

Abstract
We describe a novel pair of nested genes, CDA12 and CDA13, from Tetrahymena thermophila. Both are implicated in membrane trafficking associated with cell division and conjugation. Green fluorescent protein localization reveals Cda12p decoration of diverse membrane-bound compartments, including mobile, subcortical tubulovesicular compartments; perinuclear vesicles; and candidates for recycling endosomes. Cda13p decorates intracellular foci located adjacent to cortically aligned mitochondria and their neighboring Golgi networks. The expression of antisense CDA12 RNA in transformants produces defects in cytokinesis, macronuclear segregation, and the processing of pinosomes to downstream compartments. Antisense CDA13 RNA expression produces a conjugation phenotype, resulting in the failure of mating pairs to separate, as well as failures in postconjugation cytokinesis and macronuclear fission. This study offers insight into the membrane trafficking events linking endosome and Golgi network activities, cytokinesis, and karyokinesis and the unique membrane-remodeling events that accompany conjugation in the ciliate T. thermophila. We also highlight an unusual aspect of genome organization in Tetrahymena, namely, the existence of nested, antisense genes.
Ciliates represent a potentially rich system for the study of membrane dynamics (4, 39, 49). Of particular interest to this investigation is the trafficking of membrane components between the plasma membrane and the early endosome via pinocytosis and from a tubulovesicular compartment known as the recycling endosome to the cytokinetic furrow during cell division (2, 25). The recycling endosome was initially characterized as the early destination of clathrin-coated vesicles derived from pinocytosis. During peripheral sorting in the early endosome, membrane proteins destined for degradation are segregated from membrane receptors destined for recycling back to the plasma membrane (25, 26). Most of the recycled membrane proteins are directed back to the cell surface via the recycling endosome, a compartment also associated with the microtubule-organizing center (26, 51). The regulation of membrane and protein traffic to and from the various endosomal compartments (58) is intimately associated with small Rab GTPase proteins (41, 51). In particular, Rab11 is necessary for shuttling proteins from the recycling endosome back to the plasma membrane (42). Rab11 was initially reported to be associated with the trans-Golgi network, where it is involved in the transport of de novo-synthesized proteins from the Golgi apparatus to the plasma membrane (9, 52).
An important discovery was that membrane trafficking from both the Golgi network-exocytosis pathway (13, 32) and the endocytosis-endosome pathway (17, 44) contributes new membrane to the ingressing cleavage furrow during the late stages of cytokinesis, as reviewed in reference 2. The involvement of proteins from both the secretory and endocytic pathways in membrane trafficking has been demonstrated by RNA interference (RNAi) screens designed to identify components of the cytokinetic machinery in both Drosophila and Caenorhabditis species.
Rab GTPase proteins do not work alone. Rab11 recruits a diverse family of effector proteins, such as FIP3 and FIP4 (family of Rab11-interacting proteins), to the recycling endosome, where they help direct endosome-derived vesicles (18). FIP3 and FIP4 serve to dock Rab11 protein-bound vesicles via association with the Arf6 protein in the ingressing division furrow. Mutations that knock out (or knock down) Rab11 gene products or either FIP3 or FIP4 result in cytokinesis failure, especially during the final stages of furrow ingression and abscission (18, 46, 55).
The ciliate Tetrahymena thermophila is endowed with a complex system of dynamic membrane compartments (20). Furthermore, Tetrahymena undergoes distinct membrane-remodeling episodes associated with both cytokinesis and karyokinesis, occurring during vegetative growth, and also with nuclear exchange, occurring during sexual reproduction or conjugation (11). The T. thermophila genome sequence, first published in 2006 (15), has revealed a remarkable diversity of membrane trafficking genes, including genes for 8 dynamin-related proteins and 69 different Rab proteins (16). In contrast, humans express 60 Rab genes and Saccharomyces cerevisiae expresses only 16. The diverse and largely nonconserved nature of FIPs defies homology searching, making it difficult to gauge their abundance in T. thermophila.
Conventional chemical mutagenesis in Tetrahymena has led to the identification of numerous mutations affecting cell division (21, 22, 29), as well as biosynthetic pathways leading to the assembly, docking, and discharge of secretory granules (37, 38). We previously performed a genetic screen of a novel type of antisense library in T. thermophila while searching for cell division phenotypes (47). This antisense method is based on the fact that the ribosomal DNA (rDNA) locus in the Tetrahymena micronuclear germ line is present in a single copy, making it a viable target for modification. In addition, antisense RNAs can be inserted into the variable region of the 26S rRNA without compromising ribosome function (47). By exploiting these features, Chilcoat et al. have been able to construct a library of plasmids carrying 26S rRNAs containing short antisense insertions of cDNA derived from vegetatively expressed T. thermophila transcripts (10). This library was used successfully by Chilcoat et al. to screen for mutants expressing defects in dense-core granule secretion. We used this same antisense ribosome library to generate and screen for mutations affecting cell division.
Using this approach, we have identified and characterized the nested genes CDA12 and CDA13 (CDA12/CDA13). We provide evidence suggesting that Cda12p is involved in membrane trafficking associated with the recycling endosome. More specifically, we suggest that this protein participates in pinocytosis, cleavage furrow ingression, macronuclear segregation during cytokinesis, and micronuclear exchange during conjugation. Curiously, the CDA12 locus is entirely nested within a second, slightly larger gene. The second gene, CDA13, also appears to be involved in membrane trafficking, possibly via the trans-Golgi network. These results are consistent with the hypothesis that both Cda12p and Cda13p resemble Rab11 GTPase effector molecules related to the FIPs. The plethora of membrane events associated with these two gene products and the unusual organization of the corresponding genes are discussed below.
MATERIALS AND METHODS
Strains, growth conditions, and cell cycle synchronization.
Strains CU428 (Peter J. Bruns, Cornell University) and SB1969 (Eduardo Orias, University of California, Santa Barbara) were grown at 31°C in Neff medium (0.25% protease peptone, 0.25% yeast extract, 0.5% glucose, and 0.033 M ferric chloride). Cell divisions were synchronized by centrifugal elutriation (see reference 34).
General molecular techniques.
RNA was isolated from Tetrahymena using either a micro-poly(A) kit (Ambion) or the RNeasy total RNA kit (Qiagen). RNA samples (2 to 10 μg) were separated on glyoxal-dimethyl sulfoxide-1% agarose gels as described in reference 43 prior to capillary transfer onto Nytran filters. The radiolabeled CDA12 probe for Northern and Southern blot analyses was generated by PCR as described previously (36), using oligonucleotide primers P1(+) and P1(−) (Table 1). Southern blot analysis was performed as described previously (35). DNA sequencing was completed at the Advanced Genetic Analysis Center at the University of Minnesota, St. Paul.
TABLE 1.
Primers used
| Primera | Sequence |
|---|---|
| 5318DNF1* | 5′AAA AGG TCG ATG AGT AAG GAA ATG3′ |
| 5318DNR2* | 5′CAA TCT CAG GGT ACG CGG3′ |
| Lumpy 2 | 5′CAU CAU CAU CAU CCC CAA GCA GAA TCG TTG3′ |
| Lumpy 3 | 5′CAU CAU CAU CAU GTT ACC ACC GAG TTG TGG G3′ |
| Lumpy 6 | 5′CAU CAU CAU CAU CAC AAC TCG GTG GTA ACA AC3′ |
| Lumpy 7 | 5′GTT GTG GAG AAT TGC ATA C3′ |
| Lumpy 8 | 5′CAU CAU CAU CAU GGT TAT TTG CTG CTC TTG C3′ |
| Lumpy 9 | 5′CGA TAC CGC TCC CCA ATA TG3′ |
| Lumpy 10 | 5′CTA TAT TCT TGT CTA CAT TAG TC3′ |
| Lumpy 11-GFP | 5′ACC TCG AGA TGT GTT TTT TTC ATA ATT AAC CAT CAT ATT GG3′ |
| Lumpy 12-GFP | 5′TGG GCC CTC AAA ACA AAC TGA TAG AGT TTT AAT AAA TGC3′ |
| Lumpy 13 | 5′ACC TCG AGA TGC AAT ACG AAC AAC TAA ACC AA3′ |
| Lumpy 14 | 5′TGG GCC CTC ATA ATT AAC CAT CAT ATT GGG GAG C3′ |
| DR377** | 5′CAA AAC AAA CTG ATA GAG3′ |
| DR378** | 5′TGT TAC CAC CGA GTT GTG3′ |
| P1(+)** | TGT TAC CAC CGA GTT GTG |
| P1(−)** | CAA AAC AAA CTG ATA GAG |
| DR550** | 5′CGC ATT TAG AAGCTC TCT CAG GAT T3′ |
All Lumpy primers were synthesized by Invitrogen. *, these primers were a gift from A. Turkewitz, University of Chicago. **, these primers were synthesized at the University of Minnesota.
Transformation by electroporation and biolistic particle bombardment.
Mating cells were transformed by electroporation using the technique described in reference 24. Paromomycin was added at 100 μg/ml 24 h after electroporation to select for transformants.
The biolistic transformation of vegetative cells was performed using the technique described in reference 8. Gold particles (1.0 μm in diameter) coated with approximately 1.0 μg of HindIII-digested pCDA12/13 H4.2KO were used to bombard the cells. Following biolistic transformation, the surviving cells were incubated in 50 ml of prewarmed 2% PPYS (proteose, peptone, yeast extract, and sucrose) medium at 30°C for 3 to 4 h prior to selection with paromomycin (100 μg/ml) and distribution into 96-well microtiter plates (in volumes of 100 μl per well). Paromomycin-resistant transformants were evident after selection at 30°C for 4 to 7 days following transformation.
Screening of the antisense cDNA library for mutant phenotypes.
Transformants were diluted immediately after electroporation to ensure a Poisson distribution of single-cell isolates when 0.1-ml aliquots of the transformant suspension were transferred into 96-well microtiter plates. The resultant paromomycin-resistant clones were screened visually for gross morphological anomalies under a dissecting microscope, stained with DAPI (4′,6-diamidino-2-phenylindole), and viewed by fluorescence microscopy with an Olympus IMT-2 inverted microscope.
Antisense fragment recovery and phenotype verification.
Total DNA from the transformant expressing the mutant cda12 phenotype was isolated from whole cells by detergent lysis (57). The fragment of antisense library DNA inserted within the 26S rRNA gene was amplified with oligonucleotide primers 5318DNF1 and 5318DNR2 (Table 1) by hot-start PCR and subsequently inserted back into the rDNA-based library vector (ribosomal antisense plasmid 5318DN [48]) at the KpnI and NotI restriction sites. The resultant antisense clone was used to transform Tetrahymena by electroporation as described above.
Cloning of the CDA13 cDNA gene.
Wild-type mRNA was isolated from Tetrahymena strain CU428 by using a micro-poly(A) RNA kit (Ambion). Oligonucleotide primers based on the initial CDA12 antisense fragment were designed (Table 1) for cloning of the entire CDA13 cDNA with kits for the rapid amplification of cDNA 3′ ends (3′ RACE; Gibco/BRL) and 5′ RACE (Invitrogen). Hot starts were employed for all PCRs. All RACE products were cloned using the uracil-DNA glycosylase cloning kit from Gibco/BRL.
Cloning of the CDA12/CDA13 genomic sequence from a size-selected genomic library.
A Southern blot analysis of total DNA from Tetrahymena, digested with HindIII and probed with a radiolabeled partial CDA13 cDNA sequence, revealed specific hybridization at 4.2 kb. A HindIII-directed genomic library was constructed using a bacterial cloning vector and screened for hybridization to the radiolabeled probe as described previously (35). A positive clone was isolated, and both strands of the 4.2-kb insert were sequenced by the Advanced Genetic Analysis Center (University of Minnesota, St. Paul).
Macronuclear gene replacement and isolation of a CDA12/CDA13 null strain.
A unique SpeI site was introduced at nucleotide position 1270 (relative to the 5′-end-most HindIII site identified in CDA12) in the pCDA12 H4.2 genomic clone by oligonucleotide site-directed mutagenesis as described previously (30). Similarly, a unique SpeI site was introduced just in the 5′ direction from a neomycin (or paromomycin) resistance cassette within the Tetrahymena transformation vector p42L29B. The mutated pCDA12/13 4.2 construct was digested with SpeI and NsiI, effectively removing 2.1 kb and making it possible to subclone a 1.5-kb fragment from p42L29B that includes the entire neomycin resistance cassette. The resultant vector construct (pCDA12/13 H4.2KO) facilitates targeted disruption of the CDA12/CDA13 locus during biolistic transformation. The pCDA12/13 H4.2KO construct was digested with HindIII prior to the biolistic transformation of Tetrahymena (see above).
Total DNA from transformants was screened for the CDA12/CDA13 null allele by Southern blot analysis as described previously (34). Phenotypic assortment was achieved by the isolation of single cells, which were used for clonal expansion. Clones were selected with increasing amounts of paromomycin until a concentration of 1.0 mg/ml was reached.
Inducible expression of both GFP-Cda12p and GFP-Cda13p fusion proteins.
The construction of an inducible green fluorescent protein (GFP)-Cda12p fusion gene, targeted for integration at the T. thermophila BTU1 locus, was initiated by using site-directed mutagenesis (30) to introduce XhoI and BglII restriction sites at the initiator and stop codons, respectively, of a similarly targeted luciferase reporter construct (pRdLuc) described previously (47). The introduction of the XhoI site in pRdLuc facilitated the replacement of the 1.3-kb RAD51 promoter (47) with an inducible metallothionein 1 (MTT1)-GFP cassette (a 2.0-kb BamHI-XhoI fragment) from the T. thermophila rDNA-based expression vector pIGF-1 (56). The luciferase coding sequence (bounded by XhoI and BglII restriction sites) in this intermediate plasmid construct was replaced with the entire PCR-amplified CDA12 coding sequence (corresponding to 186 amino acids) that included XhoI and BglII sites at the termini. The resultant GFP gene-CDA12 fusion construct, expressed from the MTT1 inducible promoter and targeted for disruption of the BTU1 locus, was sequenced to confirm the absence of any missense or frameshift errors that might have arisen during the cloning process. Mating Tetrahymena cells were transformed as described above, and successfully transformed cells were selected using paclitaxel at a 20 μM final concentration.
The putative Cda13p open reading frame (ORF) sequence was amplified by PCR with primers Lumpy 11-GFP and Lumpy 12-GFP (Table 1) to introduce XhoI and ApaI restriction sites at the 5′ and 3′ termini, respectively. The resultant PCR product was TA cloned into the vector pCR2.1 (Invitrogen) and subsequently digested with EcoRI to generate a 600-bp fragment. Following gel purification, the EcoRI fragment was digested sequentially with XhoI and ApaI and ligated into the similarly digested Tetrahymena rDNA-based transformation vector pIGF-1 (a gift from Doug Chalker, Washington University in St. Louis). The XhoI and ApaI restriction sites in the pIGF-1 vector facilitate the in-frame cloning of ORFs at the position corresponding to the carboxy terminus of GFP.
The inducible expression of GFP fusion proteins from these vectors in Tetrahymena transformants is due to the MTT1 transcriptional promoter, induced in the presence of Cd2+ (45). The transformation of mating Tetrahymena cells with these vectors by electroporation was carried out as described above. Successfully transformed cells were selected using paromomycin at a 100-μg/ml final concentration. Expression was induced by exposing cells to 0.1 μg/ml CdCl2. Cadmium induction was initiated 2 to 3 h prior to observation with a fluorescence microscope.
Brefeldin A.
Brefeldin A was added to MTT-GFP-Cda13p cultures at a concentration of 10 μg/ml. Cultures were incubated at 30°C for 3 h. CdCl2 was added at 0.2 μg/ml at the 2-h time point. Cells were then examined for the localization of the GFP-tagged product.
Fluorescence assay of pinocytosis.
Cells were prepared and labeled with the probe FM 4-64 (Molecular Probes) as described by Elde et al. (16) and imaged using an Olympus BX-50 microscope with a SPOT RT Slider digital camera.
Fluorescence microscopy.
DAPI nuclear staining was performed as described previously (12), with the modifications described in reference 53. Preparations were viewed using an Olympus B-MAX fluorescence microscope, and photos were captured using an Olympus PM-30 camera. GFP images were captured using a digital SPOT RT camera. Immunofluorescence analysis was performed using a primary monoclonal antiserum specific to centrin (monoclonal antibody 17E10, a gift of J. Salisbury) and a secondary (goat anti-mouse) antiserum labeled with Cy5 (Amersham Life Science Inc., Cleveland, OH). Nuclei were labeled with Sytox (Molecular Probes). Cells were scanned using a Bio-Rad MRC1000/1024 laser confocal microscope at the University of Minnesota Imaging Center (director, Mark Sanders) or using our Olympus BX-50 microscope outfitted with a Fluoview FV300 confocal system.
Immunogold labeling for transmission electron microscopy (TEM).
Tetrahymena cells were washed in 150 mM mannitol (cryopreservant) for 1 h, pelleted, frozen under high pressure in a BAL-TEC HPM-010 high-pressure freezer, freeze substituted in 0.25% gluteraldehyde-0.1% uranyl acetate, and embedded in Lowicryl HM20 medium. Sections (55 nm thick) were taken and labeled with 1:200 dilutions of primary antibody in 1% nonfat milk-phosphate-buffered saline-Tween 20 solution that included an affinity-purified rabbit anti-GFP primary antibody (a gift from Jason Kahana and Pamela Silver, Dana-Farber Cancer Institute, Boston, MA). A 1:20 dilution of 15-nm gold particle-conjugated goat anti-rabbit secondary antibody (Ted Pella, Redding, CA) was used. Grids were stained with uranyl acetate and lead citrate and were visualized with a Philips CM10 electron microscope.
Nucleotide sequence accession number.
The nucleotide sequence data for the T. thermophila CDA12 gene (nested within CDA13) have been registered with the EMBL and GenBank databases and the DDBJ under the accession number AY337263.
RESULTS
Identifying the CDA12/CDA13 gene locus.
A novel method of antisense mutagenesis for Tetrahymena had been developed previously (10, 50). Members of the Turkewitz laboratory have constructed a library of plasmids carrying 26S ribosomal subunits that contain short, 5′-end antisense insertions from a large number of vegetatively expressed Tetrahymena genes. When expressed in Tetrahymena, these antisense ribosomes presumably result in a reduction of translation from the targeted gene transcripts. We screened transformants generated using this antisense library for mutations affecting cell division. The screen was visual, relying on our ability to discern abnormal cell division phenotypes under the compound microscope. In our first screen, we identified a clone of mutant cells exhibiting a lumpy multicellular aggregate phenotype. The antisense insert from cells expressing the lumpy phenotype was PCR amplified and sequenced, and the sequence information was used to identify the gene locus by searching the Tetrahymena Genome Database (see below). Multiple rounds of transformation using this specific antisense construct consistently yielded the lumpy phenotype.
Sequence analysis.
Sequence analysis of the CDA12/CDA13 locus revealed ORFs on complementary strands of genomic DNA (Fig. 1A). The CDA12 gene is 552 bp long and encodes a 183-amino-acid protein with no strong homolog in other organisms. CDA12 has been assigned reference number TTHERM00013410 in the Tetrahymena Genome Database (http://www.ciliate.org/). Protein sequence analysis predicts four alpha-helices potentially serving as membrane-spanning domains (Fig. 1D). For CDA12, the closest BLAST hit (with an E value of 5e−18) is a neighboring hypothetical gene in the Tetrahymena genome (TTHERM00013400). The ORF from the cDNA strand (CDA13) is 591 nucleotides long and encodes a 196-amino-acid protein. Neither gene includes introns, and the CDA12 ORF sequence is completely nested within the complementary CDA13 ORF sequence in an antisense orientation. A BLAST search for similarities to the predicted product of CDA13 also found no clear homologies, and sequence analysis predicts a pair of alpha-helices constituting potential membrane-spanning domains (Fig. 1D).
FIG. 1.

Analysis of transcripts from the CDA12/CDA13 gene locus. (A) ORFs at the CDA12/CDA13 locus (predicted by the NCBI ORF finder program). The diagram shows complete nesting of the smaller CDA12 ORF (CDA12-2) within the larger CDA13 ORF (CDA13-1). (B) Northern blot analysis using a double-stranded probe, showing transcription from the CDA12/CDA13 locus at various stages of cell division. Transcript levels drop as cells enter mitosis. (C) Northern blot analysis using a double-stranded probe, showing transcription from the CDA12/CDA13 locus at various stages during conjugation. Transcript levels rise during meiosis (3 to 4 h into conjugation) and again late in conjugation as pairs prepare to separate. MI, meiosis I; MII, meiosis II; 3rd PZD, third prezygotic division. (D) Protein topology predicted for CDA12 and CDA13 products (TopPred software prediction). Arabic numerals indicate amino acid positions. (E) Strand-specific RT-PCR analysis revealing that only CDA12 transcripts (lane 2) are present during vegetative growth. (Lane 1) PCR size markers (Promega; 1 kb and 750, 500, 300, and 150 bp). (Lane 2) RT-PCR product obtained using vegetative RNA, a unidirectional primer complementary to the CDA12 transcript, and RT. The PCR mixture was subjected to RNase treatment and heat inactivation of the RT enzyme, and then both CDA12 and CDA13 primers and Taq polymerase were added. The visible product is indicated by the arrowhead. (Lane 3) The same procedure described in the legend to lane 2 was performed, with the CDA12 unidirectional primer replaced with one complementary to the CDA13 transcript (no product is visible). (Lane 4) Control lane in which both primers were added only after RNase treatment and heat inactivation (demonstrates the effectiveness of RNase and heat treatment in terminating RT activity). (Lane 5) Both primers were added at the start but with only Taq polymerase, no RT, present (demonstrating the lack of PCR activity from Taq polymerase operating off of either RNA or contaminating DNA templates). Strand-specific primers were Lumpy 13 and Lumpy 14 (Table 1). (F) Results of an RT-PCR experiment similar to that described in the legend to panel E, except using RNA from conjugating cells at the 4-h time point rather than RNA from vegetative cells. Products from both CDA12 and CDA13 transcripts (arrowhead; lanes 2 and 3, respectively) are detectable. Lanes correspond to those in panel E.
Transcription analysis.
Using conventional double-stranded probes for Northern analysis, we detected cell cycle regulation from the CDA12/CDA13 locus (Fig. 1B). Unfortunately, double-stranded probes cannot distinguish between transcripts from individual partners of nested genes. We performed strand-specific, reverse transcriptase (RT)-PCR to probe for expression from each gene independently. This analysis revealed that during vegetative growth, only CDA12 produces a detectable (1.07-kb) product (Fig. 1E), leading us to conclude that CDA12 was solely responsible for the cycling pattern of gene expression seen during vegetative growth.
During conjugation, conventional Northern analysis revealed both an early period (around the 3-h time point) and a late period (after 10 h) of increased expression from the CDA12/CDA13 locus (Fig. 1F). Strand-specific RT-PCR revealed that both CDA12 and CDA13 are actively transcribed (Fig. 1F). To confirm the existence of a Cda13 transcript, we performed 3′ and 5′ RACE procedures. This strategy resulted in the cloning of a cDNA segment of the appropriate length from polyadenylated mRNA complementary to the CDA13 ORF.
cda12 antisense phenotype.
Antisense ribosomes were engineered to suppress expression from CDA12 and CDA13 independently. The cda12 antisense transformants were grown under drug selection conditions for 6 to 8 days, fixed, and stained to visualize nuclei and basal bodies. Cells were visualized using both conventional and scanning laser confocal microscopy. Wild-type cells adopt a classic hourglass shape just minutes before cytokinesis (Fig. 2A). Our cda12 hypomorphs occasionally exhibited macronuclear fission failure (Fig. 2B) but more frequently possessed multiple nuclei and numerous oral structures, suggesting multiple failed rounds of cytokinesis (Fig. 2C). Cells that had initiated cytokinesis (and adopted an hourglass shape) were isolated into drops of growth medium and observed. Live wild-type and cda12 mutant cells were collected for observation. The average time for hourglass wild-type cells to complete division was 5.1 min. The time needed for hourglass cda12 mutant cells to divide was greatly increased. Of 20 clones, only 6 divided after 4 h of isolation. For these 6, the average duration of cytokinesis was 16 min. The other 14 failed to divide even after 24 h, although they were still alive, as indicated by ciliary beating. These results support the conclusion that cda12 mutants are arrested during cytokinesis. Individuals expressing the antisense cda12 phenotype also exhibited a failure of macronuclear segregation (Fig. 2B). Hence, the principle phenotype involves problems in both cytokinesis and macronuclear segregation.
FIG. 2.

Immunofluorescence imaging of cda12 and cda13 hypomorph phenotypes. (A) Wild-type cell undergoing mitosis. Blue is the result of fluorescence tagging of antibody to β-tubulin, and green is Sytox labeling of macronuclei (Mac) and micronuclei (mic). (B) cda12 antisense mutant showing blue anticentrin labeling, revealing cortical basal bodies, and green nuclear Sytox labeling. The cleavage furrow has formed but has not undergone abscission. Neither micronuclei nor macronuclei (which when viewed by through focusing, appeared to have divided) have segregated into division products and aggregated. (C) Later-stage cda12 mutant showing multiple (red) nuclei labeled with propidium iodide and multiple oral structures (basal bodies labeled with anticentrin antibody), indicating numerous rounds of karyokinesis and cortical predivision patterning coupled with numerous failed rounds of cytokinesis. Images in panels A to C involved the use of confocal scanning laser microscopy. (D) cda13 antisense mutants as failed exconjugants. Pink (originating from rabbit antiserum, obtained from the Millipore Corporation, specific to phosphorylated histone H3) indicates condensed micronuclear DNA, green indicates cortical basal bodies, and blue is DAPI labeling of vacuolated macronuclei. Pairs have failed to dissociate after mating and remain parabiotically fused. The cell on the left has entered mitosis, showing micronuclear division and replication of the cortical pattern with neither macronuclear fission nor cleavage furrow formation. (E) A second cda13 postconjugation cell fusion product showing multiple micronuclei (pink), as well as macronuclear and cytoplasmic fission failures. (At this stage of cortical development, wild-type cells show complete macronuclear fission and well-developed cytokinetic furrows.) Images in panels D and E involved the use of conventional fluorescence microscopy. (F) Confocal image showing cortical details of a large exconjugant fusion mass exhibiting the effects of numerous rounds of cytokinetic fission failure (basal bodies were labeled with antibody to centrin and fluorescein isothiocyanate secondary antiserum). Red arrows indicate supernumerary oral apparatuses. Scale bar, 10 μm. (G) Differential interference contrast image of a giant living postconjugation fission mass (scale bar, 20 μm). (H) Same parabiotic aggregate seen in panel G, showing nuclei (blue) with DAPI fluorescence. The mass possesses only two macronuclei, though there are nearly 20 micronuclei, many in anaphase configurations.
GFP-Cda12p fusion protein localization (monitored by fluorescence, TEM, and live movies).
An inducible fusion gene encoding N-terminally GFP-tagged Cda12p was constructed and stably introduced into Tetrahymena by targeted disruption of the nonessential btu1-1 (β-tubulin) locus (23). Transformants expressing the fusion gene were selected using paclitaxel as described previously (47). Vegetative cells exhibited GFP-Cda12p fluorescence in several locations. Punctate foci were distributed in longitudinal cortical rows (Fig. 3A). Larger vesicles were associated with the macronucleus just prior to karyokinesis (Fig. 3B). During cytokinesis, the fluorescent signal became concentrated at the developing fission zone (Fig. 3C and G). This pattern was most conspicuous in cells that had just completed cytokinesis (Fig. 3C).
FIG. 3.

Imaging of GFP-tagged Cda12p. Panels A to F are conventional fluorescence micrographs. (A) Live-cell fluorescence image of GFP-Cda12p showing a view of the cell cortex. The signals show up in longitudinal rows of elongated vesicles docked at the cell cortex or mobile “lozenges” traveling both along and between ciliary rows (see the movie in the supplemental material). Scale bar, 10 μm. (B) Deep view showing a dividing cell with GFP-Cda12p-labeled vesicles aggregating at two poles of the macronucleus (pointers). Faint autofluorescence of food vacuoles appears in the background. (C) Postmitotic daughter cell (posterior division product) showing GFP-Cda12p-labeled vesicles concentrated at the fission zone (arrow). (D to F) Mating pairs in which the right partner is labeled with GFP-Cda12p. (D) Fluorescent vesicles are concentrated around the selected postmeiotic micronucleus (arrow). (E) As the selected micronucleus undergoes a third, prezygotic division, the GFP signal labels an anchoring capture cup (arrow). (F) GFP-Cda12p is concentrated around the migratory nucleus (arrow) during pronuclear exchange. (G) Confocal image of a living cell undergoing mitosis. The fluorescent signal is most intense in an area just posterior to the fission zone (arrow), where GFP-Cda12p-decorated vesicles are aggregating. (H) TEM image of GFP-Cda12p-positive vesicles after immunogold labeling. The section through a basal body (bb) shows a rich mixture of membrane-bound vesicles and tubulovesicular compartments. Gold particles appear to decorate noncoated vesicles (single black arrowhead) and tubulovesicular compartments (double black arrowheads). The white arrowhead indicates a coated vesicle.
Live mating pairs were also examined for GFP-Cda12p. Again, foci were abundant in longitudinal cortical rows. Internally, we saw fluorescent vesicles aggregating around micronuclei that were participating in nuclear exchange (Fig. 3D to F). These vesicles appeared during micronuclear selection (Fig. 3D), gametogenic mitosis (Fig. 3E), and nuclear exchange (Fig. 3F).
In live vegetative cells, most GFP-Cda12p-labeled structures appeared to be stationary, forming linear rows of punctate foci (0.12 μm in diameter), while others, especially elongated tubular vesicles (approximately 0.5 μm long), appeared to be highly mobile, traversing the cell cortex (see the movie in the supplemental material). These elongated vesicles moved, on average, 0.7 μm per s, consistent with fast transport along microtubule tracts (40). Microtubule bundles, organized into both longitudinal microtubule (LM) bands and transverse microtubule (TM) bands, are abundant in the Tetrahymena cortex (3, 19). Tubular vesicles were seen to cross both laterally, between ciliary rows, and longitudinally, along ciliary rows, predominantly in a posterior-anterior direction, although directional reversals were seen. Other tubular vesicles were observed to anchor along the ciliary rows and send narrow, transient processes out in multiple directions.
Immunogold labeling allowed us to probe the ultrastructural localization of GFP-Cda12p within the cell cortex (Fig. 3H). Vesicles with both circular and tubular profiles were decorated with gold, and these were located near basal bodies and parasomal sacs (sites of active pinocytosis). Gold was most often associated with smooth-walled, noncoated vesicles (Fig. 3H, single black arrowhead) and appeared less commonly over coated vesicles (Fig. 3H, white arrowhead). The latter compartments have previously been seen to form during pinocytosis (16).
Pinocytosis assay.
Having observed the GFP-Cda12p fusion protein associated with cortical vesicles near sites of active endocytosis, we examined pinocytosis in transformants expressing the cda12 antisense phenotypes, as well as in wild-type cells. We first exposed live cells to the FM 4-64 probe.
This water-soluble dye is nontoxic and nonfluorescent in aqueous media. It is believed that dye molecules insert into the outer leaflet of the plasma membrane, where they become fluorescent. This method of membrane labeling has been used previously to study endocytosis in a variety of eukaryotic cells (16, 54). Initially, pinocytosis looked normal in both mutant and wild-type cells. However, after 20 to 30 min, the FM 4-64 probe had moved from tiny cortical pinosomes into larger posterior vacuoles within wild-type cells while remaining in swarms of tiny vesicles within the cda12 mutants (Fig. 4A and B).
FIG. 4.

Endosomes in wild-type and cda12 mutant cell lines. (A and B) Pair of Tetrahymena cells, a cda12 mutant (left) and a wild-type cell (+) viewed with conventional fluorescence microscopy. Cells can be distinguished by MitoTracker (green) labeling of the wild-type cell in panel B. Cells were exposed to FM 4-64 for 25 min to load pinosomes and compartments downstream in the endocytic pathway. After 25 min, the cda12 mutant showed persistent, tiny vesicles (indicated by red arrows in panel A) while in the wild-type cell FM 4-64 had moved into larger vacuoles (indicated by white arrows in panel A). (C to G) Confocal images of live cells labeled with GFP-Cda12p (green) and FM 4-64 (red). (C) Whole cell imaged at the level of the cell cortex within minutes of FM 4-64 probe addition. Red pinosomes are clearly distinct from green Cda12p vesicles. (D) Magnified view of the cortical region boxed in panel C. Again, Cda12p (indicated by green-and-white arrows) does not overlap with the early pinosome population (indicated by red-and-white arrows). Scale bar, 3.0 μm. (E to G) Confocal optical section of GFP-Cda12p-labeled cells exposed to FM 4-64 for a longer period than the cell in panels C and D. (E) Red areas show FM 4-64 primarily in larger vacuoles (arrows). Scale bar, 2.0 μm. (F) Same field of view showing GFP-Cda12p. Small vesicles remain docked at the cortex, while larger vacuoles (arrows) reside in a deeper cortical location. (G) Double exposure shows colocalization of GFP-Cda12p and late-stage FM 4-64 probe in larger, more interior vacuoles (arrows).
We next examined the colocalization of the FM 4-64 probe (red) with the GFP-Cda12p fusion protein (green). Early during FM 4-64 loading, it was clear that FM 4-64-labeled pinosomes were discrete from GFP-Cda12p-labeled compartments, although they were quite close to each other in the cell cortex (Fig. 4C and D). After 25 min, however, we began to see larger vacuoles (approximately 0.4 μm) labeled with both the FM 4-64 probe and GFP (Fig. 4E to G). These results suggest that pinocytotic cargo and Cda12p-labeled compartments, while initially segregated, eventually converge at the cell cortex into larger, Cda12p-positive vesicles in wild-type cells.
cda13 antisense phenotype.
The cda13 antisense mutant shows a modest vegetative phenotype (slow growth and sensitivity to cold and/or shaking during incubation). When cda13 mutants were mated to one another (in mutant × mutant pairings), we observed a lethal conjugation phenotype with 74% penetrance. Pairs completed the schedule of nuclear events associated with conjugation but died during the transition from mating to exconjugant vegetative growth, often without completing a single cell division event. When mutants were outcrossed to wild-type partners, lethality was reduced (to 19%), but 32% of the viable clones generated following feeding were cell division “monsters,” giant multinucleate cells. For 24 h after mating and after transfer to nutritional medium, we found many of the outcross partners unable to dissociate from each other. Despite this block in dissociation, one partner frequently launched into mitosis, replicating its oral apparatus and its micronucleus. Despite multiple rounds of cortical development (stomatogenesis, the building of new oral structures) and micronuclear division, these cells appeared to be unable to replicate their macronuclei (Fig. 2D and E). After prolonged culture in growth medium, a number of these multicellular aggregates showed evidence of having executed numerous rounds of micronuclear division (Fig. 2D and E) and cortical development (Fig. 2F) without concomitant macronuclear or cytoplasmic fission (Fig. 2G and H). Macronuclei appeared to be enlarged and highly vacuolated. Some of these fission-arrested monsters were quite extraordinary, reaching dimensions of 0.5 mm (typical cells are 50 μm in length).
We were curious as to whether the lethality and fission arrest phenotypes were provoked simply by cell pairing and pair separation or whether the complete conjugation program of postzygotic development was required. To test this issue, we took advantage of the existence of sterile, aneuploid cell lines (star cells) to provoke pairing and unilateral nuclear transfer, with premature pair separation, retention of the parental macronucleus, and omission of postzygotic nuclear events (genomic exclusion [5]). When cda13 antisense mutants were mated to star cells (specifically A*III cells), most pairs (87%) yielded viable exconjugant clones. Of these pairs, 5% produced slow-growing clones with indications of fission failures. Significantly, about 5% of matings between wild-type and A*III cells result in bilateral nuclear exchange and completion of the postzygotic nuclear program through a process known as short-circuit genomic exclusion (7). These results strongly suggest that the cda13 exconjugant fission arrest is provoked as a result, not merely of a cycle of pairing and pair separation, but of the completion of bilateral nuclear exchange and the execution of the subsequent program of postzygotic development.
GFP-Cda13p fusion protein localization: fluorescence and TEM.
An inducible CDA13 gene construct encoding an N-terminally GFP-tagged fusion protein was made. GFP-Cda13p was localized along cortical rows and in cytoplasmic punctae (irregularly shaped structures) (Fig. 5A and B). These results can be contrasted with patterns of background autofluorescence (Fig. 5C [optical gain has been increased]). When GFP-Cda13p was expressed in mating cells, we found a transient pattern of localization in which the fusion protein appeared to decorate a ring associated with the nuclear exchange junction (Fig. 5D to F). This association coincided with periods just prior to and after nuclear exchange, when presumably the nuclear exchange junction is being resealed. Using immunogold labeling with GFP tagging and TEM, we found localization over two structures: one associated with a cytoplasmic matrix adjacent to the (presumed) Golgi apparatus and another associated with structures resembling multivesicular bodies (Fig. 5G and H). The former pattern of GFP localization is consistent with published descriptions of the Tetrahymena Golgi structures (31). It should be noted that our transcription analysis suggested that Cda13p is expressed predominantly during conjugal development, with no detectable expression during vegetative growth. Hence, inducing the expression of a GFP-tagged Cda13p fusion product during vegetative growth produces an artificial situation. We need to be cautious in interpreting the results. We should also note that, so far, we have been unsuccessful in developing a cryopreservation procedure that allows us to examine immunogold localization during conjugation. The cryopreservants we have tried disrupt mating.
FIG. 5.

Imaging of GFP-tagged Cda13p. Panels A to F are conventional fluorescence micrographs; panels A to C depict vegetative cells, and panels D to F depict conjugating cells. The TEM image in panel H shows vegetative cells. (A) Live-cell fluorescence image of GFP-Cda13p showing a view of the cell cortex. The signal shows up in longitudinal rows of irregularly shaped vesicles docked at the cell cortex. (B) Deep view showing elongated, irregular compartments. (C) Faint autofluorescence of food vacuoles in the background (optical gain was increased to highlight the pattern of autofluorescence). (D) Conjugating cells during nuclear exchange. Nuclei are blue (DAPI stained), and GFP-Cda13p is green. (E and F) Postexchange pairs show residual GFP signals at the nuclear exchange junctions. (G) TEM image showing a wild-type cortex with mitochondrion (mit), plasma membrane (pm), rough endoplasmic reticulum (rer), and Golgi structures. (Copyright Arno Tiedtke.) (H) TEM image of immunogold-labeled GFP-Cda13p-positive material. Gold particles are heavily concentrated over structures resembling multivesicular bodies (mvb) and in a region, positioned away from the rough endoplasmic reticulum-Golgi system, which we hypothesize to be downstream from the trans-Golgi network (TGN).
To test whether or not the vegetative GFP-Cda13p localization is dependent upon an intact Golgi apparatus, we compared GFP-Cda13p localization in cells treated with brefeldin A (a Golgi apparatus-disrupting agent) to that in untreated cells. Brefeldin A did indeed disrupt GFP-Cda13p localization in the cells, resulting in diffuse cytoplasmic labeling (Fig. 6). We emphasize that the GFP-Cda13p product appears not over the cisternae of the Golgi body directly but nearby, over a region of electron-dense cytoplasm adjacent to both the endoplasmic reticulum-Golgi system and the associated cortical mitochondria.
FIG. 6.

GFP-Cda13p localization in vegetative Tetrahymena cells with and without brefeldin A treatment. (A) Control cell induced to express GFP-Cda13p, showing both cortical and cytoplasmic GFP localization. (B) Cells treated with brefeldin A lose all cortical GFP localization and most cytoplasmic foci. (The photographic exposure time for panel B was twice as long as that for panel A.)
CDA12/CDA13 is an essential locus.
A disrupted CDA12/CDA13 locus was constructed and introduced into T. thermophila biolistically, and transformants were selected by their resistance to the antibiotic paromomycin. The macronuclear gene dose was driven higher by increasing levels of the drug, and clones were monitored both for the expression of the cda12/cda13 hypomorph phenotype (by DAPI staining and fluorescence microscopy) and for relative levels of the wild-type locus versus the disrupted gene locus (by Southern blot analysis).
Cultures produced cells exhibiting a predominantly wild-type phenotype despite high levels of the drug (up to 1 mg/ml). When these cultures were examined more closely, cells expressing the hypomorph phenotype (multinucleate monsters) were discovered to be accumulating on the bottoms of the culture chambers. Cells were misshapen and exhibited the characteristic multinucleate condition. Clonal assortment followed by Southern blot analysis demonstrated that we were unable to drive these transformants to complete loss of the wild-type allele (Fig. 7). Live cells maintained a dose of about 35% of the wild-type allele, despite increasing selective pressure. This finding is consistent with expectations for an essential gene: cells in which the dose of the wild-type locus drops below a critical level will express the fission arrest phenotype and drop out of the growing culture. Since a knockout created in this way disrupts both the CDA12 and CDA13 genes, it is not obvious whether one or both genes are essential. The observation that CDA13 produces no detectable transcript during vegetative growth argues that CDA12 is in fact essential for vegetative growth and development, while the lethal antisense phenotype of cda13 mutants suggests that CDA13 is essential for making the transition from conjugal to vegetative development.
FIG. 7.
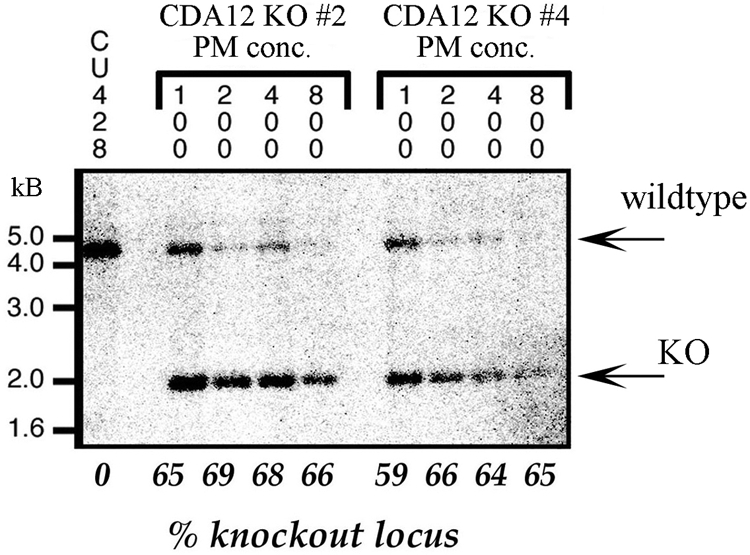
Loss of the wild-type CDA12/CDA13 locus following drug selection for the gene knockout. The CDA12/CDA13 locus was disrupted by the insertion of a selectable neomycin (or paromomycin) resistance gene driven by the histone H4 promoter. Tetrahymena cells were then transformed with this construct, which recombined into the endogenous CDA12/CDA13 locus within the macronucleus. The first lane corresponds to DNA from strain CU428, a wild-type control. Transformant (knockout [KO]) clones 2 and 4 were transferred into media with increasing concentrations (conc.; 100, 200, and 400 μg/ml, etc.) of paromomycin (PM). Numbers across the bottom indicate knockout/wild-type locus ratios from densitometric scans. The wild-type allele runs at 4.2 kb, and the knockout allele runs at 2.3 kb. Despite high drug concentrations (800 μg/ml paromomycin), cells never lost more than 30% of the wild-type CDA12/CDA13 allele.
DISCUSSION
The antisense ribosome technique.
The biology of Tetrahymena makes possible a remarkable form of antisense mutagenesis. rDNA exists as a single-copy gene within the germ line nucleus. This characteristic is unique among eukaryotes and allows for ribosomal manipulations that affect most or all copies of the somatic rDNA, resulting in structural changes within the ribosomes themselves. By coupling insertional mutagenesis with the use of 5′-end cDNA fragments oriented in the antisense configuration, Sweeney et al. showed that loss-of-function phenotypes can be generated by expressing antisense ribosomes (48). Chilcoat et al. (10) further demonstrated that this technique can be used to create cDNA antisense libraries. This protocol does have limitations. It is not certain to what degree gene expression is silenced following antisense transformation. Demonstrating this requires either Western blotting or some form of reporter gene assay. Lacking these data, we cannot assess the degree of gene silencing in our antisense transformants. A second problem is that we cannot rule out the possibility that our antisense insert is interacting with a transcript from another locus with homology to our antisense insert. (This scenario seems unlikely given that Southern blotting of various genomic digests revealed only a single hybridization signal [data not shown].) We would also argue that our ability to correlate protein localization with the antisense phenotypes strengthens the case that we have correctly identified the genes responsible for the phenotypes.
CDA12 and membrane trafficking.
We screened an antisense ribosome library (courtesy of Aaron Turkewitz, University of Chicago) for mutations affecting cell division. This process resulted in the identification of CDA12, a novel gene whose product is expressed during both vegetative growth and conjugation. Its product, Cda12p, associates with numerous membrane-bound compartments and appears to be necessary for both cytokinesis and macronuclear segregation. More specifically, GFP-Cda12p localizes over subcortical compartments near sites of active pinocytosis. By monitoring GFP-Cda12p in live cells, we observed the FM 4-64 probe to enter via vesicles that did not exhibit GFP-Cda12p labeling. After 20 to 30 min, FM 4-64 probe material returned to the cell cortex in larger, GFP-Cda12p-labeled compartments. These results suggest that Cda12p is localized over the Tetrahymena correlate of a recycling endosome (as well as other endocytic compartments). We also noticed GFP-Cda12p-labeled vesicles aggregating (i) near the macronucleus just prior to karyokinesis, (ii) at the fission zone during cytokinesis, and (iii) around the migratory gametic pronucleus during fertilization. These subcellular loci may represent targets of active membrane remodeling. It is to be expected that the macronucleus requires considerable membrane expansion to support karyokinesis, that the cytokinetic furrow requires membrane addition to support furrow ingression, and that the process of pronuclear exchange involves dramatic membrane remodeling to accompany the formation and resolution of the nuclear exchange junction during mating. It may also be possible that membrane-bound compartments aggregate near the dividing macronucleus as a prelude to cytoplasmic assortment during cytokinesis (T. Sugai, personal communication).
We were also able to characterize the movements of Cda12p-decorated vesicles along the cell cortex. The fastest motions involved long runs along cortical rows at 0.7 μm per s, the speed of “fast” motion via microtubule-based organelle motility seen in other eukaryotes (40). These motions were most frequently oriented in a posterior-anterior direction, although we also observed brief directional reversals and lateral traversing between ciliary rows. It is tempting to conclude that these membrane-bound compartments are utilizing both LM and TM bands, as seen in the cortical cytoskeleton for intracellular transport (3, 19). It has also been brought to our attention that in Paramecium species, similar bands of microtubules assemble just prior to cytokinesis (14, 27). In Paramecium, these bands are referred to as the cytospindle, and it has been suggested previously that they participate in cytokinesis. If the LM bands of Tetrahymena are comparable to the cytospindle of Paramecium, then we might propose that both structures play important roles in membrane trafficking and, more specifically, in delivering components to the ingressing cleavage furrow during late stages of cytokinesis.
CDA12 encodes a small protein with no identifiable homologs. Nevertheless, the predicted protein structure includes numerous membrane-spanning domains, and we have a body of evidence suggesting that the protein participates in membrane trafficking within the Tetrahymena cell. Functionally, Cda12p resembles members of a family of proteins that interact with Rab11, a small GTPase that functions in other eukaryotes to direct vesicle trafficking to and from the recycling endosome (41). Significantly, mutations of Rab11, or its associated FIPs, also result in defects during late stages of cytokinesis. These findings demonstrate that membrane from the recycling endosome is necessary for cleavage furrow ingression in a variety of metazoan models. It is tempting to propose that Cda12p may be functionally similar to FIPs and is involved in both trafficking associated with the recycling endosome and furrow ingression.
CDA13 and the trans-Golgi network.
CDA13 is another novel gene encoding a small protein, this time with an essential role in postconjugation pair separation, macronuclear karyokinesis, and exconjugant cytokinesis. The Cda13p protein is localized over a region adjacent to cortically aligned mitochondria and away from the nearby trans-Golgi network. It is also found within structures that resemble multivesicular bodies. Kang et al. recognized a similar region in plant cells (28), and that region has been named the membrane assembly matrix. This plant structure is also associated with multivesicular bodies and contains Rab proteins and their associated adaptors (A. L. Staehelin, personal communication). Consistent with the hypothesis that Cda13p is associated with the Golgi apparatus, GFP-Cda13p localization can be disrupted by the application of brefeldin A. Functionally, we find parallels between CDA13 and its sister gene, CDA12. Both genes encode proteins implicated in membrane trafficking, one potentially associated with the Golgi system and the other potentially associated with the recycling endosome. Both appear to be necessary for cytokinesis and macronuclear karyokinesis or segregation, though at distinct times within the cell's life history. Cda12p involvement occurs during vegetative growth and cytokinesis, whereas Cda13p exerts its influence during the specific period following sexual exchange, when mated pairs dissociate and reenter the vegetative cell division pathway. It has been shown previously that Rab11 is involved not only in endosome recycling, but also in membrane trafficking associated with the trans-Golgi network (9). Materials from the Golgi network have also been shown to participate in cytokinetic furrow ingression (13, 32). It is possible, and indeed seems likely, that both CDA12 and CDA13 encode proteins that are involved in membrane trafficking events required for cytokinesis and karyokinesis in Tetrahymena. Also, it seems likely that both participate in events associated with remodeling of the nuclear exchange junction during conjugation.
The nested-gene phenomenon.
The number of discoveries of overlapping genes has seen an exponential rise since the 2000 publication of the Drosophila genome sequence (1). This trend has gone hand-in-hand with the subsequent publication of over 2,300 other organismic genome sequences. In the most common overlap arrangements, genes have antiparallel orientations and are either convergent (with overlapping 3′ ends) or divergent (with overlapping 5′ ends); most such overlaps involve noncoding sequences (untranslated regions, introns, and promoter elements) (6). In the comparatively rare cases in which one gene is entirely encompassed by another in antisense orientation, the antisense transcripts frequently play a role in the posttranscriptional control of the principle transcript via RNAi mechanisms and are not themselves translated into functional proteins. This pattern casts our discovery into a rather unique light. CDA12 and CDA13 possess no introns and show complete overlapping of ORFs, and each appears to be expressed as a polyadenylated message. GFP tagging of the encoded proteins reveals specific, reproducible intracellular localization patterns, consistent with functions associated with the respective hypomorph phenotypes. The antisense ribosome approach that has allowed us to create hypomorphs specifically targets the expression of either the plus or minus transcript population and not that of the other. Hence, the phenotypes generated are specific to the oligonucleotide sequences targeted and not their complements. The simplest interpretation is that antiparallel, nested genes CDA12 and CDA13 each encode a unique protein. As yet, we have no evidence addressing whether or not an RNAi mechanism may also be involved. Is this an isolated example? At the time of this writing, there is a single published account of a nested, antisense gene pair in a eukaryote: the description of the yeast genes NAG1 and YGR031W, of which NAG1 is nested within YGR031W (33). Preliminary (unpublished) data suggest that such examples may be relatively common in the Tetrahymena genome. Indeed, the results of these studies argue for the existence of greater genome complexity than has been yet recognized.
Supplementary Material
Acknowledgments
This work was supported by NSF RUI grants MCB0131175, MCB0444700, and MCB0817993 awarded to E.S.C., as well as NSF grant no. MCB0091194 and no. MCB0220085 awarded to D.R. and NIH R01 GM074746 to M.W.
We thank Aaron Turkewitz (University of Chicago) for the generous gift of his antisense library, Douglas Chalker (Washington University in St. Louis, St. Louis, MO) for the gift of an inducible GFP expression plasmid, Stephan Zweifel (Carleton College) for editorial help with the manuscript, and Alex Stemm-Wolf for assistance with sample preparation for TEM. Arno Tiedtke (University of Muenster) generously provided us with Fig. 5G, highlighting the presumed Tetrahymena Golgi apparatus. We also thank Andrew Stahelein, University of Colorado—Boulder, for conversations illuminating the involvement of Rab and Rab-interacting proteins in membrane trafficking.
Footnotes
Published ahead of print on 13 March 2009.
Supplemental material for this article may be found at http://ec.asm.org/.
REFERENCES
- 1.Adams, M. D., S. E. Celniker, R. A. Holt, et al. 2000. The genome sequence of Drosophila melanogaster. Science 2872185-2195. [DOI] [PubMed] [Google Scholar]
- 2.Albertson, R., B. Riggs, and W. Sullivan. 2005. Membrane traffic: a driving force in cytokinesis. Trends Cell Biol. 1592-101. [DOI] [PubMed] [Google Scholar]
- 3.Allen, R. D. 1967. Fine structure reconstruction, and possible functions of components of the cortex of Tetrahymena pyriformis. J. Protozool. 14553-565. [DOI] [PubMed] [Google Scholar]
- 4.Allen, R. D., and A. K. Fok. 2000. Membrane trafficking and processing in Paramecium. Int. Rev. Cytol. 198277-318. [DOI] [PubMed] [Google Scholar]
- 5.Allen, S. L., S. K. File, and S. L. Koch. 1967. Genomic exclusion in Tetrahymena. Genetics 55823-837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boi, S., G. Soldà, and M. L. Tenchini. 2004. Shedding light on the dark side of the genome: overlapping genes in higher eukaryotes. Curr. Genomics 5509-524. [Google Scholar]
- 7.Bruns, P. J., T. B. Brussard, and A. B. Kavka. 1976. Isolation of homozygous mutants after induced self-fertilization in Tetrahymena. Proc. Natl. Acad. Sci. USA 733243-3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bruns, P. J., and D. Cassidy-Hanley. 2000. Biolistic transformation of macro- and micronuclei. Methods Cell Biol. 62501-512. [DOI] [PubMed] [Google Scholar]
- 9.Chen, W., Y. Feng, D. Chen, and A. Wandinger-Ness. 1998. Rab11 is required for trans-Golgi network-to-plasma membrane transport and a preferential target for GDP dissociation inhibitor. Mol. Biol. Cell 93241-3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chilcoat, N. D., N. C. Elde, and A. P. Turkewitz. 2001. An antisense approach to phenotype-based gene cloning in Tetrahymena. Proc. Natl. Acad. Sci. USA 988709-8713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cole, E. S. 2006. The Tetrahymena conjugation junction, p. 39-62. In F. Baluska, D. Volkmann, and P. W. Barlow (ed.), Cell-cell channels. Springer, New York, NY.
- 12.Cole, E. S., and T. A. Soelter. 1997. A mutational analysis of conjugation in Tetrahymena themophila. 2. Phenotypes affecting middle and late development: third prezygotic division, pronuclear exchange, pronuclear fusion and postzygotic development. Dev. Biol. 189233-245. [DOI] [PubMed] [Google Scholar]
- 13.Danilchik, M. V., S. D. Bedrick, E. E. Brown, and K. Ray. 2003. Furrow microtubules and localized exocytosis in cleaving Xenopus laevis embryos. J. Cell Sci. 116273-283. [DOI] [PubMed] [Google Scholar]
- 14.Delgado, P., M. R. Romero, and A. Torres. 1990. Microtubular systems of Paramecium in division: pattern of cytospindle assembly. J. Protozool. 37182-186. [Google Scholar]
- 15.Eisen, J. A., R. S. Coyne, M. Wu, et al. 2006. Macronuclear genome sequence of the ciliate Tetrahymena thermophila, a model eukaryote. PLoS Biol. 4e286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Elde, N. C., G. Morgan, M. Winey, L. Sperling, and A. P. Turkewitz. 2005. Elucidation of clathrin-mediated endocytosis in Tetrahymena reveals an evolutionarily convergent recruitment of dynamin. PLoS Genet. 1e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng, B., H. Schwarz, and S. Jesuthasan. 2002. Furrow-specific endocytosis during cytokinesis of zebrafish blastomeres. Exp. Cell Res. 27914-20. [DOI] [PubMed] [Google Scholar]
- 18.Fielding, A. B., E. Schonteich, J. Matheson, G. Wilson, X. Yu, G. R. Hickson, S. Srivastava, S. A. Baldwin, R. Prekeris, and G. W. Gould. 2005. Rab11-FIP3 and FIP4 interact with Arf6 and the exocyst to control membrane traffic in cytokinesis. EMBO J. 243389-3399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frankel, J. 1989. Pattern formation: ciliate studies and models. Oxford University Press, New York, NY.
- 20.Frankel, J. 1999. Cell biology of Tetrahymena thermophila, p. 27-125. In D. J. Asai and J. D. Forney (ed.), Methods in cell biology, vol. 62. Academic Press, San Diego, CA. [DOI] [PubMed] [Google Scholar]
- 21.Frankel, J., L. M. Jenkins, E. M. Nelsen, and F. P. Doerder. 1976. Mutations affecting cell division in Tetrahymena pyriformis. I. Selection and genetic analysis. Genetics 83489-506. [PMC free article] [PubMed] [Google Scholar]
- 22.Frankel, J., E. M. Nelsen, and L. M. Jenkins. 1977. Mutations affecting cell division in Tetrahymena pyriformis, syngen 1. II. Phenotypes of single and double homozygotes. Dev. Biol. 58255-275. [DOI] [PubMed] [Google Scholar]
- 23.Gaertig, J., L. Gu, B. Hai, and M. A. Gorovsky. 1994. High frequency vector-mediated transformation and gene replacement in Tetrahymena. Nucleic Acids Res. 225391-5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gaertig, J., and G. Kapler. 1999. Transient and stable DNA transformation of Tetrahymena thermophila by electroporation. Methods Cell Biol. 62485-500. [DOI] [PubMed] [Google Scholar]
- 25.Ghosh, R. N., D. L. Gelman, and F. R. Maxfield. 1994. Quantification of low density lipoprotein and transferrin endocytic sorting in HEp2 cells using confocal microscopy. J. Cell Sci. 1072177-2189. [DOI] [PubMed] [Google Scholar]
- 26.Hopkins, C. R., A. Gibson, M. Shipman, D. K. Strickland, and I. S. Trowbridge. 1994. In migrating fibroblasts, recycling receptors are concentrated in narrow tubules in the pericentriolar area, and then routed to the plasma membrane of the leading lamella. J. Cell Biol. 1251265-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iftode, F., J. Cohen, F. Ruiz, A. Torres-Rueda, L. Chen-Shan, A. Adoutte, and J. Beisson. 1989. Development of surface pattern during division in Paramecium. I. Mapping of duplication and reorganization of cortical cytoskeletal structures in the wild-type. Development (Cambridge, United Kingdom) 105191-211. [Google Scholar]
- 28.Kang, B.-H., J. R. Austin, J. M. Segui-Simarro, M. Otegui, E. Nielsen, and A. L. Staehelin. 2005. The membrane assembly matrix, a novel, non-membranous “organelle” that facilitates vesicle-mediated membrane assembly and transformation in the endomembrane system. Abstr. Am. Soc. Plant Biol. Minisymp., abstr. 29.
- 29.Krzywicka, A., M. Kiersnowska, D. Wloga, and J. Kaczanowska. 1999. Analysis of the effects of the cdaK1 mutation of Tetrahymena thermophila on the morphogenesis of the fission line. Eur. J. Protistol. 35342-352. [Google Scholar]
- 30.Kunkel, T. A., J. D. Roberts, and R. A. Zakour. 1987. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 154367-382. [DOI] [PubMed] [Google Scholar]
- 31.Kurz, S., and A. Tiedtke. 1993. The Golgi apparatus of Tetrahymena thermophila. J. Eukaryot. Microbiol. 4010-13. [DOI] [PubMed] [Google Scholar]
- 32.Lecuit, T., and E. Wieschaus. 2000. Polarized insertion of new membrane from a cytoplasmic reservoir during cleavage of the Drosophila embryo. J. Cell Biol. 150849-860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma, J., C. J. Dobry, D. J. Krysan, and A. Kumar. 2008. Unconventional genomic architecture in the budding yeast Saccharomyces cerevisiae masks nested antisense gene NAG1. Eukaryot. Cell 71289-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marsh, T. C., E. S. Cole, K. R. Stuart, C. Campbell, and D. P. Romero. 2000. RAD51 is required for propagation of the germinal nucleus in Tetrahymena thermophila. Genetics 1541587-1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McCormick-Graham, M., and D. P. Romero. 1995. Ciliate telomerase RNA structural features. Nucleic Acids Res. 231091-1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCormick-Graham, M., and D. P. Romero. 1996. A single telomerase RNA is sufficient for the synthesis of variable telomeric DNA repeats in ciliates of the genus Paramecium. Mol. Cell. Biol. 161871-1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Melia, S. M., E. S. Cole, and A. P. Turkewitz. 1998. Mutational analysis of regulated exocytosis in Tetrahymena. J. Cell Sci. 111131-140. [DOI] [PubMed] [Google Scholar]
- 38.Orias, E., M. Flacks, and B. H. Satir. 1983. Isolation and ultrastructural characterization of secretory mutants of Tetrahymena thermophila. J. Cell Sci. 6449-67. [DOI] [PubMed] [Google Scholar]
- 39.Plattner, H., and R. Kissmehl. 2003. Molecular aspects of membrane trafficking in Paramecium. Int. Rev. Cytol. 232185-216. [DOI] [PubMed] [Google Scholar]
- 40.Prahlad, V., B. T. Helfand, G. M. Langford, R. D. Vale, and R. D. Goldman. 2000. Fast transport of neurofilament protein along microtubules in squid axoplasm. J. Cell Sci. 1133939-3946. [DOI] [PubMed] [Google Scholar]
- 41.Prekeris, R. 2003. Rabs, Rips, FIPs, and endocytic membrane traffic. ScientificWorldJournal 3870-880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ren, M., G. Xu, J. Zeng, C. De Lemos-Chiarandini, M. Adesnik, and D. D. Sabatini. 1998. Hydrolysis of GTP on rab11 is required for the direct delivery of transferrin from the pericentriolar recycling compartment to the cell surface but not from sorting endosomes. Proc. Natl. Acad. Sci. USA 956187-6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, vol. 3. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY.
- 44.Schweitzer, J. K., E. E. Burke, H. V. Goodson, and C. D'Souza-Schorey. 2005. Endocytosis resumes during late mitosis and is required for cytokinesis. J. Biol. Chem. 28041628-41635. [DOI] [PubMed] [Google Scholar]
- 45.Shang, Y., B. Li, and M. A. Gorovsky. 2002. Tetrahymena thermophila contains a conventional gamma-tubulin that is differentially required for the maintenance of different microtubule-organizing centers. J. Cell Biol. 1581195-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Skop, A. R., H. Liu, J. Yates III, B. J. Meyer, and R. Heald. 2004. Dissection of the mammalian midbody proteome reveals conserved cytokinesis mechanisms. Science 30561-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith, J. J., E. S. Cole, and D. P. Romero. 2004. Transcriptional control of RAD51 expression in the ciliate Tetrahymena thermophila. Nucleic Acids Res. 324313-4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sweeney, R., Q. Fan, and M.-C. Yao. 1996. Antisense ribosomes: rRNA as a vehicle for antisense RNAs. Proc. Natl. Acad. Sci. USA 938518-8523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Turkewitz, A. P. 2004. Out with a bang! Tetrahymena as a model system to study secretory granule biogenesis. Traffic 563-68. [DOI] [PubMed] [Google Scholar]
- 50.Turkewitz, A. P., E. Orias, and G. Kapler. 2002. Functional genomics: the coming of age for Tetrahymena thermophila. Trends Genet. 1835-40. [DOI] [PubMed] [Google Scholar]
- 51.Ullrich, O., S. Reinsch, S. Urbe, M. Zerial, and R. G. Parton. 1996. Rab11 regulates recycling through the pericentriolar recycling endosome. J. Cell Biol. 135913-924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Urbe, S., L. A. Huber, M. Zerial, S. A. Tooze, and R. G. Parton. 1993. Rab11, a small GTPase associated with both constitutive and regulated secretory pathways in PC12 cells. FEBS Lett. 334175-182. [DOI] [PubMed] [Google Scholar]
- 53.Virtue, M. A., and E. S. Cole. 1999. A cytogenetic study of development in mechanically disrupted pairs of Tetrahymena thermophila. J. Eukaryot. Microbiol. 46597-605. [DOI] [PubMed] [Google Scholar]
- 54.Wiederkehr, A., S. Avaro, C. Prescianotto-Baschong, R. Haguenauer-Tsapis, and H. Riezman. 2000. The F-box protein Rcy1p is involved in endocytic membrane traffic and recycling out of an early endosome in Saccharomyces cerevisiae. J. Cell Biol. 149397-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wilson, G. M., A. B. Fielding, G. C. Simon, X. Yu, P. D. Andrews, R. S. Hames, A. M. Frey, A. A. Peden, G. W. Gould, and R. Prekeris. 2005. The FIP3-Rab11 protein complex regulates recycling endosome targeting to the cleavage furrow during late cytokinesis. Mol. Biol. Cell 16849-860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yao, M. C., C. H. Yao, L. M. Halasz, P. Fuller, C. H. Rexer, S. H. Wang, R. Jain, R. S. Coyne, and D. L. Chalker. 2007. Identification of novel chromatin-associated proteins involved in programmed genome rearrangements in Tetrahymena. J. Cell Sci. 1201978-1989. [DOI] [PubMed] [Google Scholar]
- 57.Yu, G.-L., and E. H. Blackburn. 1990. Amplification of tandemly repeated origin control sequences confers a replication advantage on rDNA replicons in Tetrahymena thermophila. Mol. Cell. Biol. 102070-2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu, X., R. Prekeris, and G. W. Gould. 2007. Role of endosomal Rab GTPases in cytokinesis. Eur. J. Cell Biol. 8625-35. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.