

Abstract
Methamphetamine abuse in young adults has long-term deleterious effects on brain function that are associated with damage to monoaminergic neurons. Administration of glial cell line-derived neurotrophic factor (GDNF) protects dopamine neurons from the toxic effects of methamphetamine in animal models. Therefore, we hypothesized that a partial GDNF gene deletion would increase the susceptibility of mice to methamphetamine neurotoxicity during young adulthood and possibly increase age-related deterioration of behavior and dopamine function. Two weeks after a methamphetamine binge (4 × 10 mg/kg, i.p., at 2 h intervals), GDNF+/− mice had a significantly greater reduction of tyrosine hydroxylase immunoreactivity in the medial striatum, a proportionally greater depletion of dopamine and 3,4-dihydroxyphenylacetic acid (DOPAC) levels in the striatum, and a greater increase in activated microglia in the substantia nigra than wild-type mice. At 12 months of age, methamphetamine-treated GDNF+/− mice exhibited less motor activity and lower levels of tyrosine hydroxylase-immunoreactivity, dopamine, DOPAC, and serotonin than wild-type mice. Greater striatal dopamine transporter activity in GDNF+/− mice may underlie their differential response to methamphetamine. These data suggest the possibility that methamphetamine use in young adults, when combined with lower levels of GDNF throughout life, may precipitate the appearance of parkinsonian-like behaviors during aging.
Keywords: aging, GDNF, methamphetamine, mice, neurotoxicity, Parkinsonism
Introduction
Use of methamphetamine (METH), a powerfully addictive psychostimulant, is highest among young adults ages 18–25 in the United States (Substance Abuse and Mental Health Services Administration Office of Applied Statistics National Survey on Drug Use and Health Report, 2006, Figure 5.4, p 50). METH binds to monoamine transporters and is transported into monoaminergic terminals, causing increased cytosolic release and, ultimately, depletion of dopamine (DA) and serotonin (5-HT) as well as their synthetic enzymes, tyrosine hydroxylase (TH) and tryptophan hydroxylase, respectively (Hotchkiss and Gibb, 1980; Wagner et al., 1980; Bakhit et al., 1981; Kita et al., 2003). High doses of METH damage monoaminergic terminals and transporters in the striatum of humans (Volkow et al., 2001) and experimental animals (Ricuarte et al., 1982; Sonsalla et al., 1992). METH-induced hyperthermia (Bowyer et al., 1992; O'Callaghan and Miller, 1994), glutamate excitotoxicity (Sonsalla et al., 1986, 1992; Mark et al., 2004), increased production of reactive oxygen species (Yamamoto and Zhu, 1998; LaVoie et al., 1999), and microglia activation (LaVoie et al., 2004) all contribute to the acute toxic effects of METH. Despite some functional recovery over time, suppressed striatal metabolism and psychomotor impairment are prolonged in abstinent human METH abusers (Wang and Volkow, 2004). Furthermore, because the function of the nigrostriatal dopamine system decreases with age, it has been suggested that people who abuse METH early in life may be more at risk for developing parkinsonian-like motor deficits as they age (Volkow et al., 2001).
Glial cell line-derived neurotrophic factor (GDNF) is required for the survival, high-affinity DA uptake, and neurite outgrowth of midbrain DA and 5-HT neurons (Lin et al., 1993; Krieglstein et al., 1995; Galter and Unsicker, 1999; Ducray et al., 2006). Importantly, GDNF is decreased in the substantia nigra (SN) of Parkinson's disease patients (Jenner and Olanow, 1998) and also in normal aged rodents (Yurek and Fletcher-Turner, 2001), suggesting its involvement in motor dysfunction and DA neuronal degeneration. Nigral GDNF reduction in Parkinson's disease patients is not just caused by the loss of DA neurons per se, but also by a reduction of GDNF expression in the remaining DA neurons, suggesting that a lifelong deficit in GDNF may contribute to DA cell loss and motor dysfunction during aging. Conversely, GDNF promotes recovery of behavioral and morphological deficits in experimental models of Parkinson's disease (Bowenkamp et al., 1995; Mandel et al., 1997; Jenner and Olanow, 1998; Cass and Manning, 1999; Kordower et al., 2000; Melega et al., 2000; Yurek and Fletcher-Turner, 2001) and after METH toxicity (Cass, 1996).
In GDNF knock-out mice, prenatal development of mesencephalic DA neurons is GDNF independent (Moore et al., 1996; Pichel et al., 1996), whereas postnatal maturation of DA neurons is GDNF dependent (Granholm et al., 2000). Furthermore, at 12 months of age, GDNF+/− mice have fewer TH-positive neurons in the SN and exhibit less motor activity than wild-type (WT) mice (Boger et al., 2006). Therefore, we hypothesized that METH-induced DAergic toxicity during adolescence would be exacerbated in GDNF+/− mice and that this “dual hit” would have long-term behavioral and structural consequences in aging mice.
Materials and Methods
Animals.
GDNF+/− mice were compared with WT littermates in all experiments. The nonfunctional allele for the GDNF gene was originally generated by replacing part of the third exon that encodes the GDNF protein with a cassette expressing the selectable marker neomycin phosphotransferase, as described previously in detail (Pichel et al., 1996). After introducing this construct into embryonic stem cells, six clones were identified with the predicted mutant allele. CD1 or C57BL/6 recipient strains were used to obtain germline transmission of the targeted allele. Heterozygous offspring are viable and fertile, whereas mice homozygous for the mutant GDNF allele (GDNF−/−) die within 24 h of birth. The mice for this study were bred locally at the Medical University of South Carolina on a C57BL/6J background, weaned, and genotyped as described previously (Boger et al., 2006), according to National Institutes of Health (NIH)-approved protocols. The mice were housed in groups of three or four to a cage and had ad libitum access to food and water. They were maintained on a 12 h light/dark cycle at an ambient temperature of 20–22°C.
Experimental design.
When the GDNF+/− and WT mice reached 2.5 months of age, they were injected with METH (10 mg/kg, i.p., four times at 2 h intervals) or saline (0.2 ml). Rectal temperatures were recorded 5 min before the first injection and 20 min after each of the four injections. Motor activity was recorded in photocell chambers for 20 min after the first and fourth injection, and 2 weeks postinjection when the mice were 3 months old. In one experiment, the selective DA uptake inhibitor, nomifensine (7.5 mg/kg, i.p.), was injected immediately before locomotor testing 2 weeks after METH or saline injections. In experiments in which the mice lived until they were 12 months old, their spontaneous locomotor activity was recorded for 20 min in the photocell chambers at 6, 9, and 12 months of age.
Body temperature measurements.
Rectal temperature was measured before the first injection and then 20 min after each intraperitoneal injection with a TH-5 Thermalert Monitor Thermometer (Physitemp Instruments, Clinton, NJ) by holding each mouse at the base of the tail and inserting a probe (RET-3) 2.0 cm past the rectum and into the colon until a rectal temperature was maintained for 3 s.
Locomotor testing.
GDNF+/− and WT mice were tested after the first and fourth injections of either saline or METH, 2 weeks postinjection and, in the longevity studies at 6, 9, and 12 months of age. Locomotor activity (total distance traveled) was assessed in a Digiscan Animal Activity Monitor system [model RXYZCM(8) TAO; Omnitech Electronics, Columbus, OH]. The details of the apparatus have been described previously (Halberda et al., 1997). On the day of testing, the mice were transferred from the animal colony into the laboratory in groups of six and tested in a darkened environment. Data were collected in 1 min intervals for 20 min at the same time of day (8 A.M. to 12 P.M.) for each test period.
Immunohistochemistry.
At 3 (2 weeks postinjection) and 12 months of age, mice were anesthetized with halothane and perfused transcardially with saline followed by 4% paraformaldehyde in phosphate buffer (0.1 m, pH 7.4). The brains were removed, postfixed in 4% paraformaldehyde for 24 h, and then transferred to 30% sucrose in 0.1 m PBS for at least 24 h before sectioning for histochemical analysis. The striatum and midbrain were sectioned on a cryostat (Microm; Zeiss, Thornwood, NY) at 45 μm. Sections through the striatum and SN were processed for free-floating immunohistochemistry using a rabbit polyclonal antibody against TH (1:5000; Pel-Freeze Biologicals, Roger, AZ, USA) or a rat anti-mouse antibody against the cluster differentiation (CD) marker, CD45 (MCA1388; 1:1000; Serotec, Raleigh, NC). Immunodetection was performed using the avidin-biotin-immunoperoxidase method (Choe and McGinty, 2000). Briefly, after a 5 min pretreatment with 2% Triton-X to allow penetration into the tissue, a subset of sections from each mouse was incubated simultaneously in the primary antisera against TH or CD45 for 24 h at 4°C. The sections were then rinsed and incubated for 1 h in biotin-conjugated goat anti-rabbit (TH) or goat anti-rat (CD45) IgG, rinsed, and incubated for 1 h in avidin-biotin-peroxidase reagents (Elite Vectastain kit; Vector Laboratories, Burlingame, CA). The reaction was developed by staining with VIP (Vector Laboratories), which yields a purple reaction product. Each of the above steps was separated by three 10 min washes in PBS. Sections were mounted on glass slides and coverslipped with distrene plasticizer xylene.
Semiquantitation of immunostaining.
Analysis of medial and lateral striatal subregions and the nucleus accumbens was performed using the NIH Image program as described previously (Choe and McGinty, 2000). Briefly, background was subtracted and the Look Up Table scale was adjusted using density slicing. This approach captures all labeled profiles above a threshold density and interactively discriminates them from density values below the threshold. The software then automatically measures the mean optical density and number of pixels per area of the extracted profiles in the selected medial or lateral striatal regions. Two parameters were obtained from this procedure: the area covered by the specific profile population (field area) and the mean density of the specific profiles. The integrated density was obtained by multiplying the field area times the mean density value.
Stereological cell counts.
Quantitative estimates of the total number of TH-immunoreactive neurons in the SN were achieved using an unbiased, stereological cell-counting method as described previously (Granholm et al., 2002). Briefly, the optical fractionator system consists of a computer-assisted image-analysis system including a Nikon (Tokyo, Japan) Eclipse E-600 microscope hard-coupled to a Prior H128 computer-controlled x-y-z motorized stage, an Olympus (Tokyo, Japan) 750 video camera system, a Micron Pentium III 450 computer (Nampa, ID), and stereological software (Stereoinvestigator; MicroBrightField, Colchester, VT). The SN was outlined under low magnification (10×) on every third section through the rostrocaudal extent of the midbrain and the outlined region was measured with a systematic random design of dissector counting frames (100 × 100 μm). Actual mounted section thickness was found to be 35–37 μm, and a 2 μm guard zone was set for the top and bottom of each section. A 40× objective lens with a 1.4 numerical aperture was used to count cells within the counting frames.
Measurement of 5-HT and DA in brain tissue.
At 3 (2 weeks postinjection) and 12 months of age, mice were killed and the striata were dissected unilaterally and frozen at −80°C. The tissue samples were placed in 300 μl of mobile phase containing 0.2 μm isoproterenol as an internal standard, sonicated, and centrifuged (2 min at 13,000 rpm). The concentration of DA and 5-HT in the supernatant was measured by injecting 5 μl of the supernatant into the high-performance liquid chromatography (HPLC). An ESA (Chelmsford, MA) pump (model 582) was used to deliver 0.5 ml/min of mobile phase (0.1 m trichloroacetic acid, 0.01 m sodium acetate, 0.1 mm EDTA, and 16% MetOH, pH = 4.1) to a reversed phase column (ESA HR-80). After separation from the column, the biogenic amines were oxidized/reduced using coulometric detection (Coulochem II; ESA). Three electrodes were used: a guard cell (+0.40 V), a reduction analytical electrode (E1, −0.15 V), and an oxidation analytical electrode (E2, +0.325 V). The peak areas were measured with an ESA 501 Chromatography Data System and the values were compared with an external standard curve for quantification. The final data were evaluated as nanograms per gram of wet tissue weight.
Striatal and plasma METH concentrations.
Striatal tissue (9–31 mg) and blood plasma were analyzed for METH concentrations 30, 60, 120, and 180 min after the last of four 10 mg/kg intraperitoneal METH injections in 2.5-month-old WT and GDNF+/− mice (n = 5 for each time point) using gas chromatography–mass spectrometry. Briefly, when the appropriate time from the last injection was reached (30, 60, 120, 180 min), mice were anesthetized with Equithesin (1 mg/kg, i.p.) and a retro-orbital sinus blood sample was immediately obtained. Blood was initially collected in 44.7 μl of heparinized microcapillary pipettes before being transferred to a 0.5 ml Eppendorf (Hamburg, Germany) tube for storage on ice at 4°C. Each mouse then underwent a rapid decapitation and the whole striatum was removed and stored on dry ice. Once dissections were complete, blood samples were spun at room temperature at 10,000 rpm for 10 min in an Eppendorf model 5415C centrifuge. The plasma was aspirated from the red blood cell pellet and assayed.
A novel method using NiCl-selected ion monitoring of ions 275 amu (METH minus hydrogen fluoride) and 283 amu [nonadeutero (D9)-METH minus deuterium fluoride] was developed to measure METH concentrations in plasma and brain. Samples were extracted by a process modified from a process described previously (Johnson-Davis et al., 2004). Frozen striatal tissue was thawed and suspended in distilled water, fortified with 100 ng of D9-METH (Cerilliant, Round Rock, TX) as an internal standard, homogenized (Polytron), sonicated, alkalinized with concentrated NH4OH (0.1 ml), and then extracted with butyl chloride/acetonitrile (4:1). After centrifugation, the organic phase was transferred to a silanized vial and pentafluoropropionic anhydride (PFPA; 40 μl) was added by vortexing to generate acid in situ for trapping METH as the nonvolatile salt. After 10 min, nitrogen was used to evaporate the sample to dryness and again PFPA (50) was added, followed by capping and derivatization at 42°C for 30 min. The sample was cooled to 25°C, evaporated to dryness under nitrogen, and then the residue was dissolved in heptane (50 μl) and transferred to a silanized microvial insert for gas chromatographic (GC) autoinjection. Calibrator samples used blank mouse brain tissue (25 mg) spiked with METH (Cerilliant) and were processed in parallel with the unknowns, yielding a highly linear standard plot (r = 0.9996). The GC–mass spectrometric (MS) method yielded ion chromatograms devoid of chemical interferences.
Synaptosome preparation.
Mice were decapitated and the striatum was immediately homogenized in 10 volumes (w/v) of cold 0.32 m sucrose. The crude synaptosomal pellet (P2 fraction) was further purified as described previously (Samuvel et al., 2005). The purified synaptosomes were suspended in regular Krebs–Ringer HEPES buffer saturated with 95% O2/5% CO2. Protein concentration was determined by a DC protein assay (Bio-Rad, Hercules, CA) using bovine serum albumin as the standard.
DA uptake.
Purified synaptosomes (10–20 μg) were incubated in 250 μl of assay buffer containing 0.1 mm ascorbic acid, 0.1 mm pargyline, and 20 nm [3H]DA for 3 min as described previously (Samuvel et al., 2005). Nonspecific [3H]DA uptake was defined as the accumulation in the presence of 100 μm cocaine and was subtracted from total uptake. Uptake was terminated by addition of 3 ml of ice-cold stop buffer (PBS containing 100 μm nomifensine) followed by rapid filtration over 0.3% polyethyleneimine-coated Glass Fiber/B filters using a Brandel (Gaithersburg, MD) Cell Harvester. Filters were washed rapidly with 5 ml of cold stop buffer and radioactivity bound to the filter was counted by liquid scintillation spectrometry. All uptake assays were performed in triplicate and mean values of specific uptake ± SEM from at least three separate experiments were determined.
DAT immunoblotting.
Dopamine transporter (DAT) immunoblotting was performed on detergent extracts of synaptosomes using an affinity purified DAT-specific antibody raised as described previously (Samuvel et al., 2005). Protein samples (25 μg) were separated using 4–15% linear gradient SDS-PAGE, transferred to a polyvinylidene difluoride membrane and probed with the DAT antibody as indicated in the figure legends. Immunoreactive bands were visualized by ECL plus reagent. Subsequently, the blots were striped and reprobed with calnexin antibodies to validate transfer and the total amount of protein used. DAT band densities were normalized using levels of calnexin to preclude errors accompanying with sample loading/transfer and values were averaged across three experiments. Band densities were quantified by scanning and analyzed using NIH Image software.
Statistical analysis.
Body temperature, locomotor activity, HPLC, stereological cell counts, immunohistochemical densitometry, DAT activity, and immunoreactivity data were analyzed by mixed-factor ANOVAs followed by Student–Neuman–Keuls tests for individual group comparisons. METH concentration data for GDNF+/− versus WT mice were analyzed with a two-tailed Student's t test.
Results
METH effects on behavior are exacerbated in GDNF+/− mice
METH, administered in a frequently used binge paradigm (10 mg/kg, i.p., four times at 2 h intervals) (Sonsalla et al., 1992; Yamamoto and Zhu, 1998) increased body temperatures and altered motor activity in 2.5-month-old GDNF+/− and WT mice (Fig. 1) without causing any mortality. A mixed-factor ANOVA revealed a significant effect of treatment, but not genotype on body temperatures after the second (F(1,16) = 49.5; p < 0.001), third (F(1,16) = 46.6; p < 0.001), and fourth (F(1,16) = 93.0; p < 0.001) injections (Fig. 1A). Similarly, METH treatment, regardless of genotype, significantly increased motor activity after the first injection (F(1,16) = 10.6; p < 0.001) (Fig. 1B) and reduced motor activity after the fourth injection (F(1,16) = 30.2; p < 0.001) (Fig. 1C) to the same extent in both genotypes. Two weeks after METH or saline, a separate cohort of GDNF+/− and WT mice was challenged with saline or the DAT uptake inhibitor, nomifensine (Fig. 1D). The locomotor activity of all mice challenged with saline did not differ from each other whereas those challenged with nomifensine demonstrated a significant increase in locomotor activity (F(1,24) = 284.7; p < 0.001). However, there was a significant METH by nomifensine interaction (F(1,24) = 21.99; p < 0.001) whereby nomifensine elevated activity of saline-pretreated mice to a greater extent than that of mice that had undergone the METH binge (Fig. 1D). Thus, the DAT uptake inhibition challenge revealed a METH-induced motor activity deficit not apparent without the drug challenge.
Figure 1.
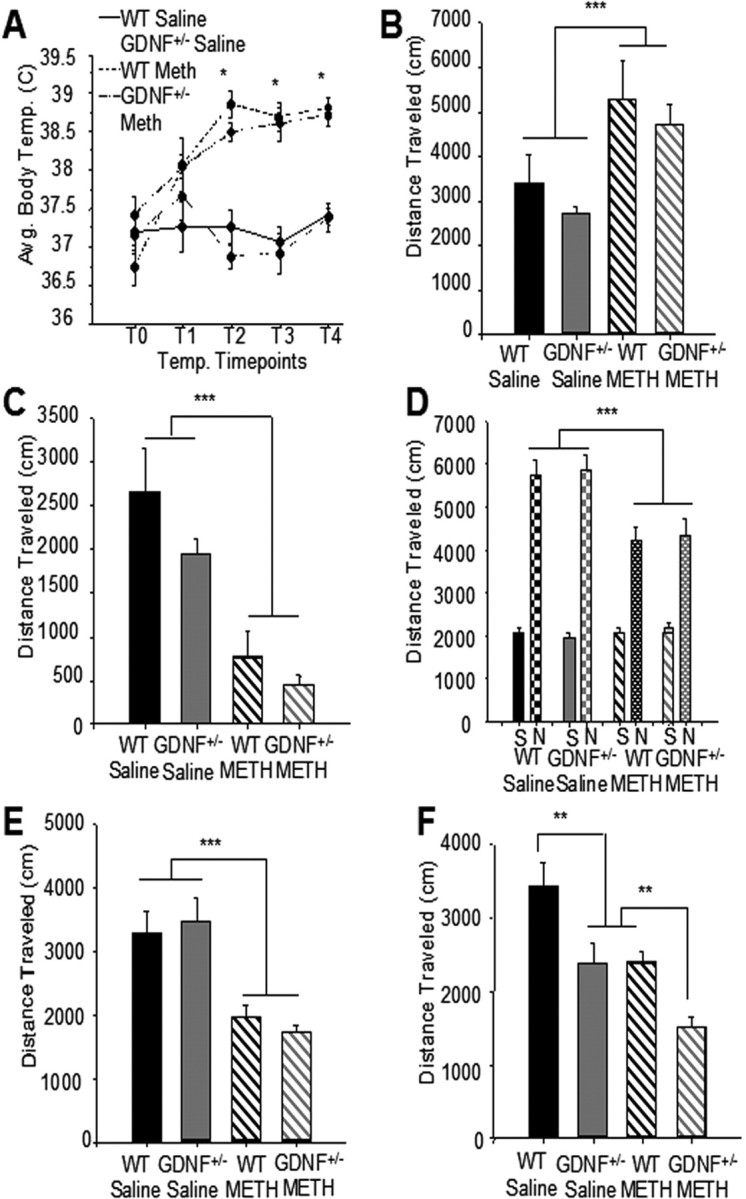
METH-induced hyperthermia and locomotor activity did not differ between GDNF+/− and WT mice initially, but differences in locomotor activity were revealed as the mice aged. A, A significant increase in body temperature occurred in METH versus saline-treated mice that did not differ between genotypes (*p < 0.05 vs WT Saline and GDNF+/− Saline). n = 5 per group. B, After the first injection, regardless of genotype, mice treated with METH were significantly more active than saline-treated mice (***p < 0.001). n = 5 per group. C, After the fourth injection, mice treated with METH, regardless of genotype, were hypoactive when compared with saline-treated mice (***p < 0.001). n = 5 per group. D, Two weeks after the injections (3 months of age), there were no differences in spontaneous motor activity among the groups challenged with saline. In contrast, METH binge-pretreated mice challenged with nomifensine 2 weeks later exhibited significantly less activity than those pretreated with saline (***p < 0.001). E, At 9 months of age, mice treated with METH, regardless of genotype, exhibited less locomotor activity than saline-treated mice (***p < 0.001). n = 8 per group. F, At 12 months of age, saline-pretreated GDNF+/− mice displayed less motor activity than WT mice (**p < 0.01). METH-treated GDNF+/− mice exhibited less motor activity than GDNF+/− mice treated with saline or METH-treated WT mice (*p = 0.05; **p = 0.01; ***p = 0.001). n = 8 per group. Error bars indicate SEM.
Another cohort of mice, treated similarly at 2.5 months of age with METH or saline, lived until 12 months of age and their locomotor activity was tested every 3 months without additional drug challenge. At 9 months of age, but not 6 months of age (data not shown), mice of both genotypes that had undergone the METH binge as young adults were less active than saline-treated mice (F(1,26) = 33.5; p < 0.001) (Fig. 1E). In contrast, a two-factor between-groups ANOVA on locomotor activity data at 12 months of age revealed a significant interaction of METH by genotype (F(1,26) = 18.0; p < 0.001). Comparison of means across the four groups with a one-way ANOVA followed by Student–Neuman–Keuls tests indicated that saline-injected GDNF+/− mice were significantly less active than age-matched WT saline controls (p < 0.01) at 12 months of age, confirming our previous results (Boger et al., 2006). Moreover, 12-month-old GDNF+/− mice were significantly less active than age-matched WT mice exposed to METH as young adults (p < 0.01) (Fig. 1F). These data suggest that partial GDNF depletion coupled with METH exposure produced an additive behavioral deficit during aging that emerged many months after the original insult.
METH effects on TH in the striatum are exacerbated in GDNF+/− mice
On preliminary visual inspection, there were apparent differences in the intensity of TH-immunoreactivity (IR) in the medial and lateral regions of the dorsal striatum 2 weeks after METH or saline injections (Fig. 2). Therefore, medial and lateral striatal areas were analyzed independently by densitometry. ANOVA revealed an overall treatment effect (F(1,16) = 1525.4; p < 0.001), but no genotypic effect or interaction, for TH-IR values in the lateral striatum (Fig. 2E, right). METH reduced TH-IR in the lateral striatum 97% (WT) and 99% (GDNF+/−) compared with saline controls (Fig. 2C–E, right). In contrast, in the medial striatum, there were significant treatment (F(1,16) = 24.1; p < 0.001) and genotype (F(1,16) = 16.8; p < 0.001) effects, but no interaction (F(1,16) = 0.146; p > 0.05) of the two factors. A one-way ANOVA followed by pairwise comparisons revealed that TH-IR in the medial striatum of saline-injected GDNF+/− mice was significantly lower (32%) than that of saline-treated WT mice (p < 0.05) (Fig. 2A,B,E, left) and METH produced a significantly greater decrease in TH-IR in GDNF+/− (89%) than in WT (55%) mice (Fig. 2C–E, left). There was no significant effect of METH on TH-IR in the nucleus accumbens shell or core (supplemental Table 1, available at www.jneurosci.org as supplemental material). Striatal TH-IR in 12-month-old GDNF+/− and WT mice after administration of a toxic regimen of METH at 2.5 months of age is quantified in Figure 2F. At this age, in the lateral striatum, ANOVA revealed overall genotypic (F(1,20) = 28.3; p = 0.001) and treatment (F(1,20) = 12.5; p = 0.01) effects, but no interaction, indicating a deficit in recovery from the METH-induced insult in the lateral striatum in both genotypes. Comparison across groups with a one-way ANOVA (F(3,20) = 15.0; p < 0.001) and Student–Neuman–Keuls pairwise comparisons indicated that TH-IR in the lateral striatum was significantly less in saline-treated GDNF+/− mice than in WT mice (p < 0.05) (Fig. 2F, right) and that TH-IR was significantly less in both genotypes treated with METH when they were young (p < 0.05). Moreover, TH-IR in GDNF+/− mice was less than in WT mice treated with METH (p < 0.05). In the medial striatum, ANOVA revealed a genotypic effect (F(1,20) = 13.7; p < 0.001), but no treatment effect or interaction. Additional analysis using a one-way ANOVA (F(3,20) = 5.8; p < 0.05) and Student–Neuman–Keuls pairwise comparisons indicated that TH-IR in the medial striatum was significantly less in GDNF+/− versus WT mice treated with saline, but not METH (Fig. 2F, left). At 12 months of age, no significant difference existed between groups with TH-IR in the nucleus accumbens shell or core (supplemental Table 1, available at www.jneurosci.org as supplemental material).
Figure 2.
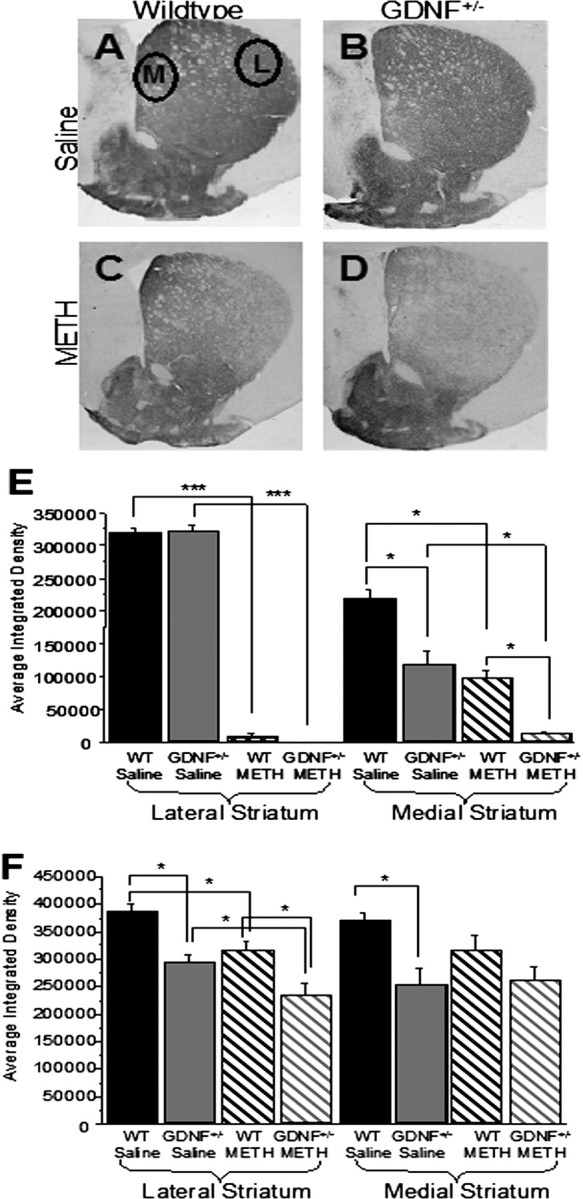
After METH treatment, 3- and 12-month-old GDNF+/− mice demonstrated a greater loss of striatal TH-IR than WT mice. A–D, Photomicrographs of TH-IR in coronal hemisections of the striatum (medial to the left; lateral to the right) from 3-month-old saline-treated WT mice (A), saline-treated GDNF+/− mice (B), METH-treated WT mice (C), and METH-treated GDNF+/− mice (D). E, Quantitation of the average integrated density from the four treatment groups at 3 months of age confirmed that there were no differences in the lateral region (right) of the striatum between the GDNF+/− and WT mice treated with saline, and METH induced a similar TH-IR depletion in the lateral striatum of GDNF+/− and WT mice when compared with saline-treated mice (***p < 0.001). In the medial striatum (left), GDNF+/− mice expressed less TH-IR than WT mice treated with saline (*p < 0.05), and METH induced a greater depletion in the medial striatum of GDNF+/− than WT mice (*p < 0.05). F, Quantitation of the average integrated density of TH-immunoreactive sections from the four treatment groups at 12 months of age demonstrated that TH-IR levels were significantly less in both the lateral and medial striatum of GDNF+/− mice versus WT mice treated with saline (*p < 0.05), that there was a residual deficit in TH-IR in METH- versus saline-treated WT mice in the lateral region of the striatum (right; *p < 0.05), and that there was significantly less TH-IR in the lateral striatum of GDNF+/− versus WT mice treated with METH (*p = 0.05). Circles denote measurement areas of the lateral (L) and medial (M) regions of the striatum. n = 8 per group. Error bars indicate SEM.
Stereological cell counts revealed no difference in the number of SN TH-positive neurons among the four groups at 2 weeks postinjection (data not shown), suggesting that the effects of the METH-binge paradigm were restricted to striatal terminals in both genotypes at this age. In contrast to these results in young adults, a two-way ANOVA on data collected from 12-month-old mice revealed an overall genotypic effect (F(1,24) = 7.3; p < 0.01), but no treatment effect or interaction. Additional analysis using a one-way ANOVA (F(3,24) = 3.938; p < 0.05) and Student–Neuman–Keuls pairwise comparisons indicated that the number of TH-positive neurons in the SNs of 12-month-old, saline-treated GDNF+/− mice (2884 ± 26 neurons) was significantly fewer (p < 0.05) than the number in saline-treated WT mice (3872 ± 25 neurons). However, the number of TH-positive neurons did not differ between saline-treated and METH-treated mice at this age. Thus, METH treatment did not affect the number of SN DA cell bodies in either of the genotypes tested, either 2 weeks or 10 months after the METH binge.
METH exacerbates SN microglial activation in 3-month-old GDNF+/− mice
Reactive microglia are characterized by increased cell body volume; short, thick processes, and increased intensity of staining for cell surface markers, such as CD antigens (LaVoie et al., 2004). Activated microglia detected with CD45 immunostaining in the striatum and SN are illustrated in Figure 3. A two-way ANOVA revealed significant genotypic (F(1,16) = 28.4; p < 0.05) and treatment (F(1,16) = 10.4; p < 0.05) effects with no interaction. Additional analysis using pairwise comparisons of means after a one-way ANOVA (F(3,16) = 13.2; p < 0.01) indicated that the SNs of GDNF+/− mice treated with saline had significantly more CD45 immunostaining than WT mice (p < 0.05), suggesting increased microglial activation (Fig. 3A,B,I). In WT mice, METH treatment did not cause a significant increase in CD45-IR (Fig. 3A,C,I) whereas in GDNF+/− mice, METH treatment caused a significantly greater increase in CD45-IR in the SN than in either saline-treated GDNF+/− mice or METH-treated WT mice (p < 0.05) (Fig. 3C,D,I). In contrast, no differences were detected in CD45-IR in the striatum 2 weeks after injection (Fig. 3E–H,J). In 12-month-old mice, no alterations in CD45-IR were detected in the striatum or SN of any of the four treatment groups (data not shown). These data indicate that GDNF+/− mice have activated microglia in the SN in the absence of a challenge and that METH exacerbates this activity transiently.
Figure 3.
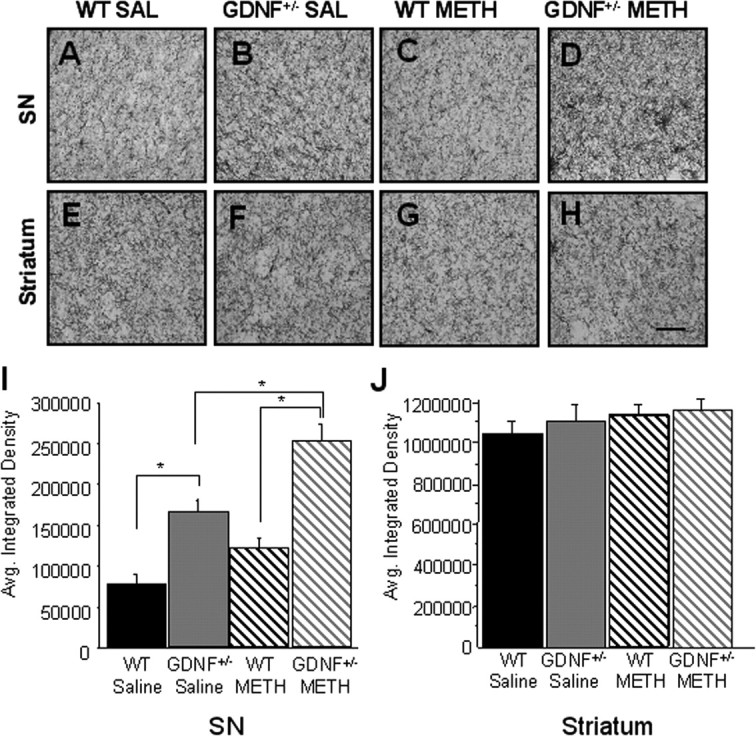
A–H, GDNF+/− mice treated with METH exhibited an inflammatory response in the SN 2 weeks after injection. CD45-IR in the SN (A–D) and striatum (E–H) in saline (SAL)-treated WT mice (A, E), saline-treated GDNF+/− mice (B, F), METH-treated WT mice (C, G), and METH-treated GDNF+/− mice (D, H). Scale bar, 0.5 mm. I, J, Quantitation of the average integrated density of CD45-ir in the SN (I) and striatum (J). Saline-treated GDNF+/− mice had more CD45-IR in the SN than saline-treated WT mice (*p < 0.05). METH treatment caused a significantly greater increase in CD45-IR in the SN of GDNF+/− mice than in saline-treated GDNF+/− mice or METH-treated WT mice (*p < 0.05). n = 5 per group. Error bars indicate SEM.
METH effects on DA and 5-HT levels are exacerbated in GDNF+/− mice
A two-by-two ANOVA on data from 3-month-old mice revealed an overall genotype-by-treatment interaction for striatal DA levels (F(1,16) = 15.1; p < 0.01) and for striatal 3,4-dihydroxyphenylacetic acid (DOPAC) levels (F(1,16) = 9.1; p < 0.01). However, only METH treatment, but not genotype differences were revealed for the DA/DOPAC ratio (F(1,16) = 40.3; p < 0.001) and striatal 5-HT levels (F(1,16) = 50.1; p < 0.001) (Fig. 4C,D). Pairwise comparisons indicated that striatal DA and DOPAC levels were significantly higher (p < 0.05) in GDNF+/− mice than in WT mice treated with saline (Fig. 4A,B). Striatal DA and DOPAC levels were depleted to a similar extent in METH-treated GDNF+/− and WT mice 2 weeks after the injections (p < 0.05) (Fig. 4A,B), resulting in a proportionally greater decrease for GDNF+/− (DA, 88%; DOPAC, 75%) than for WT mice (DA, 76%; DOPAC, 60%). The DA/DOPAC ratios were significantly lower in METH-treated than in saline-treated mice, but did not differ by genotype (Fig. 4C). METH significantly decreased striatal 5-HT, but less severely than DA, and the effect was similar in both genotypes (p < 0.05; 31% in WT and 38% in GDNF+/−) (Fig. 4D).
Figure 4.

METH treatment resulted in a significant reduction in striatal tissue levels of DA and 5-HT that did not differ between genotypes at 3 months of age (*p < 0.05). A, B, D, Saline (SAL)-treated GDNF+/− mice had higher tissue levels of DA (A) and DOPAC (B) than WT mice (p < 0.05), but there was no difference in 5-HT (D) tissue levels. METH treatment caused a significant reduction in DA, DOPAC, and 5-HT tissue levels in WT and GDNF+/− mice (p < 0.05). C, The DA/DOPAC ratio was not different between the genotypes treated with saline, and the ratio was reduced similarly in both genotypes by METH treatment (p < 0.05). D, 5-HT tissue levels were similar in saline-treated GDNF+/− and WT mice and were decreased to the same extent by METH (*p = 0.05; ***p = 0.001). n = 5 per group. Error bars indicate SEM.
At 12 months of age, there was a treatment (F(1,28) = 20.1; p < 0.001) and genotypic (F(1,28) = 15.7; p < 0.001) effect, but no interaction, for striatal levels of DA in saline-treated GDNF+/− versus WT mice (Fig. 5A). There also was a treatment (F(1,28) = 10.8; p < 0.01) and genotypic (F(1,28) = 11.8; p < 0.01) effect, but no interaction, for the striatal levels of DOPAC (Fig. 5B), and a significant treatment (F(1,28) = 6.9; p < 0.05) but no genotypic effect on the DA/DOPAC ratio (Fig. 5C). Furthermore, there was a treatment (F(1,28) = 12.1; p < 0.01) and genotypic (F(1,28) = 15.6; p < 0.001) effect, but no interaction, for striatal levels of 5-HT (Fig. 5D). Additional analysis using a one-way ANOVA (F(3,28) = 9.2; p < 0.001) and pairwise comparisons revealed that 5-HT was significantly less in GDNF+/− than in WT mice that were saline-treated (p < 0.05) (Fig. 5A–D). Importantly, 12-month-old, METH-treated GDNF+/− mice had significantly lower levels of DA (46%), DOPAC (29%), and 5-HT (35%), but similar DA/DOPAC ratios compared with METH-treated WT mice (Fig. 5A–D), suggesting that lasting striatal damage to the DA system after the adolescent METH binge is greater for the combined effect of a GDNF deficiency and METH than for either insult alone.
Figure 5.

METH-induced reductions in striatal DA, DOPAC, and 5-HT tissue levels are still present at 12 months of age. A–D, Saline (SAL)-treated GDNF+/− mice have less striatal tissue levels of DA (A) and 5-HT (D) than WT mice, but the striatal levels of DOPAC (B) and the DA/DOPAC ratio (C) do not differ by genotype. METH-treated WT mice have less tissue DA, DOPAC, DA/DOPAC, and 5-HT than saline-treated WT mice (p < 0.05). Furthermore, DA, DOPAC, and 5-HT levels were significantly lower in METH-treated GDNF+/− mice versus WT mice (*p = 0.05; ***p = 0.001). n = 8 per group. Error bars indicate SEM.
Striatal and plasma METH concentrations do not differ between GDNF+/− and WT mice
In the course of exploring mechanistic bases for the exacerbation of METH-induced neurotoxicity in GDNF+/− versus WT mice, the concentration of METH in the striatum and blood plasma was determined 30, 60, 120, and 180 min after the last of four METH injections in a separate set of mice. Figure 6A illustrates a representative GC–MS–negative ion chemical ionization (NiCl)-selected ion-monitoring chromatogram of METH (top ion profile; tR = 4.32 min) and the deuterated METH internal standard (bottom ion profile, tR = 4.28 min) extracted from a striatal microsample. NiCl yielded selected ion chromatograms free of chemical interferences. Fortified striatal tissue calibrators demonstrated highly linear responses. This method was applied to striatal tissue and blood plasma collected 30, 60, 120, and 180 min after the last of four METH injections. No significant difference in METH concentrations was found between genotypes at any of the time points, either in striatal tissue or in blood plasma (Fig. 6B,C). Thus, enhanced METH toxicity cannot be attributed to differences in METH concentrations in the brains of GDNF+/− mice.
Figure 6.

A, Representative GC–MS–NiCl-selected ion-monitoring chromatogram of METH (top; retention time, 4.32 min) and internal standard deuterated METH (bottom; 4.28 min) extracted from a striatal microsample. B, C, Striatal METH concentration (micrograms per milligram of tissue) (B) and plasma METH concentration (microgram per milliliter) (C) did not differ between WT and GDNF+/− mice at 30, 60, 120, or 180 min after the fourth METH injection. n = 5 per group per time point. Error bars indicate SEM.
Greater DAT activity may underlie enhanced METH toxicity in GDNF+/− mice
A two-by-two ANOVA indicated an age-by-genotype interaction (F(1,20) = 23.2; p < 0.001) for DAT activity data. A one-way ANOVA (F(3,20) = 83.8; p < 0.001) followed by Student–Neuman–Keuls comparisons indicated that DAT activity was significantly higher in striatal synaptosomes from untreated GDNF+/− versus WT mice at both ages and that DAT activity was greater in GDNF+/− mice at 12 months than at 3 months of age (p < 0.05) (Fig. 7A). In contrast, DAT-IR in the whole striatum did not differ according to genotype at either age (Fig. 7B,C). Although GDNF+/− mice were not available for DAT activity measurements after METH treatment in this study, future studies will determine DAT activity in both genotypes after a METH binge.
Figure 7.

GDNF+/− mice have greater DAT activity, but not DAT protein levels in the striatum than WT mice at 3 and 12 months of age. A, DAT activity in striatal synaptosomes from 3- and 12-month-old WT and GDNF+/− mice. DA uptake was assayed in purified synaptosomes (n = 6) and the results are expressed as percentage of uptake in synaptosomes derived from WT mice (mean ± SEM). *p < 0.05 versus WT; +p < 0.05 versus 3 month GDNF+/−. B, DAT protein expression in striatal synaptosomes from 3- and 12-month-old WT and GDNF+/− mice. Purified striatal synaptosomes were subjected to SDS-PAGE followed by immunoblotting with DAT antibody. Subsequently, the blots were stripped and reprobed with calnexin antibody. A representative DAT and calnexin immunoblot is shown. C, Quantitative analysis of DAT band densities. The density of DAT protein bands was quantified using NIH image (mean ± SEM) after normalization with calnexin. n = 6 per group. Error bars indicate SEM.
Discussion
GDNF+/− mice were more susceptible to METH toxicity and age-related decline in brain function than WT mice. Furthermore, long-term consequences of METH toxicity, which emerged as the mice aged, were more severe in GDNF+/− mice. Twelve-month-old GDNF+/− mice treated with saline when young had a greater deficit in motor activity, lower striatal TH-IR, DA, DOPAC, and 5-HT tissue levels, higher DAT activity, and fewer TH-positive SN DA neurons than age-matched WT mice. In addition, a toxic METH binge in young adult mice caused a greater reduction in TH-IR in the lateral striatum, DA, and 5-HT markers and motor activity in GDNF+/− mice than in WT mice at 12 months of age. These data substantiate previous reports that GDNF is an important regulator of DA and motor-system function during aging (Hebert and Gerhardt, 1998; Boger et al., 2006). Furthermore, this study clearly establishes that damage to the nervous system as a consequence of METH may be prolonged and that the absence of immediate behavioral changes does not preclude the possibility that deficits will be manifested at a later time in life. These data are consistent with reports that significant loss of DAT activity in the striatum of humans endures and accompanies psychomotor impairments for many months after the end of METH use (Volkow et al., 2001).
Although precise biological mechanisms were not identified, the present study suggests that greater DAT activity, but not differential METH accumulation in the brain, may underlie the increased vulnerability of GDNF+/− mice to METH. The greater DAT activity of GDNF+/− mice may have increased METH uptake by striatal DAergic terminals resulting in more severe and/or prolonged damage. The combination of less dense TH-IR innervation in medial striatum coupled with greater striatal DA and DOPAC tissue concentrations in GDNF+/− mice suggests that greater DAT activity is an adaptive mechanism that may keep release and uptake in balance under resting conditions when the mice are young. However, striatal levels of DA and DOPAC in GDNF+/− mice were less than in WT mice at 12 months of age, despite persistent elevation in DAT activity, suggesting an imbalance in DA function that emerges as GDNF+/− mice age.
Greater loss of TH-IR in the lateral versus the medial striatum of wild-type mice after METH treatment confirms previous reports (Joyce et al., 2004; O'Callaghan and Miller, 1994; Koike et al., 2005) and extends this observation to the striatum of mice 9.5 months after METH treatment. The reason for less TH-IR loss in the medial versus lateral striatum of WT mice is unknown, but may be attributable to a greater density of DAT normally in the lateral striatum of rodents (Ciliax et al., 1995; Watanabe et al., 2004; Zhu et al., 2005). In contrast, TH-IR loss in the medial striatum was greater in GDNF+/− mice than in WT mice, consistent with an overall increase in striatal DAT activity in the GDNF+/− mice. In 12-month-old mice, however, the more severely affected lateral striatum showed less recovery in both genotypes. Functionally, the lateral striatum, which receives input from the sensorimotor cortex and lateral substantia nigra, is more highly correlated with most aspects of motor function (Gerfen, 1984; McGeorge and Faull, 1989; Voorn et al., 2004) and dysfunction in Parkinson's disease (PD) (Brooks, 2003) than is the medial striatum.
Three-month-old, saline-treated GDNF+/− mice had greater microglial activation than WT mice, but GDNF+/− mice did not demonstrate a loss of SN DAergic neurons until 12 months of age when microgliosis was not detected in the SN. Microglial activation has been associated with DAergic neurodegeneration observed in PD patients and in animal models of the disease (Francis et al., 1995; Czlonkowska et al., 1996; Langston et al., 1999). In addition, reactive microgliosis has been demonstrated in the striatum, but not in the SN, within 1–6 d after a toxic METH binge (LaVoie et al., 2004; Thomas et al., 2004a,b). In this study, however, 2 weeks after mice underwent a METH binge, there was no evidence of activated microglia in the striatum, but activated microglia were present in the SN, with the greatest activation seen in METH-treated GDNF+/− mice. It is likely that the increase in CD45-IR in the SN 2 weeks after injection represented a slow retrograde signal from striatal terminals to the cell body region of DA neurons. These data indicate that subnormal GDNF levels may allow a low level of inflammation that becomes exacerbated when CNS injury occurs in vulnerable brain regions.
It is important to note that, despite an 80–86% reduction in striatal tissue DA levels and a 52–58% decrease in 5-HT levels 2 weeks after the METH injections, spontaneous activity in an open field was not altered in these mice. However, a motor deficit was revealed when METH-treated mice were challenged with the DA uptake blocker, nomifensine, suggesting that the DA system was sufficiently intact to maintain basic motor function, but was unable to perform normally in response to a challenge. An alternative explanation for the absence of a motor deficit despite the substantial striatal DA reduction in these young adult mice is that nucleus accumbens DA was not influenced at 2 weeks post-METH (supplemental Table 1, available at www.jneurosci.org as supplemental material). Because the nucleus accumbens is also involved in mediating locomotor activity (Mogenson and Nielsen, 1983), this undisturbed pathway may also have contributed to the normal open field behavior in 3-month-old mice.
At 12 months of age, a reduction in motor activity emerged in saline-treated GDNF+/− mice that was positively correlated with a reduction in striatal DA, DOPAC, and 5-HT levels, TH-IR in lateral striatum, and a 26% cell loss in the SN not present at 3 months of age. Furthermore, a reduction in spontaneous motor activity emerged in 9- and 12-month-old METH-treated mice that was exacerbated in GDNF+/− mice when they reached 12 months of age. Interestingly, decreases in striatal DA, DOPAC, and 5-HT were also present in METH-treated mice that were exacerbated in GDNF+/− mice at this age, implying a connection. Thus, slow recovery of TH, DA, and 5-HT systems from METH, coupled with the age-related decline in DA and 5-HT systems in GDNF+/− mice, may underlie the exacerbated motor deficits in GDNF+/− mice at 12 months of age. Exacerbated reduction in the function of the nigrostriatal DA system has consistently been associated with greater decline in motor function with aging in both humans (Suhara et al., 1991; Bannon et al., 1992; Volkow et al., 1998) and animals (Emborg et al., 1998; Hebert and Gerhardt, 1998; Cass et al., 2005; Yurek and Fletcher-Turner, 2001).
Alterations in other neurotransmitter and signaling systems may have contributed to the age-related motor deficits. Aging-related decline in motor function has been associated with noradrenergic (NE) deficits (Chan-Palay and Asan, 1989; Gesi et al., 2000). Furthermore, 18-month-old GDNF+/− mice have lower NE concentrations and morphological alterations of NE cell bodies in the locus ceruleus, lower NE transporter activity in the cerebellum and brainstem, and fewer TH-positive fibers in the hippocampus, cerebellum, and frontal cortex compared with age-matched WT mice (Zaman et al., 2003). In addition, preliminary microarray studies from our group indicate that both α-synuclein and the DA D2 receptor mRNA are upregulated in the striatum of 12-month-old GDNF+/− mice (Boger et al., 2004), indicating multiple possible neurobiological substrates of aging-related decline in these mice.
In summary, combining chronically lower GDNF levels with a DA neurotoxin in young adult mice resulted in exacerbation of DAergic system dysfunction during aging. Greater striatal DAT activity in GDNF+/− mice may underlie these genotypic differences in METH toxicity. Thus, lowered growth factor levels may predispose individuals to greater acute and/or chronic consequences of exposure to DA neurotoxins. Furthermore, METH intoxication in any young adult may have deleterious behavioral and neuronal consequences later in life, although a sustained behavioral profile may not be apparent for many months (or decades in humans) after the exposure. These studies speak directly to the possibility of protracted public health consequences resulting from the current epidemic of METH abuse among young adults.
Footnotes
This work was supported by National Institutes of Health Grants R01 DA03982 (J.F.M.), PO1 AG023630 (A.-C.G.), RO1 MH62612 (S.R.), and C06 RR015455 from the Extramural Research Facilities Program of the National Center for Research Resources, and Department of the Army Medical Command Grant 17-99-1-9480 (A.-C.G.). H.A.B. performed the behavioral experiments, immunohistochemical staining, and data analysis; L.D.M. supervised the behavioral data analysis; S.R. and Devadoss Samuel performed the DAT assays; K.S.P. and H.Z. measured brain methamphetamine concentrations; and A.M.P. and H.A.B. performed the HPLC measurements; A.-C.G. and J.F.M. supervised the project and provided the reagents; J.F.M. designed the project; A.-C. G. supported H.A.B., performed statistical analyses, and provided the mice breeding support. H.A.B. wrote the paper with editorial input from A.-C.G., L.D.M., and J.F.M. We thank Alfred Moore, Channon Reid, Devadoss Samuvel, and Vandana Zaman for technical assistance. We thank Dr. Peter Kalivas for HPLC reagents, equipment, and manuscript review, Dr. Greg Gerhardt for reviewing the HPLC data, and Dr. Barry Hoffer for providing the mouse strain and valuable advice.
References
- Bakhit C, Morgan ME, Peat MA, Gibb JW. Long-term effects of methamphetamine on the synthesis and metabolism of 5-hydroxytryptamine in various regions of the rat brain. Neuropharmacology. 1981;20:1135–1140. doi: 10.1016/0028-3908(81)90053-8. [DOI] [PubMed] [Google Scholar]
- Bannon MJ, Poosch MS, Xia Y, Goebel DJ, Cassin B, Kapatos G. Dopamine transporter mRNA content in human substantia nigra decreases precipitously with age. Proc Natl Acad Sci USA. 1992;89:7095–7099. doi: 10.1073/pnas.89.15.7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boger HA, Granholm AC, Jin L, Nelson ME, Page GP, McGinty JF. Striatal gene expression profile of 12-month-old GDNF heterozygous mice. Soc Neurosci Abstr. 2004;30:725–17. [Google Scholar]
- Boger HA, Middaugh LD, Huang P, Zaman V, Smith AC, Hoffer BJ, Tomac AC, Granholm AC. A partial GDNF depletion leads to earlier age-related deterioration of motor function and tyrosine hydroxylase expression in the substantia nigra. Exp Neurol. 2006;202:336–347. doi: 10.1016/j.expneurol.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Bowenkamp KE, Hoffman AF, Gerhardt GA, Henry MA, Biddle PT, Hoffer BJ, Granholm AC. Glial cell line-derived neurotrophic factor supports survival of injured midbrain dopaminergic neurons. J Comp Neurol. 1995;355:479–489. doi: 10.1002/cne.903550402. [DOI] [PubMed] [Google Scholar]
- Bowyer JF, Tank AW, Newport CD, Slikker W, Jr, Ali SF, Holson RR. The influence of environmental temperature on the transient effects of methamphetamine on DA levels and DA release in the striatum. J Pharmacol Exp Ther. 1992;260:817–824. [PubMed] [Google Scholar]
- Brooks DJ. Imaging endpoints for monitoring neuroprotection in PD. Ann Neurol. 2003;53(Suppl 3):S110–S118. doi: 10.1002/ana.10480. [DOI] [PubMed] [Google Scholar]
- Cass WA. GDNF selectively protects dopamine neurons over serotonin neurons against the neurotoxic effects of methamphetamine. J Neurosci. 1996;16:8132–8139. doi: 10.1523/JNEUROSCI.16-24-08132.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cass WA, Manning MW. GDNF protection against 6-OHDA-induced reductions in potassium-evoked overflow of striatal dopamine. J Neurosci. 1999;19:1416–1423. doi: 10.1523/JNEUROSCI.19-04-01416.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cass WA, Peters LE, Smith MP. Reductions in spontaneous locomotor activity in aged male, but not female, rats in a model of early Parkinson's disease. Brain Res. 2005;1034:153–161. doi: 10.1016/j.brainres.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Chan-Palay V, Asan E. Quantitation of catecholamine neurons in the locus coeruleus in human brains of normal young and older adults and in depression. J Comp Neurol. 1989;287:357–372. doi: 10.1002/cne.902870307. [DOI] [PubMed] [Google Scholar]
- Choe ES, McGinty JF. NMDA receptors and p38MAP kinase are required for cAMP-dependent CREB and Elk-1 phosphorylation in the striatum of rats. Neuroscience. 2000;101:607–617. doi: 10.1016/s0306-4522(00)00379-1. [DOI] [PubMed] [Google Scholar]
- Ciliax BJ, Heilman C, Demchyshyn LL, Pristupa ZB, Ince E, Hersch SM, Niznik HP, Levey AI. The dopamine transporter: immunochemical characterization and localization in the brain. J Neurosci. 1995;15:1714–1723. doi: 10.1523/JNEUROSCI.15-03-01714.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czlonkowska A, Kohutnicka M, Kurkowska-Jastrzebska I, Czlonkowski A. Microglial reaction in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) induced Parkinson's disease mice model. Neurodegeneration. 1996;5:137–143. doi: 10.1006/neur.1996.0020. [DOI] [PubMed] [Google Scholar]
- Ducray A, Krebs SH, Schaller B, Seiler RW, Meyer M, Widmer HR. GDNF family ligands display distinct action profiles on cultured GABAergic and serotonergic neurons of rat ventral mesencephalon. Brain Res. 2006;1069:104–112. doi: 10.1016/j.brainres.2005.11.056. [DOI] [PubMed] [Google Scholar]
- Emborg ME, Ma SY, Mufson EJ, Levey AI, Taylor MD, Brown WD, Holden JE, Kordower JH. Age-related declines in nigral neuronal function correlate with motor impairments in rhesus monkeys. J Comp Neurol. 1998;401:253–265. [PubMed] [Google Scholar]
- Francis JW, Von Visger J, Markelonis GJ, Oh TH. Neuroglial responses to the dopaminergic neurotoxicant 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mouse striatum. Neurotoxicol Teratol. 1995;17:7–12. doi: 10.1016/0892-0362(94)00048-i. [DOI] [PubMed] [Google Scholar]
- Galter D, Unsicker K. Regulation of the transmitter phenotype or rostral and caudal groups of cultured serotonergic raphe neurons. Neuroscience. 1999;88:549–559. doi: 10.1016/s0306-4522(98)00224-3. [DOI] [PubMed] [Google Scholar]
- Gerfen CR. The neostriatal mosaic: compartmentalization of corticostriatal input and striatonigral output systems. Nature. 1984;311:461–464. doi: 10.1038/311461a0. [DOI] [PubMed] [Google Scholar]
- Gesi M, Soldani P, Giorgi FS, Santinami A, Bonaccorsi I, Fornai F. The role of the locus coeruleus in the development of Parkinson's disease. Neurosci Biobehav Rev. 2000;24:655–668. doi: 10.1016/s0149-7634(00)00028-2. [DOI] [PubMed] [Google Scholar]
- Granholm AC, Reyland M, Albeck D, Sanders L, Gerhardt G, Hoernig G, Shen L, Westphal H, Hoffer B. Glial cell line-derived neurotrophic factor is essential for postnatal survival of midbrain dopamine neurons. J Neurosci. 2000;20:3182–3190. doi: 10.1523/JNEUROSCI.20-09-03182.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granholm AC, Ford KA, Hyde LA, Bimonte HA, Hunter CL, Nelson M, Albeck D, Sanders LA, Mufson EJ, Crnic LS. Estrogen restores cognition and cholinergic phenotype in an animal model of Down syndrome. Physiol Behav. 2002;77:371–385. doi: 10.1016/s0031-9384(02)00884-3. [DOI] [PubMed] [Google Scholar]
- Halberda JP, Middaugh LD, Gard BE, Jackson BP. DAD1- and DAD2-like agonist effects on motor activity of C57 mice: differences compared to rats. Synapse. 1997;26:81–92. doi: 10.1002/(SICI)1098-2396(199705)26:1<81::AID-SYN9>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Hebert MA, Gerhardt GA. Normal and drug-induced locomotor behavior in aging: comparison to evoked DA release and tissue content in Fischer 344 rats. Brain Res. 1998;797:42–54. doi: 10.1016/s0006-8993(98)00370-9. [DOI] [PubMed] [Google Scholar]
- Hotchkiss A, Gibb JW. Blockade of methamphetamine-induced depression of tyrosine hydroxylase by GABA transaminase inhibitors. Eur J Pharmacol. 1980;66:201–205. doi: 10.1016/0014-2999(80)90143-0. [DOI] [PubMed] [Google Scholar]
- Jenner P, Olanow CW. Understanding cell death in Parkinson's disease. Ann Neurol. 1998;443(Suppl 1):S72–S84. doi: 10.1002/ana.410440712. [DOI] [PubMed] [Google Scholar]
- Johnson-Davis KL, Truong KL, Fleckenstein AE, Wilkins DG. Alterations in vesicular dopamine uptake contribute to tolerance to the neurotoxic effects of methamphetamine. J Pharmacol Exp Ther. 2004;309:578–586. doi: 10.1124/jpet.103.062695. [DOI] [PubMed] [Google Scholar]
- Joyce JN, Renish L, Osredkar T, Walro JM, Kucera J, Dluzen DE. Methamphetamine-induced loss of striatal dopamine innervation in BDNF heterozygote mice does not further reduce D3 receptor concentrations. Synapse. 2004;52:11–19. doi: 10.1002/syn.10309. [DOI] [PubMed] [Google Scholar]
- Kita T, Wagner GC, Nakashima T. Current research on methamphetamine-induced neurotoxicity: animal models of monoamine disruption. J Pharmacol Sci. 2003;92:178–195. doi: 10.1254/jphs.92.178. [DOI] [PubMed] [Google Scholar]
- Koike K, Hashimoto K, Fukami G, Okamara N, Zhang L, Ohgake S, Koizumi H, Matsuzaka D, Kawamura N, Shimizu E, Iyo M. The immunophilin ligand FK506 protects against methamphetamine-induced dopaminergic neurotoxicity in mouse striatum. Neuropharmacol. 2005;48:391–397. doi: 10.1016/j.neuropharm.2004.10.015. [DOI] [PubMed] [Google Scholar]
- Kordower JH, Emborg ME, Bloch J, Ma SY, Chu Y, Leventhal L, McBride J, Chen EY, Palfi S, Roitberg BZ, Brown WD, Holden JE, Pyzalski R, Taylor MD, Carvey P, Ling Z, Trono D, Hantraye P, Deglon N, Aebischer P. Neurodegeneration prevented by lentiviral vector delivery of GDNF in primate models of Parkinson's disease. Science. 2000;290:767–773. doi: 10.1126/science.290.5492.767. [DOI] [PubMed] [Google Scholar]
- Krieglstein K, Suter-Crazzolara C, Unsicker K. Development of mesencephalic dopaminergic neurons and the transforming growth factor-beta superfamily. J Neural Transm. 1995;46(Suppl):209–216. [PubMed] [Google Scholar]
- Langston JW, Forno LS, Tetrud J, Reeves AG, Kaplan JA, Karluk D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann Neurol. 1999;46:598–605. doi: 10.1002/1531-8249(199910)46:4<598::aid-ana7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- LaVoie MJ, Hastings TG. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. J Neurosci. 1999;19:1484–1491. doi: 10.1523/JNEUROSCI.19-04-01484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVoie MJ, Card JP, Hastings TG. Microglial activation preceded dopamine terminal pathology in methamphetamine-induced neurotoxicity. Exp Neurol. 2004;187:47–57. doi: 10.1016/j.expneurol.2004.01.010. [DOI] [PubMed] [Google Scholar]
- Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science. 1993;260:1130–1132. doi: 10.1126/science.8493557. [DOI] [PubMed] [Google Scholar]
- Mandel RJ, Spratt SK, Snyder RO, Leff SE. Midbrain injection of recombinant adeno-associated virus encoding rat glial cell line-derived neurotrophic factor protects nigral neurons in a progressive 6-hydroxydopamine-induced degeneration model of Parkinson's disease in rats. Proc Natl Acad Sci USA. 1997;94:14083–14088. doi: 10.1073/pnas.94.25.14083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark K, Soghomonian JJ, Yamamoto BK. High-dose methamphetamine acutely activates the striatonigral pathway to increase striatal glutamate and mediate long-term dopamine toxicity. J Neurosci. 2004;24:11449–11456. doi: 10.1523/JNEUROSCI.3597-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeorge AJ, Faull RL. The organization of the projections from the cerebral cortex to the striatum in the rat. Neuroscience. 1989;29:503–537. doi: 10.1016/0306-4522(89)90128-0. [DOI] [PubMed] [Google Scholar]
- Melega WP, Lacan G, Desalles AA, Phelps ME. Long-term methamphetamine-induced decreases of [11C]WIN 35,428 binding in striatum are reduced by GDNF: PET studies in the vervet monkey. Synapse. 2000;35:243–249. doi: 10.1002/(SICI)1098-2396(20000315)35:4<243::AID-SYN1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Mogenson GJ, Nielsen MA. Evidence that an accumbens to subpallidal GABAergic projection contributes to locomotor activity. Brain Res Bull. 1983;11:309–314. doi: 10.1016/0361-9230(83)90166-1. [DOI] [PubMed] [Google Scholar]
- Moore MW, Klein RD, Farinas I, Sauer H, Armanini M, Phillips H, Reichardt LF, Ryan AM, Carver-Moore K, Rosenthal A. Renal and neuronal abnormalities in mice lacking GDNF. Nature. 1996;382:76–79. doi: 10.1038/382076a0. [DOI] [PubMed] [Google Scholar]
- O'Callaghan JP, Miller DB. Neurotoxicity profiles of substituted amphetamines in the C57BL/6J mouse. J Pharmacol Exp Ther. 1994;270:741–751. [PubMed] [Google Scholar]
- Pichel JG, Shen L, Sheng HZ, Granholm AC, Drago J, Grinberg A, Lee EJ, Huang SP, Saarma M, Hoffer BJ, Sariola H, Westphal H. Defects in enteric innervation and kidney development in mice lacking GDNF. Nature. 1996;382:73–76. doi: 10.1038/382073a0. [DOI] [PubMed] [Google Scholar]
- Ricuarte GA, Guillery RW, Seiden LS, Schuster CR, Moore RY. Dopamine nerve terminal degeneration produced by high doses of methylamphetamine in the rat brain. Brain Res. 1982;235:93–103. doi: 10.1016/0006-8993(82)90198-6. [DOI] [PubMed] [Google Scholar]
- Samuvel DJ, Jayanthi LD, Bhat NR, Ramamoorthy S. A role for p38 mitogen-activated protein kinase in the regulation of the serotonin transporter: evidence for distinct cellular mechanisms involved in transporter surface expression. J Neurosci. 2005;25:29–41. doi: 10.1523/JNEUROSCI.3754-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonsalla PK, Nicklas WJ, Heikkila RE. Role for excitatory amino acids in methamphetamine-induced toxicity. Science. 1986;243:398–400. doi: 10.1126/science.2563176. [DOI] [PubMed] [Google Scholar]
- Sonsalla PK, Giovanni A, Sieber BA, Donne KD, Manzino L. Characteristics of dopaminergic neurotoxicity produced by MPTP and methamphetamine. Ann NY Acad Sci. 1992;648:229–238. doi: 10.1111/j.1749-6632.1992.tb24542.x. [DOI] [PubMed] [Google Scholar]
- Suhara T, Fukuda H, Inoue O, Itoh T, Suzuki K, Yamasaki T, Tateno Y. Age-related changes in human D1 dopamine receptors measured by positron emission tomography. Psychopharmacology (Berl) 1991;103:41–45. doi: 10.1007/BF02244071. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Dowgiert J, Geddes TJ, Francescutti-Verbeem D, Liu X, Kuhn DM. Microglial activation is a pharmacologically specific marker for the neurotoxic amphetamines. Neurosci Lett. 2004a;267:349–354. doi: 10.1016/j.neulet.2004.06.065. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Walker PD, Benjamins JA, Geddes TJ, Kuhn Methamphetamine neurotoxicity in dopamine nerve endings of the striatum is associated with microglial activation. J Pharmacol Exp Ther. 2004b;311:1–7. doi: 10.1124/jpet.104.070961. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS, Ding YS, Gur RC, Gatley J, Logan J, Moberg PJ, Hitzemann R, Smith G, Pappas N. Parallel loss of presynaptic and postsynaptic dopamine markers in normal aging. Ann Neurol. 1998;44:143–147. doi: 10.1002/ana.410440125. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Chang L, Wang GJ, Fowler JS, Leonido-Yee M, Franceschi D, Sedler MJ, Gatley SJ, Hitzemann R, Ding Y-S, Logan J, Wong C, Miller EN. Association of dopamine transporter reduction with psychomotor impairment in methamphetamine abusers. Am J Psychiatry. 2001;158:377–382. doi: 10.1176/appi.ajp.158.3.377. [DOI] [PubMed] [Google Scholar]
- Voorn P, Vanderschuren LJ, Groenewegen HJ, Robbins TW, Pennartz CM. Putting a spin on the dorsal-ventral divide of the striatum. Trends Neurosci. 2004;27:468–474. doi: 10.1016/j.tins.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Wagner GC, Ricuarte GA, Seiden LS, Schuster CR, Miller RJ, Westley J. Long-lasting depletions of striatal dopamine and loss of dopamine uptake sites following repeated administration of methamphetamine. Brain Res. 1980;181:151–160. doi: 10.1016/0006-8993(80)91265-2. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Volkow ND. Partial recovery of brain metabolism in methamphetamine abusers after protracted abstinence. Am J Psychiatry. 2004;161:242–248. doi: 10.1176/appi.ajp.161.2.242. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Muramatsu Y, Kurosaki R, Michimata M, Matsubara M, Imai Y, Araki T. Protective effects of neuronal nitric oxide synthase inhibitor in mouse brain against MPTP neurotoxicity: an immunohistological study. Eur Neuropsychopharmacol. 2004;14:93–104. doi: 10.1016/S0924-977X(03)00065-8. [DOI] [PubMed] [Google Scholar]
- Yamamoto BK, Zhu W. The effects of methamphetamine on the production of free radicals and oxidative stress. J Pharmacol Exp Ther. 1998;287:107–114. [PubMed] [Google Scholar]
- Yurek DM, Fletcher-Turner A. Differential expression of GDNF, BDNF, and NT-3 in the aging nigrostriatal system following a neurotoxic lesion. Brain Res. 2001;891:228–235. doi: 10.1016/s0006-8993(00)03217-0. [DOI] [PubMed] [Google Scholar]
- Zaman V, Li Z, Middaugh L, Ramamoorthy S, Rohrer B, Nelson ME, Tomac AC, Hoffer BJ, Gerhardt GA, Granholm AC. The noradrenergic system of aged GDNF heterozygous mice. Cell Transplant. 2003;12:291–303. [PubMed] [Google Scholar]
- Zhu JPQ, Xu W, Angulo JA. Disparity in the temporal appearance of methamphetamine-induced apoptosis and depletion of dopamine terminal markers in the striatum of mice. Brain Res. 2005;1049:171–181. doi: 10.1016/j.brainres.2005.04.089. [DOI] [PMC free article] [PubMed] [Google Scholar]