

Abstract
Summary: The viral infectivity factor (Vif) is dispensable for human immunodeficiency virus type 1 (HIV-1) replication in so-called permissive cells but is required for replication in nonpermissive cell lines and for pathogenesis. Virions produced in the absence of Vif have an aberrant morphology and an unstable core and are unable to complete reverse transcription. Recent studies demonstrated that human APOBEC-3G (hA3G) and APOBEC-3F (hA3F), which are selectively expressed in nonpermissive cells, possess strong anti-HIV-1 activity and are sufficient to confer a nonpermissive phenotype. Vif induces the degradation of hA3G and hA3F, suggesting that its main function is to counteract these cellular factors. Most studies focused on the hypermutation induced by the cytidine deaminase activity of hA3G and hA3F and on their Vif-induced degradation by the proteasome. However, recent studies suggested that several mechanisms are involved both in the antiviral activity of hA3G and hA3F and in the way Vif counteracts these antiviral factors. Attempts to reconcile the studies involving Vif in virus assembly and stability with these recent findings suggest that hA3G and hA3F partially exert their antiviral activity independently of their catalytic activity by destabilizing the viral core and the reverse transcription complex, possibly by interfering with the assembly and/or maturation of the viral particles. Vif could then counteract hA3G and hA3F by excluding them from the viral assembly intermediates through competition for the viral genomic RNA, by regulating the proteolytic processing of Pr55Gag, by enhancing the efficiency of the reverse transcription process, and by inhibiting the enzymatic activities of hA3G and hA3F.
INTRODUCTION
As a complex retrovirus, human immunodeficiency virus type 1 (HIV-1) encodes several proteins that enhance infectivity. An intriguing feature of these so-called auxiliary proteins is that they present distinct activities at seemingly unrelated steps of viral replication (148). For instance, Vpu is responsible for CD4 receptor degradation, induction of apoptosis, and enhancement of viral particle release (21, 166), and recent work suggested that it could also counteract a cellular restriction factor (233). Nef is involved in T-cell receptor activation, cell surface CD4 endocytosis, and apoptosis, and it has been proposed to act as an auxiliary factor of HIV-1 reverse transcription (61). Vpr contributes to the nuclear import of the preintegration complex (131) and is responsible for cell cycle arrest (169). As we will describe in this review, Vif makes no exception to this list, as the inactivation of the vif gene results in defects in both early and late steps of the HIV-1 life cycle. The accumulation of several activities in a single small protein is the combined result of multiple selection pressures on viral replication, leading to viral hijacking of diverse cellular functions and physical constraints on the retroviral genome length. Given the modest size of auxiliary proteins, domains that are responsible for interactions with diverse partners are thought to overlap and to be exposed by conformational switches at different steps of the viral cycle.
Virion infectivity factor (Vif) is a small, highly basic protein encoded by all lentiviruses except equine infectious anemia virus. Early studies showed that the requirement for the vif gene in HIV-1 replication is cell type dependent (66, 67, 269) and that this gene is essential for in vivo infectivity and pathogenesis. Indeed, HIV-1 lacking vif (HIV-1 Δvif) can replicate in permissive cell lines but not in nonpermissive cells, including macrophages and monocytes, which are the main targets of HIV-1 infection in vivo. Early studies revealed the aberrant morphology and reduced core stability of Δvif HIV-1 particles and suggested that Vif plays a role in virion assembly (20, 94, 234). These defects of Δvif virions are correlated with defects in proviral DNA synthesis (76, 163, 219, 234).
Evaluation of the infectivity phenotype of HIV-1 Δvif produced from transient heterokaryons formed between permissive and nonpermissive cells indicated the presence of antiviral activity in the nonpermissive producer cells (145, 213). However, the function(s) of Vif in HIV-1 replication remained elusive until Sheehy and coworkers identified this restriction factor as CEM-15, now usually known as APOBEC-3G (human APOBEC-3G [hA3G]) (203). A second antiviral factor belonging to the same family of cytidine deaminases, APOBEC-3F, was discovered soon thereafter (136, 241, 266). Since the identification of these antiviral factors, an impressive amount of work unveiled the mechanisms by which hA3G and hA3F can inhibit HIV-1 replication (81, 85, 120, 136, 141, 149, 167, 188, 223, 227, 259, 262). The fundamental idea stemming from these studies is that eukaryotic cells possess ancient, intracellular mechanisms protecting their genome from mobile elements replicating via a reverse transcription step (38, 170, 198, 263).
Thus, it became clear that the main, if not the only, function of Vif is to counteract the innate antiretroviral defense mediated by the cytidine deaminases hA3G and hA3F expressed in nonpermissive cells (38, 203, 241). Vif was soon found to interact with hA3G (150) and hA3F (241) and to induce their degradation by the proteasome (138, 152, 204, 221, 256). However, recent work on the inhibition of HIV-1 replication by hA3G and hA3F and on the neutralization of these antiviral factors by Vif suggested that these phenomena are more complex than previously thought and involve several pathways. On one hand, the effects of these restriction factors on HIV-1 replication appear to be at least partially independent from their enzymatic activity that leads to viral hypermutation (15, 167). On the other hand, the production of infectious virus and degradation of hA3G were found to be separable functional properties of HIV-1 Vif (113), and Vif was able to prevent packaging and counteract the antiviral activity of a degradation-resistant hA3G variant (175).
The aim of this review is to build bridges between the studies involving Vif in virus assembly and the recent findings showing that Vif counteracts the antiviral activity of hA3G and hA3F. After presenting the general properties of Vif and the structural and functional defects of HIV-1 Δvif virions, we ask whether these defects can be explained in the light of our new knowledge about the antiviral mechanisms of hA3G and hA3F. Finally, we discuss the multiple mechanisms by which Vif can counteract these restriction factors. This analysis suggests that hA3G and hA3F interfere with the correct assembly of the HIV-1 viral core, which would be destabilized by these proteins. The defects in proviral DNA synthesis could thus partially result from the destabilization of the viral core. We suggest that Vif binding to the genomic RNA in the assembly intermediates could play a major role in neutralizing the effects of hA3G and hA3F on HIV-1 assembly.
GENERAL PROPERTIES OF Vif
Vif Is Ubiquitous among Lentiviruses
The Vif protein is encoded by all lentiviruses, with the exception of equine infectious anemia virus (117). Such ubiquity implies an important function in viral replication. However, vif sequences are highly divergent, and sequence analysis did not reveal any known protein structural motif (172). Consistently, no cellular homolog of Vif has been identified. Supposing that vif functions are conserved among lentiviruses and keeping in mind that the Vif structure remains to be experimentally determined (10, 12, 144), one can find it intriguing that Vif protein folding might be conserved despite massive amino acid variations. However, multiple alignments of lentiviral Vif proteins highlighted invariant residues located mainly in the central region of the sequence. This region contains a noncanonical zinc-coordinating structure (156, 245) and a short peptide (S/T-L-Q-Y/R-L-A consensus) conserved in all lentiviral orthologs (Fig. 1). Zinc binding to the HCCH motif induces a conformational change that induces Vif multimerization and aggregation (180), whereas zinc chelation inhibits HIV-1 activity and liberates the antiviral function of the restriction factor expressed in nonpermissive cells (see below) (244).
FIG. 1.

Schematic representation of HIV-1 Vif domains required for interactions with viral and cellular molecules. The N-terminal region of Vif is green, the central zinc binding domain is orange, and the C-terminal region is red, turquoise, and Matisse blue.
Distribution of Vif in Cell Compartments and Effects on Cytoskeletal Organization
Early studies of cellular functions of Vif characterized its intracellular localization. Upon HIV-1 infection and after Rev-mediated mRNA export (7, 201), Vif is present in the cytoplasm in substantial amounts, corresponding to about half of the Pr55Gag amount (60, 211). Although Vif is localized predominantly in the cytoplasm, some studies proposed a regulated nuclear localization. Lymphocyte cells transiently expressing vif independently of any retroviral context showed a transient nuclear localization of Vif and a significant colocalization with vimentin (212). Importantly, this localization pattern correlated with increased intracellular levels of Vif, as the nuclear localization pattern was not observed when low levels of Vif were obtained through proviral expression. Interestingly, it has been reported that Vif can associate with Sp140, a component of nuclear bodies that was selectively detected in nonpermissive cells (146).
Alternatively, Vif could be retained in the cytoplasm by a specific association with viral and/or cellular partners. Huvent and coworkers showed that baculovirus-expressed Vif localized mainly in the nucleus when expressed in Sf9 insect cells but observed an important cytoplasmic redistribution upon HIV-1 Pr55Gag coexpression (104). This relocalization was likely due to the direct interaction between Vif and Pr55Gag, which is mediated by the C-terminal 22 amino acids of Vif and two domains in Pr55Gag: the NCp7 domain and a region located between the MA and the C-terminal domain of CA (23). At least two putative nuclear localization motifs have been identified, 88WRKRRY94 and 157KKIK160, located in the N- and C-terminal regions of the protein, respectively. On the other hand, a nuclear transport-inhibitory signal has been proposed in the basic region 90RKKR93 (62), consistent with data from a recent study showing that a Vif variant isolated from a long-term-asymptomatic individual and containing a mutation in this motif localized in the nucleus (57). Interestingly, Vif of feline immunodeficiency virus (FIV) is present in both the cytoplasm and the nucleus, but its association with FIV Gag remains controversial (34, 212). The functional significance of regulated nuclear localization of Vif in HIV-1 replication as well as the nature of the subnuclear compartments where FIV and HIV-1 Vif localize remain to be established (267).
The interaction of Vif with the cytoskeleton was first addressed by Karczewski and Strebel, who observed that the transfection of vif in HeLa cells led to a reorganization of the intermediate filament (IF) network in a perinuclear aggregate, where a significant portion of cellular Vif could be found (116). Attempts to extract Vif from cells showed that it was distributed between a detergent-soluble fraction and a fraction that was resistant to high salts and detergents, suggesting a cytoskeletal association. Similar fractionation patterns were observed in systems using H9 (65, 211) and COS-7 (91) cells expressing Vif. However, in the latter case, it was proposed that the presence of Vif in detergent-resistant fraction could be explained by its interaction with cellular membranes or other insoluble structures (75, 77, 211, 212). Supporting this idea, Karczewski and Strebel showed that Vif expressed in SW13 cells devoid of cytoplasmic IFs still associated with the detergent-resistant fraction (116).
Substituting alanines for basic residues in the C-terminal region of Vif prevented membrane targeting and blocked HIV-1 replication in nonpermissive cells, suggesting that the biological activity of Vif requires membrane targeting (77). Although Vif could interact with artificial membranes directly (75, 77), membrane association ex vivo seemed to be mediated by a membrane-binding protein (77, 211). The Pr55Gag polyprotein that is addressed to cellular membranes during HIV-1 assembly and budding could fulfill this role, since the C-terminal region of Vif is also involved in Pr55Gag binding (23) (Fig. 1). However, a naturally occurring and C-terminally deleted Vif variant is fully functional despite its weak interaction and lack of colocalization with Pr55Gag (91). Importantly, this Vif variant was still found in the fractions containing cellular membrane proteins and induced IF network reorganization (91). Taken together, these studies suggest that an important proportion of intracellular Vif might be associated with specific subcellular structures, which, as we will see in the last sections of this review, might be related to hA3G neutralization by Vif.
Control of Vif Expression Level
The steady-state expression level of Vif in both permissive and nonpermissive cells is low compared to those of other HIV-1 auxiliary proteins (235), and residues 88WRKKR93 have been shown to enhance its steady-state level (65). The rapid turnover of cytosolic Vif (half-life of ∼20 min) (3) involves proteasomal degradation, since the treatment of Vif-expressing cells with the proteasome inhibitor MG-132 resulted in polyubiquitinated Vif with a longer half-life (65). While those studies (3, 65, 235) were looking at soluble cytosolic Vif, a very high proportion of Vif (90%) has been found to associate with membranes (212). Surprisingly, cytoskeleton (65) and Pr160Gag-Pol (261) associations stabilize Vif, which accumulates during steady-state expression. This observation raises the idea that the association of Vif with distinct proteins and/or subcellular compartments might be required to fulfill its functions.
In contrast, high levels of expression of Vif are detrimental to HIV-1 infectivity. In H9 cells transfected with HIV-1 Δvif, the infectivity of the viral particles could be rescued in trans with increasing amounts of Vif-expressing vector until a threshold from which a further increase in the intracellular Vif level caused a dose-dependent decrease of infectivity was reached (3). The inhibition of viral infectivity at higher levels of Vif was cell type independent and was associated with an accumulation of Gag-processing intermediates. Thus, low steady-state expression levels of Vif may be maintained to prevent interference with HIV-1 assembly and maturation. However, mutations in putative β-sheet structures that reduced Vif expression levels render H9-derived viral particles noninfectious, suggesting that the wild-type (WT) Vif expression level is optimal (65).
STRUCTURAL AND FUNCTIONAL DEFECTS OF HIV-1 Δvif VIRIONS
Requirement for Vif in HIV-1 Replication Is Cell Type Dependent
Studies of animal models have shown that the vif gene is required for retroviral replication in vivo (4, 48, 83, 84, 105). The expression of Vif also correlates with a cytopathic effect and might play an important role in pathogenesis (4, 122, 194, 195). Vif is essential for HIV-1 replication in a subset of host cells, termed nonpermissive cells. These lineages comprise primary human T cells, macrophages, and monocytes, which are the main reservoirs of HIV-1 in humans, and some lymphocyte-derived cell lines such as CEM, H9, PM1, and HUT78. In contrast, HIV-1 Δvif can replicate in permissive T-cell lines including MT4, CEM-SS, supT1, C8166, and Jurkat cells and in nonlymphoid cell lines such as HeLa cells (20, 42, 66, 67, 216, 234).
The Δvif phenotype depends on the producer cell but not on the infected cell: Δvif virions produced from nonpermissive cells are defective whether they infect nonpermissive or permissive cells, and Δvif virions produced from permissive cells can complete a single replication round in nonpermissive cells (42, 66, 224, 234). Accordingly, Δvif viruses can be complemented by the expression of Vif in the producer but not in the target cell (214, 234). However, a biochemical basis for the block to replication in nonpermissive cells remained elusive until recently (49, 68). The cell dependency of the Δvif phenotype indicates that Vif interacts with a cellular factor(s). Either nonpermissive cells express a negative factor or they lack a positive factor expressed in permissive cells. Heterokaryons resulting from the fusion of permissive and nonpermissive cells displayed the nonpermissive phenotype, strongly suggesting that nonpermissive cells express a negative factor that is overcome by Vif (145, 213).
Morphological and Biochemical Defects of HIV-1 Δvif Virions
Following the budding of viral particles, mature core formation is triggered by the activation of the viral protease within the Pr160Gag-Pol precursor, resulting in proteolytic cleavage and the release of structural (MAp17, CAp24, NCp7, and p6) and enzymatic (PRp12, RTp66/p51, and INp32) proteins. Concomitant conformational rearrangements of the viral genome lead to an increased thermal stability of the dimer of genomic RNA (93, 205). Conversely, the genomic RNA conformation might regulate polyprotein processing (206, 207). While a recent cryoelectron microscopy study provided a dynamic view of core assembly (25), molecular determinants that are responsible for the characteristic cone-shaped mature core of lentiviruses remain poorly understood (78). Despite some studies showing that HIV-1 Δvif virions produced from nonpermissive cell lines have an aberrant composition in envelope and mature structural proteins (20, 193, 210), it is now generally acknowledged that Vif does not affect the protein (24, 60, 68) or RNA (20, 68) content of viral particles. However, a typical morphological defect of Δvif virions produced from nonpermissive cells has been observed by several groups, suggesting that these particles are qualitatively rather than quantitatively defective (20, 24, 94, 173). This defect consists of a nonhomogeneous packing of the core, with electron-dense material concentrated at the flat end of the cone-shaped core or residing in lateral bodies (Fig. 2). This defect suggests that the maturation process is altered in Δvif virions, resulting in an aberrant, nonfunctional ribonucleoparticle (RNP).
FIG. 2.

Electron micrographs of WT and Δvif HIV-1 virions from nonpermissive (H9 and HUT78) or semipermissive (CEMx174) cells. (Panels for CEMx174, HUT78, and H9 cells are adapted from references 20, 173, and 24, respectively, with permission.)
Even though similar morphological defects have been observed with Δvif virions produced from different nonpermissive cells (Fig. 2), an intrinsic difficulty with the interpretation of the electron microscopy pictures stems from the heterogeneity of the morphology of HIV-1 virions and the small fraction of virions yielding a productive infection. Indeed, it is usually considered that fewer than 1 in 1,000 viral particles complete one round of infection (51, 121). However, it is probably erroneous to consider that >99.9% of viral particles are defective, as the infection process is essentially stochastic, and the cellular defenses can randomly “kill” identical virions. In addition, pseudotyping HIV-1 particles with the vesicular stomatitis virus Env protein increases infectivity by up to 2 orders of magnitude, and the infectivity of HIV-1 particles is 102 to 103 times greater during cell-to-cell transmission than the infectivity of cell-free virus stocks (51), suggesting that the limiting steps in HIV-1 infection are attachment, fusion, and/or entry of the viral particles. As Δvif virions are not defective at these steps, the information brought by electron microscopy is most likely relevant, especially as biochemical analysis also revealed a dramatically reduced stability of Δvif compared to that of WT virions produced from nonpermissive cells. Indeed, when treated with alkaline buffer, increasing salt concentrations, detergents, S100 extracts, or deoxynucleotide triphosphates (dNTPs), Δvif virions are prone to release a significant amount of reverse transcriptase and NCp7 (173).
Reverse Transcription Defects in HIV-1 Δvif Virions
Several studies demonstrated that the replication defect of Δvif virions in nonpermissive cells correlates with a defect in intracellular proviral DNA synthesis (Table 1). When viruses produced by a permissive cell line were used to infect another permissive cell line, reverse transcription of WT and Δvif viruses proceeded with the same efficiency (20, 214, 219). Similarly, when Δvif virions produced in permissive cells were used to infect nonpermissive cells, reverse transcription proceeded normally during the first round of viral infection (42, 219, 234). On the other hand, reverse transcription of Δvif virions produced by restrictive cells was dramatically affected irrespective of the target cells being permissive or not (20, 42, 55, 219, 234). These observations confirmed that the nonpermissive phenotype was linked to the identity of the producer cell. At the time when these experiments were performed, i.e., before the identification of the negative factors expressed in restrictive cells, it was assumed that the aberrant viral core of Δvif virions produced in nonpermissive cells was defective at the level of disassembly, stability, processing, and/or transport, ultimately and indirectly resulting in a defect in reverse transcription.
TABLE 1.
Effects of inactivating vif on intracellular reverse transcriptiona
| Producer cell lineb | Target cellb | Effect(s) of vif inactivation on DNA synthesis | Reference |
|---|---|---|---|
| CEM-SS | C8166 (multiple cycles) | No effect | 219 |
| MT4 | P4 (one cycle) | No effect | 20 |
| CEM-SS | C8166 | No effect | 214 |
| SupT1 or Jurkat | H9 | No effect during the first 24 h | 234 |
| CEM-SS | MT2 or H9 (multiple cycles) | Lower level of DNA that remained constant between 10 h and 10 days | 219 |
| CEMc | PBMCs | No effect (4 to 30 h) | 42 |
| CEMx174 | P4 (one cycle) | No DNA synthesis | 20 |
| H9 | C8166 | Synthesis of minus-strand and plus-strand DNA normal up to 8 h; degradation at longer time points; no integration | 214 |
| H9 | C8166 | Synthesis of minus-strand and plus-strand DNA normal up to 24 h; degradation at 48 h | 55 |
| H9 | Jurkat | Strong inhibition except for minus-strand strong-stop DNA | 55 |
| CEM | H9 | Very little synthesis; no final product | 234 |
| PBMCs | PBMCs | No DNA synthesis | 42 |
| Different chronically infected H9 clones with different levels of restrictiond | Strong global defect in DNA synthesis correlated with loss of infectivity | 163 |
Virions produced by permissive or nonpermissive cells were used to infect either permissive or nonpermissive target cells, and reverse transcription of vif+ and Δvif viruses in the target cells was compared.
Permissive and nonpermissive cells are indicated in boldface type and with underlining, respectively.
Even though CEM cells are usually considered nonpermissive for HIV-1 Δvif replication, the cells used by those authors did support the replication of HIV-1 Δvif and were classified by these authors as being semipermissive (42).
Those authors compared DNA synthesis by isogenic HIV-1 Vif− mutants produced by different chronically infected H9 clones, which exhibit different degrees of impairment in their replicative capacity (163).
Interestingly, whereas proviral DNA synthesis of Δvif virions produced by nonpermissive cells was completely or strongly inhibited in most target cells (Table 1), it was almost unaffected in permissive C8166 cells (55, 214). In these cells, a normal amount of full-length double-stranded Δvif DNA was synthesized, but it was unstable and was not integrated. These results might be explained by a lower level of expression of the cytoplasmic factors known to destabilize Δvif cores (173) in C8166 cells. Thus, even though the nonpermissive phenotype is linked primarily to the identity of the producer cell, it can be differentially revealed depending on the target cell.
Reverse transcription of genomic RNA is initiated in HIV-1 virions (99), and under specific conditions, small amounts of full-length proviral DNA can be detected in virions (260). This natural endogenous reverse transcription (NERT) can be enhanced by incubating purified virions with high dNTP concentrations and polyamines in the absence of permeabilizing agents (260), but more-efficient endogenous reverse transcription (ERT) was observed after treating virions with melittin, an amphipathic peptide, or mild detergents. In the presence of detergent, ERT of Δvif virions produced by nonpermissive cells was strongly reduced compared to that of WT virions, while a limited or no difference was observed in NERT or when ERT was performed in the presence of melittin (49, 55, 68, 76, 163, 173, 234) (Table 2). No difference in (N)ERT between WT and Δvif virions was detected when they were produced by permissive cells (49, 76). Thus, the inactivation of vif had more-pronounced effects under conditions that destabilized the viral core. Two groups performed in vitro reverse transcription with recombinant reverse transcriptase and a virion-extracted primer-template complex. Unfortunately, those authors obtained divergent results, with Dettenhofer et al. reporting that the effects of vif inactivation on DNA synthesis were a strong defect in the initiation of reverse transcription and little effect on tRNA annealing for the producer cell line H9 and with Gaddis et al. reporting that there was no effect on tRNA annealing and no effect on the initiation of reverse transcription with the producer cell line HUT78, preventing any firm conclusion (49, 68). It might be significant that the group who observed a defect in in vitro reverse transcription used more-denaturing conditions to extract the Δvif primer-template complex from nonpermissive cells.
TABLE 2.
Effects of inactivating vif on endogenous reverse transcriptiona
| Producer cellb | Permeabilizing agent | Effect(s) of vif inactivation on DNA synthesis | Reference |
|---|---|---|---|
| SupT1 | 1 mM β-octylglucoside | No effect | 76 |
| Jurkat | None; analysis of cDNA in purified virions | No effect | 49 |
| CEM | None; analysis of cDNA in purified virions | No effect | 234 |
| CEM | 1 mM β-octylglucoside | >5-fold decrease in levels of early as well as full length products | 76 |
| Different chronically infected H9 clones with different levels of restrictionc | 0.01 to 0.5% Nonidet P-40 | Strong global defect correlated with the defect in intracellular reverse transcription | 163 |
| H9 | None | No or little effect | 55 |
| H9 | Melittin | Slight defect | 55 |
| H9 | 0.01% Triton X-100 | Strong defect; no long double-stranded DNA products | 55 |
| H9 | 0.02% Triton X-100 | 8-fold decrease of nucleotide incorporation | 49 |
| H9 | None; analysis of cDNA in purified virions | ∼8-fold decrease in reverse transcription (similar effect on products of different lengths) | 49 |
| H9 or HUT78 | Melittin | ∼1.8-fold reduction | 173 |
| H9 or HUT78 | 0.01-0.04% Triton X-100 or Nonidet P-40 | ∼1.8-fold reduction | 173 |
| HUT78 | Melittin | ∼1.5-fold reduction | 68 |
Virions produced by permissive and nonpermissive cells were purified, and the efficiencies of reverse transcription inside HIV-1 vif+ and Δvif virions were compared. Virions were either untreated or treated with different permeabilizing agents to facilitate the penetration of the dNTPs.
Permissive and nonpermissive cells are indicated in boldface type and with underlining, respectively.
Those authors compared levels of DNA synthesis by isogenic HIV-1 Vif− mutants produced by different chronically infected H9 clones, which exhibit different degrees of impairment in their replicative capacity (163).
All studies mentioned so far suggest that the inactivation of vif has no effect on reverse transcription in permissive cells. However, a recent study showed that when permissive cells were pretreated with a thymidylate synthetase inhibitor prior to infection with viruses from permissive cells, the replication of HIV-1 Δvif was inhibited to a greater degree than WT HIV-1, suggesting compromised reverse transcription in the absence of Vif (30). In keeping with this interpretation, the same group showed that Vif is an integral component of the reverse transcription complexes (30, 31). An in vitro study showed that Vif may behave as a cofactor of HIV-1 reverse transcriptase by enhancing its association with nucleic acids, stimulating polymerase activity and facilitating the bypass of abasic sites (28). However, it is paradoxical that the C-terminal region that was required for the stimulation of the polymerization activity is cleaved in virion-associated Vif (119) (see below).
Vif AND VIRAL ASSEMBLY
Vif Associates with Intermediate Assembly Complexes
HIV-1 assembly takes place in the cytoplasm to produce viral particles that exit infected cells by budding from cellular membranes (71). The Pr55Gag polyprotein is thought to be the main driver of viral assembly, since Gag can spontaneously assemble in vitro into virus-like particles (VLPs) closely matching assembly intermediates observed in infected cells. Thus, the formation of Pr55Gag-Pr55Gag complexes is the first event that is likely to occur in the assembly pathway. HIV-1 genomic RNA seems to be associated early within these complexes (184), and its recruitment into viral particles relies on the binding of Pr55Gag to encapsidation signals located in its 5′ untranslated region (132, 178, 192). There are some hints that genomic RNA already exists as dimers in the cytoplasm (63, 64, 178, 179), and it would serve as a molecular scaffold for viral assembly (92, 162, 174). During the assembly process, the size of the assembly complexes increases (137) as cellular factors (32, 33, 268) and viral proteins are recruited.
Insect Sf9 cells expressing HIV-1 Pr55Gag can produce enveloped VLPs similar to natural HIV-1 virions in size and morphology, and upon the coexpression of the Vif protein, Vif can be detected in these VLPs (104). Sequences of Pr55Gag spanning the second zinc finger of the NC domain and the p6 peptide were required for Vif encapsidation. Further investigation showed that Vif was also incorporated into cytoplasmic VLPs formed by nonmyristoylated Pr55Gag proteins, indicating that membrane targeting is not a prerequisite for Vif encapsidation (11). In addition, Vif incorporation was enhanced when chimeric VLPs were made from Pr55Gag and Pr160Gag-Pol (11).
By exploiting differences in the solubilities of mature and immature HIV-1 cores, it was shown that in mammalian cells expressing HIV-1, Vif is incorporated mainly into particles containing unprocessed Pr55Gag (220). However, one cannot completely exclude the possibility that viral core preparations were contaminated with immature VLPs from lysed cells. This result correlates with electron microscopy observations of immunolabeled cells by Bardy et al. showing that Vif seemed to be gradually excluded from Pr55Gag VLPs during the time course of their assembly (11). Thus, the interaction of Vif with Pr55Gag and Pr160Gag-Pol precursors may allow its transient association with cytoplasmic assembly intermediates during the HIV-1 assembly process (130, 184). These assembly intermediates were likely present in the insoluble fractions containing Vif observed in cell fractionation studies (211). A further indication of the role of Vif in HIV-1 assembly comes from the observation that the inhibition of assembly of HIV-1 VLPs in Sf9 cells by a betulinic acid derivative is counteracted by Vif (43). In addition, while VLPs produced by cells expressing Pr55Gag alone assembled and budded at the plasma membrane, those produced by cells coexpressing P55Gag and WT Vif were redirected to a vesicular compartment and egressed via the exocytic pathway (43).
A recent work shed light on the dynamics of assembly by demonstrating that HP68, a cellular ATP-binding protein, is required for the conversion of assembly intermediates to higher-molecular-weight, conformationally distinct intermediates. The recruitment of HP68 is mediated by Pr55Gag, and interestingly, Vif was found to coimmunoprecipitate with complexes containing HP68 (268). Altogether, these studies showed that Vif plays an active role during HIV-1 assembly in infected cells.
Interactions between Vif and Viral Genomic RNA
Vif is an RNA-binding protein (49, 89, 261) that interacts with HIV-1 genomic RNA in the cytoplasm of infected cells (49, 261). Indeed, Vif can be copurified with viral unspliced RNA from infected cells using rate-zonal sedimentation (261), immunoprecipitation (49, 261), and UV cross-linking (261). An interaction with the genomic RNA seems to be required to recruit Vif into virions, since the abrogation of RNA encapsidation by mutating either the NC domain of Pr55Gag or the main packaging signal (Ψ) of the genomic RNA led to a complete absence of Vif in purified virions (118). This finding suggests a mechanism of encapsidation that is distinct from that for the Pr55Gag-mediated incorporation of Vif into assembly intermediates. Early in vitro experiments have shown that Vif exhibits nonspecific RNA binding, as shown by its association with several viral RNA fragments, RNA homopolymers (261), and bulk RNA when translated in rabbit reticulocyte lysates (49). Moreover, recent work showed a 30% diminution in the total RNA content of Δvif virions but no change in genomic RNA packaging (79). It is thus conceivable that its high content of basic residues gives Vif a gross affinity for RNA, resulting in nonspecific binding. However, while a deletion of the N-terminal region of Vif abrogated its in vitro RNA-binding properties and was detrimental to viral replication (261), and a deletion of amino acids 75 to 114 impaired RNA-mediated packaging of Vif, the Vif C-terminal region, which harbors many positively charged residues, only weakly contributed to RNA binding (118). Within the N-terminal region of Vif, the W11A, Y30A, and Y40A substitutions strongly reduced binding to both poly(G) and an RNA probe corresponding to nucleotides 5104 to 5287 of the HIV-1 genome, and viruses harboring any of these substitutions were noninfectious in restrictive cells, suggesting that RNA binding is crucial for Vif function (261).
We recently showed by a combination of biochemical and biophysical approaches that Vif can bind RNA with some specificity and has a preferential affinity for the 5′ region of HIV-1 genomic RNA (14, 89). Vif binds the 5′ end of HIV-1 genomic RNA in a cooperative manner with two types of binding sites: (i) a few high-affinity binding sites, including those which we identified in the transactivation region apical loop and in the poly(A) stem, and (ii) lower-affinity binding sites encompassing the primer-binding site (PBS) and the whole leader region. This binding pattern implies that at low concentrations, only a few Vif molecules will bind to each copy of genomic RNA, whereas a partial coverage of the 5′ region is expected at a higher Vif concentration. These observations fit with the previously reported correlation between the intracellular concentration of Vif and its encapsidation rate (215), although the relevant concentrations required for Vif function at different steps of the viral cycle and for its association with distinct molecular partners remain to be determined.
Biological Relevance of Vif Packaging
Many attempts have been made to quantify Vif incorporation into HIV-1 virions. Viral particle preparations are quite sensitive to contamination by secreted proteins or membrane microvesicles derived from cells, often leading to conflicting reports regarding the presence of virion-associated proteins (73). In the first comprehensive study of Vif encapsidation, Liu and coworkers treated cell-derived virions with proteinase K to get rid of proteins unspecifically attached to the membrane of virions and then disrupted the viral envelope and cell-derived debris by mild detergent treatment (140). By using this procedure, they recovered viral cores with significant amounts of Vif (60 to 100 Vif copies per virion). However, other authors detected reduced amounts of Vif in virion cores (7 to 20 copies per virion) (27) and argued for a nonspecific encapsidation correlated with intracellular Vif expression levels (215). Indeed, HIV-1 Vif can be encapsidated in murine leukemia virus (MLV) particles (27). However, this nonspecific incorporation could also be explained by a conservation of encapsidation determinants between MLV and HIV-1. Vif proteins of HIV-2 and sooty mangabey simian immunodeficiency virus (SIVSM) are also encapsidated in these closely related lentiviruses (140), underlining the relevance of this property. On the other hand, contamination by microvesicles can be partially prevented by using an Optiprep purification procedure. Even though this method can yield highly purified virions compared to routinely used purification protocols using sucrose gradients, conflicting results were obtained: Sova et al. detected significant amounts of Vif in Optiprep-purified virions (220), while Dettenhofer and Yu found an almost complete absence of Vif in virions by using the same procedure (50).
Differences in the protocols used to express and detect Vif could explain these discrepancies. First, one can question the precision obtained with immunological methods when trying to quantify proteins encapsidated at low levels. The efficiency of epitope recognition and the loss of immunoreactive domains by protein degradation may lead to an underestimation of protein levels. Such a possibility is reinforced by the fact that virion-associated Vif is unstable (114). Second, high levels of intracellular Vif could favor nonspecific encapsidation and mask the fact that limited although functionally relevant amounts of Vif could be specifically encapsidated (118). Interestingly, Vif is more efficiently encapsidated in virions derived from acutely infected cells than from chronically infected cells (112), consistent with earlier reports (27, 50). This difference may reflect a higher rate of de novo synthesis of Vif observed, for instance, upon the superinfection of chronically infected cells (112). Higher synthesis rates may affect the intracellular trafficking of Vif and associations with subcellular structures (such as the cytoskeleton) and may direct Vif toward encapsidation. Remarkably, despite its low packaging efficiency, Vif produced in cells chronically infected by a Vif-expressing virus was biologically active, indicating that the packaging of large amounts of Vif is not required for viral infectivity (112).
Complex Interplay between Vif, Viral Protease, Pr55Gag, and RNA
Zhang et al. showed that the interaction between Vif and viral RNA fragments corresponding to nucleotides 3627 to 3925 and 5104 to 5287 could be displaced by the formation of a Pr55Gag-RNA complex. Thus, Pr55Gag could prevent the packaging of nonspecific RNA-associated Vif (261). These observations could explain the limited amounts of Vif detected in virions, which could correspond to Vif bound to high-affinity sites on genomic RNA. We showed that multiple copies of Vif can bind the 5′ end of viral genomic RNA (14, 89), partly mimicking RNA-binding properties of the HIV-1 NCp7 protein (90, 186, 254). Either Vif and Pr55Gag compete for common RNA-binding sites or viral RNA adopts a conformation no longer recognized by Vif due to the RNA chaperone properties of Pr55Gag (58).
The function of virion-associated Vif, if any, is still a matter of debate, but most in vitro and ex vivo data suggest that Vif is able to modulate Pr55Gag processing by HIV-1 protease at MA-CA and CA-NC junctions (3, 11), even though this idea has been questioned (68) (Fig. 3, step 4). Sequences that are responsible for the modulation of p2 processing lie between amino acids 4 and 23 and 45 and 74 at the N terminus of Vif, overlapping with the peptide at amino acids 30 to 65 (18, 124), which has been shown to inhibit the viral protease in vitro, and with the peptide at amino acids 68 to 81, which is involved in Pr55Gag binding (23) (Fig. 1). Peptide p2 at the CA-NC junction is important for the sequential processing of Pr55Gag and is a key determinant of virion assembly and morphogenesis (72, 78, 181). In addition, p2 is also involved in the maturation of the HIV-1 RNA dimer, suggesting a complex and dynamic interplay between genomic RNA and Pr55Gag subdomains, which could be modulated by Vif (205).
FIG. 3.

Schematic representation of Vif and hA3G functions in HIV-1 assembly and replication. During viral particle production, Vif is found in the cytoplasm of infected cells and is encapsidated in small amounts into virions. Vif neutralizes hA3G and hA3F in virus-producing cells by different mechanisms. (1) Vif has been shown to impair the translation of hA3G mRNA, probably through an mRNA-binding mechanism. (2) Vif binding to hA3G protein recruits an E3 ubiquitin ligase that mediates the polyubiquitylation of hA3G and its degradation. (3) Vif competes with hA3G for binding to viral components like the nucleocapsid domain of Gag and/or viral genomic RNA. Taken together, these three different actions of Vif on translation, degradation, and packaging not only deplete hA3G from virus-producing cells but also prevent hA3G from being incorporated in virions. (4 and 5) Intracellular Vif may also influence viral assembly through the modulation of viral protease-mediated cleavage of Gag precursors (4) and its chaperoning activities (5), thus allowing late events such as precursor maturation, initiation of reverse transcription, and RNA dimer maturation to occur after viral budding. (6) Finally, Vif might be able to directly inhibit the activity of the few hA3G molecules that are packaged in WT virions.
Vif as a Temporal Regulator of Pr55Gag and NCp Activities
From the above-described data, it is tempting to speculate that a possible function of Vif is to prevent premature processing in the cytoplasm of infected cells and/or to modulate the activity of the viral protease after budding of the viral particles (3, 11, 18, 124) (Fig. 3, step 4). It may do so either by direct interactions with viral protease and Pr55Gag domains involved in proteolysis (103, 135, 207, 242) or by modulating Pr55Gag-RNA interactions (135, 207, 242), even though interactions between Pr55Gag and Vif may be transient or mediated by a third partner (211).
In this context, it is remarkable that Vif has recently been shown to be an RNA chaperone that modulates some but not all activities of NC proteins (90) (Fig. 3, step 5). On one hand, Vif promotes dose-dependent 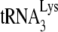 annealing to the PBS and the formation of a loose dimer of genomic RNA (90), two early steps in the process of assembly and budding (147, 178). On the other hand, Vif inhibits the NCp7-mediated conversion of the RNA kissing complex into a tight dimer and the initiation of reverse transcription (90), two steps that normally take place after budding (100, 178). Importantly, the inhibitory effects of Vif could be relieved as the Vif/NC (or Pr55Gag) ratio decreases from about 1/2 in the assembly complexes to about 1/40 in the viral particles (90).
annealing to the PBS and the formation of a loose dimer of genomic RNA (90), two early steps in the process of assembly and budding (147, 178). On the other hand, Vif inhibits the NCp7-mediated conversion of the RNA kissing complex into a tight dimer and the initiation of reverse transcription (90), two steps that normally take place after budding (100, 178). Importantly, the inhibitory effects of Vif could be relieved as the Vif/NC (or Pr55Gag) ratio decreases from about 1/2 in the assembly complexes to about 1/40 in the viral particles (90).
Taken together, these data indicate that Vif could be a temporal regulator of viral assembly. As this regulatory function of Vif would be required only in nonpermissive cells, this suggests that the restriction factor(s) expressed in these cells might interfere with the sequence of events taking place during assembly that normally leads to a functional viral RNP (see below). Importantly, this hypothesis, based mainly on biochemical data, is consistent with the decreased stability of Δvif viral cores (see above).
Pivotal Role for the C-Terminal Tail of Vif
Individual domains in the Vif amino acid sequence have been poorly characterized experimentally, with the exception of the C-terminal region downstream of the conserved S/T-L-Q-Y/R-L-A motif (Fig. 1). This region, which is proteolytically processed in virion-associated Vif (119), is involved in Pr55Gag interactions (24) and Vif self-association and multimerization (250, 251) (Fig. 1). Multimerization may be required for cooperative binding to RNA (14, 89) and pathogenesis (250), since a deletion of the multimerization domain (161PPLP164) impaired viral infectivity in H9 cells (251), and a Vif dimerization antagonist peptide suppresses HIV-1 infectivity (158). Mass spectroscopy analysis revealed an increase in the ordered structure of the C-terminal domain upon Vif oligomerization (8). This C-terminal region is rich in basic residues, and alanine-scanning studies showed that membrane targeting and Pr55Gag binding were negatively affected by the substitution of these residues, while the level of encapsidation of Vif was increased (77, 104). Whether these mutations affect the proteolytic processing of Vif is presently unknown, but the above-described studies suggest that this region could be used as a binding platform, regulating Vif packaging and interactions with various partners. The association of these molecules and/or Vif multimerization could be modulated by the phosphorylation of threonine or serine residues in the C-terminal region of Vif by kinases such as mitogen-activated protein kinase (252).
APOBEC-3G AND APOBEC-3F RESTRICTION FACTORS AND THE Δvif PHENOTYPE
APOBEC-3G and APOBEC-3F Are the Main Restriction Factors Neutralized by Vif
The first attempts to characterize restriction factors counteracted by Vif led to the identification of the tyrosine kinase Hck, whose expression is detrimental to the replication of HIV-1 Δvif (87). More recently, in a seminal paper, Sheehy and coworkers demonstrated that the expression of the cytidine deaminase hA3G, initially known as CEM-15, is specific to nonpermissive cells and sufficient to abolish the replication of HIV-1 Δvif (203). The identification of hA3G as the restriction factor counteracted by Vif (203) was soon followed by the characterization of another member of the APOBEC protein family with anti-HIV activity, hA3F, which is extensively coexpressed with hA3G in nonpermissive cells (136, 241, 266). Both the hA3G and hA3F proteins are neutralized by Vif (see below) (203, 241).
The N- and C-terminal parts of hA3G and hA3F possess significant similarity to APOBEC-1, the catalytic subunit of the mammalian apolipoprotein B mRNA-editing enzyme (203). The APOBEC family includes the cellular cytidine deaminases APOBEC-1, APOBEC-2, APOBEC-3, and APOBEC-4 and the activation-induced deaminase. In humans, there are seven APOBEC-3 genes (APOBEC-3A to APOBEC-3H; APOBEC-3E, initially considered to be a pseudogene, is now considered to be the 3′ part of an APOBEC-3DE gene), whereas rodents have a single APOBEC-3 gene that corresponds to APOBEC-3G (110; for reviews, see references 38 and 97). APOBEC-3B, APOBEC-3DE, APOBEC-3F, and APOBEC-3G contain a duplication of the catalytic site, which contains a Cys-His Zn2+ coordination motif characteristic of cytidine deaminases (36, 95, 110).
hA3A restricts some retrotransposons and another virus, but not HIV-1, due to inefficient targeting to the viral core (2, 74). Several studies reported that hA3B is able to inhibit HIV-1 replication in a Vif-independent manner (16, 52, 53). However, this protein is normally not expressed in nonpermissive cells, and thus, its activity does not seem to contribute to the restriction of HIV-1 replication in vivo (16, 53). APOBEC-3C expressed in target cells can cause relatively infrequent G-to-A mutations of some Vif-expressing HIV-1 clones, but they do not prevent spreading infection (22). On the other hand, hA3DE inhibits HIV-1 in a Vif-dependent manner, and its expression pattern is similar to that of hA3G (46), but its effect is much weaker. Finally, hA3H mRNA levels are low, and the corresponding protein is poorly expressed in cell culture (44). Thus, hA3G and hA3F are considered to be the main restriction factors neutralized by Vif (16), even though hA3F is less potent than hA3G (258). Intriguingly, hA3G only moderately restricts the replication of HIV-2 Δvif, and only peripheral blood mononuclear cells (PBMCs) display a nonpermissive phenotype in the absence of HIV-2 Vif (187).
Deamination of HIV-1 DNA by hA3G, hA3F, and hA3DE
Endogenous hA3G is incorporated into Δvif HIV-1 particles produced by nonpermissive cells (221), and similarly, when overexpressed in permissive cells, hA3G (150, 152, 203, 204, 256), hA3F (241, 266), and hA3DE (46) are incorporated into HIV-1 Δvif virions (Fig. 4).
FIG. 4.

Multiple antiviral mechanisms of virion-incorporated and endogenous hA3G. In the absence of Vif, hA3G is efficiently incorporated into budding viruses. After liberation of the viral capsid into a new (nonpermissive) target cell, hA3G impedes reverse transcription and integration in a deaminase-independent antiviral action, probably through its RNA-binding properties. Once reverse transcription has started, hA3G mediates the extensive deamination of dC to dU during minus-strand DNA synthesis. This enzymatic reaction blocks HIV-1 replication due to (i) the accumulation of dG-to-dA hypermutations in the synthesized plus-stranded viral DNA (giving rise to aberrant proteins) and (ii) the degradation of viral DNA, because the U-rich DNA can be recognized by the cellular repair machinery (uracil DNA glycosylase and apurinic-apyrimidic endonuclease). Endogenous h3AG also functions as a potent restriction factor for HIV-1 in resting CD4+ T cells. hA3G exist as HMM and LMM complexes. Only the LMM complexes function as a postentry restriction factor by blocking the accumulation of reverse transcripts of incoming viruses. HMM complexes impair the transposition of Alu elements by sequestering Alu RNA transcripts away from the nuclear transposition machinery.
Sequencing of reverse transcription products of Δvif virions produced by permissive cells transfected with an hA3G-expressing vector revealed a hypermutation of HIV-1 DNA (85, 149, 208, 255, 262). Almost all mutations were G-to-A transitions when read on the plus DNA strand. Similar results were obtained when the products of endogenous or intracellular reverse transcription of Δvif virions produced by nonpermissive cells were analyzed (129, 262). These mutations are the result of cytosine deamination in the minus DNA strand, which convert cytosines into uracils, leading to G-to-A mutations in the plus strand (Fig. 4). In contrast, hA3G does not edit HIV-1 genomic RNA (129, 149, 255). hA3G is catalytically active on single-stranded DNA but not on DNA-DNA and DNA-RNA hybrids or on single-stranded RNA (227, 255). Indeed, hA3G incorporated into virions is inhibited by HIV-1 genomic RNA and activated during plus-strand DNA synthesis as the RNA is degraded by the RNase activity of reverse transcriptase (218). The probability that a given minus-strand cytosine is deaminated depends on the time it remains single stranded (226, 255) and is increased by resistance mutations reducing the processivity of reverse transcriptase (123). hA3G proceeds processively from 3′ to 5′ of minus-strand DNA (35), and the dispersed hypermutation of the genome is the result of rapid transfers of hA3G between segments of the minus-strand HIV-1 DNA (171).
Cytosine deamination by hA3G, hA3F, and hA3DE is not random. TGGG (as read in the plus strand) is the tetranucleotide that is most frequently modified in vitro and in cell culture by hA3G, and the hot-spot consensus sequence of this enzyme is HGGR (where H is C, T, or A and R is A or G) (the mutated residue is underlined) (13, 85, 227, 255). Similarly, hA3F and hA3DE preferentially introduce mutations in the sequences WGAA (where W is A or T) (16, 136, 241) and MGWWR (where M is C or A) (46), respectively. Noticeably, hypermutation has been detected in clinical isolates of HIV-1 (29, 59, 109, 125, 177, 217, 232). Analysis of these sequences, combined with the target preference and the expression profile of these proteins, strongly suggests that hA3G, hA3F, and hA3DE are responsible for HIV-1 hypermutation in vivo (16, 46, 136, 177, 217). It has been proposed that in the absence of a fully functional Vif protein, the genomes are mutated to such an extent that they cannot produce infectious progeny viruses (85, 149, 177, 217, 262). Remarkably, cell culture experiments revealed that WT Vif does not completely inhibit deamination: low levels of catalytically active hA3G (0.3 to 0.8 molecules/virion) were detected in WT HIV-1 (171), and hA3G induced nonlethal mutagenesis in WT HIV-1 passaged in long-term culture (262). This low level of hA3G-induced mutagenesis might have a positive effect on viral infectivity by facilitating drug resistance (161) and/or immune evasion (182).
Importantly, APOBEC-3G-, APOBEC-3F-, and APOBEC-3DE-induced hypermutation certainly contributes to restricting HIV-1 replication, but it does not per se explain the inability of Δvif virions produced by nonpermissive cells to complete a single round of reverse transcription, as shown by the data summarized in Table 1. However, the expression of hA3G and hA3F in the producer cell does impair reverse transcription (Table 3). Quantitative PCR analysis suggested that the deamination of the minus-strand DNA by hA3G does not interfere with the completion of reverse transcription but that the presence of uracil results in the degradation of the DNA prior to integration (150) (Fig. 4). Indeed, fixing mutations as G-to-A transitions is not the only possible outcome of cytosine deamination: uridines might be recognized by cellular DNA repair enzymes and excised to create abasic sites that would ultimately lead to DNA degradation. The major cellular DNA glycosidase, hUNG2, has been proposed to be selectively incorporated into HIV-1 particles and essential to the HIV-1 life cycle (185, 243). However, while some studies suggested that hUNG2 contributed to the loss of infectivity of Δvif virions produced in the presence of hA3G (249), other reports indicated that hUNG2 is dispensable for hA3G antiviral activity (111, 128, 199). Thus, the mechanism underlying DNA degradation as a consequence of cytosine deamination remains elusive. In addition, several studies reported that hA3G and hA3F affect all steps of reverse transcription with similar efficiencies, except tRNA annealing (Table 2), whereas the larger DNA products should be more affected by deamination-induced DNA degradation, as found by others (5, 142, 153) (Table 2).
TABLE 3.
Effects of hA3G and hA3F on the efficiency of intracellular reverse transcription of a Δvif HIV-1- or HIV-1-derived vector
| Reverse transcription step | Fold change (reduction of products)
|
Reference | |||
|---|---|---|---|---|---|
| WT hA3G | Deaminase-minus hA3G | WT hA3F | Deaminase-minus hA3F | ||
| tRNA priming | 2 (MT2, H9, or 293T) | 1.5 (SupT1) | 79 | ||
| 2 (SupT1 and 293T) | 1.4 (SupT1) | 253 | |||
| Minus-strand strong-stop synthesis | >10 (SupT1) | 2 (SupT1) | 5 (SupT1) | 5 (SupT1) | 96 |
| >20 (SupT1) | 4 (SupT1) | 15 | |||
| 2 (SuptT1) | 79 | ||||
| 2 (SupT1) | 253 | ||||
| 2 (SupT1) | 134 | ||||
| 17. (HeLa) | 1.7 (HeLa) | 2 (HeLa) | 142 | ||
| First-strand transfer | 3 (SupT1) | SupT1: 3-fold | 134 | ||
| 1.15 (293T) | 153 | ||||
| Minus-strand DNA synthesis | 20 (SupT1) | 1.4 (SupT1) | 79 | ||
| 20 (SupT1) | 253 | ||||
| 20 (SupT1) | 134 | ||||
| 3 (HeLa) | 3 (HeLa) | 5 (HeLa) | 142 | ||
| Second-strand transfer | 10 (SupT1) | 10 (SupT1) | >10 (SupT1) | 10 (SupT1) | 134 |
| 2-3 (293T) | 153 | ||||
| Proviral DNA synthesis | 5-10 (293T) | 153 | |||
| 3 (HeLa) | 149 | ||||
| Integration | 5 (293T) | No effect (293T) | 153 | ||
| 3 (HeLa) | 3 (HeLa) | >10 (HeLa) | 142 | ||
| 7 (HOS) | 150 | ||||
Deamination-Independent Activity of hA3F and hA3G
There is increasing evidence indicating that hA3G is able to inhibit reverse transcription and the replication of HIV-1 Δvif in the absence of deaminase activity (Table 2). Shindo et al. (208) first pointed out that hA3G catalytic activity is not the sole determinant of its antiviral activity. Further investigations of the enzymatic activity of hA3G showed that of the two repeated catalytic domains of the protein, only the C-terminal one is active, while the N-terminal domain is involved in RNA binding, multimerization, and encapsidation (133, 165, 176). Interestingly, the expression of the N-terminal domain alone is sufficient to inhibit HIV-1 replication, showing that enzymatic activity is not absolutely required for antiviral function (133). Indeed, the point mutations H257R, E259Q, and C291S in the C-terminal catalytic domain strongly reduce cytosine deaminase activity but have little effect on the vif-dependent antiviral activity of hA3G (167). Using hA3G and hA3F chimeric proteins, it was shown that the antiviral phenotype of these proteins correlates with their ability to prevent the accumulation of reverse transcripts but not with their ability to induce hypermutation (15). Indeed, an important fraction (38% ± 18%) of env sequences derived from a Δvif HIV-1 strain did not contain a single mutation after two rounds of replication in PBMCs, strongly suggesting that another mechanism(s) contributes to the restriction of Δvif HIV-1 in these cells (123). Interestingly, a recent study showed that murine APOBEC-3 (mA3) is efficiently packaged into MLV particles but fails to cause a hypermutation of viral DNA (26). Similarly, several studies indicated that the inhibition of hepatitis B virus by A3G is independent of the catalytic activity of this antiviral factor (168, 230), even though HBV DNA is edited in vivo during natural infections albeit at a low level (225). In the case of hA3F, deamination-independent restriction of HIV-1 is apparently even more important, with hypermutation playing only a minor role (96).
Even though there is general agreement that deaminase activity is required for the optimal anti-HIV-1 activity of hA3G (15, 79, 108, 159, 165, 167, 200, 208), it is likely that the defects in the reverse transcription of Δvif virions produced by nonpermissive cells (Table 1) correspond mostly to the deamination-independent activities of hA3F and hA3G since they were observed during a single reverse transcription cycle, while the effects of deamination are cumulative (149). Indeed, Δvif virions exhibit a diminished ERT efficiency in the presence of a catalytically inactive hA3G mutant (133) that is reminiscent of ERT defects with Δvif virions produced by nonpermissive cells (49). In that same study (133), a variant hA3G mutated in the RNA-binding domain had no effect on ERT, suggesting that the recognition of viral RNA by hA3G is required to inhibit reverse transcriptase activity.
Recently, Kleiman and coworkers performed a thorough analysis of reverse transcription defects in Δvif virions produced from permissive cells transiently expressing hA3G or hA3F (79, 253). In these virions, the priming of reverse transcription, i.e., the addition of the first 6 nucleotides to the primer 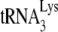 , was reduced to ∼50% of WT levels, which coincided with a strong diminution of minus-strand strong-stop and late DNA products (Table 3). Interestingly, N- and C-terminal deletion mutants of hA3G that were unable to deaminate viral DNA were still able to inhibit reverse transcription priming and extension. That same group also showed that hA3G inhibits the first-strand transfer occurring during reverse transcription and reverse transcriptase RNase H activity in vivo independently of its editing properties (134). In vitro studies aimed at unveiling the editing-independent mechanisms by which hA3G and hA3F inhibit reverse transcription yielded conflicting results. One group reported that hA3G inhibits
, was reduced to ∼50% of WT levels, which coincided with a strong diminution of minus-strand strong-stop and late DNA products (Table 3). Interestingly, N- and C-terminal deletion mutants of hA3G that were unable to deaminate viral DNA were still able to inhibit reverse transcription priming and extension. That same group also showed that hA3G inhibits the first-strand transfer occurring during reverse transcription and reverse transcriptase RNase H activity in vivo independently of its editing properties (134). In vitro studies aimed at unveiling the editing-independent mechanisms by which hA3G and hA3F inhibit reverse transcription yielded conflicting results. One group reported that hA3G inhibits 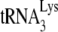 annealing to the PBS, the RNase H activity of reverse transcriptase, and strand transfers without affecting the elongation step of DNA synthesis (80, 134), while another one reported that hA3G had no effect on
annealing to the PBS, the RNase H activity of reverse transcriptase, and strand transfers without affecting the elongation step of DNA synthesis (80, 134), while another one reported that hA3G had no effect on 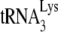 annealing, RNase H, and minus-strand transfer but did inhibit DNA elongation by reverse transcriptase (107). hA3G was also observed to inhibit the elongation of reverse transcripts during NERT, i.e., in the absence of any target cell factors (17). Thus, more work will be required to decipher the editing-independent mechanisms of inhibition of HIV-1 reverse transcription by hA3G and hA3G.
annealing, RNase H, and minus-strand transfer but did inhibit DNA elongation by reverse transcriptase (107). hA3G was also observed to inhibit the elongation of reverse transcripts during NERT, i.e., in the absence of any target cell factors (17). Thus, more work will be required to decipher the editing-independent mechanisms of inhibition of HIV-1 reverse transcription by hA3G and hA3G.
It is important that some of the deaminase-dependent and/or deaminase-independent effects of hA3G and hA3F described in this section and in the previous section might be due to the artificially high concentrations of these restriction factors used in some studies. Indeed, most cell culture experiments described in these sections relied on the transfection of plasmids expressing hA3G and hA3F, and the in vitro experiments usually required fairly high concentrations of purified proteins. However, a recent quantitative study showed that virions produced during transfection with large amounts of expression plasmids incorporate up to 30-fold more hA3G molecules than Δvif virions produced by human PBMCs (247). Therefore, it is important to compare these studies with those obtained using nonpermissive cells, which express physiological concentrations of hA3G and hA3F, and with in vivo studies.
Importantly, analysis of the HIV-1 sequences from therapy-naive infected individuals indicated that the levels of hA3G-induced G-to-A mutations did not correlate with viral load and suggested that hA3G might be restricting HIV-1 replication in vivo through a mechanism that is independent of its catalytic activity (231). Han and coworkers recently demonstrated that hA3G and hA3F require a cellular cofactor to block HIV-1 replication (82). There is no doubt that the identification of this cofactor would allow a major step forward in our understanding of the antiviral mechanisms of hA3G and hA3F.
Indirect Evidence of an Effect of hA3G and hA3F on HIV-1 Assembly
Aside from defective reverse transcription, the main feature of Δvif virions is an aberrant core morphology correlated with a reduced stability (Fig. 2). As this defect has been observed exclusively in Δvif virions produced from nonpermissive cells, it cannot be due to the absence of Vif in these virions but is likely a (direct or indirect) consequence of the expression of the antiretroviral factors hA3G and hA3G in these cells, even though we cannot totally exclude the possibility that it might be the result of an additional, as-yet-unidentified, restriction factor. The dramatic morphological defects observed in Δvif virions are not the result of a deamination of nascent proviral DNA, as they can be observed in budding virions. Moreover, quantitative analysis revealed that only 7 ± 4 molecules of hA3G are incorporated per Δvif virion produced by human PBMCs (247), and it is difficult to imagine how these few molecules could produce such dramatic effects on core morphology and stability. Therefore, we propose that intracellular hA3G and hA3F may interfere with viral core assembly, causing the observed morphological defect and contributing to the subsequent deaminase-independent impairment of endogenous or intracellular reverse transcription.
In a recent work, Arhel and coworkers (6) suggested that reverse transcription might take place into an intact capsid shell that associates with the nuclear pore after intracytoplasmic routing and that decapsidation would be triggered by the last stage of reverse transcription. Another study showed that the intracytoplasmic maturation of the HIV-1 reverse transcription complex determines its capacity to integrate into chromatin (106). Thus, cellular factors that promote the formation of unstable viral cores in the producer cells are expected to have dramatic effects during reverse transcription in the infected cells. The fact that the inhibition of the proviral DNA synthesis of Δvif virions produced by nonpermissive cells also depends on the identity of the infected cell (C8166 versus most permissive cells) (Table 1) suggests that this defect does not reflect a direct inhibition of reverse transcriptase by encapsidated hA3G and hA3F but rather reflects a destabilization of the abnormal viral cores produced in nonpermissive cells by cellular factors present in the infected cell. Indeed, lysates from different cell types have been shown to differently affect endogenous reverse transcription (239).
It would be interesting to directly look for morphological defects or an instability of the cores of Δvif virions produced in permissive cells expressing physiological levels of hA3G or hA3F, but to the best of our knowledge, such studies have not been published. Detailed analysis of the interactions between hA3G and hA3F and components of the viral assembly complexes would also likely improve our understanding of the deamination-independent antiviral activities of these factors.
Vif COUNTERACTS THE ANTIVIRAL ACTIVITIES OF hA3G AND hA3F
Vif Decreases the Intracellular Concentration of hA3G and hA3F
HIV-1 Vif dramatically reduces the packaging of hA3G into virions (114, 133, 139, 150, 152, 155, 204), and conversely, biologically inactive Vif mutants are unable to prevent hA3G incorporation (114, 152, 155, 204, 208). Vif reduces the steady-state level of hA3G, and numerous studies showed an important decrease in the hA3G half-life in the presence of Vif (41, 139, 152, 155, 204, 221, 256). Accordingly, several groups showed that HIV-1 Vif suppresses hA3G antiviral function by hijacking the cellular Cullin5 (Cul5)-ElonginB (EloB)-ElonginC (EloC) E3 ubiquitin ligase to induce the degradation of hA3G and hA3F by the proteasome (41, 138, 152, 154, 204, 221, 256, 266) (Fig. 3, step 2). Vif was indeed shown to bridge hA3G with a cellular E3 ligase (256). First, the C-terminal region of Vif possesses a 144SLQ(Y/F)LA155 motif that binds to EloC (Fig. 1). This motif is similar to a conserved sequence found in the BC box of the suppressors of cytokine signaling (SOCS) protein (152, 154, 257). Second, a novel zinc-binding motif in Vif with the consensus sequence 108Hx2YFxCFx4Φx2AΦx7-8Cx5H133 binds to Cul5, the core subunit of a Cul5-based E3 ligase (143, 156, 244-246) (Fig. 1). Finally, via these two domains, Vif recruits an active E3 ubiquitin ligase complex composed of EloC, EloB, Cul5, Nedd8, and Rbx2 (256, 257) that induces the polyubiquitylation of hA3G and hA3F and subsequently directs them to the 26S proteasome for degradation (Fig. 3). However, a recent report suggested that hA3G needs Vif polyubiquitylation, rather than its own polyubiquitylation, to be degraded (45).
The interaction of HIV-1 Vif with hA3G and hA3F maps to its N-terminal region, but different motifs are required for interactions with these two restriction factors (Fig. 1) (265). Residues 40 to 71 contain a nonlinear binding site for hA3G in which the highly conserved His42 and His43 residues are of crucial importance; however, a mutation of these residues does not affect the binding and degradation of hA3F (157). In keeping with these results, Vif residues 40YRHHY44 and 14DRMR17 were demonstrated to be essential for binding to hA3G and hA3F, respectively (189, 248). Furthermore, conserved Trp residues 11 and 79 are required for the efficient suppression of the antiviral activity of hA3F but not that of hA3G (229). However, the 40YRHHY44 and 14DRMR17 motifs are not conserved among the HIV-2, SIVmac, and SIVAgm Vif proteins (88). He and coworkers identified two motifs, 55VxIPLx4-5LxΦx2YWxL72 and 74TGERxW79, in HIV-1 Vif that are highly conserved among HIV-1, HIV-2, and various SIV proteins but not in the bovine immunodeficiency virus, FIV, or visna virus Vif proteins (88). The 55VxIPLx4-5LxΦx2YWxL72 motif is required for efficient interactions with and the suppression of both hA3G and hA3F, whereas the 74TGERxW79 motif is important mainly for hA3F interactions and suppression (88, 248). Those studies indicated that multiple motifs in the N-terminal region of Vif are involved in interactions with hA3G and hA3F. In addition, mutations in the 161PPLP164 motif, which is located in the C-terminal region of Vif (Fig. 1), reduce Vif binding to hA3G and the degradation of this restriction factor without affecting the interaction of Vif with Elongin C and Cullin5 (54). Interestingly, the level of incorporation of hA3G into WT HIV-1 particles can be increased by a peptide mimicking the Vif PPLP dimerization domain, and this peptide displays anti-HIV-1 activity (158). In addition, a small Vif antagonist that increases the cellular level of hA3G and its incorporation into virions in a Vif-dependent manner, without being a general inhibitor of the proteasome-mediated protein degradation, has been identified (164). This compound enhances the degradation of Vif in an hA3G-dependent manner.
In hA3G, the 128DPD130 motif plays a crucial role in the interaction with HIV-1 Vif, and the immediately adjacent residues 124 to 127 are important for the packaging of hA3G into progeny virions (102). This region of hA3G interacts with the 40YRHHY44 motif of Vif, while the region of hA3F interacting with the 14DRMR17 Vif motif was mapped between amino acids 283 and 300 (190). However, while amino acids 105 to 156 of hA3G were found to be sufficient for its interaction with Vif, amino acids 157 to 245 were required for its degradation (264). In addition, the phosphorylation of residue Thr32 of hA3G by protein kinase A reduces its binding to Vif and its subsequent degradation and thus promotes its anti-HIV-1 activity (209).
RNA-binding properties of Vif are essential to HIV-1 replication in nonpermissive cells, and the fact that Vif is associated exclusively with unspliced genomic RNA is a strong indication that the interaction of Vif with viral RNA is crucial for its function (49, 261). It is still unknown whether Vif binding to RNA might play a role in the degradation of hA3G and hA3F. However, both proteins are part of a common RNP localized in the processing bodies and/or stress granules of infected cells (40, 69, 70, 126, 151, 240). Indeed, several analyses have indicated that hA3G is associated mainly with mRNAs (including hA3G and HIV-1 gag mRNAs) that shuttle between translationally active polyribosomes, mRNA-processing bodies, dormant stress granules, and Staufen granules (40, 69, 70, 126, 151, 240). Interestingly, Vif localizes in processing bodies only in the presence of hA3G and hA3F, suggesting that Vif is recruited to processing bodies due to its interactions with these proteins (69, 70, 126, 151, 240). Regarding Vif functions, it is possible that the recruitment of hA3G and hA3F takes place in the processing bodies prior to proteasomal degradation. On the other hand, during viral infection, the vast majority of mRNAs and proteins present in RNPs are not found in viral particles, suggesting that competition operates within these various RNA granules with viral genomic RNA and that HIV-1 RNA and APOBEC protein trafficking converge to these compartments (40, 69, 70, 126, 151, 240).
Interestingly, hA3G has been detected in high-molecular-mass (HMM) (5- to 15-MDa) ribonucleic complexes in the cytoplasm of H9 T-cell lines and activated T cells (39, 127, 222) (Fig. 4). In these complexes, the deaminase activity of hA3G has been shown to be inactivated. Deaminase activity can be artificially restored by treating HMM complexes with ribonuclease A, thus converting the HMM complexes to low-molecular-mass (LMM) complexes, suggesting that RNA plays an important role in HMM complexes assembly (39). Additionally, HMM complexes have been shown to protect against Alu retrotransposition by sequestering Alu RNA away from its replication machinery (19, 40, 101) (Fig. 4). On the other hand, hA3G proteins present in LMM complexes have been shown to function as a potent postentry restriction factor that inhibits the replication of incoming viruses (39, 183) (Fig. 4), explaining why resting T cells that contain mainly LMM complexes are resistant to HIV-1 infection. Conversely, the activation of resting T cells converts hA3G-containing LMM complexes into inactivated HMM complexes (39, 127, 222).
Degradation is not the only mechanism leading to a reduction of the intracellular hA3G concentration. Mariani and coworkers reported a 4.6-fold reduction in levels of hA3G synthesis in the presence of Vif (150), and Vif has been shown to inhibit the translation of hA3G mRNA (114, 221). The mechanism underlying this inhibition is unknown, but it might involve the RNA-binding properties of Vif (see above) (Fig. 3, step 1).
Vif Prevents Incorporation of hA3G in HIV-1 Particles
Mutations of conserved phosphorylation sites in Vif that impair viral replication do not affect hA3G degradation, indicating that Vif has another function(s) (155). Accordingly, HIV-1 Vif is able to inhibit the packaging and antiviral activity of a degradation-resistant hA3G variant (175). Furthermore, several studies suggested that Vif prevents the packaging of hA3G into HIV-1 independently of the reduction of its intracellular concentration (114, 115, 150, 204) (Fig. 3, step 3). By cotransfecting WT or Δvif HIV-1 and various amounts of hA3G-expressing vector, it is possible to compare WT and Δvif virions produced from cells with similar hA3G levels. Under these conditions, WT virions incorporated less hA3G than Δvif virions, suggesting that Vif can directly exclude hA3G from virions (114, 204). Interestingly, the Bet protein of foamy viruses inhibits APOBEC-3 proteins without affecting their steady-state levels in the producer cells even though it prevents their packaging (191). In the same line, SIVagm can inhibit hA3G activity without inducing its degradation (228).
It has been proposed that the incorporation of hA3G into virions is dependent on the recognition of the stem-loop of the dimerization initiation site in the leader region (120). During our analysis of Vif binding to viral RNA, we found that the dimerization initiation site is one of the secondary binding sites in the leader region that are bound at high Vif concentrations (89). Strikingly, Vif seems to have a better affinity for RNA than Gag (261), and one can imagine that at early stages of assembly and prior to the multimerization of Pr55Gag on the viral RNA scaffold, the leader region would be covered with Vif, masking RNA elements otherwise recognized by hA3G. Quite interestingly, while the bindings of Vif and Gag on RNA seem to be mutually exclusive, a recent study showed that Gag and hA3G can form a common complex with viral RNA (108), with cooperative binding to the RNA-packaging signal (Psi). In addition, several recent studies indicated that the incorporation of hA3G and hA3F requires the packaging of 7SL RNA (237, 238), the small RNA normally found in the signal recognition particle that is selectively packaged in HIV-1 particles (98), but this finding was questioned by another group (9). To gain a better understanding of the interplay between Vif, hA3G, and RNA, it would be interesting to know whether Vif destabilizes the interaction between hA3G and viral or 7SL RNAs.
Moreover, it has been proposed that the inhibition of the annealing of 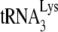 to the PBS mediated by the NC domain of Pr55Gag is (one of) the main deaminase-independent antiviral mechanism(s) of hA3G and hA3F (80, 134, 253). This antiviral mechanism might be compensated for by Vif-mediated
to the PBS mediated by the NC domain of Pr55Gag is (one of) the main deaminase-independent antiviral mechanism(s) of hA3G and hA3F (80, 134, 253). This antiviral mechanism might be compensated for by Vif-mediated 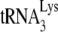 annealing (90).
annealing (90).
Vif Inhibits the Activity of Intravirion hA3G
Finally, significant amounts of A3G were detected in virions produced in 293T cells transiently expressing WT HIV-1 and A3G, showing that under certain conditions, Vif does not lead to the complete depletion of A3G from viral cores (203, 204). In every case, virion infectivity was restored, suggesting that Vif functions both in the cytoplasm to prevent the encapsidation of bulk A3G and in the virion to counteract core-associated A3G (Fig. 3). This idea is corroborated by a study showing that the production of infectious Vif does not require a depletion of hA3G from virus-producing cells (115).
Vif associated with viral nucleic acids during reverse transcription could inhibit the cytidine deamination of nascent proviral DNA by hA3G. Indeed, Santa-Marta and coworkers showed that hA3G-mediated hypermutation in Escherichia coli was inhibited by Vif (196). The fact that the ubiquitin-proteasome system is absent in bacterial cells and the fact that inhibition by Vif was correlated with binding to hA3G strongly argue in favor of a direct effect of Vif on hA3G enzymatic activity, which could involve the masking of nucleic acid substrates that are tightly associated with Vif (Fig. 3, step 6). Indeed, an important fraction of the Vif-binding sites that we identified in the 5′ region of HIV-1 genomic RNA correspond to hypermutated sites in Δvif viruses (89), and Vif also binds with significant affinity (Kd [dissociation constant] of 34 to 38 nM) to the consensus hA3G and hA3F DNA target sites (14). Thus, Vif could interfere with hA3G by limiting its access to the neosynthesized viral DNA. Alternatively, it might locally limit the RNase H activity of HIV-1 reverse transcriptase that is necessary to restore the deaminase activity of hA3G, which is otherwise inhibited upon binding to HIV-1 genomic RNA (218).
CONCLUSIONS
Despite a considerable amount of work demonstrating that the cell-dependent Δvif phenotype correlated with aberrant morphology of the viral core and defects in reverse transcription, the molecular mechanism(s) underlying the functions of Vif remained mysterious for a long time. The identification of hA3G by Sheehy and coworkers (203), soon followed by the identification of hA3F, as a cellular antiviral factor neutralized by Vif finally provided the missing piece of the puzzle. Most of the subsequent work focused on the hypermutation of HIV-1 induced by these cytidine deaminases and on the mechanism allowing Vif to induce the degradation of these antiviral factors by the proteasome.
Biochemical studies of the Vif protein characterized its association with a number of molecular machineries (11, 49, 116, 261, 268), but attention was focused mostly on the ability of Vif to form E3-like ubiquitin ligase, since a clear correlation was made between this property and the rescue of viral infectivity in nonpermissive cells (256). However, the complex intracellular trafficking and regulation of expression of Vif (65, 235, 240) as well as its uncharacterized effects on the cytoskeleton and the cell cycle (116, 194, 236) suggest that its functions are more diverse than previously thought. Consistently, properties of Vif linked to viral assembly and the increasing number of results reporting that hA3G degradation or virion exclusion is not a universal mechanism of viral resistance (114, 115, 196, 221, 228) strongly suggest that Vif functions against hA3G and hA3F may involve a modulation of HIV-1 assembly by interacting with RNA or protein components of the viral core (3, 11, 23, 28, 89, 104, 118, 119, 212, 261). It is widely recognized that HIV-1 virions incorporate a small quantity of the Vif protein (37, 68, 215), but its precise function in RNP assembly or in reverse transcription remains to be demonstrated. Thus, if subtle interactions take place between Vif and the viral RNP, their impact on assembly could be difficult to prove experimentally or to interpret given our limited knowledge of the structural rearrangements occurring during HIV-1 assembly and maturation. With this perspective, in vitro studies aiming at characterizing the interaction between Vif, viral RNA, polyprotein precursors, and mature components of the core would lead to a better understanding of the function of Vif in cytoplasmic viral assembly complexes and in virions.
A general trend in innate immunity is the recognition of a pathogen-associated molecular pattern by cellular restriction factors rather than specific structural determinants (86). Given the broad range of viral genetic information susceptible to being affected by APOBEC-3 family factors (1, 47, 56, 149, 160, 197, 202, 223), future investigations should aim at characterizing what are the common features of viral RNPs targeted by these restriction factors and mediating their encapsidation (126, 218).
Acknowledgments
We are indebted to J. Mak for helpful suggestions and comments.
This work was supported by a grant from the Agence Nationale de Recherches sur le SIDA. S.H. was a fellow of Sidaction. G.M. is supported by fellowships from the French Ministry of Research and Technology and Sidaction.
Biography
 Simon Henriet was born in Noumea, New Caledonia, on June 10, 1980. He began to study in New Caledonia and then he moved to France to attend Louis Pasteur University in Strasbourg, France. In 2002, he received a Master's degree in Molecular and Cellular Biology. The same year, he started to work on the human immunodeficiency virus type 1 (HIV-1) viral infectivity factor (Vif) protein in the laboratory of Chantal and Bernard Ehresmann. During his Ph.D. work, under the supervision of Roland Marquet (CNRS, Strasbourg, France), he studied the binding of Vif on HIV-1 RNA and the role of the interaction in the viral replication and assembly. He defended his thesis in 2006, and he is now working as a postdoctoral researcher in the laboratory of Daniel Chourrout at the Sars Centre (Bergen, Norway). His current research aims at understanding the evolution of Gypsy-like retroviruses in a host with a compact genome, the tunicate Oikopleura dioica.
Simon Henriet was born in Noumea, New Caledonia, on June 10, 1980. He began to study in New Caledonia and then he moved to France to attend Louis Pasteur University in Strasbourg, France. In 2002, he received a Master's degree in Molecular and Cellular Biology. The same year, he started to work on the human immunodeficiency virus type 1 (HIV-1) viral infectivity factor (Vif) protein in the laboratory of Chantal and Bernard Ehresmann. During his Ph.D. work, under the supervision of Roland Marquet (CNRS, Strasbourg, France), he studied the binding of Vif on HIV-1 RNA and the role of the interaction in the viral replication and assembly. He defended his thesis in 2006, and he is now working as a postdoctoral researcher in the laboratory of Daniel Chourrout at the Sars Centre (Bergen, Norway). His current research aims at understanding the evolution of Gypsy-like retroviruses in a host with a compact genome, the tunicate Oikopleura dioica.
 Gaëlle Mercenne was born in Louviers, France, on March 17, 1982. She obtained her Master's degree in Molecular and Cellular Biology in 2005 at Louis Pasteur University (Strasbourg, France). She is now a Ph.D. student working on the team of Roland Marquet (IBMC, Strasbourg, France), where she studies the translational regulation of hA3G by HIV-1 Vif. She will defend her Ph.D. thesis in a few months and plans to pursue a postdoctoral fellowship in the United States.
Gaëlle Mercenne was born in Louviers, France, on March 17, 1982. She obtained her Master's degree in Molecular and Cellular Biology in 2005 at Louis Pasteur University (Strasbourg, France). She is now a Ph.D. student working on the team of Roland Marquet (IBMC, Strasbourg, France), where she studies the translational regulation of hA3G by HIV-1 Vif. She will defend her Ph.D. thesis in a few months and plans to pursue a postdoctoral fellowship in the United States.
 Serena Bernacchi was born in Milan, Italy, in 1973. She attended Università degli Studi di Milano, where she obtained a Master's degree in Physics in 1998. She then joined the group of Yves Mely in Louis Pasteur University (Strasbourg, France). She worked on molecular analysis of the chaperone activity of the HIV-1 nucleocapsid p7 protein with transactivation region sequences by fluorescence techniques, and in 2002 earned a Ph.D. in Biophysics. She spent two years in the laboratory of Joerg Langowski at the German Cancer Research Center (DKFZ; Heidelberg, Germany), studying conformational equilibrium of HIV-1 genomic RNA dimerization initiation site and the infection process of simian virus 40 by fluorescence correlation spectroscopy. She then joined the laboratory of Roland Marquet (IBMC-CNRS, Strasbourg, France), where she obtained a Junior Scientist position. She currently studies the role of HIV-1 Vif in viral assembly, replication, and inhibition of the APOBEC-3G antiviral factor.
Serena Bernacchi was born in Milan, Italy, in 1973. She attended Università degli Studi di Milano, where she obtained a Master's degree in Physics in 1998. She then joined the group of Yves Mely in Louis Pasteur University (Strasbourg, France). She worked on molecular analysis of the chaperone activity of the HIV-1 nucleocapsid p7 protein with transactivation region sequences by fluorescence techniques, and in 2002 earned a Ph.D. in Biophysics. She spent two years in the laboratory of Joerg Langowski at the German Cancer Research Center (DKFZ; Heidelberg, Germany), studying conformational equilibrium of HIV-1 genomic RNA dimerization initiation site and the infection process of simian virus 40 by fluorescence correlation spectroscopy. She then joined the laboratory of Roland Marquet (IBMC-CNRS, Strasbourg, France), where she obtained a Junior Scientist position. She currently studies the role of HIV-1 Vif in viral assembly, replication, and inhibition of the APOBEC-3G antiviral factor.
 Jean-Christophe Paillart was born in Montargis, France, on January 17, 1968. He attended Louis Pasteur University (Strasbourg, France), where he received his Master's degree in Molecular and Cellular Biology in 1991. He then began his graduate career in the laboratory of Bernard and Chantal Ehresmann, studying the dimerization of HIV-1 genomic RNA. After a two-year postdoctoral fellowship conducted in the laboratory of Heinrich Göttlinger at the Dana Farber Cancer Institute (Boston, MA), in which the role of matrix protein in HIV-1 assembly was studied, he joined the laboratory of Roland Marquet (IBMC-CNRS, Strasbourg, France), where he is currently Senior Researcher. His research activities cover many aspects of molecular virology and in particular the role of the dimerization of HIV-1 genomic RNA in viral replication, as well as the relationship between the Vif and APOBEC-3G antiviral factor in regards to viral assembly.
Jean-Christophe Paillart was born in Montargis, France, on January 17, 1968. He attended Louis Pasteur University (Strasbourg, France), where he received his Master's degree in Molecular and Cellular Biology in 1991. He then began his graduate career in the laboratory of Bernard and Chantal Ehresmann, studying the dimerization of HIV-1 genomic RNA. After a two-year postdoctoral fellowship conducted in the laboratory of Heinrich Göttlinger at the Dana Farber Cancer Institute (Boston, MA), in which the role of matrix protein in HIV-1 assembly was studied, he joined the laboratory of Roland Marquet (IBMC-CNRS, Strasbourg, France), where he is currently Senior Researcher. His research activities cover many aspects of molecular virology and in particular the role of the dimerization of HIV-1 genomic RNA in viral replication, as well as the relationship between the Vif and APOBEC-3G antiviral factor in regards to viral assembly.
 Roland Marquet was born in Belgium in 1961. He earned a B.Sc. in Chemistry in 1983 and a Ph.D. in Chemistry in 1989 from the University of Liège (Belgium), where he worked with Claude Houssier on the interaction of small ligands with DNA and chromatin. He then joined the IBMC (CNRS, Strasbourg, France), where he started to study the dimerization and the initiation of reverse transcription of HIV-1 genomic RNA in the laboratory of Bernard and Chantal Ehresmann. He was recruited by the CNRS as Senior Scientist (1992) and promoted to Director of Research in 1999 and to First Class Director of Research in 2006. In 2008, he spent 8 months as a visiting scientist at the Burnet Institute (Melbourne, Victoria, Australia). He is interested in the selection, packaging, and replication of viral RNAs and in the inhibition of these processes. His laboratory combines chemical, biophysical, biochemical, and cellular approaches.
Roland Marquet was born in Belgium in 1961. He earned a B.Sc. in Chemistry in 1983 and a Ph.D. in Chemistry in 1989 from the University of Liège (Belgium), where he worked with Claude Houssier on the interaction of small ligands with DNA and chromatin. He then joined the IBMC (CNRS, Strasbourg, France), where he started to study the dimerization and the initiation of reverse transcription of HIV-1 genomic RNA in the laboratory of Bernard and Chantal Ehresmann. He was recruited by the CNRS as Senior Scientist (1992) and promoted to Director of Research in 1999 and to First Class Director of Research in 2006. In 2008, he spent 8 months as a visiting scientist at the Burnet Institute (Melbourne, Victoria, Australia). He is interested in the selection, packaging, and replication of viral RNAs and in the inhibition of these processes. His laboratory combines chemical, biophysical, biochemical, and cellular approaches.
REFERENCES
- 1.Abudu, A., A. Takaori-Kondo, T. Izumi, K. Shirakawa, M. Kobayashi, A. Sasada, K. Fukunaga, and T. Uchiyama. 2006. Murine retrovirus escapes from murine APOBEC3 via two distinct novel mechanisms. Curr. Biol. 161565-1570. [DOI] [PubMed] [Google Scholar]
- 2.Aguiar, R. S., N. Lovsin, A. Tanuri, and B. M. Peterlin. 2008. Vpr.A3A chimera inhibits HIV replication. J. Biol. Chem. 2832518-2525. [DOI] [PubMed] [Google Scholar]
- 3.Akari, H., M. Fujita, S. Kao, M. A. Khan, M. Shehu-Xhilaga, A. Adachi, and K. Strebel. 2004. High level expression of human immunodeficiency virus type-1 Vif inhibits viral infectivity by modulating proteolytic processing of the Gag precursor at the p2/nucleocapsid processing site. J. Biol. Chem. 27912355-12362. [DOI] [PubMed] [Google Scholar]
- 4.Alexander, L., M. J. Aquino-DeJesus, M. Chan, and W. A. Andiman. 2002. Inhibition of human immunodeficiency virus type 1 (HIV-1) replication by a two-amino-acid insertion in HIV-1 Vif from a nonprogressing mother and child. J. Virol. 7610533-10539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Anderson, J. L., and T. J. Hope. 2008. APOBEC3G restricts early HIV-1 replication in the cytoplasm of target cells. Virology 3751-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arhel, N. J., S. Souquere-Besse, S. Munier, P. Souque, S. Guadagnini, S. Rutherford, M. C. Prevost, T. D. Allen, and P. Charneau. 2007. HIV-1 DNA Flap formation promotes uncoating of the pre-integration complex at the nuclear pore. EMBO J. 263025-3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arrigo, S. J., and I. S. Chen. 1991. Rev is necessary for translation but not cytoplasmic accumulation of HIV-1 vif, vpr, and env/vpu 2 RNAs. Genes Dev. 5808-819. [DOI] [PubMed] [Google Scholar]
- 8.Auclair, J. R., K. M. Green, S. Shandilya, J. E. Evans, M. Somasundaran, and C. A. Schiffer. 2007. Mass spectrometry analysis of HIV-1 Vif reveals an increase in ordered structure upon oligomerization in regions necessary for viral infectivity. Proteins 69270-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bach, D., S. Peddi, B. Mangeat, A. Lakkaraju, K. Strub, and D. Trono. 2008. Characterization of APOBEC3G binding to 7SL RNA. Retrovirology 554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Balaji, S., R. Kalpana, and P. Shapshak. 2006. Paradigm development: comparative and predictive 3D modeling of HIV-1 virion infectivity factor (Vif). Bioinformation 1290-309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bardy, M., B. Gay, S. Pebernard, N. Chazal, M. Courcoul, R. Vigne, E. Decroly, and P. Boulanger. 2001. Interaction of human immunodeficiency virus type 1 Vif with Gag and Gag-Pol precursors: co-encapsidation and interference with viral protease-mediated Gag processing. J. Gen. Virol. 822719-2733. [DOI] [PubMed] [Google Scholar]
- 12.Barraud, P., J. C. Paillart, R. Marquet, and C. Tisne. 2008. Advances in the structural understanding of Vif proteins. Curr. HIV Res. 691-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beale, R. C., S. K. Petersen-Mahrt, I. N. Watt, R. S. Harris, C. Rada, and M. S. Neuberger. 2004. Comparison of the differential context-dependence of DNA deamination by APOBEC enzymes: correlation with mutation spectra in vivo. J. Mol. Biol. 337585-596. [DOI] [PubMed] [Google Scholar]
- 14.Bernacchi, S., S. Henriet, P. Dumas, J. C. Paillart, and R. Marquet. 2007. RNA and DNA binding properties of HIV-1 Vif protein: a fluorescence study. J. Biol. Chem. 28226361-26368. [DOI] [PubMed] [Google Scholar]
- 15.Bishop, K. N., R. K. Holmes, and M. H. Malim. 2006. Antiviral potency of APOBEC proteins does not correlate with cytidine deamination. J. Virol. 808450-8458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bishop, K. N., R. K. Holmes, A. M. Sheehy, N. O. Davidson, S. J. Cho, and M. H. Malim. 2004. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 141392-1396. [DOI] [PubMed] [Google Scholar]
- 17.Bishop, K. N., M. Verma, E. Y. Kim, S. M. Wolinsky, and M. H. Malim. 2008. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 4e1000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blumenzweig, I., L. Baraz, A. Friedler, U. H. Danielson, C. Gilon, M. Steinitz, and M. Kotler. 2002. HIV-1 Vif-derived peptide inhibits drug-resistant HIV proteases. Biochem. Biophys. Res. Commun. 292832-840. [DOI] [PubMed] [Google Scholar]
- 19.Bogerd, H. P., H. L. Wiegand, A. E. Hulme, J. L. Garcia-Perez, K. S. O'Shea, J. V. Moran, and B. R. Cullen. 2006. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc. Natl. Acad. Sci. USA 1038780-8785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Borman, A. M., C. Quillent, P. Charneau, C. Dauguet, and F. Clavel. 1995. Human immunodeficiency virus type 1 Vif− mutant particles from restrictive cells: role of Vif in correct particle assembly and infectivity. J. Virol. 692058-2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bour, S., and K. Strebel. 2003. The HIV-1 Vpu protein: a multifunctional enhancer of viral particle release. Microbes Infect. 51029-1039. [DOI] [PubMed] [Google Scholar]
- 22.Bourara, K., T. J. Liegler, and R. M. Grant. 2007. Target cell APOBEC3C can induce limited G-to-A mutation in HIV-1. PLoS Pathog. 31477-1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bouyac, M., M. Courcoul, G. Bertoia, Y. Baudat, D. Gabuzda, D. Blanc, N. Chazal, P. Boulanger, J. Sire, R. Vigne, and B. Spire. 1997. Human immunodeficiency virus type 1 Vif protein binds to the Pr55Gag precursor. J. Virol. 719358-9365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bouyac, M., F. Rey, M. Nascimbeni, M. Courcoul, J. Sire, D. Blanc, F. Clavel, R. Vigne, and B. Spire. 1997. Phenotypically Vif− human immunodeficiency virus type 1 is produced by chronically infected restrictive cells. J. Virol. 712473-2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Briggs, J. A., K. Grunewald, B. Glass, F. Forster, H. G. Krausslich, and S. D. Fuller. 2006. The mechanism of HIV-1 core assembly: insights from three-dimensional reconstructions of authentic virions. Structure 1415-20. [DOI] [PubMed] [Google Scholar]
- 26.Browne, E. P., and D. R. Littman. 2008. Species-specific restriction of Apobec3-mediated hypermutation. J. Virol. 821305-1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Camaur, D., and D. Trono. 1996. Characterization of human immunodeficiency virus type 1 Vif particle incorporation. J. Virol. 706106-6111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cancio, R., S. Spadari, and G. Maga. 2004. Vif is an auxiliary factor of the HIV-1 reverse transcriptase and facilitates abasic site bypass. Biochem. J. 383475-482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caride, E., R. M. Brindeiro, E. G. Kallas, C. A. de Sa, W. A. Eyer-Silva, E. Machado, and A. Tanuri. 2002. Sexual transmission of HIV-1 isolate showing G→A hypermutation. J. Clin. Virol. 23179-189. [DOI] [PubMed] [Google Scholar]
- 30.Carr, J. M., C. Coolen, A. J. Davis, C. J. Burrell, and P. Li. 2008. Human immunodeficiency virus 1 (HIV-1) virion infectivity factor (Vif) is part of reverse transcription complexes and acts as an accessory factor for reverse transcription. Virology 372147-156. [DOI] [PubMed] [Google Scholar]
- 31.Carr, J. M., A. J. Davis, C. Coolen, K. Cheney, C. J. Burrell, and P. Li. 2006. Vif-deficient HIV reverse transcription complexes (RTCs) are subject to structural changes and mutation of RTC-associated reverse transcription products. Virology 35180-91. [DOI] [PubMed] [Google Scholar]
- 32.Chatel-Chaix, L., L. Abrahamyan, C. Fréchina, A. J. Mouland, and L. DesGroseillers. 2007. The host protein Staufen1 participates in human immunodeficiency virus type 1 assembly in live cells by influencing pr55Gag multimerization. J. Virol. 816216-6230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chatel-Chaix, L., J. F. Clement, C. Martel, V. Beriault, A. Gatignol, L. DesGroseillers, and A. J. Mouland. 2004. Identification of Staufen in the human immunodeficiency virus type 1 Gag ribonucleoprotein complex and a role in generating infectious viral particles. Mol. Cell. Biol. 242637-2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chatterji, U., C. K. Grant, and J. H. Elder. 2000. Feline immunodeficiency virus Vif localizes to the nucleus. J. Virol. 742533-2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chelico, L., P. Pham, P. Calabrese, and M. F. Goodman. 2006. APOBEC3G DNA deaminase acts processively 3′→5′ on single-stranded DNA. Nat. Struct. Mol. Biol. 13392-399. [DOI] [PubMed] [Google Scholar]
- 36.Chen, K. M., E. Harjes, P. J. Gross, A. Fahmy, Y. Lu, K. Shindo, R. S. Harris, and H. Matsuo. 2008. Structure of the DNA deaminase domain of the HIV-1 restriction factor APOBEC3G. Nature 452116-119. [DOI] [PubMed] [Google Scholar]
- 37.Chertova, E., O. Chertov, L. V. Coren, J. D. Roser, C. M. Trubey, J. W. Bess, Jr., R. C. Sowder II, E. Barsov, B. L. Hood, R. J. Fisher, K. Nagashima, T. P. Conrads, T. D. Veenstra, J. D. Lifson, and D. E. Ott. 2006. Proteomic and biochemical analysis of purified human immunodeficiency virus type 1 produced from infected monocyte-derived macrophages. J. Virol. 809039-9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chiu, Y. L., and W. C. Greene. 2008. The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu. Rev. Immunol. 26317-353. [DOI] [PubMed] [Google Scholar]
- 39.Chiu, Y. L., V. B. Soros, J. F. Kreisberg, K. Stopak, W. Yonemoto, and W. C. Greene. 2005. Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells. Nature 435108-114. [DOI] [PubMed] [Google Scholar]
- 40.Chiu, Y. L., H. E. Witkowska, S. C. Hall, M. Santiago, V. B. Soros, C. Esnault, T. Heidmann, and W. C. Greene. 2006. High-molecular-mass APOBEC3G complexes restrict Alu retrotransposition. Proc. Natl. Acad. Sci. USA 10315588-15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Conticello, S. G., R. S. Harris, and M. S. Neuberger. 2003. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr. Biol. 132009-2013. [DOI] [PubMed] [Google Scholar]
- 42.Courcoul, M., C. Patience, F. Rey, D. Blanc, A. Harmache, J. Sire, R. Vigne, and B. Spire. 1995. Peripheral blood mononuclear cells produce normal amounts of defective Vif− human immunodeficiency virus type 1 particles which are restricted for the preretrotranscription steps. J. Virol. 692068-2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dafonseca, S., P. Coric, B. Gay, S. S. Hong, S. Bouaziz, and P. Boulanger. 2008. The inhibition of assembly of HIV-1 virus-like particles by 3-O-(3′,3′-dimethylsuccinyl) betulinic acid (DSB) is counteracted by Vif and requires its zinc-binding domain. Virol. J. 5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dang, Y., L. M. Siew, X. Wang, Y. Han, R. Lampen, and Y. H. Zheng. 2008. Human cytidine deaminase APOBEC3H restricts HIV-1 replication. J. Biol. Chem. 28311606-11614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dang, Y., L. M. Siew, and Y. H. Zheng. 2008. APOBEC3G is degraded by the proteasomal pathway in a Vif-dependent manner without being polyubiquitylated. J. Biol. Chem. 28313124-13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dang, Y., X. Wang, W. J. Esselman, and Y. H. Zheng. 2006. Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J. Virol. 8010522-10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Delebecque, F., R. Suspene, S. Calattini, N. Casartelli, A. Saib, A. Froment, S. Wain-Hobson, A. Gessain, J. P. Vartanian, and O. Schwartz. 2006. Restriction of foamy viruses by APOBEC cytidine deaminases. J. Virol. 80605-614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Desrosiers, R. C., J. D. Lifson, J. S. Gibbs, S. C. Czajak, A. Y. Howe, L. O. Arthur, and R. P. Johnson. 1998. Identification of highly attenuated mutants of simian immunodeficiency virus. J. Virol. 721431-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dettenhofer, M., S. Cen, B. A. Carlson, L. Kleiman, and X. F. Yu. 2000. Association of human immunodeficiency virus type 1 Vif with RNA and its role in reverse transcription. J. Virol. 748938-8945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dettenhofer, M., and X. F. Yu. 1999. Highly purified human immunodeficiency virus type 1 reveals a virtual absence of Vif in virions. J. Virol. 731460-1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dimitrov, D. S., R. L. Willey, H. Sato, L. J. Chang, R. Blumenthal, and M. A. Martin. 1993. Quantitation of human immunodeficiency virus type 1 infection kinetics. J. Virol. 672182-2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Doehle, B. P., A. Schafer, and B. R. Cullen. 2005. Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to HIV-1 Vif. Virology 339281-288. [DOI] [PubMed] [Google Scholar]
- 53.Doehle, B. P., A. Schafer, H. L. Wiegand, H. P. Bogerd, and B. R. Cullen. 2005. Differential sensitivity of murine leukemia virus to APOBEC3-mediated inhibition is governed by virion exclusion. J. Virol. 798201-8207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Donahue, J. P., M. L. Vetter, N. A. Mukhtar, and R. T. D'Aquila. 2008. The HIV-1 Vif PPLP motif is necessary for human APOBEC3G binding and degradation. Virology 37749-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dornadula, G., S. Yang, R. J. Pomerantz, and H. Zhang. 2000. Partial rescue of the Vif-negative phenotype of mutant human immunodeficiency virus type 1 strains from nonpermissive cells by intravirion reverse transcription. J. Virol. 742594-2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Esnault, C., J. Millet, O. Schwartz, and T. Heidmann. 2006. Dual inhibitory effects of APOBEC family proteins on retrotransposition of mammalian endogenous retroviruses. Nucleic Acids Res. 341522-1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Farrow, M. A., M. Somasundaran, C. Zhang, D. Gabuzda, J. L. Sullivan, and T. C. Greenough. 2005. Nuclear localization of HIV type 1 Vif isolated from a long-term asymptomatic individual and potential role in virus attenuation. AIDS Res. Hum. Retrovir. 21565-574. [DOI] [PubMed] [Google Scholar]
- 58.Feng, Y. X., S. Campbell, D. Harvin, B. Ehresmann, C. Ehresmann, and A. Rein. 1999. The human immunodeficiency virus type 1 Gag polyprotein has nucleic acid chaperone activity: possible role in dimerization of genomic RNA and placement of tRNA on the primer binding site. J. Virol. 734251-4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fitzgibbon, J. E., S. Mazar, and D. T. Dubin. 1993. A new type of G→A hypermutation affecting human immunodeficiency virus. AIDS Res. Hum. Retrovir. 9833-838. [DOI] [PubMed] [Google Scholar]
- 60.Fouchier, R. A., J. H. Simon, A. B. Jaffe, and M. H. Malim. 1996. Human immunodeficiency virus type 1 Vif does not influence expression or virion incorporation of gag-, pol-, and env-encoded proteins. J. Virol. 708263-8269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fournier, C., J. C. Cortay, C. Carbonnelle, C. Ehresmann, R. Marquet, and P. Boulanger. 2002. The HIV-1 Nef protein enhances the affinity of reverse transcriptase for RNA in vitro. Virus Genes 25255-269. [DOI] [PubMed] [Google Scholar]
- 62.Friedler, A., N. Zakai, O. Karni, D. Friedler, C. Gilon, and A. Loyter. 1999. Identification of a nuclear transport inhibitory signal (NTIS) in the basic domain of HIV-1 Vif protein. J. Mol. Biol. 289431-437. [DOI] [PubMed] [Google Scholar]
- 63.Fu, W., R. J. Gorelick, and A. Rein. 1994. Characterization of human immunodeficiency virus type 1 dimeric RNA from wild-type and protease-defective virions. J. Virol. 685013-5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fu, W., and A. Rein. 1993. Maturation of dimeric viral RNA of Moloney murine leukemia virus. J. Virol. 675443-5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fujita, M., H. Akari, A. Sakurai, A. Yoshida, T. Chiba, K. Tanaka, K. Strebel, and A. Adachi. 2004. Expression of HIV-1 accessory protein Vif is controlled uniquely to be low and optimal by proteasome degradation. Microbes Infect. 6791-798. [DOI] [PubMed] [Google Scholar]
- 66.Gabuzda, D. H., K. Lawrence, E. Langhoff, E. Terwilliger, T. Dorfman, W. A. Haseltine, and J. Sodroski. 1992. Role of vif in replication of human immunodeficiency virus type 1 in CD4+ T lymphocytes. J. Virol. 666489-6495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gabuzda, D. H., H. Li, K. Lawrence, B. S. Vasir, K. Crawford, and E. Langhoff. 1994. Essential role of vif in establishing productive HIV-1 infection in peripheral blood T lymphocytes and monocyte/macrophages. J. Acquir. Immune Defic. Syndr. 7908-915. [PubMed] [Google Scholar]
- 68.Gaddis, N. C., E. Chertova, A. M. Sheehy, L. E. Henderson, and M. H. Malim. 2003. Comprehensive investigation of the molecular defect in vif-deficient human immunodeficiency virus type 1 virions. J. Virol. 775810-5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gallois-Montbrun, S., R. K. Holmes, C. M. Swanson, M. Fernandez-Ocana, H. L. Byers, M. A. Ward, and M. H. Malim. 2008. Comparison of cellular ribonucleoprotein complexes associated with the APOBEC3F and APOBEC3G antiviral proteins. J. Virol. 825636-5642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gallois-Montbrun, S., B. Kramer, C. M. Swanson, H. Byers, S. Lynham, M. Ward, and M. H. Malim. 2007. Antiviral protein APOBEC3G localizes to ribonucleoprotein complexes found in P bodies and stress granules. J. Virol. 812165-2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ganser-Pornillos, B. K., M. Yeager, and W. I. Sundquist. 2008. The structural biology of HIV assembly. Curr. Opin. Struct. Biol. 18203-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gay, B., J. Tournier, N. Chazal, C. Carriere, and P. Boulanger. 1998. Morphopoietic determinants of HIV-1 Gag particles assembled in baculovirus-infected cells. Virology 247160-169. [DOI] [PubMed] [Google Scholar]
- 73.Gluschankof, P., I. Mondor, H. R. Gelderblom, and Q. J. Sattentau. 1997. Cell membrane vesicles are a major contaminant of gradient-enriched human immunodeficiency virus type-1 preparations. Virology 230125-133. [DOI] [PubMed] [Google Scholar]
- 74.Goila-Gaur, R., M. A. Khan, E. Miyagi, S. Kao, and K. Strebel. 2007. Targeting APOBEC3A to the viral nucleoprotein complex confers antiviral activity. Retrovirology 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Goncalves, J., P. Jallepalli, and D. H. Gabuzda. 1994. Subcellular localization of the Vif protein of human immunodeficiency virus type 1. J. Virol. 68704-712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Goncalves, J., Y. Korin, J. Zack, and D. Gabuzda. 1996. Role of Vif in human immunodeficiency virus type 1 reverse transcription. J. Virol. 708701-8709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Goncalves, J., B. Shi, X. Yang, and D. Gabuzda. 1995. Biological activity of human immunodeficiency virus type 1 Vif requires membrane targeting by C-terminal basic domains. J. Virol. 697196-7204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gross, I., H. Hohenberg, T. Wilk, K. Wiegers, M. Grattinger, B. Muller, S. Fuller, and H. G. Krausslich. 2000. A conformational switch controlling HIV-1 morphogenesis. EMBO J. 19103-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
79.Guo, F., S. Cen, M. Niu, J. Saadatmand, and L. Kleiman. 2006. The inhibition of
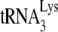 -primed reverse transcription by human APOBEC3G during human immunodeficiency virus type 1 replication. J. Virol. 8011710-11722. [DOI] [PMC free article] [PubMed] [Google Scholar]
-primed reverse transcription by human APOBEC3G during human immunodeficiency virus type 1 replication. J. Virol. 8011710-11722. [DOI] [PMC free article] [PubMed] [Google Scholar] -
80.Guo, F., S. Cen, M. Niu, Y. Yang, R. J. Gorelick, and L. Kleiman. 2007. The interaction of APOBEC3G with human immunodeficiency virus type 1 nucleocapsid inhibits
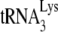 annealing to viral RNA. J. Virol. 8111322-11331. [DOI] [PMC free article] [PubMed] [Google Scholar]
annealing to viral RNA. J. Virol. 8111322-11331. [DOI] [PMC free article] [PubMed] [Google Scholar] - 81.Hache, G., M. T. Liddament, and R. S. Harris. 2005. The retroviral hypermutation specificity of APOBEC3F and APOBEC3G is governed by the C-terminal DNA cytosine deaminase domain. J. Biol. Chem. 28010920-10924. [DOI] [PubMed] [Google Scholar]
- 82.Han, Y., X. Wang, Y. Dang, and Y. H. Zheng. 2008. APOBEC3G and APOBEC3F require an endogenous cofactor to block HIV-1 replication. PLoS Pathog. 4e1000095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Harmache, A., P. Russo, F. Guiguen, C. Vitu, M. Vignoni, M. Bouyac, C. Hieblot, M. Pepin, R. Vigne, and M. Suzan. 1996. Requirement of caprine arthritis encephalitis virus vif gene for in vivo replication. Virology 224246-255. [DOI] [PubMed] [Google Scholar]
- 84.Harmache, A., P. Russo, C. Vitu, F. Guiguen, J. F. Mornex, M. Pepin, R. Vigne, and M. Suzan. 1996. Replication in goats in vivo of caprine arthritis-encephalitis virus deleted in vif or tat genes: possible use of these deletion mutants as live vaccines. AIDS Res. Hum. Retrovir. 12409-411. [DOI] [PubMed] [Google Scholar]
- 85.Harris, R. S., K. N. Bishop, A. M. Sheehy, H. M. Craig, S. K. Petersen-Mahrt, I. N. Watt, M. S. Neuberger, and M. H. Malim. 2003. DNA deamination mediates innate immunity to retroviral infection. Cell 113803-809. [DOI] [PubMed] [Google Scholar]
- 86.Harris, R. S., and M. T. Liddament. 2004. Retroviral restriction by APOBEC proteins. Nat. Rev. Immunol. 4868-877. [DOI] [PubMed] [Google Scholar]
- 87.Hassaine, G., M. Courcoul, G. Bessou, Y. Barthalay, C. Picard, D. Olive, Y. Collette, R. Vigne, and E. Decroly. 2001. The tyrosine kinase Hck is an inhibitor of HIV-1 replication counteracted by the viral vif protein. J. Biol. Chem. 27616885-16893. [DOI] [PubMed] [Google Scholar]
- 88.He, Z., W. Zhang, G. Chen, R. Xu, and X. F. Yu. 2008. Characterization of conserved motifs in HIV-1 Vif required for APOBEC3G and APOBEC3F interaction. J. Mol. Biol. 3811000-1011. [DOI] [PubMed] [Google Scholar]
- 89.Henriet, S., D. Richer, S. Bernacchi, E. Decroly, R. Vigne, B. Ehresmann, C. Ehresmann, J. C. Paillart, and R. Marquet. 2005. Cooperative and specific binding of Vif to the 5′ region of HIV-1 genomic RNA. J. Mol. Biol. 35455-72. [DOI] [PubMed] [Google Scholar]
- 90.Henriet, S., L. Sinck, G. Bec, R. J. Gorelick, R. Marquet, and J. C. Paillart. 2007. Vif is a RNA chaperone that could temporally regulate RNA dimerization and the early steps of HIV-1 reverse transcription. Nucleic Acids Res. 355141-5153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Henzler, T., A. Harmache, H. Herrmann, H. Spring, M. Suzan, G. Audoly, T. Panek, and V. Bosch. 2001. Fully functional, naturally occurring and C-terminally truncated variant human immunodeficiency virus (HIV) Vif does not bind to HIV Gag but influences intermediate filament structure. J. Gen. Virol. 82561-573. [DOI] [PubMed] [Google Scholar]
- 92.Hibbert, C. S., J. Mirro, and A. Rein. 2004. mRNA molecules containing murine leukemia virus packaging signals are encapsidated as dimers. J. Virol. 7810927-10938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hill, M. K., M. Shehu-Xhilaga, S. M. Crowe, and J. Mak. 2002. Proline residues within spacer peptide p1 are important for human immunodeficiency virus type 1 infectivity, protein processing, and genomic RNA dimer stability. J. Virol. 7611245-11253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hoglund, S., A. Ohagen, K. Lawrence, and D. Gabuzda. 1994. Role of vif during packing of the core of HIV-1. Virology 201349-355. [DOI] [PubMed] [Google Scholar]
- 95.Holden, L. G., C. Prochnow, Y. P. Chang, R. Bransteitter, L. Chelico, U. Sen, R. C. Stevens, M. F. Goodman, and X. S. Chen. 2008. Crystal structure of the anti-viral APOBEC3G catalytic domain and functional implications. Nature 456121-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Holmes, R. K., F. A. Koning, K. N. Bishop, and M. H. Malim. 2007. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J. Biol. Chem. 2822587-2595. [DOI] [PubMed] [Google Scholar]
- 97.Holmes, R. K., M. H. Malim, and K. N. Bishop. 2007. APOBEC-mediated viral restriction: not simply editing? Trends Biochem. Sci. 32118-128. [DOI] [PubMed] [Google Scholar]
- 98.Houzet, L., J.-C. Paillart, F. Smagulova, S. Maurel, Z. Morichaud, R. Marquet, and M. Mougel. 2007. HIV controls the selective packaging of genomic, spliced viral and cellular RNAs into virions through different mechanisms. Nucleic Acids Res. 352695-2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Huang, Y., A. Khorchid, J. Wang, M. A. Parniak, J. L. Darlix, M. A. Wainberg, and L. Kleiman. 1997. Effect of mutations in the nucleocapsid protein (NCp7) upon Pr160gag-pol and tRNALys incorporation into human immunodeficiency virus type 1. J. Virol. 714378-4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
100.Huang, Y., J. Wang, A. Shalom, Z. Li, A. Khorchid, M. A. Wainberg, and L. Kleiman. 1997. Primer
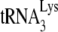 on the viral genome exists in unextended and two-base extended forms within mature human immunodeficiency virus type 1. J. Virol. 71726-728. [DOI] [PMC free article] [PubMed] [Google Scholar]
on the viral genome exists in unextended and two-base extended forms within mature human immunodeficiency virus type 1. J. Virol. 71726-728. [DOI] [PMC free article] [PubMed] [Google Scholar] - 101.Hulme, A. E., H. P. Bogerd, B. R. Cullen, and J. V. Moran. 2007. Selective inhibition of Alu retrotransposition by APOBEC3G. Gene 390199-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Huthoff, H., and M. H. Malim. 2007. Identification of amino acid residues in APOBEC3G required for regulation by human immunodeficiency virus type 1 Vif and virion encapsidation. J. Virol. 813807-3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hutoran, M., E. Britan, L. Baraz, I. Blumenzweig, M. Steinitz, and M. Kotler. 2004. Abrogation of Vif function by peptide derived from the N-terminal region of the human immunodeficiency virus type 1 (HIV-1) protease. Virology 330261-270. [DOI] [PubMed] [Google Scholar]
- 104.Huvent, I., S. S. Hong, C. Fournier, B. Gay, J. Tournier, C. Carriere, M. Courcoul, R. Vigne, B. Spire, and P. Boulanger. 1998. Interaction and co-encapsidation of human immunodeficiency virus type 1 Gag and Vif recombinant proteins. J. Gen. Virol. 791069-1081. [DOI] [PubMed] [Google Scholar]
- 105.Inoshima, Y., T. Miyazawa, and T. Mikami. 1998. In vivo functions of the auxiliary genes and regulatory elements of feline immunodeficiency virus. Vet. Microbiol. 60141-153. [DOI] [PubMed] [Google Scholar]
- 106.Iordanskiy, S., R. Berro, M. Altieri, F. Kashanchi, and M. Bukrinsky. 2006. Intracytoplasmic maturation of the human immunodeficiency virus type 1 reverse transcription complexes determines their capacity to integrate into chromatin. Retrovirology 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Iwatani, Y., D. S. Chan, F. Wang, K. S. Maynard, W. Sugiura, A. M. Gronenborn, I. Rouzina, M. C. Williams, K. Musier-Forsyth, and J. G. Levin. 2007. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 357096-7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Iwatani, Y., H. Takeuchi, K. Strebel, and J. G. Levin. 2006. Biochemical activities of highly purified, catalytically active human APOBEC3G: correlation with antiviral effect. J. Virol. 805992-6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Janini, M., M. Rogers, D. R. Birx, and F. E. McCutchan. 2001. Human immunodeficiency virus type 1 DNA sequences genetically damaged by hypermutation are often abundant in patient peripheral blood mononuclear cells and may be generated during near-simultaneous infection and activation of CD4+ T cells. J. Virol. 757973-7986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jarmuz, A., A. Chester, J. Bayliss, J. Gisbourne, I. Dunham, J. Scott, and N. Navaratnam. 2002. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics 79285-296. [DOI] [PubMed] [Google Scholar]
- 111.Kaiser, S. M., and M. Emerman. 2006. Uracil DNA glycosylase is dispensable for human immunodeficiency virus type 1 replication and does not contribute to the antiviral effects of the cytidine deaminase Apobec3G. J. Virol. 80875-882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kao, S., H. Akari, M. A. Khan, M. Dettenhofer, X. F. Yu, and K. Strebel. 2003. Human immunodeficiency virus type 1 Vif is efficiently packaged into virions during productive but not chronic infection. J. Virol. 771131-1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kao, S., R. Goila-Gaur, E. Miyagi, M. A. Khan, S. Opi, H. Takeuchi, and K. Strebel. 2007. Production of infectious virus and degradation of APOBEC3G are separable functional properties of human immunodeficiency virus type 1 Vif. Virology 369329-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kao, S., M. A. Khan, E. Miyagi, R. Plishka, A. Buckler-White, and K. Strebel. 2003. The human immunodeficiency virus type 1 Vif protein reduces intracellular expression and inhibits packaging of APOBEC3G (CEM15), a cellular inhibitor of virus infectivity. J. Virol. 7711398-11407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kao, S., E. Miyagi, M. A. Khan, H. Takeuchi, S. Opi, R. Goila-Gaur, and K. Strebel. 2004. Production of infectious human immunodeficiency virus type 1 does not require depletion of APOBEC3G from virus-producing cells. Retrovirology 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Karczewski, M. K., and K. Strebel. 1996. Cytoskeleton association and virion incorporation of the human immunodeficiency virus type 1 Vif protein. J. Virol. 70494-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kawakami, T., L. Sherman, J. Dahlberg, A. Gazit, A. Yaniv, S. R. Tronick, and S. A. Aaronson. 1987. Nucleotide sequence analysis of equine infectious anemia virus proviral DNA. Virology 158300-312. [DOI] [PubMed] [Google Scholar]
- 118.Khan, M. A., C. Aberham, S. Kao, H. Akari, R. Gorelick, S. Bour, and K. Strebel. 2001. Human immunodeficiency virus type 1 Vif protein is packaged into the nucleoprotein complex through an interaction with viral genomic RNA. J. Virol. 757252-7265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Khan, M. A., H. Akari, S. Kao, C. Aberham, D. Davis, A. Buckler-White, and K. Strebel. 2002. Intravirion processing of the human immunodeficiency virus type 1 Vif protein by the viral protease may be correlated with Vif function. J. Virol. 769112-9123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Khan, M. A., S. Kao, E. Miyagi, H. Takeuchi, R. Goila-Gaur, S. Opi, C. L. Gipson, T. G. Parslow, H. Ly, and K. Strebel. 2005. Viral RNA is required for the association of APOBEC3G with human immunodeficiency virus type 1 nucleoprotein complexes. J. Virol. 795870-5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kimpton, J., and M. Emerman. 1992. Detection of replication-competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integrated β-galactosidase gene. J. Virol. 662232-2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kishi, M., Y. Nishino, M. Sumiya, K. Ohki, T. Kimura, T. Goto, M. Nakai, M. Kakinuma, and K. Ikuta. 1992. Cells surviving infection by human immunodeficiency virus type 1: vif or vpu mutants produce noninfectious or markedly less cytopathic viruses. J. Gen. Virol. 7377-87. [DOI] [PubMed] [Google Scholar]
- 123.Knoepfel, S. A., N. C. Salisch, P. M. Huelsmann, P. Rauch, H. Walter, and K. J. Metzner. 2008. Comparison of G-to-A mutation frequencies induced by APOBEC3 proteins in H9 cells and peripheral blood mononuclear cells in the context of impaired processivities of drug-resistant human immunodeficiency virus type 1 reverse transcriptase variants. J. Virol. 826536-6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kotler, M., M. Simm, Y. S. Zhao, P. Sova, W. Chao, S. F. Ohnona, R. Roller, C. Krachmarov, M. J. Potash, and D. J. Volsky. 1997. Human immunodeficiency virus type 1 (HIV-1) protein Vif inhibits the activity of HIV-1 protease in bacteria and in vitro. J. Virol. 715774-5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Koulinska, I. N., B. Chaplin, D. Mwakagile, M. Essex, and B. Renjifo. 2003. Hypermutation of HIV type 1 genomes isolated from infants soon after vertical infection. AIDS Res. Hum. Retrovir. 191115-1123. [DOI] [PubMed] [Google Scholar]
- 126.Kozak, S. L., M. Marin, K. M. Rose, C. Bystrom, and D. Kabat. 2006. The anti-HIV-1 editing enzyme APOBEC3G binds HIV-1 RNA and messenger RNAs that shuttle between polysomes and stress granules. J. Biol. Chem. 28129105-29119. [DOI] [PubMed] [Google Scholar]
- 127.Kreisberg, J. F., W. Yonemoto, and W. C. Greene. 2006. Endogenous factors enhance HIV infection of tissue naive CD4 T cells by stimulating high molecular mass APOBEC3G complex formation. J. Exp. Med. 203865-870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Langlois, M. A., and M. S. Neuberger. 2008. Human APOBEC3G can restrict retroviral infection in avian cells and acts independently of both UNG and SMUG1. J. Virol. 824660-4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lecossier, D., F. Bouchonnet, F. Clavel, and A. J. Hance. 2003. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science 3001112. [DOI] [PubMed] [Google Scholar]
- 130.Lee, Y. M., B. Liu, and X. F. Yu. 1999. Formation of virus assembly intermediate complexes in the cytoplasm by wild-type and assembly-defective mutant human immunodeficiency virus type 1 and their association with membranes. J. Virol. 735654-5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Le Rouzic, E., A. Mousnier, C. Rustum, F. Stutz, E. Hallberg, C. Dargemont, and S. Benichou. 2002. Docking of HIV-1 Vpr to the nuclear envelope is mediated by the interaction with the nucleoporin hCG1. J. Biol. Chem. 27745091-45098. [DOI] [PubMed] [Google Scholar]
- 132.Lever, A. M. 2007. HIV-1 RNA packaging. Adv. Pharmacol. 551-32. [DOI] [PubMed] [Google Scholar]
- 133.Li, J., M. J. Potash, and D. J. Volsky. 2004. Functional domains of APOBEC3G required for antiviral activity. J. Cell. Biochem. 92560-572. [DOI] [PubMed] [Google Scholar]
- 134.Li, X. Y., F. Guo, L. Zhang, L. Kleiman, and S. Cen. 2007. APOBEC3G inhibits DNA strand transfer during HIV-1 reverse transcription. J. Biol. Chem. 28232065-32074. [DOI] [PubMed] [Google Scholar]
- 135.Liang, C., L. Rong, E. Cherry, L. Kleiman, M. Laughrea, and M. A. Wainberg. 1999. Deletion mutagenesis within the dimerization initiation site of human immunodeficiency virus type 1 results in delayed processing of the p2 peptide from precursor proteins. J. Virol. 736147-6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Liddament, M. T., W. L. Brown, A. J. Schumacher, and R. S. Harris. 2004. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr. Biol. 141385-1391. [DOI] [PubMed] [Google Scholar]
- 137.Lingappa, J. R., R. L. Hill, M. L. Wong, and R. S. Hegde. 1997. A multistep, ATP-dependent pathway for assembly of human immunodeficiency virus capsids in a cell-free system. J. Cell Biol. 136567-581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Liu, B., P. T. Sarkis, K. Luo, Y. Yu, and X. F. Yu. 2005. Regulation of Apobec3F and human immunodeficiency virus type 1 Vif by Vif-Cul5-ElonB/C E3 ubiquitin ligase. J. Virol. 799579-9587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Liu, B., X. Yu, K. Luo, Y. Yu, and X. F. Yu. 2004. Influence of primate lentiviral Vif and proteasome inhibitors on human immunodeficiency virus type 1 virion packaging of APOBEC3G. J. Virol. 782072-2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Liu, H., X. Wu, M. Newman, G. M. Shaw, B. H. Hahn, and J. C. Kappes. 1995. The Vif protein of human and simian immunodeficiency viruses is packaged into virions and associates with viral core structures. J. Virol. 697630-7638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Luo, K., B. Liu, Z. Xiao, Y. Yu, X. Yu, R. Gorelick, and X. F. Yu. 2004. Amino-terminal region of the human immunodeficiency virus type 1 nucleocapsid is required for human APOBEC3G packaging. J. Virol. 7811841-11852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Luo, K., T. Wang, B. Liu, C. Tian, Z. Xiao, J. Kappes, and X. F. Yu. 2007. Cytidine deaminases APOBEC3G and APOBEC3F interact with human immunodeficiency virus type 1 integrase and inhibit proviral DNA formation. J. Virol. 817238-7248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Luo, K., Z. Xiao, E. Ehrlich, Y. Yu, B. Liu, S. Zheng, and X. F. Yu. 2005. Primate lentiviral virion infectivity factors are substrate receptors that assemble with cullin 5-E3 ligase through a HCCH motif to suppress APOBEC3G. Proc. Natl. Acad. Sci. USA 10211444-11449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lv, W., Z. Liu, H. Jin, X. Yu, L. Zhang, and L. Zhang. 2007. Three-dimensional structure of HIV-1 VIF constructed by comparative modeling and the function characterization analyzed by molecular dynamics simulation. Org. Biomol. Chem. 5617-626. [DOI] [PubMed] [Google Scholar]
- 145.Madani, N., and D. Kabat. 1998. An endogenous inhibitor of human immunodeficiency virus in human lymphocytes is overcome by the viral Vif protein. J. Virol. 7210251-10255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Madani, N., R. Millette, E. J. Platt, M. Marin, S. L. Kozak, D. B. Bloch, and D. Kabat. 2002. Implication of the lymphocyte-specific nuclear body protein Sp140 in an innate response to human immunodeficiency virus type 1. J. Virol. 7611133-11138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mak, J., M. Jiang, M. A. Wainberg, M.-L. Hammarskjöld, D. Rekosh, and L. Kleiman. 1994. Role of Pr160gag-pol in mediating the selective incorporation of tRNALys into human immunodeficiency virus type 1 particles. J. Virol. 682065-2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Malim, M. H., and M. Emerman. 2008. HIV-1 accessory proteins—ensuring viral survival in a hostile environment. Cell Host Microbe 3388-398. [DOI] [PubMed] [Google Scholar]
- 149.Mangeat, B., P. Turelli, G. Caron, M. Friedli, L. Perrin, and D. Trono. 2003. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 42499-103. [DOI] [PubMed] [Google Scholar]
- 150.Mariani, R., D. Chen, B. Schrofelbauer, F. Navarro, R. Konig, B. Bollman, C. Munk, H. Nymark-McMahon, and N. R. Landau. 2003. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 11421-31. [DOI] [PubMed] [Google Scholar]
- 151.Marin, M., S. Golem, K. M. Rose, S. L. Kozak, and D. Kabat. 2008. Human immunodeficiency virus type 1 Vif functionally interacts with diverse APOBEC3 cytidine deaminases and moves with them between cytoplasmic sites of mRNA metabolism. J. Virol. 82987-998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Marin, M., K. M. Rose, S. L. Kozak, and D. Kabat. 2003. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat. Med. 91398-1403. [DOI] [PubMed] [Google Scholar]
- 153.Mbisa, J. L., R. Barr, J. A. Thomas, N. Vandegraaff, I. J. Dorweiler, E. S. Svarovskaia, W. L. Brown, L. M. Mansky, R. J. Gorelick, R. S. Harris, A. Engelman, and V. K. Pathak. 2007. Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J. Virol. 817099-7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mehle, A., J. Goncalves, M. Santa-Marta, M. McPike, and D. Gabuzda. 2004. Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev. 182861-2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mehle, A., B. Strack, P. Ancuta, C. Zhang, M. McPike, and D. Gabuzda. 2004. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J. Biol. Chem. 2797792-7798. [DOI] [PubMed] [Google Scholar]
- 156.Mehle, A., E. R. Thomas, K. S. Rajendran, and D. Gabuzda. 2006. A zinc-binding region in Vif binds Cul5 and determines cullin selection. J. Biol. Chem. 28117259-17265. [DOI] [PubMed] [Google Scholar]
- 157.Mehle, A., H. Wilson, C. Zhang, A. J. Brazier, M. McPike, E. Pery, and D. Gabuzda. 2007. Identification of an APOBEC3G binding site in human immunodeficiency virus type 1 Vif and inhibitors of Vif-APOBEC3G binding. J. Virol. 8113235-13241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Miller, J. H., V. Presnyak, and H. C. Smith. 2007. The dimerization domain of HIV-1 viral infectivity factor Vif is required to block APOBEC3G incorporation with virions. Retrovirology 481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Miyagi, E., S. Opi, H. Takeuchi, M. Khan, R. Goila-Gaur, S. Kao, and K. Strebel. 2007. Enzymatically active APOBEC3G is required for efficient inhibition of human immunodeficiency virus type 1. J. Virol. 8113346-13353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Muckenfuss, H., M. Hamdorf, U. Held, M. Perkovic, J. Lower, K. Cichutek, E. Flory, G. G. Schumann, and C. Munk. 2006. APOBEC3 proteins inhibit human LINE-1 retrotransposition. J. Biol. Chem. 28122161-22172. [DOI] [PubMed] [Google Scholar]
- 161.Mulder, L. C., A. Harari, and V. Simon. 2008. Cytidine deamination induced HIV-1 drug resistance. Proc. Natl. Acad. Sci. USA 1055501-5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Muriaux, D., J. Mirro, D. Harvin, and A. Rein. 2001. RNA is a structural element in retrovirus particles. Proc. Natl. Acad. Sci. USA 985246-5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Nascimbeni, M., M. Bouyac, F. Rey, B. Spire, and F. Clavel. 1998. The replicative impairment of Vif− mutants of human immunodeficiency virus type 1 correlates with an overall defect in viral DNA synthesis. J. Gen. Virol. 791945-1950. [DOI] [PubMed] [Google Scholar]
- 164.Nathans, R., H. Cao, N. Sharova, A. Ali, M. Sharkey, R. Stranska, M. Stevenson, and T. M. Rana. 2008. Small-molecule inhibition of HIV-1 Vif. Nat. Biotechnol. 261187-1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Navarro, F., B. Bollman, H. Chen, R. Konig, Q. Yu, K. Chiles, and N. R. Landau. 2005. Complementary function of the two catalytic domains of APOBEC3G. Virology 333374-386. [DOI] [PubMed] [Google Scholar]
- 166.Neil, S. J., S. W. Eastman, N. Jouvenet, and P. D. Bieniasz. 2006. HIV-1 Vpu promotes release and prevents endocytosis of nascent retrovirus particles from the plasma membrane. PLoS Pathog. 2e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Newman, E. N., R. K. Holmes, H. M. Craig, K. C. Klein, J. R. Lingappa, M. H. Malim, and A. M. Sheehy. 2005. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr. Biol. 15166-170. [DOI] [PubMed] [Google Scholar]
- 168.Nguyen, D. H., S. Gummuluru, and J. Hu. 2007. Deamination-independent inhibition of hepatitis B virus reverse transcription by APOBEC3G. J. Virol. 814465-4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Nishizawa, M., T. Myojin, Y. Nishino, Y. Nakai, M. Kamata, and Y. Aida. 1999. A carboxy-terminally truncated form of the Vpr protein of human immunodeficiency virus type 1 retards cell proliferation independently of G(2) arrest of the cell cycle. Virology 263313-322. [DOI] [PubMed] [Google Scholar]
- 170.Noguchi, C., H. Ishino, M. Tsuge, Y. Fujimoto, M. Imamura, S. Takahashi, and K. Chayama. 2005. G to A hypermutation of hepatitis B virus. Hepatology 41626-633. [DOI] [PubMed] [Google Scholar]
- 171.Nowarski, R., E. Britan-Rosich, T. Shiloach, and M. Kotler. 2008. Hypermutation by intersegmental transfer of APOBEC3G cytidine deaminase. Nat. Struct. Mol. Biol. 151059-1066. [DOI] [PubMed] [Google Scholar]
- 172.Oberste, M. S., and M. A. Gonda. 1992. Conservation of amino-acid sequence motifs in lentivirus Vif proteins. Virus Genes 695-102. [DOI] [PubMed] [Google Scholar]
- 173.Ohagen, A., and D. Gabuzda. 2000. Role of Vif in stability of the human immunodeficiency virus type 1 core. J. Virol. 7411055-11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Onafuwa-Nuga, A. A., S. R. King, and A. Telesnitsky. 2005. Nonrandom packaging of host RNAs in Moloney murine leukemia virus. J. Virol. 7913528-13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Opi, S., S. Kao, R. Goila-Gaur, M. A. Khan, E. Miyagi, H. Takeuchi, and K. Strebel. 2007. Human immunodeficiency virus type 1 Vif inhibits packaging and antiviral activity of a degradation-resistant APOBEC3G variant. J. Virol. 818236-8246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Opi, S., H. Takeuchi, S. Kao, M. A. Khan, E. Miyagi, R. Goila-Gaur, Y. Iwatani, J. G. Levin, and K. Strebel. 2006. Monomeric APOBEC3G is catalytically active and has antiviral activity. J. Virol. 804673-4682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Pace, C., J. Keller, D. Nolan, I. James, S. Gaudieri, C. Moore, and S. Mallal. 2006. Population level analysis of human immunodeficiency virus type 1 hypermutation and its relationship with APOBEC3G and vif genetic variation. J. Virol. 809259-9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Paillart, J.-C., M. Shehu-Xhilaga, R. Marquet, and J. Mak. 2004. Dimerization of retroviral RNA genomes: an inseparable pair. Nat. Rev. Microbiol. 2461-472. [DOI] [PubMed] [Google Scholar]
- 179.Paillart, J. C., M. Dettenhofer, X. F. Yu, C. Ehresmann, B. Ehresmann, and R. Marquet. 2004. First snapshots of the HIV-1 RNA structure in infected cells and in virions. J. Biol. Chem. 27948397-48403. [DOI] [PubMed] [Google Scholar]
- 180.Paul, I., J. Cui, and E. L. Maynard. 2006. Zinc binding to the HCCH motif of HIV-1 virion infectivity factor induces a conformational change that mediates protein-protein interactions. Proc. Natl. Acad. Sci. USA 10318475-18480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Pettit, S., M. Moody, R. Wehbie, A. Kaplan, P. Nantermet, C. Klein, and R. Swanstrom. 1994. The p2 domain of human immunodeficiency virus type 1 Gag regulates sequential proteolytic processing and is required to produce fully infectious virions. J. Virol. 688017-8027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Pillai, S. K., J. K. Wong, and J. D. Barbour. 2008. Turning up the volume on mutational pressure: is more of a good thing always better? A case study of HIV-1 Vif and APOBEC3. Retrovirology 526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Pion, M., A. Granelli-Piperno, B. Mangeat, R. Stalder, R. Correa, R. M. Steinman, and V. Piguet. 2006. APOBEC3G/3F mediates intrinsic resistance of monocyte-derived dendritic cells to HIV-1 infection. J. Exp. Med. 2032887-2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Poole, E., P. Strappe, H. P. Mok, R. Hicks, and A. M. Lever. 2005. HIV-1 Gag-RNA interaction occurs at a perinuclear/centrosomal site; analysis by confocal microscopy and FRET. Traffic 6741-755. [DOI] [PubMed] [Google Scholar]
- 185.Priet, S., N. Gros, J. M. Navarro, J. Boretto, B. Canard, G. Querat, and J. Sire. 2005. HIV-1-associated uracil DNA glycosylase activity controls dUTP misincorporation in viral DNA and is essential to the HIV-1 life cycle. Mol. Cell 17479-490. [DOI] [PubMed] [Google Scholar]
- 186.Rein, A., L. E. Henderson, and J. G. Levin. 1998. Nucleic-acid-chaperone activity of retroviral nucleocapsid proteins: significance for viral replication. Trends Biochem. Sci. 23297-301. [DOI] [PubMed] [Google Scholar]
- 187.Ribeiro, A. C., A. Maia e Silva, M. Santa-Marta, A. Pombo, J. Moniz-Pereira, J. Goncalves, and I. Barahona. 2005. Functional analysis of Vif protein shows less restriction of human immunodeficiency virus type 2 by APOBEC3G. J. Virol. 79823-833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Rose, K. M., M. Marin, S. L. Kozak, and D. Kabat. 2004. Transcriptional regulation of APOBEC3G, a cytidine deaminase that hypermutates human immunodeficiency virus. J. Biol. Chem. 27941744-41749. [DOI] [PubMed] [Google Scholar]
- 189.Russell, R. A., and V. K. Pathak. 2007. Identification of two distinct human immunodeficiency virus type 1 Vif determinants critical for interactions with human APOBEC3G and APOBEC3F. J. Virol. 818201-8210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Russell, R. A., J. Smith, R. Barr, D. Bhattacharyya, and V. K. Pathak. 2009. Distinct domains within APOBEC3G and APOBEC3F interact with separate regions of human immunodeficiency virus type 1 Vif. J. Virol. 831992-2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Russell, R. A., H. L. Wiegand, M. D. Moore, A. Schafer, M. O. McClure, and B. R. Cullen. 2005. Foamy virus Bet proteins function as novel inhibitors of the APOBEC3 family of innate antiretroviral defense factors. J. Virol. 798724-8731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Russell, R. S., C. Liang, and M. A. Wainberg. 2004. Is HIV-1 RNA dimerization a prerequisite for packaging? Yes, no, probably? Retrovirology 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Sakai, H., R. Shibata, J. Sakuragi, S. Sakuragi, M. Kawamura, and A. Adachi. 1993. Cell-dependent requirement of human immunodeficiency virus type 1 Vif protein for maturation of virus particles. J. Virol. 671663-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Sakai, K., J. Dimas, and M. J. Lenardo. 2006. The Vif and Vpr accessory proteins independently cause HIV-1-induced T cell cytopathicity and cell cycle arrest. Proc. Natl. Acad. Sci. USA 1033369-3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Sakai, K., X. Y. Ma, I. Gordienko, and D. J. Volsky. 1991. Recombinational analysis of a natural noncytopathic human immunodeficiency virus type 1 (HIV-1) isolate: role of the vif gene in HIV-1 infection kinetics and cytopathicity. J. Virol. 655765-5773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Santa-Marta, M., F. Aires da Silva, A. Fonseca, and J. Goncalves. 2005. HIV-1 Vif can directly inhibit APOBEC3G-mediated cytidine deamination by using a single amino acid interaction and without protein degradation. J. Biol. Chem. 2808765-8775. [DOI] [PubMed] [Google Scholar]
- 197.Sasada, A., A. Takaori-Kondo, K. Shirakawa, M. Kobayashi, A. Abudu, M. Hishizawa, K. Imada, Y. Tanaka, and T. Uchiyama. 2005. APOBEC3G targets human T-cell leukemia virus type 1. Retrovirology 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sawyer, S. L., M. Emerman, and H. S. Malik. 2004. Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol. 2E275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Schrofelbauer, B., Q. Yu, S. G. Zeitlin, and N. R. Landau. 2005. Human immunodeficiency virus type 1 Vpr induces the degradation of the UNG and SMUG uracil-DNA glycosylases. J. Virol. 7910978-10987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Schumacher, A. J., G. Haché, D. A. MacDuff, W. L. Brown, and R. S. Harris. 2008. The DNA deaminase activity of human APOBEC3G is required for Ty1, MusD, and human immunodeficiency virus type 1 restriction. J. Virol. 822652-2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Schwartz, S., B. K. Felber, and G. N. Pavlakis. 1991. Expression of human immunodeficiency virus type 1 vif and vpr mRNAs is Rev-dependent and regulated by splicing. Virology 183677-686. [DOI] [PubMed] [Google Scholar]
- 202.Seppen, J. 2004. Unedited inhibition of HBV replication by APOBEC3G. J. Hepatol. 411068-1069. [DOI] [PubMed] [Google Scholar]
- 203.Sheehy, A. M., N. C. Gaddis, J. D. Choi, and M. H. Malim. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418646-650. [DOI] [PubMed] [Google Scholar]
- 204.Sheehy, A. M., N. C. Gaddis, and M. H. Malim. 2003. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 91404-1407. [DOI] [PubMed] [Google Scholar]
- 205.Shehu-Xhilaga, M., H. G. Kraeusslich, S. Pettit, R. Swanstrom, J. Y. Lee, J. A. Marshall, S. M. Crowe, and J. Mak. 2001. Proteolytic processing of the p2/nucleocapsid cleavage site is critical for human immunodeficiency virus type 1 RNA dimer maturation. J. Virol. 759156-9164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Sheng, N., and S. Erickson-Viitanen. 1994. Cleavage of p15 protein in vitro by human immunodeficiency virus type 1 protease is RNA dependent. J. Virol. 686207-6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Sheng, N., S. C. Pettit, R. J. Tritch, D. H. Ozturk, M. M. Rayner, R. Swanstrom, and S. Erickson-Viitanen. 1997. Determinants of the human immunodeficiency virus type 1 p15NC-RNA interaction that affect enhanced cleavage by the viral protease. J. Virol. 715723-5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Shindo, K., A. Takaori-Kondo, M. Kobayashi, A. Abudu, K. Fukunaga, and T. Uchiyama. 2003. The enzymatic activity of CEM15/Apobec-3G is essential for the regulation of the infectivity of HIV-1 virion but not a sole determinant of its antiviral activity. J. Biol. Chem. 27844412-44416. [DOI] [PubMed] [Google Scholar]
- 209.Shirakawa, K., A. Takaori-Kondo, M. Yokoyama, T. Izumi, M. Matsui, K. Io, T. Sato, H. Sato, and T. Uchiyama. 2008. Phosphorylation of APOBEC3G by protein kinase A regulates its interaction with HIV-1 Vif. Nat. Struct. Mol. Biol. 151184-1191. [DOI] [PubMed] [Google Scholar]
- 210.Simm, M., M. Shahabuddin, W. Chao, J. S. Allan, and D. J. Volsky. 1995. Aberrant Gag protein composition of a human immunodeficiency virus type 1 vif mutant produced in primary lymphocytes. J. Virol. 694582-4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Simon, J. H., E. A. Carpenter, R. A. Fouchier, and M. H. Malim. 1999. Vif and the p55Gag polyprotein of human immunodeficiency virus type 1 are present in colocalizing membrane-free cytoplasmic complexes. J. Virol. 732667-2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Simon, J. H., R. A. Fouchier, T. E. Southerling, C. B. Guerra, C. K. Grant, and M. H. Malim. 1997. The Vif and Gag proteins of human immunodeficiency virus type 1 colocalize in infected human T cells. J. Virol. 715259-5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Simon, J. H., N. C. Gaddis, R. A. Fouchier, and M. H. Malim. 1998. Evidence for a newly discovered cellular anti-HIV-1 phenotype. Nat. Med. 41397-1400. [DOI] [PubMed] [Google Scholar]
- 214.Simon, J. H., and M. H. Malim. 1996. The human immunodeficiency virus type 1 Vif protein modulates the postpenetration stability of viral nucleoprotein complexes. J. Virol. 705297-5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Simon, J. H., D. L. Miller, R. A. Fouchier, and M. H. Malim. 1998. Virion incorporation of human immunodeficiency virus type-1 Vif is determined by intracellular expression level and may not be necessary for function. Virology 248182-187. [DOI] [PubMed] [Google Scholar]
- 216.Simon, J. H., D. L. Miller, R. A. Fouchier, M. A. Soares, K. W. Peden, and M. H. Malim. 1998. The regulation of primate immunodeficiency virus infectivity by Vif is cell species restricted: a role for Vif in determining virus host range and cross-species transmission. EMBO J. 171259-1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Simon, V., V. Zennou, D. Murray, Y. Huang, D. D. Ho, and P. D. Bieniasz. 2005. Natural variation in Vif: differential impact on APOBEC3G/3F and a potential role in HIV-1 diversification. PLoS Pathog. 1e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Soros, V. B., W. Yonemoto, and W. C. Greene. 2007. Newly synthesized APOBEC3G is incorporated into HIV virions, inhibited by HIV RNA, and subsequently activated by RNase H. PLoS Pathog. 3e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Sova, P., and D. J. Volsky. 1993. Efficiency of viral DNA synthesis during infection of permissive and nonpermissive cells with vif-negative human immunodeficiency virus type 1. J. Virol. 676322-6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Sova, P., D. J. Volsky, L. Wang, and W. Chao. 2001. Vif is largely absent from human immunodeficiency virus type 1 mature virions and associates mainly with viral particles containing unprocessed gag. J. Virol. 755504-5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Stopak, K., C. de Noronha, W. Yonemoto, and W. C. Greene. 2003. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol. Cell 12591-601. [DOI] [PubMed] [Google Scholar]
- 222.Stopak, K. S., Y. L. Chiu, J. Kropp, R. M. Grant, and W. C. Greene. 2007. Distinct patterns of cytokine regulation of APOBEC3G expression and activity in primary lymphocytes, macrophages, and dendritic cells. J. Biol. Chem. 2823539-3546. [DOI] [PubMed] [Google Scholar]
- 223.Strebel, K. 2005. APOBEC3G & HTLV-1: inhibition without deamination. Retrovirology 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Strebel, K., D. Daugherty, K. Clouse, D. Cohen, T. Folks, and M. A. Martin. 1987. The HIV ‘A’ (sor) gene product is essential for virus infectivity. Nature 328728-730. [DOI] [PubMed] [Google Scholar]
- 225.Suspene, R., D. Guetard, M. Henry, P. Sommer, S. Wain-Hobson, and J. P. Vartanian. 2005. Extensive editing of both hepatitis B virus DNA strands by APOBEC3 cytidine deaminases in vitro and in vivo. Proc. Natl. Acad. Sci. USA 1028321-8326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Suspene, R., C. Rusniok, J. P. Vartanian, and S. Wain-Hobson. 2006. Twin gradients in APOBEC3 edited HIV-1 DNA reflect the dynamics of lentiviral replication. Nucleic Acids Res. 344677-4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Suspene, R., P. Sommer, M. Henry, S. Ferris, D. Guetard, S. Pochet, A. Chester, N. Navaratnam, S. Wain-Hobson, and J. P. Vartanian. 2004. APOBEC3G is a single-stranded DNA cytidine deaminase and functions independently of HIV reverse transcriptase. Nucleic Acids Res. 322421-2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Takeuchi, H., S. Kao, E. Miyagi, M. A. Khan, A. Buckler-White, R. Plishka, and K. Strebel. 2005. Production of infectious SIVagm from human cells requires functional inactivation but not viral exclusion of human APOBEC3G. J. Biol. Chem. 280375-382. [DOI] [PubMed] [Google Scholar]
- 229.Tian, C., X. Yu, W. Zhang, T. Wang, R. Xu, and X. F. Yu. 2006. Differential requirement for conserved tryptophans in human immunodeficiency virus type 1 Vif for the selective suppression of APOBEC3G and APOBEC3F. J. Virol. 803112-3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Turelli, P., B. Mangeat, S. Jost, S. Vianin, and D. Trono. 2004. Inhibition of hepatitis B virus replication by APOBEC3G. Science 3031829. [DOI] [PubMed] [Google Scholar]
- 231.Ulenga, N. K., A. D. Sarr, D. Hamel, J. L. Sankale, S. Mboup, and P. J. Kanki. 2008. The level of APOBEC3G (hA3G)-related G-to-A mutations does not correlate with viral load in HIV type 1-infected individuals. AIDS Res. Hum. Retrovir. 241285-1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Vartanian, J. P., M. Henry, and S. Wain-Hobson. 2002. Sustained G→A hypermutation during reverse transcription of an entire human immunodeficiency virus type 1 strain Vau group O genome. J. Gen. Virol. 83801-805. [DOI] [PubMed] [Google Scholar]
- 233.Varthakavi, V., R. M. Smith, S. P. Bour, K. Strebel, and P. Spearman. 2003. Viral protein U counteracts a human host cell restriction that inhibits HIV-1 particle production. Proc. Natl. Acad. Sci. USA 10015154-15159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.von Schwedler, U., J. Song, C. Aiken, and D. Trono. 1993. Vif is crucial for human immunodeficiency virus type 1 proviral DNA synthesis in infected cells. J. Virol. 674945-4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Wang, H., A. Sakurai, B. Khamsri, T. Uchiyama, H. Gu, A. Adachi, and M. Fujita. 2005. Unique characteristics of HIV-1 Vif expression. Microbes Infect. 7385-390. [DOI] [PubMed] [Google Scholar]
- 236.Wang, J., J. M. Shackelford, C. R. Casella, D. K. Shivers, E. L. Rapaport, B. Liu, X. F. Yu, and T. H. Finkel. 2007. The Vif accessory protein alters the cell cycle of human immunodeficiency virus type 1 infected cells. Virology 359243-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Wang, T., C. Tian, W. Zhang, K. Luo, P. T. Sarkis, L. Yu, B. Liu, Y. Yu, and X. F. Yu. 2007. 7SL RNA mediates virion packaging of the antiviral cytidine deaminase APOBEC3G. J. Virol. 8113112-13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Wang, T., C. Tian, W. Zhang, P. T. Sarkis, and X. F. Yu. 2008. Interaction with 7SL RNA but not with HIV-1 genomic RNA or P bodies is required for APOBEC3F virion packaging. J. Mol. Biol. 3751098-1112. [DOI] [PubMed] [Google Scholar]
- 239.Warrilow, D., L. Meredith, A. Davis, C. Burrell, P. Li, and D. Harrich. 2008. Cell factors stimulate human immunodeficiency virus type 1 reverse transcription in vitro. J. Virol. 821425-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Wichroski, M. J., G. B. Robb, and T. M. Rana. 2006. Human retroviral host restriction factors APOBEC3G and APOBEC3F localize to mRNA processing bodies. PLoS Pathog. 2e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Wiegand, H. L., B. P. Doehle, H. P. Bogerd, and B. R. Cullen. 2004. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J. 232451-2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Wiegers, K., G. Rutter, H. Kottler, U. Tessmer, H. Hohenberg, and H. G. Krausslich. 1998. Sequential steps in human immunodeficiency virus particle maturation revealed by alterations of individual Gag polyprotein cleavage sites. J. Virol. 722846-2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Willetts, K. E., F. Rey, I. Agostini, J. M. Navarro, Y. Baudat, R. Vigne, and J. Sire. 1999. DNA repair enzyme uracil DNA glycosylase is specifically incorporated into human immunodeficiency virus type 1 viral particles through a Vpr-independent mechanism. J. Virol. 731682-1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Xiao, Z., E. Ehrlich, K. Luo, Y. Xiong, and X. F. Yu. 2007. Zinc chelation inhibits HIV Vif activity and liberates antiviral function of the cytidine deaminase APOBEC3G. FASEB J. 21217-222. [DOI] [PubMed] [Google Scholar]
- 245.Xiao, Z., E. Ehrlich, Y. Yu, K. Luo, T. Wang, C. Tian, and X. F. Yu. 2006. Assembly of HIV-1 Vif-Cul5 E3 ubiquitin ligase through a novel zinc-binding domain-stabilized hydrophobic interface in Vif. Virology 349290-299. [DOI] [PubMed] [Google Scholar]
- 246.Xiao, Z., Y. Xiong, W. Zhang, L. Tan, E. Ehrlich, D. Guo, and X. F. Yu. 2007. Characterization of a novel Cullin5 binding domain in HIV-1 Vif. J. Mol. Biol. 373541-550. [DOI] [PubMed] [Google Scholar]
- 247.Xu, H., E. Chertova, J. Chen, D. E. Ott, J. D. Roser, W. S. Hu, and V. K. Pathak. 2007. Stoichiometry of the antiviral protein APOBEC3G in HIV-1 virions. Virology 360247-256. [DOI] [PubMed] [Google Scholar]
- 248.Yamashita, T., K. Kamada, K. Hatcho, A. Adachi, and M. Nomaguchi. 2008. Identification of amino acid residues in HIV-1 Vif critical for binding and exclusion of APOBEC3G/F. Microbes Infect. 101142-1149. [DOI] [PubMed] [Google Scholar]
- 249.Yang, B., K. Chen, C. Zhang, S. Huang, and H. Zhang. 2007. Virion-associated uracil DNA glycosylase-2 and apurinic/apyrimidinic endonuclease are involved in the degradation of APOBEC3G-edited nascent HIV-1 DNA. J. Biol. Chem. 28211667. [DOI] [PubMed] [Google Scholar]
- 250.Yang, B., L. Gao, L. Li, Z. Lu, X. Fan, C. A. Patel, R. J. Pomerantz, G. C. DuBois, and H. Zhang. 2003. Potent suppression of viral infectivity by the peptides that inhibit multimerization of human immunodeficiency virus type 1 (HIV-1) Vif proteins. J. Biol. Chem. 2786596-6602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Yang, S., Y. Sun, and H. Zhang. 2001. The multimerization of human immunodeficiency virus type I Vif protein: a requirement for Vif function in the viral life cycle. J. Biol. Chem. 2764889-4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Yang, X., and D. Gabuzda. 1998. Mitogen-activated protein kinase phosphorylates and regulates the HIV-1 Vif protein. J. Biol. Chem. 27329879-29887. [DOI] [PubMed] [Google Scholar]
- 253.Yang, Y., F. Guo, S. Cen, and L. Kleiman. 2007. Inhibition of initiation of reverse transcription in HIV-1 by human APOBEC3F. Virology 36592-100. [DOI] [PubMed] [Google Scholar]
- 254.You, J. C., and C. S. McHenry. 1993. HIV nucleocapsid protein. J. Biol. Chem. 26816519-16527. [PubMed] [Google Scholar]
- 255.Yu, Q., R. Konig, S. Pillai, K. Chiles, M. Kearney, S. Palmer, D. Richman, J. M. Coffin, and N. R. Landau. 2004. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 11435-442. [DOI] [PubMed] [Google Scholar]
- 256.Yu, X., Y. Yu, B. Liu, K. Luo, W. Kong, P. Mao, and X. F. Yu. 2003. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 3021056-1060. [DOI] [PubMed] [Google Scholar]
- 257.Yu, Y., Z. Xiao, E. S. Ehrlich, X. Yu, and X. F. Yu. 2004. Selective assembly of HIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complex through a novel SOCS box and upstream cysteines. Genes Dev. 182867-2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Zennou, V., and P. D. Bieniasz. 2006. Comparative analysis of the antiretroviral activity of APOBEC3G and APOBEC3F from primates. Virology 34931-40. [DOI] [PubMed] [Google Scholar]
- 259.Zennou, V., D. Perez-Caballero, H. Gottlinger, and P. D. Bieniasz. 2004. APOBEC3G incorporation into human immunodeficiency virus type 1 particles. J. Virol. 7812058-12061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Zhang, H., G. Dornadula, and R. J. Pomerantz. 1996. Endogenous reverse transcription of human immunodeficiency virus type 1 in physiological microenviroments: an important stage for viral infection of nondividing cells. J. Virol. 702809-2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Zhang, H., R. J. Pomerantz, G. Dornadula, and Y. Sun. 2000. Human immunodeficiency virus type 1 Vif protein is an integral component of an mRNP complex of viral RNA and could be involved in the viral RNA folding and packaging process. J. Virol. 748252-8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Zhang, H., B. Yang, R. J. Pomerantz, C. Zhang, S. C. Arunachalam, and L. Gao. 2003. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 42494-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Zhang, J., and D. M. Webb. 2004. Rapid evolution of primate antiviral enzyme APOBEC3G. Hum. Mol. Genet. 131785-1791. [DOI] [PubMed] [Google Scholar]
- 264.Zhang, L., J. Saadatmand, X. Li, F. Guo, M. Niu, J. Jiang, L. Kleiman, and S. Cen. 2008. Function analysis of sequences in human APOBEC3G involved in Vif-mediated degradation. Virology 370113-121. [DOI] [PubMed] [Google Scholar]
- 265.Zhang, W., G. Chen, A. M. Niewiadomska, R. Xu, and X. F. Yu. 2008. Distinct determinants in HIV-1 Vif and human APOBEC3 proteins are required for the suppression of diverse host anti-viral proteins. PLoS ONE 3e3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Zheng, Y. H., D. Irwin, T. Kurosu, K. Tokunaga, T. Sata, and B. M. Peterlin. 2004. Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J. Virol. 786073-6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Zimber, A., Q. D. Nguyen, and C. Gespach. 2004. Nuclear bodies and compartments: functional roles and cellular signalling in health and disease. Cell. Signal. 161085-1104. [DOI] [PubMed] [Google Scholar]
- 268.Zimmerman, C., K. C. Klein, P. K. Kiser, A. R. Singh, B. L. Firestein, S. C. Riba, and J. R. Lingappa. 2002. Identification of a host protein essential for assembly of immature HIV-1 capsids. Nature 41588-92. [DOI] [PubMed] [Google Scholar]
- 269.Zou, J. X., and P. A. Luciw. 1996. The requirement for Vif of SIVmac is cell-type dependent. J. Gen. Virol. 77427-434. [DOI] [PubMed] [Google Scholar]