

Abstract
The inability to predict the pharmacology and toxicology of drug candidates in preclinical studies has led to the decline in the number of new drugs which make it to market and the rise in cost associated with drug development. Identifying molecular interactions associated with therapeutic and toxic drug effects early in development is a top priority. Traditional mechanism elucidation strategies are narrow, often focusing on the identification of solely the molecular target. Methods which can offer additional insight into wide-ranging molecular interactions required for drug effect and the biochemical consequences of these interactions are in demand. Genomic strategies have made impressive advances in defining a more global view of drug action are expected to increasingly be used a complimentary tool in drug discovery and development.
Keywords: Mechanism of action, drug discovery, genomics, drug target, small molecule drugs, molecular interactions, pharmacogenomics
1. Predicting Drug Variability Requires Knowledge of Drug Action
There is significant interpatient variability in response to anticancer agents; different patients may experience therapeutic benefit, no effect, or even life-threatening side effects from identical doses of the same drug. Very few methods are available to prospectively distinguish those who will benefit from those who may be harmed. Consequently, the number of adverse events associated with cancer therapy remains high. While clinical and environmental variables (e.g., age, gender, diet, organ function, concurrent medications) have been associated with variation in drug response, genetics has been estimated to account for as much as 20–95% of the variability.1 A drug’s activity is the result of interactions with proteins involved in absorption, distribution, metabolism, elimination (so called ADME) and cellular targets. Genetic variations in any one of these proteins can have a significant affect drug response.
Pharmacogenomics examines the inherited variations in genes that dictate drug response. It seeks to identify those variations associated with differential responses between patients. In the past, pharmacogenomic studies used the candidate gene approach to identify factors responsible for variable response. These studies required some a priori knowledge about a drug’s mechanism of action and the proteins it interacts with to elicit a pharmacological or toxic effect. For example, many cancers overexpress the epidermal growth factor receptor (EGFR) which, when ligand bound triggers, cell proliferation. Gefitinib was developed specifically to inhibit EGFR and suppress tumor growth. Early clinical trials revealed that most patients administered gefitinib saw no therapeutic effect.2 However, 10% of the patients had a dramatic positive response to therapy.3 It was subsequently discovered that the tumors of patients experiencing therapeutic benefit had specific activating mutations in the EGFR gene that made them susceptible to the chemotherapeutic agent. Understandably, it was concluded that administering this drug to patients whose tumors did not possess the EGFR mutations is neither medically or financially practical. Knowledge of the mechanism and required protein interactions is crucial for the development of safe and efficacious anticancer therapies.
Unfortunately, many drugs currently in use were developed without knowledge of their underlying molecular mechanisms. Predicting the mechanism of action has proven very difficult for both old and new drugs for several reasons. In many cases the target is unknown; as a result, the biochemical consequences of the drug-target interaction remain elusive. The cellular consequences continue to be vague when the target is known. Moreover, drugs are often capable of binding more than one target (considered off-target proteins), many of which have not been characterized. The end product is concurrent changes in many different biochemical pathways. Our limited understanding is even further confounded by unpredictable drug absorption, distribution, and metabolism. Drug action rapidly becomes a very complex process and understandably difficult to untangle. Our inability to elucidate a drug’s mechanism is a significant cause for the high failure rates and high costs associated with drug development. Methods that can provide information on direct targets, indirect targets, affected cellular pathways, and proteins involved in the ADME of a drug would be powerful tools in drug discovery and development.
2. Limitations of Current Methods of Mechanism Elucidation
While there are many systems available for identifying the molecular targets of small molecules, they do have weaknesses (Table 1). Affinity chromatography is a powerful and classic method used to identify target proteins.4,5 In this approach, a protein extract is passed over a packed column consisting of drug immobilized to a solid support. Following repeated washing to remove unbound proteins, the bound protein is eluted using denaturing conditions. However, this method requires high affinity ligands and a high abundance of the target protein in the cell extract suitable for detection. Compounds isolated following high throughput screens are typically not very potent, and low abundance target proteins are difficult to detect over background non-specific binders.
Table 1.
Local versus Global Methods for Mechanism of Action Studies
| Class | Technique | Description | Advantages | Disadvantages | Ref |
|---|---|---|---|---|---|
| Proteomic | Affinity chromatography | Target proteins maintains the 3D strucutre necessary for protein binding | Requires high affinity small molecules and an abundance of target protein | ||
| Yeast 3 hybrid system | Small molecule-protein target interactions occur within living cells rather than in vitro | False positives may be identified if the reporter gene is activated without the small molecule-protein interaction. | |||
| Phage display | Bacteriophage expressing a library of proteins are exposed to immobilized small molecules. The solid support is washed extensively to remove nonspecific binders, bound proteins eluted, and the process repeated for further enrichment of binding proteins and isolation of high-affinity sequences. | Repeated cycles of selection allow the detection of low abundance proteins. | Library must be sufficiently large and diverse or it may not contain the protein target responsible for investigated drug activity. | ||
| mRNA display | A protein is chemically attached to its own mRNA at the 3′ end through a puromycin linker. mRNA-protein fusions are subjected to repeated cycles of in vitro selection using an immobilized small molecule affinity column. Binding mRNA-protein fusions will be amplified to a cDNA library that codes for fewer proteins but with greater binding affinity for the small molecule. | Early rounds of selection can identify protein targets with low affinity binding | Target proteins may not be in their native form or possess post-translational modifications. | ||
| Protein microarrays | Proteins immobolized to chips are incubated with a labeled small molecule and repeatedly wased. Labeled drug remains bound to target proteins. | Proteins expressed at low levels natively receive equal exposure to the small molecule on the microarray | If target proteins must complex with other proteins for drug interaction or require post-translational modification, the target may not be identified | ||
| Biological | NCI60 IVCLSP using COMPARE | The cytotoxic response of 60 cancer cell lines to an agent is used in a pattern recognition algorithm to relate small molecules and their mechanisms. | Chemosensitivity profiles do not always match existing small molecules with defined mechanisms of action. | ||
| Genomic | Yeast deletion library screening | Compounds active in mammalian organisms may not be active in yeast | |||
| DNA microarray | Not all changes in gene expression associated with drug effect are under transcriptional control | ||||
| Murine haplotype based genetic mapping | Can directly identify numerous individual genes responsible for drug action | ||||
| Ex vivo famililial genetics | CEPH cell lines have been extensively genotyped and the data is publicly available allowing the identification of many loci influencing drug action | Tissue specific phenotypic effects such as hepatotoxicity can not be studied in these lymphoblastoid cell lines |
The yeast three hybrid system, phage display and mRNA display are three relatively new protein based methods for small molecule target discovery. They all utilize affinity chromatography but were developed to counter problems with low affinity ligands and the low abundance of the target protein in extracts. These methods involve the generation of a library or a pool of proteins which are then submitted to a selection process that entails repeated amplification and enrichment to isolate proteins of interest. This approach has been most often exemplified by the phage display system.6 However, the phage display system requires the in vitro bacterial transformation of the library DNA. This significantly limits the possible size of the protein library.7 The probability of finding a binding protein with high affinity increases as the library size increases. In addition, that probability is also influenced by both the ability to diversify the library, and then isolate and characterize selected proteins from the library. A library lacking diversity or possessing underrepresented binding proteins may inadvertently be missing the target. Libraries prepared for mRNA display can be generated containing more than 1013 different protein sequences.8 However, since these proteins are expressed in E. coli, they may fold improperly and form insoluble inactive aggregates or inclusion bodies, which can not be easily purified. The steps needed to solubilize and refold the fusion protein can be highly variable and may not always result in high yields of active protein.
Another very powerful approach to predict the molecular targets of anticancer agents was developed at the NCI and evaluates the cytotoxicity of compounds in a panel of 60 human cancer cell lines. The COMPARE algorithm then matches the cell growth inhibition (fingerprint) of a test compound with one or more of the thousands of other compounds in the NCI database.9 A high degree of correlation between the two fingerprints suggests that the compounds share a molecular target. This model identified the novel drug kenpaullone as a cyclin dependent kinase inhibitor when its fingerprint matched with other CDK inhibitors that have been through the screen previously.10 The model is unsuccessful in assigning a mechanistic classification when the fingerprint for a drug candidate is too distinct from the patterns of established agents, a scenario suggesting a novel molecular target.
3. Genomics broadens understanding of drug action
To meet the challenging problem of identifying the mechanisms of action of drug candidates, novel methods are constantly being developed and old methods increasingly improved upon. Some impressive successes have been attributed to the use of genetics as a tool in the identification of mechanisms of action for drugs. The innovative genetic models that follow have several advantages over the target identification assays described above. They require no a priori knowledge about the compound mode of action, which allows truly novel drug activities to be determined in a systematic and unbiased method. These processes allow the discovery of biological pathways involved in drug action (including proteins associated with metabolism, distribution, and off target effects) in addition to the precise mechanism of action to be determined. Traditional methods of elucidating mechanism are restricted by a static view of drug action: they oversimplify and focus the search on a single molecular target. By allowing the biology to reveal the genes influencing activity, genomic tools offer a more dynamic and global perspective of a drug’s mechanism.
Enhanced knowledge of yeast genomics have enabled the use of the budding yeast Saccharomyces cerevisiae as a powerful tool for mechanistic discovery. Libraries of genome-wide yeast haploid deletion strains with molecular barcodes have been developed. When these libraries are grown in the presence of drug, the deletions that sensitize cells to a particular drug will cause a decrease in cell growth relative to control.11 The barcode associated with each strain is used to quantitate growth and identify genes involved in the drug’s mechanism. This method has been used to explore the cellular pathways and processes for a collection of compounds with known and unknown modes of action. 11,12 Hierarchal clustering of compounds with similar genetic profiles suggests common molecular targets and pathways.12 For example, the genetic profiles of amiodarone, an antiarrythmic drug, and the chemotherapeutic agent tamoxifen which targets the estrogen receptor, were quite similar. Amiodarone acts through perturbation of calcium homeostasis. In three independent validation assays, tamoxifen was also shown to disrupt calcium homeostatis confirming published evidence that the drug increases calcium concentrations in a variety of mammalian cells.12 Moreover, the system could be used to identify unknown targets for novel agents. The target of the antifungal, papuamide B (PapB), was identified by assessing both drug resistant and sensitive mutants. Sensitive mutants affected by exposure to PapB had gene deletions related to cell wall organization. A single gene, the enzyme required for synthesis of phosphatidylserine (PS) in yeast cell membranes, was associated with the growth of resistant wild type cells in the presence of PapB. Investigators proposed that papB binds PS and acts on membrane integrity and permeability. A comparison of the genetic profile of PapB with known membrane permeabilizers and agents which bind other phospholipids revealed a match. Yeast genetic profiling is exceptional because it can be used to identify primary and secondary targets via sensitivity as well as loss-of-function mutations that result in drug-resistance.13
Another noteworthy example involves the use of mouse haplotype computational genetic analysis to identify genes that affect drug metabolism or response. This method was recently used to identify genes and the resulting proteins affecting the overall metabolism of warfarin in mice (Figure 1).14 Warfarin, a commonly prescribed anticoagulant, is metabolized by many different pathways and by a variety of enzymes into different metabolites. Warfarin was administered to a number of inbred mouse strains and both parent compound and metabolites quantified in plasma for up to 24 h following dosing. Strain specific differences were observed in the production of warfarin metabolites. Computational genetic analysis was used to look for patterns of genetic variation that correlated with the observed phenotypic differences across mouse strains. The two strains of mice with the lowest rates of warfarin metabolism differed from the other strains in a region on chromosome 19 that encodes for the metabolizing enzyme Cyp2c. To confirm the role of Cyp2c in murine warfarin metabolism, the formation of the major metabolite 7-hydroxywarfarin was inhibited in murine liver microsomes following the administration of a Cyp2c specific inhibitor.14 Moreover, the expression of Cyp2c29 in liver extracts were 2–7.4 fold higher than in the two strains with the low rate of metabolite generation. The impact of this tool extends beyond evaluating drug metabolism. For example, this approach has also been used to link the beta-2-adrenergic receptor to increased pain sensitization, a side effect associated with the administration and subsequent withdrawal of opiods.15,16
Figure 1. Murine haplotype computational pharmacogenetic analysis.
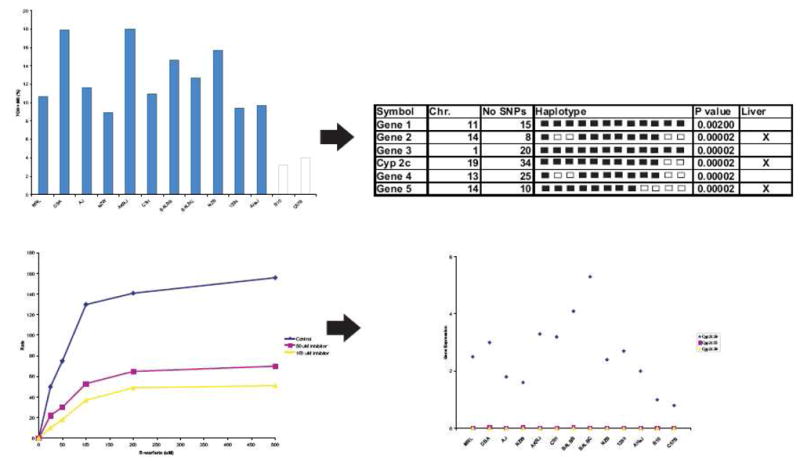
This computational method allows for the rapid identification of genes involved in drug metabolism. A single IP dose of 10 mg/kg 14C-R-warfarin was administered to thirteen mouse strains and parent compound and metabolites analyzed in pooled blood samples. Strain specific differences were observed in the formation of the major warfarin metabolites 7-hydroxywarfarin (7-OH) and its glucoronidated metabolite (M8). The correlation between the measure phenotype variation and genomic variation between strains was evaluated using haplotype blocks. Computational analysis identified strains who have the same phenotypic values and haplotype blocks. Haplotype blocks are then analyzed to identify a list of genes potentially influencing warfarin metabolism. The list is reduced to genes expressed in the liver. Strain groupings for Cyp2c which is expressed in the liver best correlated to the observed phenotypes for warfarin metabolism. To confirm Cyp2c involvement, the effect of a Cyp2c specific inhibitor on the rate of formation of 7-OH and M8 was examined. The genomic region associated with the Cyp2c haplotype block suggested that genetic variation in the CYP450 enzymes Cyp2c55, Cyp2c39, or Cyp2c29 may be responsible for the observed phenotypic variation. Gene expression levels of the Cyp enzymes were evaluated in the livers of the 13 mouse strains. Cyp2c55 and Cyp2c39 were not expressed in the livers, and Cyp2c29 gene expression varied greatly among strains. Finally, differences in protein expression for Cyp2c29 were shown to correlate to the differences in warfarin metabolism.
Recently studies have employed ex vivo human familial genetic models to investigate drug action.17,18 The genes influencing the cytotoxicity of chemotherapeutic agents have been studied using immortalized lymphoblastoid cell lines (LCLs) derived from Centre d’Etude du Polymorphisme Humain (CEPH) populations. The CEPH cell lines are a collection of multigenerational families that have been extensively genotyped. Cells from these families are phenotyped for response to a given anticancer agent then linkage analysis is used to correlate variation in response to variation in regions of the genome referred to as quantitative trait loci or QTLs (Figure 2). Waters phenotyped sensitivity to increasing concentrations of 5-fluorouracil in 427 CEPH cell lines across 38 families.18 Significant variation was noted across individual cell lines at each dose. Heritability, the degree to which a trait can be explained by genetic factors, ranged from 26% at the lowest concentration of 5-fluorouracil to 65 % at the highest concentration. QTLs associated with 5-fluorouracil cytotoxicity were observed on chromosomes 5 and 9. The cytotoxic responses to cisplatin19, etoposide20, docetaxel18, and daunorubicin21 have also been discovered to be heritable traits in human families with identified QTLs. Future works are expected to narrow these QTLs down to specific genes which influence cytotoxicity.
Figure 2. Discovery of genetic loci involved in the cytotoxic effect of chemotherapeutic agents.
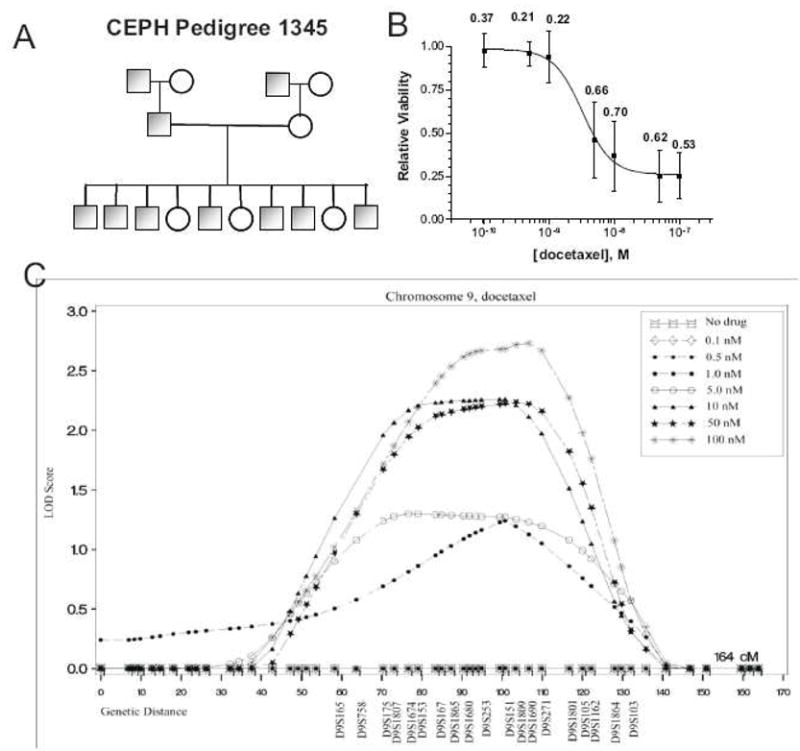
An ex vivo familial study was used to identify genes associated with docetaxel cytotoxicity. A) Variability at increasing concentrations of docetaxel was assessed I a collection of lymphoblastoid cell lines derived from CEPH pedigrees. Each pedigree consists of 5–10 offspring per family. B) A dose-response curve shows significant variation in cell viability at all concentrations across the entire CEPH population. Data points are mean cell viability and bars are standard deviations across the entire population. The degree to which observed variation in cell viability can be explained by genetic factors (heritability) is represented by numbers. C) Linkage analyis correlated regions of chromosome 9 with the observed variability in cytotoxic response. Furthermore, as drug dose increased the LOD score (probability the observed phenotype is related to the variation in a specific region) also increased.
It should be acknowledged that genomics will not solve all of the challenges encountered with mechanism elucidation. Genomic studies in yeast have their own limitations. Yeast are primitive organisms whose intracellular conditions such as protein folding and post-translational modifications can differ significantly from mammalian cells. Similarly, genes and the resulting proteins identified as contributors to drug action in murine models may not always reflect events in humans. For example, drug metabolism in rodents can differ from humans due to major differences in P450 isoforms, expression, and catalytic activity. Studies using human lymphoblasts can bring us closer to an understanding of the biochemical events involved in drug activity. However, LCls are derived from lymphoid tissue and are not suitable for the study of all phenotypes such as hepatotoxicity. In addition, the genes of the Epstein-Barr virus (EBV) are expressed in LCLs following immortalization with the virus which may confound results.
4. Conclusion
Drug activity is clearly a tangled and complex process. Its elucidation remains a daunting task for investigators. It is also a costly dilemma. Nearly one-third of the capital lost on all drug failures, $8 billion, was due to the inability to accurately predict drug pharmacology and toxicology early on in the development process.22 Genomics is an under utilized tool that can strengthen current efforts in identifying mechanisms of action. The successful integration of genomics and other biochemical assays will reveal potential novel drug targets, and hopefully then facilitate the development of safer drugs and help identify new therapeutic uses of existing drugs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Evans WE, McLeod HL. N Engl J Med. 2003;348:538–49. doi: 10.1056/NEJMra020526. [DOI] [PubMed] [Google Scholar]
- 2.Cohen MH, Williams GA, Sridhara R, Chen G, McGuinn WD, Jr, Morse D, Abraham S, Rahman A, Liang C, Lostritto R, Baird A, Pazdur R. Clin Cancer Res. 2004;10:1212–8. doi: 10.1158/1078-0432.ccr-03-0564. [DOI] [PubMed] [Google Scholar]
- 3.Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, Louis DN, Christiani DC, Settleman J, Haber DA. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 4.Takayuki Yamaguchi TYRKJKYHTNKHHAKTYMMKSYYSTYNTT. Cancer Science. 2007;98:1809–1816. doi: 10.1111/j.1349-7006.2007.00604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshida M, Kabe Y, Wada T, Asai A, Handa H. Mol Pharmacol. 2008;73:987–94. doi: 10.1124/mol.107.043307. [DOI] [PubMed] [Google Scholar]
- 6.Rodi DJ, Makowski L, Kay BK. Curr Opin Chem Biol. 2002;6:92–6. doi: 10.1016/s1367-5931(01)00287-3. [DOI] [PubMed] [Google Scholar]
- 7.Sidhu SS, Lowman HB, Cunningham BC, Wells JA. Methods Enzymol. 2000;328:333–63. doi: 10.1016/s0076-6879(00)28406-1. [DOI] [PubMed] [Google Scholar]
- 8.Liu R, Barrick JE, Szostak JW, Roberts RW. Methods Enzymol. 2000;318:268–93. doi: 10.1016/s0076-6879(00)18058-9. [DOI] [PubMed] [Google Scholar]
- 9.Paull KD, Shoemaker RH, Hodes L, Monks A, Scudiero DA, Rubinstein L, Plowman J, Boyd MR. J Natl Cancer Inst. 1989;81:1088–92. doi: 10.1093/jnci/81.14.1088. [DOI] [PubMed] [Google Scholar]
- 10.Zaharevitz DW, Gussio R, Leost M, Senderowicz AM, Lahusen T, Kunick C, Meijer L, Sausville EA. Cancer Res. 1999;59:2566–9. [PubMed] [Google Scholar]
- 11.Lum PY, Armour CD, Stepaniants SB, Cavet G, Wolf MK, Butler JS, Hinshaw JC, Garnier P, Prestwich GD, Leonardson A, Garrett-Engele P, Rush CM, Bard M, Schimmack G, Phillips JW, Roberts CJ, Shoemaker DD. Cell. 2004;116:121–37. doi: 10.1016/s0092-8674(03)01035-3. [DOI] [PubMed] [Google Scholar]
- 12.Parsons AB, Lopez A, Givoni IE, Williams DE, Gray CA, Porter J, Chua G, Sopko R, Brost RL, Ho CH, Wang J, Ketela T, Brenner C, Brill JA, Fernandez GE, Lorenz TC, Payne GS, Ishihara S, Ohya Y, Andrews B, Hughes TR, Frey BJ, Graham TR, Andersen RJ, Boone C. Cell. 2006;126:611–25. doi: 10.1016/j.cell.2006.06.040. [DOI] [PubMed] [Google Scholar]
- 13.Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H. Science. 1999;285:901. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- 14.Guo Y, Weller P, Farrell E, Cheung P, Fitch B, Clark D, Wu SY, Wang J, Liao G, Zhang Z, Allard J, Cheng J, Nguyen A, Jiang S, Shafer S, Usuka J, Masjedizadeh M, Peltz G. Nat Biotechnol. 2006;24:531–6. doi: 10.1038/nbt1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo Y, Weller P, Allard J, Usuka J, Masjedizadeh M, Wu SY, Fitch B, Clark D, Clark JD, Shafer S, Wang J, Liao G, Peltz G. Proc Am Thorac Soc. 2006;3:409–12. doi: 10.1513/pats.200601-014AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liang DY, Liao G, Wang J, Usuka J, Guo YY, Peltz G, Clark JD. Anesthesiology. 2006;104:1054–62. doi: 10.1097/00000542-200605000-00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dolan ME, Newbold KG, Nagasubramanian R, Wu X, Ratain MJ, Cook EH, Jr, Badner JA. Cancer Res. 2004;64:4353–6. doi: 10.1158/0008-5472.CAN-04-0340. [DOI] [PubMed] [Google Scholar]
- 18.Watters JW, Kraja A, Meucci MA, Province MA, McLeod HL. Proc Natl Acad Sci U S A. 2004;101:11809–14. doi: 10.1073/pnas.0404580101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang RS, Duan S, Shukla SJ, Kistner EO, Clark TA, Chen TX, Schweitzer AC, Blume JE, Dolan ME. Am J Hum Genet. 2007;81:427–37. doi: 10.1086/519850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang RS, Duan S, Bleibel WK, Kistner EO, Zhang W, Clark TA, Chen TX, Schweitzer AC, Blume JE, Cox NJ, Dolan ME. Proc Natl Acad Sci U S A. 2007;104:9758–63. doi: 10.1073/pnas.0703736104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang RS, Duan S, Kistner EO, Bleibel WK, Delaney SM, Fackenthal DL, Das S, Dolan ME. Cancer Res. 2008;68:3161–8. doi: 10.1158/0008-5472.CAN-07-6381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee AC, Shedden K, Rosania GR, Crippen GM. J Chem Inf Model. 2008;48:1379–88. doi: 10.1021/ci800097k. [DOI] [PMC free article] [PubMed] [Google Scholar]