

Abstract
DB921 has a benzimidazole-biphenyl system with terminal amidines that gives the compound a linear conformation with a radius of curvature that does not match the DNA minor groove shape. Surprisingly, the compound binds in the groove with an unusually high equilibrium constant [Miao, Y.; Lee, M. P. H.; Parkinson, G. N.; Batista-Parra, A.; Ismail, M. A.; Neidle, S.; Boykin, D. W.; Wilson, W. D. Biochemistry 2005, 44, 14701-14708]. X-ray crystallographic analysis of DB921 bound to -AATT- in d(CGCGAATTCGCG)2 showed that the benzimidazole is in position to directly interact with bases at the floor of the groove while the phenylamidine of DB921 forms indirect contacts with the bases through an interfacial water. The DB921-water pair forms a curved, flexible module with a high Ka (or a low Kd) value of binding. To better understand the dynamics of the DB921-DNA complex and how water can be used in the design of compounds to recognize DNA, a 100 ns molecular dynamics simulation of the complex was conducted. In addition to the X-ray conformation some significantly variant, dynamic conformations, which had additional interfacial water molecules between DB921 and DNA, appeared in the MD simulation. The benzimidazole contacts remained relatively constant through the entire simulation. The biphenyl-amidine end of the bound molecule, however, undergoes much larger changes in orientation relative to the floor of the groove as well as variations in the type of water interactions. The results provide an understanding of how water couples the linear DB921 compound to the minor groove for tight binding, without a large unfavorable contribution to the entropy of binding.
1 Introduction
Dicationic heterocyclic systems that bind in the DNA minor groove are promising agents against the kinetoplastid parasites that cause deadly infectious diseases1. Compounds such as furamidine (DB75, Figure 1-Top), which have a curvature that complements that of the DNA minor groove, bind strongly to parasite DNA and cause rapid destruction of the mitochondrial kinetoplast2, 3. In contrast to the curved compounds, the binding of linear molecules has been investigated less frequently. The classical model for minor groove binding emphasizes the complementary curvature of DNA and the binding compound4, 5. This model indicates that compounds, which are linear or have a large radius of curvature, should bind weakly to the minor groove. DB921 (Figure 1-Middle), a linear heterocyclic diamidine, however, binds strongly to the DNA minor groove, even more strongly than curved compounds6. The discovery of such linear molecules that bind to the minor groove necessitates the reevaluation of traditional views with regard to the curvature requirement for binding6, 7, 8, 9.
Figure 1.
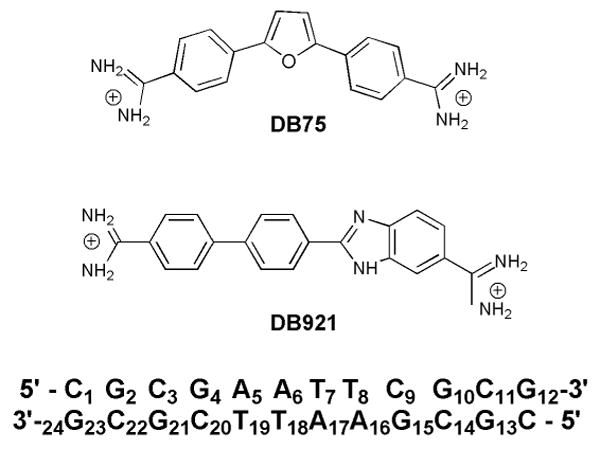
Top: DB75, a classical minor groove binder with a curvature that is complementary to the minor groove. Middle: DB921, a linear benzimidazole, biphenyl diamidine. The amidine on the phenyl is the one described in detail in the text and figures. Bottom: DNA segment with the number scheme used in the text and figure.
An X-ray crystal structure of the DB921-d(CGCGAATTCGCG)2 complex has been solved (PDB ID: 2B0K) with DB921 bound at the -AATT- site6. The structure shows that a water-mediated, non-covalent interaction between the phenylamidine of DB921 and DNA, in addition to direct benzimidazole-amidine-DNA non-bonded interactions, can serve to complete the curvature of the bound system and yield a very energetically favorable complex. It should be emphasized that the complexed water is quite different from the more loosely bound and quite dynamic water that externally hydrates most DNA minor groove complexes10, 11. The specific water that links the phenyl-amidine of DB921 to the bases at the floor of the groove is different from stabilizing water molecules observed in minor groove complexes previously7. The linking water forms hydrogen bonds between the amidine of DB921, which does not directly interact with DNA bases, and an adenine base. On the other hand, complexes that involve the binding of traditional crescent shaped compounds in the minor groove are frequently stabilized by water in or out of the groove, but these are external to the compound-base interactions and the waters are localized at the ends of the complex or at the outer edge of the minor groove7.
Water molecules, such as the one in the DB921-DNA complex, are often observed to mediate intermolecular interactions within the binding cavities of macromolecules. Interfacial water molecules connect two solute molecules by forming hydrogen bonds and/or other interactions with each of them. These interfacial waters are the subject of active research since they are essential components in many macromolecular complexes. In many cases, a compound binding to a macromolecular site is accompanied by the release of localized water molecules that previously occupied the site12. The gain in entropy by the release of these water molecules is generally larger than the loss of enthalpy due to breaking hydrogen bonds between the water molecules and the binding site as they are released into the relatively more disordered bulk water environment. The conservation of water molecules in an unoccupied binding site results in a relative entropy loss that may be compensated by the favorable enthalpic contributions of these interfacial waters12, 13.
The ability of the relatively flexible water molecules to mediate dynamic, noncovalent interactions by acting as adaptable hydrogen bond donors or acceptors to stabilize ligand-macromolecule complexes has been observed in both protein and nucleic acid complexes (recently reviewed12, 14, 15). There are interesting cases when the expulsion of a water molecule from the binding cavity by ligands actually decreases the binding affinity16. In the case of the Concanavalin A - trimannoside complex, an analog of trimannoside was designed to displace the bound water molecule and this resulted in a decrease in binding energy16. In another study, the modulation of the network of water molecules in the binding cavity of OppA by various tripeptides was observed to result in higher binding constants by tripeptide variants that were able to conserve water molecules as opposed to variants that displaced the water17. The curved compound DB911 binds to the -AATT- minor groove site in DNA significantly more weakly than the DB921-water complex6. These observations illustrate a critical point – water molecules in the binding cavity can orient themselves to provide favorable interactions between the ligand and bioreceptor and hence improve the binding affinity of the compound.
Although bound waters that mediate protein-ligand interactions are well known, the DB921-DNA complex provides a unique and well-defined system for analysis of water-mediated binding in the context of a DNA complex. The system also provides valuable new information for incorporation of water towards the design of new lead scaffolds. To better understand the dynamics of the bound water observed in the X-ray crystal structure of the DB921 complex as well as the dynamics of the entire complex, molecular dynamics (MD) simulations have been conducted. MD simulations can capture dynamic conformational variation of biomolecular complexes as a complement to the structural information provided by X-ray and NMR structures. Further, MD provides a detailed molecular characterization of multiple binding modes, transitions and their dynamic interconversions that are difficult to obtain by other methods10, 18, 19. The DB921 simulation detected multiple modes of binding and revealed that bound waters with different residence times lead to several low energy conformational states. Additionally, we identified one water molecule that resides adjacent to a nonpolar pocket between the biphenyl system of DB921 and the -AATT- DNA minor groove site. Simulation also shows that the biphenyl-amidine part of the DB921 complex is dynamic and involves multiple conformational modes stabilized by relatively mobile bound waters. Details of a 100 ns simulation of the DB921-DNA complex are reported here.
2 Methods
A. Parametrization of DB921 for use with the Cornell et al. force field
The Cornell force field20 used in the AMBER 9.021 suite of programs contains parameters for many atom types to reproduce bonded and non-bonded interactions of atoms present in various chemical bonding environments. These parameters have been extensively used in simulating the dynamic behavior of nucleic acids, proteins, inter-molecular interactions between macromolecules, as well as ligand-macromolecular interactions.
Atom types used in force field parametrizations represent a particular atom in a specific chemical bonding context. In many cases, analogous atom types can be found that represent the chemical bonding environments for each atom in the compound under consideration. Though most atom types required to parametrize DB921 were present in the Cornell force field, the dihedral parameters that suitably reproduce torsional profiles were absent, and a new set of force field parameters were derived for the DB921 molecule. A detailed discussion of force field parameters for DB921 is presented in the Supplementary Material (Section 1).
The parametrization procedures we have followed to derive the new set of parameters reflect standard techniques used in AMBER ff9922 and have been previously outlined by Hopfinger and co-workers23. An appropriate set of atom types (see the MASS section of Figure S2) was used to generate these force field parameters with most atom types taken from the AMBER ff99 parameter set. To ensure reproducibility, the DB921 charges were derived using the R.E.D (RESP ESP charge Derive) algorithm24 that, sequentially, optimizes geometry using ab initio methods at the HF/6-31G* level (via GAUSSIAN25), and then uses the RESP26, 27 methodology to obtain atom-centered charges.
B. Molecular dynamics protocol
The starting DNA structure (d(CGCGAATTCGCG)2 duplex) for MD simulations was taken from the PDB file6 (Figure 2). DB921 was optimized using the new force field parameters (see Table S1 and Figure S2) and was then docked into the central -AATT- minor groove region by superposing it on the X-ray crystal structure using SYBYL 6.9.228. The duplex with the minimized, docked ligand was then stored as a PDB file for use with the AMBER suite. The DNA complex was placed in a truncated octahedral water box, using the TIP3P29 water model, of approximately 4000 water molecules by using the LEaP module in the AMBER 9.0 suite of programs. The image separation distance for the periodic boundary conditions was 19 Å. A salt concentration of approximately 0.15 M was maintained by adding 28 Na+ ions and 8 Cl- ions. The MD simulation was performed with the SANDER module of the AMBER 9.0 suite on 16 CPUs of an Apple Xserve G5 cluster. The simulation progressed at a rate of 26181 s/ns (∼3.3 ns/day). The experiment was performed in an NPT ensemble with periodic boundary conditions at a constant temperature of 300 K and a pressure of 1 bar. SHAKE30 algorithm was applied to all bonds involving hydrogen atoms with an integration time step of 2 fs. The Particle Mesh Ewald31 (PME) method was used to handle coulombic interactions and a 9 Å cut-off was applied on all van der Waals interactions.
Figure 2.
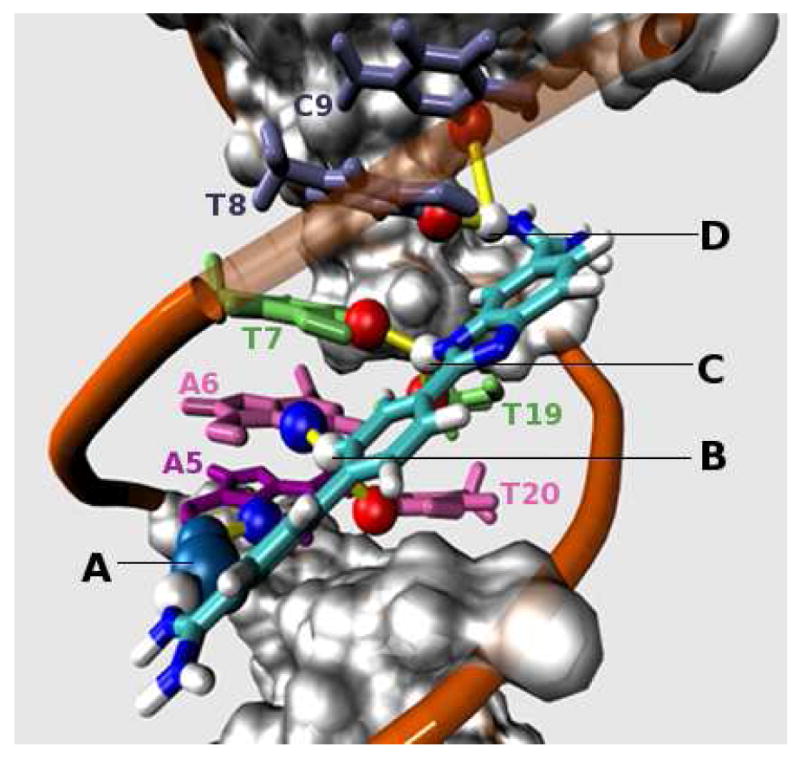
The DB921-d(CGCGAATTCGCG)2 X-ray structure (PDB ID: 2B0K) with added protons. Yellow colored cylindrical lines represent hydrogen bonds formed between groups at the start and end points of the line. Hydrogen bonds are defined as interactions between acceptor and donor atoms that are less than 3.4 Å apart. Legends: (A) Water-mediated, hydrogen-bonding interaction between the phenylamidine of DB921 and A5-N3 (see numbering scheme in Figure 1-Bottom) of the DNA minor groove. (B) Two short C – H•••N/O type interaction of –CH of the central phenyl of DB921 with A6-N3 and T20-O2. (C) Bifurcated H-bonded interactions of the inner benzimidazole nitrogen (-NH) with T7-O2 and T19-O2. (D) Bifurcated hydrogen bond between benzimidazole side amidine and T8-O2 and C9-O2.
In the multistage equilibration protocol32, the DNA-DB921 complex was restrained with a 500 kcal/mol Å2 restraint and the solvent structure of the system was relaxed over 1000 cycles of potential energy minimization. This was followed by a 25 ps MD to warm up the system from 100 K to 300 K, with the DNA/ligand held fixed with a 100 kcal/mol Å2 restraint, using a 1 fs time step. The system was further equilibrated by 25 ps of MD at 300 K, with the restraint on the complex reduced to 50 kcal/mol Å2, using a 2 fs time step. A five stage minimization protocol, each of 1000 steps, was then used with the restraint gradually reduced by 5 kcal/mol Å2 progressively while starting with a 25 kcal/mol Å2 restraint. Finally, since the multiple minimization steps cool down the system, it was re-heated from 100 K to 300 K using a 10 ps MD simulation with no harmonic restraints on the complex.
It has been recently reported that MD simulations of 10 ns or more can produce erroneous representations of α/γ concerted rotations in DNA structures when the AMBER ff99 (or older) force field is used33. The study shows that large populations of α/γ = (g+, t) were observed due to single, irreversible α/γ transitions. This bias was found to be particularly obvious in long range simulations. To correct this, new parameters for the α/γ torsional terms were derived while retaining the remaining parameters and we have, thus, used the PARMBSC0 force field. As observed in the original study33, we found very stable values for the α/γ angles in the DB921 complex through the entire 100 ns simulation (see Figure S8 for plots of α/γ angles observed in this study). Finally, a recent study34 suggests the use of Smith and Dang34 parameters for Na+ and Cl- ions to avoid aggregation at concentrations greater than 1.0 M. We have used a lower salt concentration of 0.15 M and have not observed any salt aggregation up to 100 ns of simulation.
C. Trajectory analysis and characterization of binding mode
VMD35 was used for visualization and analysis of molecular trajectories. For DB921, multiple binding modes were observed in the simulations and were characterized by Tcl scripts. The first script constructed a geometric reference plane that was used to partition the trajectory into the different binding conformations. As shown in Figures 3 and 4 (Label A), three phosphorous atoms on the DNA-backbone were used to define the reference plane. This plane maintains an approximately parallel orientation to the floor of the DNA and, importantly, provides a relatively rigid and visually consistent reference to measure the angle formed by the amidine group of DB921 (see Supplementary Material, Section 2, for a detailed discussion about the construction and usage of the reference plane). The angle formed between the line joining the two nitrogens of the amidine (Label B, Figures 3 and 4), on the phenyl side, to the reference plane was approximately either 0° or 90° in each frame (see Supplementary Material, Section 2, for a discussion on this approximation). The partitioning of trajectories was based on this angle measurement of the phenylamidine group of DB921 with respect to the plane (see Labels A and B in Figures 3 and 4). Further, individual programs were written to calculate the respective distances and intermolecular interactions that characterize the DNA-DB921 complex in each partitioned trajectory. The programs captured the exclusive interactions that stabilize the respective conformations in each of the modes. Finally, to capture the residence time of the water molecule between the DNA minor groove and DB921 (Label C, Figures 3 and 4), we counted the dwell time distribution as described previously36. The time distribution was then fit to an exponential to get the residence time.
Figure 3.
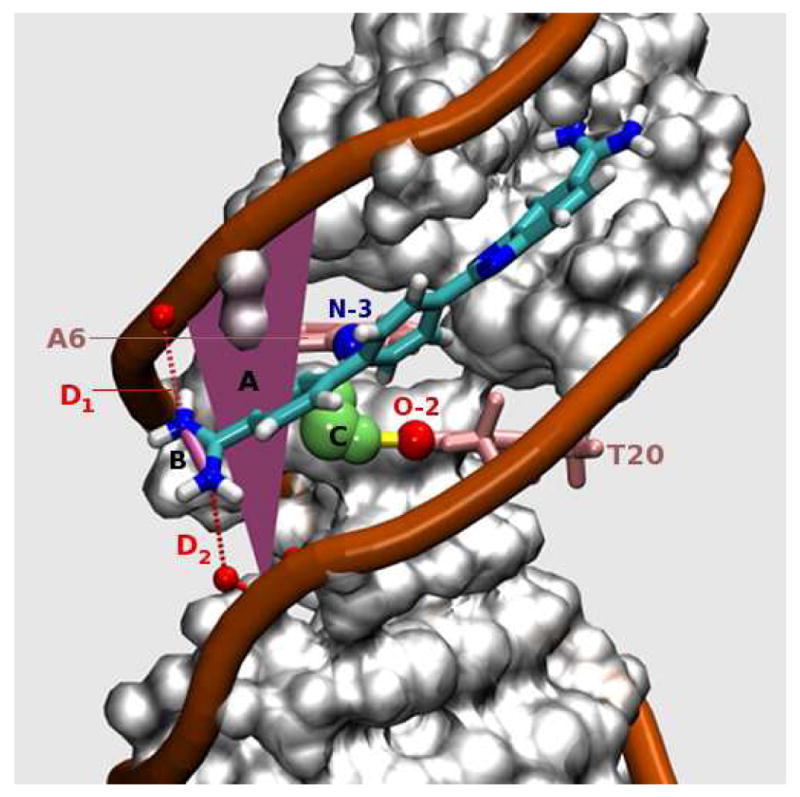
DB921-DNA complex in the parallel conformation (‖). The angle from the line, labeled B, (connecting N-N of the phenylamidine) to reference plane, labeled A, is close to 0° (+/- 20°), and thus the figure represents one of the parallel conformations (‖) observed in the MD simulation. The yellow cylindrical lines represent H-bonds. Legends: (A) Plane of reference constructed to computationally classify each frame (20 ps snapshot) into ‖ or ⊥ modes of interaction. The plane is approximately parallel to the floor of the DNA minor groove (see text for details). (B) Line used to calculate the angle made by the amidine group to the reference plane (and hence, approximately to the floor of the groove). This angle is calculated for each snapshot of the MD and is used as an index to classify the frame into its respective binding mode. (C) Long residence time water observed consistently in the simulation. The persistent DNA H-bonding partners for this water are A6-N3 (labeled in blue) and T20-O2 (labeled in red). (D) The phosphate-amidine electrostatic interactions are denoted by D1 and D2 (red dotted lines).
Figure 4.
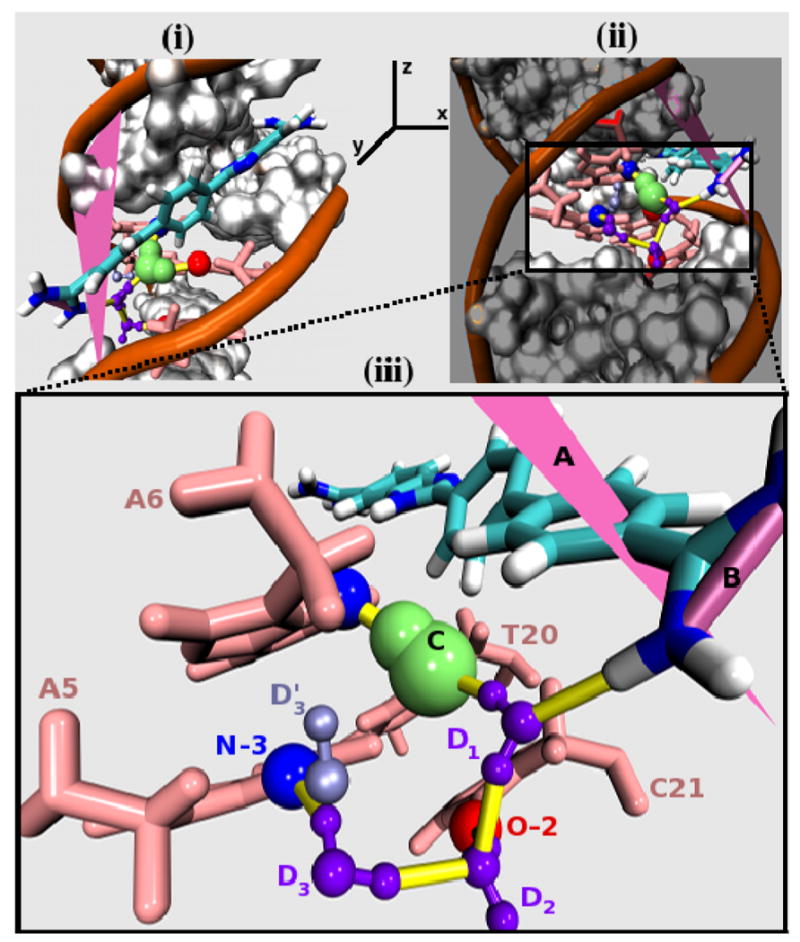
A DB921-DNA complex in the perpendicular mode (⊥-Major). (i) DB921-DNA complex in a comparable orientation as Figures 2 and 3. (ii) With respect to the axis shown in (i), this orientation is obtained by rotating (i) by approximately 90° anti-clockwise about the z-axis and approximately 40° clockwise about the x-axis. (iii) A magnified view of non-covalent interactions that are typical of the ⊥-Major mode of binding. The angle made by the line, labeled B, to the reference plane, labeled A, is close to 90° (+/- 20) and thus the figures represent one of the perpendicular conformations (⊥-Major) observed in the MD simulation that is different from the X-ray crystal structure. The yellow cylindrical lines represent H-bonds. Legends: (A) Plane of reference; (B) Line used to calculate the angle between the amidine groups to the reference plane; (C) Long residence time water, observed consistently in the simulation. The persistent H-bonding partners for this water are A6-N3 and T20-O2. (D1 through D3) Water molecules that mediate transient H-bonding interactions involving A5-N3 and C21-O2 and stabilize the binding of DB921 to the minor groove (see text for description). (D3′) The alternative orientation of the water molecule D3 associated with A5-N3.
3 Results and Discussion
The goal of the project is to investigate the indirect water-mediated binding of DB921 to the minor groove as indicated by the X-ray structure (Figure 2), and to describe the dynamics of the water molecule that mediates the DNA-ligand interaction. Our initial visual inspection of the trajectories indicated that, in addition to the binding mode observed in the X-ray crystal structure, two other distinct and persistent conformations were identified. Even though these three binding modes were distinguishable by visual inspection, the specific interactions that contribute to facilitate them were not as visually apparent and we conducted a more detailed analysis of all three structural families.
A. Analysis and Classification of MD trajectories
The X-ray crystal structure of DB921 (Figure 2) provides a reference for the conformational states seen in the molecular dynamics simulation. Figure 2 describes four interactions of the ligand (Labels A, B, C and D) that provide multiple non-covalent contacts with the DNA. These stabilizing interactions include an indirect H-bond network (Label A, Figure 2) connecting A5-N3 to the amidinium group at the biphenyl side of DB921 that is mediated by a water molecule. The MD simulation was carried out to further investigate the dynamics of these interactions.
As described in the Supplementary Material (Section 1), two sets of parameters were tested to determine how well they could reproduce dihedral profiles derived from ab initio calculations. The first set was from GAFF37 and the second was a new set we have derived. In general, GAFF has parameters for most commonly used organic molecules and provides a convenient framework to extend this set to parametrize new molecules. In the case of DB921, atom types and dihedral parameters present in GAFF were not sufficient to generate the potential energy profile obtained using the HF/6-31G* basis set (see Figures S5(b), S6(b) and S7(b)). As observed in other studies38, GAFF may give inaccurate potential barriers for compounds with aromatic conjugated systems that have torsional freedom. If used directly, they can be expected to significantly and incorrectly restrain conformational variation of the four dihedrals across the DB921 molecule during simulation. On the other hand, the modified parameters, which were partly derived by analogy to the AMBER ff99 force field, induce appropriate torsional flexibility in the small molecule and, thus, enable sampling of conformational space that facilitate the diverse intermolecular interactions with the minor groove. The modified set of parameters is comprised of derived dihedral constants along with parameters (electrostatics and van der Waals) and atom types that were adapted from the AMBER ff99 force field (see Supplementary Material, Section 1, for a detailed discussion of the DB921 force field).
In the 100 ns simulation, using these new force field parameters, it was observed that the conformation of DB921 and its interaction with DNA remain essentially constant on the benzimidazole side of the molecule throughout the simulation due to strong hydrogen bonds and close interaction with the DNA minor groove. The interactions on this side are essentially those observed in the X-ray structure (Labels C and D in Figure 2). On the other hand, there are large torsional dynamics across the biphenyl system of DB921 and the dihedral formed between the terminal phenyl and amidine group. These fluctuations sample torsional angles within the range defined by the respective torsional profiles (see Figures S5(a), S6(a) and S7(a)). The simulation identified interfacial water molecules that are critical to the structural variations. A particularly interesting water molecule was present in a dynamic cavity between the biphenyl group of DB921 and the minor groove of the DNA. Further, the variations are stabilized by dynamic non-bonded interactions between the amidine on the biphenyl side, water and DNA. These structural fluctuations in the biphenyl-amidine system are in agreement with the large B-factors for this area of the DB921-DNA crystal structure6. It was observed that the amidine (phenyl side) preferred to adopt either a parallel (‖, Figure 3) or a perpendicular (⊥, Figure 4) conformation with respect to the floor of the minor groove. As described in the Methods, a reference plane was used to categorize the orientation of the phenylamidine with respect to the DNA minor groove. The reference plane, which was defined by three atoms of the DNA (Label A in Figures 3 and 4), allowed partitioning of the trajectories into the various binding conformations, counting their respective populations and capturing structural and chemical features that distinguished the binding modes (see Supplementary Material, Section 2, for a description of the construction of the plane and details about the classification into the two main binding modes).
A total of three binding modes that had distinct non-bonded interactions were observed in the simulation. The ‖(Figure 3) conformation is characterized by electrostatic interactions and/or hydrogen bonds between one or two oxygens of the DNA backbone and the phenylamidine of DB921 (Label D, Figure 3). Label D in Figure 3 shows lines (in red) drawn from the amidine to the oxygens in the backbone and highlights participating partners in non-bonded interactions. Scripts were used to monitor the distances between these substituents. In the perpendicular conformation (Figure 4) it was observed that there are two possible modes of interaction, namely, ⊥-Major and ⊥-Xray. The first of these involves the formation of a hydrogen bonding network involving two or more water molecules between DB921 and DNA (Label D, Figure 4), and is referred to as ⊥-Major. Finally, the ⊥-Xray mode of binding involved interactions of only one interfacial water molecule (the snapshot of this observed conformation is not shown separately in any figure since it is very similar to the X-ray crystal conformation shown in Figure 2). The primary feature of differentiation between ⊥-Major and ⊥-Xray was the nature of non-bonded interactions due to differences in the number of waters involved in stabilizing the complex. Note that both the ⊥-Major and ⊥-Xray adopt a conformation that is approximately perpendicular to the floor of the minor groove (see Figure 4).
B. Bimodal separation between the parallel and perpendicular modes of binding
The scatter plot of the angle made by the phenylamidine nitrogens (considered to be equivalent) to the reference plane in each frame across the length of simulation is shown in Figure 5. Since the aim was to check if the angle made by the amidine (Label B, Figures 3 and 4) was either around 90° (⊥ mode) or 0° (‖ mode) only the absolute values were used. Hence the sign (+ or -) of the angles is ignored.
Figure 5.
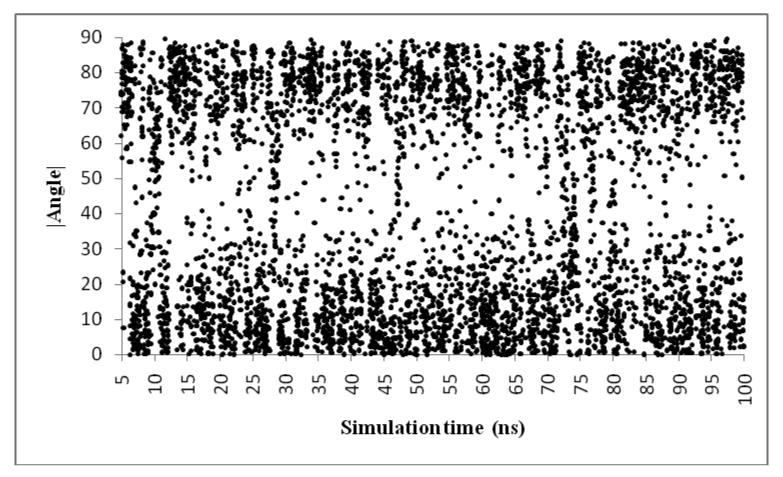
Scatter plot of angle made by the phenylamidine of DB921 (Label B in Figures 3 and 4) and the reference plane (Label A in Figures 3 and 4) across the length of the simulation. The y-axis is the absolute value of this angle, and the x-axis is the time during the simulation (5-100 ns). It is observed that there is a concentration of values around 0° (+/- 20°) and 90c (+/- 20°). The populations on the bottom and top of the plot are the groups of ‖ and ⊥ conformations respectively.
As seen in Figure 5, there is a concentration of data points within the range of +/- 20° of 90° and of 0°. This shows a bimodal split in populations between the parallel and perpendicular modes of binding with significant concentration of the observed MD conformations into the two clusters. Since we have used 20 ps snapshots across 100 ns for this analysis, the trajectory consisted of a total of 5000 frames. The first 250 frames (5 ns) consisted of a large number of transitory frames and were not used in the analysis. This was considered as the time required for the system to equilibrate. Across the 4750 frames (95 ns) that were analyzed, the populations were closely balanced with 2313 frames (46.2 ns) parallel (‖) and 1948 frames (38.9 ns) perpendicular (⊥). Points that lie in the region between the two clusters in Figure 5 represent 489 frames (9.7 ns) and were classified as transitory conformations. While the X-ray structure suggested that the binding of DB921 to the DNA minor groove (Figure 2) is facilitated by a hydrogen bond network that is mediated by a single water molecule (Label A, Figure 2) connecting A5-N3 and the phenylamidine of DB921, the MD simulations agree and highlight two other modes of binding denoted by ‖ and ⊥-Major (explained above). The complex stayed in the ⊥-Xray conformation for an aggregate of 2.4 ns during the simulation. Note that the percentage population of each of the modes depends on various factors and could be represented by a higher or lower fraction subject to environment conditions. The ability to jump from the ⊥ mode seen in the X-ray structure to the ‖ mode increases the conformational states of the bound DB921. These conformational states explain how the compound binds without a large loss of entropy as compared to curved molecules6.
C. Interfacial water
An unanticipated observation in the MD simulation was the presence of a long residence-time interfacial water detected in the region between the biphenyl system of DB921 and the floor of the minor groove (Label C, in Figures 3 and 4). This water molecule resides in a cavity with polar and nonpolar regions that is created by the dynamic motions of DB921, water and, to a less extent, DNA. The DNA that is directly opposite the biphenyl system of DB921 forms the polar region of the cavity. Specifically, the bases A6 and T20 facilitate the formation of hydrogen bonds with this water molecule, while the biphenyl system of DB921 forms a nonpolar wall in the cavity. The oxygen of the water is in the proximity of a partially positively charged -CH group of this biphenyl system to enable favorable electrostatic interactions.
This observation raises the question of how common a similar water environment is in other macromolecular complexes. The first crystal structure to show a critical role for water in completing the interface in a protein-DNA interaction was with the trp repressor-operator complex39, 40, 41 (PDB ID: 1TRO). Similar to the water molecule (Label C, Figures 3 and 4) in the DB921-DNA complex, a number of water molecules make contacts between nonpolar protein amino acid side chains with H-bonds to DNA bases. Water 397 in PDB 1TRO, for example, makes nonpolar contacts to the T6 methyl (O to C distance of 3.44 Å) and the Ala60-CH3 (O-C of 4.06 Å). It also has polar contacts to the T6 O4 (O-O of 2.67 Å) and other water molecules.
A recent example, in which water molecules are present in a binding cavity that has polar-hydrogen bonding regions opposite nonpolar walls, is the P22 repressor-operator complex42. Water molecules at each end of the DNA binding sequence have interactions similar to that of the DB921 biphenyl-DNA-water contacts. In the P22 complex (PDB ID: 2R1J), water 70 is 3.47 Å from the C of the T3 methyl and 3.70 Å from the T4 methyl C on the nonpolar face of the water cavity. Hydrogen bonds are made from water 70 to the Gln 21 amide O (2.72 Å), the Asn 32 amide N (2.85 Å), and to water 27 (2.83 Å) on the polar side of the binding cavity. Very similar contacts are made by water 71 on the opposite end of the DNA binding sequence (T 23 and 24 methyl contacts). This asymmetric polar-nonpolar cavity for water is quite similar to the cavity with a nonpolar face on DB921 and H-bonds to DNA bases at the floor of the minor groove.
The various types and energetics of water-mediated protein-DNA interactions have been recently reviewed15. This type of water interaction must add a favorable Gibbs energy term to the complex through a combination of terms that include H-bonds, van der Waals and electrostatic (partial atomic charges) interactions. The interstitial water with its flexible orientations and properties (particularly as an isolated molecule) fills what would otherwise be an empty space and provides favorable interactions.
As mentioned in the Methods section, a survival probability function was used to quantitatively evaluate the dynamics of the long-residence time water in the DB921-DNA complex. The time distribution fit a first order kinetics exponential function with a correlation coefficient of 0.98 (Figure 6) and the residence time of this dynamic water molecule was found to be 1.6 ns. The release of this water molecule in transient frames facilitated the shift of the conformation to the ⊥-Xray, crystal-like, conformation of DB921. The presence of this water molecule acts like a fulcrum, which pushes the phenyl side of DB921 away from the floor of the minor groove, facilitating conformational fluctuations that would include torsional fluctuations of the phenyl system across the phenyl-phenyl bond and the rotation of the amidine group across the phenyl-amidine bond. The high occupancy rate of this water molecule suggests that the DB921 complex prefers to maintain this conformational flexibility that results in a favorable contribution to the system entropy.
Figure 6.
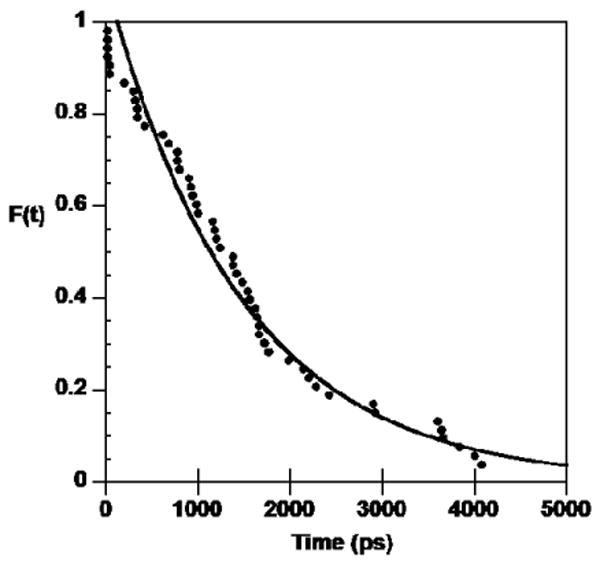
Exponential decay of the probability distribution of the residence time of water molecule labeled C in Figures 3 and 4.
This water molecule is pivotal in facilitating the complex to explore the conformational space available to the biphenyl-amidine of DB921. It should be noted that the X-ray structure (PDB ID: 2B0K) reported the B-value of the phenylamidine nitrogens to be 58.2 (average) and the trigonal planar carbon connecting the amidine groups to have a B-value of 44.9. Comparatively, the B-values of the corresponding atoms on the benzmidazole side amidine are much lower [nitrogens: 41.6 (average) and carbon: 27.9]. Thus, the X-ray structure indicates that the co-ordinates of the constituent atoms of amidine on the phenyl side should be expected to vary more than the benzimidazole counterpart. The dynamic nature of the bound DB921 expands on the X-ray structure but does not disagree with it since the B-factors at that end of DB921 in its -AATT- complex are quite large and suggest a relatively mobile structure.
D. Parallel Mode of Interaction
The interaction that characterizes the ‖ mode of interaction is the electrostatic association of the amidine group with the anionic DNA backbone (Labels D1 and D2 in Figure 3). Both these distances, D1 (blue) and D2 (red) are plotted for the frames that were detected as the parallel mode of interaction in Figure 7. Note that the distances that are plotted in Figure 7 are from the phosphate oxygens to the closest amidine nitrogen. Due to the torsional rotation that is possible, the nitrogens that are associated with D1 or D2 may interchange as the simulation progresses. The D1 distances range between 1.6 Å to 6.9 Å with a standard deviation of 1.1 Å and a mean of 2.9 Å. The values of D2 range between 1.6 Å to 7.5 Å with a standard deviation of 0.9 Å and a mean of 4 Å. The plot suggests that at most times in the ‖ mode the amidine group maintains optimal distances that are favorable for specific electrostatic interactions with the oxygens of the phosphate backbone on either strand or at least a strong interaction on one strand. In other words, the values of D1 and D2 are close to each other and the amidines are approximately equidistant from the respective oxygens of the phosphate backbone on opposite strands or, either D1 or D2 is a significantly smaller value, i.e. one of the amidines is very close to the backbone oxygen on one of the strands. In the case where only one of the amidine groups is interacting with the oxygen the distances are in the range of 1.6 Å to 2.5 Å.
Figure 7.
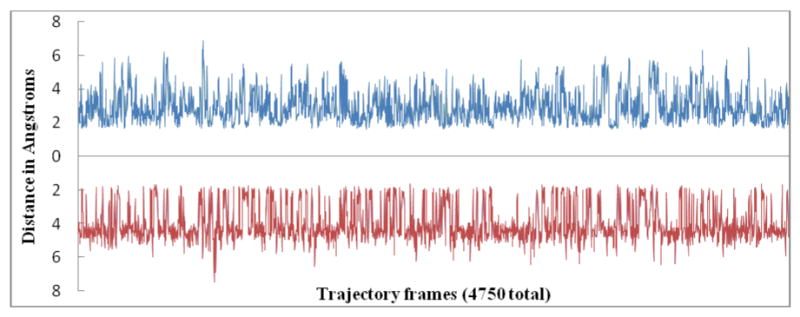
Top (blue plot)- Distance between the phosphate that connects DNA bases A5 and T6 and the closest amidine nitrogen of DB921. This is the same as D1 in Figure 3. Each point is the distance for that particular frame. Bottom (red plot)- Distance between the phosphate that connects DNA bases T20 and C21 and the closest amidine nitrogen of the DB921 molecule. This is the same as D2 in Figure 3. An inverted plot is used for D2. This enables the direct comparison of distances D2 and D1 at corresponding frames. Each point is the distance for that particular frame. There are a total of 2313 frames i.e 46.2 ns.
E. Perpendicular Mode of Interaction
As compared to the ‖ mode, additional dynamic water interactions couple DB921 to the bases at the floor of the minor groove in the ⊥ conformation. Apart from A6-N3 and T20-O2 that interact with the long residence time water (Label C, Figure 4), two other DNA atoms, A5-N3 and C21-O2 also stabilize the complex through water mediated networks. Water plays a principal role in stabilizing the two perpendicular modes of binding. A set of distance-based criteria was used to identify the transient water molecules that contributed to the stability of the ⊥ mode of binding (see Supplementary Material, Section 2).
In the ⊥-Major mode of interaction, C21-O2 forms a two water network in which the water interacting with C21 (Label D2 in Figure 4) has residence times ranging from a few pico seconds to 0.5 ns. D2 further interacts with D1, which mediates interactions between the interfacial water molecule, labeled C in Figure 4, and DB921. It can be seen that D2 forms hydrogen bonds with C21-O2 and also interacts with D1 that in turn interacts with -NH of the biphenyl amidine of DB921. Compared to D2, D1 is spatially restrained and has better stabilizing contacts. Hence, it has higher residence times ranging from a few pico seconds to 1.2 ns and an average of 0.6 ns. Water molecule D3 (Figure 4) mediates structure-stabilizing interactions between A5-N3 and C21-O2 (via D2) and, alternatively, A5-N3 and a sugar oxygen (Label D3′, Figure 4) of the DNA backbone. This A5-N3 bound water has residence times ranging from 60 ps to 0.8 ns. Transient variations of this set of interactions were also observed.
As described earlier, the ⊥ mode includes the ⊥-Xray conformation in which a single water molecule H-bonds with A5-N3 and forms a direct H-bond interaction with DB921. Unlike the ⊥-Major mode of interaction, this set of conformations does not involve any direct participation of T20-O2, C21-O2 or A6-N3 with respect to forming stabilizing water mediated networks. Apart from the single water-mediated interaction that is exclusive to ⊥-Xray conformation, as mentioned in Section 3.C, the interfacial water (Label C in Figures 3 and 4) is expelled from the cavity that is located near the biphenyl region of DB921 in this conformation.
The nature of interactions between the phenylamidine and DNA in the perpendicular binding mode is the usual type of minor groove interactions and is also seen at the benzimidazole end of DB921. The linear shape of DB921, however, still requires amidine-water-DNA indirect interactions at the phenylamidine. The dynamic results illustrate the variety of water interactions that can be used to couple DB921 to the floor of the minor groove in the ⊥ mode.
4 Conclusions
For development of new agents that target DNA in chemical biology and therapeutic applications, it is essential to design compounds that interact with DNA sequences in new and functionally effective modes of binding. The DB921 complex represents a new minor groove interaction paradigm that does not fit the classical binding model. The compound has a benzimidazole-biphenyl system with terminal amidines that result in a linear conformation without the appropriate radius of curvature to match the minor groove shape. Given these structural features, it was initially surprising that DB921 binds in the groove with an unusually high binding constant6. X-ray results from the Neidle laboratory clearly showed that the lack of curvature of the compound for minor groove binding can be solved by incorporating a water molecule into the complex. DB921 when paired with water forms a curved noncovalent minor groove binding module that matches the shape of the groove. In the 100 ns molecular dynamics simulation of the system, the expected, limited thermal motions in the X-ray conformation of the complex were observed but also some significantly rearranged conformations appeared that have additional water molecules to couple DB921 to the DNA bases at the floor of the groove. The benzimidazole -NH and amidine contacts with the bases remain relatively constant through the entire simulation. The biphenyl-amidine end of the bound molecule, which is connected to DNA through water, however, undergoes much larger dynamic changes in orientation relative to the floor of the groove.
The MD results provide an expanded understanding of how DB921 binds to DNA without a large unfavorable entropy relative to more classical curved minor groove compounds6. Both the DB921 phenylamidine and the water molecules that couple it to the minor groove are quite dynamic. The flexible interaction capability of water is used to full advantage in the different complexes to accept H-bonds from the amidine and to donate H-bonds to bases at the floor of the groove. In the most unusual complex observed in the MD trajectories, a water is between the central phenyl group of DB921 and DNA. The water H-bonds with bases and other water molecules and has van der Waals and electrostatic interactions with the phenyl of DB921. As described above, a similar interaction of water that H-bonds to amino acid side chains and other waters but sits in a pocket with one nonpolar side is also seen in some protein-DNA complexes. Clearly interaction modes with an interstitial water molecule in the polar-nonpolar cavity should be included in the array of favorable modules involved in DNA complexes. The additional DB921-water complexes observed in the MD simulation suggest that modified derivatives to replace or enhance the water-phenyl interaction should be possible. For example, replacement of the central phenyl with a pyridine or pyrimidine can provide a nitrogen to accept an H-bond from the internal water and stabilize the complexes shown in Figures 3 and 4. On the other hand, change of a -H on the internal face of the central phenyl with a -OH can stabilize the water networks shown in Figures 3 and 4 while replacing the long residence time water with the appropriately placed -OH. Synthesis of such modified compounds is in progress.
Supplementary Material
Acknowledgments
This research was supported by NIH Grant AI064200. We gratefully acknowledge the contributions of Professors Stephen Neidle and David W. Boykin and coworkers for crystallography, and synthesis of DB921. We also acknowledge Dr. Donald Hamelberg and Dr. Thomas Cheatham for discussions and very valuable suggestions that made the research reported here possible.
Footnotes
Supplementary Material Available: Procedure for defining new force field parameters for DB921, AMBER frcmod file with the complete force field parameters for DB921, plots of energy versus dihedral angle for DB921 calculated from ab initio methods, GAFF and the new force field parameters, procedures for the construction of the reference plane that defines the various binding modes of DB921, criteria used for tracking water molecules in the different binding modes and analysis of α/γ angles of bases comprising the binding region.
References
- 1.Tidwell RR, Boykin DW. Dicationic DNA Minor Groove Binders as Microbial Agents. In: Demeunynck M, Bailly C, Wilson WD, editors. DNA and RNA Binders: From Small Molecules to Drugs. Wiley-VCH Verlag GmbH & Co KGaA; Weinheim (Germany): 2003. pp. 414–460. [Google Scholar]
- 2.Wilson WD, Tanious FA, Mathis A, Tevis D, Hall JE, Boykin DW. Biochimie. Vol. 90. 2008. pp. 999–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mathis AM, Bridges AS, Ismail MA, Kumar A, Francesconi I, Anbazhagan M, Hu Q, Tanious FA, Wenzler T, Saulter J, Wilson WD, Brun R, Boykin DW, Tidwell RR, Hall JE. Antimicrob Agents Chemother. 2007;51:2801–2810. doi: 10.1128/AAC.00005-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goodsell D, Dickerson RE. J Med Chem. 1986;29:727–733. doi: 10.1021/jm00155a023. [DOI] [PubMed] [Google Scholar]
- 5.Fairley TA, Tidwell RR, Donkor I, Naiman NA, Ohemeng KA, Lombardy RJ, Bentley JA, Cory M. J Med Chem. 1993;36:1746–1753. doi: 10.1021/jm00064a008. [DOI] [PubMed] [Google Scholar]
- 6.Miao Y, Lee MPH, Parkinson GN, Batista-Parra A, Ismail MA, Neidle S, Boykin DW, Wilson WD. Biochemistry. 2005;44:14701–14708. doi: 10.1021/bi051791q. [DOI] [PubMed] [Google Scholar]
- 7.Nguyen B, Neidle S, Wilson WD. Acct Chem Res. 2009;42:11–21. doi: 10.1021/ar800016q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nguyen B, Hamelberg D, Bailly C, Colson P, Stanek J, Brun R, Neidle S, Wilson WD. Biophys J. 2004;86:1028–1041. doi: 10.1016/s0006-3495(04)74178-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nguyen B, Lee MP, Hamelberg D, Joubert A, Bailly C, Brun R, Neidle S, Wilson WD. J Am Chem Soc. 2002;124:13680–13681. doi: 10.1021/ja027953c. [DOI] [PubMed] [Google Scholar]
- 10.Furse KE, Corcelli SA. J Am Chem Soc. 2008;130:13103–13109. doi: 10.1021/ja803728g. [DOI] [PubMed] [Google Scholar]
- 11.Pal SK, Zhao LA, Zewail AH. Proc Natl Acad Sci USA. 2003;100:8113–8118. doi: 10.1073/pnas.1433066100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hamelberg D, McCammon JA. Dealing with Bound Waters in a Site: Do they Leave or Stay. In: Stroud RM, Finer-Moore J, editors. Computational and Structural Approaches to Drug Design. Royal Society of Chemistry; Cambridge (UK): 2007. pp. 95–110. [Google Scholar]
- 13.Cooper A. Biophys Chem. 2005;115:89–97. doi: 10.1016/j.bpc.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 14.Raschke MT. Curr Opin Struc Biol. 2006;16:152–159. doi: 10.1016/j.sbi.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 15.Jayaram B, Jain T. Annu Rev Biophys Biomol Struct. 2004;33:343–361. doi: 10.1146/annurev.biophys.33.110502.140414. [DOI] [PubMed] [Google Scholar]
- 16.Clarke C, Woods JR, Gluska J, Cooper A, Nutley AM, Boons GJ. J Am Chem Soc. 2001;123:12238–12247. doi: 10.1021/ja004315q. [DOI] [PubMed] [Google Scholar]
- 17.Ladbury JE. Chem Biol. 1996;3:973–980. doi: 10.1016/s1074-5521(96)90164-7. [DOI] [PubMed] [Google Scholar]
- 18.Spackova N, Cheatham TE, 3rd, Ryjacek F, Lankas F, van Meervelt L, Hobza P, Sponer J. J Am Chem Soc. 2003;125:1759–1769. doi: 10.1021/ja025660d. [DOI] [PubMed] [Google Scholar]
- 19.Bostock-Smith CE, Harris SA, Laughton CA, Searle MA. Nucleic Acids Res. 2001;29:693–702. doi: 10.1093/nar/29.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Jr, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. J Am Chem Soc. 1995;117:5179–5197. [Google Scholar]
- 21.Case DA, et al. AMBER 9.0. University of California; San Fransisco: 2006. [Google Scholar]
- 22.Wang JM, Cieplak P, Kollman PA. J Comput Chem. 2000;21:1049–1074. [Google Scholar]
- 23.Hopfinger AJ, Pearlstein RA. J Comput Chem. 1984;5:486–499. [Google Scholar]
- 24.Pigache A, Cieplak P, Dupradeau FY. Automatic and Highly Reproducible RESP and ESP Charge Derivation: Application to the Development of Programs RED and XRED. 227th ACS National Meeting of the American Chemical Society; Anaheim, CA. March 27 - April 1, 2004; Washington, DC: American Chemical Society; 2004. [Google Scholar]
- 25.Frisch MJ, et al. Gaussian 03, Revision C.02. Gaussian, Inc.; Wallingford CT: 2004. [Google Scholar]
- 26.Bayly CI, Cieplak P, Cornell WD, Kollman PA. J Phys Chem. 1993;97:10269–10280. [Google Scholar]
- 27.Cornell WD, Cieplak P, Bayly CI, Kollman PA. J Am Chem Soc. 1993;115:9620–9631. [Google Scholar]
- 28.SYBYL 6.9.2. Tripos International; 1699 South Hanley Rd., St. Louis, Missouri, 63144, USA: [Google Scholar]
- 29.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 30.Ryckaert JP, Ciccotti G, Berendsen HJC. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 31.Darden T, York D, Pedersen L. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 32.Hamelberg D, McCammon AJ. J Am Chem Soc. 2004;126:7683–7689. doi: 10.1021/ja0377908. [DOI] [PubMed] [Google Scholar]
- 33.Pérez A, Marchán I, Svozil D, Sponer J, Cheatham TE, 3rd, Laughton CA, Orozco M. Biophys J. 2007;92:3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smith DE, Dang LX. J Chem Phys. 1994;100:3757–3766. [Google Scholar]
- 35.Humphrey W, Dalke A, Schulten K. J Molec Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 36.Hamelberg D, Shen T, McCammon AJ. J Chem Phys. 2005;122:241103. doi: 10.1063/1.1942487. [DOI] [PubMed] [Google Scholar]
- 37.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- 38.Pophristic V, Vemparala S, Ivanov I, Liu Z, Klein ML, DeGrado WF. J Phys Chem B. 2006;110:3517–3726. doi: 10.1021/jp054306+. [DOI] [PubMed] [Google Scholar]
- 39.Otwinowski Z, Schevitz RW, Zhang RG, Lawson CL, Joachimiak A, Marmorstein RQ, Luisi BF, Sigler PB. Nature. 1988;335:321–329. doi: 10.1038/335321a0. [DOI] [PubMed] [Google Scholar]
- 40.Ladbury JE, Wright JG, Sturtevant JM, Sigler PB. J Mol Biol. 1994;238:669–681. doi: 10.1006/jmbi.1994.1328. [DOI] [PubMed] [Google Scholar]
- 41.Morton CJ, Ladbury JE. Protein Sci. 1996;5:2115–2118. doi: 10.1002/pro.5560051018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Watkins D, Hsiao C, Woods KK, Koudelka GB, Williams LD. Biochemistry. 2008;47:2325–2338. doi: 10.1021/bi701826f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.