

Abstract
Mortality due to multidrug-resistant Staphylococcus aureus infection is predicted to surpass that of human immunodeficiency virus/AIDS in the United States. Despite the various treatment options for S. aureus infections, it remains a major hospital- and community-acquired opportunistic pathogen. With the emergence of multidrug-resistant S. aureus strains, there is an urgent need for the discovery of new antimicrobial drug targets in the organism. To this end, we reconstructed the metabolic networks of multidrug-resistant S. aureus strains using genome annotation, functional-pathway analysis, and comparative genomic approaches, followed by flux balance analysis-based in silico single and double gene deletion experiments. We identified 70 single enzymes and 54 pairs of enzymes whose corresponding metabolic reactions are predicted to be unconditionally essential for growth. Of these, 44 single enzymes and 10 enzyme pairs proved to be common to all 13 S. aureus strains, including many that had not been previously identified as being essential for growth by gene deletion experiments in S. aureus. We thus conclude that metabolic reconstruction and in silico analyses of multiple strains of the same bacterial species provide a novel approach for potential antibiotic target identification.
Staphylococcus aureus is a major hospital/community-acquired opportunistic pathogen. It causes bacteremia, pneumonia, endocarditis, meningitis, and toxic-shock syndrome in adult humans; skin lesions, impetigo, and abscesses in children; and mastitis in cattle (7, 22, 27). In general, S. aureus infections are treated with β-lactam antibiotics, sulfa drugs, tetracycline, and clindamycin. However, drug-resistant strains, such as methicillin-resistant S. aureus (MRSA) and vancomycin-resistant S. aureus (VRSA), have emerged from both hospital and community infections in recent years. To date, only one new drug candidate, platensimycin, has been found to be effective against some strains of MRSA and VRSA (30). A recent meta-analysis suggested that mortality due to multidrug-resistant S. aureus in the United States may exceed that from human immunodeficiency virus infections and AIDS (19). This has resulted in a renewed interest in identifying new targets and molecules effective against multidrug-resistant strains of bacteria, and S. aureus in particular.
Based on whole-genome sequence comparisons, S. aureus strains can be divided into three divergent groups arising from a common lineage (11). Significant sequence variations between animal and human S. aureus strains have also been identified (15). Though many virulence and drug resistance markers have been studied, the cause of the continuous emergence of multidrug-resistant strains remains elusive, as the resistance phenotype is not attributable to a few studied genes. Combining the data from multilocus sequence typing, microarray analysis, sequence relationships, homologous recombination, and phages of S. aureus, two major groups of clonal strains have been identified (11). A similar conclusion was reached when the S. aureus Newman genome sequence was compared to those of 11 other S. aureus strains (3). These studies not only confirm the clonality of the genome, but also reveal that nearly 20% of the sequence variations are due to prophages and pathogenicity islands.
In order to further refine a generic antimicrobial drug target identification scheme (2), we performed metabolic reconstructions of multidrug-resistant and sensitive strains of S. aureus. This was feasible, as the genome sequences of 13 S. aureus strains are now available. They include strain N315 (a MRSA strain), Mu50 (a VRSA strain), JH9 (a vancomycin-nonsusceptible MRSA strain), JH1 (a vancomycin-susceptible, hospital-acquired MRSA strain), COL (a hospital-acquired MRSA strain), 252 (a hospital-acquired MRSA strain), USA 300 (a community-acquired MRSA strain), MW2 (a community-acquired MRSA strain), and RF122 (a bovine mastitis strain).
Previous efforts in the metabolic reconstruction and subsequent flux balance analysis (FBA) of a single S. aureus strain (N315) provided valuable but limited insight into the metabolic capabilities of the bacterium (4, 14). Using this strain (20), Becker and Palsson predicted 518 metabolic reactions and 571 metabolites based on a limited set of genes (enzymes) (4). Their study also identified the components of minimal growth medium for S. aureus. Of the six required amino acids, only four (l-alanine, l-arginine, l-proline, and l-glycine) were common to both experimental and computational studies. Glucose (carbon source), phosphate, sulfate, nicotinamide, and thiamine were both experimentally utilized and computationally verified. However, other substrates, such as the nucleosides cytidine and uridine, were predicted not to be required in their metabolic model. A second genome-scale reconstruction of the same strain based on the KEGG ligand database was carried out and yielded 774 metabolic reactions catalyzed by 394 unique enzymes (13). Heinemann et al. (14) also validated their reconstruction using published experimental data and further defined a biomass composition for S. aureus.
To reconcile the results of these two previous reconstructions and to address the differences in the metabolic capabilities of various S. aureus strains, we employed comprehensive genomic and metabolic reconstruction methodologies using the ERGO bioinformatics suite (24). This approach enabled us to identify the functional pathways, metabolic reactions, and transport reactions of several sequenced strains of S. aureus. The identified metabolic pathways and their individual reactions were systematically compared with those archived in the KEGG ligand database (17). FBA of such reconstructed metabolic networks have allowed in silico single and double gene deletion experiments, e.g., in Escherichia coli (2, 8, 23, 29). The application of these methods has led us to the identification of single enzymes and synthetic enzyme pairs that are unconditionally required for the growth (biomass production) of all S. aureus strains.
MATERIALS AND METHODS
Metabolic reconstruction of multidrug-resistant S. aureus strains.
In this study, we carried out the metabolic reconstruction of multiple S. aureus genomes using the ERGO bioinformatics suite (24) and the KEGG ligand/reaction database as of 2007 (http://www.genome.jp/ligand) (17). Initially, we focused on the MRSA strain N315 as a model organism for metabolic reconstruction and FBA studies (20). Its whole-genome sequence (NC_002745) and the sequence for plasmid pN315 (NC_003140) were obtained from NCBI (GenBank) and downloaded into ERGO, a genome informatics platform that is developed and maintained by Integrated Genomics, Inc. (24). Similar processes were performed on several other sequenced genomes, including S. aureus Mu50 (NC_002758), S. aureus MW2 (NC_003923), S. aureus subsp. aureus COL (NC_002951), S. aureus EMRSA-16 strain 252 (NC_002952), S. aureus methicillin-sensitive strain 476 (NC_002953), S. aureus subsp. aureus JH1 (NC_009632), S. aureus subsp. aureus JH9 (NC_009487), S. aureus RF122 (NC_007622), S. aureus subsp. aureus COL (NC_002951), S. aureus subsp. aureus USA300 (NC_007793), S. aureus subsp. aureus USA300_ TCH1516 (NC_010079), and S. aureus subsp. aureus Newman (NC_009641).
In general, the whole-genome DNA sequences, along with associated open reading frames (ORFs)/protein sequences, were integrated into the ERGO genome database. Automated and manual annotations of the ORFs were carried out as described previously (18). Briefly, automatic ORF calling was performed using a combination of programs, including GLIMMER and Critica. The protein similarities were computed by BLAST “all against all,” with over 3.5 million protein sequences present in the nonredundant database from over 1,362 genomes. This was followed by functional assignments, relationship computation, pathway analyses, and automated metabolic reconstruction (metabolic and nonmetabolic subsystems). These automated annotations are largely based on the existence of ortholog and protein family clusters. Manual annotation included a review of every gene in the genome. Once the functional annotations were assigned, we performed additional manual pathway curations of the functions in order to identify their roles in appropriate metabolic subsystems. The automatically assigned functions were validated using both proprietary and publicly available tools to make new functional assignments. During manual annotation, we also considered and reconciled motif/domain database results (for example, from COGS, Pfam, and INTERPRO) to make functional assignments. Missing steps in pathways were identified, and genes that could fulfill these functions were sought in order to determine if the pathways should be included in the organism's metabolic catalog. This process resulted in the identification of functions for unannotated or erroneously annotated proteins. Metabolic/cellular reconstructions were finally derived by interconnecting the entire set of pathways identified in a given strain. These pathways in ERGO were grouped into metabolic and nonmetabolic systems that were interconnected into a metabolic network between subsystems, such as amino acids, carbohydrates, lipids, secondary metabolism, and sulfur and phosphorus metabolism. The nonmetabolic pathways contained lists of genes/functions that represented subsystems, such as protein secretion, virulence, secretion, and phages (24). All 13 S. aureus genomes were simultaneously annotated and analyzed. Gap-filling reactions were not performed because the ORFs corresponding to the missing enzymes were not present. Unlike in the work of Becker and Palsson (4), where the gap-filling reactions were performed, the aim of this study was to identify the genes that could be experimentally tested for essentiality. In cases of missing steps within a given pathway, we searched for orthologs or published biochemical activities (for example, for the histidine biosynthesis pathway). ORFs with enzymes as functions with complete or incomplete Enzyme Commission (EC) numbers were identified from the functional categories of the ERGO genome analysis suite. The associated biochemical reactions for each of the enzymes were selected in the KEGG reaction database. The biochemical reactions with substrates that did not have an identified transport or uptake system and those that were not supported by experimental studies of S. aureus were considered “unlikely reactions” and were not included in the FBA. Individual transport reactions were added from the ERGO pathway collection. Universal (SwissProt) names were used for enzymes to avoid nomenclature conflict for the FBA studies. All the reactions and their corresponding KEGG identifiers (reaction IDs and compound IDs) were used in the FBA computations.
FBA of S. aureus metabolic networks.
The metabolic networks were constructed individually for all 13 S. aureus strains, and the reaction data from each of the species were used for FBA computations. The total numbers of enzymes of all the species ranged from 522 to 547. The biochemical reactions varied from 1,444 to 1,497, with total metabolites ranging from 1,399 to 1,437. The metabolic reactions were classified into three categories: cellular (reactions in the cytoplasm), transport (reactions involving both the intra- and extracellular metabolites), and exchange (reactions involving either uptake or excretion of the extracellular metabolites). The stoichiometries of these metabolic reactions were adapted from the KEGG ligand database and the ERGO bioinformatics suite, yielding the stoichiometry matrix S, with its element Sij indicating the stoichiometric coefficient of the metabolite i in the reaction j. The FBA states that in the stationary state, the fluxes {vj} of the metabolic reactions are those that maximize vbiomass subject to:
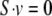 |
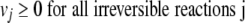 |
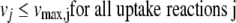 |
(1) |
where vbiomass indicates the flux of the reaction that produces biomass, acting as a drain of those biomass compounds (1, 2, 4, 8, 23, 25, 26, 29). The biomass-generating reaction is given by 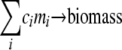 in which mi is each of the biomass compounds and ci is the coefficient.
in which mi is each of the biomass compounds and ci is the coefficient.
Currently, there are two predicted biomass compositions proposed for S. aureus strain N315 (4, 14): one is based on and similar to that of a related gram-positive bacterium, Bacillus subtilis (4), with 58 biomass compounds, and the second was established from an extensive literature search on S. aureus resulting in 50 biomass compounds (14). For each set of biomass components, we first checked whether each of the biomass compounds, mi, was synthesized in our metabolic-network reconstructions by performing the FBA assuming the biomass-generating reaction to be mi→biomass for each mi. If this reaction had a nonzero flux, we considered the biomass compound to be synthesized in the given strain of S. aureus. For the biomass composition from reference 4, we found that for nine S. aureus strains (TCH1516, USA300, Mu3, JH9, JH1, RF122, Mu50, MRSA252, and N315), 47 of the 58 biomass components could be synthesized, that is, the flux of the modified biomass-generating reaction for each of these compounds was nonzero. In contrast, the other 11 components could not be synthesized, suggesting absence of the biosynthetic pathways to generate them or absence of the uptake transporters for them. Three S. aureus strains, Newman, COL, and MW2, could synthesize only 44 biomass components, whereas 43 components could be synthesized by the methicillin-sensitive strain 476. For the biomass composition of reference (14), each of the 13 strains synthesized 41 compounds of the proposed 51. For the missing components, one could assume that unknown gene products catalyzed the corresponding biosynthetic pathways and therefore add them to the metabolic network. Those added pathways would be likely to be essential for the growth of S. aureus. However, we elected to retain only the biomass components synthesized in the current form of the metabolic networks of S. aureus strains to obtain definitely and unconditionally essential enzymes.
These computational studies predicted the synthesis of 47 and 41 biomass compounds, respectively, with 25 common compounds (see Dataset 1 in the supplemental material). We considered both biomass compositions from Becker and Palsson (4) and Heinemann (4, 14) in our study for comparison. Therefore, we used two metabolic-reconstruction versions for each S. aureus strain. The two versions were identical except for the biomass generation reaction involving biomass compounds from either Becker and Palsson (4) or Heinemann et al. (14).
In this study, we present the results obtained from the metabolic reconstructions with the biomass composition based on the studies of Becker and Palsson (4). The results using the biomass composition from Heinemann et al. (14) can be found in the supplemental material. For the FBA computations, we used ideally rich medium in which all the uptake reactions could occur without limitation on their fluxes, since we aimed to identify unconditionally essential enzymes, which we define below. The FBA is a linear programming problem and was carried out using the GNU Linear Programming Kit.
Identification of single unconditionally essential and synthetic-lethal enzymes of S. aureus strains.
In the FBA scheme, an enzyme is considered to be essential (2) if an additional constraint,
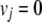 |
(2) |
for all reactions j catalyzed by the selected enzyme lead to no growth (vbiomass = 0). If inactivation of an enzyme leads to no growth in an ideally rich environment (medium) in which all the uptake reactions are allowed to have unlimited fluxes, it would also lead to no growth in any other environment. Therefore, the unconditionally essential enzymes, i.e., essential under all growth conditions, can be obtained from the FBA equation 1 combined with equation 2 and setting all upper limits of uptake reactions as follows:
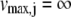 |
(3) |
for all uptake reactions j. Similarly, an unconditionally synthetic-lethal pair of enzymes can be identified by monitoring the biomass generation reaction while blocking the reactions catalyzed by the considered pair of enzymes in an ideally rich environment. That is, the removal of two enzymes, E1 and E2, is considered to be unconditionally lethal if the removal of just one of them is not lethal and the FBA (equation 1) in the ideally rich environment (equation 3) leads to no growth (vbiomass = 0) (8, 29) under the following constraints:
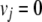 |
(4) |
for all uptake reactions j catalyzed by the enzyme E1 or E2.
Design and analysis of growth media.
Using the complete metabolic analysis of all the strains of S. aureus and the reference minimal medium, we derived a medium that supports growth on a minimal medium, using metabolic-reconstruction methods, as described for Xylella fastidiosa (5). The compounds from the published minimal medium (AAM+) for S. aureus (26) were verified with the metabolic reconstruction of S. aureus genomes. A modified synthetic minimum medium was predicted that did not contain l-arginine, l-glutamic acid, l-valine, l-threonine, l-phenlyalanine, and biotin (AAM−). The AAM+ and AAM− media were calibrated to pH 7.4 to prevent salt precipitation, and LB was prepared using standard-grade chemicals. Three strains, including two MRSA strains (S. aureus Mu50 and USA300) and a methicillin-sensitive patient isolate (from the University of Pittsburg Medical Center), were tested for growth in the minimal media under identical medium and growth conditions. These strains were inoculated into a sterile glass tube containing 3 ml LB and were grown at 37°C at 200 rpm for 16 h. For each strain, 900 μl of the overnight culture was pipetted into three separate 1.5-ml microcentrifuge tubes, centrifuged, and washed with LB, AAM+, or AAM− medium, respectively. The individual pellets were then resuspended in 200 μl of LB, AAM+, or AAM− medium, and 10 μl of each suspension was then inoculated into 2 ml of the corresponding medium for culture in a 24-well plate with uninoculated LB, AAM+, or AAM− medium as a control. The cultures were placed on a shaker rotating at 225 rpm at 37°C. Each experiment was done in triplicate and was repeated three times, and the growth was measured for 25 h at 600 nm using a microplate reader.
RESULTS
Analysis of metabolic reconstruction of S. aureus.
We initially studied the MRSA N315 strain as a model for reconstruction and FBA studies and then included all 12 of the other strains. From the ERGO genomic database, we identified 2,593 ORFs in the S. aureus N135 chromosome and 31 ORFs on its plasmid, of which 2,505 ORFs were assigned a functional role. A total of 906 ORFs were identified to have an enzymatic activity (i.e., they had EC numbers), and 668 ORFs had unique complete EC numbers (including two full EC numbers on the plasmid). We identified a list of 61 functions with incomplete or partial EC numbers. For all the complete and incomplete EC number annotations, the associated reactions were identified from the ERGO pathway collections and the KEGG pathway database. A total of 1,493 reactions were generated from the two databases (see Table S1 in the supplemental material for details), after reactions with substrates that were not likely to be metabolized by S. aureus and those that did not have a corresponding transport system were removed.
We compared this genome-scale reconstruction with two previous reconstructions of S. aureus N315 (4, 14). Becker and Palsson identified a total of 640 reactions from 619 genes, including 59 gap-filling reactions and 84 transport reactions (4). We found that 75 reactions in their reconstruction did not have a corresponding ORF in the S. aureus N315 genome, and several reactions were catalyzed strictly by eukaryotic enzymes. In a similar study by Heinemann et al. (14), only 774 reactions representing only 23% of the ORFs with 394 unique enzymes were considered. In contrast, we identified a total of 546 enzymes representing 1,497 reactions. The number of identified transport reactions in our study was 164 compared to 84 in the Becker and Palsson (4) and 59 in the Heinemann et al. (14) studies. We also found that 51 of the 59 transport reactions had an associated ORF, and 47 of them (92%) are also found in our reconstruction. The comparisons of our reconstruction with the two previous reconstructions are summarized in Table S1 in the supplemental material.
Using annotations based on protein similarities, comparative genomics, and pathways for all the strains, we obtained a comprehensive list of enzymes, reactions, and metabolites for all the S. aureus strains (Table 1). We found that over 90% of the enzymes, metabolic reactions, and metabolites were common to all strains, suggesting the presence of a common “core of metabolic reactions” among all the strains studied (2). It is not surprising to identify similar sets of single essential enzymes, especially with a higher degree of metabolic reconstruction similarities among strains. However, for biomass production, synthetic-lethality analyses showed greater variation in synthetic-lethal pairs of enzymes for lethality, particularly in S. aureus N315.
TABLE 1.
Comparative statistics for enzymes and metabolites that were used for the metabolic reconstructions of 13 S. aureus strainsa
| Parameter | No. for strain:
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Newman | TCH1516 | USA300 | Mu3 | JH9 | JH1 | COL | RF122 | Mu50 | MW2 | MSSA476 | MRSA252 | N315 | |
| ORFs | 2,641 | 2,683 | 2,604 | 2,698 | 2,726 | 2,681 | 2,618 | 2,515 | 2,714 | 2,632 | 2,597 | 2,655 | 2,593 |
| ORFs with functions | 1,915 | 1,896 | 2,002 | 2,031 | 2,087 | 2,096 | 1,953 | 1,915 | 2,091 | 2,021 | 1,975 | 2,024 | 2,031 |
| unique full EC no. | 650 | 646 | 671 | 663 | 670 | 663 | 666 | 666 | 667 | 665 | 671 | 675 | 668 |
| Dual-fusion enzymes | 51 | 49 | 50 | 51 | 51 | 51 | 50 | 50 | 50 | 50 | 49 | 49 | 51 |
| Triple-fusion enzymes | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 |
| Quadruple-fusion enzymes | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| Very likely reactions | 1,231 | 1,213 | 1,255 | 1,247 | 1,254 | 1,247 | 1,254 | 1,248 | 1,251 | 1,251 | 1,258 | 1,258 | 1,253 |
| Less likely reactions | 215 | 220 | 218 | 218 | 220 | 219 | 214 | 225 | 219 | 215 | 216 | 221 | 220 |
| Likely reactions | 20 | 19 | 20 | 20 | 20 | 19 | 20 | 16 | 20 | 20 | 20 | 20 | 20 |
| EC no. with reactions | 534 | 522 | 544 | 543 | 547 | 541 | 538 | 537 | 545 | 540 | 540 | 544 | 546 |
| Cellular reactions | 1,208 | 1,186 | 1,224 | 1,222 | 1,227 | 1,220 | 1,214 | 1,213 | 1,225 | 1,219 | 1,217 | 1,223 | 1,227 |
| Transport reactions | 141 | 146 | 148 | 144 | 146 | 145 | 150 | 151 | 145 | 147 | 152 | 152 | 146 |
| Exchange reactions | 117 | 120 | 121 | 119 | 121 | 120 | 124 | 125 | 120 | 120 | 125 | 125 | 120 |
| Metabolites | 1,412 | 1,399 | 1,428 | 1,422 | 1,431 | 1,422 | 1,425 | 1,414 | 1,427 | 1,421 | 1,432 | 1,437 | 1,431 |
The numbers of ORFs, functions, cellular reaction metabolites, and transport reactions identified for all the strains were used in the FBA and synthetic-lethal studies.
Subsystem analysis of S. aureus strains.
Previous whole-genome sequencing of S. aureus genomes had identified the majority of pathogenic and metabolic features. However, a comprehensive system-wide analysis of metabolism and nonmetabolic reaction networks is essential for both FBA and in silico gene deletion studies (1). Therefore, we have systematically examined and computed the metabolic capabilities of all S. aureus strains and used these results for FBA, including gene deletion and growth medium design studies. Select examples of these are presented below.
Most of the amino acid biosynthetic pathways have been identified in S. aureus N315, as in all the other strains. However, synthetic pathways for 4 amino acids, i.e., for l-histidine, l-serine, l-homocysteine, and l-asparagine, are absent in all 13 strains. Histidine biosynthesis in E. coli and B. subtilis is encoded by the hisGIAFHBCD operon. However, the ORF for the enzyme histidinol phosphatase, which converts l-histidinol phosphate to l-histidinol, is absent in all the S. aureus genomes. Instead, there are three ORFs (SA0513, SA0530, and phoB) that code for alkaline phosphatases, which can convert l-histidinol phosphate to l-histidinol, as in Saccharomyces cerevisiae (13).
Enzymes that convert l-homoserine to l-homocysteine (either by transulfurylation or by direct sulfurylation in the presence of succinyl-coenzyme A [CoA] or acetyl-CoA) are absent in all the S. aureus genomes. However, l-serine can be synthesized either by phosphorylation using 3-phosphoglycerate or by a nonphosphorylated pathway using 2-phosphoglycerate. An alanine biosynthetic pathway is present but is not required to be supplemented in the minimal growth medium. The l-asparagine pathway is absent in all the S. aureus genomes, along with the capability to convert l-aspartate to asparagine, using l-glutamine, by asparagine synthase or aspartate-ammonia ligase.
Genes for fatty acid biosynthesis, such as fabG, fabA, fabZ, fabI, fabK, fabH, and fabF, are uniformly present in all the S. aureus genomes. All except the gene for FabI (EC 1.3.1.9) were identified as essential. ORFs for the synthesis of phosphatidylcholine, phosphatidylethanolamine, or phosphatidylserine are absent, but pathways for the synthesis of phosphatidylglycerol and cardiolipin are present in all the S. aureus strains. None of the strains have ORFs for enzymes that modify fatty acids.
Pathways for the synthesis of several cofactors, such as biotin, FMN, riboflavin, FAD, folate, DHF, THF, and molybdenum, are absent in all the strains of S. aureus. Other cofactors, such as PQQ, ubiquinone, tocopherol, and plastoquinone synthesis, are absent as well.
Identification of single unconditionally essential enzymes and synthetic-lethal pairs.
In an effort to identify antibiotic drug targets, we performed in silico enzyme deletion studies for each S. aureus strain (2, 4, 8, 23, 29). With a metabolic reconstruction in hand, we built a flux balance-based model of S. aureus. The vulnerability of the reconstructed metabolic-network models can be utilized to derive the candidates for antibiotic targets. In the case of S. aureus N315, of the 1,497 identified metabolic reactions, only 23% were active, i.e., carrying nonzero fluxes (2). The FBA, as described in Materials and Methods, identified only 337 reactions that were active in the rich growth medium, in which all the included transport reactions can occur without limitation. For comparison, about 39% of the reactions were active in the reconstruction by Becker and Palsson (4). Figure 1a shows the metabolic network of active reactions for the N315 strain, in which a node is an enzyme for cellular, transport, or exchange reactions, and a link denotes a common metabolite that is processed by the reactions. Furthermore, to validate the identified potential antibiotic drug targets, we initially performed an in silico single-enzyme deletion study for growth in (simulated) rich medium for each S. aureus strain. In various natural environments, in which uptake reactions are limited, more enzymes are essential than in the rich medium. In contrast, the unconditionally essential enzymes are at the intersection of the sets of such essential enzymes in all possible environments. We found that 70 enzymes proved essential for growth in one or more of the 13 S. aureus strains, while 44 of these were essential in all the strains (Table 2; see Dataset 2 in the supplemental material). In strain N315, we identified 63 essential enzymes compared to the 135 single essential enzymes reported by Heinemann et al. (14). Thirty essential enzymes were common to both reconstructions, and 27 were strain-independent essential enzymes in our study. The larger number of single essential enzymes found in the FBA by Heinemann et al. (14) may be partially attributable to the smaller number of metabolic reactions identified in that study and to the directionality (irreversibility) of the reactions. Figure 1a shows the network structure of the common unconditionally essential enzymes. For example, two enzymes, EC 2.5.1.7 and EC 1.1.1.158, that are involved in the conversion of UDP-N-acetyl-d-glucosamine to UDP-N-acetylmuramate through UDP-N-acetyl-3-O-(1-carboxyvinyl)-d-glucosamine are predicted to be unconditionally essential. Deletion (inactivation) of either of these enzymes leads to failure in generating the biomass component UDP-N-acetylmuramate, since these two consecutive reactions constitute the only pathway generating UDP-N-acetylmuramate. It should be noted that the enzyme essentiality crucially depends on the selection of biomass components. For example, there is a pathway generating the “cross-linked peptidoglycan” in the study by Heinemann et al. (14), which starts from UDP-N-acetylmuramate and ends at the peptidoglycan. There are several enzymes acting on the pathway, which are all essential, since the pathway is the only one leading to the specific biomass compound.
FIG. 1.

Single essential and synthetic-lethal enzymes of the S. aureus metabolic network and their strain dependence. (a) Subnetworks of 44 strain-independent single essential enzymes of multiple S. aureus strains. Two nodes (enzymes) are connected if their reactions involve common metabolites. The EC number and the associated genes are presented for each node. The node colors correspond to the metabolic pathways the enzymes belong to. (b) Subset of the amino sugar pathway involving two single essential enzymes, EC 2.5.1.7 and EC1.1.1.158. Three pairs of enzymes, EC 2.3.1.157-EC 5.1.3.14, EC 2.7.7.23-EC 5.1.3.14, and EC 5.4.2.10-EC 5.1.3.14, are synthetic-lethal pairs in all strains. The enzymes in each pair individually do not affect growth (biomass production), but their simultaneous inactivation/deletion causes lethality. (c) Distribution of the numbers of strains in which an enzyme is singly essential. Altogether, 70 enzymes were identified as singly essential in at least one strain. Of these, more than 60% are found in all the S. aureus strains. (d) Distribution of the numbers of strains in which the deletion of a pair of enzymes is synthetic lethal. A total of 54 synthetic-lethal pairs were identified in at least one strain. About 40% of them are found only in one strain, reflecting the strain specificity of synthetic-lethal pairs, with less than 20% being present in all the strains.
TABLE 2.
Single essential enzymes in all strains of S. aureusa
| Pathway | EC no. | Gene | Enzyme name | Published experimental evidence |
|---|---|---|---|---|
| Amino sugar metabolism | 1.1.1.158 | murB | UDP-N-acetylmuramate dehydrogenase | Salmonella enterica serovar Typhimurium |
| 2.5.1.7 | murA | UDP-N-acetylglucosamine 1-carboxyvinyltransferase | Streptococcus pneumoniae, Mycobacterium tuberculosis H37Rv, S. aureus | |
| Fatty acid biosynthesis | 1.1.1.100 | fabG | 3-Oxoacyl-[acyl carrier protein] reductase | |
| 2.3.1.39 | fabD | [Acyl carrier protein] S-malonyltransferase | ||
| 2.3.1.41 | fabB/fabH | Beta-ketoacyl-acyl-carrier-protein synthase I | ||
| 3.1.2.14 | Oleoyl-[acyl carrier protein] hydrolase | |||
| 4.2.1.58 | fabZ | Crotonoyl-[acyl carrier protein] hydratase | ||
| 4.2.1.60 | fabA | 3-Hydroxydecanoyl-[acyl carrier protein] dehydratase | E. coli | |
| 4.2.1.61 | 3-Hydroxypalmitoyl-[acyl carrier protein] dehydratase | |||
| 6.4.1.2 | accA/accD | Acetyl-CoA carboxylase | B. subtilis, Haemophilus influenzae, S. pneumoniae, S. aureus | |
| Histidine metabolism | 1.1.1.23 | hisD | Histidinol dehydrogenase | M. tuberculosis H37Rv |
| 2.4.2.17 | hisG | ATP phosphoribosyltransferase | M. tuberculosis H37Rv, H. influenzae | |
| 3.1.3.15 | hisB | Histidinol phosphatase | ||
| 3.5.4.19 | hisI | Phosphoribosyl-AMP cyclohydrolase | M. tuberculosis H37Rv | |
| 3.6.1.31 | hisI | Phosphoribosyl-ATP diphosphatase | ||
| 4.2.1.19 | hisB | Imidazoleglycerol-phosphate dehydratase | M. tuberculosis H37Rv, H. influenzae | |
| 5.3.1.16 | hisA | 1-(5-Phosphoribosyl)-5-[(5-phosphoribosylamino)methylideneamino] imidazole-4-carboxamide isomerase | ||
| 2.6.1.9 | hisC | Histidinol-phosphate transaminase | ||
| Amino acid (phenylalanine, tyrosine and tryptophan) biosynthesis | 1.1.1.25 | aroE | Shikimate dehydrogenase | |
| 2.4.2.18 | trpD | Anthranilate phosphoribosyltransferase | M. tuberculosis H37Rv | |
| 2.5.1.19 | aroA | 3-Phosphoshikimate 1-carboxyvinyltransferase | ||
| 2.5.1.54 | aroP/aroH | 3-Deoxy-7-phosphoheptulonate synthase | ||
| 2.7.1.71 | aroL/aroK | Shikimate kinase | M. tuberculosis H37Rv | |
| 4.1.1.48 | trpC | Indole-3-glycerol-phosphate synthase | M. tuberculosis H37Rv | |
| 4.1.3.27 | trpE | Anthranilate synthase | M. tuberculosis H37Rv, H. influenzae | |
| 4.2.1.10 | aroQ | 3-Dehydroquinate dehydratase | M. tuberculosis H37Rv | |
| 4.2.1.20 | trpA/trpB | Tryptophan synthase | M. tuberculosis H37Rv, H. influenzae | |
| 4.2.1.51 | pheC | Prephenate dehydratase | H. influenzae | |
| 4.2.3.4 | aroB | 3-Dehydroquinate synthase | M. tuberculosis H37Rv, H. influenzae | |
| 4.2.3.5 | aroC | Chorismate synthase | M. tuberculosis H37Rv, H. influenzae, Helicobacter pylori | |
| 5.3.1.24 | trpF | Phosphoribosylanthranilate isomerase | ||
| 5.4.99.5 | aroQ | Chorismate mutase | H. influenzae | |
| Porphyrin metabolism | 2.1.1.107 | cysG | C-Methyltransferase, uroporphyrinogen-III | |
| 2.5.1.61 | hemC | Hydroxymethylbilane synthase | S. aureus | |
| 4.2.1.24 | hemB | Porphobilinogen synthase | ||
| 4.2.1.75 | hemD | Uroporphyrinogen-III synthase | ||
| 4.99.1.4 | cysG | Sirohydrochlorin ferrochelatase | ||
| Methionine metabolism | 2.5.1.6 | metK | Methionine adenosyltransferase | S. aureus |
| 3.2.2.9 | Adenosylhomocysteine nucleosidase | |||
| 4.4.1.21 | luxS | S-Ribosylhomocysteine lyase | ||
| Pentose phoshpate pathway | 2.2.1.1 | tktA | Transketolase | B. subtilis, S. aureus, H. influenzae, M. tuberculosis H37Rv |
| Multiple | 2.7.1.69 | yadI | Protein-N(pi)-phosphohistidine-sugar phosphotransferase | S. aureus |
| Unknown | 3.5.1.- | pcnA | Pyrazinamidase | M. tuberculosis H37Rv |
| 4.1.3.- | hisF | Imidazole glycerol phosphate synthase, cyclase subunit |
Forty-four enzymes functions were identified to be “singly essential” in all 13 strains of S. aureus.
While there is no comprehensive list of experimentally confirmed essential genes for any strain of S. aureus, we used the list of available ORFs that were identified to be essential for S. aureus strains RN450 and RN4220 (12, 16) (using the DEG database [http://tubic.tju.edu.cn/deg/]) to determine if our prediction matched any experimentally verified genes. Of the 44 common single essential enzymes that were identified from FBA, we found at least six enzymes, including transketolase (EC 2.2.1.1), hydroxyl-methylbilane synthase (EC 2.5.1.61), methionine adenosyltransferase (EC 2.5.1.6), UDP-N-acetylglucosamine 1-carboxyvinyltransferase (EC 2.5.1.7), protein N (pi)-phospho-histidine-sugar phosphortransferase (EC 2.7.1.69), and acetyl-CoA carboxylase (EC 6.4.1.2), to be experimentally verified in S. aureus (12). In addition, orthologs for 17 enzymes were found to be experimentally determined to be essential in other bacteria (Table 2).
Many enzymes were predicted to be dispensable for growth, i.e., biomass could be generated in the presence of alternate biosynthetic routes, along with their corresponding ORFs. We hypothesized that if pairs of enzymes independently play a seminal role in metabolism, then deleting both enzymes could lead to lethality (6, 21). Such pairs were identifiable by monitoring the biomass production reactions on inactivation of two enzymes simultaneously in the FBA computations. We identified 54 synthetic-lethal pairs of enzymes in all 13 strains whose deletions could completely abolish biomass generation (see Table S3 in the supplemental material for details). Interestingly, only 10 of these 54 synthetic-lethal pairs were common to all 13 strains (Table 3). Six lethal pairs belong to amino sugars/peptidoglycan pathways that can be grouped under the cell wall metabolic subsystem. Two lethal pairs belong to the amino acid subsystem, and a single pair of enzymes belongs to amino acid and 1-carbon metabolic pathways. Only one pair of enzymes is grouped in the nucleotide and carbohydrate subsystems. An example of a common lethal pair is shown in amino sugar metabolism (Fig. 1b). In S. aureus, there are two pathways that can generate UDP-N-acetylglucosamine, the precursor of UDP-N-acetylmuramate. One is from d-glucosamine-1P through N-acetyl-d-glucosamine-1P, and the other is from N-acetyl-d-mannosamine. The first pathway consists of three enzymatic reactions catalyzed by EC 5.4.2.10, EC 2.3.1.157, and EC 2.7.7.23, and the second has EC 5.1.3.14. Therefore, we found three synthetic-lethal pairs (EC 5.3.1.14-EC 5.4.2.10, EC 5.1.3.14-EC 2.3.1.157, and EC 5.1.3.14-EC 2.7.7.23). The subset of the amino sugar biosynthetic pathway involving these enzymes is shown in Fig. 1b.
TABLE 3.
Common synthetic-lethal pairs of enzymes required for growth identified in all 13 S. aureus strainsa
| Sl | Enzyme pair | Gene | Reaction | Pathway | Subsystem |
|---|---|---|---|---|---|
| 1 | Prephenate dehydrogenase EC 1.3.1.12 | tyrA | Prephenate + NAD+ ↔ 3-(4 hydroxyphenyl) pyruvate + CO2 + NADH + H+ | Tyrosine biosynthesis | Amino acid biosynthesis |
| Arogenate dehydrogenase EC 1.3.1.43 | adH | l-Arogenate + NAD+ ↔ l-tyrosine + CO2 + NADH | Tyrosine biosynthesis via arogenate | Amino acid biosynthesis | |
| 2 | Methylenetetrahydrofolate reductase EC1.5.1.20 | mthfR | 5-Methyltetrahydrofolate + NADP+ ↔ 5,10-methylenetetrahydrofolate + NADPH + H+ | Tetrahydrofolate synthesis | One-carbon metabolism |
| Cystathionine gamma-synthase EC 4.4.1.8 | metF | l-Cysteine + H2O ↔ hydrogen sulfide + pyruvate + NH3 | Cysteine metabolism | Amino acid biosynthesis | |
| 3 | 5-Methyltetrahydrofolate-homocysteine methyl-transferase EC 2.1.1.13 | metH | 5-Methyltetrahydrofolate + l-homocysteine ↔ tetrahydrofolate + l-methionine | l-Methionine biosynthesis | Amino acid biosynthesis |
| Cystathionine gamma-synthase EC 4.4.1.8 | metF | l-Cysteine + H2O ↔ hydrogen sulfide + pyruvate + NH3 | Cysteine metabolism | Amino acid biosynthesis | |
| 4 | UDP-N-acetylglucosamine pyrophosphorylase EC 2.3.1.157 | gcaD/ | Acetyl-CoA + d-glucosamine 1-phosphate ↔ CoA + N-acetyl-d-glucosamine 1-phosphate | Peptidoglycan and lipopolysaccharide biosynthesis | Cell wall metabolism |
| UDP-N-acetylglucosamine pyrophosphorylase EC 2.7.7.23 | glmU | UTP + N-acetyl-d-glucosamine 1-phosphate ↔ pyrophosphate + UDP-N-acetyl-d-glucosamine | Amino sugar biosynthesis | Cell wall metabolism | |
| UTP + N-acetyl-alpha-d-glucosamine 1-phosphate ↔ pyrophosphate + UDP-N-acetyl-d-glucosamine | |||||
| 5 | UDP-N-acetylglucosamine pyrophosphorylase EC 2.3.1.157 | gcaD/ | Acetyl-CoA + d-glucosamine 1-phosphate ↔ CoA + N-acetyl-d-glucosamine 1-phosphate | Peptidoglycan and lipopolysaccharide biosynthesis | Cell wall metabolism |
| UDP-N-acetylglucosemine 2-epimerase EC 5.1.3.14 | rffE | UDP-N-acetyl-d-glucosamine ↔ UDP-N-acetyl-d-mannosamine | Amino sugar biosynthesis | Cell wall metabolism | |
| UDP-N-acetyl-d-glucosamine + H2O ↔ N-acetyl-d-mannosamine + UDP | |||||
| 6 | UDP-N-acetylglucosamine pyrophosphorylase EC 2.3.1.157 | gcaD | Acetyl-CoA + d-glucosamine 1-phosphate ↔ CoA + N-acetyl-d-glucosamine 1-phosphate | Peptidoglycan and lipopolysaccharide biosynthesis | Cell wall metabolism |
| Phosphoglucosamine mutase EC 5.4.2.10 | glmM | d-Glucosamine 1-phosphate ↔ d-glucosamine 6-phosphate | Amino sugar biosynthesis | Cell wall metabolism | |
| 7 | UDP-N-acetylglucosamine pyrophosphorylase EC 2.7.7.23 | glmU | UTP + N-acetyl-d-glucosamine 1-phosphate ↔ pyrophosphate + UDP-N-acetyl-d-glucosamine | Amino sugar biosynthesis | Cell wall metabolism |
| UTP + N-acetyl-alpha-d-glucosamine 1-phosphate ↔ pyrophosphate + UDP-N-acetyl-d-glucosamine | |||||
| UDP-N-acetylglucosemine 2-epimerase EC 5.1.3.14 | rffE | UDP-N-acetyl-d-glucosamine ↔ UDP-N-acetyl-d-mannosamine | Amino sugar biosynthesis | Cell wall metabolism | |
| UDP-N-acetyl-d-glucosamine + H2O ↔ N-acetyl-d-mannosamine + UDP | |||||
| 8 | UDP-N-acetylglucosamine pyrophosphorylase EC 2.7.7.23 | glmU | UTP + N-acetyl-d-glucosamine 1-phosphate ↔ pyrophosphate + UDP-N-acetyl-d-glucosamine | Amino sugar biosynthesis | Cell wall metabolism |
| UTP + N-acetyl-alpha-d-glucosamine 1-phosphate ↔ pyrophosphate + UDP-N-acetyl-d-glucosamine | |||||
| Phosphoglucosamine mutase EC 5.4.2.10 | glmM | d-Glucosamine 1-phosphate ↔ d-glucosamine 6-phosphate | Amino sugar biosynthesis | Cell wall metabolism | |
| 9 | UDP-N-acetylglucos-amine 2-epimerase EC 5.1.3.14 | rffE | UDP-N-acetyl-d-glucosamine ↔ UDP-N-acetyl-d-mannosamine | Amino sugar biosynthesis | Cell wall metabolism |
| UDP-N-acetyl-d-glucosamine + H2O ↔ N-acetyl-d-mannosamine + UDP | |||||
| Phosphoglucosamine mutase EC 5.4.2.10 | glmM | d-Glucosamine 1-phosphate ↔ d-glucosamine 6-phosphate | Amino sugar biosynthesis | Cell wall metabolism | |
| 10 | Carbamoyl-phosphate synthase EC 6.3.5.5 | carA | 2 ATP + l-glutamine + HCO3- + H2O ↔ 2 ADP + orthophosphate + l-glutamate + carbamoyl phosphate | Pyrimidine and glutamate metabolism | Nucleotide amino acid metabolism |
| Pyruvate carboxylase EC 6.4.1.1 | pycA/pycB | ATP + pyruvate + HCO3− ↔ ADP + orthophosphate + oxaloacetate | Gluconeogenesis | Carbohydrate metabolism |
When deleted individually these enzymes do not affect growth (biomass formation); however, simultaneous inactivation of both enzyme activities in each synthetic-lethal pair is predicted to inhibit growth.
The number of single essential enzymes varies by strain. As seen in Fig. 1c, more than 90% of the identified single essential enzymes are essential in nine strains. The strain dependence of the single essential enzymes is, however, stronger than that of the metabolic reconstructions, as 63% of the single essential enzymes are found in all the strains, while more than 90% of the metabolic reconstructions overlap. Synthetic-lethal pairs exhibit even stronger strain dependence (Fig. 1d); less than 20% are found in all the strains, and 40% are found in only one specific strain. The same analyses have been performed for the single and synthetic-lethal enzymes derived from the FBA with the biomass composition in reference 14, and the results are detailed in Tables S2 and S3 in the supplemental material. In spite of the use of different biomass compositions, the single and synthetic-lethal enzymes obtained exhibited characteristics in their strain dependence similar to those mentioned above.
Analysis of the components of minimal medium for S. aureus.
The minimal medium for the growth of the laboratory strain S. aureus NCTC 8385, requiring a combination of salts, carbon source, amino acids, and vitamins, was deduced by trial and error in the early 1970s (28). However, with the availability of complete genome sequences, annotations, pathway analyses, and metabolic reconstructions, one can rationally design an optimum growth medium (4, 5). We compared the published minimal-medium components (27) with the reconstructions and calculated that six components could be synthesized by S. aureus, and therefore, these components are not required for growth of S. aureus (Table 4). Specifically, for several salt components of the medium, a dedicated transport system(s) has been identified. For amino acids, such as l-arginine, l-glutamic acid, l-valine, l-threonine, and l-phenylalanine, both biosynthetic pathways and transport systems are present in all genomes. We also identified the ORFs coding for synthesizing and transporting enzymes for cofactors, such as biotin and pantothenate. Interestingly, the ORF for pantothenate uptake (Na/pantothenate symporter) is not found in any of the S. aureus genomes (Table 4). We conclude that S. aureus can synthesize biotin and does not require its exogenous addition to the medium.
TABLE 4.
Analysis of minimal growth medium (28) for drug-resistant S. aureus
| Sl | Ingredient | Amt/liter | Reconstruction-based analysis | Comments |
|---|---|---|---|---|
| 1 | KCl | 3.0 g | Potassium uptake sodium symport ktrAB | K is transported by a dedicated transport system |
| 2 | NaCl | 9.5 g | Potassium uptake sodium symport ktrAB | At least three symport systems are identified |
| N-Acetylneuraminate uptake sodium symport | ||||
| l-Proline uptake sodium symport putP | ||||
| 3 | MgSO4·7H2O | 1.3 g | Mg phosphate uptake proton symport pit | At least three symport systems are identified |
| Magnesium uptake protein mgtE | ||||
| Magnesium uptake via corA | ||||
| 4 | (NH4)2 SO4 | 4.0 g | Ammonium uptake uniport via amtB | One dedicated uniport system has been identified |
| 5 | CaCl2·2H2O | 22 mg | Ca phosphate uptake proton symport pit | One dedicated uptake system has been identified |
| 6 | KH2PO4 | 140 mg | Potassium uptake sodium symport ktrAB | K is transported by a dedicated transport system |
| FeSO4·7H2O | 6 mg | Ferrous iron uptake feoB Ferrous iron uptake FTR1 family | One dedicated and a generic “family” transport system have been identified | |
| 7 | MnSO4·H2O | 10 mg | Mn phosphate uptake proton symport pit | One dedicated uptake system has been identified |
| 8 | Citiric acid | 6 mg | Taken in by citrate-Fe3 dicitrate ABC transport system; transports iron, as well | One dedicated uptake system has been identified |
| 9 | Tris | 12.1 g | Buffer | |
| 10 | Glucose | 5 g | Glucose uptake glcBC PTS forming glucose-6-phosphate | At least three uptake systems are identified |
| d-Glucose uptake glucose transporter glcU | ||||
| Glucose uptake glcABC PTS forming glucose-6-phosphate | ||||
| 11 | l-Arginine | 125 mg | 3 enzymes in the synthesis of l-arginine from carobmyl phosphate and ornithine are present (EC 2.1.3.3, EC 6.3.4.5, EC 4.3.2.1). Ornithine is made from 5-glutmate semialdehyde and 1-pyrroline 5-carboxylate from proline | Arginine need not be added in the medium, as the biosynthetic pathway is present |
| Arginine uptake proton symport Arg permease | Two uptake systems are identified; one is a permease, and the second is an ABC transporter | |||
| l-Arginine uptake arginine ABC transporter | ||||
| 12 | l-Proline | 200 mg | The organism is missing two enzymes (EC 2.7.2.11 and EC 1.2.1.41) involved in l-proline synthesis from l-glutamate. However, it possesses a bypass producing l-proline from l-ornithine using two other enzymes (EC 2.6.1.13 and EC 1.5.1.2) | Proline is to be added in the medium. In addition, two protein symport systems, proP and putP, have been identified |
| l-proline uptake proton symport proP | ||||
| l-proline uptake sodium symport putP | ||||
| 13 | l-Glutamic acid | 250 mg | 3 enzymes producing l-glutamate from 2-oxoglutarate are present | Glutamic acid need not be added in the medium, as the biosynthetic pathway is present |
| l-glutamate uptake sodium symport gltS | Two uptake systems using Na+ and proton symport have been identified | |||
| l-glutamate uptake proton sodium symport gltT | ||||
| 14 | l-Valine | 150 mg | Synthetic pathway present | Valine need not be added in the medium, as the biosynthetic pathway is present |
| l-Valine uptake proton symport (Leu-Ile-Val) transporter | A dedicated transporter has been identified | |||
| 15 | l-Threonine | 150 mg | Synthetic pathway present | Threonine need not be added in the medium, as the biosynthetic pathway is present |
| l-Threonine export proton antiport Thr-Ser exporter | A dedicated transporter has been identified | |||
| 16 | l-Phenylalanine | 150 mg | Synthetic pathway present | Phenlyalanine need not be added in the medium, as the biosynthetic pathway is present |
| No uptake transporter present | A dedicated phenylalanine transporter has been not identified | |||
| 17 | l-Leucine | 150 mg | The organism is missing an ORF for leucine transaminase (EC 2.6.1.6) and leucine dehydrogenase (EC 1.4.1.9). However, it is able to synthesize l-leucine due to the presence of an ORF for branched-chain amino acid transaminase (EC 2.6.1.42) | One specific and one general leucine transporter have been identified |
| l-Isoleucine export proton antiport AzlCD | ||||
| l-Leucine uptake proton symport (Leu-Ile-Val) transporter | ||||
| 18 | l-Cysteine | 80 mg | Two enzymes (EC 2.3.1.30 and EC 2.5.1.47) involved in the l-cysteine biosynthesis pathway from l-serine are present. However, the l-serine pathway is broken, so l-cysteine will be synthesized in the presence of exogenous l-serine. In addition, the l-cysteine biosynthesis pathway from l-homocysteine is also broken due to the absence of cystatione beta-synthase (EC 4.2.1.22). Cysteine uptake by ABC transporter present | One ABC transporter system has been identified |
| 19 | Biotin | 0.1 mg | Biotin synthetic pathway present, primarily from pimelate. Pimelate is converted to pimeloyl CoA by 6.2.1.14. In the presence of alanine and pimeloyl-CoA, biotin is made with the four enzymes (EC 2.3.1 0.47, EC 2.6.1.62, EC 6.3.3.3, and EC 2.8.16) that are present in the N315 genome | Biotin need not be added in the medium, as the biosynthetic pathway is present. However, ORFs for biotin accessory proteins, such as BioH, BioC, and BioY, are absent |
| 20 | Thiamine | 2 mg | Four enzymes out of six required for de novo thiamine biosynthesis are absent. Therefore, thiamine should be added in the medium for growth | |
| 21 | Nicotinic acid | 2 mg | De novo synthesis from aspartate is absent; however, salvage reactions from NAD or nicotinic acid are complete. Salvage pathway enzymes that convert nicotinamide to nicotinase (EC. 3.5.1.19) and nicotinase to β-nicotinate d-ribonucletoide (EC. 2.4.2.11) are present. Further conversion to amido NAD (by EC 2.7.7.18) and to NAD by DNA synthase (EC. 6.3.5.1) is present | Addition of either NAD or nicotinic acid is essential for growth on minimal medium |
| 22 | Calcium pantothenate | 2 mg | d-Pantothenate can be synthesized from keto-alaine and THF with the use of enzymes (EC.2.1.2.11, EC.1.1.1.69 and EC 6.3.2.1). The genes for these enzymes are present, as well | Exogenously provided pantothenate may not be taken in due to the absence of Na/pantothenate symporter |
To experimentally confirm the minimal-medium prediction, we compared the growth of three different S. aureus strains. These included 2 of the 13 reconstructed S. aureus strains (Mu50 and USA300), as well as a patient S. aureus isolate. We tested the growth abilities of these strains in rich LB medium (control), in the standard minimal medium (AAM+) (27), and in minimal medium (AAM−) predicted from this study. The growth patterns of all three strains were the highest in LB medium compared to growth in either minimal medium and followed a typical sigmoid growth curve (Fig. 2). However, the growth rates in the predicted minimal growth medium (AAM−) without the addition of seven compounds were similar to those in the standard minimal medium (AAM+). These results thus support the minimal-medium predictions, and hence, the validity of our metabolic-pathway reconstructions.
FIG. 2.

Experimental validation of S.aureus growth in metabolic-reconstruction-based minimal medium. Shown are the growth curves of S. aureus Mu50 (a), S. aureus USA300 (b), and an S. aureus patient isolate (c). AAM− is a modified minimal medium derived from metabolic reconstruction; AAM+ is a published minimal medium (27).
DISCUSSION
The availability of whole-genome sequences not only provides the possibility of identifying gene variability and pathogenic features, but is also valuable for identifying in silico metabolic networks, FBA, etc. Although several virulence and pathogenic genes in S. aureus have been identified, they tend not to be feasible drug target candidates, as they are usually strain or species specific or prophage borne. On the other hand, metabolic enzymes tend to be common to all strains and offer broad-spectrum target candidates irrespective of the bacteria or their host specificities.
In this study, we performed metabolic-network reconstructions of fully sequenced S. aureus strains via protein similarity calculation, automated annotation, and manual curation. Compared to the previous reconstructions made for a single strain of S. aureus (N315), our reconstructions contain a larger data set of both biochemical reactions and transport compounds. Moreover, based on the functional annotation of several S. aureus strains, we completed strain-dependent metabolic reconstructions, which help us to understand the wide range of virulence and drug resistance among the S. aureus strains. As the first step, we carried out FBA on the metabolic reconstructions and derived unconditionally essential single enzymes and pairs of enzymes in each strain.
The variation of the reconstructed metabolic networks among the strains does not appear to be great, i.e., over 90% of the metabolic enzymes and reactions are similar in the 13 strains. This was expected, owing to the clonality of the S. aureus genomes, with more than 80% overall DNA sequence similarity (3, 11). However, in order to describe the metabolic diversity of various drug-resistant phenotypes of S. aureus, we performed the FBA for these metabolic networks. In particular, we identified unconditionally essential metabolic enzymes by in silico enzyme deletion studies, which in turn represent potential antimicrobial drug targets. These essential enzymes partly overlap among all the strains to about 60% for single essentiality and about 20% for synthetic lethality, suggesting strong strain specificity of the functional states of the metabolic networks. We therefore conclude that development of antimicrobial drugs should take into consideration the strain dependency of the vulnerability of metabolic networks in S. aureus. We presented a list of the single essential and synthetic-lethal enzymes for each drug-resistant S. aureus strain. Previously published experimental results show that more than half of the single essential enzymes were identified experimentally in various bacteria, including S. aureus.
Two interesting findings of this study are the identification of new single essential enzymes and paired synthetic-lethal enzymes as antibiotic targets common to all the strains of S. aureus. A surprising finding is that a majority (six pairs) of synthetic-lethal pairs of enzymes are in the amino sugar biosynthesis pathways, which are involved in cell wall metabolism. Three lethal enzyme pairs were identified in amino acid metabolic pathways. Among the single essential enzymes, eight were identified as playing roles in fatty acid biosynthesis.
It should be noted that the metabolic reconstructions performed in this work are not complete. First, incorrect ORF calling led to inevitable errors in the reconstructions for several reasons (10). Second, in contrast to well-studied model organisms, such as E. coli and B. subtilis, there are limited biochemical and physiological data for S. aureus, which hinders testing and improving the metabolic networks reconstructed in this study in spite of literature-based curation (10). For instance, protein complexes and isoenzymes in the metabolic network of S. aureus have not been identified as extensively as in E. coli (9). Given this limitation, we assumed that each of the genes associated with a reaction was needed to catalyze the reaction, and this might have created false positives in our essentiality predictions. Moreover, the biomass components of S. aureus have not yet been fully described, forcing us to use those of the related gram-positive bacterium B. subtilis or those derived from literature searches. Future biochemical experiments on S. aureus will improve and confirm the results obtained in this study. It would also be informative to perform experimental gene deletion studies to test our findings.
Supplementary Material
Acknowledgments
Support for this work was provided by NIH grant 5U01AI070499-02.
Footnotes
Published ahead of print on 17 April 2009.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Almaas, E., B. Kovacs, T. Vicsek, Z. N. Oltvai, and A.-L. Barabási. 2004. Global organization of metabolic fluxes in the bacterium Escherichia coli. Nature 427839-843. [DOI] [PubMed] [Google Scholar]
- 2.Almaas, E., Z. N. Oltvai, and A.-L. Barabási. 2005. The activity reaction core and plasticity of metabolic networks. PLoS Comp. Biol. 10557-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baba, T., T. Bae, O. Schneewind, F. Takeuchi, and K. Hiramatsu. 2008. Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of staphylococcal genomes: polymorphism and evolution of two major pathogenicity islands. J. Bacteriol. 190300-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Becker, S. A., and B. O. Palsson. 2005. Genome-scale reconstruction of the metabolic network in Staphylococcus aureus N315: an initial draft to the two dimensional annotation. BMC Microbiol. 58-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhattacharyya, A., S. Stilwagen, G. Reznik, H. Feil, W. Feil, I. Anderson, A. Bernal, M. D' Souza, N. Ivanova, V. Kapatral, N. Larsen, T. Los, A. Lykidis, E. J. Selkov, T. L. Walunas, A. Purcell, R. A. Edwards, T. Hawkins, R. Haselkorn, R. Overbeek, N. C. Kyrpides, and P. Predki. 2002. Draft sequencing and comparative genomics of Xylella fastidiosa subspecies reveal novel biological insights. Genome Res. 121556-1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chait, R., A. Craney, and R. Kishnoy. 2007. Antibiotic interactions that select against resistance. Nature 446668-671. [DOI] [PubMed] [Google Scholar]
- 7.Cheung, A. L., S. J. Projan, and H. Gresham. 2002. The genomic aspect of virulence, sepsis and resistance to killing mechanisms in Staphylococcus aureus. Curr. Infect. Dis. Rep. 4400-410. [DOI] [PubMed] [Google Scholar]
- 8.Deutscher, D., I. Meilijson, M. Kupiec, and E. Ruppin. 2006. Multiple knockout analysis of genetic robustness in the yeast metabolic network. Nat. Genet. 38993-998. [DOI] [PubMed] [Google Scholar]
- 9.Feist, A. M., C. S. Henry, J. L. Reed, M. Krummenacker, A. R. Joyce, P. D. Karp, L. J. Broadbelt, V. Hatzimanikatis, and B. O. Palsson. 2007. A genome-scale metabolic reconstruction for Escherichia coli K-12 MG1655 that accounts for 1260 ORFs and thermodynamic information. Mol. Syst. Biol. 3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feist, A. M., M. J. Herrgard, I. Thiele, J. L. Reed, and P. B. Ø. Palsson. 2009. Reconstruction of biochemical networks in microorganisms. Nat. Rev. Microbiol. 7129-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng, Y., C. J. Chen, L. H. Su, S. Hu, J. Yu, and C. H. Chiu. 2008. Evolution and pathogenesis of Staphylococcus aureus: lessons learned from genotyping and comparative genomics. FEMS. Microbiol. Rev. 3223-37. [DOI] [PubMed] [Google Scholar]
- 12.Forsyth, R. A., R. J. Haselbeck, K. L. Ohlsen, R. T. Yamamoto, H. Xu, J. D. Trawick, D. Wall, L. Wang, V. Brown-Driver, J. M. Froelich, G. C. Kedar, P. King, M. McCarthy, C. Malone, B. Misiner, D. Robbins, Z. Tan, Z.-Y. Zhu, G. Carr, D. A. Mosca, C. Zamudio, J. G. Foulkes, and J. W. Zyskind. 2002. A genome-wide strategy for the identification of essential genes in Staphylococcus aureus. Mol. Microbiol. 431387-1400. [DOI] [PubMed] [Google Scholar]
- 13.Gormian, J. A., and A. S. L. Hu. 1969. The separation and partial characterization of l-histinol phosphatase and alkaline phosphatase of Saccharomyces cerevisiae. J. Biol. Chem. 2441645-1650. [PubMed] [Google Scholar]
- 14.Heinemann, M., A. Kümmel, R. Ruinatscha, and S. Panke. 2005. In silico genome-scale reconstruction and validation of the Staphylococcus aureus metabolic network. Biotechnol. Bioeng. 92850-864. [DOI] [PubMed] [Google Scholar]
- 15.Herron, L. L., R. Chakravarty, C. Dwan, J. R. Fitzgerald, J. M. Musser, E. Retzel, and V. Kapur. 2002. Genome sequence survey identifies unique sequences and key virulence genes with unusual rates of amino-acid substitution in bovine Staphylococcus aureus. Infect. Immun. 703978-3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ji, Y., B. Zhang, S. F. V. Horn, P. Warren, G. Woodnutt, M. K. Burnham, and M. Rosenberg. 2001. Identification of critical staphylococcal genes using conditional phenotypes generated by antisense RNA. Science 2932266-2269. [DOI] [PubMed] [Google Scholar]
- 17.Kanehisa, M., S. Goto, M. Hattori, K. F. Aoki-Kinoshita, M. Itoh, S. Kawashima, T. Katayama, M. Araki, and M. Hirakawa. 2006. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 34354-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kapatral, V., I. Anderson, N. Ivanova, G. Reznik, T. Los, A. Lykidis, A. Bhattacharyya, A. Bartman, W. Gardner, G. Grechkin, L. Zhu, O. Vasieva, L. Chu, Y. Kogan, O. Chaga, E. Goltsman, A. Bernal, N. Larsen, M. D'Souza, T. Walunas, G. Pusch, R. Haselkorn, M. Fonstein, N. Kyrpides, and R. Overbeek. 2002. Genome sequence and analysis of the oral bacterium Fusobacterium nucleatum strain ATCC 25586. J. Bacteriol. 1842005-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klevens, R. M., M. A. Morrison, J. Nadle, S. Petit, K. Gershman, S. Ray, L. H. Harrison, R. Lynfield, G. Dumyati, J. M. Townes, A. S. Craig, E. R. Zell, G. E. Fosheim, L. K. McDougal, R. B. Carey, and S. K. Fridkin. 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 2981763-1771. [DOI] [PubMed] [Google Scholar]
- 20.Kuroda, M., T. Ohta, I. Uchiyama, T. Baba, H. Yuzawa, I. Kobayashi, L. Cui, A. Oguchi, K. Aoki, Y. Nagai, J. Lian, T. Ito, M. Kanamori, H. Matsumaru, A. Maruyama, H. Murakami, A. Hosoyama, Y. Mizutani-Ui, N. Kobayashi, T. Tanaka, T. Sawano, R. Inoue, C. Kaito, K. Sekimizu, H. Hirakawa, S. Kuhara, S. Goto, J. Yabuzaki, M. Kanehisa, A. Yamashita, K. Oshima, K. Furuya, C. Yoshino, T. Shiba, M. Hattori, N. Ogasawara, H. Hayashi, and K. Hiramatsu. 2001. Whole genome sequencing of methicillin-resistant Staphylococcus aureus. Lancet 3571225-1240. [DOI] [PubMed] [Google Scholar]
- 21.Lehar, J., B. R. Stockwell, G. Giaever, and C. Nislow. 2008. Combination chemical genetics. Nat. Chem. Biol. 4674-681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lowy, F. 1998. Staphylococcus aureus infections. N. Engl. J. Med. 339520-532. [DOI] [PubMed] [Google Scholar]
- 23.Motter, A. E., N. Gulbahce, E. Almaas, and A.-L. Barabasi. 2008. Predicting synthetic rescues in metabolic networks. Mol. Syst. Biol. 4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Overbeek, R., N. Larsen, T. Walunas, M. D'Souza, G. Pusch, E. J. Selkov, K. Liolios, V. Joukov, D. Kaznadzey, I. Anderson, A. Bhattacharyya, H. Burd, W. Gardner, P. Hanke, V. Kapatral, N. Mikhailova, O. Vasieva, A. Osterman, V. Vonstein, M. Fonstein, N. Ivanova, and N. Kyrpides. 2003. The ERGO genome analysis and discovery system. Nucleic Acids Res. 31164-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Palsson, B. O. 2006. Systems biology: properties of reconstructed networks. Cambridge University Press, New York, NY.
- 26.Price, N. D., J. L. Reed, and B. O. Palsson. 2004. Genome-scale models of microbial cells: evaluating the consequences of constraints. Nat. Rev. Microbiol. 2886. [DOI] [PubMed] [Google Scholar]
- 27.Projan, S. I., and R. P. Novick. 1997. The molecular basis of pathogenicity. In K. B. Crossley and G. L. Archer (ed.), The staphylococci in human diseases. Churchill Livingstone, New York, NY.
- 28.Rudin, L., J. E. Sjöström, M. Lindberg, and L. Philipson. 1974. Factors affecting competence for transformation in Staphylococcus aureus. J. Bacteriol. 118155-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thiele, I., T. D. Vo, N. D. Price, and B. Ø. Palsson. 2005. An expanded metabolic reconstruction of Helicobacter pylori (iIT341 GSM/GPR): An in silico genome-scale characterization of single and double deletion mutants. J. Bacteriol. 1875818-5830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang, J., S. Kodali, S. H. Lee, A. Galgoci, R. Painter, K. Dorso, F. Racine, M. Motyl, L. Hernandez, E. Tinney, S. L. Colletti, K. Herath, R. Cummings, O. Salazar, I. González, A. Basilio, F. Vicente, O. Genilloud, F. Pelaez, H. Jayasuriya, K. Young, D. F. Cully, and S. B. Singh. 2007. Discovery of platencin, a dual FabF and FabH inhibitor with in vivo antibiotic properties. Proc. Natl. Acad. Sci. USA 1047612-7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.