

Abstract
Mapping DNase I hypersensitive sites is an accurate method of identifying the location of gene regulatory elements, including promoters, enhancers, silencers and locus control regions. Although Southern blots are the traditional method of identifying DNase I hypersensitive sites, the conventional manual method is not readily scalable to studying large chromosomal regions, much less the entire genome. Here we describe DNase-chip, an approach that can rapidly identify DNase I hypersensitive sites for any region of interest, or potentially for the entire genome, by using tiled microarrays. We used DNase-chip to identify DNase I hypersensitive sites accurately from a representative 1% of the human genome in both primary and immortalized cell types. We found that although most DNase I hypersensitive sites were present in both cell types studied, some of them were cell-type specific. This method can be applied globally or in a targeted fashion to any tissue from any species with a sequenced genome.
To understand how the genome is regulated in different cell types, several genome-wide strategies to identify functional elements are being developed and applied, including comparative genomics, identification of origins of replication, chromatin immunoprecipitation using microarray detection (ChIP-chip), DNA methylation, histone mapping and the use of various computational methods1. One classic method that has been somewhat slower to make its way into the genomics arena is the mapping of DNase I hypersensitive sites. For the last 25 years, the mapping of DNase I hypersensitive sites by Southern blotting has been a highly accurate method for identifying the location of promoters, silencers, enhancers and locus control regions2-4. Since the technique is experimentally demanding and requires many manual steps, however, this method has generally been limited to examining only one small region of the genome at a time.
Recently, we and others have developed sequencing and real-time PCR strategies to scale up the search for DNase I hypersensitive sites5-8. One method we have described uses massively parallel signature sequencing (MPSS) to sequence a library of DNase I hypersensitive sites8. With a total of 270,000 sequence tags, this method identified about 20% of all DNase I hypersensitive sites in human CD4+ T cells8. Thus, major advances in sequencing technologies will be needed to identify all DNase I hypersensitive sites within an individual cell type at an affordable cost. Furthermore, for researchers interested in studying targeted regions of the genome, whole-genome sequencing of DNase I hypersensitive sites represents an excess of effort and cost. An affordable, targeted approach to identifying DNase I hypersensitive sites is needed.
Here we describe DNase-chip, a high-resolution method, which can be used to identify DNase I hypersensitive sites by hybridizing captured DNase I–digested ends to tiled microarrays. This method is easy to perform, is readily adaptable to any region of a sequenced genome, is less costly than whole-genome DNase-I library sequencing and uses many of the same techniques used in standard ChIP-chip assays9. We used DNase-chip to accurately identify DNase I hypersensitive sites within the 1% of the human genome selected by the ENCODE (Encyclopedia Of DNA Elements) consortium1. Using DNase-chip, we successfully identified DNase I hypersensitive sites within the ENCODE regions from both primary CD4+ T cells and a cycling B lymphoblastoid cell line, indicating that this method is applicable to any primary or immortalized cell type.
RESULTS
DNase-chip protocol and data analysis
The DNase-chip protocol builds on traditional Southern blot strategies to identify regulatory sites in intact chromatin. DNase I is an enzyme, which at low levels preferentially digests nucleosome-depleted DNA, whereas tightly packaged chromatin is more resistant to cleavage. To perform DNase-chip, we digested nuclei from CD4+ T cells and GM06990 lymphoblastoid cells with three different concentrations of DNase I (Fig. 1a). We performed all DNase I digestions on three biological replicates. Careful manipulation, including embedding the DNA in low-melt gel agarose plugs was used to reduce the frequency of randomly sheared DNA ends, which could introduce background noise. As with traditional Southern blots, valid DNase I hypersensitive sites should be detected with more than one concentration of DNase I. Therefore, by generating DNase-chip libraries from three different concentrations of DNase I, we expected to detect valid DNase I hypersensitive sites along a spectrum of DNase I concentrations. Furthermore, as not all DNase I hypersensitive sites are equally hypersensitive to DNase I digestion, assaying with multiple DNase I concentrations will distinguish those that are extremely hypersensitive to DNase I digestion from those that are moderately hypersensitive.
Figure 1. DNase-chip protocol.
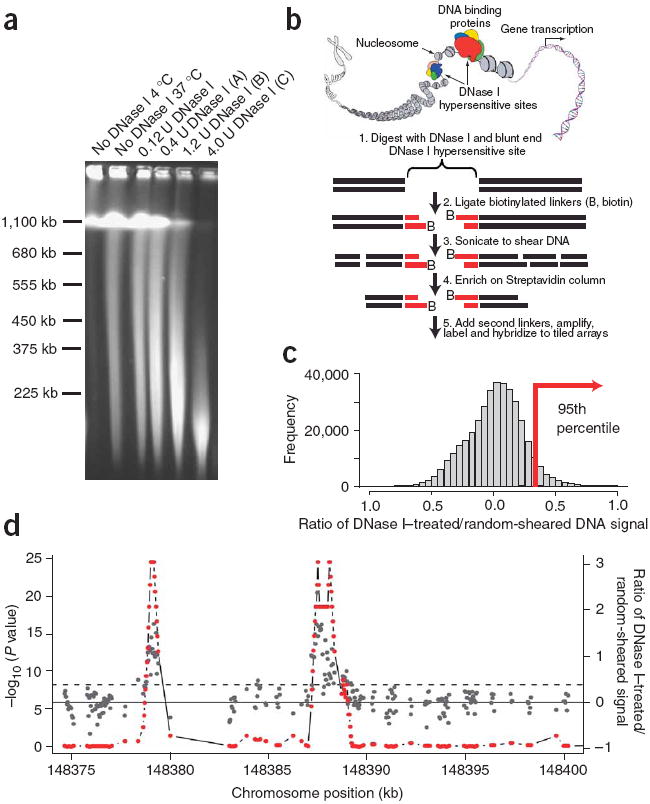
(a) Pulsed field gel electrophoresis of DNase I–digested nuclear DNA. The concentrations of DNase I used for DNase-chip are labeled as A, B and C. (b) Outline of DNase-chip protocol. (c) Histogram of signal ratios of DNase I–treated versus random-sheared DNA. Tiled oligos that displayed the top 5% ratios are located to the right of the red bar. (d) Identification of regions with significant P values. The raw ratio data are plotted in gray, with the y-axis label on the right; the top 5% cutoff is displayed as a dotted horizontal gray line. The P value data for sliding 500-bp windows are plotted in red, with the y-axis label on the left.
We blunt-ended DNase I–digested fragments (as well as randomly sheared ends from control DNA), ligated the resulting fragments to a biotinylated linker, sonicated the reactions, incubated them on a streptavidin column to enrich for the DNase I–digested ends, and ligated the fragments to a second nonbiotinylated linker (Fig. 1b). We PCR-amplified the DNase I–digested and randomly sheared captured material, labeled it with Cy3 and Cy5 dyes, and cohybridized it to tiled microarrays that span the ENCODE regions.
To rule out sequence-based bias of DNase I digestion, we digested naked DNA with multiple concentrations of DNase I. We labeled digested and undigested naked DNA (using no amplification) with Cy3 and Cy5 dyes. DNA from reactions with four different DNase I concentrations was cohybridized with undigested DNA to four tiled microarrays that span the ENCODE regions. We did not detect any sequence-based bias of DNase I digestion (Supplementary Fig. 1 online).
We developed a program called ‘algorithm for capturing microarray enrichment’ (ACME) to identify regions of the genome enriched for DNase I–captured material10. This program works by identifying oligonucleotides that displayed the top 5% of DNase I cleavage signal to random shear control signal ratios that were averaged from three biological replicates (Fig. 1c). Next we performed a χ2 test on sliding 500 base pair (bp) windows to identify regions that had a significant (P < 0.001) overrepresentation of DNase I–captured material (Fig. 1d). We have performed extensive testing using different window sizes and found that windows larger than 500 bp resulted in decreased resolution, whereas smaller window sizes did not contain enough probes to generate statistical significance11. Owing to the multiple-testing problem and because data points are not independent, caution must be used when interpreting the resulting P values. Therefore, the following three sections are dedicated to extensive experimental validation.
Validation of DNase-chip: positive predictive value
The positive predictive value is defined as TP / (TP + FP), where TP is true positives and FP is false positives. To determine the positive predictive value of DNase-chip, we tested primer sets that flank significant (P < 0.001) DNase-chip signals or “peaks” by a real-time PCR assay to confirm whether the region detected was a true DNase I hypersensitive site5,12. True positives are defined here as DNase-chip peaks for which the corresponding real-time PCR assay falls above a stringent threshold for background noise. We set this threshold at a point at which ~95% of real-time PCR results using random primer sets fell below it. False positives represent DNase-chip peaks that are below the same threshold.
As traditional Southern blot strategies identify DNase I hypersensitive sites at multiple concentrations of DNase I, we predicted that valid DNase-chip peaks would be detected at multiple concentrations, whereas false positives would mostly be detected at a single concentration of the enzyme. We separated DNase-chip peaks into seven mutually exclusive categories: those peaks that occurred in reactions with all three DNase I concentrations (ABC), with two out of three DNase I concentrations (AB, BC and AC), or only present in a single concentration of DNase I (A only, B only and C only).
Based on real-time PCR validation, the positive predictive value for DNase-chip peaks detected at all three DNase I concentrations was 84% for CD4+ T cells and 68% for the GM06990 cells (Fig. 2a,b and Supplementary Table 1 online). The positive predictive value for DNase-chip peaks present in two out of three concentrations was 40–70% for CD4+ T cells and 5–30% for the GM06990 cells (Fig. 2c,d and Supplementary Table 1). For both cell types, DNase-chip peaks detected in the two highest concentrations (BC) were better at identifying valid DNase I hypersensitive sites than the DNase-chip peaks that included the lowest concentration (AB and AC). In CD4+ T cells, we validated 30% of the DNase-chip peaks detected in concentration ‘C only’, whereas ‘A only’ and ‘B only’ were less accurate (Fig. 2e and Supplementary Table 1). Single-concentration (A only, B only and C only) DNase-chip peaks from GM06990 cells did not result in the same number of validated DNase I hypersensitive sites as CD4+ T cells, indicating that GM06990 cells may have an elevated background level of DNase I digestion (Fig. 2f and Supplementary Table 1).
Figure 2. Confirmatory real-time PCR analysis.
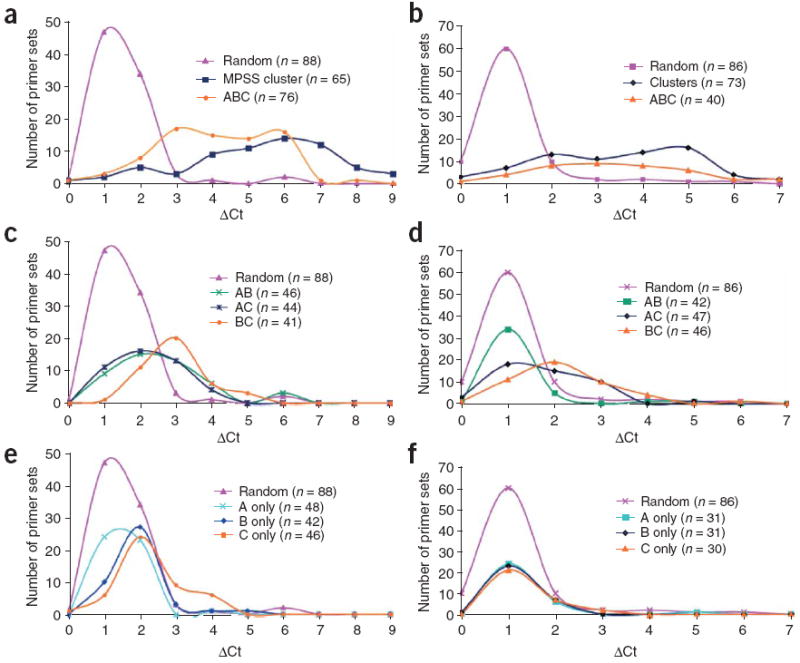
(a–f) DNase-chip–identified regions were confirmed by real-time PCR for CD4+ T cells (a,c,e) and GM06990 cells (b,d,f). ΔCt values represent the number of additional cycles to achieve threshold levels of amplification between DNase I–treated and non-digested nuclear DNA. Higher ΔCt values represent elevated levels of DNase I–digestion. Control primer sets were designed around random regions of the genome, as well as known DNase I hypersensitive sites generated from a separate study8 (MPSS cluster). Real-time PCR using primer sets flanking DNase-chip peaks that are present with all three DNase I concentrations (a,b). Real-time PCR using primers sets flanking DNase-chip peaks that are present with two out of three DNase I concentrations (c,d). Real-time PCR using primer sets flanking DNase-chip peaks that are present with only a single DNase I concentration (e,f).
Validation of DNase-chip: sensitivity
Sensitivity is defined as TP / (TP + FN), where TP is true positives and FN is false negatives. As another means of assessing performance of DNase-chip, we compared these data with well validated DNase I hypersensitive sites, which we previously identified using MPSS in CD4+ T cells8. True DNase I hypersensitive sites were identified by repeated observation of MPSS sequences clustered within a 500-bp region. By visual inspection, there appears to be a high correlation of DNase-chip and MPSS cluster data (Fig. 3). To determine true sensitivity of the DNase-chip assay, we performed real-time PCR on clusters of two or more MPSS tags that mapped to the ENCODE regions, to validate those MPSS clusters that represented true DNase I hypersensitive sites. For CD4+ T cells, we validated 130 such sites, and for GM06990 cells, we validated 81. These sites represented our ‘gold-standard’ DNase I hypersensitive sites.
Figure 3. Comparison of DNase-chip and MPSS data for CD4+ T cells.
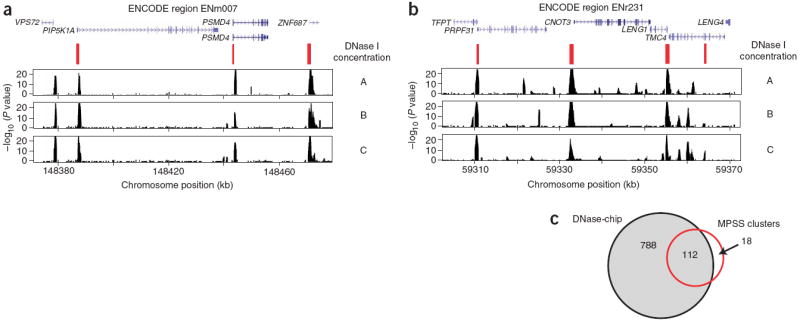
(a,b) DNase-chip P value data from two different ENCODE regions shows peaks that are detected using multiple concentrations of DNase I. MPSS clusters (in red) show DNase I hypersensitive sites that were previously identified using a sequence-based approach and clustering of tags. Note that there are regions detected by DNase-chip that have not been detected by MPSS, as expected because of limitations in the number of sequence tags. Many DNase-chip peaks that are only detected with a single DNase I concentration appear to be false positives. (c) Venn diagram shows the number of DNase I hypersensitive sites identified by DNase-chip (black; the number of DNase-chip regions are normalized using positive predictive values) and validated MPSS clusters (red).
As the positive predictive value indicated that DNase-chip peaks detected using at least two DNase I concentrations, as well as the highest DNase I concentration (ABC, AB, AC, BC, and C only) were likely to be enriched for true DNase I hypersensitive sites, we first determined sensitivity of DNase-chip for these combined categories. Using DNase-chip, we detected approximately 78% and 75% of the gold-standard DNase I sites from these combined categories for CD4+ T cells and GM06990 cells, respectively (Table 1). An alternative approach to choosing DNase-chip categories is to average DNase-chip data from experiments with all three DNase I concentrations and all three biological replicates. Tested against the gold-standard set of DNase I hypersensitive sites, the averaged DNase-chip data from CD4+ T cells and GM06990 cells detected 86% and 89% of these sites, respectively (Table 1). DNase-chip detected not only the majority of MPSS clusters, but also additional DNase I hypersensitive sites, which were missed using the present level of sequencing by MPSS (Fig. 3c).
Table 1.
Sensitivity of DNase-chip, as assessed against gold-standard DNase I hypersensitive sites derived from MPSS clusters (n = 130 for CD4+ T cells, n = 81 for GM06990 cells)
| Cell line | DNase I concentrations | Total number of sites detected by DNase-chip | Gold-standard sites detected by DNase-chip | Sensitivity |
|---|---|---|---|---|
| CD4+ T cells | ABC + AB + AC + BC + C | 1,423 | 101/130 | 78% |
| Averaged | 1,264 | 112/130 | 86% | |
| GM06990 cells | ABC + AB + AC + BC + C | 1,463 | 61/81 | 75% |
| Averaged | 1,100 | 72/81 | 89% |
Validation of DNase-chip: specificity
Specificity is defined as TN / (TN + FP), where TN is true negatives and FP is false positives. True negatives are represented by real-time PCR primer sets (designed around randomly selected coordinates from ENCODE regions), which are negative by DNase-chip and whose values are below a stringent threshold value determined by real-time PCR. We tested 177 randomly chosen primer sets from the ENCODE regions on both CD4+ T cells and GM06990 cells. Of these, 134 showed no evidence of marking a DNase I hypersensitive site (ΔCt < 1.5; ΔCt values represent the number of additional cycles to achieve threshold levels of amplification between DNase I-treated and nondigested nuclear DNA) in CD4+ T cells, and 146 were negative in GM06990 cells. These became our gold-standard for non–DNase I hypersensitive sites. We defined a false positive from the DNase-chip data as a signal that fell within one of these non–DNase I hypersensitive sites. We calculate that specificity for DNase-chip peaks that are present when using at least two DNase I concentrations is > 99% for both cell types. Specificity for DNase-chip peaks that are present only when using a single DNase I concentration is 97% for CD4+ T cells and 93% for GM06990 cells. Specificity for averaged data from all three DNase I concentrations and all three biological replicates was 97% for both cell types (Table 2).
Table 2.
Specificity of DNase-chip, as assessed against gold-standard negative DNase I hypersensitive sites derived by real-time PCR of random segments (n = 134 for CD4+ T cells, n = 146 for GM06990 cells)
| Cell line | DNase I concentrations | Total number of sites detected by DNase-chip | Number of sites detected by DNase-chip that fall within a gold-standard negative site | Specificity |
|---|---|---|---|---|
| CD4+ T cells | ABC | 378 | 0/134 | ~100% |
| AB | 134 | 1/134 | 99% | |
| AC | 174 | 0/134 | ~100% | |
| BC | 200 | 0/134 | ~100% | |
| A only | 981 | 2/134 | 99% | |
| B only | 836 | 0/134 | ~100% | |
| C only | 547 | 3/134 | 98% | |
| Averaged | 1,264 | 4/134 | 97% | |
| GM06990 cells | ABC | 172 | 0/146 | ~100% |
| AB | 64 | 1/146 | 99% | |
| AC | 112 | 1/146 | 99% | |
| BC | 159 | 0/146 | ~100% | |
| A only | 712 | 9/146 | 94% | |
| B only | 777 | 4/146 | 97% | |
| C only | 504 | 2/146 | 99% | |
| Averaged | 1,100 | 5/146 | 97% |
Identification of cell type specific DNase I hypersensitive sites
Although many DNase-chip peaks were present in both CD4+ T cells and GM06990 cells, some were detected in only one of the two cell types. We performed confirmatory real-time PCR analysis with primers designed to flank ubiquitous and cell type–specific DNase-chip peaks. In most instances, we confirmed the tissue pattern of DNase-chip (Fig. 4). As a whole, DNase I hypersensitive sites that appeared to be cell type–specific tended to be less sensitive to DNase I (lower ΔCt values), which is in agreement with our previous report8. A small number of the DNase-chip peaks that appeared to be cell type–specific by DNase-chip were present in both cell types, as determined by real-time PCR, indicating that this method has a low but non-zero false negative rate.
Figure 4. Identification of cell type–specific DNase I hypersensitive sites.
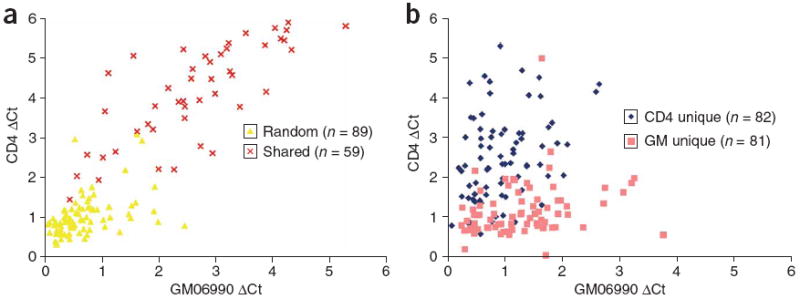
(a,b) Real-time PCR was performed on both CD4+ T cells and GM06990 cells. Real-time PCR using primer sets that flank random regions of the genome or DNase-chip peaks that are present for both cell types (a). Real-time PCR using primer sets that flank DNase-chip peaks that are present in only CD4+ T cells (CD4) or GM06990 cells (GM; b).
Correlation of DNase-chip to the annotated genome
Thirty of the ENCODE regions consist of randomly selected 500-kb segments stratified by different levels of gene density (low, average, and high) and sequence conservation (low, average, and high)1. This 3 × 3 matrix of ENCODE stratified regions therefore contains an overall representation of the entire genome (for example, regions that are gene dense and highly conserved, gene dense and not conserved, etc.) DNase-chip peaks detected in at least two DNase I concentrations from CD4+ T cells and the GM06990 cell line were most highly enriched in ENCODE regions that were gene rich and highly conserved (Fig. 5a,b). The second highest enrichment was in ENCODE regions that were gene rich and moderately conserved. Notably, the lowest number of DNase I hypersensitive sites for both cell types was in gene poor regions that had the highest levels of conservation. Cell type–specific DNase I hypersensitive sites (present in CD4+ T cells but not in GM06990 cells and vice-versa) also were enriched in similar stratified ENCODE regions (data not shown).
Figure 5. Location of DNase I hypersensitive sites relative to the annotated genome.
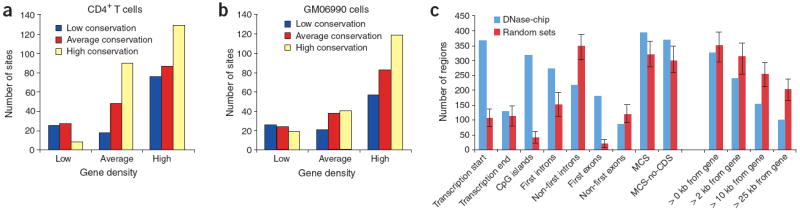
(a,b) DNase-chip peaks were mapped to ENCODE regions stratified by gene density and human-mouse sequence conservation for both CD4+ T cells (a) and the GM06990 lymphoblastoid cell line (b). (c) The genomic locations of DNase I hypersensitive sites (detected with at least two concentrations of DNase I) and computationally generated random controls (n = 1,000) were compared to Gencode transcription start and end sites (within a 2-kb window), CpG islands, first introns, non-first introns, first exons, non-first exons, conserved sequences (MCS), conserved sequences minus coding exons (MCS-no-CDS). The number of DNase I hypersensitive sites at different distances (0 kb, 2 kb, 10 kb, and 25 kb) from Gencode genes was also determined. Error bars represent the entire range of values seen randomly generated mock datasets (n = 1,000). Compared to the random controls, the locations of the DNase-chip peaks are significantly (Monte Carlo P < 0.001) over- or under-represented at all positions except transcription end and > 0 kb from gene.
We mapped DNase-chip peaks that were present with at least two DNase I concentrations from both cell types to the annotated genome. Compared to 1,000 randomly generated datasets, there was a significant enrichment (P < 0.001) of DNase I hypersensitive sites detected within 2 kb of transcriptional start sites, first exons, first introns, CpG islands and regions that are highly conserved between species. There was less enrichment of DNase I hypersensitive sites within 2 kb of transcriptional end sites. We detected a significant reduction (P < 0.001) of DNase I hypersensitive sites at non-first exons or introns and regions of the genome greater than 2 kb from the nearest gene (Fig. 5c).
Gene expression analysis
To determine whether DNase I hypersensitive sites correlate with elevated levels of gene expression, we compared the expression level of each gene transcript by the distance of each transcription start site to the nearest DNase I hypersensitive site (Fig. 6a,b). As expected (from the above analysis), a high percentage of transcription start sites have a DNase I hypersensitive site nearby (<1 kb). Additionally, for both CD4+ T cells and GM06990 lymphoblastoid cells, we observed higher levels of expression more commonly when there is a nearby DNase I hypersensitive site. Genes that do not have a DNase I hypersensitive site nearby (>1 kb) are more likely to have lower gene expression. But the presence of a nearby DNase I hypersensitive site is not sufficient for elevated levels of expression. We detected a significant change in average gene expression for genes that had a DNase I hypersensitive site nearby versus those that did not for both CD4+ T cells (P < 1.2 × 10−55) and GM06990 cells (P < 1.4 × 10−38; Fig. 6c). For genes that were near a DNase I hypersensitive site unique to one cell type, however, we did not detect a significant change in average gene expression between the two cell types (data not shown).
Figure 6. Expression of genes relative to proximity to DNase I hypersensitive sites.
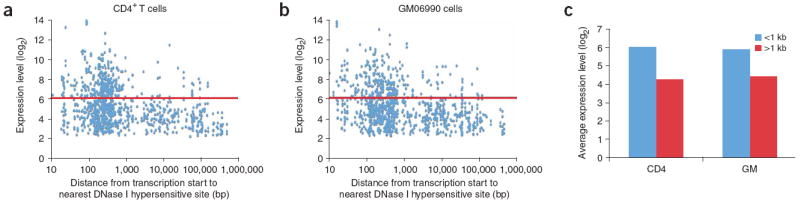
(a,b) The distance of each transcription start site (blue dots) to the nearest DNase I hypersensitive site was compared to the gene expression values of each transcript for both CD4+ T cells (a) and the GM06990 lymphoblastoid cell line (b). Horizontal red lines mark the expression level that separates most genes that have a DNase I hypersensitive site nearby (<1,000 bp) versus those that do not (>1,000 bp). (c) Average expression values of genes that have a DNase I hypersensitive site within 1 kb versus those that do not were determined for both cell types. Average expression values of genes that have a DNase I hypersensitive site are significantly different from genes that do not (P < 1 × 10−38). CD4+ T cells, CD4; or GM06990 cells, GM.
DISCUSSION
DNase-chip is a robust method to identify DNase I hypersensitive sites by hybridizing DNase I–treated, end-captured material to tiled microarrays. This method has many of the same strengths of traditional Southern blot strategies for identifying regulatory regions. By using three DNase I concentrations, the ability to distinguish DNase I hypersensitive sites that display different levels of sensitivity is enhanced. The protocol for DNase-chip is straightforward, sensitive and specific at identifying valid DNase I hypersensitive sites.
Although we have validated the approach in the ENCODE regions, this method can make use of custom tiled microarrays to readily focus on any portion of the genome from any organism. The method is fully scalable and should be amenable to whole-genome scans, as was recently done with ChIP-chip13.
One of our original concerns regarding mapping DNase I hypersensitive sites in replicating cells was that replicating DNA forks would introduce background5,14. This was an early justification for using CD4+ T cells, which are nonreplicating when derived by aphoresis. Even though there were slightly elevated background levels of DNase I digestion in the cycling GM06990 lymphoblastoid cell line, this method appears to be quite effective regardless of the status of cell division. But to reduce the background even further, it may be helpful to synchronize or block cell division.
As with any array-based method, DNase-chip has limitations. First, the resolution size for DNase-chip is limited by the extent of sonication-based shearing (about 200–500 bp). But because DNase I hypersensitive sites are typically around 250 bp in size, we believe this level of resolution is acceptable. Second, tiled microarrays exclude repetitive DNA. Therefore, combining DNase-chip with other methods, such as MPSS with long sequence reads, might be needed to discover DNase I hypersensitive sites within repetitive elements. Third, presently DNase-chip currently requires a large number of cells (~5 × 107). Finally, the cost of performing DNase-chip is considerable, if performed on the whole genome, but will become more affordable as arrays become less expensive.
The genomic coordinates of all DNase I hypersensitive sites described in this manuscript are publicly available (http://research.nhgri.nih.gov/DNaseHS/chip_2006 and http://genome.ucsc.edu/ENCODE/). The locations of these DNase I hypersensitive sites correlate well with other annotated regions of the genome known to mark gene regulatory elements, such as 5′ ends of genes, CpG islands and highly conserved sequences. Notably, the lowest density of DNase I hypersensitive sites mapped to gene-poor regions of the genome that were highly conserved between human and mouse. Future studies aimed at this latter set of conserved elements will determine what type of functional elements these regions may represent. Genes with a DNase I hypersensitive site nearby were more likely to have elevated gene expression, but the presence of a DNase I hypersensitive site was not sufficient for higher expression levels. The small number of outliers (genes that have elevated expression levels, but do not have a nearby DNase I hypersensitive site) could be due to the false negative rate of DNase-chip, the presence of repetitive elements nearby the transcription start (thereby not included on the arrays), or an incorrectly mapped transcription start site. We were also surprised that we were unable to detect significant changes in gene expression between CD4+ and GM06990 cells for genes that had a nearby DNase-chip signal present in only one of the two cell types. One explanation for this could be the similar gene expression patterns of these two lymphocyte cell types. Future expression studies using more diverse cell types may help clarify how chromatin structure and gene expression are related.
In the future, whole-genome DNase-chip from several cell types can be used to identify ubiquitous versus cell type–specific DNase I hypersensitive sites. It should also be possible to apply this approach genome-wide to different states of the same tissue, including normal versus diseased, resting versus stimulated, undifferentiated versus differentiated, and untreated versus drug-treated, to identify global changes in regulation. But even though methods such as DNase-chip will help identify the functional regions of the genome, determining the type of regulatory function for each DNase I hypersensitive site remains a daunting challenge. Clues can be gleaned from correlating DNase I hypersensitive sites with sequence conservation, transcription factor binding sites and histone modifications (ChIP-chip), motif discovery, promoter or enhancer activity, DNA methylation and more detailed gene expression analysis. Groups such as the ENCODE consortium are beginning to compare and contrast these global data sets in an effort to better understand how the genome is regulated1. DNase I hypersensitive site identification can be an important component of such ground breaking efforts to understand the complete function of the genome.
METHODS
Preparation of DNase I–treated nuclei
We obtained primary human CD4+ T cells (>95% purity) from aphoresed human blood from three individuals (National Institutes of Health Blood Bank, Institutional Review Board exemption issued by National Institutes of Health Office of Human Subjects) using a negative purification method (Miltenyi Biotech). We grew GM06990 cells in culture in RPMI with 15% fetal bovine serum (Coriell Cell Repositories). We isolated intact nuclei from three biological replicates from at least 5 × 107 cells by methods previously described5. We digested DNA from these nuclei with six different concentrations of DNase I (0–12 units) for 10 min at 37 °C (Roche). We stopped digests with 0.1 M EDTA, and imbedded DNA in 1% InCert low-melt gel agarose plugs (BioWhittaker). We incubated DNA plugs overnight at 37 °C in LiDS buffer (1% lithium dodecyl sulfate, 10 mM Tris-HCl (pH 7.5), 100 mM EDTA), washed them four times in 50 mM EDTA, and stored them in 50 mM EDTA at 4 °C. DNase I–digested fragments were made blunt in gel using T4 DNA polymerase (New England Biolabs). Then we purified DNA from the agarose by melting the agarose at 65 °C, phenol-chloroform extracting the DNA, and ethanol precipitating it.
We used pulsed field gel electrophoresis (pulse time, 20–60 s for 18 h) to identify the three concentrations of DNase I to be used for DNase-chip (Bio-Rad). After digestion with concentration ‘A’, most DNA was very high molecular weight (>1,000 kb), but there was some digestion resulting in smaller fragments (50–1,000 kb). Nuclear DNA digested with concentration ‘B’ had an average size of 50–1,000 kb. Digestion with concentration ‘C’ generally resulted in fragments less than 200 kb.
We purified genomic DNA used for the random-sheared control from CD4+ T cells and GM06990 cells (Gentra). We vigorously pipetted DNA from each cell type and vortexed it to generate randomly sheared DNA fragments approximately the same size as after treatment with DNase I concentration ‘B’. These fragments were blunt ended using T4 DNA polymerase and purified by phenol-chloroform extraction and ethanol precipitation.
Capture of DNase I digested ends
The complete in-depth DNase-chip protocol is available online (Supplementary Protocol online). We ligated biotinylated linkers (5′-Biotin-GCGGTGACCCGGGAGATCTGAATTC3-′ and 5′-Phosphate-GAATTCAGATC-3AmM-3′) onto blunt-ended fragments from DNase I–treated or randomly sheared DNA. We resuspended the ligation mix in TE buffer and sonicated it to generate 200–500-bp fragments (Branson). We captured biotinylated fragments with streptavidin-coated beads (Dynal). Sheared ends were made blunt using T4 DNA polymerase and ligated to nonbiotinylated linkers (same as linkers above but without the biotin). DNase I–captured ends were amplified using ligation-mediated PCR (using primer 5′-GCGGTGACCCGGGAGATCTGAATTC-3′).
Hybridization to tiled ENCODE microarrays and data analysis
We randomly labeled ligation-mediated PCR products from DNase I–treated and random-sheared DNA with Cy3-dUTP and Cy5-dUTP, and removed unincorporated nucleotides from the samples using Centrisep columns (Princeton Separations). We mixed the purified Cy3- and Cy5-labeled samples, supplemented them with blocking cocktail (tRNA, Cot-1 DNA, poly(A)+ RNA and poly(T)+ RNA), and ethanol-precipitated them. We resuspended the pellet in water and an equal volume of 2× hybridization buffer (50% formamide, 10× SSC and 0.4% SDS) and hybridized the samples to Nimblegen ENCODE tiled arrays for >20 h (Maui). The ENCODE array has approximately 385,000 50-mer oligos spaced approximately every 38 bp of unique sequence (Nimble-Gen). We washed the slides and scanned them (Agilent); signals were normalized using Nimblescan software (NimbleGen). Normalized ratio data (DNase:random) was averaged either from all nine hybridizations (three DNase I concentrations and three biological replicates) or from each DNase I concentration separately (including three biological replicates). These ratios were used to perform a χ2 test on sliding 500-bp windows to identify regions with a higher than expected number of oligos in the top 5% of the log-ratio distribution (P < 0.001). Software was written in R (http://www.r-project.org) and is available upon request.
Additional methods
A description of computational, expression and PCR-validation analyses is available in Supplementary Methods online.
Accession codes
Gene Expression Omnibus (GEO): GSE4406.
Supplementary Material
Acknowledgments
We thank E. Margulies and the Multiple Species Alignment group from the ENCODE consortium for kindly providing the sequence conservation track. We also thank S. Anderson, A. Elkahloun, and the National Human Genome Research Institute Microarray core for excellent technical assistance. This research was supported by the Intramural Research Program of the US National Human Genome Research Institute, National Institutes of Health.
Footnotes
COMPETING INTERESTS STATEMENT The authors declare competing financial interests (see the Nature Methods website for details).
Note: Supplementary information is available on the Nature Methods website.
References
- 1.The Encode Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
- 2.Wu C. The 5′ ends of Drosophila heat shock genes in chromatin are hypersensitive to DNase I. Nature. 1980;286:854–860. doi: 10.1038/286854a0. [DOI] [PubMed] [Google Scholar]
- 3.Gross DS, Garrard WT. Nuclease hypersensitive sites in chromatin. Annu Rev Biochem. 1988;57:159–197. doi: 10.1146/annurev.bi.57.070188.001111. [DOI] [PubMed] [Google Scholar]
- 4.Wu C, Wong YC, Elgin SC. The chromatin structure of specific genes: II. Disruption of chromatin structure during gene activity. Cell. 1979;16:807–814. doi: 10.1016/0092-8674(79)90096-5. [DOI] [PubMed] [Google Scholar]
- 5.Crawford GE, et al. Identifying gene regulatory elements by genome-wide recovery of DNase hypersensitive sites. Proc Natl Acad Sci USA. 2004;101:992–997. doi: 10.1073/pnas.0307540100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dorschner MO, et al. High-throughput localization of functional elements by quantitative chromatin profiling. Nat Methods. 2004;1:219–225. doi: 10.1038/nmeth721. [DOI] [PubMed] [Google Scholar]
- 7.Sabo PJ, et al. Discovery of functional noncoding elements by digital analysis of chromatin structure. Proc Natl Acad Sci USA. 2004;101:16837–16842. doi: 10.1073/pnas.0407387101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crawford GE, et al. Genome-wide mapping of DNase hypersensitive sites using massively parallel signature sequencing (MPSS) Genome Res. 2006;16:123–131. doi: 10.1101/gr.4074106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ren B, et al. E2F integrates cell cycle progression with DNA repair, replication and G(2)/M checkpoints. Genes Dev. 2002;16:245–256. doi: 10.1101/gad.949802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scacheri PC, et al. Genome-wide analysis of menin binding provides insights to MEN1 tumorigenesis. PLoS Genet. 2006;2:406–419. doi: 10.1371/journal.pgen.0020051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scacheri PC, Crawford GE, Davis S. Statistics for ChIP-chip and DNase hypersensitivity experiments on NimbleGen arrays. Methods Enzymol. doi: 10.1016/S0076-6879(06)11014-9. in the press. [DOI] [PubMed] [Google Scholar]
- 12.McArthur M, Gerum S, Stamatoyannopoulos G. Quantification of DNase I-sensitivity by real-time PCR: quantitative analysis of DNase I-hypersensitivity of the mouse beta-globin LCR. J Mol Biol. 2001;313:27–34. doi: 10.1006/jmbi.2001.4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim TH, et al. A high-resolution map of active promoters in the human genome. Nature. 2005;436:876–880. doi: 10.1038/nature03877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jantzen K, Fritton HP, Igo-Kemenes T. The DNase I sensitive domain of the chicken lysozyme gene spans 24 kb. Nucleic Acids Res. 1986;14:6085–6099. doi: 10.1093/nar/14.15.6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.