

Abstract
In anaerobic bacteria using aromatic growth substrates, glutaryl-coenzyme A (CoA) dehydrogenases (GDHs) are involved in the catabolism of the central intermediate benzoyl-CoA to three acetyl-CoAs and CO2. In this work, we studied GDHs from the strictly anaerobic, aromatic compound-degrading organisms Geobacter metallireducens (GDHGeo) (Fe[III] reducing) and Desulfococcus multivorans (GDHDes) (sulfate reducing). GDHGeo was purified from cells grown on benzoate and after the heterologous expression of the benzoate-induced bamM gene. The gene coding for GDHDes was identified after screening of a cosmid gene library. Reverse transcription-PCR revealed that its expression was induced by benzoate; the product was heterologously expressed and isolated. Both wild-type and recombinant GDHGeo catalyzed the oxidative decarboxylation of glutaryl-CoA to crotonyl-CoA at similar rates. In contrast, recombinant GDHDes catalyzed only the dehydrogenation to glutaconyl-CoA. The latter compound was decarboxylated subsequently to crotonyl-CoA by the addition of membrane extracts from cells grown on benzoate in the presence of 20 mM NaCl. All GDH enzymes were purified as homotetramers of a 43- to 44-kDa subunit and contained 0.6 to 0.7 flavin adenine dinucleotides (FADs)/monomer. The kinetic properties for glutaryl-CoA conversion were as follows: for GDHGeo, the Km was 30 ± 2 μM and the Vmax was 3.2 ± 0.2 μmol min−1 mg−1, and for GDHDes, the Km was 52 ± 5 μM and the Vmax was 11 ± 1 μmol min−1 mg−1. GDHDes but not GDHGeo was inhibited by glutaconyl-CoA. Highly conserved amino acid residues that were proposed to be specifically involved in the decarboxylation of the intermediate glutaconyl-CoA were identified in GDHGeo but are missing in GDHDes. The differential use of energy-yielding/energy-demanding enzymatic processes in anaerobic bacteria that degrade aromatic compounds is discussed in view of phylogenetic relationships and constraints of overall energy metabolism.
In anaerobic bacteria, most aromatic growth substrates are channeled to the central intermediate benzoyl-coenzyme A (CoA), which is then degraded in the so-called benzoyl-CoA degradation pathway (for reviews, see references 8, 12, 17, and 18). The upper part of this pathway comprises the reductive dearomatization by benzoyl-CoA reductase (BCR), modified β-oxidation reactions, and hydrolytic ring cleavage (Fig. 1). In the lower part, the aliphatic thiol ester of a dicarboxylic acid, in most cases 3-hydroxypimelyl-CoA, is metabolized by β-oxidation reactions including a decarboxylation step, yielding three acetyl-CoAs and CO2. The benzoyl-CoA degradation pathway appears to proceed in most facultatively and obligately anaerobic bacteria via identical intermediates; an exception is Rhodopseudomonas palustris, which uses a slightly modified pathway (18). However, there are two enzymatic reactions in the benzoyl-CoA degradation pathway, which appear to be catalyzed by different enzymes in obligate and facultative anaerobes: (i) the dearomatization of benzoyl-CoA and (ii) the decarboxylation of glutaryl-CoA to crotonyl-CoA. The rationale for these differences can be explained by the differing overall energy metabolisms; e.g., the ΔG°′ for the complete oxidation of benzoate to CO2 is 15 times more negative in denitrifying than in sulfate-reducing bacteria (32).
FIG. 1.

Benzoyl-CoA degradation pathway in anaerobic bacteria. Benzoyl-CoA is reduced to cyclohexa-1,5-diene-1-carboxyl-CoA by an ATP-dependent (facultative anaerobes) or ATP-independent (obligate anaerobes) benzoyl-CoA reductase. Further reactions of the upper degradation pathway include a hydratase, dehydrogenases, and a ring-opening hydrolase. The lower pathway comprises modified β-oxidation reactions. In facultative anaerobes, glutaryl-CoA is dehydrogenated and decarboxylated to crotonyl-CoA by a single GDH; in some obligate anaerobes, these reactions are catalyzed by separated nondecarboxylating GDH and membrane-bound glutaconyl-CoA decarboxylase.
First, dearomatizing benzoyl-CoA reductases catalyzing the initial step of the benzoyl-CoA degradation pathway differ fundamentally in facultatively and obligately anaerobic bacteria. Benzoyl-CoA reductases from the former group are iron-sulfur enzymes that couple electron transfer to the aromatic ring to stoichiometric ATP hydrolysis (5-7). Instead, obligate anaerobes use a so-far merely studied, most possibly ATP-independent, benzoyl-CoA reductase complex consisting of Fe/S-, Mo- or W-, and Se-containing protein components (32, 40). Despite these differences, both classes of benzoyl-CoA reductases appear to reduce the substrate benzoyl-CoA by two electrons to cyclohexa-1,5-diene-1-carboxyl-CoA (Fig. 1) (25, 33).
Second, in the lower part of the benzoyl-CoA degradation pathway, a decarboxylation step is required for the β-oxidation of the C7 compound 3-hydroxypimelyl-CoA to three acetyl-CoAs and CO2 (12, 17, 18). In both facultative and obligate anaerobes, glutaryl-CoA (C5) is considered to be oxidatively decarboxylated to crotonyl-CoA by glutaryl-CoA dehydrogenases (GDH). However, as discussed below, there is initial evidence that obligately but not facultatively anaerobic bacteria couple this decarboxylation to an energy-conserving process.
GDH enzymes belong to the family of flavin adenine dinucleotide (FAD)-containing acyl-CoA dehydrogenases, but all GDHs studied so far are unique in decarboxylating the dehydrogenated, enzyme-bound intermediate glutaconyl-CoA (2,3-dehydroglutaryl-CoA) to crotonyl-CoA (3, 4, 19, 20, 22-24, 26, 28, 30, 31). Electrons are usually transferred to an electron-transferring flavoprotein. Next to the role in β-oxidation of dicarboxylic acids, GDH plays a role in the catabolism of lysine, hydroxylysine, and tryptophan in many organisms. The human enzyme has been studied extensively, and a number of mutations in the gene coding for human GDH have been identified, which have been associated with cerebral aciduria type I (21). Meanwhile, details about the structure-function relationship and the catalytic mechanism have been elucidated in studies with human GDH (14, 16, 34, 35). A conserved catalytic glutamate residue abstracts the α-proton from the substrate, which is followed by hydride transfer from the β-carbon to the flavin cofactor. The subsequent decarboxylation of the intermediate glutaconyl-CoA is initiated by cleavage of the Cγ-Cδ bond, which yields a crotonyl-CoA dienolate anion and CO2. A number of conserved amino acids have been identified in the crystal structure of human GDH, which are supposed to be involved in substrate binding and in polarizing the Cγ-Cδ bond of the substrate to facilitate the decarboxylation step (16).
So far, GDH enzymes involved in the anaerobic degradation of aromatic compounds have been studied in denitrifying, facultatively anaerobic Thauera and Azoarcus species (4, 23). In contrast, much less is known about GDHs in obligately anaerobic bacteria. In vitro assays with glutarate-, benzoate-, and 3-hydroxybenzoate-fermenting bacteria suggested that glutaconyl-CoA is the product released from glutaryl-CoA dehydrogenase without a concomitant decarboxylation to crotonyl-CoA. The subsequent decarboxylation is then catalyzed by a membrane-bound, sodium ion-pumping glutaconyl-CoA decarboxylase containing a biotin cofactor (15, 28, 30). In accordance, the genes coding for such a decarboxylase are present in the genome of the benzoate-fermenting organism Syntrophus aciditrophicus (29). Similar energy-conserving enzymes have been studied intensively in amino acid-fermenting clostridia (2, 9, 10, 38). The low energy yield has been suggested to necessitate fermenting bacteria to conserve the energy of the exergonic decarboxylation step (ΔG°′ = −30 kJ mol−1) (9, 13).
In this work, two GDH enzymes involved in the benzoyl-CoA degradation pathways of the obligately anaerobic bacteria Geobacter metallireducens (Fe[III] reducing) and Desulfococcus multivorans (sulfate reducing) were studied. Although both organisms are phylogenetically related and belong to the Deltaproteobacteria, G. metallireducens used a decarboxylating GDH, whereas the sulfate-reducing organism D. multivorans used a nondecarboxylating GDH/membrane-bound glutaconyl-CoA decarboxylase in benzoate catabolism. The genes of both GDH enzymes were identified, and their products were purified and characterized in view of the different reactions catalyzed. The rationale for using decarboxylating and nondecarboxylating GDHs in benzoyl-CoA degradation pathways of different Deltaproteobacteria appears to be governed by the overall energy metabolism rather than by the phylogenetic relationship.
MATERIALS AND METHODS
Cultivation of bacteria and preparation of cell extracts.
Geobacter metallireducens (DSMZ-Nr. 7210) cells were cultured anaerobically in modified freshwater acetate medium (27) in which the routinely added acetate was replaced by benzoate (3 mM). Desulfococcus multivorans (DSMZ-Nr. 2059) cells were grown in a bicarbonate-buffered (pH 7 to 7.3) mineral salt medium under strictly anaerobic conditions at 30°C (39). Benzoate (3 mM), cyclohexanecarboxylate (3 mM), or lactate (20 mM) was used as the sole source of carbon and energy, and sodium sulfate (30 mM) served as the electron acceptor. For the expression of the genes coding for the GDH of G. metallireducens (GDHGeo) and the GDH of D. multivorans (GDHDes), Escherichia coli One Shot BL21(DE3) cells were cultured in Luria-Bertani medium with ampicillin (100 μg ml−1) and riboflavin (10 μg ml−1). Cells were harvested in the exponential growth phase by centrifugation (13,700 × g) and stored at −80°C until further use. For the preparation of cell extracts, frozen cells were suspended in buffer as described below. Cell lysates were obtained by passage through a French pressure cell at 137 MPa followed by centrifugation at 100,000 × g (1 h at 4°C).
Construction and screening of a cosmid gene library from D. multivorans.
A cosmid gene library was constructed and screened for the gene coding for GDHDes. For the construction of an appropriate labeled probe for the screening procedure, highly conserved nucleotide sequences in the genes coding for benzoate-CoA ligases from Thauera aromatica (gi23450983), Azoarcus evansii (gi18369667), “Aromatoleum aromaticum” EbN1 (gi56314492), Magnetospirillum magnetotacticum (gi23015258), Rhodopseudomonas palustris (gi77690072), S. aciditrophicus (gi85860705), and G. metallireducens (gi78194605) were identified. The oligonucleotide primer pairs for the amplification of a labeled internal fragment of the putative benzoate-CoA ligase gene from D. multivorans were CTATCATCCGGGGAGACCGAC (forward primer) and CTCGCTTCGTATCCCGGAACGATC (reverse primer). For the preparation of the labeled DNA probe, the 400-bp internal fragment was randomly labeled with a digoxigenin DNA labeling kit (digoxigenin-11-dUTP label; Roche). Screening of the cosmid library was carried out by hybridization as described below.
The construction of the cosmid library of D. multivorans was performed as described previously (36). Thirty micrograms of genomic DNA from D. multivorans was partially digested with Sau3AI, and fragments of 30 to 45 kb were ligated into a XbaI- and BamHI-digested and alkaline phosphatase-treated SuperCos1 vector (Stratagene Cloning Systems). Gigapack III XL packaging extract (Stratagene) was used to package the recombinant phage, and phage particles were obtained by infecting E. coli SURE cells according to the manufacturer's instructions. Positive plaques were detected with a digoxigenin luminescence detection kit (Roche), and cosmid DNA was purified with a NucleoBond PC 20 or PC 500 (Macherey-Nagel) plasmid isolation kit according to the manufacturer's instructions. Sequencing was performed at Seqlab (Sequence Laboratories Göttingen GmbH).
Reverse transcription-PCR.
Total RNA from D. multivorans cells grown on benzoate, cyclohexanecarboxylate, and lactate was used for reverse transcription-PCR. RNA was isolated from cells harvested in the exponential growth phase. After DNase I treatment, RNA was purified with the RNeasy minikit (Qiagen). One microgram of purified RNA was used to prepare cDNA using a RevertAid H Minus first-strand cDNA synthesis kit (Fermentas) with the provided random hexamer primer. Gene expression was studied using undiluted, 10-fold-diluted, and 100-fold-diluted cDNA. For internal sequences, a primer pair for an Na+-pumping decarboxylase β-subunit (gcd) (see Fig. 4) was used with the primer sequences CCTGTTTTCCCTGTTCAAGACCTCCG (forward primer) and CGTTGTCGGGCCGTCGGCGCCGCCG (reverse primer). For intergenic regions, the following primer pairs were used: CCTGTTGGGCATTTCCTTGC (forward primer) and CGGCCTTCACGAATGTTGC (reverse primer) for the intergenic region between rre and bcl, CCCGAACTGGAGAAGGAACTCA (forward primer) and GCCGTACTCCTCGGGAATCAC (reverse primer) for the intergenic region between bcl and gdh, and GGAGGGCTCGGCCAATATCTGC (forward primer) and CCGGAGGTCTTGAACAGGGAAAAC (reverse primer) for the intergenic region between gdh and gcd. Genomic DNA of D. multivorans was used as a positive control. PCR products were visualized by agarose gel electrophoresis. Standard protocols were used for DNA amplification.
FIG. 4.
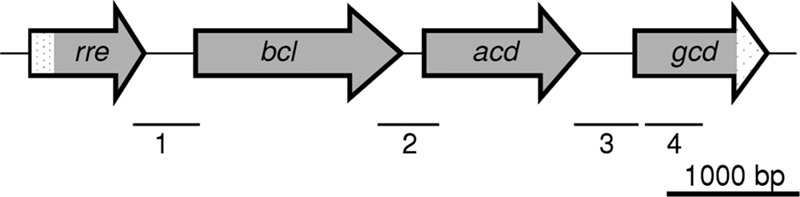
Organization of a benzoate-induced gene cluster obtained from a cosmid gene library of D. multivorans. A gene probe deduced from the conserved region of benzoate-CoA ligase was used to screen the cosmid gene library. The open reading frames bcl (1,551 bp) and acd (1,173 bp) were sequenced completely, while rre (>688 bp) and gcd (>1,118 bp) were partially sequenced. Assignments of the putative open reading frames to gene products are as follows: rre, DNA-binding response regulator; bcl, benzoate-CoA ligase; gdh, glutaryl-CoA dehydrogenase/acyl-CoA dehydrogenase; gcd, Na+-pumping decarboxylase, β-subunit. Bars 1 to 4 indicate the regions of the gene cluster, which were amplified using cDNA obtained from total RNA using appropriate oligonucleotide primers. The corresponding amplificates were obtained only with cDNA from cells grown on benzoate but not with cDNA from cells grown on cyclohexane carboxylate or lactate.
Synthesis of CoA-esters.
Glutaryl-CoA, crotonyl-CoA, acetyl-CoA, and 3-hydroxyglutaryl-CoA were synthesized from CoA (trilithium salt) and the corresponding anhydrides by a method described previously by Simon and Shemin (37). Glutaconyl-CoA was a kind gift of Wolfgang Buckel, University of Marburg.
Enzyme assays. (i) Spectrophotometric assay.
GDH activity was measured with a continuous assay at 30°C in a cuvette (1-cm diameter) containing 50 mM Tris-HCl (pH 7.8), 250 mM KCl, and 0.2 mM ferrocenium hexafluorophosphate; the reaction was started by addition of 200 μM glutaryl-CoA to the mixture. The time-dependent reduction of ferrocenium hexafluorophosphate was monitored at 300 nm (Δɛ300 = 3,600 M−1 cm−1 [self-determined]). For the oxidation of 1 mol glutaryl-CoA, 2 mol ferrocenium was required.
(ii) HPLC assay.
For product analysis and identification, the same assay described above was used. For the discontinuous assay, 90-μl samples were taken at different time points and mixed with 10 μl 10% formic acid. After centrifugation (10 min at 13,700 × g), the supernatant was diluted with a 10-fold volume of water, and 100 μl was analyzed by C18 reversed-phase high-performance liquid chromatography (HPLC) (Waters 2695) with a Eurospher 100-5 C18 column (Knauer) using acetonitrile in 50 mM potassium phosphate buffer (pH 6.8) at a flow rate of 0.75 ml min−1. The applied gradient was 4% to 9% at 0 to 10 min and 9% to 40% at 10 to 17 min. Products were identified by comparing retention times and UV/Vis spectra with standards (2996 photodiode array detector; Waters) and by mass spectrometry (see below).
Mass spectrometric analysis. (i) CoA ester analysis.
Products of interest obtained by HPLC analysis (see above) were analyzed by matrix-assisted laser desorption-ionization time-of-flight mass spectrometry (Ultraflex TOF/TOF III; Bruker Daltonics, Bremen, Germany). After the HPLC assays, the collected products were lyophilized and suspended in 100 μl 20 mM ammonium acetate buffer (pH 7). Subsequently, products were desalted by HPLC using the same gradient described above but with 20 mM ammonium acetate buffer (pH 7) instead of potassium phosphate buffer. After lyophilization, products were suspended in 10 μl 0.1% trifluoroacetic acid, and 0.5 μl of the products was mixed with 1.5 μl 2,5-dihydroxybenzoic acid (4% dihydroxybenzoic acid, 33% acetonitrile, and 0.5% trifluoroacetic acid in ultrapure water) as a matrix solution. All samples were prepared on AnchorChip target plates (Bruker Daltonics) by using a dried droplet preparation. Measuring was performed by matrix-assisted laser desorption-ionization tandem time-of-flight mass spectrometry in a positive mode, and the m/z values were given as [M + H]+1. The mass spectra were acquired with external calibration using Peptide Calibration Standard II (Bruker Daltonics), which covers a mass range from m/z ∼700 to 3,200.
(ii) Protein identification.
For the protein identification of purified GDHGeo, 20 μg of protein was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The band containing GDHGeo was excised and digested overnight with trypsin according to a protocol described previously by Jehmlich et al. (24a). Cleaved peptides were eluted, concentrated by vacuum centrifugation, and thereafter separated by reversed-phase nano-liquid chromatography (LC1100 series; Agilent Technologies) with a Zorbax 300SB-C18 column (3.5 mm by 150 μm by 0.075 mm) (eluent, 0.1% formic acid and 0% to 60% acetonitrile) and analyzed by tandem mass spectrometry (MS/MS) (LC/MSD Trap XCT mass spectrometer; Agilent Technologies). Database searches were carried out with an MS/MS ion search by Mascot In-House version 2.2 (Matrix Science, London, United Kingdom) against all taxonomy entries of the NCBInr Database (National Center for Biotechnology Information). The search was restricted to peptides containing peptide charges +2 and +3, carbamidomethyl at cysteines and oxidized methionine were given as variable modifications, and the following accuracies were conducted: peptide tolerance of ±0.8 Da and MS/MS tolerance of ±0.6 Da.
Purification of GDHGeo from the wild type.
Frozen cells of G. metallireducens grown on benzoate were suspended in extraction buffer containing 20 mM Tris-HCl (pH 7.8), 5 mM MgCl2, 50 mM KCl, lysozyme, dithioerythritol, and DNase I, with 0.1 mg ml−1 each (1 g cells in 1.5 ml buffer). Cell lysates were obtained as described above, and 10% (vol/vol) glycerol was added before dialysis for 12 h at 4°C against extraction buffer (molecular mass cutoff of 6,000 to 8,000 Da; Spectrum Laboratories, Inc.). GDHGeo was purified from this crude extract in three chromatographic steps. The purification of GDHGeo was monitored by UV/Vis detection at 280 nm (protein) and 450 nm (FAD). The fractions obtained were tested for enzyme activity with the continuous spectrophotometric assay. For the first chromatographic step, a DEAE-Sepharose anion exchange column (19-ml DEAE Sepharose Fast Flow column with a diameter of 2.6 cm; GE Healthcare) was applied equilibrated with 20 mM Tris-HCl (pH 7.8)-5 mM MgCl2 (referred to as basic buffer) (1 ml min−1). A KCl gradient from 50 to 500 mM in basic buffer was applied, and GDHGeo was eluted between 150 and 175 mM. The fractions containing GDHGeo activity were concentrated to 2.5 ml (molecular mass cutoff of 10,000 Da; VivaScience), desalted (PD-10 desalting column; GE Healthcare), and loaded onto a Mono Q ion exchange column (Mono Q 5/50 GL, 1 ml; GE Healthcare). The column was equilibrated with basic buffer plus 10% (vol/vol) glycerol. A KCl gradient from 50 to 300 mM was applied to the buffer. GDHGeo was eluted at 250 mM. After concentration to 0.5 ml, GDHGeo was applied to a gel filtration column as described below (determination of native mass).
Heterologous expression and purification of His-tagged GDHDes.
The putative gene for GDHDes identified in this work was heterologously cloned in E. coli with an additional sequence coding for six histidines at the C terminus. The gene was amplified using the primer pair ATGGATTTCAACTTATCCAAAGAA (forward primer) and CCGGTTGCCCTTTCTGACC (reverse primer). The transfer of the amplified gene into expression vector pEXP5-CT/TOPO, the transformation into One Shot RTOP10F′ for propagation and into One Shot R BL21(DE3) for expression, as well as the analysis of the fusion protein were carried out according to the instructions of the manufacturer (Invitrogen GmbH, Karlsruhe, Germany).
Expression of His-tagged GDHDes in E. coli was achieved at 18°C for 16 h after induction with isopropyl-β-d-thiogalactopyranoside (IPTG) (0.5 mM). Cells were harvested at an optical density of 2.4 by centrifugation at 13,700 × g for 10 min at 4°C and stored at −80°C until further use. For purification of His-tagged GDHDes, frozen cells were suspended in buffer containing 20 mM Tris-HCl (pH 7.8), 250 mM KCl, 10% (vol/vol) glycerol, and 0.1 mg ml−1 DNase I (1 g cells in 1 ml buffer). Cell lysates were obtained as described above. The expressed His-tagged gene product was purified from the supernatant at 4°C by a Ni-Sepharose high-performance affinity column (HisTrap HP 1.6-cm-diameter, 1-ml-volume column; GE Healthcare). After equilibration of the column with the suspension buffer described above without DNase I, the extract was applied to the column at a flow rate of 0.5 ml min−1 and washed with 10 column volumes, followed by a linear gradient over 30 column volumes from 0 to 500 mM imidazole in buffer.
Heterologous expression and purification of His-tagged GDHGeo.
bam cluster IA of G. metallireducens contains the bamM gene (gi78194537), annotated as acyl-CoA dehydrogenase. It was heterologously expressed in E. coli cells with an additional sequence coding for six histidines at the C-terminal end. The bamM gene was amplified using primer pair CACCATGAGTGTTACTACAGAACTGACG (forward primer) and CCGGAAGGCCGAAATGCC (reverse primer). The amplified gene was transferred into expression vector pET101/D-TOPO. The construct was transformed into One Shot RTOP10F′ for propagation and into One Shot R BL21(DE3) for expression and analysis of the fusion protein. All steps were carried out according to the instructions provided by the manufacturer (Invitrogen GmbH, Karlsruhe, Germany). Expression of the fusion protein, cell harvesting, and purification by nickel affinity chromatography were carried out as described above for GDHDes.
Determination of kinetic parameters.
For determinations of Km and Vmax values, the continuous spectrophotometric assay was used at 30°C. For this purpose, the glutaryl-CoA concentration was varied from 1 to 500 μM (double to triple determinations). Km values were determined by fitting the data to Michaelis-Menten curves using the GraphPad Prism software package (GraphPad Software, Inc., La Jolla, CA). For testing the inhibition of glutaconyl-CoA, 16 and 40 μM of the inhibitor were added to the assay mixture before the reaction was started by adding glutaryl-CoA (80 μM). Due to the limited amount of glutaconyl-CoA available, the Ki value could be only roughly estimated.
Determination of native molecular mass.
The native molecular masses of GDHGeo and GDHDes were determined by gel filtration (Superdex 200 HR 25-ml column; GE Healthcare). After equilibration with 10 mM Tris-HCl (pH 7.5) and 100 mM KCl, 2.5 mg in 100 μl was loaded at a flow rate of 0.2 ml min−1. Mass standards (lysozyme, carboanhydrase, bovine serum albumin, lactate dehydrogenase, and catalase) were used for establishing a calibration curve.
Determination of the flavin cofactor.
One hundred microliters of purified GDHGeo and GDHDes (approximately 1 mg ml−1) was incubated with 5 μl 10% (wt/vol) perchloric acid for 1 h in the dark at 0°C. After centrifugation (10 min at 13,700 × g), the pH was adjusted to 6.5 with 2 M K2CO3, and the mixture was centrifuged again. One hundred microliters of the supernatant was analyzed by HPLC (Waters 2695 apparatus) using an Eurospher RP C18 column (Knauer), which was isocratically run with 11% acetonitrile (by volume) in 20 mM aqueous ammonium acetate (pH 6). The flavin cofactor was identified by comparing retention times and visible absorption spectra with standards (FAD, flavin mononucleotide, and riboflavin). The amount of the FAD cofactor was determined by comparison of the peak areas with FAD standards.
UV/Vis spectroscopy.
UV/Vis spectra of purified GDHGeo and GDHDes were taken from 240 to 700 nm (UV-1650PC; Shimadzu). For this purpose, spectra of 0.1 to 1 mg purified protein in basic buffer were taken before and 5 min after the addition of 0.2 mM glutaryl-CoA.
Further determinations.
Protein concentrations were determined according to a method described previously by Bradford (8a). Proteins on gels were stained with Simply Blue Safe Stain Coomassie G-250 (Invitrogen).
Nucleotide sequence accession number.
The newly sequenced DNA from D. multivorans was deposited in the GenBank database under accession number FJ688103.
RESULTS
While many details about the structure-function relationship and the mechanism of decarboxylating GDHs were obtained from studies with human GDH, nondecarboxylating GDHs have not been isolated and characterized so far. The presence of the latter was so far deduced from in vitro enzyme assays with a few fermenting bacteria (15, 28, 30). To elucidate the properties and distribution of decarboxylating/nondecarboxylating glutaryl-CoA dehydrogenases in obligately anaerobic bacteria, we studied the GDH enzymes involved in the aromatic catabolism of the Fe(III)-reducing organism G. metallireducens and the sulfate-reducing organism D. multivorans.
GDH activities in cell extracts of G. metallireducens and D. multivorans.
Extracts from cells of G. metallireducens and D. multivorans grown on benzoate were tested for GDH activity using ferrocenium hexafluorophosphate as an electron acceptor; HPLC analysis was used for the determination of the amount of glutaryl-CoA consumed and the products formed.
Using extracts from G. metallireducens cells grown on benzoate, glutaryl-CoA was readily consumed (specific activity, 0.3 μmol min−1 mg−1). The major products formed coeluted with 3-hydroxybutyryl-CoA and crotonyl-CoA (Fig. 2). Mass spectrometric analysis of the isolated products revealed masses of 853.15 Da and 835.14 Da ([M + H]+1), which confirmed that the products formed were 3-hydroxybutyryl-CoA and crotonyl-CoA in a 2:1 ratio. Obviously, the crotonyl-CoA formed was immediately hydrated by crotonase. The crotonase reaction favors the formation of the hydrated product (1). As NAD+ was absent in the assay mixture, no further conversion of 3-hydroxybutyryl-CoA to acetoacetyl-CoA and, finally, acetyl-CoA was observed.
FIG. 2.

HPLC analysis of enzymatic glutaryl-CoA conversion. (B and C, left) HPLC diagram of glutaryl-CoA conversion by soluble crude extracts of D. multivorans (B) or G. metallireducens after 30 min (C). (A, left) Glutaryl-CoA standard. (Right) HPLC diagram of glutaryl-CoA conversion after 20 min of incubation with recombinant GDHDes (B) and with native GDHGeo (C) and at 0 min without enzyme (A). The individual peaks were analyzed by mass spectrometry, and the assignment to proposed CoA esters is presented: peak 1, 882.3 Da ([M + H]+1), glutaryl-CoA; peak 2, 880.1 Da ([M + H]+1), glutaconyl-CoA; peak 3, 898.1 Da ([M + H]+1), 3-hydroxyglutaryl-CoA; peak 4, 768.3 Da ([M + H]+1), CoA; peak 5, 853.15 Da ([M + H]+1), 3-hydroxybutyryl-CoA; peak 6, 836.14 Da ([M + H]+1), crotonyl-CoA.
Using soluble extracts from D. multivorans cells grown on benzoate, glutaryl-CoA was also rapidly converted (0.2 μmol min−1 mg−1), but the reaction always leveled off when maximally 30% of the glutaryl-CoA was consumed (Fig. 2). This observation suggested that GDHDes was possibly inhibited by the glutaconyl-CoA formed (see below). Mass spectrometric analysis of a product (Fig. 2) revealed a mass of 880.1 Da ([M + H]+1), which was 2 mass units below that of glutaryl-CoA, with 882.3 Da ([M + H]+1). This finding strongly suggested that this product was glutaconyl-CoA, which was formed by the dehydrogenation of the substrate. A further product had a mass of 898.1 Da ([M + H]+1), which can be explained by the addition of water to glutaconyl-CoA most possibly yielding 3-hydroxyglutaryl-CoA. This assumption was confirmed by coelution with a chemically synthesized 3-hydroxyglutaryl-CoA standard (not shown). When the membrane protein fraction was added to the assay mixture, glutaryl-CoA, glutaconyl-CoA, and 3-hydroxyglutaryl-CoA were readily and completely converted to 3-hydroxybutyryl-CoA (not shown). This conversion was at least threefold higher in the presence of 20 mM NaCl than with no extra NaCl added. Taken together, all these observations suggest that D. multivorans contains a nondecarboxylating, glutaconyl-CoA-forming GDH. As glutaconyl-CoA decarboxylases are membrane enzymes, the formation of crotonyl-CoA was observed only after the addition of the membrane protein fraction. The formation of 3-hydroxyglutaryl-CoA from glutaconyl-CoA may be an unusual side reaction of a hydratase such as crotonase. This assumption is confirmed by the finding that the hydration of glutaconyl-CoA could be partially inhibited by adding large amounts of crotonyl-CoA (>0.5 mM) (not shown). The addition of the membrane fraction in the presence of Na+ enabled the decarboxylation and further degradation of the crotonyl-CoA formed.
Using extracts of cells grown on acetate (G. metallireducens) or lactate (D. multivorans), no glutaryl-CoA conversion was observed, suggesting that GDH activity was induced by the aromatic growth substrate.
Purification and properties of GDHGeo.
GDHGeo was purified from soluble extracts from cells grown on benzoate/Fe(III) by chromatography using DEAE-Sepharose, MonoQ-Sepharose, and gel filtration. The FAD cofactor was partially lost during purification; however, GDHGeo could be partially reactivated by incubation with FAD (see below). Thus, the numbers given for activities during the purification always refer to reactivated GDH. The enzyme was highly enriched (purity of >95%) by a factor of 41, with a yield of 12%. Using ferrocenium hexafluorophosphate as an electron acceptor, the purified enzyme had a maximal specific activity of 12 μmol min−1 mg−1.
GDHGeo consisted of a single protein band demonstrated by SDS-PAGE analysis (Fig. 3A). The native mass determined by gel filtration was 167 ± 10 kDa, suggesting an α4 composition. Mass spectrometric analysis of a tryptic digest of the 43-kDa band revealed only one hit, with a score of 119 (search against the complete MASCOT database with a score of >77), which was the product of the bamM gene (gi78194537) of the so-called benzoic acid metabolism (bam) gene cluster of G. metallireducens (sequence coverage was 50%). Further similarities were obtained with a number of other deduced gene products annotated as glutaryl-CoA dehydrogenases/acyl-CoA dehydrogenases.
FIG. 3.

SDS-PAGE analysis of protein fractions obtained during purification of native GDHGeo (A) and heterologous expression/purification of recombinant GDHDes (B) and GDHGeo (C). (A) Lane 1, soluble extract after ultracentrifugation; lane 2, soluble extract after dialysis; lanes 3 to 5, fraction containing GDH activity after DEAE-Sepharose chromatography (lane 3), after MonoQ-Sepharose chromatography (lane 4), and after gel filtration (lane 5). (B) Lanes 1 and 2, soluble protein fraction of E. coli cells containing a plasmid coding for GDHDes after (lane 1) and before (lane 2) induction by IPTG; lanes 3 and 4, membrane protein fraction of E. coli cells containing a plasmid coding for GDHDes after (lane 3) and before (lane 4) induction with 0.5 mM IPTG; lane 5, purified His-tagged GDHDes (approximately 43 kDa) after purification by Ni-chelating affinity chromatography. (C) Lanes 1 and 2, soluble protein fraction of E. coli cells containing a plasmid coding for GDHGeo after (lane 1) and before (lane 2) induction by IPTG; lanes 3 and 4, membrane protein fraction of E. coli cells containing a plasmid coding for GDHGeo after (lane 3) and before (lane 4) induction with 0.5 mM IPTG; lane 5, purified His-tagged GDHGeo (approximately 46.5 kDa including a V5 epitope) after purification by Ni-chelating affinity chromatography. M, molecular mass standards. Proteins were visualized by staining with Simply Blue Safe Stain Coomassie G-250.
Flavin cofactor analysis of yellowish GDHGeo revealed a content of 0.4 FADs per subunit. Upon incubation with 0.5 mM FAD for 60 min at 30°C, the FAD content increased to 0.6 FADs per subunit (after removal of excess FAD by a desalting column); in parallel, the specific activity increased by a factor of 1.4. Thus, the loss of activity was indeed due to a loss of FAD cofactor and was at least partially reversible. The UV/Vis spectrum of GDHGeo was typical for a flavoprotein, with absorption maxima at 369 nm and 448 nm. The addition of 0.2 mM glutaryl-CoA completely bleached the spectrum at both maxima.
HPLC analysis unambiguously revealed that purified GDHGeo converted glutaryl-CoA completely to crotonyl-CoA with ferrocenium hexafluorophosphate as an electron acceptor; virtually no formation of glutaconyl-CoA was observed (Fig. 2C). The Km and Vmax for glutaryl-CoA were determined by the spectrophotometric assay to be 30 ± 2 μM and 3.2 ± 0.2 μmol min−1 mg−1, respectively, by fitting the initial rates obtained in the presence of various glutaryl-CoA concentrations to a Michaelis-Menten curve.
Identification and heterologous expression of the gene coding for GDHGeo.
For the unambiguous identification of the gene coding for the decarboxylating GDHGeo and the characterization of the properties of the isolated gene product, bamM was cloned into E. coli cells and heterologously expressed with a sixfold His tag/V5 epitope tag at the C terminus. The expression of the soluble fusion protein was achieved, and purification by nickel affinity chromatography resulted in a single band with a molecular mass of 45.5 kDa, which corresponded to the mass predicted from the bamM sequence (46.7 kDa, including tags) (Fig. 3C). The molecular and kinetic properties of recombinant GDHGeo are summarized in Table 1. Recombinant and wild-type GDHGeo apparently exhibited highly similar properties. HPLC analysis revealed that the recombinant enzyme catalyzed a complete conversion of glutaryl-CoA to crotonyl-CoA; no other product was formed.
TABLE 1.
Properties of wild-type and recombinant GDHGeo and recombinant GDHDesa
| Potein | Substrate | Product(s) | Molecular mass (kDa) | Mean native mass (kDa) ± SD | Suggested composition | Mean Km of glutaryl-CoA (μM) ± SD | Mean Vmax (μmol min−1 mg−1) ± SD | Absorption maxima (nm) | No. of FADs/monomer | Inhibitor |
|---|---|---|---|---|---|---|---|---|---|---|
| GDHGeo WT | Glutaryl- CoA | Crotonyl-CoA, CO2 | 43.1 | 170 ± 10 | α4 | 30 ± 2 | 3.2 ± 0.2 | 370, 450 | 0.6 | None |
| GDHGeo RC | Glutaryl | Crotonyl-CoA, CO2 | 43.1 | 180 ± 10 | α4 | 31 ± 2 | 3.6 ± 0.2 | 369, 448 | 0.62 | None |
| GDHDes RC | Glutaryl | Glutaconyl-CoA | 43.4 | 180 ± 10 | α4 | 52 ± 5 | 11 ± 1 | 370, 447 | 0.65 | Glutaconyl-CoA Ki of 5-10 μM |
The molecular masses deduced from the amino acid sequence are presented. WT, wild type; RC, recombinant.
Identification of the gene coding for GDHDes.
The heterologous expression of the gene coding for GDHDes was carried out. As the genome was not sequenced, a cosmid gene library of D. multivorans was constructed. Using a DNA probe which was deduced from conserved amino acid sequence regions of benzoate-CoA ligases from several organisms, a cosmid was identified, 5 kb of which was sequenced. Analysis of the DNA sequence revealed two complete (bcl and gdh) and two incomplete (rre and gcd) open reading frames (Fig. 4). The deduced amino acid sequence of the bcl gene product was highly similar to amino acid sequences from benzoate-CoA ligases from Geobacter species that use aromatic growth substrates (62 to 64% amino acid sequence identity), and the N terminus was identical to that of the previously isolated benzoate-CoA ligase from D. multivorans (32). The gdh gene product showed high-level amino acid sequence similarities to glutaryl-CoA dehydrogenases/acyl-CoA dehydrogenases from sulfate-reducing and fermenting bacteria (up to 73% identity). The deduced partial sequence of the gcd product showed significant similarities to the β-subunit of Na+-transporting decarboxylases (carboxybiotin decarboxylase subunit, 50% identity); the rre product showed similarities with DNA-binding transcriptional regulators (the highest similarities were found with BamW [gi78194607], which is involved in benzoate metabolism in G. metallireducens).
Induction of the genes coding for GDHGeo (bamM) and GDHDes (gdh).
The induction of the bamM gene, coding for GDHGeo, during growth on aromatic compounds was demonstrated previously (40). We also tested whether gdh, putatively coding for GDHDes, was induced during growth on aromatic compounds. For this purpose, total RNA from D. multivorans cells grown on benzoate, cyclohexanecarboxylate, and lactate was isolated, transcribed into cDNA by reverse transcription, and finally subjected to PCRs for amplifying fragments from the rre, bcl, acd (gdh), and gcd open reading frames/intergenic regions (Fig. 4). With the four primer pairs used, the corresponding fragments were obtained only with cDNA from cells grown on benzoate (using undiluted DNA and 1:10 and 1:100 dilutions) but were virtually absent even using undiluted cDNA obtained from cells grown on cyclohexane carboxylate or lactate. Thus, the cluster of the four open reading frames was induced during growth on benzoate.
Heterologous expression of gdh from D. multivorans.
The gdh gene was cloned and heterologously expressed in E. coli cells with a His6 tag at the C terminus. The expression of a soluble 43-kDa protein was achieved; the molecular mass corresponded to that predicted from the gdh gene sequence (44.4 kDa, including the His tag) (Fig. 3B). After purification via Ni-chelating chromatography, a soluble yellow protein was obtained, consisting of a single 43-kDa band.
Molecular properties of recombinant GDHDes.
The purified gdh gene product obtained showed a tendency to precipitate at a low ionic strength. Therefore, it was routinely kept at 200 mM KCl, where it was stable for weeks at −20°C. The native molecular mass of GDHDes was determined by gel filtration to be 180 ± 10 kDa, suggesting an α4 composition. The amount of flavin was determined by HPLC analysis to be 0.45 FADs per subunit. Incubation with 0.5 mM FAD for 60 min at 30°C resulted in an increase to 0.65 FADs per subunit (after the removal of excess FAD by a desalting column). The UV/Vis spectrum of GDHDes was highly similar to that of GDHGeo, with absorption maxima at 370 nm and 447 nm. Similar to GDHGeo, the spectrum of GDHDes was bleached upon the addition of the substrate glutaryl-CoA.
Kinetic properties of recombinant GDHDes.
HPLC analysis with ferrocenium hexafluorophosphate as the electron acceptor revealed that GDHDes converted glutaryl-CoA to glutaconyl-CoA; crotonyl-CoA was not detected (Fig. 2). However, as already observed in experiments with cell extracts, only ∼30% of glutaryl-CoA was converted to glutaconyl-CoA. The reason for this finding is assigned to an inhibitory effect of the product formed rather than to a thermodynamic equilibrium. When 0.015 or 0.04 mM glutaconyl-CoA was added to the assay mixture, the initial rate of glutaryl-CoA conversion (0.1 mM) decreased to 25% or 10%, respectively, in comparison with the activity in the absence of extra glutaconyl-CoA added. Due to the limiting amounts of glutaconyl-CoA available, the Ki value for the apparent competitive inhibition could only roughly be estimated to be 5 to 10 μM. Glutaconyl-CoA had no inhibitory effect on GDHGeo. The Km and Vmax of GDHDes for glutaryl-CoA were determined by the spectrophotometric assay to be 52 ± 5 μM and 11 ± 1 μmol min−1 mg−1, respectively, by fitting the initial rates obtained in the presence of various glutaryl-CoA concentrations to a Michaelis-Menten curve.
DISCUSSION
Properties of GDH from G. metallireducens and D. multivorans.
The properties of GDHGeo (wild type and recombinant) and GDHDes (recombinant) are summarized in Table 1. While many properties of the GDH enzymes are similar, there are two marked differences: (i) GDHGeo catalyzed both the dehydrogenation and decarboxylation of the substrate, while GDHDes catalyzed only the dehydrogenation of the substrate, and (ii) GDHGeo completely converted the substrate to crotonyl-CoA, while the enzyme from D. multivorans converted only 30% of the substrate (0.2 mM) to glutaconyl-CoA. The latter finding is explained by the inhibition of GDHDes by the product, whereas glutaconyl-CoA had no inhibitory effect on GDHGeo. This finding cannot be explained by a thermodynamic equilibrium, as electron transfers from glutaryl-CoA to ferrocenium and phenazine methosulfate-2,6-dichlorophenol indophenol are both considered to be irreversible. In the case of GDHGeo, the glutaconyl-CoA added was probably immediately converted to crotonyl-CoA by the decarboxylase activity of the enzyme. As GDHDes has no such activity, product inhibition was observed.
Amino acids involved in the decarboxylation reaction of GDHs.
The crystal structure of human GDH with a bound substrate analogue revealed insights into the mechanism of decarboxylation (16). A number of amino acids were found to be involved in binding the substrate and polarizing the Cδ-Cɛ bond.
The amino acid sequence identities of GDHGeo compared to human GDH and GDHDes are 54% and 33%, respectively. Thus, with the amino acid sequence identified in this work, it is now possible to reveal conserved amino acids that are specifically required for substrate binding (should be present in decarboxylating GDH and nondecarboxylating GDH) and for decarboxylation (should be present only in the former GDH). A careful alignment allowed a comparison; the numberings of amino acids are according to the human enzyme. The conserved arginine R94, which is involved in binding the terminal carboxylate of the substrate (16), is indeed present in all GDH enzymes. In contrast, E87, S95, T170, and Y369, which were all proposed to polarize Cδ-Cɛ, are conserved in known decarboxylating GDH enzymes but are missing in nondecarboxylating GDHs (Table 2). This finding supports that the proposed amino acids play a specific role in the decarboxylation reaction rather than in substrate binding and provides a rational for the decarboxylating/nondecarboxylating activities of GDHGeo and GDHDes. However, initial studies with two molecular mutants of GDHDes, V367→Y and A80→E, have not been successful so far (our unpublished results). Both mutants lost more than 99% of the activity, probably due to an irreversible loss of the flavin cofactor. The residual activity did not decarboxylate the glutaconyl-CoA formed. This finding suggests that single amino acid mutations may not be sufficient to convert the nondecarboxylating GDHDes to a decarboxylating one.
TABLE 2.
Alignment of conserved amino acid residues at the active site involved in decarboxylation of the intermediate glutaconyl-CoA in decarboxylating and nondecarboxylating GDHsa
| Conserved amino acid in GDHHum | Conserved amino acid in:
|
||||
|---|---|---|---|---|---|
| GDHGeo | GDHAro | GDHRho | GDHDes | GDHSyn | |
| E87 | E89 | E92 | E100 | A80 | S78 |
| R94 | R96 | R99 | R107 | R87 | R85 |
| S95 | S97 | S100 | S108 | V88 | V86 |
| T170 | T171 | T173 | S181 | S161 | S159 |
| Y369 | Y370 | Y372 | Y382 | V367 | V364 |
All GDHs listed below (except human GDH) refer to aromatic-compound-degrading bacteria. The alignment was carried out with the MultAlin platform (11). GDHHum, GDH from Homo sapiens (gi55669741); GDHAro, GDH from Aromatoleum aromaticum EbN1 (gi56477122); GDHRho, GDH from Rhodopseudomonas palustris CGA009 (gi39934169); GDHSyn, GDH from S. aciditrophicus SB (gi85859865). Amino acids in boldface type indicate marked differences.
Adaptation of anaerobic aromatic degradation pathways to energetic constraints.
The benzoyl-CoA degradation pathway comprises a possible energy-demanding reaction (ATP-dependent benzoyl-CoA dearomatization) and a possible energy-yielding reaction (glutaconyl-CoA decarboxylation). The latter is possible only when nondecarboxylating GDH enzymes are used in the pathway. The relative overall energy yield in anaerobic bacteria using benzoate as a growth substrate is as follows: denitrifying > iron-reducing > sulfate-reducing > fermenting bacteria. Thus, a tendency to avoid energy-consuming reactions and to employ energy-conserving reactions wherever possible is expected for bacteria with a low energy yield, and the opposite should be observed for bacteria with a clearly higher energy yield. In Table 3, the distribution of different types of BCR and GDH enzymes in different physiological classes of aromatic-compound-degrading anaerobes is summarized. Indeed, phototrophic and denitrifying bacteria obviously can afford an ATP-dependent dearomatization process and have no need to couple glutaryl-CoA dehydrogenation to the generation of a membrane potential, whereas in fermenting and sulfate-reducing bacteria, the opposite is the case. The benzoyl-CoA degradation pathway of Fe(III)-reducing Geobacter species seems to represent a chimeric type, which avoids an ATP-dependent BCR enzyme but uses a decarboxylating GDH (highly similar decarboxylating GDH homologues are present in the genomes of all aromatic-compound-degrading Geobacter species). The advantage for aromatic-compound-degrading denitrifying/phototrophic bacteria may be that the use of aromatic growth substrates becomes independent of enzymes that require numerous additional cofactors (Mo- or W-pterin and selenocysteine for BCR; biotin and glutaconyl-CoA for decarboxylase).
TABLE 3.
Distribution of BCR and GDH enzymes in anaerobic bacteria that degrade aromatic compoundsa
| Organism | Type of catabolism | Anaerobe type | BCR type | GDH type |
|---|---|---|---|---|
| Rhodopseudomonas palustris | Phototrophic | Facultative | ATP dependent | Decarboxylating |
| Thauera aromatica | Denitrifying | Facultative | ATP dependent | Decarboxylating |
| Azoarcus sp. | Denitrifying | Facultative | ATP dependent | Decarboxylating |
| Geobacter sp. | Iron(III) reducing | Obligate | Mo/W-SeCys | Decarboxylating |
| Desulfococcus multivorans | Sulfate reducing | Obligate | Mo/W-SeCys | Nondecarboxylating |
| Syntrophus aciditrophicus | Fermenting | Obligate | Mo/W-SeCys | Nondecarboxylating |
The Mo/W-selenocysteine (SeCys)-containing BCR enzymes from obligate anaerobes are considered to be ATP-independent enzymes. The presence of a nondecarboxylating GDH is always associated with the presence of a sodium-pumping glutaconyl-CoA decarboxylase.
Acknowledgments
This work was funded by the Deutsche Forschungsgemeinschaft (BO 1565/6-1).
Footnotes
Published ahead of print on 24 April 2009.
REFERENCES
- 1.Agnihotri, G., and H. W. Liu. 2003. Enoyl-CoA hydratase. Reaction, mechanism, and inhibition. Bioorg. Med. Chem. 119-20. [DOI] [PubMed] [Google Scholar]
- 2.Beatrix, B., K. Bendrat, S. Rospert, and W. Buckel. 1990. The biotin-dependent sodium ion pump glutaconyl-CoA decarboxylase from Fusobacterium nucleatum (subsp. nucleatum). Comparison with the glutaconyl-CoA decarboxylases from gram-positive bacteria. Arch. Microbiol. 154362-369. [DOI] [PubMed] [Google Scholar]
- 3.Besrat, A., C. E. Polan, and L. M. Henderson. 1969. Mammalian metabolism of glutaric acid. J. Biol. Chem. 2441461-1467. [PubMed] [Google Scholar]
- 4.Blazquez, B., M. Carmona, J. L. Garcia, and E. Diaz. 2008. Identification and analysis of a glutaryl-CoA dehydrogenase-encoding gene and its cognate transcriptional regulator from Azoarcus sp. CIB. Environ. Microbiol. 10474-482. [DOI] [PubMed] [Google Scholar]
- 5.Boll, M. 2005. Dearomatizing benzene ring reductases. J. Mol. Microbiol. Biotechnol. 10132-142. [DOI] [PubMed] [Google Scholar]
- 6.Boll, M. 2005. Key enzymes in the anaerobic aromatic metabolism catalysing Birch-like reductions. Biochim. Biophys. Acta 170734-50. [DOI] [PubMed] [Google Scholar]
- 7.Boll, M., and G. Fuchs. 1995. Benzoyl-coenzyme A reductase (dearomatizing), a key enzyme of anaerobic aromatic metabolism. ATP dependence of the reaction, purification and some properties of the enzyme from Thauera aromatica strain K172. Eur. J. Biochem. 234921-933. [DOI] [PubMed] [Google Scholar]
- 8.Boll, M., G. Fuchs, and J. Heider. 2002. Anaerobic oxidation of aromatic compounds and hydrocarbons. Curr. Opin. Chem. Biol. 6604-611. [DOI] [PubMed] [Google Scholar]
- 8a.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72248-254. [DOI] [PubMed] [Google Scholar]
- 9.Buckel, W. 2001. Sodium ion-translocating decarboxylases. Biochim. Biophys. Acta 150515-27. [DOI] [PubMed] [Google Scholar]
- 10.Buckel, W. 2001. Unusual enzymes involved in five pathways of glutamate fermentation. Appl. Microbiol. Biotechnol. 57263-273. [DOI] [PubMed] [Google Scholar]
- 11.Corpet, F. 1988. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 1610881-10890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diaz, E. 2004. Bacterial degradation of aromatic pollutants: a paradigm of metabolic versatility. Microbiology 7173-180. [PubMed] [Google Scholar]
- 13.Dimroth, P., and B. Schink. 1998. Energy conservation in the decarboxylation of dicarboxylic acids by fermenting bacteria. Arch. Microbiol. 17069-77. [DOI] [PubMed] [Google Scholar]
- 14.Dwyer, T. M., K. S. Rao, S. I. Goodman, and F. E. Frerman. 2000. Proton abstraction reaction, steady-state kinetics, and oxidation-reduction potential of human glutaryl-CoA dehydrogenase. Biochemistry 3911488-11499. [DOI] [PubMed] [Google Scholar]
- 15.Elshahed, M. S., V. K. Bhupathiraju, N. Q. Wofford, M. A. Nanny, and M. J. McInerney. 2001. Metabolism of benzoate, cyclohex-1-ene carboxylate, and cyclohexane carboxylate by “Syntrophus aciditrophicus” strain SB in syntrophic association with H2-using microorganisms. Appl. Environ. Microbiol. 671728-1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fu, Z., M. Wang, R. Paschke, K. S. Rao, F. E. Frerman, and J. J. Kim. 2004. Crystal structures of human glutaryl-CoA dehydrogenase with and without an alternate substrate: structural bases of dehydrogenation and decarboxylation reactions. Biochemistry 439674-9684. [DOI] [PubMed] [Google Scholar]
- 17.Fuchs, G. 2008. Anaerobic metabolism of aromatic compounds. Ann. N. Y. Acad. Sci. 112582-99. [DOI] [PubMed] [Google Scholar]
- 18.Gibson, J., and C. S. Harwood. 2002. Metabolic diversity in aromatic compound utilization by anaerobic microbes. Annu. Rev. Microbiol. 56345-369. [DOI] [PubMed] [Google Scholar]
- 19.Gomes, B., G. Fendrich, and R. H. Abeles. 1981. Mechanism of action of glutaryl-CoA and butyryl-CoA dehydrogenases. Purification of glutaryl-CoA dehydrogenase. Biochemistry 201481-1490. [DOI] [PubMed] [Google Scholar]
- 20.Goodman, S. I., L. E. Kratz, K. A. DiGiulio, B. J. Biery, K. E. Goodman, G. Isaya, and F. E. Frerman. 1995. Cloning of glutaryl-CoA dehydrogenase cDNA, and expression of wild type and mutant enzymes in Escherichia coli. Hum. Mol. Genet. 41493-1498. [DOI] [PubMed] [Google Scholar]
- 21.Goodman, S. I., D. E. Stein, S. Schlesinger, E. Christensen, M. Schwartz, C. R. Greenberg, and O. N. Elpeleg. 1998. Glutaryl-CoA dehydrogenase mutations in glutaric acidemia (type I): review and report of thirty novel mutations. Hum. Mutat. 12141-144. [DOI] [PubMed] [Google Scholar]
- 22.Harrison, F. H., and C. S. Harwood. 2005. The pimFABCDE operon from Rhodopseudomonas palustris mediates dicarboxylic acid degradation and participates in anaerobic benzoate degradation. Microbiology 151727-736. [DOI] [PubMed] [Google Scholar]
- 23.Härtel, U., E. Eckel, J. Koch, G. Fuchs, D. Linder, and W. Buckel. 1993. Purification of glutaryl-CoA dehydrogenase from Pseudomonas sp., an enzyme involved in the anaerobic degradation of benzoate. Arch. Microbiol. 159174-181. [DOI] [PubMed] [Google Scholar]
- 24.Husain, M., and D. J. Steenkamp. 1985. Partial purification and characterization of glutaryl-coenzyme A dehydrogenase, electron transfer flavoprotein, and electron transfer flavoprotein-Q oxidoreductase from Paracoccus denitrificans. J. Bacteriol. 163709-715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24a.Jehmlich, U., F. Schmidt, M. von Bergen, M.-M. Richnow, and C. Vogt. 2008. Protein-based stable isotope probing (protein-SIP) reveals active site species within anoxic mixed cultures. ISME J. 21122-1133. [DOI] [PubMed] [Google Scholar]
- 25.Kuntze, K., Y. Shinoda, H. Moutakki, M. J. McInerney, C. Vogt, H. H. Richnow, and M. Boll. 2008. 6-Oxocyclohex-1-ene-1-carbonyl-coenzyme A hydrolases from obligately anaerobic bacteria: characterization and identification of its gene as a functional marker for aromatic compounds degrading anaerobes. Environ. Microbiol. 101547-1556. [DOI] [PubMed] [Google Scholar]
- 26.Lenich, A. C., and S. I. Goodman. 1986. The purification and characterization of glutaryl-coenzyme A dehydrogenase from porcine and human liver. J. Biol. Chem. 2614090-4096. [PubMed] [Google Scholar]
- 27.Lovley, D. R., and E. J. Phillips. 1988. Novel mode of microbial energy metabolism: organic carbon oxidation coupled to dissimilatory reduction of iron or manganese. Appl. Environ. Microbiol. 541472-1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matthies, C., and B. Schink. 1992. Fermentative degradation of glutarate via decarboxylation by newly isolated strictly anaerobic bacteria. Arch. Microbiol. 157290-296. [DOI] [PubMed] [Google Scholar]
- 29.McInerney, M. J., L. Rohlin, H. Mouttaki, U. Kim, R. S. Krupp, L. Rios-Hernandez, J. Sieber, C. G. Struchtemeyer, A. Bhattacharyya, J. W. Campbell, and R. P. Gunsalus. 2007. The genome of Syntrophus aciditrophicus: life at the thermodynamic limit of microbial growth. Proc. Natl. Acad. Sci. USA 1047600-7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Müller, J. A., and B. Schink. 2000. Initial steps in the fermentation of 3-hydroxybenzoate by Sporotomaculum hydroxybenzoicum. Arch. Microbiol. 173288-295. [DOI] [PubMed] [Google Scholar]
- 31.Numa, S., Y. Ishimura, T. Nakazawa, T. Okazaki, and O. Hayaishi. 1964. Enzymic studies on the metabolism of glutarate in Pseudomonas. J. Biol. Chem. 2393915-3926. [PubMed] [Google Scholar]
- 32.Peters, F., M. Rother, and M. Boll. 2004. Selenocysteine-containing proteins in anaerobic benzoate metabolism of Desulfococcus multivorans. J. Bacteriol. 1862156-2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peters, F., Y. Shinoda, M. J. McInerney, and M. Boll. 2007. Cyclohexa-1,5-diene-1-carbonyl-coenzyme A (CoA) hydratases of Geobacter metallireducens and Syntrophus aciditrophicus: evidence for a common benzoyl-CoA degradation pathway in facultative and strict anaerobes. J. Bacteriol. 1891055-1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rao, K. S., M. Albro, T. M. Dwyer, and F. E. Frerman. 2006. Kinetic mechanism of glutaryl-CoA dehydrogenase. Biochemistry 4515853-15861. [DOI] [PubMed] [Google Scholar]
- 35.Rao, K. S., Z. Fu, M. Albro, B. Narayanan, S. Baddam, H. J. Lee, J. J. Kim, and F. E. Frerman. 2007. The effect of a Glu370Asp mutation in glutaryl-CoA dehydrogenase on proton transfer to the dienolate intermediate. Biochemistry 4614468-14477. [DOI] [PubMed] [Google Scholar]
- 36.Redenbach, M., H. M. Kieser, D. Denapaite, A. Eichner, J. Cullum, H. Kinashi, and D. A. Hopwood. 1996. A set of ordered cosmids and a detailed genetic and physical map for the 8 Mb Streptomyces coelicolor A3(2) chromosome. Mol. Microbiol. 2177-96. [DOI] [PubMed] [Google Scholar]
- 37.Simon, E., and D. Shemin. 1953. The preparation of S-succinyl-CoA. J. Am. Chem. Soc. 19532520. [Google Scholar]
- 38.Wendt, K. S., I. Schall, R. Huber, W. Buckel, and U. Jacob. 2003. Crystal structure of the carboxyltransferase subunit of the bacterial sodium ion pump glutaconyl-coenzyme A decarboxylase. EMBO J. 223493-3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Widdel, F., and N. Pfennig. 1981. Studies on dissimilatory sulfate-reducing bacteria that decompose fatty acids. I. Isolation of new sulfate-reducing bacteria enriched with acetate from saline environments. Description of Desulfobacter postgatei gen. nov., sp. nov. Arch. Microbiol. 129395-400. [DOI] [PubMed] [Google Scholar]
- 40.Wischgoll, S., D. Heintz, F. Peters, A. Erxleben, E. Sarnighausen, R. Reski, A. Van Dorsselaer, and M. Boll. 2005. Gene clusters involved in anaerobic benzoate degradation of Geobacter metallireducens. Mol. Microbiol. 581238-1252. [DOI] [PubMed] [Google Scholar]