

Abstract
A new type of manganese-oxidizing enzyme has been identified in two alphaproteobacteria, “Aurantimonas manganoxydans” strain SI85-9A1 and Erythrobacter sp. strain SD-21. These proteins were identified by tandem mass spectrometry of manganese-oxidizing bands visualized by native polyacrylamide gel electrophoresis in-gel activity assays and fast protein liquid chromatography-purified proteins. Proteins of both alphaproteobacteria contain animal heme peroxidase and hemolysin-type calcium binding domains, with the 350-kDa active Mn-oxidizing protein of A. manganoxydans containing stainable heme. The addition of both Ca2+ ions and H2O2 to the enriched protein from Aurantimonas increased manganese oxidation activity 5.9-fold, and the highest activity recorded was 700 μM min−1 mg−1. Mn(II) is oxidized to Mn(IV) via an Mn(III) intermediate, which is consistent with known manganese peroxidase activity in fungi. The Mn-oxidizing protein in Erythrobacter sp. strain SD-21 is 225 kDa and contains only one peroxidase domain with strong homology to the first 2,000 amino acids of the peroxidase protein from A. manganoxydans. The heme peroxidase has tentatively been named MopA (manganese-oxidizing peroxidase) and sheds new light on the molecular mechanism of Mn oxidation in prokaryotes.
Mn(III,IV) oxides (Mn oxides) and soluble Mn(III) complexes are the strongest oxidizing agents in the environment after oxygen and play an important role in many biogeochemical cycles (25). At pH 7, they can oxidize metals, catalyze the formation of humic substances and organic nitrogen complexes, and oxidatively degrade humic and fulvic acids to bioavailable low-molecular-weight organic compounds (6, 38, 40). Geochemical cycling of Mn oxides can also control the distribution of many trace elements, as Mn minerals are highly charged and can adsorb and concentrate metals (22). In the pH range of aerobic natural waters (pH 6 to 8), chemical oxidation of Mn(II) is slow, but in the presence of Mn(II)-oxidizing microorganisms, the rate can be 4 to 5 orders of magnitude higher (30, 39, 48).
Multicopper oxidases (MCOs) are the only identified proteins from bacteria capable of manganese oxidation. These enzymes are a class of proteins that utilize copper as a cofactor to perform four one-electron substrate oxidations, thereby reducing molecular oxygen to H2O (37). Generally, MCOs oxidize organic compounds such as phenolics, but some fungal MCOs (laccases) that can oxidize Mn(II) to Mn(III) and Mn(IV) have been described previously (21, 36, 28). Bacterial MCOs involved with Mn oxidation have been genetically identified in Pseudomonas putida strains MnB1 and GB1 (cumA), Leptothrix discophora SS-1 (mofA), Bacillus sp. strain SG-1 (mnxG), and the alphaproteobacterium Pedomicrobium sp. strain ACM 3067 (moxA) (35). None of these MCOs share significant homology except for their copper binding motifs (35), and only in Bacillus sp. (11) have MCOs been directly linked to Mn oxidation.
Another class of proteins known in eukaryotes to oxidize manganese, but not commonly identified to be involved in bacterial Mn oxidation, are heme-containing manganese peroxidases (MnPs) (5, 33). These enzymes are extremely important for the degradation activities of lignin-degrading fungi. The MnP from the basidiomycete Phanerochaete chrysosporium has a single Mn(II) binding site near the heme and produces two Mn(III) equivalents at the expense of one H2O2 equivalent (18, 34, 45). MnPs and MCOs are able to work in concert, with the MnP utilizing H2O2 produced by the MCO-catalyzed Mn(II) oxidation (36). Both types of protein produce Mn(III). While MnPs are best known to occur in fungi, a similar mechanism has been reported for a catalase/peroxidase from Mycobacterium (27), and a catalase-peroxide mechanism was suggested to be involved in Fe and Mn oxidation in Arthrobacter (13).
A search for MCO-like genes in the draft genome sequence of the Mn(II)-oxidizing marine alphaproteobacterium “Aurantimonas manganoxydans” strain SI85-9A1 revealed duplicate copies of moxA-like genes (12). Initial attempts to isolate the Mn oxidase enzyme focused on the secreted proteins. Two regions were identified by a native polyacrylamide gel electrophoresis (PAGE) in-gel activity assay to have active Mn(II) oxidation, one at >250 kDa and the other at approximately 50 kDa. The active areas were excised from the gel, digested with trypsin, and analyzed with tandem mass spectrometry (MS/MS) but no peptides could be identified (12). In the same study, five bands from a corresponding Coomassie-stained sodium dodecyl sulfate (SDS)-PAGE gel returned identifications of peptides from a putative Ca2+ binding heme peroxidase, but no attempt was made to connect this protein to the active bands from the in-gel activity assay because they would have migrated differently (Coomassie bands were denatured protein, the in-gel activity assay native proteins).
Based on the genome data, the expected size of the MoxA-like proteins was approximately 50 kDa, similar to the size of MoxA from Pedomicrobium sp. strain ACM 3067 and similar in size to the most active region from the in-gel activity assay. It was then inferred that the Mn oxidase from A. manganoxydans strain SI85-9A1 could be a Mox ortholog with an estimated size of 50 kDa that may be part of a larger >250-kDa holoenzyme (12). The experimentally identified Ca2+ binding heme peroxidase was suggested to be involved with the biodegradation of complex organics utilizing H2O2 abiotically generated by the Mn(III) produced by the Mn(II)-oxidizing MoxA-like protein (after the mechanism described by Schlosser and Höfer [36]) (12).
In Erythrobacter sp. strain SD-21, the Mn oxidase enzyme was found in both the soluble and excreted fractions, suggesting that the activity may be loosely associated with the cell surface (23). Protein chromatography was employed to identify the Mn oxidase in this organism but did not conclusively implicate an MCO. The enzyme was partially purified and was found to be associated with a quinone cofactor, PQQ, that stimulated manganese oxidation in partially pure protein extracts and rescued the manganese oxidation activity in a mutant strain of Pseudomonas putida MnB1. Mn oxidation was not stimulated in vitro when copper was added, and activity was vastly decreased in the presence of MCO and quinone inhibitors. The addition of o-phenanthroline, a copper chelator and potent inhibitor of Mn oxidation in P. putida GB-1 (32), inhibited Mn oxidation only partially at concentrations far in excess of those required for GB-1. The absorbance spectrum of the partially purified protein extract did not show characteristics of an MCO, and the activity of the cell extract was between 7- and 30-fold higher than the activity measured for Mn-oxidizing organisms containing MCOs. Although the evidence pointed away from MCO involvement, H2O2 did not stimulate activity as expected if the enzyme was an MnP (23).
A. manganoxydans strain SI85-9A1 is not easily amenable to genetic techniques, and thus, isolation of the manganese oxidase was performed through protein chromatography techniques. Since early studies failed to conclusively identify the Mn oxidase, it was decided to fractionate the proteins in the organism to localize the activity and purify the protein from the active fraction. In this work, we report the significant purification of the Mn-oxidizing protein leading to its identification as an animal heme peroxidase with multiple calcium binding motifs, and localization of the protein to the outer membrane as a peripheral membrane protein. We revisit the Mn oxidase from Erythrobacter sp. strain SD-21 (also genetically recalcitrant) and identify the protein from an active Mn-oxidizing band with a native PAGE in-gel activity assay. This protein is also an animal heme peroxidase with calcium binding motifs.
MATERIALS AND METHODS
Strain isolation.
Aurantimonas manganoxydans strain SI85-9A1 was collected in 1985 from Sannich Inlet, Vancouver Island, British Columbia, Canada. This strain was isolated from a water sample taken from a depth of 125 m and has recently been described (1a). Erythrobacter sp. strain SD-21 was isolated in 2000 from surface sediments in San Diego Bay, CA. Both SI85-9A1 and SD-21 strains were originally isolated on K medium (41).
Cultivation.
A. manganoxydans strain SI85-9A1 was grown at room temperature in 1-liter liquid culture batches in either M medium or J medium made with artificial seawater (41) supplemented with a mixture of sterile filtered 10 mM glycerol, 10 mM Na-formate, 100 μM MnCl2, and 100 μl of 3 mg ml−1 ferric ammonium citrate. Ferric ammonium citrate and MnCl2 were always made fresh prior to inoculation. Control flasks without MnCl2 added were always cultured simultaneously. The inoculum was a 1-ml volume of a 3-ml starter culture that was grown for 48 h in media without Mn. The inoculum for the starter cultures was always a single isolated colony from a K medium plate that was known to be oxidizing manganese. Flasks were shaken on an orbital shaker at 150 rpm. Cells were harvested within 24 h after the onset of manganese oxidation (visible darkening of the growth medium due to particulate oxides being produced). Erythrobacter sp. strain SD-21 was cultured as previously described (23).
Protein fractionation and isolation from A. manganoxydans.
A. manganoxydans cells were fractionated using a combination of protocols from Myers and Myers (29), Gaspard et al. (17), and DiChristina et al. (10). The following five different protein fractions were recovered: secreted, loosely bound outer membrane (LBOM), soluble (periplasmic and cytoplasmic), inner membrane, and outer membrane proteins.
Cells were harvested by centrifugation at 10,000 × g at 4 °C. The spent culture medium was decanted into sterile 1-liter Erlenmeyer flasks, prefiltered through sterile Whatman filter paper, filtered through a 0.2-μm Millipore Stericup filter, and then ultrafiltered to a volume of ∼5 ml, using a 400-ml Amicon stirred filtration cell with a 10-kDa nominal molecular mass limit (NMWL) filter. This fraction was considered the secreted protein fraction.
The cell pellet was resuspended in 50 ml of 100 mM HEPES buffer with 100 μM ascorbate added to reduce any Mn oxides to Mn(II). Cells were pelleted by centrifugation, and the supernatant was collected. After centrifugation, the cell pellet was washed in 100 ml of phosphate-buffered saline, centrifuged again, and finally resuspended and stirred vigorously in 100 ml of a high-salt Tris buffer (100 mM Tris [pH 7.5], 1 M KCl, 1 mM dithioerythritol) for 4 h at 4°C; then, it was centrifuged again. The high-salt buffer supernatant was collected and added to the supernatant from the ascorbate wash and the phosphate-buffered saline wash solutions. These combined supernatants were prefiltered through sterile Whatman paper, filtered through a 0.2-μm Millipore Stericup filter, and then ultrafiltered to a volume of ∼5 ml, using the Amicon stirred filtration cell with a 10-kDa NMWL filter. The resulting concentrated solution was considered the LBOM protein fraction.
The pellet was then resuspended and washed in 6 ml of 10 mM Tris-HCl (pH 8.1) buffer and then treated with three rounds of lysis using a French press at 20,000 lb/in2. The membrane and soluble proteins were then separated by ultracentrifugation at 200,000 × g for 1 h at 4°C, using a Beckman TLA 100.1 rotor. The supernatant was collected and was considered the soluble (periplasmic and cytoplasmic) protein fraction.
The pelleted outer and inner membrane fractions were then separated using Triton X-100 to solubilize the inner membrane proteins. The pellet was first resuspended in 3 ml of 10 mM HEPES buffer (pH 7.5), with 10 mM MgCl2, and then equilibrated overnight at 4°C. The resuspended pellets were then equilibrated to 22°C, Triton X-100 was added to a final concentration of 2.0% (vol/vol), and then the pellets were incubated for 10 min at room temperature. The tubes were put on ice and then ultracentrifuged at 100,000 × g for 2 h at 4°C. The supernatant was collected and was considered the inner membrane protein fraction.
The resulting pellet, containing the Triton X-100 insoluble material, was then resuspended in 3 ml of 10 mM Tris-HCl solution (pH 8.1). A 1/10 volume equivalent of lysozyme (final concentration, 6.4 mg ml−1) was then added to the solution and allowed to incubate for 20 min. The Triton X-100 insoluble material was again pelleted by ultracentrifugation as previously described. The resulting pellet was resuspended in 10 mM HEPES buffer (pH 7.5) and was considered the outer membrane protein fraction.
Once all of the fractions were collected, they were dialyzed overnight against 10 mM HEPES buffer (pH 7.5), using 6,000- to 8,000-molecular-weight-cutoff dialysis tubing. After dialysis, each fraction was then concentrated further to a final volume of ∼2 ml, using Millipore Biomax Ultra centrifugal filters with a 10-kDa NMWL cutoff. This 2-ml volume was used for fraction activity assays, fast protein liquid chromatography (FPLC) protein purification, SDS-PAGE, and native PAGE in-gel activity assays.
Functional assays and protein localization (crude protein extracts).
The protein concentration in each subcellular fraction was normalized to 100 μg ml−1 using Coomassie Plus (Pierce) and then assayed over 48 h for the formation of Mn(IV) oxides and Mn(III)-pyrophosphate complexes [Mn(III)-PPi] (41, 47). The assay mix consisted of 10 mM HEPES (pH 7.5), 50 mM NaCl, 10 μg of protein, ±5 mM PPi, and ±100 μM MnCl2. Mn oxides and Mn(III)-PPi complexes were quantified using a colorimetric leucoberbelin blue (LBB) microplate assay, where 50 μl of each sample was reacted with 250 μl of LBB solution (41). LBB reacts with Mn oxides and Mn(III)-PPi, forming a blue color that absorbs at 620 nm. In tubes without PPi, the MnCl2 was oxidized to Mn(IV) oxides, whereas in tubes with PPi, the MnCl2 was oxidized only to Mn(III), as PPi captures Mn(III), forming a stable complex (46, 47). Real-time formation of Mn(III)-PPi complexes (growth of an absorption peak at 256 nm) was monitored with a spectral scan between 200 and 350 nm for a period of 48 h.
Differences in activity due to the cations available were investigated by substituting NaCl with 0 to 100 mM concentrations of CaCl2 and MgCl2. The effect of peroxide, pyrroloquinoline quinone (PQQ), and NAD+ addition was determined by adding final concentrations of 0 to 100 μM H2O2, 0 to 50 μM PQQ, and 0 to 200 μM NAD+. Laccase-like activity was determined, as described in reference 16, by adding 100 μl of a 9.1 mM ABTS [2,2′-azinobis(3-ethylbenzthiazolinesulfonic acid)] solution to 10 μg of nonpurified proteins (per the NaCl assay described above) and visualizing a green color change.
Protein extracts and Mn oxidation assays used for Erythrobacter sp. strain SD-21 have been previously described (23). The effect of peroxide addition was determined by adding 10 μM hydrogen peroxide to the assay.
PAGE and manganese oxidation in-gel activity assay.
Crude protein extracts and FPLC-purified proteins were separated by native PAGE using 10% Tris-HCl resolving gels. Native PAGE was carried out in the absence of β-mercaptoethanol, SDS, and sample boiling. Two sets of samples were run side by side, and after electrophoresis, the gel was sliced in two, with one half stained with Coomassie blue or silver (Pierce) and the other assayed for Mn oxidase activity. For the Mn oxidation assay, the gel was first immersed in prewash solution (10% glycerol with 0.5% Triton X-100) for 30 min, with the solution being changed after 15 min. The prewash solution is then replaced with 10 mM HEPES buffer (pH 7.6) and incubated at room temperature for 10 min. The HEPES buffer is then replaced with fresh buffer containing 100 μM MnCl2 and incubated overnight. Active protein forms a brown band of Mn oxides, and the position of this band can be compared to the Coomassie blue/silver-stained gel.
SDS-PAGE was performed under the conditions specified by the manufacturer (Bio-Rad). SDS and β-mercaptoethanol were used, and protein samples were boiled prior to loading. Precast 4 to 15% Tris-HCl gels (Bio-Rad) were used for determining protein purity.
FPLC purification of the A. manganoxydans Mn oxidase protein.
The Mn oxidase protein was partially purified from LBOM proteins extracted from four 1-liter batches of A. manganoxydans SI85-9A1 isolates. Purification was performed with Äkta FPLC, with all chromatography columns from GE Healthcare, at a temperature of 4°C. Dialyzed (10 mM HEPES [pH 7.5]) LBOM proteins were initially separated by size exclusion chromatography (see below for conditions) and analyzed with MS/MS. After initial analysis, subsequent LBOM protein extractions were separated with up to three types of chromatography. The first purification step used a 5-ml HiTrap Q fast-flow anion exchange column in 20 mM Tris buffer (pH 8.5), with a linear NaCl elution gradient from 0 to 1 M. Each eluted fraction (1 ml) was assayed for Mn(II) oxidation activity by adding 10 μl of 1 mM MnCl2 to 90-μl aliquots of the eluted fractions (100 μM final Mn2+ concentration). After overnight incubation at room temperature, 100 μl of LBB was added to detect Mn oxide production. The remainder of the eluted fractions that tested positive for Mn oxides was then pooled and concentrated using 10-kDa NMWL spin filters (Amicon Ultra and Millipore Biomax) before either being tested by a quantitative Mn oxidation activity assay or being reinjected for further purification. Subsequent purification steps used a 5-ml HiTrap phenyl high-performance hydrophobic interaction column (phenyl HP) [in 20 mM HEPES buffer (pH 7.5) with a decreasing linear gradient of 1.7 M (NH4)2SO4], and then size exclusion chromatography used a 120-ml HiPrep 16/60 Sephacryl S-200 high-resolution column (in 20 mM HEPES with 150 mM NaCl). Each injection volume was ∼1.5 ml, with a 2-ml injection loop used.
Samples of LBOM proteins and purified active fractions were then analyzed using MS/MS at the Proteomic Shared Resource Facility at the Oregon Health & Science University (OHSU).
MS/MS and genome confirmation of the Aurantimonas manganoxydans Mn oxidase identity.
MS/MS was used to identify the proteins from the active fraction(s) pooled by FPLC. The protein concentration in each sample was estimated using a Nanodrop ND-1000 spectrophotometer (assuming that an A280 of 1 is 1 mg ml−1) and was then diluted and dried in a Speedvac (Savant) to a final dry mass of ∼20 μg. These dried samples were then resuspended and digested using trypsin at the Shared Protein Resource Facility at OHSU. The samples were rehydrated in digestion buffer, and 2 μl of dithiothreitol (DTT) solution was added. The samples were incubated for 15 min at 50°C, and then 1 μl of isoamyl alcohol solution was added. The samples were then incubated in the dark at room temperature for 15 min, and then an additional 4 μl of DTT solution was added; the samples were then incubated at room temperature for another 15 min. Enough trypsin was then added to ensure a 1:25 enzyme-to-substrate ratio in the solution along with water to achieve a final volume of 40 μl. The samples were then centrifuged and left to incubate overnight at 37°C. After incubation, formic acid (2 μl of 88% acid) was added to halt the digestion process, and the samples were frozen until time of analysis.
All MS/MS samples were analyzed using Mascot (version Mascot; Matrix Science, London, United Kingdom). Mascot was set up to search NCBI assuming that the digestion enzyme is trypsin. Mascot was searched with a fragment ion mass tolerance of 0.60 Da and a parent ion tolerance of 1.2 to 1.4 Da. U−1 of selenocysteine, b+1 of asparagine/aspartic acid, z+1 of glutamine/glutamic acid, and an iodoacetamide derivative of cysteine were specified in Mascot as fixed modifications. Oxidation of methionine was specified in Mascot as a variable modification.
Scaffold (version Scaffold_2_01_00; Proteome Software, Inc., Portland, OR) was used to validate MS/MS-based peptide and protein identifications. Peptide identifications were accepted if they could be established at >50.0% probability as specified by the Peptide Prophet algorithm (24) as well as if the identifications could be established at >95.0% probability and contained at least two identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm (31). Proteins that contained similar peptides but could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. Protein fragments were then queried against the draft genome database of Aurantimonas sp. strain SI85-9A1, and full gene sequences were then translated and queried using PROSITE to identify domains.
Protein isolation from Erythrobacter sp. strain SD-21.
Multiple native PAGE Mn-oxidizing in-gel activity assays and Coomassie blue-stained gels were used to purify and subsequently identify the manganese-oxidizing protein as described below. Mn stained bands were prepared from soluble cell extract and the excreted protein fraction. The soluble cell extract was prepared as previously described (23). The excreted fraction was prepared from a 2-liter culture grown on K medium. The Mn oxides were reduced with 3 mM ascorbic acid, and the cells were removed by centrifugation at 10,000 × g for 20 min at 4°C. The supernatant was passed through a 0.2-μm filter, concentrated in a stirred cell with a 30-kDa NMWL membrane, and finally concentrated to 3 ml by ultrafiltration (50-kDa NMWL). Coomassie-stained protein bands which migrated an equal distance as the corresponding Mn-oxidizing band were also used for Mn-oxidizing protein identification. These samples were enriched in Mn-oxidizing activity by a phenyl HP as described above. The Mn-oxidizing activity was not bound to the column.
Purification and identification of the Erythrobacter sp. strain SD-21 Mn oxidase.
The proteins were separated by one-dimensional SDS-PAGE using 12% polyacrylamide Tris-glycine gels. The gel was stained with Coomassie blue by standard methods. The Mn-oxidizing in-gel activity assays were prepared as described above. Excised gel bands were washed twice with 200 μl of 50% acetonitrile and 50% 5 mM DTT-25 mM NH4HCO3 with vortexing for 10 min and finally washed with 200 μl acetonitrile. The dehydrated gel piece was rehydrated by adding 20 μl of ice-cold 10 ng μl−1 trypsin (Promega) in 5 mM DTT-25 mM NH4HCO3 and then was incubated on ice for 30 min, and the remaining trypsin solution was removed and replaced with fresh 5 mM DTT-25 mM NH4HCO3. The digestion was allowed to continue at 37°C overnight. The peptide mixture was then acidified with 2 μl of 2% trifluoroacetic acid and vortexed for 30 min, and the supernate was extracted. Finally, 20 μl of 20% acetonitrile-0.1% trifluoroacetic acid was added, and the mixture was vortexed to extract the remaining peptides and then was combined with the previous fraction. The combined extractions are analyzed directly by nanobore liquid chromatography (LC)-MS/MS.
LC-MS/MS data were acquired in a data-dependent fashion by selecting the most intense peak with charge state of 2 to 4 that exceeds 40 counts, with exclusion of former target ions set to “always” and the mass tolerance for exclusion set to 100 ppm. Time-of-flight MS were acquired at m/z 500 to 1,800 Da for 0.5 s with 20 time bins to sum. MS/MS data were acquired from m/z 65 to 2,000 Da by using “enhance all” and 20 time bins to sum, dynamic background subtract, automatic collision energy, and automatic MS/MS accumulation with the fragment intensity multiplier set to 12 and maximum accumulation set to 3 s before returning to the survey scan.
The MS/MS peptide fragments were analyzed by Analyst 2.0 (Applied Biosystems) and subjected to a database search using Mascot 2.2.1 (Matrix Science) with Mascot Daemon 2.2 (Matrix Science) data import filter parameters set as follows: default precursor charge state from 2 to 4; precursor and MS/MS data centroiding using 50% height and 0.05 amu merge distances. MS/MS peaks with intensity of <1% of the base peak were discarded, as were MS/MS spectra with fewer than 22 peaks remaining. Data were searched against the Swissprot database obtained at ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/, containing 237,168 sequences. The search identified tryptic peptides with up to two missed cleavages and used mass tolerances of 100 ppm (MS) and 0.10 Da (MS/MS), with variable modifications as follows: deamidation (NQ), oxidation (M), pyro-Glu (N-term Q). The search results indicated that individual ion scores of >22 indicate identity or extensive homology (P < 0.05). Protein fragments were screened against the incomplete Erythrobacter sp. strain SD-21 genome sequence.
Heme peroxidase assay.
Native PAGE was performed as described previously. The procedure for staining described by Thomas et al. (43) and Hagan and Mobley (19) was performed as follows. Prior to use, a 6.3 mM (15 mg ml−1) solution of 3,3′,5,5′-tetramethylbenzidine (TMBZ; Fisher) was prepared in methanol and cooled to 4°C once the TMBZ was fully dissolved. This was mixed with prechilled 250 mM sodium acetate (NaAc; pH 5.0) at a ratio of 3 parts TMBZ solution to 7 parts NaAc buffer. After a brief wash with water, the gel was allowed to equilibrate in the 3:7 solution for ∼1.5 h at 4°C and covered from light. Hydrogen peroxide was then added to a final concentration of 30 mM, and the bands were allowed to develop for 30 min (light-blue bands were seen almost immediately after the addition of hydrogen peroxide). The gel was imaged without further clarification of the background. The bands were related to native PAGE silver-stained activity gels as described above.
RESULTS AND DISCUSSION
Biogeochemical cycling of manganese has important implications for the cycling of carbon, nitrogen, and trace metals (11, 40, 42). The cycling of manganese is slow abiotically but is vastly increased in the presence of manganese-oxidizing and -reducing organisms (30, 39, 48). On the oxidative side of this cycle, many phylogenetically diverse prokaryotic microorganisms that can oxidize manganese have been described, and a common family of proteins that have been implicated in Mn oxidation are the MCOs. Mn oxidation mechanisms in eukaryotes also involve MCOs, but heme-containing Mn peroxidases are common as well. These proteins are extremely important for the lignin degradation capabilities of litter-decaying basidiomycetes (45).
This study investigates two members of the alphaproteobacteria, Aurantimonas manganoxydans strain SI85-9A1 and Erythrobacter sp. strain SD-21. As with other manganese-oxidizing organisms, the Mn oxides produced by these bacteria are found extracellularly with colonies of Erythrobacter, even demonstrating a diffuse Mn oxide halo suggestive of a diffusible manganese-oxidizing factor or perhaps a diffusible Mn(III) complex, which is then oxidized or disproportionated, forming Mn(IV) oxides away from the cell. Accompanying the microscopic and macroscopic colony scale evidence is biochemical evidence that supports an outer membrane or extracellular location for bacterial manganese oxidases (1, 4, 9, 15, 26). For Erythrobacter, it is already known that the Mn oxidase protein is soluble, but due to interfering polysaccharides, it is still not known if the protein is transported out of the cell and is freely diffusible or if it attaches to the outer membrane (23). For A. manganoxydans strain SI85-9A1, the Mn oxidase protein was localized to the LBOM fraction by using a colorimetric assay that measures the concentration of Mn oxides produced (41). The LBOM fraction accounted for 90% of the activity, with the remaining activity associated with nonbound secreted proteins (Fig. 1A).
FIG. 1.

Manganese oxidation in subcellular protein fractions from Aurantimonas manganoxydans strain SI85-9A1 where the MnO2 and/or Mn(III)-PPi complexes produced over 48 h were quantified with LBB (A). Error bars represent 1 standard deviation (n = 2). Extracts of LBOM proteins from A. manganoxydans grown in J medium monitored for Mn(III)-PPi complex formation over 48 h (B). Appearance and growth of an absorption peak at 258 nm indicates Mn(III)-PPi complex formation. Dotted lines represent 0 h, dashed lines 12 to 24 h, and solid lines 48 h. Unlike M medium, J medium does not contain any Mn(II) impurities.
The formation of Mn(III) intermediates can be measured using a PPi capture assay where any Mn(III) produced is complexed and stabilized by PPi (47). The formation of this complex can be monitored in real time by observation of the adsorption at 256 nm. For LBOM protein extracts from A. manganoxydans, most of the Mn(II) oxidation occurred over the first 24 h, with very little difference in the amount of Mn(III)-PPi produced between 24 and 48 h (Fig. 1B). Erythrobacter also produces an Mn(III) intermediate that can be captured by PPi with rapid initial production for about 5 h with very little increase in concentration after 12 h (23).
In A. manganoxydans, the protein involved in Mn oxidation is induced by the presence of manganese. Figure 1A represents data collected from cells grown in M medium, which has yeast extract and peptone added and is therefore associated with Mn impurities. As shown in Fig. 1A, the manganese oxidase is present but at a lower level, with the controls having approximately one-third the activity of the Mn-supplemented medium. Figure 1B represents data produced from cells grown in J medium, which has no yeast extract or peptone added. There is little or no enzyme present unless MnCl2 is added to the growth medium.
The LBOM proteins were characterized further by their ability to oxidize the artificial laccase (or MCO) substrate ABTS. This compound turned a characteristic green color, suggesting that laccase activity was also associated with the LBOM fraction. Mn-oxidizing activity in the LBOM fraction was further enriched by size exclusion chromatography, and 29 proteins were identified by MS/MS analysis (matched to the draft genome database of Aurantimonas sp. strain SI85-9A1). Within these 29 proteins, there was the putative hemolysin-type Ca2+ binding heme peroxidase previously identified by Dick et al. (12) and an MCO (NCBI accession number ZP_01225909) (data not shown). This MCO was not one of the MoxA homologs identified in the A. manganoxydans genome by Dick et al. (12), but it is closely associated with an operon encoding homologs of type II protein secretion machinery. Flanking the operon at one end is a transposon and then the Ca2+ binding heme peroxidase gene, and at the other end is the MCO gene (12). It has been demonstrated before that the export of an Mn(II)-oxidizing MCO in Pseudomonas putida strain GB-1 requires a type II secretion system, so it is conceivable that the MCO and heme peroxidase in A. manganoxydans are also transported via a type II secretion pathway (2, 4). Analysis of LBOM protein samples using native PAGE in-gel activity assays confirmed the results of Dick et al. (12) that the main active protein was approximately 50 kDa in size, but it also revealed other weakly active bands that were between 50 and 250 kDa in size (Fig. 2). This phenomenon has been observed with this protein before by Dick et al. (9), who identified the Ca2+ binding heme peroxidase in five different Coomassie-stainable bands in a denaturing PAGE gel in a size range from ∼60 kDa to >250 kDa.
FIG. 2.
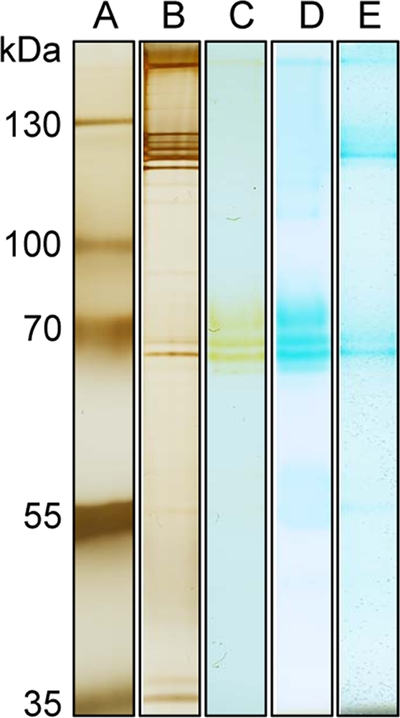
Native PAGE gel of the LBOM protein fraction from Aurantimonas manganoxydans strain SI85-9A1. Lane A, size standard; lane B, silver stained; lane C, in-gel Mn oxidation assay; lane D, in-gel Mn oxidation assay stained with LBB; lane E, heme-staining assay.
In Erythrobacter, the nonpurified manganese oxidase was stimulated by PQQ, NAD, and Ca2+ (23). The addition of 25 mM CaCl2 instead of 50 mM NaCl to assays containing A. manganoxydans LBOM proteins resulted in a 4.3-fold rate increase, and the addition of 10 μM H2O2 caused a further 1.6-fold increase. The addition of NAD+ or PQQ alone had detrimental effects on the Mn(II) oxidation rate, but the addition of NAD+ and PQQ together increased the oxidation rate 1.44-fold (Table 1). The maximum in vitro Mn(II) oxidation rate (measured using dialyzed LBOM protein fraction) was calculated to be 700 μM min−1 mg−1 (Vmax of ∼0.09 μM min−1; Km of ∼10 μM).
TABLE 1.
Effect of various additives on the in vitro Mn(II)-oxidizing activity of the Mn oxidase protein from Aurantimonas manganoxydans strain SI85-9A1a
| Additive(s)b | Fold difference (compared to control) |
|---|---|
| Salts (control, 10 mM HEPES [pH 7.5]) | |
| NaCl (50 mM) | 1.81 |
| CaCl2 (25 mM) | 4.26 |
| MgCl2 (25 mM) | 2.14 |
| Cofactors (control, 10 mM HEPES [pH 7.5] with 25 mM CaCl2) | |
| H2O2 (10 μM) | 1.60 |
| PQQ (10 μM) + NAD+ (100 μM) | 1.44 |
| H2O2 (10 μM) + PQQ (10 μM) | 0.93 |
| H2O2 (10 μM) + NAD+ (100 μM) | 0.14 |
| PQQ (10 μM) | 0.84 |
| NAD+ (75 μM) | 0.20 |
| H2O2 (10 μM) + PQQ (10 μM) + | |
| NAD+ (75 μM) | 1.07 |
Assays were performed on the LBOM protein fraction.
Numbers in parentheses represent the concentration that produced maximum activities.
A three-step purification strategy was used to purify the Mn oxidase from the LBOM protein fraction. This included ion exchange, hydrophobic interaction, and size exclusion chromatography. After each step, each eluted fraction was tested for active manganese oxidation, and any fractions that were active were pooled together, quantified, and analyzed with SDS-PAGE (Fig. 3). Purity and yield were also determined by reacting subsamples from each step with 100 μM Mn2+ and 25 mM Ca2+, with protein activity detected by the LBB method (Table 2).
FIG. 3.

SDS-PAGE gel of the LBOM protein fraction from Aurantimonas manganoxydans strain SI85-9A1 after successive purification by FPLC. Lane A, size standard; lane B, LBOM protein fraction; lane C, post-ion exchange chromatography; lane D, post-hydrophobic interaction chromatography; lane E, post-gel-filtration; lane F, size standard. The protein was silver stained with 1 μg total protein loaded into each well, except the size standard, for which 1 μl was used.
TABLE 2.
Purification of Mn(II)-oxidizing protein from LBOM proteinsb
| Stepa | Volume (ml) | Total protein (mg) | Total activity (U) | Sp act (U/mg) | % Yield | Fold purification |
|---|---|---|---|---|---|---|
| LBOM proteins | 1.500 | 0.381 | 1,503 | 3,946 | 100 | 1.0 |
| IEX (Q FF) | 0.500 | 0.058 | 319.6 | 5,510 | 21 | 1.4 |
| HIC (phenyl HP) | 0.200 | 0.015 | 183.5 | 12,240 | 12 | 3.1 |
| SEC (S-200) | 0.100 | 0.004 | 62.29 | 15,570 | 4 | 3.9 |
IEX, ion exchange; HIC, hydrophobic interaction; SEC, size exclusion chromatography; Q FF, HiTrap Q fast flow; S-200, HiPrep 16/60 Sephacryl S-200.
U, nmol MnO2 produced/hour.
Preliminary MS/MS investigation of peptides from proteins contained in the pooled active fractions after ion exchange chromatography identified 15 proteins, with the top hit being the putative Ca2+ binding heme peroxidase. The other proteins identified were five flagellum-associated proteins (both “hook-associated” and flagellin proteins themselves), three proteins from ABC-type amino acid transport systems (transporters and substrate binding), a high-affinity urea/thiourea/hydroxyurea porter, an IMP dehydrogenase/GMP reductase, a porin, a TRAP-type dicarboxylate transporter, and an acetyl coenzyme A C-acyltransferase protein. The MCO identified previously could not be identified in this preparation. A closer MS/MS investigation of native proteins extracted from a native PAGE gel in the vicinity of the most active band (∼50 kDa) returned similar results, with identifications of the putative Ca2+ binding heme peroxidase protein, ABC-type branched-chain amino acid transport system, a periplasmic substrate binding protein, the putative porin, and an ABC transporter periplasmic binding protein (data not shown).
Following the three levels of chromatography previously described, enough concentrated active protein was recovered so as to be able to analyze it on an SDS-PAGE gel and subject the fraction to MS/MS analysis directly. Peptides were identified from only five proteins, with 70% of the identifications matching the putative Ca2+ binding heme peroxidase and the rest derived from flagellin proteins (Table 3). The peptide sequences for the 69 spectra that identified the peroxidase were manually aligned against the full sequence from the genome and were distributed across the entire length of the 350-kDa protein, covering 27% of the protein. It is unknown why this 350-kDa protein was eluted during size exclusion chromatography with other proteins between 31 and 34 kDa.
TABLE 3.
Proteins identified by MS/MS in the purified LBOM proteins from Aurantimonas manganoxydans strain SI85-9A1a
| Protein | Total no. of spectra | % Coverage | Accession no. | Predicted molecular mass (kDa) |
|---|---|---|---|---|
| Ca2+ binding heme peroxidase | 69 | 27 | ZP_01225898 | 347 |
| Flagellin | 20 | 78 | ZP_01226291 | 34 |
| Flagellin | 3 | 34 | ZP_01226296 | 33 |
| Flagellin | 3 | 57 | ZP_01226292 | 31 |
| Flagellin | 3 | 25 | ZP_01226294 | 34 |
MS/MS was performed on the pooled active fractions eluted from the final FPLC size exclusion purification step. Prior purification steps included ion-exchange chromatography and hydrophobic interaction chromatography.
This is conclusive evidence that peroxidases are indeed involved in prokaryotic Mn oxidation, especially considering that the four other copurified proteins are associated with flagella. Further support is shown by the MS/MS identification of a heme peroxidase from the second Mn-oxidizing alphaproteobacterium in this study, Erythrobacter sp. strain SD-21. The protein was identified from Mn-oxidizing bands in in-gel activity assays of soluble and excreted fractions and from a Coomassie-stained band after the soluble fraction was enriched by hydrophobic interaction chromatography (Fig. 4 and Table 4). The Mn-oxidizing protein in Erythrobacter sp. strain SD-21 ran as an ∼250-kDa protein in SDS-PAGE and as a similarly sized protein in a gel filtration column. In all cases, the majority of the peptides provided a match to a 225-kDa animal heme peroxidase (Table 4). These data provided 16% coverage of the animal heme peroxidase protein.
FIG. 4.

SDS-PAGE gel of soluble protein fraction from Erythrobacter sp. strain SD-21. Lane A, size standard; lane B, soluble cell extract; lane C, phenyl HP-enriched sample; lane D, Mn-oxidizing in-gel activity assay.
TABLE 4.
Proteins identified by MS/MS in bands from native PAGE gels separating soluble and excreted protein fractions from Erythrobacter sp. strain SD-21 (see Fig. 4)
| Protein | Total no. of spectra | Accession no. | Predicted molecular mass (kDa) |
|---|---|---|---|
| Soluble proteins in Mn in-gel activity assay | |||
| Animal heme peroxidase | 7 | ZP_01865380 | 225 |
| Peptidoglycan-associated protein | 1 | ZP_01863706 | 39 |
| TonB-dependent receptor | 1 | ZP_01864457 | 97 |
| Excreted proteins in Mn in-gel activity assay | |||
| Animal heme peroxidase | 30 | ZP_01865380 | 225 |
| Peptidoglycan-associated protein | 3 | ZP_01863706 | 39 |
| Curli production assembly | 2 | ZP_01864079 | 35 |
| TonB siderophore receptor | 2 | ZP_01865221 | 77 |
| Protein of unknown function | 2 | ZP_01865209 | 119 |
| Phenyl HP-enriched Coomassie-stained bands | |||
| Animal heme peroxidase | 31 | ZP_01865380 | 225 |
| Protein of unknown function | 1 | ZP_01862549 | 35 |
The identification of the putative Ca2+ binding heme peroxidase in Aurantimonas manganoxydans strain SI85-9A1 is quite certain, as the automated annotation of the draft genome database of Aurantimonas sp. strain SI85-9A1 was exhaustively checked manually, with the results published by Dick et al. in 2008 (12). Furthermore, the heme peroxidases from both A. manganoxydans strain SI85-9A1 and Erythrobacter sp. strain SD-21 have been investigated by PROSITE motif-based searches (8). The peroxidase from A. manganoxydans contains two peroxidase domains related to the animal heme peroxidase superfamily (Pfam accession number PF03098, PROSITE accession number PDOC00394), and following each of these domains are hemolysin-type Ca2+ binding regions which contain a total of 10 predicted Ca2+ binding sites (Fig. 5). There are a number of other features, such as phosphorylation sites and cell attachment sequences. Interestingly, the heme peroxidase from Erythrobacter sp. strain SD-21 also contains an animal heme peroxidase domain followed by a hemolysin-type Ca2+ binding domain consisting of seven predicted Ca2+ binding sites. This protein has 2,138 amino acids and shares homology with the first 2,000 amino acids of the A. manganoxydans protein. The Erythrobacter sp. strain SD-21 protein does not contain the additional C-terminal heme peroxidase domain or the second Ca2+ binding region that the Aurantimonas protein has (Fig. 5). Although these additional two domains are missing in Erythrobacter sp. strain SD-21, they have been found in homologous proteins from other Mn-oxidizing species, such as Pseudomonas putida GB-1.
FIG. 5.

Manganese-oxidizing peroxidase (MopA) proteins from Aurantimonas manganoxydans strain SI85-9A1 and Erythrobacter sp. strain SD-21 with predicted animal heme peroxidase domains and Ca2+ binding regions labeled. The Erythrobacter peroxidase is homologous to the first ∼2,000 amino acids of the Aurantimonas peroxidase.
Further evidence to support the identification as a Ca2+ binding heme peroxidase is the 4.3-fold increase in Mn oxidation activity when CaCl2 is used as a base salt in assays as opposed to NaCl, and a further 1.6-fold increase when H2O2 is added and a heme-staining assay where heme-containing proteins appear in the exact same locations as do the active Mn-oxidizing proteins in A. manganoxydans (Table 1 and Fig. 2). Heme staining of Erythrobacter sp. strain SD-21 soluble cell extract did not yield a heme-containing protein that corresponded to Mn-oxidizing bands. The partially purified proteins of A. manganoxydans do not have laccase-like activity like the crude LBOM extract, thereby decreasing the likelihood that an MCO is involved. This does not preclude MCO involvement in the native mechanism as MCOs can produce the H2O2 needed for the peroxidase activity (36). The presence of Mn(III) intermediates for both A. manganoxydans (Fig. 1) and Erythrobacter (23) also fits with the chemistry previously described for heme-containing MnPs although it is unknown what the stoichiometry is between H2O2 equivalents and Mn(III) equivalents. In a coupled system, both laccase (MCO) and MnP produce Mn(III) (36).
Are MnPs the missing link in the mechanism of prokaryotic manganese oxidation? MnPs are well known to be involved in Mn(II) oxidation in fungi, but this is the first report of this type of MnP (i.e., hemolysin-type Ca2+ binding animal heme peroxidase) being involved in Mn(II) oxidation in prokaryotes. There are also a number of inconsistencies in the previous reports invoking MCOs as the manganese oxidase enzymes. It could very well be that MCOs are working in concert with MnP. For example, in a recent report on iron requirements for Mn(II) oxidation by Leptothrix discophora, there is an ∼75% reduction in Mn oxidation in iron-limited growth media, yet no decrease in the mofA (MCO) transcript (14). The authors speculate that low levels of iron may affect the heme-containing c-type cytochrome MofC (the mofC gene is in the same operon as mofA). Using the same thread, we could speculate instead that an undiscovered heme peroxidase is required. A protein similar to the heme peroxidases described here has been identified in the genome of Leptothrix cholodnii SP-6 (YP_001791329.1). The involvement of c-type cytochromes in Mn oxidation has been suggested for A. manganoxydans as well, but this is based on proximity (i.e., genes for c-type cytochromes appear in close proximity to the putative MoxA homologs) (12).
There is also uncertainty in the results for alphaproteobacterium Pedomicrobium sp. strain ACM 3067 implicating the MCO MoxA as the manganese oxidase enzyme. Ridge et al. (35) report that 12 Kmr mutants were produced by knocking out the moxA gene by plasmid insertion. Of these 12 knockouts, only one, ML2, was characterized further, as it did not produce the characteristic Mn oxide precipitates on plates containing Mn(II). It is unclear whether any of the other 11 mutants could or could not oxidize manganese. If they could, this would suggest that the lack of Mn oxidation in ML2 could be due to moxA being knocked out, as indicated, but also a random mutation elsewhere in the genome that could be disrupting an MnP or associated regulatory gene.
The discovery of an Mn-oxidizing heme peroxidase in A. manganoxydans may refute the assumptions made by Dick et al. (12) implicating MoxA-like MCOs in Aurantimonas Mn(II) oxidation, although it does appear from these results that another MCO could be involved in laccase-like activity. The confirmation that a heme peroxidase is also responsible for Mn(II) oxidation in Erythrobacter lends weight to the argument that heme peroxidases are important, and it is consistent with previous studies of Erythrobacter sp. strain SD-21 that also questioned the role of an MCO in Mn oxidation in this strain (23). Although peroxide addition to soluble cell extracts of Erythrobacter sp. strain SD-21 did not stimulate Mn(II) oxidation in this strain, we were subsequently able to identify a catalase in these preparations that may explain the lack of stimulation (data not shown). In addition, Hiraku and Kawanishi (20) have shown that PQQ and NADH can react to generate superoxide and peroxide, and this reaction could be a source of peroxide for Mn(II) oxidation in cell extracts of Erythrobacter sp. strain SD-21 and the cause of stimulation of Mn(II) oxidation by Aurantimonas manganoxydans SI85-9A1 in the presence of NAD+ and PQQ (Table 1).
Although the role of MCOs in Mn(II) oxidation in Bacillus sp. strain SG-1, Leptothrix discophora SS-1, Pseudomonas putida strain GB-1, and Pedomicrobium sp. strain ACM 3067 is currently undisputed (3, 7, 35, 44), it is still unclear what role MCOs may play in these different microbes. Multiple MCOs have been identified in Aurantimonas manganoxydans strain SI85-9A1 (12), and Erythrobacter sp. strain SD-21 contains five annotated MCOs in the genome sequence. Currently none of the MCOs have been directly linked to Mn(II) oxidation in these two strains. In contrast, our work has illustrated an important role for heme peroxidase in bacterial Mn(II) oxidation and the distinct possibility that fungal-like laccase/peroxidase systems may exist in bacteria. We tentatively suggest the name MopA (for manganese-oxidizing peroxidase) for the putative Ca2+ binding heme peroxidase identified in this study.
Acknowledgments
This work was supported by the Proteomics Shared Resource, which is generously funded by the Oregon Opportunity, and NIH center grants 5P30CA069533 and 5P30EY010572. This study was partially supported by the National Science Foundation grant OCE-0635493 and by grant number ES010337 from the National Institute of Environmental Health Sciences (NIEHS), NIH.
The contents of the manuscript are solely the responsibility of the authors and do not necessarily represent the official views of the NIEHS, NIH.
We thank Jim Whittaker for helpful advice and Kati Geszvain for stimulating discussion and for reviewing our manuscript.
Footnotes
Published ahead of print on 1 May 2009.
REFERENCES
- 1.Adams, L. F., and W. C. Ghiorse. 1987. Characterization of an extracellular Mn2+-oxidizing activity and isolation of Mn2+-oxidizing protein from Leptothrix discophora SS-1. J. Bacteriol. 169:1279-1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1a.Anderson, C. R., G. J. Dick, M.-L. Chu, S. Bräuer, J.-C. Cho, and B. M. Tebo. 2009. Aurantimonas manganoxydans, sp. nov. and Aurantimonas litoralis, sp. nov.: representatives of a globally distributed clade of Mn(II)-oxidizing Alphaproteobacteria from the order Rhizobiales. Geomicrobiol. J. 26:189-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bobik, C., E. Meilhoc, and J. Batut. 2006. FixJ: a major regulator of the oxygen limitation response and late symbiotic functions of Sinorhizobium meliloti. J. Bacteriol. 188:4890-4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brouwers, G. J., J. P. M. de Vrind, P. L. A. M. Corstjens, P. Cornelis, C. Baysse, and E. W. de Vrind-de Jong. 1999. cumA, a gene encoding a multicopper oxidase, is involved in Mn2+ oxidation in Pseudomonas putida GB-1. Appl. Environ. Microbiol. 65:1762-1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brouwers, G. J., E. W. J. P. M. de Vrind, P. L. A. M. Corstjens, and E. W. de Vrind-De Jong. 1998. Involvement of genes of the two-step protein secretion pathway in the transport of the manganese-oxidizing factor across the outer membrane of Pseudomonas putida strain GB-I. Am. Mineral. 83:1573-1582. [Google Scholar]
- 5.Camarero, S., F. J. Ruiz-Duenas, S. Sarkar, M. J. Martinez, and A. T. Martinez. 2000. The cloning of a new peroxidase found in lignocellulose cultures of Pleurotus eryngii and sequence comparison with other fungal peroxidases. FEMS Microbiol. Lett. 191:37-43. [DOI] [PubMed] [Google Scholar]
- 6.Canfield, D. E., B. Thamdrup, and J. W. Hansen. 1993. The anerobic degradation of organic matter in Danish coastal sediments: iron reduction, manganese reduction, and sulfate reduction. Geochim. Cosmochim. Acta 57:3867-3885. [DOI] [PubMed] [Google Scholar]
- 7.Corstjens, P. L. A. M., J. P. M. de Vrind, T. Goosen, and E. W. de Vrind-De Jong. 1997. Identification and molecular analysis of the Leptothrix discophora SS-1 mofA gene, a gene putatively encoding a manganese-oxidizing protein with copper domains. Geomicrobiol. J. 14:91-108. [Google Scholar]
- 8.de Castro, E., C. J. A. Sigrist, A. Gattiker, V. Bulliard, P. S. Langendijk-Genevaux, E. Gasteiger, A. Bairoch, and N. Hulo. 2006. ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucleic Acids Res. 34:W362-W365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Vrind, J., A. de Groot, G. J. Brouwers, J. Tommassen, and E. de Vrind-De Jong. 2003. Identification of a novel Gsp-related pathway required for secretion of the manganese-oxidizing factor of Pseudomonas putida strain GB-1. Mol. Microbiol. 47:993-1006. [DOI] [PubMed] [Google Scholar]
- 10.DiChristina, T. J., C. M. Moore, and C. A. Haller. 2002. Dissimilatory Fe(III) and Mn(IV) reduction by Shewanella putrefaciens requires ferE, a homolog of the pulE (gspE) type II protein secretion gene. J. Bacteriol. 184:142-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dick, G. J., J. W. Torpey, T. J. Beveridge, and B. M. Tebo. 2008. Direct identification of a bacterial manganese(II) oxidase, the multicopper oxidase MnxG, from spores of several different marine Bacillus species. Appl. Environ. Microbiol. 74:1527-1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dick, G. J., S. Podell, H. A. Johnson, Y. Rivera-Espinoza, R. Bernier-Latmani, J. K. McCarthy, J. W. Torpey, B. G. Clement, T. Gaasterland, and B. M. Tebo. 2008. Genomic insights into Mn(II) oxidation by the marine alphaproteobacterium Aurantimonas sp. strain SI85-9A1. Appl. Environ. Microbiol. 74:2646-2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dubinina, G. A. 1978. Mechanism of the oxidation of divalent iron and manganese by iron bacteria developing in a neutral acidic medium. Mikrobiologiia 47:591-599. [PubMed] [Google Scholar]
- 14.El Gheriany, I. A., D. Bocioaga, A. G. Hays, W. C. Ghiorse, M. L. Shuler, and L. W. Lion. 2009. Iron requirement for Mn(II) oxidation by Leptothrix discophora SS-1. Appl. Environ. Microbiol. 75:1229-1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Francis, C. A., K. L. Casciotti, and B. M. Tebo. 2002. Localization of Mn(II)-oxidizing activity and the putative multicopper oxidase, MnxG, to the exosporium of the marine Bacillus sp. strain SG-1. Arch. Microbiol. 178:450-456. [DOI] [PubMed] [Google Scholar]
- 16.Francis, C. A., and B. M. Tebo. 2001. Diversity of cumA multicopper oxidase genes from Mn(II)-oxidizing and non-Mn(II)-oxidizing Pseudomonas strains. Appl. Environ. Microbiol. 67:4272-4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaspard, S., F. Vazquez, and C. Holliger. 1998. Localization and solubilization of the iron(III) reductase of Geobacter sulfurreducens. Appl. Environ. Microbiol. 64:3188-3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glenn, J. K., L. Akileswaran, and M. H. Gold. 1986. Mn(II) oxidation is the principal function of the extracellular Mn-peroxidase from Phanerochaete chrysosporium. Arch. Biochem. Biophys. 251:688-696. [DOI] [PubMed] [Google Scholar]
- 19.Hagan, E. C., and H. L. T. Mobley. 2009. Haem acquisition is facilitated by a novel receptor Hma and required by uropathogenic Escherichia coli for kidney infection. Mol. Microbiol. 71:79-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hiraku, Y., and S. Kawanishi. 1996. NADH-mediated DNA damage induced by a new coenzyme, pyrroloquinoline quinone, in the presence of copper(II) ion. FEBS Lett. 393:317-320. [DOI] [PubMed] [Google Scholar]
- 21.Höfer, C., and D. Schlosser. 1999. Novel enzymatic oxidation of Mn2+ to Mn3+ catalyzed by a fungal laccase. FEBS Lett. 451:186-190. [DOI] [PubMed] [Google Scholar]
- 22.Huang, P. M. 1991. Kinetics of redox reactions on manganese oxides and its impact on environmental quality, p. 191-230. In D. L. Sparks and D. L. Suarez (ed.), Rates of soil chemical processes. Soil Science Society of America, Inc., Madison, WI.
- 23.Johnson, H. A., and B. M. Tebo. 2008. In vitro studies indicate a quinone is involved in bacterial Mn(II) oxidation. Arch. Microbiol. 189:59-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keller, A., A. I. Nesvizhskii, E. Kolker, and R. Aebersold. 2002. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74:5383-5392. [DOI] [PubMed] [Google Scholar]
- 25.Laha, S., and R. G. Luthy. 1990. Oxidation of aniline and other primary aromatic amines by manganese dioxide. Environ. Sci. Technol. 24:363-373. [Google Scholar]
- 26.Larsen, E. I., L. I. Sly, and A. G. McEwan. 1999. Manganese(II) adsorption and oxidation by whole cells and a membrane fraction of Pedomicrobium sp. ACM 3067. Arch. Microbiol. 171:257-264. [Google Scholar]
- 27.Magliozzo, R. S., and J. A. Marcinkeviciene. 1997. The role of Mn(II)-peroxidase activity of mycobacterial catalase-peroxidase in activation of the antibiotic isoniazid. J. Biol. Chem. 272:8867-8870. [DOI] [PubMed] [Google Scholar]
- 28.Miyata, N., Y. Tani, K. Maruo, H. Tsuno, M. Sakata, and K. Iwahori. 2006. Manganese(IV) oxide production by Acremonium sp. strain KR21-2 and extracellular Mn(II) oxidase activity. Appl. Environ. Microbiol. 72:6467-6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Myers, C. R., and J. M. Myers. 1992. Localization of cytochromes to the outer membrane of anaerobically grown Shewanella putrefaciens MR-1. J. Bacteriol. 174:3429-3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nealson, K. H., B. M. Tebo, and R. A. Rosson. 1988. Occurrence and mechanisms of microbial oxidation of manganese. Adv. Appl. Microbiol. 33:279-318. [Google Scholar]
- 31.Nesvizhskii, A. I., A. Keller, E. Kolker, and R. Aebersold. 2003. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75:4646-4658. [DOI] [PubMed] [Google Scholar]
- 32.Okazaki, M., T. Sugita, M. Shimizu, Y. Ohode, K. Iwamoto, E. W. de Vrinne de-Jong, J. P. M de Vrind, and P. L. A. M. Corstjens. 1997. Partial purification and characterization of manganese-oxidizing factors of Pseudomonas fluorescens GB-1. Appl. Environ. Microbiol. 63:4793-4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palma, C., A. T. Martinez, J. M. Lema, and M. J. Martinez. 2000. Different fungal manganese-oxidizing peroxidases: a comparison between Bjerkandera sp. and Phanerochaete chrysosporium. J. Biotechnol. 77:235-245. [DOI] [PubMed] [Google Scholar]
- 34.Perez, J., and T. W. Jeffries. 1992. Roles of manganese and organic acid chelators in regulating lignin degradation and biosynthesis of peroxidases by Phanerochaete chrysosporium. Appl. Environ. Microbiol. 58:2401-2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ridge, J. P., M. Lin, E. I. Larsen, M. Fegan, A. G. McEwan, and L. I. Sly. 2007. A multicopper oxidase is essential for manganese oxidation and laccase-like activity in Pedomicrobium sp. ACM 3067. Environ. Microbiol. 9:944-953. [DOI] [PubMed] [Google Scholar]
- 36.Schlosser, D., and C. Höfer. 2002. Laccase-catalyzed oxidation of Mn2+ in the presence of natural Mn3+ chelators as a novel source of extracellular H2O2 production and its impact on manganese peroxidase. Appl. Environ. Microbiol. 68:3514-3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Solomon, E. I., U. M. Sundaram, and T. E. Machonkin. 1996. Multicopper oxidases and oxygenases. Chem. Rev. 96:2563-2605. [DOI] [PubMed] [Google Scholar]
- 38.Sunda, W. G., and D. J. Kieber. 1994. Oxidation of humic substances by manganese oxides yields low-molecular-weight organic substrates. Nature 367:62-64. [Google Scholar]
- 39.Tebo, B. M. 1991. Manganese(II) oxidation in the suboxic zone of the Black Sea. Deep-Sea Res. 38:S883-S905. [Google Scholar]
- 40.Tebo, B. M., J. R. Bargar, B. G. Clement, G. J. Dick, K. J. Murray, D. Parker, R. Verity, and S. M. Webb. 2004. Biogenic manganese oxides: properties and mechanisms of formation. Annu. Rev. Earth Planet. Sci. 32:287-328. [Google Scholar]
- 41.Tebo, B. M., B. Clement, and G. J. Dick. 2006. Biotransformations of manganese, p. 1223-1238. In C. J. Hurst, R. L. Crawford, J. L. Garland, D. A. Lipson, A. L. Mills, and L. D. Stetzenbach (ed.), Manual of environmental microbiology, 3rd ed. ASM Press, Washington, DC.
- 42.Tebo, B. M., H. A. Johnson, J. K. McCarthy, and A. S. Templeton. 2005. Geomicrobiology of manganese(II) oxidation. Trends Microbiol. 13:421-428. [DOI] [PubMed] [Google Scholar]
- 43.Thomas, P. E., D. Ryan, and W. Levin. 1976. An improved staining procedure for the detection of the peroxidase activity of cytochrome P-450 on sodium dodecyl sulfate polyacrylamide gels. Anal. Biochem. 75:168-176. [DOI] [PubMed] [Google Scholar]
- 44.van Waasbergen, L. G., M. Hildebrand, and B. M. Tebo. 1996. Identification and characterization of a gene cluster involved in manganese oxidation by spores of the marine Bacillus sp. strain SG-1. J. Bacteriol. 178:3517-3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wariishi, H., K. Valli, and M. H. Gold. 1992. Manganese(II) oxidation by manganese peroxidase from the basidiomycete Phanerochaete chrysosporium. J. Biol. Chem. 267:23688-23695. [PubMed] [Google Scholar]
- 46.Webb, S. M., J. R. Bargar, and B. M. Tebo. 2002. Existence of intermediate Mn(III) species in the microbially mediated oxidation of Mn(II). Abstr. Pap. Am. Chem. Soc. 223:062-Geoc. [Google Scholar]
- 47.Webb, S. M., G. J. Dick, J. R. Bargar, and B. M. Tebo. 2005. Evidence for the presence of Mn(III) intermediates in the bacterial oxidation of Mn(II). Proc. Natl. Acad. Sci. USA 102:5558-5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wehrli, B., G. Friedl, and A. Manceau. 1995. Reaction rates and products of manganese oxidation at the sediment-water interface. In C. P. Huang, C. R. O'Melia, and J. J. Morgan (ed.), Aquatic chemistry: interfacial and interspecies processes. American Chemical Society, Washington, DC.