspended in monophosphoryl-lipid A and trehalose dicorynomycolate adjuvant (RIBI, Sigma, St. Louis, MO). Two weeks following final immunization with transfected CHO cells, rats were challenged subcutaneously with 10 μg TLR4/MD-2 fusion protein in RIBI. Recombinant flag-tagged TLR4/MD-2 encompassing the TLR4 extracellular domain (aa1 to aa 629) fused to MD-2 (aa 19 to aa 170) via a peptide linker (GGGGSGGGGSGGGGS, cloning vector pFastBac1, Invitrogen, Zurich, Switzerland) was purified from lysates of transduced SF9 cells by use of an anti-flag M2 affinity matrix (Sigma). Three days after final challenge, lymph node cells were fused with Sp2/0 myeloma cells. Hybridoma supernatants were screened for binding to TLR4/MD-2 CHO cells. Positive hybridomas were subcloned twice and monoclonal antibodies were purified from hybridoma supernatants via a protein G affinity column (GE Healthcare, Zurich, Switzerland). Antibodies were subsequently characterized for LPS blocking activity in RAW 264.7 cells and HEK293 cells overexpressing murine TLR4 and MD-2 (67). To ensure the reproducibility of our findings, we also employed a monoclonal mouse IgG1 antibody directed against the mouse TLR4/MD-2 complex. The data generated in parallel were similar (data not shown).
Fig. 1.

TLR4 blockade ameliorates DSS-induced colitis. A: treatment schedule for anti-TLR4 antibody and isotype control during 2.5% DSS administration. Mice were injected intraperitoneally (ip) at the indicated time points. B: mice treated with anti-TLR4 antibody had reduced severity of colitis as measured by disease activity index (DAI). Data represent means ± SE of 3 independent experiments with a total of 24 mice (n = 15 for anti-TLR4 and n = 9 for isotype control-treated mice). *P < 0.05. C: bacterial translocation as measured by culture of mesenteric lymph nodes (MLNs) on indicated media from anti-TLR4-treated (n = 4) and isotype-treated (n = 4) mice. Data represent means ± SE of 2 independent experiments. CFUs, colony-forming units; TSA, Trypticase soy agar; ANA, anaerobic with PEA; NS, not significant.
Assessment of colitis activity.
DSS-treated mice were monitored daily for body weight, stool consistency, and stool blood with disease activity index determined as previously described (12). Transfer model mice were monitored weekly for body weight and wasting disease. At euthanasia, the cecum and proximal and distal halves of the colon were removed and fixed in 10% buffered formalin, paraffin-embedded, sectioned, and stained with hematoxylin and eosin (H&E). Histological assessment was performed by a pathologist blinded to the treatment. Histological severity of acute colitis of DSS-treated mice was determined using a combined score of crypt damage (0–4), polymorphonuclear neutrophil (PMN) infiltrate (0–3), edema (0–3), erosion/ulceration (0–3), and epithelial regeneration (0–3). Histological score for transfer model mice was a combined score of crypt damage (0–4), acute inflammatory cell infiltrate (0–4), edema (0–4), erosion/ulceration (0–4), chronic inflammatory cell infiltrate (0–2), epithelial regeneration (0–3), and crypt distortion/branching (0–3).
Isolation of lamina propria cells and flow cytometry.
Colons were removed, washed in PBS with 5% penicillin- streptomycin, cut into small pieces, and incubated with Ca2+- and Mg2+-free HBSS containing 0.002 mol/l EDTA for 12 min with gentle agitation to remove epithelial cells. Tissue pieces were then incubated while being shaken in complete medium containing 5% FCS, 0.02 mol/l HEPES, l-glutamine, 5% penicillin and streptomycin with 1 mg/ml collagenase, and 1.5 mg/ml dispase in RPMI 1640 at 37°C for 60 min. The supernatant was centrifuged and the pellet was washed two times with RPMI 1640 containing 5% penicillin and streptomycin. Lamina propria cells were isolated by lymphocyte separation medium (Mediatech, Manassas, VA) density gradient centrifugation (800 g for 20 min) and collected at the interface. Dendritic cells were then isolated from lamina propria cells by positive selection magnetic sorting using anti-CD11c MACS beads (Miltenyi Biotec). Isolated CD11c+ cells were counted and stained with anti-mouse APC-Cy7-CD45, FITC-CD11b, APC-CD8, and PE-CD103 (eBioscience, San Diego, CA) for 30 min at 4°C. The purity of isolated CD11c+ dendritic cells was greater than 98% by FACS analysis. Cells were washed in PBS and analyzed by flow cytometry using FACSDiva software (BD Biosciences, San Jose, CA).
Immunofluorescent studies.
Lamina propria macrophages were identified by CD68 staining. Paraffin embedded sections were incubated with 0.1% trypsin (Sigma) CaCl2 dissolved in 0.05 M Tris·HCl, pH 7.6 for 15 min at 37°C. Subsequently sections were blocked in 5% skim milk for 1 h and then incubated with rat anti-CD68 antibody (1:50, MCA1957S, Serotec, Raleigh, NC) overnight at 4°C. After being washed in PBS, sections were incubated with TRITC-conjugated rabbit anti-rat IgG (1:200, Sigma) for 1 h at room temperature. Slides were viewed on a Nikon eclipse E600 immunofluorescence microscope (Nikon, Melville, NY) and photographs were taken with a digital camera using Spot Advanced software program (Diagnostic Instruments, Sterling Heights, MI) and imported into Adobe Photoshop software (Adobe, San Jose, CA) for analysis. The number of CD68-positive cells infiltrating the lamina propria was counted at a magnification of ×400.
Real-time PCR.
One milligram of total RNA isolated with RNA Bee (Tel-Test, Friendwood, TX) was used as the template for single-strand cDNA synthesis utilizing the Transcriptor First Strand cDNA synthesis kit (Roche, Indianapolis, IN) according to the manufacturer's instructions. Quantitative real-time PCR was performed for CCL2, CCL20, CX3CL1, COX-2, amphiregulin, and β-actin SYBR Premix Ex Taq (Takara Bio, Shiga, Japan) with the standard SYBR Green setting of 7900HT.
The primers used in this study are as follows (5′ to 3′ direction), for mouse CCL2: sense primer, GCT GGA GCA TCC ACG TGT T, antisense primer ATC TTG CTG GTG AAT GAG TAG CA; for mouse CCL20: sense primer, GGT GGC AAG CGT CTG CTC, antisense primer, GCC TGG CTG CAG AGG TGA; for mouse CX3CL1: sense primer, CAG CAG TGA CCG GAT CAT CTC, antisense primer, TGC TCT GAG GCT TAG CCG TAA; for mouse COX-2: sense primer, AAG GAA CTC AGC ACT GCA TCC, antisense primer, ACA GGG ATT GGA ACA GCA AGG A; for mouse amphiregulin: sense primer, TGT CAC TAT CTT TGT CTC TGC CAT, antisense primer, AGC CTC CTT CTT TCT TCT GTT TCT; for mouse β-actin: sense primer, ATG ACC CAG ATC ATG TTT G, antisense primer, TAC GAC CAG AGG CAT ACA. The amplification results were analyzed by use of SDS 2.2.1 software (Applied Biosystems, Foster City, CA) and the gene of interest was normalized to the corresponding β-actin results.
Enzyme-linked immunosorbent assay.
To conduct ex vivo colonic tissue cultures, colonic samples were washed in cold PBS containing penicillin, streptomycin, and Fungizone (100 U/ml each), and 100 mg of tissue fragments from each part of the colon were cultured in 12-well flat-bottom plates in 1 ml of serum-free RPMI 1640 at 37°C. Supernatants were harvested after 24 h. Enzyme-linked immunosorbent assays were performed to measure TNF-α (eBioscience), IL-6, and IFN-γ (BD Bioscience) per the manufacturer's instructions.
Assessment of epithelial cell proliferation.
Colonic tissue sections were examined for cell proliferation by bromodeoxyuridine (BrdU) labeling. Mice were injected with 120 mg/kg of BrdU (Sigma) ip, 90 min prior to euthanasia, and colonic tissues was stained for BrdU by use of a BrdU staining kit (Zymed Laboratories, South San Francisco, CA) according to the manufacturer's instructions. The number of BrdU-positive cells per well-oriented crypt was calculated in every three crypts for each colon segment at high magnification (×400) under light microscopy.
Measurement of PGE2.
Production of PGE2 in the tissue culture supernatant was determined by using a monoclonal EIA kit (Cayman, Ann Arbor, MI) according to the manufacturer's instructions and as described previously (21). Briefly, colonic samples were washed in cold PBS containing penicillin, streptomycin, and Fungizone (100 U/ml each), and 100 mg of tissue fragments from each part of the colon was cultured for 24 h in 12-well flat-bottom plates in serum free RPMI 1640. Culture supernatants were harvested for PGE2 measurement.
Bacterial cultures.
MLNs were removed under sterile conditions. Samples were weighed and prepared using a sterile grinder in 1 ml of sterile PBS, and then plated onto Trypticase soy agar with 5% sheep blood (TSA II), and MacConkey agar (BBL Becton Dickinson, Franklin Lakes, NJ). Anaerobic culture was performed using the CDC ANA BLD W/PEA plate and GasPak EZ anaerobe container system (BBL Becton Dickinson). Cultures were incubated at 37°C and examined at 24-h intervals for 3 days. Any bacterial growth was quantified and identified using The Dade MicroScan System (Dade MicroScan, West Sacramento, CA).
Statistical analysis.
Data were presented as mean values ± SE. The significance was analyzed by Student's t-test. P values were considered significant when <0.05.
RESULTS
Blocking TLR4 ameliorates acute DSS-induced colitis.
Innate immune signaling may contribute to the initiation and perpetuation of colitis. TLR4 expression is increased in mouse intestinal epithelial cells and lamina propria cells during DSS-induced colitis (51). We reasoned that TLR4 might be critical for the induction of acute inflammation. To address this question, we used a DSS model of colitis. In this model, we have observed development of clinical symptoms within 1 day of DSS treatment. We therefore looked at the effect of early administration of a blocking antibody on the earliest signs of colitis (Fig. 1A). At 1 and 2 days after induction of colitis, antagonizing TLR4 reduced the signs of colitis (Fig. 1B). Anti-TLR4-treated mice had less rectal bleeding and increased stool consistency compared with isotype-treated mice. There was no short-term difference in body weight during DSS treatment when comparing the two groups. At day 7 of DSS, there was a modest improvement in clinical signs of colitis in anti-TLR4-treated mice. These data suggest that blocking TLR4 improves the acute clinical manifestations of colitis.
We previously demonstrated that TLR4−/− mice have increased bacterial translocation to MLNs compared with wild-type (WT) mice (26). Given these data, we asked whether blocking TLR4 increased bacterial translocation in DSS-treated mice. MLN were cultured after 7 days of DSS. Although anti-TLR4 antibody-treated mice had increased gram-negative and anaerobic bacterial growth compared with isotype control-treated mice, these differences did not reach statistical significance (Fig. 1C). Bacteria isolated in both groups included Escherichia coli and Proteus mirabilis. It is important to note that the average number of colony-forming units cultured in each group was minimal (less than 103). These data demonstrate that antagonizing TLR4 did not increase bacterial translocation in the setting of colitis.
TLR4 blockade reduces proinflammatory cytokines.
TLR4 signaling results in the production of proinflammatory cytokines that are increased in the intestinal mucosa of patients with IBD, including TNF-α, IL-6, and IFN-γ (53). These cytokines are also upregulated during DSS colitis (15, 49). We sought to determine whether TLR4 blockade attenuates DSS-induced colitis by inhibiting proinflammatory cytokine production. ELISAs were used to measure the levels of TNF-α, IL-6, and IFN-γ in colon culture supernatants collected at day 7 of DSS treatment. Mice treated with anti-TLR4 antibody had decreased colonic production of TNF-α and IL-6 compared with controls (Fig. 2). Levels of TNF-α present in supernatants harvested from colon cultures were significantly lower in anti-TLR4 antibody-treated mice (141.5 ± 16.3 pg/ml vs. 336 ± 53.8 pg/ml, P < 0.01). A more modest but significant reduction in IL-6 was also seen with TLR4 blockade (4,816 ± 145.5 pg/ml vs. 5,850.4 ± 144.4 pg/ml, P < 0.01). In contrast, no significant difference was seen in the production of IFN-γ (894.2 ± 120.5 pg/ml vs. 973.8 ± 587.3, P = 0.34). These results suggest that TLR4 signaling contributes to the production of TNF-α and IL-6 but less so to IFN-γ levels during DSS-induced colitis.
Fig. 2.

Inhibition of TLR4 decreases production of proinflammatory cytokines. Mucosal production of cytokines at day 7 of DSS treatment was measured by ELISA using colon culture supernatants. 100 mg of tissue fragments from each part of the colon were cultured for 24 h in 1 ml of serum-free RPMI 1640 from anti-TLR4-treated (n = 9) and isotype-treated (n = 8) mice. Data represent mean values ± SE of supernatants measured in duplicate wells from 3 independent experiments. *P < 0.05.
Blockade of TLR4 signaling decreases the recruitment of APCs to the colon.
TLR4 signaling is a key mediator in the recruitment of inflammatory cells to the colon (24, 26, 41). DSS colitis is characterized by marked inflammation and ulceration of the colonic mucosa (12). We therefore evaluated the effect of anti-TLR4 antibody on the histology of acute colitis in H&E sections. On day 2 of DSS treatment, anti-TLR4 antibody-treated mice had no evidence of histological damage whereas animals treated with the isotype control antibody had some crypt damage and PMN infiltrate (total score: 0 vs. 0.17 ± 0.09, P = 0.08, maximum score: 16). However, by day 7 there was no difference in the colitis severity on histology. Anti-TLR4- and isotype-treated mice also had similar numbers of PMNs per high-power field (data not shown).
To understand the mechanism for the protective effect of TLR4 blockade in DSS colitis, we looked more closely at specific cell types in the colon. Our laboratory has found that TLR4−/− mice have impaired recruitment of dendritic cells (unpublished observations) and previously demonstrated that TLR4−/− mice have decreased recruitment of macrophages to the lamina propria (24, 26). Furthermore, transgenic mice that overexpress TLR4 in the intestinal epithelium have increased recruitment of dendritic cells to the lamina propria (62). We hypothesized on the basis of our previous data that inhibiting TLR4 would decrease recruitment of dendritic cells and macrophages. To answer this question, lamina propria dendritic cell and macrophages were evaluated using magnetic sorting for CD11c+ cells and immunofluorescence for CD68, respectively.
Dendritic cell infiltrate in the lamina propria was determined at day 2 and day 7 of DSS administration in mice injected with anti-TLR4 antibody or isotype control (Table 1). The total number of lamina propria cells and dendritic cells isolated from the colon increased from day 2 to day 7 of DSS colitis, suggesting increasing levels of inflammation with continued DSS treatment. TLR4 blockade significantly reduced the percentage of dendritic cells present in the lamina propria at day 2 when comparing antibody- and isotype-treated mice (3.4 ± 0.7 vs. 8.7 ± 0.4%, P < 0.05).
Table 1.
Lamina propria dendritic cell infiltrate
| Total No. LP Cells | Total No. DCs | % DCs | P value | |
|---|---|---|---|---|
| Day 2 | ||||
| Isotype | 1,945 × 103±1,088 × 103 | 159 × 103±94 × 103 | 8.7±0.4 | 0.01 |
| Anti-TLR4 | 950 × 103±750 × 103 | 36 × 103±32 × 103 | 3.4±0.7 | |
| Day 7 | ||||
| Isotype | 4,050 × 103±600 × 103 | 463 × 103±163 × 103 | 12.3±5.8 | 0.47 |
| Anti-TLR4 | 4,917 × 103±1,499 × 103 | 621 × 103±189 × 103 | 12.6±0.1 |
Data represent means ± SE of 3 mice per group. Magnetic sorting for CD11c+ cells was used to determine the number and percentage of dendritic cells (DCs) present in the total lamina propria (LP) cell population. Day 2 and day 7 correspond to the treatment schedule in Fig. 1A. P values are for % DCs in the LP.
We next evaluated for changes in macrophage infiltrate. Mice treated with anti-TLR4 antibody according to the treatment schedule in Fig. 1A had reduced numbers of lamina propria macrophages (Fig. 3, A and B). On day 2 of DSS treatment, both anti-TLR4- and isotype-treated mice had significant increases in the macrophage infiltrate compared with baseline levels at day 0. However, at days 2 and 7, anti-TLR4-treated mice had a significant reduction in macrophages in the colonic lamina propria compared with controls (Fig. 3, A and B).
Fig. 3.

Anti-TLR4 antibody treatment reduces lamina propria macrophage infiltrate. A: number of lamina propria macrophages per high-power field (HPF) at different time points of DSS administration. CD68 staining in anti-TLR4-treated (day 2: n = 3; day 7: n = 9) and isotype-treated (day 2: n = 3; day 7: n = 8) mice was evaluated in at least 5 different areas per mouse at ×400 magnification. Data are expressed as mean values ± SE from 3 independent experiments. Day 2 and day 7 correspond to the treatment schedule in Fig. 1A. *P < 0.05. B: representative photos of immunofluorescent staining of CD68-positive cells at ×400 magnification on day 7 of DSS treatment. The primary antibody was omitted for negative control slides.
These results suggest that TLR4 signaling plays an important role in the recruitment of lamina propria dendritic cells and macrophages during the induction of colitis. In contrast, no effect was seen on PMN infiltrate, which may explain the lack of bacterial translocation following anti-TLR4 treatment.
TLR4 signaling directs chemokine expression involved in APC recruitment to the colon.
To understand the mechanism for decreased APC recruitment in mice treated with anti-TLR4 antibody, we analyzed the expression of chemokines involved in the chemotaxis of monocytes. Specifically, we examined mucosal expression of CCL2, CCL20, and CX3CL1 mRNA by real-time PCR.
TLR4 blockade resulted in a decrease in the mucosal expression of CCL2 and CCL20 following DSS treatment at both day 2 and day 7 (Fig. 4). No statistically significant change was evident in the expression of CX3CL1. This dampening of the monocyte-attracting chemokines CCL2, CCL20, and CX3CL1 may underlie the reduced numbers of dendritic cells and macrophages seen in the lamina propria of anti-TLR4 antibody-treated mice. These results suggest that TLR4 signaling contributes to the production of chemokines important in the recruitment of macrophages and dendritic cells.
Fig. 4.
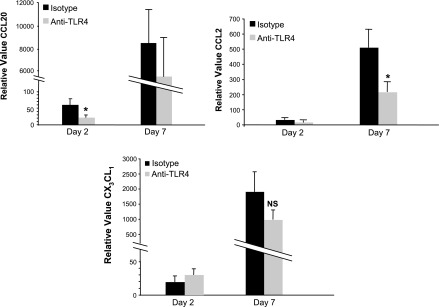
TLR4 signaling contributes to expression of chemokines involved in the recruitment of antigen presenting cells (APCs). Real-time PCR results for mucosal expression of CCL20, CCL2, and CX3CL1 in anti-TLR4 antibody-treated (day 2: n = 3; day 7: n = 9) and isotype control-treated (day 2: n = 3; day 7: n = 8) mice. Data are presented as mean ± SE relative values of expression from 2 independent experiments with duplicate samples. Day 2 and day 7 correspond to the treatment schedule in Fig. 1A. *P < 0.05.
Blockade of TLR4 during recovery from DSS-induced colitis impairs mucosal healing.
We have previously characterized the role of TLR4 in mucosal healing using TLR4−/− mice (24, 25). We sought to determine the effect of short-term blockade of TLR4 on mucosal repair. To understand how TLR4 inhibition affects mucosal healing, mice were allowed to recover from DSS-induced colitis for 7 days. Anti-TLR4 antibody or an isotype control was injected ip at day 7 and day 10 of DSS with euthanasia on day 14 (Fig. 5A). DAI was used to assess clinical recovery from colitis. Anti-TLR4- and isotype-treated mice had no significant difference in DAI during the recovery phase through day 14 (1.28 ± 0.19 vs. 1.33 ± 0.23, P = 0.42, maximum score: 4) (Fig. 5B). However, TLR4 blockade appeared to be associated with a slightly higher mortality rate during the recovery phase with survival rates of 50% in the anti-TLR4 antibody-treated group and 60% in the isotype control group at day 14.
Fig. 5.

TLR4 blockade during recovery from DSS colitis impairs mucosal healing and epithelial proliferation. A: treatment schedule for anti-TLR4 antibody and isotype control during recovery from DSS colitis. Mice were injected intraperitoneally (ip) at the indicated time points. B: there were no clinical differences between mice treated with anti-TLR4 antibody or isotype control during recovery from DSS colitis as measured by DAI. Data represent the mean ± SE of 2 independent experiments with a total of 22 mice (n = 12 for anti-TLR4 and n = 10 for isotype control-treated mice). C: histological assessment of colitis severity in anti-TLR4 antibody- and isotype control-treated mice. Data represent the mean histological score ± SE for mice from 2 independent experiments (n = 4 for anti-TLR4 and n = 4 for isotype). Anti-TLR4 antibody-treated mice failed to recover from DSS-induced damage with higher total histological damage scores at day 14. Specifically, whereas isotype control mice had begun to repair mucosal damage caused by DSS, the anti-TLR4 antibody-treated mice had persistent crypt damage and ulceration at day 14. *P < 0.05. D: representative microscopic pictures of hematoxylin and eosin (H&E) staining of colon sections from anti-TLR4 antibody and isotype control-treated mice. Arrows indicate areas of persistent crypt damage and ulceration in an anti-TLR4-treated mouse colon section. E: epithelial proliferation was measured by bromodeoxyuridine (BrdU) labeling index (number of BrdU-positive cells per crypt at ×400 magnification). Data represent means ± SE for mice from 2 independent experiments (n = 4 for anti-TLR4 and n = 4 for isotype). *P < 0.05. F: representative photo of immunohistochemical staining of incorporated BrdU into colonic epithelium. Proliferating cells labeled with BrdU have brown nuclei as indicated by the arrows.
Histological assessment of the distal colon, proximal colon, and cecum was carried out at day 14. Anti-TLR4 antibody-treated mice had significantly more mucosal damage at day 14 than isotype control-treated mice (total score: 10.96 ± 0.99 vs. 5.92 ± 1.63, P < 0.05, maximum score: 16) suggesting a defect in mucosal healing in anti-TLR4-treated mice (Fig. 5, C and D). Specifically, anti-TLR4 antibody-treated mice had significantly worse crypt damage (2.58 ± 0.25 vs. 1.17 ± 0.4, P < 0.05, maximum score: 4) and necrosis and ulceration (2.58 ± 0.25 vs. 1.7 ± 0.4, P < 0.05) compared with controls (Fig. 5, C and D).
We sought to determine whether this defect in mucosal healing was due to impaired epithelial proliferation and decreased mucosal COX-2, PGE2, and amphiregulin. The BrdU labeling index demonstrated that antibody-treated mice had significantly reduced epithelial proliferation compared with isotype-treated mice (BrdU-positive cells per crypt: 6.5 ± 0.16 vs. 8.4 ± 0.34, P < 0.01) (Fig. 5, E and F). Mucosal expression of COX-2 was significantly decreased in anti-TLR4 antibody-treated mice at day 14 (relative value: 841.7 ± 367.6 vs. 2,267.4 ± 614.7, P < 0.05) (Fig. 6A). This decrease in COX-2 expression was associated with significantly reduced mucosal levels of PGE2 (pg/mg tissue: 2,104.5 ± 632.5 vs. 7,106.4 ± 537.9, P < 0.01) (Fig. 6B). We previously demonstrated that LPS induces expression of another mediator of epithelial repair, the EGFR ligand amphiregulin, in an intestinal epithelial cell line and that this process is dependent on TLR4 signaling (25). We therefore sought to determine whether TLR4 blockade reduced mucosal levels of amphiregulin in addition to COX-2 and PGE2. Indeed, amphiregulin expression was also significantly reduced at day 14 with anti-TLR4 treatment (relative value: 74 ± 37.2 vs. 611.5 ± 256.8, P < 0.05) (Fig. 6C). These results suggest that TLR4 blockade impairs recovery from DSS-induced mucosal damage by inhibiting production of COX-2, PGE2, and amphiregulin.
Fig. 6.

Anti-TLR4 antibody treatment results in decreased expression of mediators of mucosal repair. A: real-time PCR results of mucosal expression of COX-2 in anti-TLR4 antibody-treated (n = 4) and isotype control-treated (n = 4) mice at day 14. Data are presented as mean ± SE relative values of expression from 2 independent experiments with duplicate samples. *P < 0.05. B: ELISA results for PGE2 levels in colon culture supernatants from anti-TLR4 antibody-treated (n = 4) and isotype control-treated (n = 4) mice at day 14. Data represent mean values ± SE of supernatants measured in triplicate from 2 independent experiments. *P < 0.05. C: real-time PCR results of mucosal expression of amphiregulin in anti-TLR4 antibody-treated (n = 4) and isotype control-treated (n = 4) mice at day 14. Data are presented as mean ± SE relative values of expression from 2 independent experiments with duplicate samples. *P < 0.05.
TLR4 blockade has a minimal effect in a T cell-dependent model of colitis.
The adoptive transfer model of colitis is a model of chronic colitis induced by pathogenic, effector T cells responding to luminal bacterial antigens (19). We and others have previously demonstrated that T cells express TLRs, including TLR4, and that naive T cells from MyD88−/− mice, which lack TLR signaling, are defective in their ability to induce colitis in RAG1−/− mice (10, 23, 37, 80). We therefore sought to investigate whether TLR4 blockade ameliorates colitis in the adoptive transfer model. We transferred CD4+CD62L+ T cells from WT mice into RAG1−/− mice to induce colitis and injected mice with either anti-TLR4 antibody or isotype control at the indicated time points (Fig. 7A). Transfer model mice developed signs of wasting disease compared with RAG1−/− mice that did not receive transferred T cells as measured by percent body weight loss (Fig. 7B). Although anti-TLR4-treated mice appeared to maintain a higher body weight than isotype-treated mice, this difference was not statistically significant (Fig. 7B). Histological examination of mouse colons demonstrated that transfer mice had developed mild to moderate chronic colitis. Anti-TLR4-treated mice had slightly worse mucosal damage, although the difference was modest and not statistically significant (Fig. 7, C and D). These results suggest that TLR4 blockade is minimally effective at ameliorating clinical signs of colitis in a murine model of dysregulated adaptive immunity.
Fig. 7.

Anti-TLR4 antibody treatment has minimal effect in the transfer model of chronic colitis. A: treatment schedule for anti-TLR4 antibody and isotype control during adoptive transfer model of colitis. CD4+CD62L+ T cells were injected ip into RAG1−/− mice at day 0 of week 1. Mice were then injected ip at the indicated time points with either anti-TLR4 antibody or isotype control and euthanized at the end of 5 wk. B: anti-TLR4 treatment does not result in significant clinical differences. Graph shows percent body weight change. Data represent the average ± SE of 2 independent experiments with a total of 18 mice (n = 9 for anti-TLR4 and n = 9 for isotype-treated mice). Top line represents normal growth of control RAG−/− mice that did not undergo T cell transfer (n = 4). C: histological assessment of colitis severity in anti-TLR4 antibody and isotype control-treated mice. Data represent the mean histological score ± SE for mice from 2 independent experiments (n = 4 for anti-TLR4 and n = 4 for isotype). Anti-TLR4-treated mice have slightly worse colitis on histology. D: representative microscopic pictures of H&E staining of colon sections from anti-TLR4 antibody and isotype control-treated mice.
DISCUSSION
Current treatment strategies for IBD involve broadly inhibiting the immune system. However, more targeted biological immunotherapy has recently shown great clinical efficacy in ameliorating the inflammation of IBD. Dysregulated innate immune responses to commensal bacteria contribute to the pathogenesis of IBD. The inflammatory pathways underlying these exuberant innate responses to luminal flora present a host of possible therapeutic targets for IBD. TLR4 is a key mediator of innate immune responses and its expression is upregulated in the intestinal mucosa of patients with IBD. In addition, TLR4 is upstream of the production of many inflammatory mediators important in IBD. Our findings demonstrate that blockade of TLR4 signaling during DSS-induced murine colitis can reduce intestinal inflammation by reducing APC recruitment and attenuating proinflammatory chemokine and cytokine production. However, TLR4 blockade during recovery from DSS colitis may impair mucosal healing by reducing mediators of intestinal repair and homeostasis. Lastly, anti-TLR4 treatment appears to have minimal efficacy in ameliorating inflammation in a T cell-dependent model of chronic colitis.
To inhibit TLR4 signaling, we utilized an antagonist antibody targeting the TLR4/MD-2 complex that had previously been shown to increase survival in a murine model of sepsis and reduce the production of IL-6 and TNF-α (67). Previous studies have suggested that targeting MD-2 in addition to TLR4 is likely to be the most efficacious strategy for inhibiting TLR4 signaling since MD-2 is necessary for LPS binding (73). Furthermore, early clinical trials with healthy volunteers and in patients undergoing coronary bypass or valve surgery demonstrated that the TLR4 antagonist E5564 was safe and well tolerated (5, 78). These findings suggest that TLR4 blockade is a potential clinical therapeutic option for IBD.
In our study, anti-TLR4 treatment had a protective effect in acute murine colitis, delaying the development of signs of colitis with reduced clinical severity at day 7 of DSS treatment. These results suggest that TLR4 blockade may be most effective when administered during the induction or early stages of intestinal inflammation or early in a flare of disease. Studies have suggested that TLR4 may be upregulated during inactive IBD, so anti-TLR4 treatment may therefore also be helpful in maintenance therapy (9, 22). One previous study demonstrated that blocking TLR4 signaling in vivo with a lipid A analog reduced the severity of colitis after 7 days of DSS administration (20). However, that study only examined gross clinical measures such as DAI and did not specifically characterize alterations in the inflammatory infiltrate or cytokine and chemokine production. In addition, the potential effects of TLR4 blockade on epithelial proliferation and mucosal repair were not addressed. We have now demonstrated potential mechanisms for the protective effect of TLR4 blockade in DSS colitis and investigated potential side effects of anti-TLR4 treatment.
One mechanism for the reduced clinical severity of DSS-induced colitis in anti-TLR4-treated mice appears to be decreased recruitment of APCs, specifically dendritic cells and macrophages, to the colon. APCs are pivotal in the initiation and maintenance of intestinal inflammation and have been implicated in the pathogenesis of IBD (11, 61). Patients with IBD have greater numbers of dendritic cells in the inflamed colonic mucosa (70, 74). Disease severity during IBD flares correlates with a significant drop in blood dendritic cells that are likely to have migrated to the gut (4). Furthermore, greater numbers of dendritic cells in the colonic mucosa of patients with ulcerative colitis are associated with more severe disease, as measured by crypt inflammation, crypt atrophy, and mononuclear cell infiltration (77). A study by Berndt et al. (6) used the DSS model of colitis in C57BL/6 mice to study the contribution of dendritic cells to the development of intestinal inflammation. The magnitude of the dendritic cell infiltrate appeared to have a significant impact on colitis severity. DSS colitis worsened when the percentage of dendritic cells present in the colon was increased from 0.95 to 2.3% through adoptive transfer of bone marrow-derived dendritic cells. In contrast, depletion of dendritic cells ameliorated colitis severity. Another study using the DSS model demonstrated that the calcitriol analog ZK191784 ameliorated colitis by reducing dendritic cell recruitment to the colon (68). Our results provide further evidence that the magnitude of the cell infiltrate in the lamina propria may significantly affect inflammation. The most dramatic differences in DAI in our study were during the first 2 days of DSS colitis when anti-TLR4 treatment reduced the percentage of dendritic cells in the lamina propria from 8.7 to 3.4%. Therefore, inhibition of dendritic cell recruitment to the colon appears to ameliorate intestinal inflammation.
Macrophages also appear to be significant contributors to the inflammatory process in IBD. Under normal steady-state conditions, resident intestinal macrophages display inflammatory anergy (42, 66). However, during IBD it appears that peripheral monocytes with an inflammatory profile are recruited to the intestine (1, 8, 44, 59). Previous studies utilizing mouse models of colitis have demonstrated reduced intestinal inflammation with decreased macrophage recruitment (41, 71, 76). Mouse models of colitis in which macrophages were selectively depleted have shown reduced inflammation although some studies have had conflicting results (57, 76). In our study, anti-TLR4 antibody-treated mice had fewer macrophages in the lamina propria. TNF-α, which has been shown to be major contributor to the pathogenesis of IBD, is largely produced by macrophages in the intestine (31). The decreased numbers of lamina propria macrophages seen in the anti-TLR4 group may therefore potentially account for the lower production of TNF-α seen in anti-TLR4-treated mice. Interestingly, one of the mechanisms that makes anti-TNF-α treatment effective in IBD patients is likely to be depletion of monocytes and macrophages (61).
In contrast to our previous findings in TLR4−/− mice, anti-TLR4-treated mice did not have a reduction in PMN infiltrate. The similar numbers of PMNs seen in anti-TLR4- and isotype-treated groups may account for the absence of bacterial translocation in anti-TLR4-treated mice and may therefore mitigate the risk of infection with anti-TLR4 therapy. When comparing the present study to our previous work using TLR4−/− mice we believe that differences between TLR4−/− mice and the anti-TLR4-treated mice are likely due to the degree of suppression of TLR4 signaling in the two models (24, 26). Whereas TLR4−/− mice have a complete lack of signaling, anti-TLR4 antibody treatment dampens signaling but does not abolish it. In previous experiments, 1A6 decreased TLR4 signaling by 60–70% as measured by NF-κB luciferase activity and TNF-α production in LPS stimulated murine macrophages (67). Our results may therefore point to a difference between a complete lack of TLR4 vs. dampening of TLR4 signaling. It appears that a minimal level of TLR4 signaling is needed to trigger a robust PMN response whereas higher levels of TLR4 signaling are needed to recruit greater numbers of APCs to the lamina propria.
Aberrant chemokine production may contribute to the uncontrolled influx of inflammatory cells seen in the intestine of IBD patients (81). CCL2, monocyte-chemotactic protein 1 (MCP-1), is involved in the recruitment of macrophages to the intestine and has been shown to be TLR4 dependent (30, 41, 65). CCL20 (macrophage inflammatory protein-3α) and CX3CL1 (fractalkine) contribute to the recruitment of dendritic cells to inflamed tissues (2, 13, 16, 32, 75). CCL20 may also influence the recruitment of macrophages to inflamed tissues (60). Our results suggest that TLR4 signaling contributes to dendritic cell and macrophage infiltrates by influencing the production of chemokines such as CCL2, CX3CL1, and CCL20.
TNF-α production is increased during DSS-induced colitis and TLR4 appears to be a key contributor to TNF-α expression (15, 49). IL-6 and IFN-γ are also upregulated in response to DSS treatment (14, 15, 33, 39). The importance of these cytokines has been further elucidated through knockout mouse experiments in which IL-6−/− and IFN-γ−/− mice developed less severe inflammation during DSS colitis (38, 46). Therefore, the reduced production of TNF-α and IL-6 seen in anti-TLR4-treated mice provides an additional mechanistic explanation for the amelioration of inflammation seen with TLR4 blockade. This dampened cytokine production may be due to a decrease in local APC production as well as due to fewer lamina propria APCs.
To gain an understanding of the potential limitations of anti-TLR4 therapy, we also investigated the effects of TLR4 blockade on mucosal healing. We and others have previously demonstrated that TLR4−/− mice have defects in mucosal healing with decreased epithelial proliferation (26, 58). The mechanism underlying this phenotype appears to be decreased expression of COX-2, PGE2, and amphiregulin (24, 25). Our results indicate that TLR4 inhibition during recovery from DSS-induced injury reduces the capacity for mucosal healing. Anti-TLR4-treated mice were impaired in their ability to repair the severe crypt damage and ulceration caused by DSS. This was associated with reduced mucosal expression of key mediators of cell proliferation and mucosal repair: COX-2, PGE2, and amphiregulin. These findings suggest that the timing of TLR4 blockade may be critical to dampen inflammation without negatively affecting mucosal homeostasis. For example, anti-TLR4 therapy may need to be avoided during times of intestinal healing such as periods immediately following flares.
In contrast to the DSS model results, TLR4 blockade appears to have minimal efficacy in ameliorating inflammation in the transfer model of T cell-mediated chronic colitis. This suggests that TLR4 is more important in modulating acute inflammation than it is in maintaining chronic colitis. We have previously seen that T cells from MyD88−/− mice, which lack signaling from multiple TLRs, cannot induce as robust a colitis in RAG1−/− mice as T cells from WT mice (23). The findings from our transfer model experiments suggest that signaling through other inflammatory mediators or through TLRs other than TLR4 is sufficient to maintain chronic inflammation. TLR4 activation is upstream in the inflammatory cascade so it may be that, once chronic inflammation is established, other pathways can perpetuate colitis. Our finding that anti-TLR4-treated mice appeared to maintain a higher body weight and have less wasting disease is consistent with previous reports indicating a role for TLR4 in the regulation of metabolism (63). Interestingly, anti-TLR4-treated mice in the transfer model had slightly worse mucosal damage on histology, mirroring the results seen when blocking TLR4 during recovery from DSS colitis, although the effect was not as marked.
In conclusion, our results demonstrate the potential of TLR4 as a therapeutic target in IBD. TLR4 blockade may offer improvement in the uncontrolled inflammation of IBD by reducing APC infiltrate and proinflammatory chemokine and cytokine production. However, the timing of such blockade will have to be carefully orchestrated since treatment during recovery from acute colitis may impair mucosal healing.
GRANTS
This research was supported by National Institutes of Health Grants AI052266 (M. T. Abreu) and DK069594 (M. T. Abreu), Career Development Award from CCFA (M. Fukata), and Howard Hughes Medical Institute Medical Research Training Fellowship (R. Ungaro).
DISCLOSURES
G. Elson and M. Kosco-Vilbois are employed by Nov Immune SA, Geneva, Switzerland, whose anti-mouse TLR4 mAb was studied in this work.
Acknowledgments
The authors thank Glaucia Furtado, Federica Marchesi, and Nanthakumar Thirunarayanan for technical advice and assistance.
REFERENCES
- 1.Allison MC, Poulter LW. Changes in phenotypically distinct mucosal macrophage populations may be a prerequisite for the development of inflammatory bowel disease. Clin Exp Immunol 85: 504–509, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ancuta P, Rao R, Moses A, Mehle A, Shaw SK, Luscinskas FW, Gabuzda D. Fractalkine preferentially mediates arrest and migration of CD16+ monocytes. J Exp Med 197: 1701–1707, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aranda R, Sydora BC, McAllister PL, Binder SW, Yang HY, Targan SR, Kronenberg M. Analysis of intestinal lymphocytes in mouse colitis mediated by transfer of CD4+, CD45RBhigh T cells to SCID recipients. J Immunol 158: 3464–3473, 1997. [PubMed] [Google Scholar]
- 4.Baumgart DC, Metzke D, Schmitz J, Scheffold A, Sturm A, Wiedenmann B, Dignass AU. Patients with active inflammatory bowel disease lack immature peripheral blood plasmacytoid and myeloid dendritic cells. Gut 54: 228–236, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bennett-Guerrero E, Grocott HP, Levy JH, Stierer KA, Hogue CW, Cheung AT, Newman MF, Carter AA, Rossignol DP, Collard CD. A phase II, double-blind, placebo-controlled, ascending-dose study of Eritoran (E5564), a lipid A antagonist, in patients undergoing cardiac surgery with cardiopulmonary bypass. Anesth Analg 104: 378–383, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Berndt BE, Zhang M, Chen GH, Huffnagle GB, Kao JY. The role of dendritic cells in the development of acute dextran sulfate sodium colitis. J Immunol 179: 6255–6262, 2007. [DOI] [PubMed] [Google Scholar]
- 7.Brown SJ, Mayer L. The immune response in inflammatory bowel disease. Am J Gastroenterol 102: 2058–2069, 2007. [DOI] [PubMed] [Google Scholar]
- 8.Burgio VL, Fais S, Boirivant M, Perrone A, Pallone F. Peripheral monocyte and naive T-cell recruitment and activation in Crohn's disease. Gastroenterology 109: 1029–1038, 1995. [DOI] [PubMed] [Google Scholar]
- 9.Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect Immun 68: 7010–7017, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caron G, Duluc D, Fremaux I, Jeannin P, David C, Gascan H, Delneste Y. Direct stimulation of human T cells via TLR5 and TLR7/8: flagellin and R-848 up-regulate proliferation and IFN-gamma production by memory CD4+ T cells. J Immunol 175: 1551–1557, 2005. [DOI] [PubMed] [Google Scholar]
- 11.Coombes JL, Powrie F. Dendritic cells in intestinal immune regulation. Nat Rev Immunol 8: 435–446, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cooper HS, Murthy SN, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest 69: 238–249, 1993. [PubMed] [Google Scholar]
- 13.Dichmann S, Herouy Y, Purlis D, Rheinen H, Gebicke-Harter P, Norgauer J. Fractalkine induces chemotaxis and actin polymerization in human dendritic cells. Inflamm Res 50: 529–533, 2001. [DOI] [PubMed] [Google Scholar]
- 14.Dieleman LA, Palmen MJ, Akol H, Bloemena E, Pena AS, Meuwissen SG, Van Rees EP. Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin Exp Immunol 114: 385–391, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dieleman LA, Ridwan BU, Tennyson GS, Beagley KW, Bucy RP, Elson CO. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology 107: 1643–1652, 1994. [DOI] [PubMed] [Google Scholar]
- 16.Dieu-Nosjean MC, Vicari A, Lebecque S, Caux C. Regulation of dendritic cell trafficking: a process that involves the participation of selective chemokines. J Leukoc Biol 66: 252–262, 1999. [DOI] [PubMed] [Google Scholar]
- 17.Duchmann R, Kaiser I, Hermann E, Mayet W, Ewe K, Meyer zum Buschenfelde KH. Tolerance exists towards resident intestinal flora but is broken in active inflammatory bowel disease (IBD). Clin Exp Immunol 102: 448–455, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dunne C Adaptation of bacteria to the intestinal niche: probiotics and gut disorder. Inflamm Bowel Dis 7: 136–145, 2001. [DOI] [PubMed] [Google Scholar]
- 19.Elson CO, Cong Y, McCracken VJ, Dimmitt RA, Lorenz RG, Weaver CT. Experimental models of inflammatory bowel disease reveal innate, adaptive, and regulatory mechanisms of host dialogue with the microbiota. Immunol Rev 206: 260–276, 2005. [DOI] [PubMed] [Google Scholar]
- 20.Fort MM, Mozaffarian A, Stover AG, da Silva Correia J, Johnson DA, Crane RT, Ulevitch RJ, Persing DH, Bielefeldt-Ohmann H, Probst P, Jeffery E, Fling SP, Hershberg RM. A synthetic TLR4 antagonist has anti-inflammatory effects in two murine models of inflammatory bowel disease. J Immunol 174: 6416–6423, 2005. [DOI] [PubMed] [Google Scholar]
- 21.Franchimont D, Vermeire S, El Housni H, Pierik M, Van Steen K, Gustot T, Quertinmont E, Abramowicz M, Van Gossum A, Deviere J, Rutgeerts P. Deficient host-bacteria interactions in inflammatory bowel disease? The toll-like receptor (TLR)-4 Asp299gly polymorphism is associated with Crohn's disease and ulcerative colitis. Gut 53: 987–992, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frolova L, Drastich P, Rossmann P, Klimesova K, Tlaskalova-Hogenova H. Expression of Toll-like receptor 2 (TLR2), TLR4, and CD14 in biopsy samples of patients with inflammatory bowel diseases: upregulated expression of TLR2 in terminal ileum of patients with ulcerative colitis. J Histochem Cytochem 56: 267–274, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukata M, Breglio K, Chen A, Vamadevan AS, Goo T, Hsu D, Conduah D, Xu R, Abreu MT. The myeloid differentiation factor 88 (MyD88) is required for CD4+ T cell effector function in a murine model of inflammatory bowel disease. J Immunol 180: 1886–1894, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fukata M, Chen A, Klepper A, Krishnareddy S, Vamadevan AS, Thomas LS, Xu R, Inoue H, Arditi M, Dannenberg AJ, Abreu MT. Cox-2 is regulated by Toll-like receptor-4 (TLR4) signaling: role in proliferation and apoptosis in the intestine. Gastroenterology 131: 862–877, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fukata M, Chen A, Vamadevan AS, Cohen J, Breglio K, Krishnareddy S, Hsu D, Xu R, Harpaz N, Dannenberg AJ, Subbaramaiah K, Cooper HS, Itzkowitz SH, Abreu MT. Toll-like receptor-4 promotes the development of colitis-associated colorectal tumors. Gastroenterology 133: 1869–1881, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fukata M, Michelsen KS, Eri R, Thomas LS, Hu B, Lukasek K, Nast CC, Lechago J, Xu R, Naiki Y, Soliman A, Arditi M, Abreu MT. Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am J Physiol Gastrointest Liver Physiol 288: G1055–G1065, 2005. [DOI] [PubMed] [Google Scholar]
- 27.Fukuda M, Kanauchi O, Araki Y, Andoh A, Mitsuyama K, Takagi K, Toyonaga A, Sata M, Fujiyama Y, Fukuoka M, Matsumoto Y, Bamba T. Prebiotic treatment of experimental colitis with germinated barley foodstuff: a comparison with probiotic or antibiotic treatment. Int J Mol Med 9: 65–70, 2002. [PubMed] [Google Scholar]
- 28.Furrie E, Macfarlane S, Thomson G, Macfarlane GT. Toll-like receptors-2, -3 and -4 expression patterns on human colon and their regulation by mucosal-associated bacteria. Immunology 115: 565–574, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Furuta T, Kikuchi T, Akira S, Watanabe N, Yoshikawa Y. Roles of the small intestine for induction of toll-like receptor 4-mediated innate resistance in naturally acquired murine toxoplasmosis. Int Immunol 18: 1655–1662, 2006. [DOI] [PubMed] [Google Scholar]
- 30.Gregory JL, Morand EF, McKeown SJ, Ralph JA, Hall P, Yang YH, McColl SR, Hickey MJ. Macrophage migration inhibitory factor induces macrophage recruitment via CC chemokine ligand 2. J Immunol 177: 8072–8079, 2006. [DOI] [PubMed] [Google Scholar]
- 31.Grip O, Janciauskiene S, Lindgren S. Macrophages in inflammatory bowel disease. Curr Drug Targets Inflamm Allergy 2: 155–160, 2003. [DOI] [PubMed] [Google Scholar]
- 32.Guo J, Zhang M, Wang B, Yuan Z, Guo Z, Chen T, Yu Y, Qin Z, Cao X. Fractalkine transgene induces T-cell-dependent antitumor immunity through chemoattraction and activation of dendritic cells. Int J Cancer 103: 212–220, 2003. [DOI] [PubMed] [Google Scholar]
- 33.Hans W, Scholmerich J, Gross V, Falk W. Interleukin-12 induced interferon-gamma increases inflammation in acute dextran sulfate sodium induced colitis in mice. Eur Cytokine Netw 11: 67–74, 2000. [PubMed] [Google Scholar]
- 34.Hans W, Scholmerich J, Gross V, Falk W. The role of the resident intestinal flora in acute and chronic dextran sulfate sodium-induced colitis in mice. Eur J Gastroenterol Hepatol 12: 267–273, 2000. [DOI] [PubMed] [Google Scholar]
- 35.Hart AL, Al-Hassi HO, Rigby RJ, Bell SJ, Emmanuel AV, Knight SC, Kamm MA, Stagg AJ. Characteristics of intestinal dendritic cells in inflammatory bowel diseases. Gastroenterology 129: 50–65, 2005. [DOI] [PubMed] [Google Scholar]
- 36.Hausmann M, Kiessling S, Mestermann S, Webb G, Spottl T, Andus T, Scholmerich J, Herfarth H, Ray K, Falk W, Rogler G. Toll-like receptors 2 and 4 are up-regulated during intestinal inflammation. Gastroenterology 122: 1987–2000, 2002. [DOI] [PubMed] [Google Scholar]
- 37.Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, Endres S, Hartmann G. Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol 168: 4531–4537, 2002. [DOI] [PubMed] [Google Scholar]
- 38.Ito R, Shin-Ya M, Kishida T, Urano A, Takada R, Sakagami J, Imanishi J, Kita M, Ueda Y, Iwakura Y, Kataoka K, Okanoue T, Mazda O. Interferon-gamma is causatively involved in experimental inflammatory bowel disease in mice. Clin Exp Immunol 146: 330–338, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karlsson A, Jagervall A, Pettersson M, Andersson AK, Gillberg PG, Melgar S. Dextran sulphate sodium induces acute colitis and alters hepatic function in hamsters. Int Immunopharmacol 8: 20–27, 2008. [DOI] [PubMed] [Google Scholar]
- 40.Kelsall BL Innate and adaptive mechanisms to control of pathological intestinal inflammation. J Pathol 214: 242–259, 2008. [DOI] [PubMed] [Google Scholar]
- 41.Khan MA, Ma C, Knodler LA, Valdez Y, Rosenberger CM, Deng W, Finlay BB, Vallance BA. Toll-like receptor 4 contributes to colitis development but not to host defense during Citrobacter rodentium infection in mice. Infect Immun 74: 2522–2536, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leon F, Smythies LE, Smith PD, Kelsall BL. Involvement of dendritic cells in the pathogenesis of inflammatory bowel disease. Adv Exp Med Biol 579: 117–132, 2006. [DOI] [PubMed] [Google Scholar]
- 43.Macpherson A, Khoo UY, Forgacs I, Philpott-Howard J, Bjarnason I. Mucosal antibodies in inflammatory bowel disease are directed against intestinal bacteria. Gut 38: 365–375, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mahida YR, Wu K, Jewell DP. Enhanced production of interleukin 1-beta by mononuclear cells isolated from mucosa with active ulcerative colitis of Crohn's disease. Gut 30: 835–838, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway CA Jr. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell 2: 253–258, 1998. [DOI] [PubMed] [Google Scholar]
- 46.Naito Y, Takagi T, Uchiyama K, Kuroda M, Kokura S, Ichikawa H, Yanagisawa R, Inoue K, Takano H, Satoh M, Yoshida N, Okanoue T, Yoshikawa T. Reduced intestinal inflammation induced by dextran sodium sulfate in interleukin-6-deficient mice. Int J Mol Med 14: 191–196, 2004. [PubMed] [Google Scholar]
- 47.O'Hara AM, Shanahan F. The gut flora as a forgotten organ. EMBO Rep 7: 688–693, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ohkawara T, Takeda H, Miyashita K, Nishiwaki M, Nakayama T, Taniguchi M, Yoshiki T, Tanaka J, Imamura M, Sugiyama T, Asaka M, Nishihira J. Regulation of Toll-like receptor 4 expression in mouse colon by macrophage migration inhibitory factor. Histochem Cell Biol 125: 575–582, 2006. [DOI] [PubMed] [Google Scholar]
- 49.Ohkawara T, Takeda H, Nishihira J, Miyashita K, Nihiwaki M, Ishiguro Y, Takeda K, Akira S, Iwanaga T, Sugiyama T, Asaka M. Macrophage migration inhibitory factor contributes to the development of acute dextran sulphate sodium-induced colitis in Toll-like receptor 4 knockout mice. Clin Exp Immunol 141: 412–421, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oostenbrug LE, Drenth JP, de Jong DJ, Nolte IM, Oosterom E, van Dullemen HM, van der Linde K, te Meerman GJ, van der Steege G, Kleibeuker JH, Jansen PL. Association between Toll-like receptor 4 and inflammatory bowel disease. Inflamm Bowel Dis 11: 567–575, 2005. [DOI] [PubMed] [Google Scholar]
- 51.Ortega-Cava CF, Ishihara S, Rumi MA, Kawashima K, Ishimura N, Kazumori H, Udagawa J, Kadowaki Y, Kinoshita Y. Strategic compartmentalization of Toll-like receptor 4 in the mouse gut. J Immunol 170: 3977–3985, 2003. [DOI] [PubMed] [Google Scholar]
- 52.Palsson-McDermott EM, O'Neill LA. Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology 113: 153–162, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Papadakis KA, Targan SR. Role of cytokines in the pathogenesis of inflammatory bowel disease. Annu Rev Med 51: 289–298, 2000. [DOI] [PubMed] [Google Scholar]
- 54.Perencevich M, Burakoff R. Use of antibiotics in the treatment of inflammatory bowel disease. Inflamm Bowel Dis 12: 651–664, 2006. [DOI] [PubMed] [Google Scholar]
- 55.Podolsky DK Inflammatory bowel disease. N Engl J Med 347: 417–429, 2002. [DOI] [PubMed] [Google Scholar]
- 56.Powrie F, Mauze S, Coffman RL. CD4+ T-cells in the regulation of inflammatory responses in the intestine. Res Immunol 148: 576–581, 1997. [DOI] [PubMed] [Google Scholar]
- 57.Qualls JE, Kaplan AM, van Rooijen N, Cohen DA. Suppression of experimental colitis by intestinal mononuclear phagocytes. J Leukoc Biol 80: 802–815, 2006. [DOI] [PubMed] [Google Scholar]
- 58.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118: 229–241, 2004. [DOI] [PubMed] [Google Scholar]
- 59.Rugtveit J, Brandtzaeg P, Halstensen TS, Fausa O, Scott H. Increased macrophage subset in inflammatory bowel disease: apparent recruitment from peripheral blood monocytes. Gut 35: 669–674, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ruth JH, Shahrara S, Park CC, Morel JC, Kumar P, Qin S, Koch AE. Role of macrophage inflammatory protein-3alpha and its ligand CCR6 in rheumatoid arthritis. Lab Invest 83: 579–588, 2003. [DOI] [PubMed] [Google Scholar]
- 61.Schenk M, Mueller C. Adaptations of intestinal macrophages to an antigen-rich environment. Semin Immunol 19: 84–93, 2007. [DOI] [PubMed] [Google Scholar]
- 62.Shang L, Fukata M, Thirunarayanan N, Martin AP, Arnaboldi P, Maussang D, Berin C, Unkeless JC, Mayer L, Abreu MT, Lira SA. Toll-like receptor signaling in small intestinal epithelium promotes B-cell recruitment and IgA production in lamina propria. Gastroenterology 135: 529–538, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty-acid induced insulin resistance. J Clin Invest 116: 3015–3025, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shirai Y, Hashimoto M, Kato R, Kawamura YI, Kirikae T, Yano H, Takashima J, Kirihara Y, Saito Y, Fujino MA, Dohi T. Lipopolysaccharide induces CD25-positive, IL-10-producing lymphocytes without secretion of proinflammatory cytokines in the human colon: low MD-2 mRNA expression in colonic macrophages. J Clin Immunol 24: 42–52, 2004. [DOI] [PubMed] [Google Scholar]
- 65.Singh JC, Cruickshank SM, Newton DJ, Wakenshaw L, Graham A, Lan J, Lodge JP, Felsburg PJ, Carding SR. Toll-like receptor-mediated responses of primary intestinal epithelial cells during the development of colitis. Am J Physiol Gastrointest Liver Physiol 288: G514–G524, 2005. [DOI] [PubMed] [Google Scholar]
- 66.Smythies LE, Sellers M, Clements RH, Mosteller-Barnum M, Meng G, Benjamin WH, Orenstein JM, Smith PD. Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. J Clin Invest 115: 66–75, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Spiller S, Elson G, Ferstl R, Dreher S, Mueller T, Freudenberg M, Daubeuf B, Wagner H, Kirschning CJ. TLR4-induced IFN-gamma production increases TLR2 sensitivity and drives Gram-negative sepsis in mice. J Exp Med 205: 1747–1754, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Strauch UG, Obermeier F, Grunwald N, Dunger N, Rath HC, Scholmerich J, Steinmeyer A, Zugel U, Herfarth HH. Calcitriol analog ZK191784 ameliorates acute and chronic dextran sodium sulfate-induced colitis by modulation of intestinal dendritic cell numbers and phenotype. World J Gastroenterol 13: 6529–6537, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Szebeni B, Veres G, Dezsofi A, Rusai K, Vannay A, Mraz M, Majorova E, Arato A. Increased expression of Toll-like receptor (TLR) 2 and TLR4 in the colonic mucosa of children with inflammatory bowel disease. Clin Exp Immunol 151: 34–41, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.te Velde AA, van Kooyk Y, Braat H, Hommes DW, Dellemijn TA, Slors JF, van Deventer SJ, Vyth-Dreese FA. Increased expression of DC-SIGN+IL-12+IL-18+ and CD83+IL-12-IL-18- dendritic cell populations in the colonic mucosa of patients with Crohn's disease. Eur J Immunol 33: 143–151, 2003. [DOI] [PubMed] [Google Scholar]
- 71.Tokuyama H, Ueha S, Kurachi M, Matsushima K, Moriyasu F, Blumberg RS, Kakimi K. The simultaneous blockade of chemokine receptors CCR2, CCR5 and CXCR3 by a non-peptide chemokine receptor antagonist protects mice from dextran sodium sulfate-mediated colitis. Int Immunol 17: 1023–1034, 2005. [DOI] [PubMed] [Google Scholar]
- 72.Torok HP, Glas J, Tonenchi L, Mussack T, Folwaczny C. Polymorphisms of the lipopolysaccharide-signaling complex in inflammatory bowel disease: association of a mutation in the Toll-like receptor 4 gene with ulcerative colitis. Clin Immunol 112: 85–91, 2004. [DOI] [PubMed] [Google Scholar]
- 73.Visintin A, Halmen KA, Latz E, Monks BG, Golenbock DT. Pharmacological inhibition of endotoxin responses is achieved by targeting the TLR4 coreceptor, MD-2. J Immunol 175: 6465–6472, 2005. [DOI] [PubMed] [Google Scholar]
- 74.Vuckovic S, Florin TH, Khalil D, Zhang MF, Patel K, Hamilton I, Hart DN. CD40 and CD86 upregulation with divergent CMRF44 expression on blood dendritic cells in inflammatory bowel diseases. Am J Gastroenterol 96: 2946–2956, 2001. [DOI] [PubMed] [Google Scholar]
- 75.Wang L, Liu Q, Sun Q, Zhang C, Chen T, Cao X. TLR4 signaling in cancer cells promotes chemoattraction of immature dendritic cells via autocrine CCL20. Biochem Biophys Res Commun 366: 852–856, 2008. [DOI] [PubMed] [Google Scholar]
- 76.Watanabe N, Ikuta K, Okazaki K, Nakase H, Tabata Y, Matsuura M, Tamaki H, Kawanami C, Honjo T, Chiba T. Elimination of local macrophages in intestine prevents chronic colitis in interleukin-10-deficient mice. Dig Dis Sci 48: 408–414, 2003. [DOI] [PubMed] [Google Scholar]
- 77.Watanabe S, Yamakawa M, Hiroaki T, Kawata S, Kimura O. Correlation of dendritic cell infiltration with active crypt inflammation in ulcerative colitis. Clin Immunol 122: 288–297, 2007. [DOI] [PubMed] [Google Scholar]
- 78.Wong YN, Rossignol D, Rose JR, Kao R, Carter A, Lynn M. Safety, pharmacokinetics, and pharmacodynamics of E5564, a lipid A antagonist, during an ascending single-dose clinical study. J Clin Pharmacol 43: 735–742, 2003. [PubMed] [Google Scholar]
- 79.Young Y, Abreu MT. Advances in the pathogenesis of inflammatory bowel disease. Curr Gastroenterol Rep 8: 470–477, 2006. [DOI] [PubMed] [Google Scholar]
- 80.Zanin-Zhorov A, Tal-Lapidot G, Cahalon L, Cohen-Sfady M, Pevsner- Fischer M, Lider O, Cohen IR. Cutting edge: T cells respond to lipopolysaccharide innately via TLR4 signaling. J Immunol 179: 41–44, 2007. [DOI] [PubMed] [Google Scholar]
- 81.Zhong W, Kolls JK, Chen H, McAllister F, Oliver PD, Zhang Z. Chemokines orchestrate leukocyte trafficking in inflammatory bowel disease. Front Biosci 13: 1654–1664, 2008. [DOI] [PubMed] [Google Scholar]