

Abstract
Multiple inflammatory mediators in osteoarthritis (OA) cartilage, including S100/calgranulin ligands of receptor for advanced glycation end products (RAGE), promote chondrocyte hypertrophy, a differentiation state associated with matrix catabolism. In this study, we observed that RAGE knockout was not chondroprotective in instability-induced knee OA in 8-wk-old mice. Hence, we tested the hypothesis that expression of the alternative S100/calgranulin and patterning receptor CD36, identified here as a marker of growth plate chondrocyte hypertrophy, mediates chondrocyte inflammatory and differentiation responses that promote OA. In rat knee joint destabilization-induced OA, RAGE expression was initially sparse throughout cartilage but increased diffusely by 4 wk after surgery. In contrast, CD36 expression focally increased at sites of cartilage injury and colocalized with developing chondrocyte hypertrophy and aggrecan cleavage NITEGE neoepitope formation. However, CD36 transfection in normal human knee-immortalized chondrocytes (CH-8 cells) was associated with decreased capacity of S100A11 and TNF-α to induce chondrocyte hypertrophy and ADAMTS-4 and matrix metalloproteinase 13 expression. S100A11 lost the capacity to inhibit proteoglycans synthesis and gained the capacity to induce proteoglycan synthesis in CD36-transfected CH-8 cells. Moreover, S100A11 required the p38 MAPK pathway kinase MKK3 to induce NITEGE development in mouse articular cartilage explants. However, CH-8 cells transfected with CD36 demonstrated decreased S100A11-induced MKK3 and p38 phosphorylation. Therefore, RAGE and CD36 patterning receptor expression were linked with opposing effects on inflammatory, procatabolic responses to S100A11 and TNF-α in chondrocytes.
Multiple inflammatory mediators increased in osteoarthritis (OA)3 cartilage, including IL-1β, TNF-α, CXCL1, and S100/calgranulins promote cartilage matrix catabolism, in part by inducing matrix-degrading proteases and suppressing proteoglycan (PG) synthesis by chondrocytes (1–9). Altered chondrocyte differentiation also plays a role in OA pathophysiology (1, 4, 8 –10), including transition of resting to hypertrophic cells that appear within a few weeks of the course of surgical instability-induced mouse knee OA (11). In the growth plate, the progression of chondrocyte maturation to hypertrophy follows exhaustive rounds of cell proliferation and is tightly regulated by the balance between systemic and local inhibitors (e.g., parathyroid hormone, parathyroid hormone-related protein, TGF-β) and promoters (e.g., thyroxine, runx2) of maturation of prehypertrophic cells (11–13). The growth plate zone of chondrocyte hypertrophy is characterized by profound extracellular matrix depletion associated with increased type X collagen and matrix metalloproteinase 13 (MMP-13) expression (14, 15).
The calgranulin S100A11 and the inflammatory cytokines CXCL1, CXCL8, and TNF-α all are increased in OA cartilage and induce hypertrophic differentiation in cultured chondrocytes (8, 9, 16). Physiologic intracellular roles of S100/calgranulins include shuttling of calcium between subcellular compartments, modulation of function of multiple calgranulin-binding partners including certain annexins, and service as molecular chaperones (17–21). However, multiple S100/calgranulins can be induced to undergo secretion, which allows other S100/calgranulin functions to emerge, exemplified by function of S100A8 and S100A9 as proinflammatory alarmins (22, 23) and promoters of apoptosis (24). Certain inflammatory cytokines induce chondrocyte expression and/or secretion of calgranulins including S100A4, S100A9, and S100A11 (5, 6, 8, 25). For example, TNF-α induces S100A11 expression in articular chondrocytes (8). Dependent on signaling transduced by the transmembrane patterning receptor and Ig superfamily member RAGE, exogenous S100A4 induces MMP-13 in chondrocytes (5, 6) and exogenous S100A11 induces chondrocyte hypertrophy and certain procatabolic responses (8, 9). RAGE recognizes at least five different classes of ligands (26) and RAGE increases in expression with aging in multiple tissues (27), including in human knee cartilage with established OA (6). Lesional increases in ligands of RAGE such as advanced glycation end product (AGE), β-amyloid, and HMGB1 have been observed in diabetes and a variety of other disorders associated with chronic, low-grade inflammation (26, 27). Moreover, RAGE has been demonstrated to play a major role in certain vascular, renal, neurologic, and cell proliferative disorders in studies of RAGE-deficient mice or in studies where soluble RAGE has been used to suppress RAGE signaling (26, 28). Conversely, RAGE promotes tissue repair under some circumstances, including nerve regeneration after crush injury (29, 30).
Pathways other than RAGE-dependent signaling also modulate biologic effects of RAGE ligands on cells (31). One example is the engagement of AGE by AGE-R1, which suppresses RAGE-driven proinflammatory effects (32). CD36 is a broadly expressed patterning and sequestration receptor that functions as a scavenger receptor for AGE and certain other ligands, promoting their uptake and intracellular degradation (33, 34). Hence, this study investigated CD36-mediated effects of the chondrocyte-expressed RAGE ligand S100A11. Significantly, CD36 expression by macrophages, platelets, and certain other cell types promotes a variety of inflammatory and thrombotic responses to tissue injury (34, 35). Moreover, CD36 plays a central role in driving inflammatory responses to the shared RAGE and CD36 ligand β-amyloid (36, 37).
CD36 is increased in human cartilages with established OA (38). Hence, this study, in which RAGE knockout was not chondroprotective in mouse knee OA, compared expression of RAGE and CD36 in experimental OA and RAGE and CD36 function in chondrocytic cells. We observe that CD36 is a marker of hypertrophic differentiation, but unexpectedly observe CD36 expression to be linked with suppressed catabolic responses and promotion of repair responses to inflammatory stimuli in chondrocytes.
Materials and Methods
Reagents
Unless otherwise indicated, all chemical reagents were obtained from Sigma-Aldrich. Human recombinant S100A11 was generated as previously described (8, 9). Human recombinant insulin-like growth factor I (IGF-I) and human and mouse TNF-α were from R&D Systems.
Mouse studies and mouse OA model
All mouse procedures were humanely performed and were reviewed and approved by the institutional animal care research committees. We obtained knees of mitogen-activated protein kinase kinase 3 (MKK3) knockout and congenic wild-type control mice (originally developed by Dr. R. Davis (University of Massachusetts, Worcester, MA)) (39) through a collaboration with Dr. G. Firestein (University of California San Diego, CA). We established a breeding colony of RAGE null mice on the C57BL/6 background (from Dr. A. M. Schmidt, Columbia University, New York, NY) (40) and congenic wild-type C57BL/6 mice were purchased from The Jackson Laboratory.
To induce varying degrees of knee instability by ACL disruption in anesthetized mice, as previously described (41), a 28.5-gauge needle was inserted through the patellar tendon into the groove between the femoral condyles, and the needle was then advanced (“stabbed”) three times between 10 mm and a maximum of 15 mm to prevent rupture of the popliteal artery. Eight weeks after surgery, the mice were sacrificed and preserved and decalcified mouse knees were processed through graded alcohols and a clearing agent, infiltrated and embedded (in the frontal plane) in paraffin, sectioned, and stained with toluidine blue. Joints that had histological evidence of instability induction were identified by the presence of proliferative changes in the medial synovium, marginal zones, collateral and cruciate ligaments. An instability score was recorded for each joint (0 = no instability; 1 = minimal to mild instability (minimal to mild proliferative changes in ligaments, synovium, and marginal zones); 2 = moderate instability (moderate proliferative changes in ligaments, synovium, and marginal zones); 3 = severe instability (severe proliferative changes in ligaments, synovium, and marginal zones)).
The medial and lateral femoral and tibial cartilage scores for severity of cartilage degeneration were assigned with attention to zonal (inside, middle, and outside) distribution of lesions as: 1 = superficial damage, tangential layer of collagen absent over 50% or greater of the zone surface or up to 10% loss of PG and/or chondrocytes in focal or diffuse distribution in zone; 2 = matrix loss extends into the upper one-quarter of 50% or greater area of the zone or up to 25% loss of PG and/or chondrocytes in focal or diffuse distribution in zone; 3 = matrix loss extends through one-half of the cartilage thickness over 50% or greater of the zone or there are focal areas of full- thickness loss that are up to 25% of the width of the zone or up to 50% loss of PG and/or chondrocytes in focal or diffuse distribution in zone; 4 = matrix loss extends through three-quarters of cartilage thickness over 50% or greater of the zone or there are focal areas of full-thickness loss that are 26–50% of the width of the zone or up to 75% loss of PG and/or chondrocytes in focal or diffuse distribution in zone; and 5 = matrix loss extends through entire cartilage thickness over 50% or greater of the zone or up to 100% loss of PG and/or chondrocytes in focal or diffuse distribution in zone. Scores for each third were summed for each area of the joint and then the whole joint.
Rat knee OA model
In the experimental rat OA model (42), surgery was performed on the right knees of male 300 –325 g Sprague Dawley rats (Charles River Canada). Anesthetic (50% ketamine (100 mg/ml), 25% xylazine (20 mg/ml), 10% acepromazine (10 mg/ml), and 15% saline (0.9% solution) and Trisbrissen antibiotic (Schering Canada) were both administered at a dose of 100 μl/100 g body mass. All animals were randomly assigned to one of two groups: the first underwent ACL transection (ACLT) and partial anteromedial meniscectomy via an incision on the medial aspect of the right knee joint capsule, anterior to the medial collateral ligament. The second group underwent a sham (control) operation consisting of a similar incision through the right joint capsule but without ACLT or partial anteromedial meniscectomy. Each animal underwent 30 min, three times per week, of forced mobilization as previously described (42).
Immunofluorescence and immunohistochemistry
Frozen sections (9 μm) of mouse femoral head explants were studied as previously described (9). Rabbit polyclonal Abs (10 μg/ml) to the aggrecanase neoepitope NITEGE were from GeneTex. Mouse mAb to CD36 (10 μg/ml) was from BD Pharmingen. Deparaffinized sections of mouse knee joints were incubated with 2.5% hyaluronidase for 1 h at 37°C, washed two times with PBS containing 0.05% Tween 20 (T-PBS), blocked in 1% BSA/1% casein for 30 min at 37°C, and then incubated for 1 h at 37°C with whole serum containing Ab to type X collagen (Cosmobio) diluted 1/100 in the same blocking buffer. After three washes, primary Ab was detected via the avidin-biotin conjugate method using the Histostain-Plus reagent (Invitrogen) and with Fast Green (0.001%) added for 5 min followed by two washes in water.
For rat knee samples, three sham and three OA knee joints were obtained at 2, 4, and 8 wk after surgery. Tissue samples were fixed via intracardiac perfusion with 4% paraformaldehyde and dissected. Demineralization in 0.4 M EDTA, 0.3 N NaOH, and 1.5% glycerol (pH 7.3) was conducted for 4–5 wk before paraffin embedding. Sagittal sectioning through the medial joint compartment was performed to obtain 6-μm sections for immunohistochemistry/immunofluorescence (43, 44). Primary Abs used included goat anti-CD36 (R&D Systems), rabbit anti-NITEGE (aggrecan cleavage neoepitope) (as above) (45), mouse anti-type X collagen (Abcam). and rabbit anti-RAGE (Sigma-Aldrich). Secondary Abs for immunofluorescence were donkey anti-goat IgG FITC-conjugated, donkey anti-rabbit IgG Texas Red-conjugated, donkey anti-mouse IgG Texas Red-conjugated Abs (Cedarlane Laboratories), and goat anti-mouse FITC-conjugated Abs (R&D Systems). TOTO-3 iodide (Molecular Probes) was used as fluorescent nuclear counterstain. Goat anti-rabbit HRP-conjugated and rabbit anti-goat HRP-conjugated secondary Abs and Harris’ hematoxylin counterstaining were used for immunohistochemistry (Santa Cruz Biotechnology). Negative controls without primary Ab were included for each experiment. Immunofluorescent image development was performed using a Zeiss LSM 510 META microscope and software. Colorimetric detection with diaminobenzidine substrate (DakoCytomation) was conducted for equal time periods for each section.
Studies of human chondrocytic CH-8 cells
CH-8-immortalized normal human knee articular chondrocytes (46), studied between passages 5 and 15, were carried in monolayer culture in DMEM/high glucose supplemented with 10% FCS, 1% L-glutamine, 100 U/ml penicillin, and 50 μg/ml streptomycin at 37°C with 5% CO2. Maintenance of type II collagen and aggrecan expression was confirmed by RT-PCR as described previously (45). For transfection studies, aliquots of 1 × 106 CH-8 cells were grown in 100-mm culture dishes for 18 h in DMEM/high glucose containing 10% serum. Transient transfection used Lipofectamine Plus (Invitrogen), according to manufacturer’s protocol, with a ~60% transfection efficiency (assessed by using β-galactosidase transfection as a control). At the time, chondrocytes were stimulated with agonists; serum supplementation of medium was reduced to 1%.
SDS-PAGE/Western blotting
Western blotting of samples from cell cultures analyzed aliquots of 30 μg of protein obtained from whole cell lysates or precipitated from conditioned medium using 15% trichloroacetic acid, followed by separation via 10% SDS-PAGE, with detection by ECL (9). Abs to type X collagen from Calbiochem were diluted 1/3000. Rabbit polyclonal Abs to phosphorylated p38 and total p38 from BioSource International, rabbit polyclonal Abs to phosphorylated MKK and total MKK from Gen-Way, and rabbit polyclonal Abs to ADAMTS-4 and MMP-13 from Millipore were applied at 1 μg/ml.
Studies of mouse femoral head cartilage explants
Isolated femoral head caps of 2-mo-old mice were placed in a 96-well plate containing 0.05 ml of DMEM/high glucose supplemented with 10% FCS, 1% L-glutamine, 100 U/ml penicillin, and 50 μg/ml streptomycin at 37°C in 5% CO2 for 18 h before treatments, and GAG release at 48 h was measured spectrophotometrically using 1,9-dimethylmethylene blue as previously described (9).
35S/3H incorporation assay for sulfated PG synthesis
After transfection, CH-8 cells were stimulated for 3 days and then labeled with 1 μCi/ml [35S]sulfate and [3H]proline (to control for cell protein) for 24 h before sample collection, with cpm of 35S divided by the cpm of [3H]proline in this assay (47). The medium was removed and cells were washed three times with PBS and sulfated PG, then extracted, treated, and analyzed by scintillation counting, as described elsewhere (47).
Quantitative real-time PCR
For RT-PCR, total RNA was isolated using TRIzol (Invitrogen) and reverse transcribed, and quantitative real-time PCR was performed as previously described (47), with normalized target gene to GAPDH mRNA copy ratios calculated using Roche LightCycler software (version 4.0). CD36 primers (forward 5′-CTGTCCTATTGGGAAAGTCAC-3′ and reverse 5′-ATTCTTTTCAGATTAACGTCG-3′) were designed using the LightCycler Probe Design software 2.0.
Statistical analyses
Error bars, where indicated, represent SD. Statistical analyses were performed using one-way ANOVA.
Results
RAGE knockout is not chondroprotective for instability-induced knee OA induced in 2-mo-old mice
Since RAGE modulates inflammation and synovitis could promote cartilage degradation, we selected a minimally invasive procedure to induce knee OA in mice. Specifically, we destabilized the ACL by a blind small-gauge needle “stab” (41), following which mice immediately resumed normal activity. The stab procedure was performed on both knee joints of all animals. Joints that had 0 instability score (due to insufficient ACL trauma by the needle penetration) served as the controls. PG content and surface architecture were analyzed histologically by toluidine blue staining (Fig. 1A). Cartilage degeneration was observed in both medial and lateral tibial plateaus and both femoral condyles (data not shown). However, as the severity of OA increased, PG staining decreased. Both RAGE null and wild-type animals developed severe knee OA, with cartilages demonstrating focal areas of complete chondrocyte loss and severe depletion of PG (Fig. 1A).
FIGURE 1.
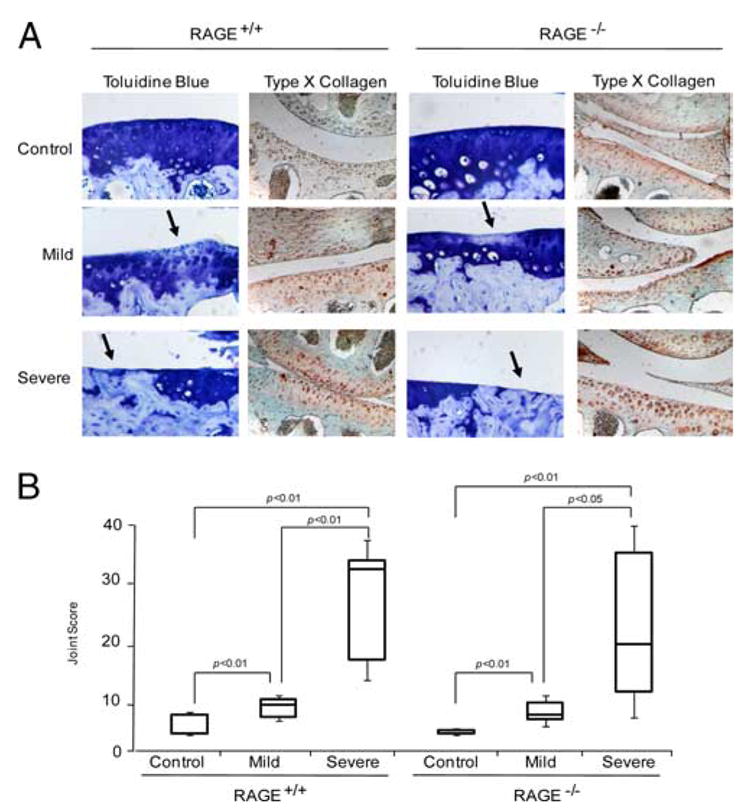
RAGE knockout is not chondroprotective for knee OA in mice. A, Two months after a “stab” procedure for minimally invasive disruption of the ACL in both knees done at 2 mo of age, congenic RAGE+/+ and RAGE−/− mouse knee joints were stained with toluidine blue and analyzed by immunohistochemistry for type X collagen. The extent of knee instability and OA were graded histologically. Knee joints with a grade of 0 for instability (due to insufficient ACL trauma by the needle penetration) served as controls and grade 1 is designated as mild OA, with grades 2 and 3 representing severe OA in A and B, respectively. Loss of blue staining (arrows) indicates loss of PG. These data are representative of at least five different mouse donors (original magnification, ×100). B, The total joint score was measured in euthanized RAGE+/+ and RAGE−/− mouse knee joints 2 mo after the stab procedure, as described in Materials and Methods. Scores for cartilage degeneration in the whole joint are represented as box and whisker plots showing the 25th through 75th percentiles boxed and the median joint score as the central horizontal line. For RAGE+/+, control n = 50, mild n = 7, and severe n = 10, and for RAGE−/−, control n = 49, mild n = 11, and severe n = 9.
RAGE and CD36 expression both were below limits of detection by immunohistochemistry in knee articular cartilages of mice at baseline and after induction of OA (data not shown). Hence, we examined the hypertrophic chondrocyte marker type X collagen, since RAGE signaling transduces chondrocyte hypertrophic differentiation in response to multiple inflammatory signals (8). Some type X collagen has been detected in normal mouse articular cartilage in situ (48). In this study, type X collagen was detected in control articular cartilages, with expression markedly up-regulated in joints with severe OA (Fig. 1A), but type X collagen expression did not appear to differ between RAGE+/+ and RAGE−/− cartilages.
To assess cartilage degeneration, scores were summed for both the tibial plateau and femoral condyle to arrive at the total joint score (Fig. 1B). As the instability score increased, the total joint score significantly increased in both the RAGE−/− and RAGE+/+ mice, but there was no significant difference in joint scores between the two genotypes. In both groups, as instability scores increased, medial osteophyte size also increased (data not shown). There was negligible lateral osteophyte formation and no osteophyte generation was detected in controls or with mild OA (data not shown).
Temporal-spatial changes in cartilage RAGE and CD36 in instability-induced rat knee OA
To define and compare temporal-spatial cartilage RAGE and CD36 expression in a larger species, we assessed moderate OA induced surgically by ACLT/partial anteromedial meniscectomy with forced mobilization in adult rats (42). Baseline RAGE and CD36 expression also were low in rat knees, but by 4 wk after surgery, RAGE expression was increased in all zones of cartilage, whereas CD36 was more focally and variably expressed (Fig. 2A). In rat growth plate cartilage, RAGE expression was detected in all zones, whereas CD36 was detected only in the hypertrophic chondrocyte zone denoted by type X collagen expression (Fig. 2B). Therefore, we tested for colocalization of CD36 and type X collagen in rat knee articular cartilage after surgical induction of OA. Four weeks after surgery, type X collagen and CD36 colocalized most robustly in the superficial zone of rat knee cartilage and by 8 wk colocalization was detected in all zones of knee cartilage (Fig. 3A). Furthermore, 8 wk after surgery, knee cartilage demonstrated colocalization of CD36 and the aggrecan NITEGE aggrecanase neoepitope in the superficial zone (Fig. 3B).
FIGURE 2.
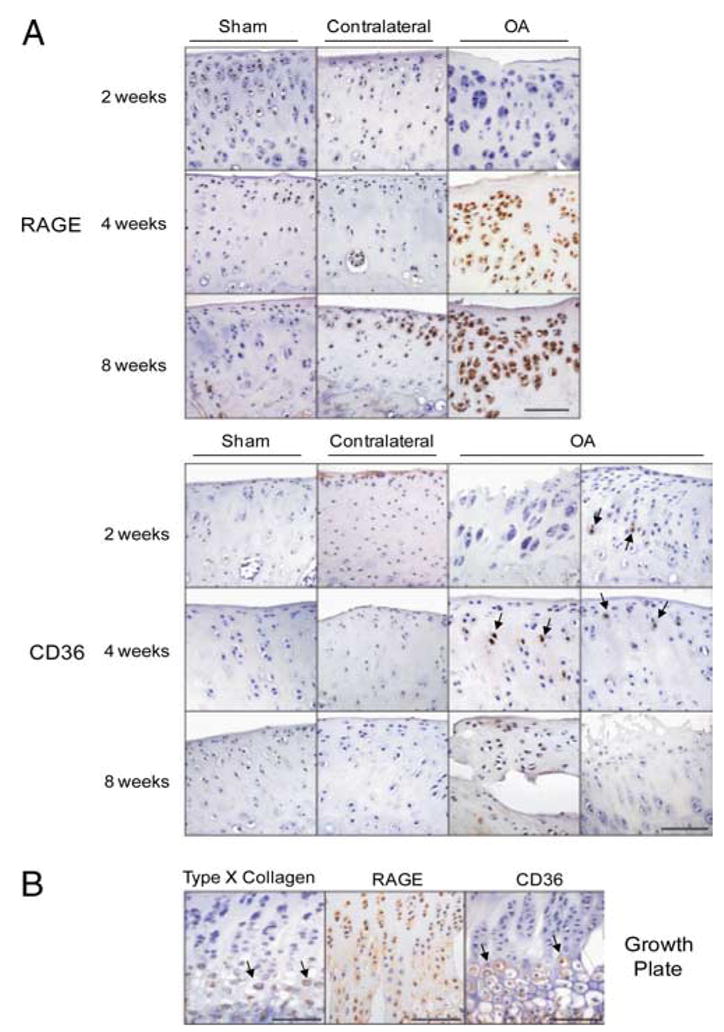
RAGE and CD36 expression increase during the evolution of surgical instability-induced knee OA in adult rats. A, OA was induced by ACLT and partial anteromedial meniscectomy in adult rats. Sham (control) and OA knee joints isolated at 2, 4, and 8 wk after surgery were fixed, paraffin embedded, and sectioned through the medial joint compartment. Histological knee joint sections were analyzed by immunohistochemistry for RAGE and CD36. Two panels of CD36 expression are shown to indicate the variability of CD36 expression (arrows). B, Histological sections of the growth plate at 4 wk after surgery were analyzed by immunohistochemistry for type X collagen (arrows), RAGE, and CD36 (arrows).
FIGURE 3.
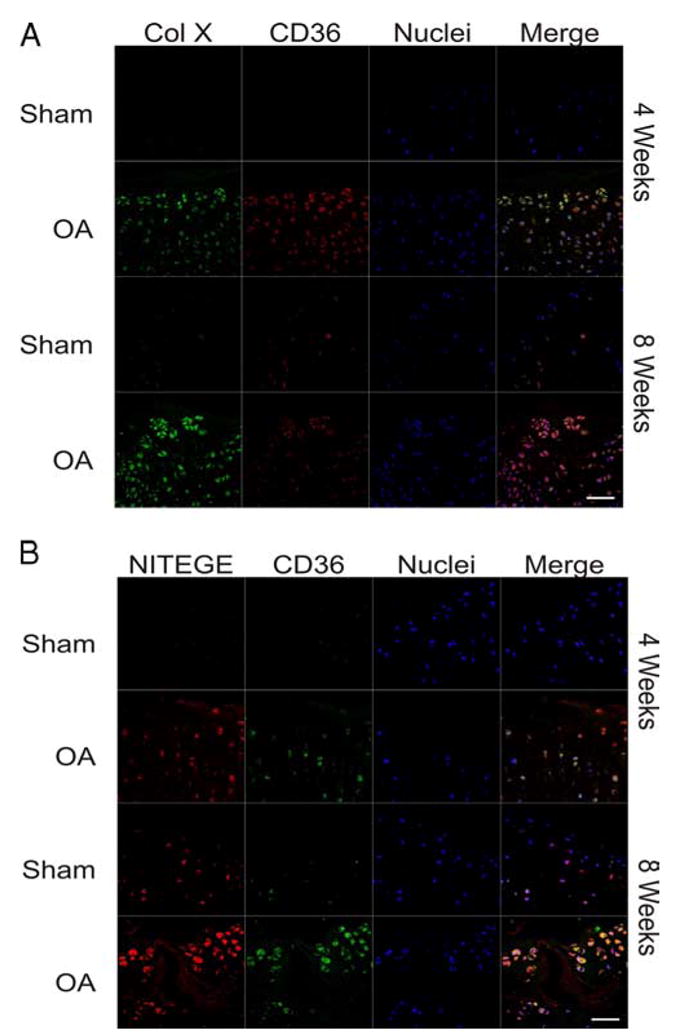
CD36 colocalizes with both type X collagen (Col X) and aggrecan cleavage as rat knee OA progresses. Sham (control) and OA knee joints isolated at 4 and 8 wk after surgery were fixed, paraffin embedded, and sectioned through the medial joint compartment. A, Histological sections were analyzed by immunofluorescence for CD36 (red) and type X collagen (green). Nuclei were counterstained with TOTO-3 iodide (blue). B, Histological sections analyzed by immunofluorescence for CD36 (green) and the aggrecan neoepitope of aggrecanase cleavage NITEGE (red), with nuclei counterstained with TOTO-3 iodide (blue signal). Scale, 50 μm.
Chondrocyte CD36 expression and effects of CD36 on chondrocytic cell differentiation and function in vitro
Since cartilage CD36 expression became focally increased in the course of rat knee OA and colocalized with markers of chondrocyte hypertrophy and matrix catabolism in vivo, we assessed factors regulating CD36 expression and tested the hypothesis that CD36 signaling, like RAGE (8, 9), promotes cartilage matrix catabolism. To do so, we used immortalized normal human knee chondrocytic CH-8 cells (46), as baseline CD36 expression was below limits of detection by quantitative PCR in these cells (Fig. 4A). Moreover, the CH-8 cells allowed for both efficient transfection and analyses of CD36 effects on signal transduction. These studies used as a positive control, the peroxisome proliferator-activated receptor γ (PPARγ) agonist GW1929, since PPARγ agonists induce CD36 in cells other than chondrocytes (49), and PPARγ agonism has been observed to be chondroprotective in OA in vivo (50, 51). GW1929, as did the chondrocyte growth and anabolic factor IGF-I, robustly induced CD36 in both CH-8 cells and normal mouse femoral head cartilage explants (Fig. 4). In contrast, both S100A11 and IL-1β, chosen for study because they stimulate cartilage matrix catabolism, were relatively weak inducers of CD36 in normal mouse femoral head cartilage explants (Fig. 4B) and S100A11 and IL-1β failed to induce a statistically significant increase in CD36 mRNA in CH-8 cells (Fig. 4A).
FIGURE 4.
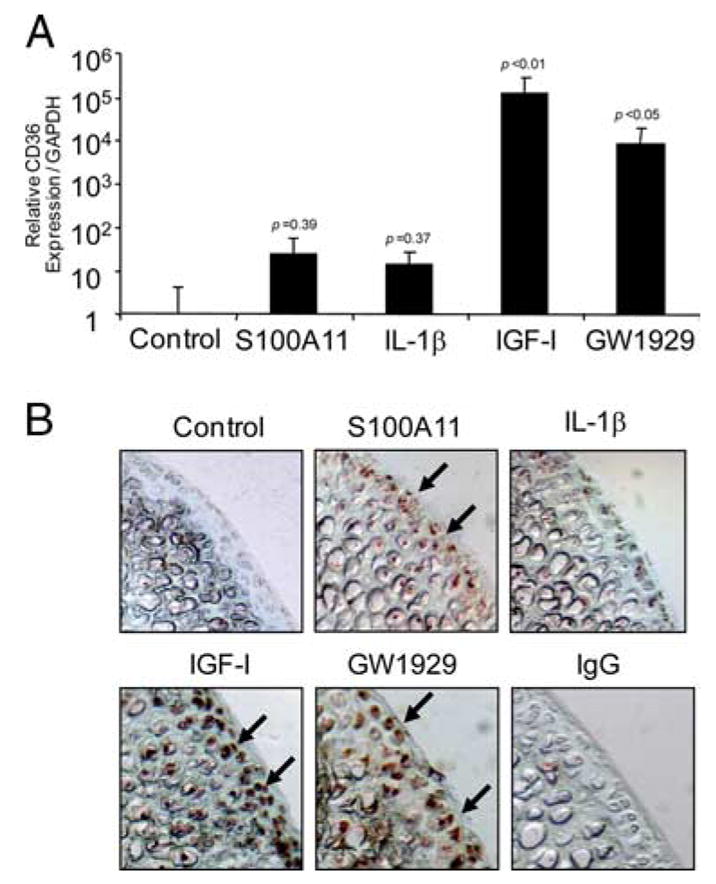
Both IGF-I and the PPARγ agonist GW1929 markedly increase CD36 expression. A, Human immortalized normal knee chondrocytes (CH-8 cells) were stimulated with 100 ng/ml S100A11, 10 ng/ml IL-1β, 10 ng/ml IGF-I, and 100 nM GW1929 for 8 h. Quantitative real-time PCR analysis of CD36 expression (as described in Materials and Methods) is shown pooled from three separate experiments. The p values indicated are compared with control, nonstimulated CH-8 cells. B, Mouse femoral head cartilage explants were stimulated for 48 h with the agonists described in A, and frozen sections were examined by immunohistochemistry for CD36 (arrows) as described in Materials and Methods. Data are representative of five different mouse donors.
Next, we transfected CD36 into CH-8 cells and confirmed induction of plasma membrane CD36 by flow cytometry (data not shown). We observed that CD36 transfection in CH-8 cells was associated with the inhibition of TNF-α- and S100A11-induced MMP-13, ADAMTS-4, and type X collagen expression (Fig. 5, A–C). Additionally in CD36-transfected CH-8 cells, S100A11 lost the capacity to inhibit PG synthesis and gained the capacity to induce PG synthesis (Fig. 5D).
FIGURE 5.
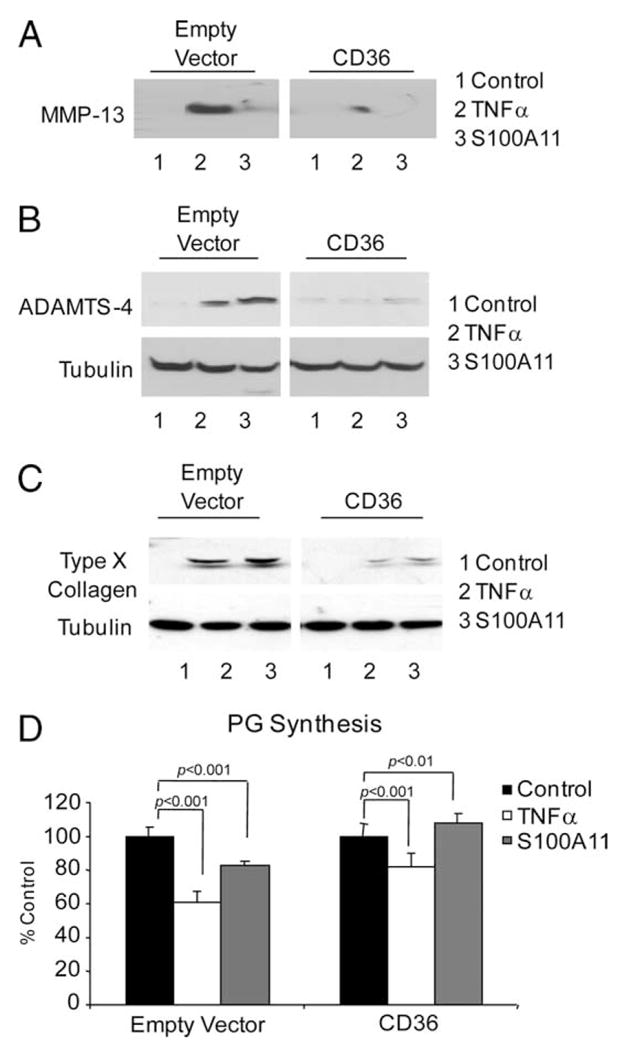
TNF-α- and S100A11-induced hypertrophic differentiation and procatabolic responses of chondrocytic cells are inhibited by transfection of CD36. CH-8 cells were plated in 96-well plates containing poly-HEME and treated with 10 ng/ml TNF-α or 100 ng/ml S100A11. After 24 h, conditioned medium was examined for MMP-13 secretion (A) and SDS-PAGE/Western blotting of cell lysates was performed to assess induction of ADAMTS-4 (B) and, at 3 days, type X collagen was assessed in cell lysates by SDS-PAGE/Western blotting (C). Tubulin was visualized as a loading control. Data are representative of three or more separate experiments for each parameter. D, CH-8 cells were plated in 24-well plates and treated with 10 ng/ml TNF-α or 100 ng/ml S100A11 for 2 days in replicates of six. One microCurie of 35S and 0.5 μCi of [3H]proline were added, and sulfated PG synthesis was assayed as described in Materials and Methods. Data are representative of three separate experiments.
CD36 transfection is associated with decreased S100A11-induced MKK3-p38 MAPK signaling essential for cartilage matrix catabolism in response to S100A11
S100A11 has been demonstrated to promote chondrocyte hypertrophy by RAGE signaling transduced by the MKK3-p38 MAPK pathway (8). Using congenic MKK3−/− and MKK3+/+ femoral head cartilage explants, we observed that cartilage GAG release and NITEGE neoepitope development were inhibited by MKK3 knockout in response to S100A11 (Fig. 6, A and B). Last, S100A11-induced MKK3 and p38 MAPK signaling were inhibited in CH-8 cells transfected with CD36 (Fig. 6C).
FIGURE 6.

S100A11-stimulated MKK3 signaling essential to S100A11-induced cartilage matrix catabolism is inhibited in CH-8 cells transfected with CD36. A, Congenic MMK3−/− and MKK3+/+ adult mouse femoral head cartilage explants were treated with 100 ng/ml S100A11. A, At 48 h, GAG release was measured in conditioned medium. Data are pooled from five different mouse donors of each genotype in replicates of three. B, Frozen sections of mouse femoral head cartilage explants were analyzed by immunohistochemistry for the aggrecanase neoepitope NITEGE at 72 h in culture. Data are representative of five different mouse donors of each genotype. C, CH-8 cells were plated in 96-well plates containing polyHEME and serum starved overnight. The cells were then treated with 100 ng/ml S100A11 for the duration indicated and SDS-PAGE/Western blotting was performed for phosphorylated and total MMK3 and p38. These data are representative of results from six separate experiments.
Discussion
Our analyses of experimental mouse and rat knee OA in this study reinforce evidence for the linkage between chondrocyte hypertrophic differentiation and matrix catabolism in OA articular cartilage (11). Chondrocyte RAGE signaling is essential for the inflammatory mediators TNF-α, CXCL8, and S100A11 to induce hypertrophy in cultured chondrocytes (8) and for certain alarmins to induce MMP-13 in chondrocytes (5). Hence, our finding that RAGE knockout was not chondroprotective for either mild or severe instability-induced mouse knee OA was unexpected. However, we focused on a model of relatively rapid OA development in young adult mice. Furthermore, baseline and OA-associated expression of RAGE by articular chondrocytes was relatively low in mouse knees in situ. RAGE expression may be too low in the earliest stages of the OA pathogenesis cascade in mice to significantly modulate OA progression. Moreover, RAGE may have a protective role in certain forms of inflammation and may promote tissue regeneration (29, 30). The loss of endogenous secretory RAGE and proteolytically released soluble RAGE in the joint also may have contributed to the lack of chondroprotection in OA of RAGE knockout mice, since soluble RAGE suppresses certain forms of inflammation, presumably by sequestering proinflammatory ligands of RAGE (26–28).
Baseline RAGE expression in adult rat knee articular cartilage (and in rat knees contralateral to surgically destabilized knees developing OA) was also observed to be sparse. Although knee chondrocyte RAGE expression remained low at 2 wk into the course of surgical instability-induced rat knee OA, RAGE progressively increased, such that cartilage RAGE expression in operated knees became robust at 4 wk after the procedure. IL-1β induces chondrocyte expression of RAGE (6) and release of at least two calgranulins (S100A11 and S100A9) (8, 25). Moreover, pericellular S100A11 is increased in human knee cartilages with advanced OA (8). As such, it remains possible that chondrocyte RAGE expression promotes cartilage degradation over longer periods of time than that studied here in OA and possibly in inflammatory arthritides. Baseline and OA-associated RAGE expression also may be different in joints other than the knee. Aging also could modulate RAGE-driven cartilage responses (6). For example, AGE ligands of RAGE increase in aging cartilage and can modulate cartilage matrix turnover in vitro (52).
We identified the alternative S100/calgranulin and patterning receptor CD36 as a marker of not only growth plate chondrocyte hypertrophy but also articular chondrocyte hypertrophy and aggrecan cleavage in rat knee OA. Baseline CD36 expression was sparse in normal rat and mouse knee articular cartilage, but in rat knee joint surgical destabilization-induced OA, CD36 expression focally increased at the sites of cartilage injury. We determined that chondrocyte expression of CD36 antagonized rather than shared the effects of RAGE in chondrocytes. In this regard, CD36 transfection resulted in suppression of the capacity of both S100A11 and TNF-α to induce chondrocyte hypertrophy and responses that promote matrix catabolism, but conversely allowed S100A11, but not TNF-α, to gain the capacity to induce PG synthesis. Suppression of the S100A11-induced MKK3-p38 pathway was implicated in anticatabolic effects of CD36 expression.
This study also established that the chondrocyte growth and anabolic factor IGF-I and PPARγ agonist treatment induced CD36 expression in both chondrocytic cells and cartilage explants. PPARγ agonists were previously observed to induce CD36 expression and reduce RAGE expression in cells other than chondrocytes (53). Moreover, PPARγ agonist treatment has reduced the severity of experimental OA (50, 51). Since we observed markedly induced CD36 in chondrocytic cells and cartilage explants, our results revealed that CD36 induction could contribute to the chondroprotective effects of PPARγ agonists in OA.
Whereas RAGE is a signaling receptor that does not internalize ligands (22, 26, 27), CD36, a transmembrane receptor with two cytosolic tails, can bind ligands, carry out ligand-induced signaling, and internalize and promote degradation of multiple ligands (33), with a variety of potential effects, alone or in combination, on cell differentiation and function (34, 35). Potential CD36 ligands in cartilage include not only certain calgranulins and AGE, but also non-RAGE ligands such as native and oxidized lipoproteins, oxidized phospholipids, and free long chain fatty acids (34, 35). In addition, CD36 is a receptor for thrombospondin-1, whose expression increases in retinoic acid-treated cartilage explants (25) and in OA cartilage in vivo (38). Hence, effects beyond the modulation of calgranulin-induced MKK3-p38 pathway signal transduction demonstrated here may contribute to the observed consequences associated with CD36 expression in chondrocytes.
Limitations of this study included that we did not address a full spectrum of mild-severe small animal OA models. Due to sparse expression of CD36 in mouse knee articular cartilage, we did not examine OA in CD36 knockout mice and did not knockdown CD36 mRNA. We have not yet explored whether RAGE-independent effects, such as modulation of cell viability by calgranulins (24, 31), may have contributed to the lack of chondroprotection by RAGE knockout in vivo. Some responses to the RAGE ligand HMGB1 are mediated by TLR2 and TLR4 (54). We have observed binding of S100A11 to CD36-expressing cells (D. Cecil and R. Terkeltaub, unpublished observations), but have not yet analyzed chondrocyte binding of other CD36 ligands. Last, we have not yet elucidated the molecular mechanism of CD36 effects in chondrocytes. Pitfalls of such work include that thorough assessment of dominant-negative forms of CD36 involves mutagenesis in both CD36 cytosolic tails, and such mutations can impair both signal transduction and ligand internalization (55). Moreover, study of potential endoproteolytic release of soluble CD36 by chondrocytes and functional autocrine and paracrine effects of soluble CD36 (56) were beyond the scope of this study.
In summary, RAGE and CD36 expression have opposing effects on responses to inflammatory stimuli in chondrocytes. Our results are further significant because CD36 has been reported to be increased in human OA cartilage (38) and CD36 genetic variation was linked with a number of human knee OA susceptibility traits in a recent longitudinal study that focused on older women (57). CD36 expression is linked with chondrocyte hypertrophy, and chondrocyte hypertrophy, which promotes OA progression (11), is induced by several inflammatory mediators (7, 8). However, CD36 is a novel example of a hypertrophic chondrocyte-expressed patterning receptor associated with cartilage repair responses to inflammatory stimuli. CD36 expression was relatively low in two models of rodent knee OA, likely contributing to a regenerative defect in articular cartilage. Factors that increase chondrocyte CD36 expression, including IGF-I and PPARγ agonism, merit further translational investigation as tools to suppress OA progression.
Footnotes
Funded by the Veterans Affairs Research Service, National Institutes of Health (HL077360, AR54135, PAG07998), a Canada Research Chair Award, and awards from the Canadian Institute for Health Research and the Canadian Arthritis Network (to F.B.).
Abbreviations used in this paper: OA, osteoarthritis; ACL, anterior cruciate ligament; ACLT, ACL transection; AGE, advanced glycation end product; GAG, glycosaminoglycan; GW1929, N-(2-benzoylphenyl)-O-[2-(methyl-2-pyridinylamino)ethyl]-L-tyrosine hydrate; HMGB1, high mobility group box protein 1; MKK3, mitogen-activated protein kinase kinase 3; PG, proteoglycan; RAGE, receptor for advanced glycation end product; TIMP3, tissue inhibitor of metalloproteinase 3; IGF-I, insulin-like growth factor I; MMP-13, matrix metalloproteinase 13; PPARγ, peroxisome proliferator-activated receptor γ.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Pelletier JP, Martel-Pelletier J, Abramson SB. Osteoarthritis, an inflammatory disease: potential implication for the selection of new therapeutic targets. Arthritis Rheum. 2001;44:1237–1247. doi: 10.1002/1529-0131(200106)44:6<1237::AID-ART214>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 2.Kobayashi M, Squires GR, Mousa A, Tanzer M, Zukor DJ, Antoniou J, Feige U, Poole AR. Role of interleukin-1 and tumor necrosis factor α in matrix degradation of human osteoarthritic cartilage. Arthritis Rheum. 2005;52:128–135. doi: 10.1002/art.20776. [DOI] [PubMed] [Google Scholar]
- 3.Borzì RM, Mazzetti I, Marcu KB, Facchini A. Chemokines in cartilage degradation. Clin Orthop Relat Res. 2004;427:S53–S61. doi: 10.1097/01.blo.0000143805.64755.4f. [DOI] [PubMed] [Google Scholar]
- 4.Loeser RF. Molecular mechanisms of cartilage destruction: mechanics, inflammatory mediators, and aging collide. Arthritis Rheum. 2006;54:1357–1360. doi: 10.1002/art.21813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yammani RR, Carlson CS, Bresnick AR, Loeser RF. Increase in production of matrix metalloproteinase 13 by human articular chondrocytes due to stimulation with S100A4: role of the receptor for advanced glycation end products. Arthritis Rheum. 2006;54:2901–2911. doi: 10.1002/art.22042. [DOI] [PubMed] [Google Scholar]
- 6.Loeser RF, Yammani RR, Carlson CS, Chen H, Cole A, Im HJ, Bursch LS, Yan SD. Articular chondrocytes express the receptor for advanced glycation end products: potential role in osteoarthritis. Arthritis Rheum. 2005;52:2376–2385. doi: 10.1002/art.21199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Merz D, Liu R, Johnson K, Terkeltaub R. IL-8/CXCL8 and growth-related oncogene α/CXCL1 induce chondrocyte hypertrophic differentiation. J Immunol. 2003;171:4406–4415. doi: 10.4049/jimmunol.171.8.4406. [DOI] [PubMed] [Google Scholar]
- 8.Cecil DL, Johnson K, Rediske J, Lotz M, Schmidt AM, Terkeltaub R. Inflammation-induced chondrocyte hypertrophy is driven by receptor for advanced glycation end products. J Immunol. 2005;175:8296–8302. doi: 10.4049/jimmunol.175.12.8296. [DOI] [PubMed] [Google Scholar]
- 9.Cecil DL, Terkeltaub R. Transamidation by transglutaminase 2 transforms S100A11 calgranulin into a procatabolic cytokine for chondrocytes. J Immunol. 2008;180:8378–8385. doi: 10.4049/jimmunol.180.12.8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldring MB, Goldring SR. Osteoarthritis. J Cell Physiol. 2007;213:626–634. doi: 10.1002/jcp.21258. [DOI] [PubMed] [Google Scholar]
- 11.Kamekura S, Kawasaki Y, Hoshi K, Shimoaka T, Chikuda H, Maruyama Z, Komori T, Sato S, Takeda S, Karsenty G, et al. Contribution of runt-related transcription factor 2 to the pathogenesis of osteoarthritis in mice after induction of knee joint instability. Arthritis Rheum. 2006;54:2462–2470. doi: 10.1002/art.22041. [DOI] [PubMed] [Google Scholar]
- 12.Guo J, Chung UI, Yang D, Karsenty G, Bringhurst FR, Kronenberg HM. PTH/PTHrP receptor delays chondrocyte hypertrophy via both Runx2-dependent and -independent pathways. Dev Biol. 2006;292:116–128. doi: 10.1016/j.ydbio.2005.12.044. [DOI] [PubMed] [Google Scholar]
- 13.Tchetina EV, Antoniou J, Tanzer M, Zukor DJ, Poole AR. Transforming growth factor-β2 suppresses collagen cleavage in cultured human osteoarthritic cartilage, reduces expression of genes associated with chondrocyte hypertrophy and degradation, and increases prostaglandin E2 production. Am J Pathol. 2006;168:131–140. doi: 10.2353/ajpath.2006.050369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown CC, Hembry RM, Reynolds JJ. Immunolocalization of metalloproteinases and their inhibitor in the rabbit growth plate. J Bone Joint Surg Am. 1989;71:580–593. [PubMed] [Google Scholar]
- 15.Stickens D, Behonick DJ, Ortega N, Heyer B, Hartenstein B, Yu Y, Fosang AJ, Schorpp-Kistner M, Angel P, Werb Z. Altered endochondral bone development in matrix metalloproteinase 13-deficient mice. Development. 2004;131:5883–5895. doi: 10.1242/dev.01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Merz D, Liu R, Johnson K, Terkeltaub R. IL-8/CXCL8 and growth-related oncogene α/CXCL1 induce chondrocyte hypertrophic differentiation. J Immunol. 2003;171:4406–4415. doi: 10.4049/jimmunol.171.8.4406. [DOI] [PubMed] [Google Scholar]
- 17.Wolf R, Howard OM, Dong HF, Voscopoulos C, Boeshans K, Winston J, Divi R, Gunsior M, Goldsmith P, Ahvazi B, et al. Chemotactic activity of S100A7 (psoriasin) is mediated by the receptor for advanced glycation end products and potentiates inflammation with highly homologous but functionally distinct S100A15. J Immunol. 2008;181:1499–1506. doi: 10.4049/jimmunol.181.2.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pietzsch J, Hoppmann S. Human S100A12: a novel key player in inflammation? Amino Acids. 2008 doi: 10.1007/s00726-008-0097-7. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 19.Santamaria-Kisiel L, Rintala-Dempsey AC, Shaw GS. Calcium-dependent and -independent interactions of the S100 protein family. Biochem J. 2006;396:201–214. doi: 10.1042/BJ20060195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bode G, Lüken A, Kerkhoff C, Roth J, Ludwig S, Nacken W. Interaction between S100A8/A9 and annexin A6 is involved in the calcium induced cell surface exposition of S100A8/A9. J Biol Chem. 2008;283:31776–31784. doi: 10.1074/jbc.M803908200. [DOI] [PubMed] [Google Scholar]
- 21.Okada M, Hatakeyama T, Itoh H, Tokuta N, Tokumitsu H, Kobayashi R. S100A1 is a novel molecular chaperone and a member of the Hsp70/Hsp90 multichaperone complex. J Biol Chem. 2004;279:4221–4233. doi: 10.1074/jbc.M309014200. [DOI] [PubMed] [Google Scholar]
- 22.Gebhardt C, Riehl A, Durchdewald M, Németh J, Fürstenberger G, Müller-Decker K, Enk A, Arnold B, Bierhaus A, Nawroth PP, et al. RAGE signaling sustains inflammation and promotes tumor development. J Exp Med. 2008;205:275–285. doi: 10.1084/jem.20070679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukocyte Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 24.Yui S, Nakatani Y, Mikami M. Calprotectin (S100A8/S100A9), an inflammatory protein complex from neutrophils with a broad apoptosis-inducing activity. Biol Pharm Bull. 2003;26:753–760. doi: 10.1248/bpb.26.753. [DOI] [PubMed] [Google Scholar]
- 25.Wilson R, Belluoccio D, Little CB, Fosang AJ, Bateman JF. Proteomic characterization of mouse cartilage degradation in vitro. Arthritis Rheum. 2008;58:3120–3131. doi: 10.1002/art.23789. [DOI] [PubMed] [Google Scholar]
- 26.Yan SF, D’Agati V, Schmidt AM, Ramasamy R. Receptor for advanced glycation endproducts (RAGE): a formidable force in the pathogenesis of the cardiovascular complications of diabetes and aging. Curr Mol Med. 2007;7:699–710. [PubMed] [Google Scholar]
- 27.Ramasamy R, Yan SF, Herold K, Clynes R, Schmidt AM. Receptor for advanced glycation end products: fundamental roles in the inflammatory response: winding the way to the pathogenesis of endothelial dysfunction and atherosclerosis. Ann NY Acad Sci. 2008;1126:7–13. doi: 10.1196/annals.1433.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamagishi S, Matsui T, Nakamura K. Kinetics, role and therapeutic implications of endogenous soluble form of receptor for advanced glycation end products (sRAGE) in diabetes. Curr Drug Targets. 2007;8:1138–1143. doi: 10.2174/138945007782151298. [DOI] [PubMed] [Google Scholar]
- 29.Rong LL, Yan SF, Wendt T, Hans D, Pachydaki S, Bucciarelli LG, Adebayo A, Qu W, Lu Y, Kostov K, et al. RAGE modulates peripheral nerve regeneration via recruitment of both inflammatory and axonal outgrowth pathways. FASEB J. 2004;18:1818–1825. doi: 10.1096/fj.04-1900com. [DOI] [PubMed] [Google Scholar]
- 30.Rong LL, Trojaborg W, Qu W, Kostov K, Yan SD, Gooch C, Szabolcs M, Hays AP, Schmidt AM. Antagonism of RAGE suppresses peripheral nerve regeneration. FASEB J. 2004;18:1812–1817. doi: 10.1096/fj.04-1899com. [DOI] [PubMed] [Google Scholar]
- 31.Ghavami S, Kerkhoff C, Chazin WJ, Kadkhoda K, Xiao W, Zuse A, Hashemi M, Eshraghi M, Schulze-Osthoff K, Klonisch T, Los M. S100A8/9 induces cell death via a novel: RAGE-independent pathway that involves selective release of Smac/DIABLO and Omi/HtrA2. Biochim Biophys Acta. 2008;1783:297–311. doi: 10.1016/j.bbamcr.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 32.Cai W, He JC, Zhu L, Chen X, Striker GE, Vlassara H. AGE-receptor-1 counteracts cellular oxidant stress induced by AGEs via negative regulation of p66shc-dependent FKHRL1 phosphorylation. Am J Physiol. 2008;294:C145–C152. doi: 10.1152/ajpcell.00350.2007. [DOI] [PubMed] [Google Scholar]
- 33.Ohgami N, Nagai R, Ikemoto M, Arai H, Miyazaki A, Hakamata H, Horiuchi S, Nakayama H. CD36 serves as a receptor for advanced glycation endproducts (AGE) J Diabetes Complications. 2002;16:56 –59. doi: 10.1016/s1056-8727(01)00208-2. [DOI] [PubMed] [Google Scholar]
- 34.Podrez EA, Febbraio M, Sheibani N, Schmitt D, Silverstein RL, Hajjar DP, Cohen PA, Frazier WA, Hoff HF, Hazen SL. Macrophage scavenger receptor CD36 is the major receptor for LDL modified by monocyte-generated reactive nitrogen species. J Clin Invest. 2000;105:1095–1108. doi: 10.1172/JCI8574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Febbraio M, Silverstein RL. CD36: implications in cardiovascular disease. Int J Biochem Cell Biol. 2007;39:2012–2030. doi: 10.1016/j.biocel.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moore KJ, El Khoury J, Medeiros LA, Terada K, Geula C, Luster AD, Freeman MW. A CD36-initiated signaling cascade mediates inflammatory effects of β-amyloid. J Biol Chem. 2002;277:47373–47379. doi: 10.1074/jbc.M208788200. [DOI] [PubMed] [Google Scholar]
- 37.El Khoury JB, Moore KJ, Means TK, Leung J, Terada K, Toft M, Freeman MW, Luster AD. CD36 mediates the innate host response to β-amyloid. J Exp Med. 2003;197:1657–1666. doi: 10.1084/jem.20021546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pfander D, Cramer T, Deuerling D, Weseloh G, Swoboda B. Expression of thrombospondin-1 and its receptor CD36 in human osteoarthritic cartilage. Ann Rheum Dis. 2000;59:448–454. doi: 10.1136/ard.59.6.448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wysk M, Yang DD, Lu HT, Flavell RA, Davis RJ. Requirement of mitogen-activated protein kinase kinase 3 (MKK3) for tumor necrosis factor-induced cytokine expression. Proc Natl Acad Sci USA. 1999;96:3763–3768. doi: 10.1073/pnas.96.7.3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weigand MA, Bierhaus A, Nicklas W, Kasper M, Hofer S, Plachky J, Grone HJ, Kurschus FC, Schmidt AM, Yan SD, et al. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest. 2004;113:1641–1650. doi: 10.1172/JCI18704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bendele A. Animal models. In: Bronner F, Farach-Carson MC, editors. Bone and Osteoarthritis. Vol. 4. Springer-Verlag; New York: 2007. pp. 149–163. [Google Scholar]
- 42.Appleton CT, McErlain DD, Pitelka V, Schwartz N, Bernier SM, Henry JL, Holdsworth DW, Beier F. Forced mobilization accelerates pathogenesis: characterization of a preclinical surgical model of osteoarthritis. Arthritis Res Ther. 2007;9:R13. doi: 10.1186/ar2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Appleton CT, Usmani SE, Bernier SM, Aigner T, Beier F. Transforming growth factor α suppression of articular chondrocyte phenotype and Sox9 expression in a rat model of osteoarthritis. Arthritis Rheum. 2007;56:3693–3705. doi: 10.1002/art.22968. [DOI] [PubMed] [Google Scholar]
- 44.Appleton CT, Pitelka V, Henry J, Beier F. Global analyses of gene expression in early experimental osteoarthritis. Arthritis Rheum. 2007;56:1854–1868. doi: 10.1002/art.22711. [DOI] [PubMed] [Google Scholar]
- 45.Hughes CE, Caterson B, Fosang AJ, Roughley PJ, Mort JS. Monoclonal antibodies that specifically recognize neoepitope sequences generated by “aggrecanase” and matrix metalloproteinase cleavage of aggrecan: application to catabolism in situ and in vitro. Biochem J. 1995;305:799–804. doi: 10.1042/bj3050799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burton DW, Foster M, Johnson KA, Hiramoto M, Deftos LJ, Terkeltaub R. Chondrocyte calcium-sensing receptor expression is up-regulated in early guinea pig knee osteoarthritis and modulates PTHrP: MMP-13, and TIMP-3 expression. Osteoarthritis Cartilage. 2005;13:395–404. doi: 10.1016/j.joca.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 47.Johnson KA, Yao W, Lane NE, Naquet P, Terkeltaub RA. Vanin-1 pantetheinase drives increased chondrogenic potential of mesenchymal precursors in ank/ank mice. Am J Pathol. 2008;172:440–453. doi: 10.2353/ajpath.2008.070753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chambers MG, Kuffner T, Cowan SK, Cheah KS, Mason RM. Expression of collagen and aggrecan genes in normal and osteoarthritic murine knee joints. Osteoarthritis Cartilage. 2002;10:51–61. doi: 10.1053/joca.2001.0481. [DOI] [PubMed] [Google Scholar]
- 49.Lim HJ, Lee S, Lee KS, Park JH, Jang Y, Lee EJ, Park HY. PPARγ activation induces CD36 expression and stimulates foam cell like changes in rVSMCs. Prostaglandins Other Lipid Mediat. 2006;80:165–174. doi: 10.1016/j.prostaglandins.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 50.Boileau C, Martel-Pelletier J, Fahmi H, Mineau F, Boily M, Pelletier JP. The peroxisome proliferator-activated receptor γ agonist pioglitazone reduces the development of cartilage lesions in an experimental dog model of osteoarthritis: in vivo protective effects mediated through the inhibition of key signaling and catabolic pathways. Arthritis Rheum. 2007;56:2288–2298. doi: 10.1002/art.22726. [DOI] [PubMed] [Google Scholar]
- 51.Kobayashi T, Notoya K, Naito T, Unno S, Nakamura A, Martel-Pelletier J, Pelletier JP. Pioglitazone, a peroxisome proliferator-activated receptor γ agonist, reduces the progression of experimental osteoarthritis in guinea pigs. Arthritis Rheum. 2005;52:479–487. doi: 10.1002/art.20792. [DOI] [PubMed] [Google Scholar]
- 52.DeGroot J, Verzijl N, Jacobs KM, Budde M, Bank RA, Bijlsma JW, TeKoppele JM, Lafeber FP. Accumulation of advanced glycation endproducts reduces chondrocyte-mediated extracellular matrix turnover in human articular cartilage. Osteoarthritis Cartilage. 2001;9:720–726. doi: 10.1053/joca.2001.0469. [DOI] [PubMed] [Google Scholar]
- 53.Wang K, Zhou Z, Zhang M, Fan L, Forudi F, Zhou X, Qu W, Lincoff AM, Schmidt AM, Topol EJ, Penn MS. Peroxisome proliferator-activated receptor γ down-regulates receptor for advanced glycation end products and inhibits smooth muscle cell proliferation in a diabetic and nondiabetic rat carotid artery injury model. J Pharmacol Exp Ther. 2006;317:37–43. doi: 10.1124/jpet.105.095125. [DOI] [PubMed] [Google Scholar]
- 54.Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, Fenton MJ, Tracey KJ, Yang H. HMGB1 signals through Toll-like receptor (TLR) 4 and TLR2. Shock. 2006;26:174–179. doi: 10.1097/01.shk.0000225404.51320.82. [DOI] [PubMed] [Google Scholar]
- 55.Lee KJ, Ha ES, Kim MK, Lee SH, Suh JS, Lee SH, Park KH, Park JH, Kim DJ, Kang D, et al. CD36 signaling inhibits the translation of heat shock protein 70 induced by oxidized low density lipoprotein through activation of peroxisome proliferators-activated receptor. γ. Exp Mol Med. 2008;40:658–668. doi: 10.3858/emm.2008.40.6.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Handberg A, Skjelland M, Michelsen AE, Sagen EL, Krohg-Sørensen K, Russell D, Dahl A, Ueland T, Oie E, Aukrust P, Halvorsen B. Soluble CD36 in plasma is increased in patients with symptomatic atherosclerotic carotid plaques and is related to plaque instability. Stroke. 2008;39:3092–3095. doi: 10.1161/STROKEAHA.108.517128. [DOI] [PubMed] [Google Scholar]
- 57.Valdes AM, Hart DJ, Jones KA, Surdulescu G, Swarbrick P, Doyle DV, Schafer AJ, Spector TD. Association study of candidate genes for the prevalence and progression of knee osteoarthritis. Arthritis Rheum. 2004;50:2497–2507. doi: 10.1002/art.20443. [DOI] [PubMed] [Google Scholar]