

Abstract
Rhizobium leguminosarum bv. viciae forms nitrogen-fixing nodules on several legumes, including pea (Pisum sativum) and vetch (Vicia cracca), and has been widely used as a model to study nodule biochemistry. To understand the complex biochemical and developmental changes undergone by R. leguminosarum bv. viciae during bacteroid development, microarray experiments were first performed with cultured bacteria grown on a variety of carbon substrates (glucose, pyruvate, succinate, inositol, acetate, and acetoacetate) and then compared to bacteroids. Bacteroid metabolism is essentially that of dicarboxylate-grown cells (i.e., induction of dicarboxylate transport, gluconeogenesis and alanine synthesis, and repression of sugar utilization). The decarboxylating arm of the tricarboxylic acid cycle is highly induced, as is γ-aminobutyrate metabolism, particularly in bacteroids from early (7-day) nodules. To investigate bacteroid development, gene expression in bacteroids was analyzed at 7, 15, and 21 days postinoculation of peas. This revealed that bacterial rRNA isolated from pea, but not vetch, is extensively processed in mature bacteroids. In early development (7 days), there were large changes in the expression of regulators, exported and cell surface molecules, multidrug exporters, and heat and cold shock proteins. fix genes were induced early but continued to increase in mature bacteroids, while nif genes were induced strongly in older bacteroids. Mutation of 37 genes that were strongly upregulated in mature bacteroids revealed that none were essential for nitrogen fixation. However, screening of 3,072 mini-Tn5 mutants on peas revealed previously uncharacterized genes essential for nitrogen fixation. These encoded a potential magnesium transporter, an AAA domain protein, and proteins involved in cytochrome synthesis.
Bacteria from the family Rhizobiaceae form species-specific symbioses with host plants of the Leguminosae, resulting in the formation of specialized root structures called nodules (21, 50). This process is initiated by plants releasing flavonoids, which induce synthesis of lipochitooligosaccharides by rhizobia. Bacteria are typically trapped by the curling of root hairs, which they enter, and grow down plant-derived infection threads. These ramify into the root cortex, where newly induced meristematic cells form the nodule (50). Bacterial cells are released from infection threads into the cytoplasm of cortical cells, where they are surrounded by a plant-derived symbiosome membrane. In indeterminate nodules of the galegoid legumes (a clade in the subfamily Papilionoideae, e.g., Medicago, Pisum, and Vicia), bacteria undergo dramatic changes in size, shape, and DNA content to become terminally differentiated bacteroids (46). N2 reduction to ammonium occurs in bacteroids, which the plant provides with dicarboxylic acids as a source of carbon, energy, and reductant (54). In return, ammonium is secreted back to the plant. However, the metabolic exchange between partners is almost certainly more complex than this, since amino acid movement is also essential for productive nitrogen fixation in pea nodules (43).
Rhizobium leguminosarum bv. viciae, which forms nodules on Pisum sativum (pea) and Vicia cracca (vetch), has been widely used as a model to study nodule biochemistry, and its genomic sequence has recently been determined (63). With the genome sequences of several rhizobia available (23, 25, 27, 36, 37), it is now possible to use microarrays to study aspects of this complex host-microbe interaction, from the survival of free-living bacteria in the often hostile rhizosphere to the development of mature N2-fixing nodules on plant roots (4, 5, 16, 51). Several studies have examined gene expression in free-living rhizobia grown under different conditions, e.g., osmotic stress (16, 19) and microoxic conditions (5). Formation of determinate nodules in the Bradyrhizobium japonicum-Glycine max symbiosis has been investigated with microarrays studying gene expression in both the bacteria (51) and the plant (8). In addition, transcriptomic analysis of Mesorhizobium loti has been performed for the M. loti-Lotus japonicus symbiosis (58). Although studies of various host-symbiont interactions forming determinate nodules have been carried out, for indeterminate nodule formation, only the Sinorhizobium meliloti-Medicago sativa symbiosis has been studied with microarrays (2, 4, 5). These studies have provided significant insight into bacteroid function and development; however, it is very difficult to interpret many of the complex metabolic changes that occur in bacteroids relative to free-living cells. Due to large-scale changes caused by bacterial differentiation, radically different cell types must be compared. A major problem is deciding which cultured bacteria should be compared to bacteroids, since a metabolic baseline is not available for laboratory-cultured free-living rhizobia. Therefore, in this study, we first constructed such a baseline before investigating the more complex problem of bacteroid development and metabolism. We used whole-genome microarrays of R. leguminosarum bv. viciae 3841 to examine gene expression in free-living bacteria grown on succinate, pyruvate, inositol, glucose, acetate, and acetoacetate. The pattern of gene expression in mature N2-fixing nodules of P. sativum was examined and compared, not only with that of free-living cells, but also with that in bacteroids from mature nodules of V. cracca. Furthermore, to investigate bacteroid development, gene expression was measured at 7, 15, and 21 days after inoculation onto peas.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
The bacterial strains and plasmids used in this study are detailed in Table S1 in the supplemental material. Rhizobium strains were grown at 28°C on either tryptone yeast extract (TY) (6), acid minimal salts medium, or acid minimal salts agar (53) with an added carbon source—glucose (10 mM), inositol (10 mM), pyruvate (30 mM), succinate (20 mM), acetate (1%), or acetoacetate (10 mM)—and 10 mM ammonium chloride. Antibiotics were used at the following concentrations (μg ml−1): ampicillin, 50; spectinomycin (Sp), 100; streptomycin, 500; kanamycin (Km), 20; neomycin (Nm), 160; and tetracycline (Tc), 5.
Plant growth.
Seeds of P. sativum cv. Avola and V. cracca were surface sterilized and grown as described previously (38). Inoculation with R. leguminosarum was performed either on seeds at planting (P. sativum and V. cracca) or on 7-day-old seedlings (P. sativum). Nodules were harvested at 28 days postinoculation (p.i.) from seed-inoculated plants and at 7, 15, and 21 days p.i. from those inoculated as seedlings.
RNA preparation from free-living cultures and bacteroids.
RNA was prepared from free-living cultures (optical density at 600 nm = 0.6) as described previously (19, 38). The yield was approximately 80 to 100 μg RNA per 25 ml of culture.
To extract RNA from pea and vetch bacteroids, nodules (1 g) were picked into liquid nitrogen and ground in 5 to 10 ml of PSM buffer (10 mM phosphate buffer, pH 7.0, 300 mM sucrose, and 2 mM MgCl2). This was centrifuged (200 relative centrifugal force; 5 min) to remove plant material and then again (3,380 relative centrifugal force; 5 min) to pellet bacteroids. Following resuspension in 10 mM Tris, pH 8.0, the bacteroid fraction was examined by light microscopy to ensure that it was substantially free from contaminating plant material, and then RNA was extracted as described above. The yield was approximately 5 to 10 μg RNA per 1 g nodules.
Preparation of labeled cDNA, amplification of bacteroid RNA, and microarray hybridization.
For microarray analysis, three to five independent free-living cultures or bacteroid harvests were used for RNA isolation and subsequent microarray analysis. The only exceptions to this were the acetate and acetoacetate samples, which were from single cultures. The results from these were not extensively analyzed but were used to obtain a general picture of gene expression during growth on short-chain organic acids.
A whole-genome DNA oligonucleotide array for R. leguminosarum 3841 was constructed from the published genome sequence (63). Unique 70-mer oligonucleotides were synthesized for all 7,344 genes of R. leguminosarum bv. viciae 3841 by Operon Biotechnologies and printed with Pronto buffer on Corning Ultragaps slides at the Functional Genomics and Proteomics Laboratory, University of Birmingham, Birmingham, United Kingdom. The microarray slides were blocked for 15 min in sodium borohydride (20 mM sodium borohydride, 140 mM succinic anhydride, dissolved in 1-methyl-2-pyrrolidinone [90% {vol/vol}]), washed in deionized water, and dried. It should be noted that the microarray slides contained duplicates or triplicates of each gene-specific oligonucleotide printed in a random pattern so that all arrays contained technical replicates.
Total RNA was labeled and purified with the Cyscribe Post-Labeling kit (GE Healthcare) according to the manufacturer's instructions. Equal amounts (30 to 35 pmol) of Cy5- and Cy3-labeled cDNA were mixed and dried (5 to 10 μl). Hybridization solution (90 μl) containing 25% formamide, 5× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate), 0.1% sodium dodecyl sulfate (SDS), and calf thymus DNA (9 μg) was added to the labeled DNA and heated at 95°C for 2 min. The mixture was incubated at room temperature for 2 min and centrifuged for 2 min. Hybridization was carried out at 42°C for 16 h using an Array Booster (Implen, United Kingdom). Microarray slides were then washed in 2× SSC, 0.1% SDS for 8 min at 42°C; twice in 0.2× SSC, 0.1% SDS for 5 min at 42°C; twice in 0.2× SSC for 4 min at room temperature; and for 1 min in 0.1× SSC. The slides were then washed with MilliQ water (5 s) and isopropanol (5 s) at room temperature, dried, and scanned for fluorescence intensity by using a GenePix 4000A microarray scanner at a resolution of 10 μm (Axon Instruments, Union City, CA). Spot recognition was performed with Bluefuse (BlueGnome Limited, Cambridge, United Kingdom), and the data were imported into GeneSpring 7.2 (Silicon Genetics, Redwood, CA). The local background value was subtracted from the intensity of each spot, and a Lowess normalization was applied to the slide. The normalized expression ratio of the experimental sample to the control was calculated by taking the natural log of the ratio for each replicate experiment, averaging this value for all replicates, and then calculating the natural antilog. To obviate dye effects, labeling with different dyes was performed on replicates (i.e., dye swap). However, no significant differences between Cy3 and Cy5 in incorporation, fluorescence yield, or stability were observed.
Since RNA from mature bacteroids labeled poorly with Cy dyes (see Results and Discussion), an amplification procedure was adopted. Total RNA (50 to 250 ng) was amplified to give “sense” RNA using T7-based amplification with the Genisphere Sense Amplification Kit (Genisphere Inc., Hatfield, PA) according to the manufacturer's instructions. This was converted to cDNA and Cy dye labeled as for unamplified RNA, as described above. Most array results were obtained by direct comparison of conditions using two-color arrays. However, an effective gene expression set of bacteroids versus a glucose-grown free-living culture was obtained by cross-multiplying a bacteroid/free-living succinate array with a free-living succinate/free-living glucose array (bacteroid/free-living succinate × free-living succinate/free-living glucose = bacteroid/free-living glucose).
qRT-PCR.
Gene expression was measured by quantitative reverse transcription-PCR (qRT-PCR) performed in triplicate using the QuantiTect SYBR green PCR kit (Qiagen) on an MJ Mini cycler MiniOpticon Real-Time PCR Detection System (Bio-Rad) as previously described (38). The primers are listed in Table S2 in the supplemental material. The data were analyzed by the relative quantification method (comparative threshold cycle method [ΔΔCT]) to calculate the expression (10, 11). Expression levels were normalized against either mdh or the absolute level of RNA using REST analysis (10). The results from the two normalization procedures were not significantly different.
β-d-Glucuronidase assays.
Measurements were made of β-d-glucuronidase (GusA) activity in bacteria extracted from 21-day pea bacteroids and compared with that in free-living cells. Strains containing gusA fusions (RU4325 and RU4326, and RU4334 to RU4339) (see Table S1 in the supplemental material) were constructed by cloning PCR products into plasmid pRU877, upstream of a promoterless gusA, and integrated into the R. leguminosarum bv. viciae 3841 chromosome as described below for pK19mob mutants. Bacterial cells were resuspended in 50 mM phosphate buffer, pH 7.0, and GusA activity was measured as previously described (41), except p-nitrophenyl-β-d-glucuronide (at a final concentration of 40 μg ml−1) was substituted as the chromogenic substrate.
Mutagenesis and assessment of mutant phenotypes on peas.
To make specific mutants in R. leguminosarum bv. viciae 3841, internal gene fragments were PCR amplified and cloned into the suicide vector pK19mob (the PCR primers are listed in Table S2 in the supplemental material). This was performed by cloning PCR products directly into HindIII-digested pK19mob using an In-Fusion Dry Down PCR Cloning Kit (Clontech) according to the manufacturer's instructions. Alternatively, PCR fragments were cloned into pCR4-TOPO, and the resultant plasmids were digested with XhoI/HindIII and ligated into pK19mob digested with the same enzymes. After confirmation that the Escherichia coli strains contained the expected plasmid, each was conjugated into strain 3841, using pRK2013 as a helper plasmid (52). Positive clones in R. leguminosarum were confirmed by PCR using a pK19mob-specific primer (either pK19A or pK19B), together with a gene-specific primer (see Table S2 in the supplemental material). To assess the phenotypes of the mutants, six P. sativum seeds were inoculated with each mutant strain (see Table S1 in the supplemental material), germinated, and grown for 21 days in 1-liter pots of vermiculite with nitrogen-free medium. The nitrogen fixation phenotype of each mutant was assessed by acetylene reduction as described previously (52). For each strain tested, 12 nodules were picked, surface sterilized, and crushed, and the bacteria were streaked on TY medium. From each TY plate, 10 individual colonies were then checked for retention of the neomycin resistance gene.
Random mutagenesis of R. leguminosarum bv. viciae 3841 using pCRS487 (containing the minitransposon mTn5-GNm) was carried out as previously described (53). Transposon mutants were selected on acid minimal salts medium/succinate/pyruvate/ammonium chloride with neomycin and individually stocked. Mutants were inoculated onto individual pea plants grown in 250-ml flasks as previously described (53), and the symbiotic phenotypes were scored after 6 weeks of growth. Yellow plants were scored as Fix−, and the corresponding bacterial strain was recovered from stock and reinoculated onto three plants to confirm the phenotype. Confirmed Fix− and Nod− strains were then transduced into R. leguminosarum bv. viciae 3841 using phage RL38 (9), and three transductants were reinoculated onto peas to confirm transposon linkage to the Fix/Nod phenotype.
Microarray data accession numbers.
Microarray data were deposited at Array Express (http://www.ebi.ac.uk/arrayexpress) with accession numbers E-MEXP-1918, E-MEXP1919, E-MEXP1924, E-MEXP1925, E-MEXP1959, E-MEXP1961, E-MEXP1962, E-MEXP1963, and E-MEXP1964.
RESULTS AND DISCUSSION
The microarray experiments were performed to provide a foundation for understanding gene expression in the Rhizobium/pea-vetch symbiosis. It was considered essential to first obtain a metabolic baseline for free-living Rhizobium, which would allow a more rational exploration of gene expression in highly differentiated bacteroids. The microarray experiments performed are summarized in Table 1. To establish the pattern of gene expression on different carbon sources, cultures grown with succinate, pyruvate, or inositol as the carbon source were compared with cultures grown on glucose. (Full microarray results for cultured free-living bacteria are shown in Table S3 in the supplemental material.) To study gene expression in R. leguminosarum during nodule formation, gene expression in bacteroids was compared with that in free-living cells grown on succinate (Table 1; full microarray results for bacteroids are shown in Table S4 in the supplemental material).
TABLE 1.
Microarray experiments performed on R. leguminosarum bv. viciae 3841
| Array Express accession no. | Condition 1a,b
|
Condition 2a
|
||
|---|---|---|---|---|
| Samplec | Cell type | Samplec | Cell type | |
| E-MEXP-1919 | Succinate | Free living | Glucose | Free living |
| E-MEXP-1959 | Pyruvate | Free living | Glucose | Free living |
| E-MEXP-1961 | Inositol | Free living | Glucose | Free living |
| E-MEXP-1963 | Acetated | Free living | Glucose | Free living |
| E-MEXP-1964 | Acetoacetated | Free living | Glucose | Free living |
| E-MEXP-1962 | Succinate (SA) | Free living | Glucose (SA) | Free living |
| E-MEXP-1918 | 7 day (SA) | Pea bacteroid | Succinate | Free living |
| E-MEXP-1918 | 15 day (SA) | Pea bacteroid | Succinate | Free living |
| E-MEXP-1918 | 21 day (SA) | Pea bacteroid | Succinate | Free living |
| E-MEXP-1925 | 28 day (SA)e | Pea bacteroid | Succinate | Free living |
| E-MEXP-1924 | 28 day (SA) | Vetch bacteroid | Succinate | Free living |
SA, sense amplification of RNA was performed. Three biological replicates were performed.
Twenty-eight-day bacteroid samples were seed inoculated; 7-, 15-, and 21-day bacteroid samples were inoculated onto 7-day-old plants.
For growth of free-living cells, the carbon source added to the medium is shown.
A single array was performed.
Five biological replicates were performed.
Structure of rRNA in bacteroids.
During the initial stages of this work, it was found that RNA isolated from mature pea bacteroids was difficult to label with Cy dye. In addition, the RNA profile obtained from mature bacteroids was reproducibly different from that obtained from free-living cells (Fig. 1). The profile of RNA from free-living cells showed clear 23S, 16S, and 5S rRNA peaks and also a doublet of approximately 1.3 kb, corresponding to processed 23S rRNA. The processing of 23S rRNA in Rhizobiaceae and R. leguminosarum has been well documented (39, 56). While rRNA samples from mature pea bacteroids had 5S and 16S peaks, they almost completely lacked a 23S peak, although the doublet (approximately 1.3 kb) was reproducibly observed, along with peaks for several smaller fragments (Fig. 1). This further processing of 23S rRNA into numerous smaller fragments appears to be specific to mature pea bacteroids, as it was found in R. leguminosarum strains 3841 and A34 from peas, but not in R. leguminosarum bv. viciae 3841 isolated from vetch nodules (data not shown). The degree of processing of 23S rRNA increased with bacteroid age, so by 15 days p.i., processing was similar to that at 28 days p.i. (data not shown).
FIG. 1.
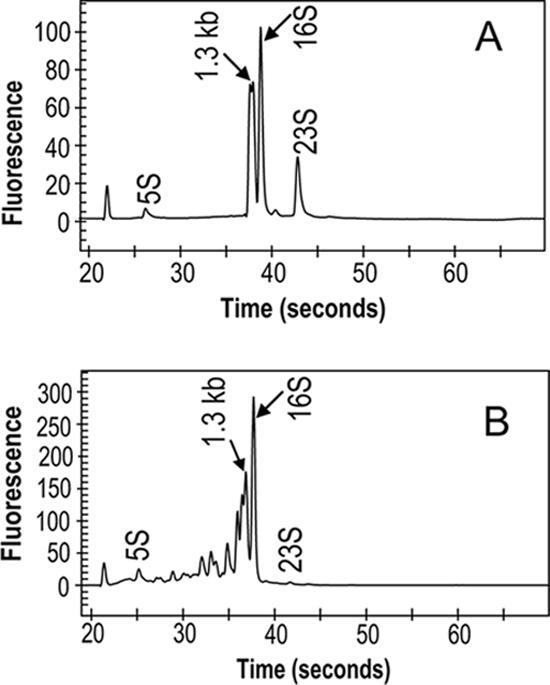
Comparison of RNA structures in free-living bacteria and mature bacteroids. (A) Free-living R. leguminosarum bv. viciae 3841. (B) Twenty-eight-day pea bacteroids of 3841. In free-living cells (A), there is some processing of 23S rRNA, as shown by the large 1.3-kb doublet; however, a 23S rRNA peak is clearly visible. In the bacteroid sample (B), there is almost complete processing of 23S rRNA into smaller fragments. A Bio-Rad Experion microfluidic analyzer was used to determine the RNA structure.
The cDNA formed by RT of bacteroid RNA was refractory to Cy dye labeling, in contrast to the ease of labeling cDNA from cultured bacteria. Due to this labeling problem and the low level of mRNA obtained in bacteroid samples, linear T7 amplification was used to increase the amount of RNA. Typically, 250 ng of total bacteroid RNA was amplified to approximately 50 μg, and after RT to cDNA, it could be easily Cy dye labeled. The relative abundance of stable RNA after T7 amplification was measured using qRT-PCR of 16S RNA and was found to decrease 8.8-fold (ΔCT, 3.14) relative to unamplified RNA. To check whether bias was introduced by amplification, three cultures were grown on media with glucose and three with succinate as carbon sources. Arrays were conducted (succinate versus glucose) on either RNA without amplification or RNA with linear T7 amplification. From 1-way analysis of variance, in which the variances were not assumed to be equal (Welch t test) with a P value of ≤0.01, only 15 expression ratios were significantly different between arrays conducted with amplified versus unamplified RNA (73 would be expected by chance). This showed that no significant bias was introduced by the amplification procedure used.
Metabolic reconstruction from microarrays performed on different carbon sources.
As direct comparison of bacteroids and free-living cells is difficult, in order to make sense of the many metabolic changes, a metabolic baseline of free-living cultures is needed. The first compound chosen for testing as a sole carbon source was succinate, because dicarboxylates are the likely carbon source for bacteroids in nodules (42).
(i) Succinate catabolism.
Predictably, the gene most highly upregulated in growth of free-living R. leguminosarum bv. viciae 3841 cells on succinate compared with glucose-grown cells was dctA (RL3424) (75-fold), which encodes the C4-dicarboxylate transport protein. Likewise, genes for the gluconeogenic enzymes phosphoenolpyruvate carboxykinase (pckA; RL0037) and fructose biphosphate aldolase (fbaA; RL4012) were induced 43- and 7-fold, respectively. In a study of succinate-grown cultures of S. meliloti, these three genes were identified as the most highly elevated (4). In strain 3841, the next most highly upregulated genes were aldA (RL1966), encoding alanine dehydrogenase (13-fold), and dadX (pRL120416) (10-fold). The gene encoding DadA (pRL120417) was fivefold upregulated. The proteins encoded by the dadAX genes are responsible for alanine catabolism, while aldA synthesizes alanine and may have a role in bacteroid metabolism (1, 41). The fact that such systems were elevated in the presence of succinate indicates that alanine metabolism has a role in the growth of 3841 on dicarboxylic acids.
(ii) Pyruvate catabolism.
Many of the genes expressed in R. leguminosarum bv. viciae 3841 grown on pyruvate or succinate may be related generally to organic acid metabolism. For example, in pyruvate-grown cells, genes encoding gluconeogenic enzymes, phosphoenolpyruvate carboxykinase (pckA; RL0037) and fructose biphosphate aldolase (fbaA; RL4012), were induced 56- and 12-fold, respectively. The gene for AldA was also induced threefold, but unlike in succinate-grown cells, dadA and dadX were not induced by pyruvate.
In R. leguminosarum bv. viciae 3841, both succinate and pyruvate strongly repressed the Entner-Doudoroff (ED) pathway relative to growth on glucose. There was a reduction in expression of RL0751 to RL0753, encoding phosphogluconate dehydratase (Edd), 6-phophogluconolactonase (Pgl), and glucose-6-phosphate-1-dehydrogenase (Zwf). This indicates that the ED pathway is more strongly expressed on sugars than on organic acids, which is consistent with its role as the principal pathway for sugar catabolism in rhizobia. Similarly, expression of the genes encoding enzymes of the ED pathway in glucose-grown cells of S. meliloti was elevated (4). As noted above, the gene for fructose biphosphate aldolase (fbaA; RL4012) was induced 7- and 12-fold on succinate and pyruvate, respectively. This enzyme normally functions in glycolysis, but in 3841, which uses the ED pathway, its role is in gluconeogenesis, and it is induced by organic acids.
Three ABC transport systems were strongly repressed by growth on either succinate or pyruvate: RL1824 to RL1827, RL4249 to RL4252, and RL3624 to RL3626. Two of these (CUT1 systems RL1824 to RL1827 and RL4249 to RL4252) are almost identical, with equivalent components showing >97% amino acid identity. The single mutants (LMB109 [RL1826::ΩTc], RU4184 [RL4249::ΩSp], and RU4174 [RL3624::pK19Km]), as well as a triple mutant (LMB110 [RL1826::ΩTc, RL3624::pK19Km, RL4250::ΩSp]), grew on glucose as the sole carbon source. Since there are 36 CUT1 and 30 CUT2 sugar transporters in R. leguminosarum bv. viciae 3841, it is likely that 1 or more of over 60 remaining systems would transport enough glucose to allow bacterial growth.
(iii) Inositol catabolism.
Inositol catabolism is important for bacterial competitiveness to form nodules on peas (20), required for rhizopine catabolism (3, 22), and essential for nitrogen fixation by Sinorhizobium fredii in soybean nodules (35).
Two clusters of divergently expressed genes, RL1490 to RL1496 and RL1497-RL1498, were strongly upregulated (up to 24-fold) by growth on inositol. The first cluster includes the known inositol catabolism genes iolCDEB (RL1495 to RL1492) (20), although in the previous study, iolC was missed, as it was at the end of a cloned fragment. Malonic semialdehyde dehydrogenase (iolA; RL0776), which is not part of the main catabolic operon, was also strongly upregulated (25-fold). The gene for inositol dehydrogenase (idhA; RL3622), catalyzing the first step of inositol degradation, was fivefold upregulated. It is known that mutants in iolA (RL0776) and iolD (RL1494) of R. leguminosarum bv. viciae 3841 are unable to grow on inositol (20), as are idhA mutants of S. meliloti and S. fredii (22). The pathway for inositol degradation has been well characterized in Bacillus subtilis (62), and it is likely that the same reactions occur in 3841, as enzymes of the pathway (IdhA [or IolG] and IolA to -E) show 26 to 48% amino acid identity between the two organisms (see Fig. S1 in the supplemental material).
Clustered with the inositol-catabolic genes iolCDEB (RL1495 to RL1492) is a downstream gene, RL1490, encoding an AraC family transcriptional regulator, and an upstream gene, RL1496, annotated as encoding a putative sugar isomerase but with high identity to RpiR-type negative transcriptional regulators. Mutations were made in RL1490 and RL1496 to test whether they control expression of the inositol-catabolic genes. Expression of genes upregulated in the presence of inositol in strain 3841 and in the two potential regulator (RL1490 and RL1496) mutant backgrounds was measured using the plasmid-borne gfp reporter gene fusions pRU1231 (iolC; RL1495) and pRU1577 (idhA; RL3622). In 3841, expression of both iolC and idhA was induced by growth on inositol compared to growth on glucose (Fig. 2). However, in the RL1496 background, both iolC and idhA were expressed even when cultures were grown on glucose (Fig. 2). Thus, RL1496 (now designated iolR) probably acts as a repressor of iolC and idhA and is inactivated in the presence of inositol or an intermediate in its metabolism.
FIG. 2.

Expression of gfp-mut3.1 gene fusions to inositol-regulated genes (iolC and idhA) in R. leguminosarum bv. viciae 3841 and iolR mutant backgrounds, grown with either glucose or inositol as the sole carbon source. The error bars indicate standard error of the means.
Growth on inositol led to upregulation of genes encoding isocitrate lyase (RL0761) (20-fold) and malate synthase (glcB; RL0054) (4-fold). This suggests that inositol degradation proceeds via the glyoxylate cycle. To test this, a glcB mutant of R. leguminosarum bv. viciae 3841 was made (RU4287 [see Table S1 in the supplemental material]), and while it grew normally on glucose, succinate, and xylose, it was unable to grow on inositol. In addition, a glcB mutant of R. leguminosarum bv. viciae VF39 (24) failed to grow on inositol. However, while inositol catabolism appears to be important for nodulation competitiveness in peas and N2 fixation in soybeans (22, 35), genes encoding enzymes in this pathway are not upregulated in bacteroids, suggesting inositol catabolism is important either in the rhizosphere or very early in infection. Interestingly, Becker et al. (5) reported the elevation of iolB in bacteroids of S. meliloti; however, this was not observed for 3841 in this study.
Growth on inositol induced expression of the characterized inositol ABC transporter (20); expression of RL4655 (intA, encoding a solute-binding protein) and RL4653 (intC, encoding an integral membrane protein) was elevated 71- and 33-fold, respectively. A second ABC system (RL4651 to RL4647) (PepT family) was upregulated approximately twofold, as was RL4652, encoding a dipeptidase. The upregulation of these genes may be due solely to their positions immediately downstream of the massively induced (>70-fold) intA (RL4655).
Central metabolism of bacteroids.
A comparison of 28-day bacteroids from P. sativum to free-living succinate-grown cells showed 276 genes that were ≥2-fold upregulated (3.8% of the annotated genes of R. leguminosarum bv. viciae 3841) (63). A detailed analysis of their distribution throughout the genome, regulation, and development is considered below. However, first, the central metabolism of mature bacteroids is compared with that of free-living bacteria, as approximately a fifth of the ≥2-fold-upregulated genes in mature bacteroids are concerned with metabolism. Although strain 3841 contains three putative citrate synthase genes (RL2234, RL2508, and RL2509), only RL2234 (annotated as ccsA) was strongly upregulated (three- to eightfold) in mature nodules. Of these three proteins, RL2234 has the highest identity to the characterized citrate synthases of S. meliloti (GltA) and the chromosomal citrate synthase of Rhizobium tropici (33, 47). The increase in transcription of RL2234 agrees with the very large increase in citrate synthase activity measured in 3841 bacteroids (45) and suggests it is a true symbiotically regulated activity. In bacteroids, the gene encoding aconitase (acnA; RL4536) was threefold upregulated and the suc operon (RL4435 to RL4438) was up to threefold upregulated; the latter is consistent with the very high rate of metabolism of succinate and malate in nodules. Thus, pea bacteroids showed strong transcriptional upregulation of the decarboxylating arm of the tricarboxylic acid (TCA) cycle (citrate synthase, aconitase, isocitrate dehydrogenase, and 2-ketoglutarate dehydrogenase), which agrees very well with the measured activities of all these enzymes (45). While these data from 3841 suggest the decarboxylating arm of the TCA cycle is very active in bacteroids, there is evidence that this part of the cycle is dispensable in soybean bacteroids. For example, 2-ketoglutarate dehydrogenase mutants of B. japonicum have far fewer bacteroids, but they fix N2 at rates similar to those of wild-type bacteria (28, 29, 31). In addition, aconitase mutants of B. japonicum that have lost 70% of enzyme activity are able to fix N2 at wild-type rates (57). In contrast, 2-ketoglutarate dehydrogenase mutants of R. leguminosarum and isocitrate dehydrogenase and citrate synthase null mutants of S. meliloti, as well as citrate synthase mutants of S. fredii, are all Fix− (40, 44, 47, 60). However, only 7% of citrate synthase activity is required in S. meliloti to obtain wild-type rates of nitrogen fixation (32). Thus, while a complete TCA cycle is almost always important for full bacteroid development and proliferation, in mature bacteroids, at least part of the decarboxylating arm is dispensable (soybean bacteroids) or its activity can be severely reduced (alfalfa bacteroids). Overall, the data indicate that the expression and activity of enzymes of the decarboxylating arm of the TCA cycle are elevated in mature bacteroids, but the metabolism of mutants may be sufficiently plastic to allow compensation by other pathways. However, it is clear there is insufficient information about metabolic flux in bacteroids to assess different pathways.
In mature nodules, genes involved in γ-aminobutyrate metabolism, gabD2 (RL0101) and gabT (RL0102), were induced two- and sevenfold, respectively. However, both were more strongly elevated at early stages of bacteroid formation, particularly at 7 days, when they were 15- and 38-fold upregulated, respectively. This is consistent with labeling and enzyme activity data showing γ-aminobutyrate metabolism is active in bacteroids (54a).
One intriguing result was strong elevation (ninefold) of the expression of glcB (RL0054), encoding malate synthase, in the absence of induction of the gene encoding isocitrate lyase (RL0761). This contrasts with inositol-grown free-living cultures, where both genes were strongly upregulated, enabling expression of the classical glyoxylate pathway. Likewise, microarrays of acetate- and acetoacetate-grown cultures of R. leguminosarum bv. viciae 3841 revealed induction of both isocitrate lyase and malate synthase, and a glcB mutant (RU4287) (see Table S1 in the supplemental material) did not grow on acetate or myo-inositol as the sole carbon source. This suggests that a novel pathway for synthesis of malate that is not dependent on lysis of isocitrate to release glyoxylate may occur in bacteroids. Indeed, while malate synthase activity has been measured in bacteroids from nodules isolated from pea, alfalfa, clover, bean, cowpea, and soybean, isocitrate lyase activity has been found only in trace amounts in bacteroids from pea, alfalfa, clover, and soybean nodules (17, 30). Given the importance of malate metabolism in N2 fixation, expression of malate synthase without concomitant expression of isocitrate lyase is an important observation which requires further investigation. However, the 3841 glcB mutant (RU4287) was Fix+, as was a glcB mutant of R. leguminosarum VF39 (24).
Acetoacetyl-coenzyme A (CoA) synthetase, RL0768, corresponds to the acsA2 gene product in S. meliloti 1021 (SMc00774), which catalyzes the activation of acetoacetate to acetoacetyl-CoA for further metabolism (12). Cells grown on acetoacetate had 2.4-fold-elevated expression of acsA2 (RL0768) compared with glucose-grown cells, suggesting its involvement in acetoacetate catabolism in strain 3841. Undifferentiated bacteria in infection threads contain polyhydroxybutyrate (PHB) granules, while in mature bacteroids they are absent, as PHB is degraded to β-hydroxybutyrate and metabolized to acetoacetate. At 28 days p.i., expression of RL0768 (acsA2) was 3.2-fold upregulated, consistent with the metabolism of acetoacetate released from the mobilization of PHB granules in bacteroids.
The effective gene expression of bacteroids relative to glucose-grown free-living cells was obtained by cross-multiplication of the relevant data sets (see Materials and Methods). In most cases, the normalized data (bacteroid versus succinate or bacteroid versus glucose) were similar, and bacteroid-specific genes (e.g., nif and fix) were the genes that showed the most induction. However, the four most strongly expressed genes in the bacteroid-versus-glucose data not present in the bacteroid-versus-succinate data set were dctA (RL3424; 35-fold), aldA (RL1966; 12-fold), pckA (RL0037; 10-fold), and dadX (pRL120416; 8-fold). Elevated expression of these genes does not appear in direct comparison of bacteroids with succinate-grown cells, because they are strongly induced by dicarboxylic acids. Thus, key aspects of bacteroid carbon metabolism resemble that of dicarboxylates.
General comparison of mature bacteroids with free-living cells.
Among the most upregulated genes in mature bacteroids compared to free-living cells are the extensive nif and fix gene clusters. Of the 276 genes that are ≥2-fold upregulated in mature bacteroids, 39% are found in putative operons containing two or more genes. The distribution of upregulated genes between chromosome and plasmids shows no bias; the chromosome, containing 65% of the genes, has 63% of those upregulated. However, when plasmids are considered separately, there is a strong bias toward pRL10 and pRL9, where there are, respectively, three and two times as many genes upregulated as expected. This is due, at least in part, to the presence of nif and fix gene clusters on these plasmids. For pRL9, excluding 10 genes of the fix cluster (pRL90012 to pRL90021) removes the bias, but for pRL10, even excluding 24 genes of the nif and fix operons, the bias remains at 1.5, which indicates further clustering of genes involved in bacteroid development. Although the functions of almost 50% of the ≥2-fold-upregulated genes are not known, approximately 20% are involved in bacteroid central metabolism (primarily nitrogen fixation), approximately 20% are involved in transport across membranes and the membranes themselves, and the remaining approximately 10% encode regulators of transcription, heat shock/stress proteins, and enzymes to break down macromolecules (e.g., peptidases and polysaccharidases). There was elevated expression of genes for the heat shock proteins HspF (RL1883; 56-fold) and HspA (RL4089; 21-fold). HspA shows 53% identity to SMb21295, identified by a proteomic study as being bacteroid specific (15). However, a mutation in either of these genes gave a Fix+ phenotype (see below).
In mature bacteroids, 478 genes were ≥2-fold downregulated (6.6% of the total genome). Of these, 44% are in operons of 2 or more genes, the most obvious being the cluster of 32 downregulated genes (RL1761 to RL1799) encoding ribosomal proteins. Slightly more genes on the chromosome were downregulated than would be expected from the gene distribution alone, which was also observed in microarray studies of S. meliloti bacteroids (4, 5). Among the ≥2-fold-downregulated genes, the functions of approximately one-third are unknown, but approximately 20% encode ribosomal proteins and approximately 20% encode proteins involved in the metabolism of small molecules, including enzymes of amino acid biosynthesis. It is clear that bacteroids do not express genes for chemotaxis, motility, cell division, and macromolecular metabolism to synthesize or replicate nucleic acids. For example, RNA polymerase, transcription termination and antitermination factors, DNA replication proteins, RNA/DNA binding proteins, RNA helicase, and DNA topoisomerase I were all downregulated, as were many genes involved in synthesis of amino-acyl tRNAs. Exceptions to this in R. leguminosarum bv. viciae 3841 are the genes encoding arginyl-tRNA synthetase (argS; RL1058) and tRNA Δ(2)-isopentenylpyrophosphate transferase (miaA; RL3249), which showed slightly elevated expression in mature bacteroids. Elevated expression of these two genes was also found in bacteroids of S. meliloti (5). Other genes ≥2-fold downregulated included those for several two-component sensor regulators and at least four encoding proteins with GGDEF or GGDEF/EAL domains. There was downregulation of the only two operons encoding subunits of ATP synthase (RL0924 to RL0927 and RL4405 to RL4410, two- to fivefold downregulated) and cytochrome oxidases (ctaC [RL1021] and ctaD [RL1022]). Despite the obvious need for ATP in nitrogen fixation, the amount required by bacteroids may be less than that of exponentially growing free-living cells. Two extracytoplasmic function sigma factors (RL3509 and pRL110007) were three- and twofold downregulated, respectively. Six genes encoding cold shock proteins (CspAs; pRL120765, pRL100052, pRL90321, RL0595, RL2102, and RL2964) were two- to fivefold downregulated. In mature nodules of S. meliloti, genes encoding the cold shock proteins CspA2, -A5, and -A6 were found to be downregulated >3-fold (5). The most downregulated (7- to 16-fold) gene cluster (pRL100104-pRL100105) encodes the monocarboxylate transport (Mct) system (34), responsible for transport of pyruvate, lactate, and alanine. The RNA chaperone Hfq was downregulated >4-fold in strain 3841, as in mature nodules of S. meliloti (5).
Bacteroid development.
Bacteroid development was examined by analyzing gene expression in nodule bacteria isolated at 7, 15, and 21 days p.i. We use the term nodule bacteria to refer to undifferentiated bacteria and bacteroids. Although young nodules contain a higher proportion of undifferentiated bacteria, such as those in infection threads and immature bacteroids, for simplicity we will refer to the overall process as bacteroid development. In comparison with free-living cultures, a total of 386 genes were induced ≥3-fold in at least one stage of bacteroid development (M ≥ 3; P < 0.05, using the multiple-testing correction factor Benjamini and Hochberg false-discovery rate). Hierarchical clustering analysis was performed on these 386 genes and on 13 individual hybridizations using P. sativum 7, 15, and 21 days p.i. of seedlings and for 28 days p.i. of seeds (Fig. 3). Key markers, including nod, nif, and fix genes, were followed. At 7 days, nod gene expression was elevated, but it dropped at 15 and 21 days, while nif gene expression was elevated at 15 and 21 days compared to 7 days. For fix genes, although the majority were already elevated at 7 days (2- to 90-fold over free-living cells, the induction continued to rise at 15 and 21 days. The hierarchical tree (Fig. 3) shows that bacteria isolated from nodules 7 days p.i. form a separate branch from those at 15, 21, and 28 days. This indicates that bacteroid development is largely complete by 15 days p.i. and that 15- to 28-day nodules are dominated by mature bacteroids. S. meliloti bacteroids from 5-day-old nodules of M. sativa were considered to be early/intermediate, whereas 8-day and older bacteroids were considered mature (13). The hierarchical tree of the expression of the 386 upregulated genes was divided into nine clusters (Fig. 3). The nod genes were in cluster 2, while nif genes and the majority of fix genes were in cluster 8. nifH was found in cluster 8, together with genes that have a copy of the nifH promoter either directly upstream or at the start of the operon (see below).
FIG. 3.

Hierarchical gene tree of expression from 13 experiments over the course of bacteroid development on peas (7, 15, and 21 days p.i. of 7 day-old plants and 28 days p.i. of seeds). The hierarchical tree was generated with the gene tree clustering algorithm in GeneSpring 7.2. using Pearson correlation and average linkage of the 386 genes that were ≥3-fold upregulated in nodule bacteria. Red indicates highly expressed genes, yellow intermediate, and blue low. Nine clusters can be distinguished and are indicated.
Classification of proteins encoded by genes ≥3-fold differentially regulated for early/intermediate (7-day) bacteroids and mature (15-day and older) bacteroids is shown in Fig. 4. Throughout bacteroid development, several groups of genes were found to be strongly downregulated. They include those encoding proteins involved in motility and chemotaxis (24 genes), nucleic acid replication/repair (21 genes), cell division (7 genes), amino acid biosynthesis (8 genes), outer membrane proteins/cell surface polysaccharides and peptidoglycan (18 genes), and the large group of ribosomal proteins (30 genes) (Fig. 4). It is apparent that the bacteroid has a reduced need for protein synthesis, concurrent with 23S rRNA being highly processed into numerous small fragments (Fig. 1). Decreased gene expression in mature bacteroids has been reported for S. meliloti (4, 13), and the pattern of genes expressed in S. meliloti bacteroids (5) is similar to that observed in this study of R. leguminosarum bv. viciae 3841. The single group in which genes were only upregulated is concerned with degradation of protein and amino acids (15 genes). Most groups had both up- and downregulated genes, and since the bacterium is undergoing a complex differentiation process, this would be expected. Genes involved in the specialized bacteroid function of nitrogen fixation were classified under nitrogen metabolism (17 upregulated; 5 downregulated) and electron transport (6 upregulated; 4 downregulated). There were changes in the expression of a large number of regulatory proteins that control genes expressed in the complex differentiation leading to the formation of mature bacteroids (26 upregulated; 24 downregulated). Thirty-nine regulators were differentially expressed at 7 days, which reflects large changes early in bacteroid formation. Furthermore, in keeping with bacteria adapting to life within the nodule, there were changes in genes involved in adaptation to atypical conditions, eight upregulated and eight downregulated genes, including those of several stress proteins. For genes concerned with transposon-related functions, 9 were downregulated early in bacteroid development, but the expression of 11 genes was elevated in mature bacteroids. This burst of transposon-related activity observed in bacteroids ≥15 days p.i., which may be starting to senesce, is likely to be part of the typical aging process and is seen in mature bacteroids of both S. meliloti (2) and M. loti (58).
FIG. 4.

Classification based on Riley codes of genes differentially regulated (≥3-fold) during bacteroid development. Genes expressed only in early/intermediate bacteroids (7 days p.i.) are represented by black bars, and genes expressed only in mature bacteroids (≥15 days p.i.) are represented by unfilled bars. Genes expressed throughout bacteroid development are represented by gray bars. (A) Upregulated genes. (B) Downregulated genes. The functional classifications were derived from annotations available via RhizoDB.
Overall, perhaps the most startling change in gene expression was in inner membrane and transport proteins. There were 25 upregulated and 18 downregulated genes of the inner membrane that were especially marked early in bacteroid development. Changes in transport across cell membranes were revealed by 33 upregulated and 19 downregulated genes, with upregulation peaking in early stages of bacteroid development (Fig. 4). There was upregulation of genes encoding the HlyD-like secretion proteins RL1454 (10-fold) and RL4274 (14-fold) early in bacteroid development (at 7 days). Expression of the gene for the HlyD-like secretion protein RmrA (pRL90059) was elevated (threefold) in developing bacteroids. In Rhizobium etli, an rmrA mutant formed 40% fewer nodules on beans and showed increased sensitivity to naringenin, coumaric acid, and salicyclic acid (26). It was suggested that RmrAB is involved in the export of natural toxic plant-derived substances in R. etli (26), and it may be that in R. leguminosarum bv. viciae 3841 the observed upregulation of genes encoding multidrug resistance (MDR) proteins (including RmrA) is required to perform a similar function. An rmrA mutant in R. etli showed a partial Nod− phenotype (26), and we can speculate that the phenotype might be more severe but for the possible redundancy in efflux/MDR proteins. In 3841, expression of SugE (RL2561), another efflux/MDR protein, was elevated sevenfold in mature bacteroids.
Protein secretion.
In mature bacteroids, RL0680 was 35-fold upregulated compared with a free-living succinate-grown culture. RL0680 encodes a putative protein of the SecDF family with 41% amino acid identity to RL2055, which had been annotated as SecD. In fact, both RL0680 and RL2055 have SecD and SecF domains fused into a single polypeptide (SecDF), as originally found in B. subtilis (7). We suggest that RL2055 should be reannotated as SecDF1 and RL0680 as SecDF2, in keeping with S. meliloti, where SMc02059 (annotated as SecD1) shows 50% identity with RL2055 and SMc02265 (SecD2) has 57% identity with RL0680. The S. meliloti proteins also have both putative SecD and SecF domains. Homologues of both SecDF1 and SecDF2 are present in R. etli and Sinorhizobium medicae, showing >85% and approximately 50% identity, respectively, to those of R. leguminosarum bv. viciae 3841. The exact role of SecDF is unclear (49), but it may have a role late in protein export, possibly clearing channels of prematurely folded or defective signal peptides (48). Based on the large induction of RL0680 expression in bacteroids, an appealing idea is that it acts as an alternative subunit in the Sec export machinery, possibly replacing SecDF1. It might permit or promote the export of proteins required for the large-scale changes in membrane structure observed here on nodule formation.
In mature bacteroids expression of TatABC (RL2046 to RL2048) was elevated (approximately 1.5 to 3-fold), but not earlier in bacteroid development, suggesting Tat-dependent export of proteins late in the developmental process.
Orthologues in rhizobia and homology within the Rhizobiaceae.
The proteins encoded by the 100 most upregulated genes (>4.83-fold) in 28-day bacteroids (Table 2) were compared by BLAST analysis of RhizoDB (http://xbase.bham.ac.uk/rhizodb/) with closely related species for which the full genome sequences have been determined: R. etli CFN 42, S. meliloti 1021, B. japonicum USDA 110, M. loti MAFF303099, S. medicae WSM419, and Agrobacterium tumefaciens C58 (23, 25, 27, 36, 37). With the exception of A. tumefaciens, these organisms all form N2-fixing nodules with specific host legumes. If genes upregulated in R. leguminosarum bv. viciae 3841 are common to this process, it might be expected that they would be found in other N2-fixing rhizobia. Approximately one-third were present (having >50% amino acid identity) in all N2-fixing rhizobia, with approximately half of these also having a homologue in A. tumefaciens. Apart from the nif-fix clusters found exclusively on pRL10 and pRL9, the remarkably few upregulated genes common to rhizobia encode the following putative proteins: acetyl-CoA synthetase (pRL100121), 4-hydroxybutyrate dehydrogenase (pRL120227), a dehydrogenase (pRL120632), UPF0261 domain protein (pRL90194), malate synthase (GlcB; RL0054), GFO/IDH/MocA dehydrogenase (RL2323), and PrkA protein kinase (RL3274). Of the proteins encoded by these 100 most upregulated genes, approximately another one-third show high amino acid identity (>70%) to a protein in a single species, and in the majority of these cases (81%), the homologue is found in R. etli, where the amino acid identity often exceeds 90%.
TABLE 2.
The 100 most highly upregulated genes in 28-day pea bacteroids compared with succinate-grown free-living cells
| Genea | Fold induction | Distributionc | Gene name | Product description |
|---|---|---|---|---|
| pRL100067b | 18 | B | Putative racemase/decarboxylase | |
| pRL100092 | 5 | PG | Putative transcriptional regulatory protein, pseudogene | |
| pRL100097b | 12 | B | Conserved hypothetical protein | |
| pRL100098 | 14 | B | Conserved hypothetical protein | |
| pRL100099b | 19 | B | Conserved hypothetical protein | |
| pRL100100 | 65 | PG | Putative nitrogenase iron protein, pseudogene (has start of nifH) | |
| pRL100101b | 105 | PG | Putative acetolactate synthase, pseudogene | |
| pRL100103b | 13 | A | Putative alcohol dehydrogenase | |
| pRL100104b | 39 | A | Conserved hypothetical protein | |
| pRL100105b | 10 | A | phaC2 | Putative polyhydroxyalkanoate synthase subunit C (shows 25% identity with residues 252-527 of PhaC (RL2094) of R. leguminosarum bv. viciae 3841) |
| pRL100106b | 21 | A | Hypothetical protein | |
| pRL100117b | 7 | PG | Putative iron-containing alcohol dehydrogenase, pseudogene | |
| pRL100119b | 34 | B | Putative propionate CoA transferase | |
| pRL100120 | 9 | A | Conserved hypothetical protein | |
| pRL100121 | 6 | C | acsA1 | Acetyl-CoA synthetase (shows 64% identity with AcsA1 (SMc04093) of S. meliloti) |
| pRL100137b | 25 | A | metX | Putative homoserine O-acetyltransferase |
| pRL100148b | 19 | PG | thiC | Putative thiamine biosynthesis protein, pseudogene |
| pRL100154 | 5 | A | Hypothetical protein | |
| pRL100156 | 9 | C | fdxB | Putative ferredoxin |
| pRL100157 | 12 | PG | NifX protein, pseudogene | |
| pRL100158 | 23 | C | nifN | Putative nitrogenase iron-molybdenum cofactor biosynthesis protein |
| pRL100159 | 46 | C | nifE | Putative nitrogenase iron-molybdenum cofactor biosynthesis protein |
| pRL100160 | 94 | C | nifK | Nitrogenase molybdenum-iron protein beta chain |
| pRL100161 | 71 | C | nifD | Nitrogenase molybdenum-iron protein alpha chain |
| pRL100162b | 60 | C | nifH | Putative nitrogenase iron protein |
| pRL100169 | 6 | A | rhiA | Rhizosphere-expressed protein |
| pRL100192 | 9 | B | Hypothetical protein | |
| pRL100193 | 9 | B | Conserved hypothetical protein | |
| pRL100194 | 9 | A | Hypothetical protein | |
| pRL100195 | 10 | C | nifB | FeMo cofactor biosynthesis protein |
| pRL100197 | 17 | C | fixX | Ferredoxin-like protein |
| pRL100198 | 24 | C | fixC | Nitrogen fixation protein |
| pRL100199 | 28 | C | fixB | Electron transfer protein |
| pRL100200 | 120 | C | fixA | Electron transfer protein |
| pRL100201b | 26 | A | Conserved hypothetical protein | |
| pRL100205 | 27 | C | fixN | Putative transmembrane cytochrome oxidase subunit |
| pRL100206 | 45 | C | fixO | Putative cytochrome oxidase subunit |
| pRL100206A | 24 | C | fixQ | Putative component of cytochrome oxidase |
| pRL100207 | 38 | C | fixP | Putative cytochrome oxidase subunit |
| pRL100210A | 7 | C | fixS | Putative component of cation pump |
| pRL100444b | 103 | B | Putative oxidoreductase | |
| pRL110199b | 62 | A | Conserved hypothetical protein | |
| pRL120027b | 15 | A | Putative aldolase | |
| pRL120227b | 10 | C | Putative 4-hydroxybutyrate dehydrogenase | |
| pRL120254 | 5 | A | Conserved hypothetical protein | |
| pRL120632b | 31 | C | Putative dehydrogenase | |
| pRL120644 | 7 | A | Conserved hypothetical protein (originally annotated as pRL120645) | |
| pRL90016 | 32 | C | fixP | Putative cytochrome oxidase component cytochrome c protein |
| pRL90016A | 27 | C | fixQ | Putative component of cytochrome oxidase |
| pRL90017 | 21 | C | fixO | Putative transmembrane cytochrome c oxidase protein |
| pRL90018 | 11 | C | fixN | Putative cytochrome oxidase transmembrane component |
| pRL90021b | 14 | C | azuP | Putative pseudoazurin protein |
| pRL90031 | 10 | B | Conserved hypothetical protein | |
| pRL90039 | 6 | B | Conserved hypothetical protein | |
| pRL90046 | 5 | A | Hypothetical protein | |
| pRL90047b | 14 | B | Putative universal stress protein | |
| pRL90194 | 5 | C | Putative UPF0261 domain protein | |
| pRL90322 | 18 | PG | Hypothetical protein, pseudogene | |
| RL0018b | 11 | C | Putative transmembrane protein | |
| RL0054b | 9 | C | glcB | Putative malate synthase |
| RL0102 | 7 | C | gabT | Putative 4-aminobutyrate aminotransferase |
| RL0540 | 7 | C | Putative two-component sensor histidine kinase transcriptional regulatory protein | |
| RL0680b | 35 | B | secDF2 | Putative transmembrane export SecD/F family protein |
| RL0702 | 7 | C | fliM | Putative flagellum motor switch protein |
| RL0913b | 11 | C | Putative PRC family protein | |
| RL1145 | 12 | A | Putative conjugated bile salt hydrolase | |
| RL1148b | 33 | A | Hypothetical protein | |
| RL1149 | 6 | B | Putative exported arylsulfatase | |
| RL1150 | 7 | A | Hypothetical protein | |
| RL1151 | 7 | B | Conserved hypothetical protein | |
| RL1173 | 10 | C | Putative transmembrane protein | |
| RL1249 | 5 | A | Hypothetical protein | |
| RL1259 | 6 | B | Conserved hypothetical protein | |
| RL1423 | 6 | C | Putative transmembrane protein | |
| RL1606 | 5 | A | Putative esterase (beta-lactamase family) | |
| RL1694b | 22 | B | Putative aldo-keto reductase | |
| RL1860 | 6 | B | phhA | Putative phenylalanine-4-hydroxylase |
| RL1868 | 9 | B | Putative universal stress-related protein | |
| RL1870 | 7 | A | dksA | Putative DNAK suppressor protein |
| RL1871 | 5 | B | Putative transmembrane cation ATPase transporter | |
| RL1873 | 5 | B | Putative pyridoxine oxidase protein | |
| RL1876b | 5 | B | adh | Putative alcohol dehydrogenase |
| RL1877 | 5 | B | Putative protease | |
| RL1878b | 13 | B | Putative peptidoglycan binding protein | |
| RL1883b | 56 | A | hspF | Putative small heat shock protein |
| RL2271b | 30 | A | Conserved hypothetical protein | |
| RL2272b | 19 | B | Conserved hypothetical protein | |
| RL2323b | 6 | C | Putative GFO/IDH/MocA dehydrogenase | |
| RL2561 | 7 | C | Putative transmembrane efflux/MDR protein | |
| RL3016b | 137 | B | pcaH | Protocatechuate 3,4-dioxygenase beta chain (3,4-pcd) |
| RL3130b | 14 | A | Putative phosphatase protein | |
| RL3274 | 5 | C | prkA | Putative PrkA protein kinase |
| RL3366b | 14 | B | Putative flavoprotein | |
| RL3495 | 5 | B | Conserved hypothetical protein | |
| RL3533 | 5 | C | gbcX | Putative solute-binding component of ABC transporter, QAT family |
| RL3682b | 9 | C | Putative transmembrane component of ABC exporter | |
| RL4089b | 21 | C | hspA | Putative heat shock protein A |
| RL4244 | 7 | A | Putative transmembrane component of ABC transporter, CUT1 family | |
| RL4279 | 5 | C | clpB | Putative chaperone ClpB (heat shock protein) |
| RL4530 | 6 | C | Putative orphan ATP-binding component of ABC transporter, family unclassified |
Genes are listed in order; the first gene in a putative operon is in boldface.
A strain with this gene mutated was isolated and tested on peas for nitrogen fixation.
Found (≥50% amino acid identity) as follows: A, unique to R. leguminosarum bv. viciae 3841; B, in one other species; C, in three or more species. PG, pseudogene (excluded from search). The species investigated were R. etli CFN42, S. meliloti 1021, B. japonicum USDA 110, A. tumefaciens C58, M. loti MAFF303099, and S. medicae WSM419.
Although, as described above, there is homology between the proteins encoded by genes highly upregulated in R. leguminosarum bv. viciae 3841 mature bacteroids and those of other rhizobia, what is surprising is that the homology is not more extensive. In the 100 proteins examined, 30 had no obvious orthologue showing >65% amino acid identity in any closely related species. Even among the closest sequenced relatives of R. leguminosarum, R. etli and S. meliloti, only 58 and 45 of these 100 proteins were present, respectively. On the Sym plasmid, pRL10, there were 13 genes that had no obvious homologue in any rhizobia, including the clusters pRL100103 to pRL100106 and pRL100119-pRL100120, and on the chromosome, there are 11 genes, including the clusters RL1148 to RL1151 and RL2271-RL2272. Although over half of the proteins encoded by genes found only in strain 3841 mature bacteroids are annotated as “hypothetical” or “conserved hypothetical,” giving no clue to their functions, others are better characterized and include the heat shock protein RL1883 (56-fold upregulated), the stress protein pRL90047 (14-fold upregulated), and RhiA (pRL100169) (6-fold upregulated).
Duplication of signals for high-level gene expression in mature bacteroids.
Pseudogenes make up 7% of the most elevated genes in 28-day bacteroids, and all of them, with one exception, are found on pRL10. In three cases, they are the first gene of a probable operon. In the case of the pseudogene pRL100100, the first 20 amino acids of the protein encoded are identical to those of NifH, and the 110 nucleotides before the start of the two genes show 90% identity, including the RpoN binding site. However outside of this small highly homologous region (170 bp), there is no significant similarity. The observed upregulation (65-fold in mature bacteroids) of this four-gene operon, pRL100100 to pRL100097, is likely to be due to its copy of the nifH promoter. The genes pRL100099 to pRL100097 encode three conserved hypothetical proteins that show 50 to 73% amino acid identity to bll1977, bll1980, and bll1981 from B. japonicum USDA 110, but there was little homology with other rhizobia. To test whether the expression of this operon is important in bacteroids, mutants in pRL100099 and pRL100097 were isolated (strains RU4027 and RU4075, respectively) (see Table S1 in the supplemental material), but both were Fix+. In all, six copies of the nifH upstream region (promoter) were found in the R. leguminosarum bv. viciae 3841 genome, and all on pRL10 and downstream genes were 5- to >100-fold upregulated in 28-day bacteroids. These genes were found together with nifH in cluster 8 of the hierarchical tree based on expression of upregulated genes (Fig. 3) and in a single group with K-means clustering analysis (into five clusters, using a similarity measure; Pearson coefficient). A summary of the expression of these genes, their operonic structure, and the mutants made in nine of the genes is provided in Table S5 in the supplemental material. Expression of this whole group of genes was highly elevated in mature bacteroids due to the presence of the nifH upstream region. However the Fix+ phenotype of all mutants tested revealed that their expression is not essential for nodule function.
Role of host specificity in nodule formation.
The large number of genes specifically induced in the R. leguminosarum bv. viciae 3841-pea symbiosis shows that there is a broad range of responses in rhizobia during bacteroid formation and nitrogen fixation. While there is a very well-established nif/fix core, this indicates great metabolic diversity in the responses of different rhizobia, possibly to different plants. The response observed in 3841 with peas was compared with that in 28-day nodules of vetch, V. cracca. There is no evidence, with the possible exception of rhiA (pRL100169), that the pattern of regulation of the 100 most upregulated genes in vetch differed from that in pea. Expression of rhiA (pRL100169) was extremely variable, and a change from 6-fold upregulated in pea to <2-fold upregulated (1.7-fold) in vetch is probably insignificant. This similarity between pea and vetch bacteroids is notable because many of these genes are not even present in the genomes of the closest relatives of R. leguminosarum. In contrast with this, the processing of the 23S rRNA appears to be pea specific.
Validation of microarray experiments.
To validate the microarray results, qRT-PCR was performed on 20 genes, and the results were compared with those obtained with microarrays for 28-day bacteroids (see Table S6 in the supplemental material). Although for 70% of the genes the results of qRT-PCR confirmed the microarray data, there were six genes for which the results differed. To examine this, gusA reporter gene fusions to pRL100067, pRL100444, pRL110199, pRL120632, RL0680, and RL3016 and to nifH (positive control) (pRU2189 to pRU2199) were constructed (see Table S1 in the supplemental material). The GusA activities of bacteroids isolated from strains containing reporter gene fusions were measured (see Table S6 in the supplemental material). In each case, the GusA activities broadly confirmed 28-day bacteroid microarray results. While the reason for discrepancies between the techniques is not known, differences between the results of microarrays and qRT-PCR are often observed (4, 13).
Mutant analysis.
In addition to the nine mutants described above, mutations were made in a further 28 genes that were highly upregulated (6- to >100-fold) in mature bacteroids (Table 2) (the mutants listed in Table S1 in the supplemental material are strains RU4022 to RU4174 and RU4301 to RU4306). Mutants in known nitrogen fixation (nifH; pRL100162) and bacteroid development (bacA; RL3557) genes were also isolated (strains RU4062 and RU4040, respectively) (see Table S1 in the supplemental material). When inoculated onto peas and assayed at 3 weeks, bacA and nifH mutants were unable to reduce acetylene (Fix−), but all other mutants showed a Fix+ phenotype, reducing acetylene at between 75 to 125% of R. leguminosarum bv. viciae 3841 levels and producing red nodules. For each mutant, Neor bacteria were isolated from nodules squashed at 3 weeks p.i., demonstrating that pK19 mutations were stable in nodules.
From the above-mentioned results, it is clear that high expression of a gene during bacteroid formation does not guarantee that it is essential for N2 fixation. We therefore conducted a small trial of randomly mutated R. leguminosarum bv. viciae 3841 strains to see whether novel mutants that were unable to fix N2 would be obtained. To do this, pea plants were individually screened with 3,072 mini-Tn5-mutated 3841 strains to assess their phenotypes. Mutants were selected on minimal medium prior to pea inoculation to deliberately exclude the numerous auxotrophs obtained on complex medium (e.g., TY), which are often Fix− because they are unable to make essential cofactors. After 6 weeks of growth on plants, one Nod− and six Fix− strains were obtained. Each of the mini-Tn5 markers was transduced into 3841, and after reinoculation onto plants, six of seven strains showed the same Nod− or Fix− phenotype, demonstrating linkage to the transposon. Each mini-Tn5 insertion was cloned and sequenced. In the single Nod− strain, RU4017, the insertion was in nodC (pRL100187). Of the five Fix− strains (RU4018, RU4067, RU4068, RU4105, and RU4107) (see Table S1 in the supplemental material), only one is mutated in a previously identified gene required for N2 fixation. RU4068 has a mutation in cycY (RL4540), encoding a cytochrome c biogenesis protein and demonstrated to be essential for symbiotic nitrogen fixation (59). The first mutation leading to a previously unknown Fix− phenotype was in RL1461, encoding a putative Mg2+ transporter (MgtE) (strain RU4107). This implies that Mg2+ transport is necessary to bacteroids and suggests that it becomes limiting during the developmental process. Since magnesium is crucial to the functions of many cellular factors, including many enzymes and ATP, it easy to speculate how this mutation could prevent the formation of N2-fixing bacteroids. However, further work is needed to verify that it does transport Mg2+ and/or other divalent metal ions. The second mutation causing a Fix− phenotype was in the large subunit of GOGAT (gltB; RL4085) (incorrectly annotated as gltA in the published sequence [63]) (strain RU4018). In R. etli, a strain mutated in gltB was reported to be unaffected in N2 fixation (14); however, it was also reported that in a strain mutated in gltD (small unit), nitrogenase activity was reduced to 25% of wild type (18). The third Fix− mutation was an insertion toward the 3′ end of pRL100036, which encodes a putative ATPase associated with diverse cellular activity (AAA ATPase) (strain RU4067). AAA ATPases are found in a wide range of prokaryotes and eukaryotes and have an equally diverse range of roles. Since pRL100036 and pRL100035 (encoding a conserved hypothetical protein) are only 15 bp apart, it is possible that the mini-Tn5 insertion interferes with the transcription of downstream pRL100035. While it is beyond the scope of this study to determine the precise role of pRL100036 in symbiosis, some AAA ATPases are already known to play an essential role in symbiotic nitrogen fixation. DctD (RL3426), the σ54-dependent enhancer binding protein, is a well-characterized example in R. leguminosarum, binding upstream of dctA (RL3424) and influencing its expression (55). The final Fix− strain was mutated in petC (RL3484), encoding a cytochrome c1 precursor (strain RU4105). Downstream genes (petB [RL3485] and petA [RL3486]), whose expression is probably affected, encode cytochrome b and a Rieske protein, an iron-sulfur protein subunit of cytochrome bc1, respectively. The cytochrome bc1 complex has been shown to be essential for nitrogen fixation (61). In R. leguminosarum bv. viciae 3841, expression of each mutated gene was not upregulated in nodules. This indicates that although there are likely to be numerous uncharacterized proteins essential for N2 fixation, targeting those with highly elevated expression in mature nodules may not lead to their discovery.
Conclusion.
We set out to examine the complex changes in bacteroid gene expression with an emphasis on metabolism. Metabolic changes in bacteroids are consistent with the use of dicarboxylic acids as the carbon source, but there are also large numbers of novel metabolic genes with unknown functions that are upregulated. Changes consistent with this include induction of dicarboxylate transport, gluconeogenesis, alanine synthesis, and the decarboxylating arm of the TCA cycle, as well as repression of the ED pathway. Moreover there are extensive changes in the expression of regulatory genes and those coding for membrane/cell surface components, including membrane transport, early in the developmental process of differentiation into bacteroids. It is surprising that so many of the most strongly induced genes in both pea and vetch nodules are not present in other rhizobia, which emphasizes the plasticity in the responses of different rhizobia to their respective hosts.
Supplementary Material
Acknowledgments
This work was supported by the Biotechnology and Biological Sciences Research Council (grant number P19406).
Footnotes
Published ahead of print on 17 April 2009.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Allaway, D., E. Lodwig, L. A. Crompton, M. Wood, T. R. Parsons, T. Wheeler, and P. S. Poole. 2000. Identification of alanine dehydrogenase and its role in mixed secretion of ammonium and alanine by pea bacteroids. Mol. Microbiol. 36508-515. [DOI] [PubMed] [Google Scholar]
- 2.Ampe, F., E. Kiss, F. Sabourdy, and J. Batut. 2003. Transcriptome analysis of Sinorhizobium meliloti during symbiosis. Genome Biol. 4R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bahar, M., J. deMajnik, M. Wexler, J. Fry, P. S. Poole, and P. J. Murphy. 1998. A model for the catabolism of rhizopine in Rhizobium leguminosarum involves a ferredoxin oxygenase complex and the inositol degradative pathway. Mol. Plant-Microbe Interact. 111057-1068. [DOI] [PubMed] [Google Scholar]
- 4.Barnett, M. J., C. J. Tolman, R. F. Fisher, and S. R. Long. 2004. A dual-genome symbiosis chip for coordinate study of signal exchange and development in a prokaryote-host interaction. Proc. Natl. Acad. Sci. USA 10116636-16641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Becker, A., H. Berges, E. Krol, C. Bruand, S. Ruberg, D. Capela, E. Lauber, E. Meilhoc, F. Ampe, F. J. de Bruijn, J. Fourment, A. Francez-Charlot, D. Kahn, H. Kuster, C. Liebe, A. Puhler, S. Weidner, and J. Batut. 2004. Global changes in gene expression in Sinorhizobium meliloti 1021 under microoxic and symbiotic conditions. Mol. Plant-Microbe Interact. 17292-303. [DOI] [PubMed] [Google Scholar]
- 6.Beringer, J. E. 1974. R factor transfer in Rhizobium leguminosarum. J. Gen. Microbiol. 84188-198. [DOI] [PubMed] [Google Scholar]
- 7.Bolhuis, A., C. P. Broekhuizen, A. Sorokin, M. L. van Roosmalen, G. Venema, S. Bron, W. J. Quax, and J. M. van Dijl. 1998. SecDF of Bacillus subtilis, a molecular Siamese twin required for the efficient secretion of proteins. J. Biol. Chem. 27321217-21224. [DOI] [PubMed] [Google Scholar]
- 8.Brechenmacher, L., M.-Y. Kim, M. Benitez, M. Li, T. Joshi, B. Calla, M. P. Lee, M. Libault, L. O. Vodkin, D. Xu, S.-H. Lee, S. J. Clough, and G. Stacey. 2008. Transcription profiling of soybean nodulation by Bradyrhizobium japonicum. Mol. Plant-Microbe Interact. 21631-645. [DOI] [PubMed] [Google Scholar]
- 9.Buchanan-Wollaston, V. 1979. Generalized transduction in Rhizobium leguminosarum. J. Gen. Microbiol. 112135-142. [Google Scholar]
- 10.Bustin, S. A. 2000. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J. Mol. Endocrinol. 25169-193. [DOI] [PubMed] [Google Scholar]
- 11.Bustin, S. A. 2002. Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. J. Mol. Endocrinol. 2923-39. [DOI] [PubMed] [Google Scholar]
- 12.Cai, G. Q., B. T. Driscoll, and T. C. Charles. 2000. Requirement for the enzymes acetoacetyl coenzyme A synthetase and poly-3-hydroxybutyrate (PHB) synthase for growth of Sinorhizobium meliloti on PHB cycle intermediates. J. Bacteriol. 1822113-2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Capela, D., C. Filipe, C. Bobik, J. Batut, and C. Bruand. 2006. Sinorhizobium meliloti differentiation during symbiosis with alfalfa: a transcriptomic dissection. Mol. Plant-Microbe Interact. 19363-372. [DOI] [PubMed] [Google Scholar]
- 14.Castillo, A., H. Taboada, A. Mendoza, B. Valderrama, S. Encarnacion, and J. Mora. 2000. Role of GOGAT in carbon and nitrogen partitioning in Rhizobium etli. Microbiology 1461627-1637. [DOI] [PubMed] [Google Scholar]
- 15.Djordjevic, M. A., H. C. Chen, S. Natera, G. Van Noorden, C. Menzel, S. Taylor, C. Renard, O. Geiger, and G. F. Weiller. 2003. A global analysis of protein expression profiles in Sinorhizobium meliloti: discovery of new genes for nodule occupancy and stress adaptation. Mol. Plant-Microbe Interact. 16508-524. [DOI] [PubMed] [Google Scholar]
- 16.Dominguez-Ferreras, A., R. Perez-Arnedo, A. Becker, J. Olivares, M. J. Soto, and J. Sanjuan. 2006. Transcriptome profiling reveals the importance of plasmid pSymB for osmoadaptation of Sinorhizobium meliloti. J. Bacteriol. 1887617-7625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dunn, M. F. 1998. Tricarboxylic acid cycle and anaplerotic enzymes in rhizobia. FEMS Microbiol. Rev. 22105-123. [DOI] [PubMed] [Google Scholar]
- 18.Ferraioli, S., R. Tate, M. Cermola, R. Favre, M. Iaccarino, and E. J. Patriarca. 2002. Auxotrophic mutant strains of Rhizobium etli reveal new nodule development phenotypes. Mol. Plant-Microbe Interact. 15501-510. [DOI] [PubMed] [Google Scholar]
- 19.Fox, M. A., R. Karunakaran, M. E. Leonard, B. Mouhsine, A. Williams, A. K. East, J. A. Downie, and P. S. Poole. 2008. Characterization of the quaternary amine transporters of Rhizobium leguminosarum bv. viciae 3841. FEMS Microbiol. Lett. 287212-220. [DOI] [PubMed] [Google Scholar]
- 20.Fry, J., M. Wood, and P. S. Poole. 2001. Investigation of myo-inositol catabolism in Rhizobium leguminosarum bv. viciae and its effect on nodulation competitiveness. Mol. Plant-Microbe Interact. 141016-1025. [DOI] [PubMed] [Google Scholar]
- 21.Gage, D. J. 2004. Infection and invasion of roots by symbiotic, nitrogen-fixing rhizobia during nodulation of temperate legumes. Microbiol. Mol. Biol. Rev. 68280-300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Galbraith, M. P., S. F. Feng, J. Borneman, E. W. Triplett, F. J. deBruijn, and S. Rossbach. 1998. A functional myo-inositol catabolism pathway is essential for rhizopine utilization by Sinorhizobium meliloti. Microbiology 1442915-2924. [DOI] [PubMed] [Google Scholar]
- 23.Galibert, F., T. M. Finan, S. R. Long, A. Puhler, P. Abola, F. Ampe, F. Barloy-Hubler, M. J. Barnett, A. Becker, P. Boistard, G. Bothe, M. Boutry, L. Bowser, J. Buhrmester, E. Cadieu, D. Capela, P. Chain, A. Cowie, R. W. Davis, S. Dreano, N. A. Federspiel, R. F. Fisher, S. Gloux, T. Godrie, A. Goffeau, B. Golding, J. Gouzy, M. Gurjal, I. Hernandez-Lucas, A. Hong, L. Huizar, R. W. Hyman, T. Jones, D. Kahn, M. L. Kahn, S. Kalman, D. H. Keating, E. Kiss, C. Komp, V. Lalaure, D. Masuy, C. Palm, M. C. Peck, T. M. Pohl, D. Portetelle, B. Purnelle, U. Ramsperger, R. Surzycki, P. Thebault, M. Vandenbol, F. J. Vorholter, S. Weidner, D. H. Wells, K. Wong, K. C. Yeh, and J. Batut. 2001. The composite genome of the legume symbiont Sinorhizobium meliloti. Science 293668-672. [DOI] [PubMed] [Google Scholar]
- 24.Garcia-de los Santos, A., A. Morales, L. Baldoma, S. R. D. Clark, S. Brom, C. K. Yost, I. Hernandez-Lucas, J. Aguilar, and M. F. Hynes. 2002. The glcB locus of Rhizobium leguminosarum VF39 encodes an arabinose-inducible malate synthase. Can. J. Microbiol. 48922-932. [DOI] [PubMed] [Google Scholar]
- 25.González, V., R. I. Santamaría, P. Bustos, I. Hernández-González, A. Medrano-Soto, G. Moreno-Hagelsieb, S. C. Janga, M. A. Ramírez, V. Jiménez-Jacinto, J. Collado-Vides, and G. Dávila. 2006. The partitioned Rhizobium etli genome: genetic and metabolic redundancy in seven interacting replicons. Proc. Natl. Acad. Sci. USA 1033834-3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gonzalez-Pasayo, R., and E. Martinez-Romero. 2000. Multiresistance genes of Rhizobium etli CFN42. Mol. Plant-Microbe Interact. 13572-577. [DOI] [PubMed] [Google Scholar]
- 27.Goodner, B., G. Hinkle, S. Gattung, N. Miller, M. Blanchard, B. Qurollo, B. S. Goldman, Y. Cao, M. Askenazi, C. Halling, L. Mullin, K. Houmiel, J. Gordon, M. Vaudin, O. Iartchouk, A. Epp, F. Liu, C. Wollam, M. Allinger, D. Doughty, C. Scott, C. Lappas, B. Markelz, C. Flanagan, C. Crowell, J. Gurson, C. Lomo, C. Sear, G. Strub, C. Cielo, and S. Slater. 2001. Genome sequence of the plant pathogen and biotechnology agent Agrobacterium tumefaciens C58. Science 2942323-2328. [DOI] [PubMed] [Google Scholar]
- 28.Green, L. S., and D. W. Emerich. 1997. Bradyrhizobium japonicum does not require alpha-ketoglutarate dehydrogenase for growth on succinate or malate. J. Bacteriol. 179194-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Green, L. S., and D. W. Emerich. 1997. The formation of nitrogen-fixing bacteroids is delayed but not abolished in soybean infected by an alpha-ketoglutarate dehydrogenase-deficient mutant of Bradyrhizobium japonicum. Plant Physiol. 1141359-1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Green, L. S., D. B. Karr, and D. W. Emerich. 1998. Isocitrate dehydrogenase and glyoxylate cycle enzyme activities in Bradyrhizobium japonicum under various growth conditions. Arch. Microbiol. 169445-451. [DOI] [PubMed] [Google Scholar]
- 31.Green, L. S., Y. Z. Li, D. W. Emerich, F. J. Bergersen, and D. A. Day. 2000. Catabolism of alpha-ketoglutarate by a sucA mutant of Bradyrhizobium japonicum: evidence for an alternative tricarboxylic acid cycle. J. Bacteriol. 1822838-2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grzemski, W., J. P. Akowski, and M. L. Kahn. 2005. Probing the Sinorhizobium meliloti-alfalfa symbiosis using temperature-sensitive and impaired-function citrate synthase mutants. Mol. Plant-Microbe Interact. 18134-141. [DOI] [PubMed] [Google Scholar]
- 33.Hernández-Lucas, I., M. A. Pardo, L. Segovia, J. Miranda, and E. Martínez-Romero. 1995. Rhizobium tropici chromosomal citrate synthase gene. Appl. Environ. Microbiol. 613992-3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hosie, A. H. F., D. Allaway, and P. S. Poole. 2002. A monocarboxylate permease of Rhizobium leguminosarum is the first member of a new subfamily of transporters. J. Bacteriol. 1845436-5448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang, G. Q., A. H. Krishnan, Y. W. Kim, T. J. Wacek, and H. B. Krishnan. 2001. A functional myo-inositol dehydrogenase gene is required for efficient nitrogen fixation and competitiveness of Sinorhizobium fredii USDA191 to nodulate soybean (Glycine max [L.] Merr.). J. Bacteriol. 1832595-2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaneko, T., Y. Nakamura, S. Sato, E. Asamizu, T. Kato, S. Sasamoto, A. Watanabe, K. Idesawa, A. Ishikawa, K. Kawashima, T. Kimura, Y. Kishida, C. Kiyokawa, M. Kohara, M. Matsumoto, A. Matsuno, Y. Mochizuki, S. Nakayama, N. Nakazaki, S. Shimpo, M. Sugimoto, C. Takeuchi, M. Yamada, and S. Tabata. 2000. Complete genome structure of the nitrogen-fixing symbiotic bacterium Mesorhizobium loti. DNA Res. 7331-338. [DOI] [PubMed] [Google Scholar]
- 37.Kaneko, T., Y. Nakamura, S. Sato, K. Minamisawa, T. Uchiumi, S. Sasamoto, A. Watanabe, K. Idesawa, M. Iriguchi, K. Kawashima, M. Kohara, M. Matsumoto, S. Shimpo, H. Tsuruoka, T. Wada, M. Yamada, and S. Tabata. 2002. Complete genomic sequence of nitrogen-fixing symbiotic bacterium Bradyrhizobium japonicum USDA110. DNA Res. 9189-197. [DOI] [PubMed] [Google Scholar]
- 38.Karunakaran, R., K. Ebert, S. Harvey, M. E. Leonard, V. Ramachandran, and P. S. Poole. 2006. Thiamine is synthesized by a salvage pathway in Rhizobium leguminosarum bv. viciae strain 3841. J. Bacteriol. 1886661-6668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Klein, F., R. Samorski, G. Klug, and E. Evguenieva-Hackenberg. 2002. Atypical processing in domain III of 23S rRNA of Rhizobium leguminosarum ATCC 10004(T) at a position homologous to an rRNA fragmentation site in protozoa. J. Bacteriol. 1843176-3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krishnan, H. B., W. S. Kim, J. Sun-Hyung, K. Y. Kim, and G. Q. Jiang. 2003. Citrate synthase mutants of Sinorhizobium fredii USDA257 form ineffective nodules with aberrant ultrastructure. Appl. Environ. Microbiol. 693561-3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lodwig, E., S. Kumar, D. Allaway, A. Bourdès, J. Prell, U. Priefer, and P. Poole. 2004. Regulation of l-alanine dehydrogenase in Rhizobium leguminosarum bv. viciae and its role in pea nodules. J. Bacteriol. 186842-849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lodwig, E., and P. Poole. 2003. Metabolism of Rhizobium bacteroids. Crit. Rev. Plant Sci. 2237-78. [Google Scholar]
- 43.Lodwig, E. M., A. H. F. Hosie, A. Bourdes, K. Findlay, D. Allaway, R. Karunakaran, J. A. Downie, and P. S. Poole. 2003. Amino-acid cycling drives nitrogen fixation in the legume-Rhizobium symbiosis. Nature 422722-726. [DOI] [PubMed] [Google Scholar]
- 44.McDermott, T. R., and M. L. Kahn. 1992. Cloning and mutagenesis of the Rhizobium meliloti isocitrate dehydrogenase gene. J. Bacteriol. 1744790-4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McKay, I. A., M. J. Dilworth, and A. R. Glenn. 1989. Carbon catabolism in continuous cultures and bacteroids of Rhizobium leguminosarum MNF3841. Arch. Microbiol. 152606-610. [Google Scholar]
- 46.Mergaert, P., T. Uchiumi, B. Alunni, G. Evanno, A. Cheron, O. Catrice, A.-E. Mausset, F. Barloy-Hubler, F. Galibert, A. Kondorosi, and E. Kondorosi. 2006. Eukaryotic control on bacterial cell cycle and differentiation in the Rhizobium-legume symbiosis. Proc. Natl. Acad. Sci. USA 1035230-5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mortimer, M. W., T. R. McDermott, G. M. York, G. C. Walker, and M. L. Kahn. 1999. Citrate synthase mutants of Sinorhizobium meliloti are ineffective and have altered cell surface polysaccharides. J. Bacteriol. 1817608-7613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nouaille, S., E. Morello, N. Cortez-Peres, Y. Le Loir, J. Commissaire, J. J. Gratadoux, E. Poumerol, A. Gruss, and P. Langella. 2006. Complementation of the Lactococcus lactis secretion machinery with Bacillus subtilis SecDF improves secretion of staphylococcal nuclease. Appl. Environ. Microbiol. 722272-2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nouwen, N., and A. J. Driessen. 2002. SecDFYajC forms a heterotetrameric complex with YidC. Mol. Microbiol. 441397-1405. [DOI] [PubMed] [Google Scholar]
- 50.Oldroyd, G. E. D., and J. A. Downie. 2008. Coordinating nodule morphogenesis with rhizobial infection in legumes. Annu. Rev. Plant Biol. 59519-546. [DOI] [PubMed] [Google Scholar]
- 51.Pessi, G., C. H. Ahrens, H. Rehrauer, A. Lindemann, F. Hauser, H.-M. Fischer, and H. Hennecke. 2007. Genome-wide transcript analysis of Bradyrhizobium japonicum bacteroids in soybean root nodules. Mol. Plant-Microbe Interact. 201353-1363. [DOI] [PubMed] [Google Scholar]
- 52.Poole, P. S., A. Blyth, C. J. Reid, and K. Walters. 1994. myo-Inositol catabolism and catabolite regulation in Rhizobium leguminosarum bv viciae. Microbiology 1402787-2795. [Google Scholar]
- 53.Poole, P. S., N. A. Schofield, C. J. Reid, E. M. Drew, and D. L. Walshaw. 1994. Identification of chromosomal genes located downstream of dctD that affect the requirement for calcium and the lipopolysaccharide layer of Rhizobium leguminosarum. Microbiology 1402797-2809. [DOI] [PubMed] [Google Scholar]
- 54.Prell, J., and P. Poole. 2006. Metabolic changes of rhizobia in legume nodules. Trends Microbiol. 14161-168. [DOI] [PubMed] [Google Scholar]
- 54a.Prell, J., A. Bourdès, R. Karunakaran, M. Lopez-Gomez, and P. Poole. 2009. Pathway of γ-aminobutyrate metabolism in Rhizobium leguminosarum 3841 and its role in symbiosis. J. Bacteriol. 1912177-2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scholl, D., and B. T. Nixon. 1996. Cooperative binding of DctD to the dctA upstream activation sequence of Rhizobium meliloti is enhanced in a constitutively active truncated mutant. J. Biol. Chem. 27126435-26442. [DOI] [PubMed] [Google Scholar]
- 56.Selenska-Pobell, S., and H. Doring. 1998. Sequences around the fragmentation sites of the large subunit ribosomal RNA in the family Rhizobiaceae. Antonie van Leeuwenhoek 7355-67. [DOI] [PubMed] [Google Scholar]
- 57.Thony-Meyer, L., and P. Kunzler. 1996. The Bradyrhizobium japonicum aconitase gene (acnA) is important for free-living growth but not for an effective root-nodule symbiosis. J. Bacteriol. 1786166-6172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Uchiumi, T., T. Ohwada, M. Itakura, H. Mitsui, N. Nukui, P. Dawadi, T. Kaneko, S. Tabata, T. Yokoyama, K. Tejima, K. Saeki, H. Omori, M. Hayashi, T. Maekawa, R. Sriprang, Y. Murooka, S. Tajima, K. Simomura, M. Nomura, A. Suzuki, Y. Shimoda, K. Sioya, M. Abe, and K. Minamisawa. 2004. Expression islands clustered on the symbiosis island of the Mesorhizobium loti genome. J. Bacteriol. 1862439-2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vargas, C., G. H. Wu, A. E. Davies, and J. A. Downie. 1994. Identification of a gene encoding a thioredoxin-like product necessary for cytochrome c biosynthesis and symbiotic nitrogen fixation in Rhizobium leguminosarum. J. Bacteriol. 1764117-4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walshaw, D. L., A. Wilkinson, M. Mundy, M. Smith, and P. S. Poole. 1997. Regulation of the TCA cycle and the general amino acid permease by overflow metabolism in Rhizobium leguminosarum. Microbiology 1432209-2221. [DOI] [PubMed] [Google Scholar]
- 61.Wu, G. H., M. J. Delgado, C. Vargas, A. E. Davies, R. K. Poole, and J. A. Downie. 1996. The cytochrome bc(1) complex but not CycM is necessary for symbiotic nitrogen fixation by Rhizobium leguminosarum. Microbiology 1423381-3388. [DOI] [PubMed] [Google Scholar]
- 62.Yoshida, K., M. Yamaguchi, T. Morinaga, M. Kinehara, M. Ikeuchi, H. Ashida, and Y. Fujita. 2008. myo-Inositol catabolism in Bacillus subtilis. J. Biol. Chem. 28310415-10424. [DOI] [PubMed] [Google Scholar]
- 63.Young, J. P., L. Crossman, A. Johnston, N. Thomson, Z. Ghazoui, K. Hull, M. Wexler, A. Curson, J. Todd, P. Poole, T. Mauchline, A. East, M. Quail, C. Churcher, C. Arrowsmith, I. Cherevach, T. Chillingworth, K. Clarke, A. Cronin, P. Davis, A. Fraser, Z. Hance, H. Hauser, K. Jagels, S. Moule, K. Mungall, H. Norbertczak, E. Rabbinowitsch, M. Sanders, M. Simmonds, S. Whitehead, and J. Parkhill. 2006. The genome of Rhizobium leguminosarum has recognizable core and accessory components. Genome Biol. 7R34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.