

Abstract
Streptococcus sanguinis is an important cause of infective endocarditis. Previous studies have identified lipoproteins as virulence determinants in other streptococcal species. Using a bioinformatic approach, we identified 52 putative lipoprotein genes in S. sanguinis strain SK36 as well as genes encoding the lipoprotein-processing enzymes prolipoprotein diacylglyceryl transferase (lgt) and signal peptidase II (lspA). We employed a directed signature-tagged mutagenesis approach to systematically disrupt these genes and screen each mutant for the loss of virulence in an animal model of endocarditis. All mutants were viable. In competitive index assays, mutation of a putative phosphate transporter reduced in vivo competitiveness by 14-fold but also reduced in vitro viability by more than 20-fold. Mutations in lgt, lspA, or an uncharacterized lipoprotein gene reduced competitiveness by two- to threefold in the animal model and in broth culture. Mutation of ssaB, encoding a putative metal transporter, produced a similar effect in culture but reduced in vivo competiveness by >1,000-fold. [3H]palmitate labeling and Western blot analysis confirmed that the lgt mutant failed to acylate lipoproteins, that the lspA mutant had a general defect in lipoprotein cleavage, and that SsaB was processed differently in both mutants. These results indicate that the loss of a single lipoprotein, SsaB, dramatically reduces endocarditis virulence, whereas the loss of most other lipoproteins or of normal lipoprotein processing has no more than a minor effect on virulence.
Streptococcus sanguinis is a member of the viridans group of streptococci and is a primary colonizer of teeth (8). The viridans species and, in particular, S. sanguinis (15, 18) are a leading cause of infective endocarditis, a serious infection of the valves or lining of the heart (48). Damage to the heart resulting from rheumatic fever or certain congenital heart defects dramatically increases the risk of developing endocarditis (48, 71). The damage is thought to result in the formation of sterile cardiac “vegetations” composed of platelets and fibrin (48) that can be colonized by certain bacteria during periods of bacteremia. This view is supported by animal studies in which formation of sterile vegetation by cardiac catheterization is required for the efficient establishment of streptococcal endocarditis (17). Prevention of infective endocarditis currently relies upon prophylactic administration of antibiotics prior to dental or other surgical procedures that are likely to produce bacteremia. The growing realization that oral bacteria such as S. sanguinis can enter the bloodstream through routine daily activities such as eating has led the American Heart Association (71) and others (57) to question the value of using antibiotic prophylaxis for dental procedures. Clearly, a better understanding of the bacterial virulence factors that contribute to endocarditis could lead to better preventive measures, such as a vaccine that could potentially afford continuous protection to high-risk patients (71).
In a previous study, we used the signature-tagged mutagenesis (STM) technique to search for endocarditis virulence factors of S. sanguinis in a rabbit model (53). This study identified a number of housekeeping enzymes that contribute to endocarditis. Because these proteins are not likely to be surface localized, they hold little promise as vaccine candidates. One class of streptococcal surface proteins that is rich in both virulence factors (4, 7, 25, 33, 38, 60) and promising vaccine candidates (6, 39, 42, 51, 70) is the lipoproteins. Lipoprotein activities that have been suggested to contribute to streptococcal virulence include adhesion (4, 7, 63), posttranslational modification (25, 29, 51), and ATP-binding cassette (ABC)-mediated transport (33, 52, 60). In the last instance, lipoproteins anchored to the cell membrane by their lipid tails appear to serve the same transport function as the periplasmic substrate-binding proteins of gram-negative bacteria (66). STM studies performed with Streptococcus pneumoniae (26, 41, 55) and Streptococcus agalactiae (34) have identified multiple lipoprotein mutants among collections of reduced virulence mutants. In an attempt to determine the cumulative contribution of streptococcal lipoproteins to virulence, some investigators have created mutations in the lgt or lspA genes, encoding lipoprotein-processing enzymes (12, 25, 27, 36). The lgt gene encodes prolipoprotein diacylglyceryl transferase, which catalyzes the transfer of a diacylglycerol lipid unit to a cysteine in the conserved N-terminal “lipobox” of lipoproteins, while lspA encodes the signal peptidase II enzyme that cleaves the signal peptide of the prolipoprotein just prior to the conserved cysteine (59, 65). While mutation of these genes has been shown to be lethal in gram-negative bacteria (21, 73), many gram-positive bacterial species have been shown to tolerate such mutations, often with only minor effects on growth (3, 12, 13, 25, 27, 36, 54). Some of these studies indicated a deleterious effect on the virulence of the lgt (25, 54) or lspA (36) mutation, but others found no effect (12) or an enhancement of virulence (27). It is clear from these and other studies (3, 13) that neither the loss of acylation due to lgt inactivation nor the loss of signal peptidase II-mediated cleavage completely eliminates lipoprotein function, necessitating alternative approaches for assessing the global contribution of lipoproteins to virulence.
We have used bioinformatic approaches to identify every putative lipoprotein encoded by S. sanguinis strain SK36. To determine the contribution of these lipoproteins to the endocarditis virulence of S. sanguinis, we have systematically mutagenized each of these genes, as well as the lgt and lspA genes, and evaluated these mutants for virulence by using STM in an animal model. Selected mutants were further examined for virulence in competitive index (CI) assays. A strain with a disrupted ssaB gene, which encodes a putative metal transport protein, was found to exhibit a profound defect in virulence that was far greater than that of any other strain tested, including the lgt or lspA mutant.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. S. sanguinis strain SK36 (37) and its derivatives were routinely grown in brain heart infusion (BHI) broth (Difco Inc., Detroit, MI) at 37°C with 6% O2 in an Anoxomat jar (Spiral Biotech), unless otherwise stated. Antibiotics employed for selection in streptococci were chloramphenicol (Cm), erythromycin (Em), spectinomycin (Spc), tetracycline (Tet), and kanamycin (Kan) at concentrations of 5 μg/ml, 10 μg/ml, 200 μg/ml, 5 μg/ml, and 500 μg/ml, respectively. For transformation, S. sanguinis cells were grown in Todd-Hewitt broth (Difco Inc.) adjusted to pH 7.6, containing 2.5% heat-inactivated horse serum. For the selection of plasmids in Escherichia coli, Em, Spc, Tet, and Kan at concentrations of 300, 100, 2.5, and 50 μg/ml, respectively, were added to Luria-Bertani (LB) medium.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Genotype(s) or phenotype(s) | Source or reference |
|---|---|---|
| S. sanguinis strains | ||
| SK36 | Human plaque isolate | 37 |
| JFP36 | Emr SSA_0169::pSerm; derived from SK36 | 68a |
| JFP76 | Tetr SSA_0169::tetM; derived from SK36 | 68a |
| JFP57 | Spcr CmrssaB::mariner2 SSA_0169::SSA_0169::ssaB-aad9 | This study |
| JFP58 | Spcr Cmr EmrssaB::mariner2/pJFP58 | This study |
| 0588k | Kanr SSA_0588::aphA-3 | This study |
| Plasmids | ||
| pVA838 | Emr; E. coli-Streptococcus shuttle plasmid | 43 |
| pJFP46 | Kanr SSA_0169::SSA_0169 locus with AscI-NotI-NcoI adapter | 68a |
| pJFP57 | Kanr Spcr; pJFP46 containing ssaB-aad9 | This study |
| pJFP58 | Emr Spcr; pVA838 containing ssaB-aad9 | This study |
| pR412 | Spcr; source of aad9 gene | 45 |
Bioinformatics.
Genes encoding putative lipoproteins in S. sanguinis were identified by two methods. Conceptual translations of the unfinished genome sequence of S. sanguinis strain SK36 (www.sanguinis.mic.vcu.edu/) were searched for matches to the gram-positive bacterial lipoprotein pattern from Sutcliffe and Harrington (65), modified to include an “L” two positions upstream from the final “C,” using the program fuzzpro (56). This search was repeated on the open reading frames (ORFs) from the final genome sequence (72). The final list of ORFs was also examined for putative lipoproteins using the LipoP web server (35). Putative Lgt and LspA ORFs were identified in the S. sanguinis sequence by BLASTp searches of SK36 proteins using ORFs annotated as such in published streptococcal genome sequences. These results were confirmed by performing hidden Markov model analyses using the hmmsearch program (http://hmmer.janelia.org/) (19), with the Pfam profiles PF01252 for LspA and PF01790 for Lgt (62).
Creation of signature-tagged mutants by in vitro transposition.
The collection of 41 tagged mariner-derived magellan2 transposons used previously for random mutagenesis of S. sanguinis SK36 (53) was employed in the present study to create directed mutations in lipoprotein-related genes. Each gene to be assessed within the same pool was mutagenized with a differently tagged transposon. Oligonucleotide primers were designed to amplify a fragment approximately 2.5 kb in length containing each gene to be mutagenized and flanking upstream and downstream DNA (see Table S1 in the supplemental material). Amplifications were performed using purified SK36 genomic DNA as a template and a high-fidelity polymerase mix (Platinum PCR supermix high fidelity; Invitrogen). In vitro transposition reactions were performed with differently tagged transposons, as described by Paik et al. (53). One-fourth of each reaction mixture was used to transform S. sanguinis SK36, as described previously (53). Cells were plated on BHI agar with Cm. In most cases, at least eight transformants were selected and screened for insertion site location by PCR using primer M-out (Table 2), which binds to the inverted repeat at either end of the transposon facing outward, in combination with each of the two primers used to create the 2.5-kb target amplicon (see Table S1 in the supplemental material). A single transformant with the transposon in the desired location within the gene of interest was selected. The exact location and orientation of each insertion were subsequently determined by amplifying each locus using the same primers used to create the original amplicon and then performing sequencing reactions with transposon-specific primers M110L14 and M1163U18 (Table 2) (11). Insertion locations are indicated in Table S1 in the supplemental material and Fig. 1. Differently tagged mutants were assembled into pools of 40 and 17 mutants each.
TABLE 2.
Primers used in this study
| Primer | Sequencea |
|---|---|
| SsaBF1 | TAAAATTTGATGGAGGACTCCC |
| SsaBR1 | ATCCACTAGTTCTAGAGCGGCCTAACCTTTTATTATTATT |
| AscI-SsaB-prom | AGTCGAGGCGCGCCGTTTTTTCAAACATTTCTTATATTAATTTTAGAAATTTGTTAAAAATTAACTTGACTTAATTTTTATTTTATAAAATTTGATGGAGGACTC |
| Spc-F1 | AATAATAATAAAAGGTTAGGCCGCTC TAGAACTAGTGGAT |
| NcoI-Spc-R1 | TCTCGACCATGGCAATTTTTTTATAATTTTTTTAATCTG |
| EcoRISsaBF1 | AGTCCAGAATTCACCAAACCAACCTTGGTCCCGT |
| EcoR1SpcR1 | TCTCGAGAATTCCAATTTTTTTATAATTTTTTTAATCTG |
| 1U20Lp33 | TTAAAGCTGGTGCCATGATG |
| 797L41Lp33 | CATTAAAAATCAAACGGGCCCTTGACAGCCGCCAGAGAGAA |
| 1163U55Lp33 | TTACTGGATGAATTGTTTTAGTACCTAGAATTCGGGAAAAATCTGGAAGTGATTG |
| 2678L19Lp33 | GAAGCGGAATTGATAACAC |
| M-out | CCGGGGACTTATCAGCCAACC |
| M110L14 | AGCCCGGGAATCAT |
| M1163U18 | CCGTTAGTTGAAGAAGGT |
| SsX_0753_F1 | GTTTTCCAATATTCGACAGGATTTT |
| SsX_0753_R1 | GCCATTTATTCCTCCTAGTTAGTCACTTCATTTTATATTGACTCCTTATTC |
| SsX_0753_F3 | GTTTTAGTACCTGGAGGGAATAATGAAAGAAGCTTCATCCTCAGCCG |
| SsX_0753_R3 | AAATTGCCTAGCATTTCAAACATAG |
| SsX_0941_F1 | ATTTTGATTTGATTGTGCTAGATGC |
| SsX_0941_R1 | GCCATTTATTCCTCCTAGTTAGTCATGTCATAGATTCACCACGGTCA |
| SsX_0941_F3 | GTTTTAGTACCTGGAGGGAATAATGAATGTCACAGCAATAGAAGGAG |
| SsX_0941_R3 | GTAAGAAGAGGATTTCCCTGTCAAG |
| SsX_1667_F1 | ACTGGAAGTCTGCTGTTACATCTTC |
| SsX_1667_R1 | GTTTTAGTACCTGGAGGGAATAATGGCAACTTCCAGTGCGTCACAGA |
| SsX_1667_F3 | GCCATTTATTCCTCCTAGTTAGTCAAGACATACGAGTCCTCCATTTA |
| SsX_1667_R3 | TCGAAGGAGAAAATAGCTGAGATTA |
Underlined amino acids denote restriction enzyme recognition sites.
FIG. 1.

Predicted S. sanguinis SK36 lipoprotein and neighboring ABC transport component genes. The number (SSA-) of each gene is indicated. Adjacent genes are connected by black lines. Nonlipoprotein ABC transport genes are depicted only when separated from a lipoprotein gene and from one another by no more than a single non-ABC component gene. Predicted component classifications are indicated by symbol shape; COG categories are indicated by color. Orientation and relative positions of transposon insertions are indicated by flags above the gene symbols; allelic exchanges are indicated by orange boxes below the symbols. Genes and intergenic regions are not drawn to scale. ATPB, ATP-binding.
Screening of mutants in a rabbit model of endocarditis.
Signature-tagged mutants were screened for virulence in a rabbit model of infective endocarditis, exactly as described previously (53). Selected strains were also examined in the rabbit model via a CI assay, as described previously (53), with the following modifications. First, mutant strains were competed against derivatives of SK36 that were resistant to Em (JFP36) or Tet (JFP76) rather than against SK36. Second, competing strains were grown separately rather than together for 3 hours prior to inoculation. Third, the layer plating technique described previously (53) employed BHI medium rather than tryptic soy broth. Antibiotic concentrations in the top layer were 10 μg/ml (Cm or Tet) or 20 μg/ml (Em). The experiments received IACUC approval and complied with all applicable federal guidelines and institutional policies.
Creation of allelic exchange mutants.
The SSA_0588k mutant was created by overlap extension PCR (30). A Kan cassette was PCR amplified using primers DAM 303 and DAM 347 (64). Primers 1U20Lp33 and 796L41Lp33 (Table 2) were used to amplify the first 45 bp of SSA_0588 along with 768 bp of upstream DNA. Primers1163U55Lp33 and 2678L19Lp33 were used to amplify the last 193 bp of the same gene along with 806 bp of downstream DNA. Both internal primers had 5′ tails complementary to the Kan cassette. All products were purified with MinElute PCR purification columns (Qiagen Inc., Valencia, CA). The final fused PCR product was used to transform SK36, selecting for Kan resistance. A transformant with the expected DNA sequence was selected for further study. A similar homologous recombination method was used for allelic exchange mutagenesis of SSA_0941, SSA_0753, and SSA_1667 (X. Ge and P. Xu, unpublished data). The primers employed are listed in Table 2.
Creation of constructs for complementation of ssaB.
Overlap extension PCR was used to create a tripartite fusion of the ssaA promoter region, the ssaB gene, and a Spc cassette. The ssaB gene was PCR amplified from SK36 genomic DNA using primers SsaBF1 and SsaBR1 (Table 2). The purified product was fused with a 71-bp sequence upstream from the ssaA gene using primers AscI-ssaB-prom and SsaBR1 in a second PCR. A Spc cassette was PCR amplified from the plasmid pR412 (45) using the primers Spc-F1 and NcoI-Spc-R1. The purified Spc cassette was fused to the ssaA-ssaB fusion sequence using the AscI-SsaB-prom and NcoI-Spc primers. This product was ligated into the SSA_0169 gene contained in the insertion vector pJFP46 (68a). The plasmid isolated from a transformant, called pJFP57, was confirmed to have the expected sequence. Transformation of the ssaB (SSA_0260) mutant with pJFP57 created strain JFP57, possessing the ssaA promoter-ssaB gene-Spc cassette fusion within the chromosomal SSA_0169 locus. This locus was amplified from the chromosome of JFP57 with primers SsaB-Spc-F EcoRI and Spc-EcoRI. The purified product was ligated into the EcoRI site of the shuttle vector pVA838 (43). The purified plasmid pJFP58 was confirmed to have the correct sequence and desired insert orientation before introduction into the ssaB mutant to create strain JFP58.
In vitro growth studies.
Competitive growth of selected strains was measured using in vitro CI assays. JFP36 and a competitor strain were grown in BHI medium and harvested as in the in vivo CI assay described above. Equal volumes of strains at the same optical density (OD) were combined, and 10 μl of the mixed culture was diluted into 10 ml of preincubated BHI medium for growth at 37°C for 20 h. The inoculum and 20-h cultures were serially diluted and subjected to layer plating with selective antibiotics, as described earlier.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting.
Cultures were grown overnight with the appropriate antibiotics, diluted tenfold, and grown for 4 h to exponential phase. Cultures were centrifuged, washed with phosphate-buffered saline, resuspended in TEP buffer (10 mM Tris, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride; pH 7.5), and transferred to a tube containing an equal volume of Lysing Matrix B (MP Biomedicals, Solon, OH). Cells were lysed in a bead beater (FastPrep FP120; MP Biomedicals). Protein concentrations of supernatants were determined using the BCA protein assay kit (Pierce, Rockford, IL). Samples were mixed with sample buffer, heated for 10 min at 70°C, and electrophoresed on a 12% polyacrylamide gel, along with 5 ml of kaleidoscope marker (Bio-Rad), and transferred to a nitrocellulose membrane by electroblotting. The membrane was blocked with 5% nonfat dry milk in TBST (10 mM Tris-HCl [pH 7.4], 100 mM NaCl, and 0.1% Tween 20) for 1 h and then incubated with rabbit polyclonal anti-FimA antibody at a dilution of 1:4,000 for 2 h before being washed and incubated with anti-rabbit immunoglobulin G horseradish peroxidase-conjugated secondary antibody (Amersham Biosciences, Pittsburgh, PA) (1:5,000 dilution) for 1 h at room temperature. The polyclonal antiserum was generated by the vaccination of rabbits with recombinant FimA protein, as described previously (70). Blots were developed using a chemiluminescence system (ECL Plus; Amersham Biosciences) and exposed to X-ray film (Blue Devil Film, San Diego, CA). Band intensities were quantified using a FluorChem imager.
Labeling of lipoproteins with [3H]palmitate.
S. sanguinis lipoproteins were labeled with [3H]palmitate, essentially as described by McNab and Jenkinson (47), except that BHI medium was used in place of BHI medium containing 0.5% yeast extract. After electrophoresis of lysates, the gel was fixed with 40% methanol, 10% glycerol, and 7.5% acetic acid for 1 h and then incubated in 10 volumes of Amplify (GE Healthcare, Piscataway, NJ) for 45 min with gentle shaking. The fluor-impregnated gel was vacuum dried onto Whatman 3MM filter paper at 60 to 70°C and exposed to X-ray film for up to 7 days at −80°C.
Statistics.
In vivo and in vitro CI values were calculated as the ratio of mutant strain colony numbers/virulent strain colony numbers in the output sample divided by the mutant/virulent strain ratio of the inoculum. Geometric mean CI values were calculated from log-transformed individual CI values. Mutant strains were determined to be significantly more or less competitive than their virulent competitors by comparing the log-transformed CI values to 0 (log1) in paired Student's t test, with α of 0.05. Log-transformed CI values from different strains or from the same strain assessed in vitro and in vivo were compared by unpaired t test with Welch correction for unequal standard deviations, with α of 0.05.
RESULTS
Identification of putative lipoprotein-related genes in S. sanguinis.
The genome sequence of S. sanguinis strain SK36 (72) was examined for putative lipoprotein genes, as described in Materials and Methods. Use of the LipoP web server (35) resulted in the identification of 59 putative lipoprotein ORFs. This number was greater than that predicted by the same method for several other streptococcal genomes, including Streptococcus mutans UA159, with 29 predicted lipoproteins; Streptococcus pyogenes M1, with 30 predicted lipoproteins; and S. pneumoniae strains D39, R6, and TIGR4, with 43, 39, and 40 predicted lipoproteins, respectively. Streptococcus gordonii strain CH1, however, had a greater number, with 63 predicted lipoproteins. Although the hidden Markov model employed by the LipoP server (35) was developed for gram-negative bacteria, the results of the S. sanguinis analysis were similar to those obtained from a regular-expression pattern search created for gram-positive bacterial lipoproteins. The modified pattern identified 52 of the ORFs identified in the LipoP analysis and none that were not identified by LipoP. The seven ORFs identified solely by the LipoP analysis were examined further. Five of these (SSA_0493, -1038, -1315, -1340, and -2143) possessed only one mismatch to the pattern and had at least one ortholog in the NCBI nonredundant protein sequence database (with an E value of <10−5 by BLASTp) that was also classified as a lipoprotein by both LipoP and the pattern search. These ORFs were therefore retained. The sixth ORF, SSA_0245, possessed no orthologs in the NCBI database, with an E value of less than 10−5. It was paralogous to the ORFs encoded by its flanking genes, SSA_0244 and SSA_0246, neither of which was recognized as encoding a lipoprotein by either algorithm. This ORF was therefore not retained. The seventh ORF, encoded by SSA_2192, possessed no orthologs in NCBI and had a predicted signal sequence that was 49 amino acids (aa) in length compared to lengths of 17 to 29 aa for the remaining candidates. This ORF was also rejected, leaving 57 candidates.
We next considered the possibility that some of the ORFs identified by both analyses might have matched by chance. As one test of this possibility, the remaining candidates were used as query sequences in BLASTp searches of the NCBI database, as described above, to identify candidates that either had no orthologs or had orthologs that were not annotated as lipoproteins. Six candidates were dropped from the list based on this analysis, as follows: SSA_0384, -0850, -1116, -1792, -1860, and -2139.
Finally, we considered the possibility that some potential lipoproteins may have been missed due to mistakes in the genomic sequence or to selection of an incorrect start codon in ORFs possessing more than one potential start site. A number of genes were examined that had homology to proteins annotated as lipoproteins or as substrate-binding proteins in the NCBI database. SSA_0941 was the first gene in an apparent five-gene operon putatively encoding an ABC-type phosphate transport system and had homology to the substrate-binding component. An in-frame potential TTG start site was discovered 32 aa upstream from the annotated ATG start site. If that site were used, the ORF would have the same start site as its 10 closest relatives in the NCBI protein database and would be identified as a lipoprotein by both LipoP and the pattern analysis applied above. SSA_0941 was therefore added to the list of putative lipoproteins, and the modified start site was added to the GenBank record. This left the final count of candidate lipoproteins at 52. Features of these proteins are listed in Table 3.
TABLE 3.
Putative S. sanguinis lipoproteins
| S. sanguinis locus tag | Signal sequence | No. of aaa | Name or description(s) |
|---|---|---|---|
| SSA_0004 | MKKNTFKTLFLSFLAVSALFVLAAC | 163 | Lipoprotein |
| SSA_0074 | MKNLKKFLFVLLTGLVALIISGC | 448 | ABC transporter substrate-binding protein, sugar transport |
| SSA_0138 | MKKISLLLAGLLSIFLVAC | 500 | Metal-binding (Zn) lipoprotein |
| SSA_0142 | MKKKALPFLLAGAALLAMTAC | 59 | Hypothetical protein |
| SSA_0260 | MKKLGFLSLLLLAVCTLFAC | 309 | SsaB |
| SSA_0375 | MMNLKKIFSVGLVGLAALGLAAC | 290 | Lipoprotein transporter |
| SSA_0493 | MKHFKKIMALALAGLALASC | 511 | ABC-type dipeptide/nickel transport system, periplasmic component |
| SSA_0508 | MRTKGIVLLCSLALLLGAC | 438 | Conserved uncharacterized protein, possible phosphoserine phosphatase |
| SSA_0584 | MKIQKITFMALTALLTLSLGAC | 267 | Hypothetical protein |
| SSA_0588 | MNLKKTLKYFSLAAVSVLAIGALVAC | 275 | Amino acid ABC transporter, substrate-binding component |
| SSA_0750 | MKKIFVIFLTIITLSSAMFLGGC | 128 | Hypothetical protein |
| SSA_0753 | MKKKIFAGAVTLLSVAVLAAC | 335 | Foldase protein PrsA |
| SSA_0892 | MNVKKIAMLVALIGISGFFLAAC | 90 | ATP-binding cassette lipoprotein |
| SSA_0893 | MFRRKKMNRKTILLGSTVILAC | 421 | ATP-binding cassette transporter-like protein |
| SSA_0918 | MTITKKAFIGILSLTAAVLLAAC | 155 | Conserved uncharacterized protein |
| SSA_0941 | MKKKQKLGIAALFLLSSMALSGC | 261 | ABC-type phosphate transport system, periplasmic component |
| SSA_1003 | MKWYKKMSLAAITGLSLLGLSAC | 419 | ABC transporter substrate-binding protein, multiple sugars |
| SSA_1038 | MNKKQWLGLGLVTVAAFGLAAC | 351 | Lipoprotein |
| SSA_1066 | MKASKWLIATGVALSAGLLLTAC | 653 | ABC-type oligopeptide transport system |
| SSA_1117 | MKKLSILTVSLLCIGLLGAC | 187 | Hypothetical protein |
| SSA_1122 | MKKFVTLLATVSAAVFLAAC | 191 | Thioredoxin family protein |
| SSA_1129 | MKKLGIVLFSTAILLTGC | 290 | Periplasmic iron transport lipoprotein |
| SSA_1198 | MMKKKHLLITLLSIALLTLSGC | 203 | ATPase (PilT family) |
| SSA_1277 | MKYKKLLLLSVAVFSLSAC | 246 | d-Alanyl-d-alanine carboxypeptidase |
| SSA_1298 | MKTWKKVVLGSVSLLAAGTLLAAC | 418 | Maltose/maltodextrin ABC transporter, sugar-binding protein MalX |
| SSA_1315 | MTMKKKYFLLSLTILAGLTLSSC | 214 | Hypothetical protein |
| SSA_1340 | MKRALKFSACLSLLALALCLWAC | 312 | Zn/Mn ABC-type porter lipoprotein |
| SSA_1382 | MKRHLFFLFMGVSLVLLAAC | 157 | Hypothetical protein |
| SSA_1454 | MKKYSKIIMSCACLAMALGLAAC | 281 | Hypothetical protein |
| SSA_1474 | MKKLCALAILLLAGTILAGC | 224 | Lipoprotein |
| SSA_1570 | MKKLTLLFITFLTLIFLSAC | 132 | Hypothetical protein |
| SSA_1581 | MKKTLSILLVATAALTMAAC | 342 | Metal-binding ABC transporter |
| SSA_1629 | MKKILLISLLSGLVLAGC | 272 | Peptidyl-prolyl cis-trans isomerase, cyclophilin type |
| SSA_1667 | MSRFKNIGSLLLALISVFILTAC | 61 | Hypothetical protein |
| SSA_1689 | MKKIFSASVALLATVTLAAC | 186 | Hypothetical protein |
| SSA_1729 | MKKKFALSLVAFASAALLAAC | 386 | ABC transporter substrate-binding protein, branched chain amino acid transport |
| SSA_1742 | MKKFLSFATLTVTIFLLVAC | 311 | Ferrichrome-binding protein |
| SSA_1948 | MKQSKVLALAGVTLLAAGFLAAC | 661 | Oligopeptide-binding lipoprotein precursor |
| SSA_1949 | MKTSKLAAAAGLVILSAGILAAC | 656 | AliA protein |
| SSA_1950 | MLMKKSKWLAITGLTVVSALILAAC | 653 | ABC-type oligopeptide transport system, periplasmic component |
| SSA_1976 | MKKFFILVISASVLLTGC | 202 | Conserved uncharacterized protein |
| SSA_1990 | MKKRTAVLLMLSILALMLGAC | 305 | Zn porter lipoprotein |
| SSA_2067 | MKKILLASACLLTLTAC | 180 | Hypothetical protein |
| SSA_2143 | MKKWIIILVGGLVALLVLVTC | 211 | Conserved uncharacterized protein |
| SSA_2165 | MRKSNILLAAGLTLLSVGLLTAC | 657 | ABC-type oligopeptide transporter, periplasmic component |
| SSA_2196 | MKKIFSLLTLAFALLLVGC | 133 | Conserved uncharacterized protein |
| SSA_2204 | MKKVSFYALSLLSLLALSAC | 254 | Hypothetical protein |
| SSA_2235 | MAISKKLIFSGLTAAAVLALAAC | 159 | Conserved uncharacterized protein |
| SSA_2242 | MRKLIVFLLAALAILTAC | 180 | Hypothetical protein |
| SSA_2243 | MKKILVGLGLSVALLAGC | 190 | Conserved uncharacterized protein |
| SSA_2327 | MKKLSKISLFLLPLIALFFLAAC | 131 | Hypothetical protein |
| SSA_2352 | MMKKSYKVLLAGLAAVSIIGLAAC | 333 | ABC-type nitrate/sulfonate/bicarbonate transporter, periplasmic component |
The length of the prolipoprotein is reported using amino acids.
The genome was next examined for genes encoding the lipoprotein-processing enzymes lipoprotein signal peptidase (LspA) and prolipoprotein diacylglyceryl transferase (Lgt). Both BLAST and hidden Markov model analyses of the S. sanguinis genome (see Materials and Methods) identified a single candidate for each ORF, SSA_1069 for LspA and SSA_1546 for Lgt.
Properties of putative S. sanguinis lipoproteins.
Predicted signal sequences of the 52 putative lipoproteins ranged in size from 17 to 26 aa in length (Table 3). The total protein length ranged from 59 to 661 aa, with the smallest proteins annotated as hypothetical proteins and the largest as oligopeptide transporters. The most common function evident in Table 3 is transport, as could be expected for proteins that are often components of ABC transport systems. As another indication of putative function, the clusters of orthologous group (COG) category (67) for each putative lipoprotein was extracted from the published genome annotation (72). In addition, whenever other apparent ABC transport system component genes were separated by no more than one gene from a putative lipoprotein gene or one another, the COG classifications of these genes were also extracted. The result is shown in Fig. 1. In agreement with expectations, most of the lipoproteins annotated as transporters (16 of 21) were adjacent to other apparent ABC transport component genes annotated with the same transport function. Four of the five remaining genes were not located in apparent transport operons, and in only one case (SSA_1990) was the putative lipoprotein assigned to a COG category different from that of its neighboring genes. Also, in agreement with expectations, the four proteins assigned to the category of posttranslational modification were not located within ABC transport operons. Almost half of the lipoproteins (25 of 52) were not assigned to any COG category.
Examination of the contribution of lipoprotein-related genes to endocarditis virulence by STM.
To determine the role of lipoproteins in virulence for endocarditis, we sought to systematically mutagenize each of the 52 genes identified as encoding putative lipoproteins. As described in Materials and Methods, each gene was mutagenized by in vitro transposition with one of 40 magellan2-derived minitransposons (26), each containing a Cm resistance gene and a unique 40-bp signature tag (53). Mutants were successfully created for 51 of the 52 genes. As described below, the final gene (SSA_0941) was successfully mutagenized by allelic exchange, indicating that none of the genes were essential. An insertion was also created in SSA_0499, encoding a predicted protein with homology to peptide transport lipoproteins in other bacteria and which is located within an apparent peptide transport operon (Fig. 1). The predicted protein lacks an N-terminal cysteine; however, introduction of a single A to a run of seven A's in the upstream nucleotide sequence would have created an ORF predicted to be a lipoprotein by the LipoP software. Additional sequencing reactions confirmed that the published sequence was correct, indicating that SSA_0499 does not encode a conventional lipoprotein. Nevertheless, the mutant was retained for virulence testing. Finally, signature-tagged transposons were also introduced into the putative lspA and lgt genes SSA_1069 and SSA_1546. As with the lipoprotein mutants, the successful creation of these mutants indicated that neither enzyme was required for growth in BHI medium.
We next wanted to assess the ability of each of the insertion mutants to cause endocarditis in the rabbit model (17). Given the relative complexity of the model and the large number of mutants, it was desirable to examine the mutants in pools rather than individually. The STM technique allows for such testing (28) and has been used previously by us with the same transposons and the same animal model to test pools of randomly generated mutants of the same S. sanguinis strain (53). With this technique, the signature tags from the pooled inoculum and from vegetation-derived cells are amplified and labeled. Any tag that produces a strong signal in a dot blot probed with the inoculum probe but a weak signal with the vegetation-derived probe indicates a strain with a potential defect in virulence (28, 53).
Forty mutants, each with a different signature tag, were assembled into a pool and screened for virulence reduction by STM. The results from one study performed with two rabbits are shown in Fig. 2, top. Strains with mutations in two genes, S. sanguinis adhesin B (ssaB) (SSA_0260) and lspA (SSA_1069), were detected in the inoculum but were essentially undetectable in the population recovered ∼20 h postinoculation from the infected heart of either rabbit. SsaB is an LraI family lipoprotein (22, 32), homologs of which have been shown to be important for endocarditis virulence in other viridans group streptococci (7, 52). The lspA gene (SSA_1069) encodes the lipoprotein-processing enzyme signal peptidase II. Four additional rabbits from two additional experiments also exhibited complete (ssaB) or partial (lspA) loss of detection for both mutants (data not shown).
FIG. 2.

Virulence assessment of lipoprotein mutants by STM. Hybridization signals from probes derived from the inoculum pool or from vegetations recovered from infected rabbits are shown on the left. The number (SSA-) or name of the mutated gene corresponding to the signature tag used as a hybridization target for each spot is indicated in the keys on the right. Mutants associated with weak signals in all the vegetation blots shown are indicated with solid lines surrounding the signals on the blots and the mutant designations in the keys. Gray outlines of designations in the keys indicate mutants that produced weak vegetation signals in experiments not shown here. Dashed outlines of designations in the keys highlight two mutants possessing independently derived, differently tagged transposon insertions within the SSA_1382 gene. Neg., negative control target derived from a signature-tagged transposon not used for mutant creation. (Top) Pool 1 experiment, testing 40 mutants; (bottom) pool 2 experiment, testing 17 mutants.
The remaining 14 lipoprotein mutants and the lgt mutant were assembled into a second pool for STM screening (pool 2) (Fig. 2). Strains with mutations in SSA_0004, SSA_1546, and SSA_1976 produced weak signals from all three rabbits. These genes are annotated as lipoprotein, lgt, and conserved uncharacterized protein, respectively. Three additional rabbits were also tested, and all three mutants produced signals that were weak but stronger than that of the negative control (data not shown). Two additional strains were also included in pool 2. The SSA_0588 mutant, which showed poor detection in rabbit 2 (Fig. 2) and in all four additional rabbits tested with pool 1 (not shown), was included in pool 2 for additional testing. This mutant produced weak signals for all three rabbits shown. It was also observed that the SSA_1382 mutant in pool 1, which produced a weak signal from rabbit 2 (Fig. 2), also produced a weak signal in both inoculum and recovery blots from four additional rabbits tested (data not shown). This suggested that there might be a technical problem with tag 15, which was used to create this mutant. Therefore, a new signature-tagged transposon insertion was created for this gene, and the mutant was included in pool 2. Changing the signature tag in this mutant gave rise to strong signals in the inoculum and recovery pools (Fig. 2 and data not shown). Finally, examination of the results from the rabbits not shown in Fig. 2 identified possible virulence defects for the SSA_1122 mutant in half of the rabbits receiving pool 1 and for the SSA_2243 mutant in one-third of rabbits tested.
Additional virulence testing.
The STM analysis identified one mutant, ssaB, with a signal intensity equivalent to that of the negative control in all animals tested and seven additional mutants (SSA_0004, -0588, -1122, -1976, -2243, lgt, and lspA) that showed reduced signal intensity in some of the animals tested. We sought to quantitate potential virulence defects in these strains in CI assays (69) in the rabbit endocarditis model (24, 53). For this assay, a mutant and an Em-resistant derivative of SK36, JFP36, which possesses wild-type virulence (68a), were coinoculated into catheterized rabbits, as in the STM assay. Serial dilutions of the inocula and the vegetation homogenates were incorporated into plates containing antibiotics selective for each strain. The CI was calculated as the ratio of the mutant to the wild-type strain in the recovered population divided by the ratio of the mutant to the wild-type strain in the inoculum (69). The results of this analysis are shown in Fig. 3A. Surprisingly, the strains with mutations in SSA_1122 and SSA_0004 possessed mean CI values slightly greater than 1, suggesting that these strains were slightly more competitive than the virulent competitor strain JFP36.
FIG. 3.
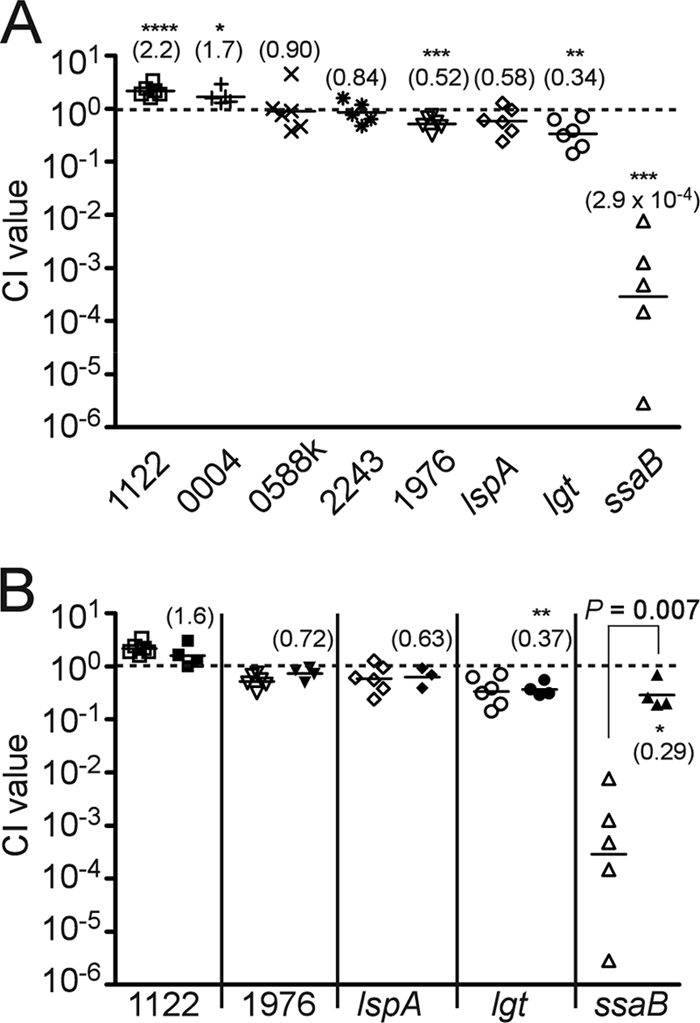
In vivo and in vitro CI analyses of selected lipoprotein mutants. The gene mutated in each test strain is indicated by the number (SSA-) or gene name at the bottom of each graph. The dashed line denotes a CI value of 1, indicating equal competitiveness. Each symbol indicates the CI value derived from a single animal or bacterial culture; solid horizontal lines indicate geometric mean values. (A) In vivo CI analysis. Mean CI values from five to six animals tested in two separate experiments each are indicated in parentheses. Those values that were significantly different from 1 are indicated, as follows: *, P of <0.05; **, P of <0.01; ***, P of <0.005; ****, P of <0.001. (B) In vivo CI analysis versus in vitro CI analysis. Open symbols, in vivo CI values taken from values shown Fig. 3A; closed symbols, in vitro CI values obtained from three to four separate cultures. In vitro CI values that were significantly different from 1 are indicated as in panel A.
In preparation for CI testing, it was observed that the SSA_0588 STM mutant was somewhat diminished during growth in broth culture and highly attenuated during growth on agar plates, producing fewer and smaller colonies than other strains, even with extended incubation (data not shown). If the mutant was incubated anaerobically rather than in reduced oxygen, colony sizes were normal. We have observed previously, however, that anaerobic incubation is incompatible with Cm selection in S. sanguinis STM mutants (53). We therefore inserted a Kan resistance cassette into the SSA_0588 gene by allelic exchange, as described in Materials and Methods. This mutant (SSA_0588k) also produced small colonies in 6% O2 but produced colonies typical of SK36 derivatives under anaerobic conditions in the presence or absence of Kan. As shown in Fig. 3A, the mean CI value for the SSA_0588k mutant was 0.90, which is not significantly different from 1.
Strains with mutations in SSA_2243 and SSA_1976 had CI values slightly less than 1, although the CI value for the former mutant was not significantly different from 1. The lspA and lgt mutants also exhibited modest reductions in competitiveness, with geometric mean CI values of 0.58 and 0.34, respectively (Fig. 3A). In contrast to the other tested mutants, the ssaB mutant was severely attenuated in this model, with a mean CI value of 2.9 × 10−4.
We next considered whether the failure to find more mutants with virulence deficits might be due to transposon insertions occurring near the 3′ ends of genes, perhaps allowing for retention of activity by truncated proteins. DNA sequence analysis was used to determine the exact location of each insertion. The results are indicated in Fig. 1 and also in Table S1 in the supplemental material. Insertions occurred within the 5′ half of 34 of the 55 genes examined. All but three of the remaining mutants possessed insertions within the 5′ two-thirds of the gene. Insertions were located in the last third of the SSA_0753 and SSA_1667 coding sequences and upstream from SSA_0941. These three genes were mutagenized by allelic exchange, replacing most of each gene's coding sequence with a Kan cassette (Fig. 1; see also Table S1 in the supplemental material). CI assays were performed as described above. The mean CI value for the SSA_0753 mutant was 1.5 (range, 1.3 to 1.8; n = 4), which was slightly, though significantly, higher than 1 (P = 0.02). The SSA_1667 mutant produced a similar result (CI value, 1.5; range, 1.4 to 1.6; n = 3; P = 0.03). The SSA_0941 mutant exhibited a mean CI value of 0.07 (range, 0.06 to 0.08; n = 3), which was significantly less than 1 (P = 0.002). We noticed, however, that although the SSA_0941 mutant reached essentially the same OD in BHI broth as JFP36 (1.025 versus 1.044, respectively) and the two cultures were combined at a 1:1 dilution ratio for inoculation, plating of the inoculum yielded an SSA_0941/JFP36 ratio of 1:21. A repeat growth and plating experiment yielded almost identical results. The results were the same when cultures and plates were incubated anaerobically. Thus, while the SSA_0941 mutant exhibits reduced competitiveness in vivo, it also displays reduced viability in vitro.
Examination of growth in broth culture.
We next wanted to determine whether the CI values obtained from the in vivo analyses of the other mutants merely reflected increased or decreased growth rates of mutants relative to the growth rate of the wild-type competitor strain. Strains were inoculated into individual wells of a microtiter plate and monitored for growth at 37°C using OD. All strains produced similar growth curves and final ODs within ±15% of that of SK36, except for the ssaB and SSA_0588 STM mutants, both of which reached final ODs that were two-thirds of that of SK36 (data not shown). As a more sensitive test of growth rate differences and to measure viability, an in vitro CI assay was performed for five of the mutants tested in Fig. 3A. For this assay, a mutant and virulent strain were prepared as for an in vivo CI assay and then coinoculated into BHI broth for a 20-h incubation. The ratio of mutants to virulent control strains in the inocula and in 20-h cultures was determined by dilution plating, as used for the in vivo CI assays. Figure 3B shows these results in comparison with the in vivo CI results obtained above. The in vitro CI values for the lgt and ssaB mutants were significantly less than 1, indicating that these mutations had a modest negative impact on competitiveness in broth culture. The in vitro CI values of the other mutants were not significantly different from 1. There was no significant difference between the in vitro and in vivo CI values for SSA_1122, SSA_1976, lspA, or lgt. In contrast, the in vivo mean CI value for the ssaB mutant was 1,000-fold lower than the in vitro CI value (2.9 × 10−4 versus 0.29, respectively), a difference that was statistically significant (P = 0.007).
Examination of lgt and lspA mutant phenotypes.
Given the finding that a mutation in a single lipoprotein gene (ssaB) produced a far more profound defect in virulence than mutation of the lgt or lspA gene, encoding an enzyme thought to be necessary for processing of all cellular lipoproteins, we asked whether the lgt and lspA mutants had the expected phenotypes. Figure 4A shows the results of an experiment in which cells were cultured with [3H]palmitate to label lipoproteins, and the lysates separated by SDS-PAGE. At least 10 proteins ranging in apparent size from 27 to 89 kDa are distinguishable in the SK36 lane. As expected, mutation of the lgt gene resulted in a complete loss of protein labeling, indicating a lack of acylation (40). The lspA mutant exhibited a pattern that was very similar to that of the wild-type parent strain SK36, except that the labeled proteins appeared slightly larger. This would be the expected result if signal peptide cleavage of lipoproteins was eliminated by lspA mutation. Finally, the ssaB mutant was also examined in this experiment. To obtain a clearer picture of SsaB processing, Western blotting was performed with the same strains. Figure 4B indicates that a single band was detected in SK36 using an antiserum raised against the SsaB ortholog from Streptococcus parasanguinis, FimA (7, 70). This band was missing in the ssaB mutant, confirming its identity. The lgt mutant exhibited a single band of low abundance, with apparent mobility that was ∼1.5 kDa larger than that of SsaB from SK36. This is consistent with the expected difference in size between a fully processed (acylated and cleaved) molecule of SsaB and its uncleaved, unacylated precursor. The lspA mutant exhibited two bands, migrating with apparent molecular masses 0.8 and 2.5 kDa larger than that of the band in SK36. The size of the larger, more abundant band is consistent with that expected of acylated SsaB retaining the signal peptide. The less abundant band will be discussed below. The combined results suggest that the lgt and lspA mutants had the expected phenotypes in relation to global lipoprotein processing and that the SsaB protein was not processed normally in either mutant.
FIG. 4.
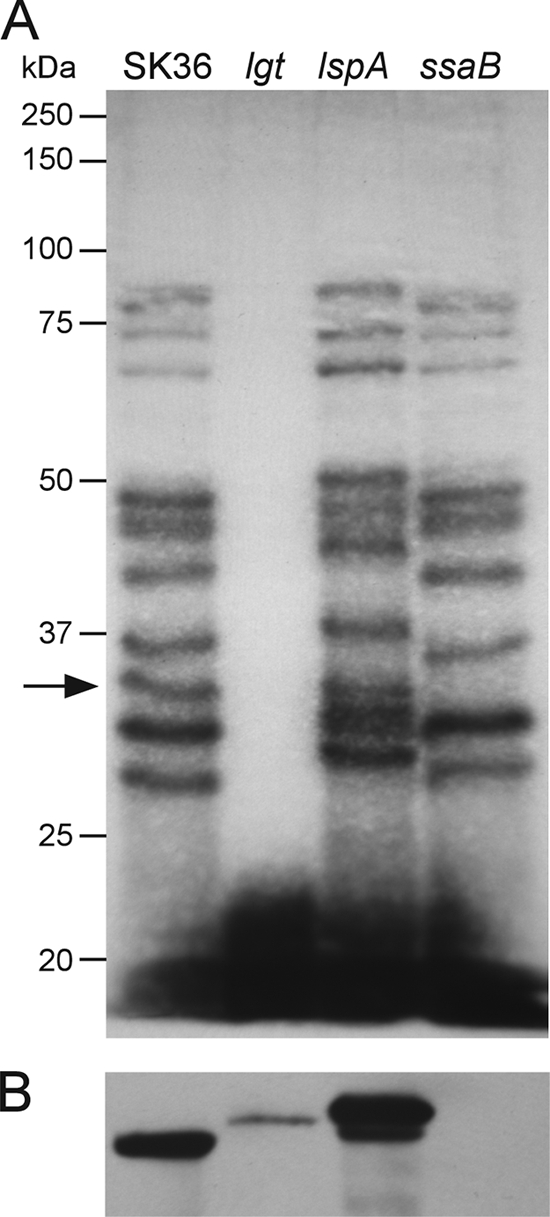
Lipoprotein processing in S. sanguinis.
(A) [3H]palmitate labeling of SK36 and selected isogenic mutants. The migration of molecular mass standards is indicated to the left. Arrow, 33-kDa band present in SK36—presumably SsaB, which is approximately 33 kDa in its mature form— but not in the ssaB mutant. (B) Western blot analysis of the same strains shown in panel A using an antiserum reactive against SsaB.
Complementation of the ssaB mutant.
Another possible explanation for the dramatic effect of the ssaB mutation on virulence, in combination with the minor effect of lgt or lspA mutation, is that the loss of ssaB is not responsible for the virulence defect in this mutant. Instead, the mutation could have created a polar effect on expression of a downstream nonlipoprotein virulence factor, or the strain could have some other unintended mutation. Complementation of the ssaB mutant was therefore attempted. The ssaB gene is preceded upstream by the ssaA and ssaC genes encoding putative ATP-binding and permease components of a metal transport system, respectively, and is followed downstream by the putative thiol peroxidase gene tpx (72) (Fig. 5). Orthologs of these genes appear in the same arrangement in S. pneumoniae (49, 58), S. parasanguinis (20), and S. gordonii (2). In each case, the three transport component genes have been shown to be cotranscribed from a promoter upstream from the first component, with the tpx gene transcribed from this same promoter as well as from a separate gene-specific promoter (2, 20, 31, 49, 50, 58). Therefore, overlap extension PCR was used to fuse the 71-bp 5′-end segment from the ssaA gene to the ssaB gene and then to fuse this fragment to the aad9 gene encoding Spc resistance. This fusion cassette was cloned into the suicide plasmid pJFP46, bearing the SSA_0169 locus (68a). This allowed for insertion of the promoter-ssaB-aad9 fusion into the ectopic SSA_0169 locus, which is the same locus into which the ermB gene is inserted in the competitor strain JFP36 (68a). Introduction of this plasmid, pJFP57, into the ssaB mutant created strain JFP57, possessing the mutant ssaB gene and a wild-type copy integrated by double-crossover recombination elsewhere on the chromosome. (Fig. 5).
FIG. 5.

Map of the ssaB locus and complementation constructs. Symbols: open horizontal arrows, genes; filled horizontal arrows, putative ssaACB promoters; flags, transposon insertion sites in the ssaB mutant. See Materials and Methods and Results for further details.
The results from a trial CI experiment in which JFP57 was coinoculated with the Em-resistant, virulent strain JFP36 are shown in Fig. 6A. The mean CI value of 0.019 was greater than that of the ssaB mutant but less than that of JFP36, although too few animals were used to allow for statistical analysis. To measure expression of SsaB in strain JFP57, Western blotting was performed using anti-FimA antiserum. Dilutions of SK36 lysate were used to create a standard curve for the determination of SsaB expression levels in JFP57. As shown in Fig. 6B, the level of SsaB expression in JFP57 was less than that in SK36 (approximately 20 to 25% of that of the wild type, as determined by densitometry of this blot and replicate blots), suggesting a possible reason for the incomplete complementation of this mutant. The ssaB fusion cassette from JFP57 was next cloned into the streptococcal shuttle vector pVA838 (43) to create pJFP58. This plasmid was introduced into the ssaB mutant strain to create strain JFP58. (Fig. 5) Densitometric analysis of the Western blot results shown in Fig. 6B and replicate blots suggested that SsaB expression in strain JFP58 was between 90 and 160% of that in SK36. It was not possible to test strain JFP58 with JFP36 in a CI assay because both strains were resistant to Em. Therefore, JFP58 was coinoculated with an SK36-derivative strain, JFP76, which possesses a Tet resistance gene in the same SSA_0169 locus into which the ermB gene is inserted in strain JFP36. This strain has been shown to have virulence equivalent to that of JFP36 in the rabbit model (68a). Figure 6A shows that JFP58 was approximately half as competitive as JFP76. The ssaB mutant was also compared to JFP76. The mean CI value of 1.7 × 10−5 was similar to that obtained in comparison to JFP36 (2.9 × 10−4) and was significantly less than that of JFP58, suggesting that the reduced virulence of the ssaB mutant can be accounted for almost entirely by the loss of SsaB expression.
FIG. 6.
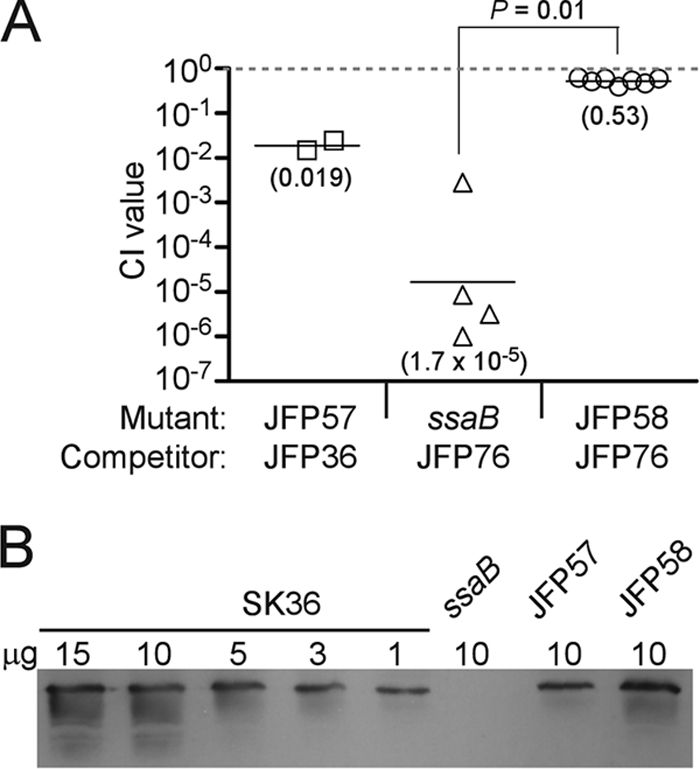
Analysis of the ssaB-complemented strains JFP57 and JFP58. (A) In vivo CI analyses of the ssaB mutant and complemented strains. Mean CI values are indicated in parentheses. Symbols are used as described in the legend to Fig. 3. JFP58 values were obtained from two separate experiments using three or four animals each, whereas the values for the other two strains were obtained from a single experiment employing two or four animals each. (B) Semiquantitative Western blot analysis of SsaB expression in JFP57 and JFP58. The indicated amount of each lysate was loaded onto an SDS-PAGE gel and analyzed by Western blotting. Replicate blots performed on different days with the same and independently derived lysates produced similar results.
DISCUSSION
So far as we are aware, this is the first study to systematically identify and mutagenize every lipoprotein gene within a bacterium and then test the effect of these mutations on virulence. We believe this is also the first study to employ STM for directed rather than random mutagenesis. STM is an efficient method for screening large numbers of mutants (69), and the directed nature of our approach also allowed us to examine essentiality. Two studies have identified a total of 246 essential genes in S. pneumoniae (61, 68). Only one of these, the SSA_1990 ortholog spr0906, appeared to be a surface lipoprotein (61). The combined results suggest that most lipoproteins are individually dispensable in rich media.
The main limitation of STM is that it provides a qualitative rather than quantitative measure of virulence. A few technical issues also arose during the course of the study. Two mutants possessing differently tagged transposons in the same gene produced noticeably different results in the STM analysis, with the SSA_1382 mutant possessing tag 15 producing poor signals in several inoculum and output blots, while the SSA_1382 mutant with tag 2 produced strong signals in all blots. Additional studies suggested this may have been due to degradation of plasmid DNA rather than an intrinsic problem with this tag (data not shown). The CI assay would not be affected by such potential artifacts, since it does not rely on hybridization for strain quantification.
It was somewhat surprising that out of eight mutants identified as potentially avirulent by the STM screen, only three were significantly less competitive when tested by CI assay, and two (SSA_0004 and SSA_1122) were significantly more competitive. Much of this discrepancy is likely due to our inclusion of mutants that would have been discarded using our previous criterion of requiring that a mutant produce signals no more intense than that of the negative control in all output blots from at least two independent experiments (53). The ssaB mutant was the only mutant to fulfill this criterion (Fig. 2 and data not shown), and its reduced competitiveness was confirmed by CI assay. In an effort to identify mutants with minor virulence deficits, we also examined mutants that produced signals that were weak but stronger than those of the negative control strain and mutants that produced undetectable signals in only some of the tested rabbits. It is likely that the weak signals were produced in some cases by the well-known bottleneck effect that can occur in STM studies (28), in which the number of cells reaching the target organ is too small to ensure representation of every virulent strain present in a complex inoculum. This causes strains of normal virulence to be lost from some animals. This likely explains results obtained with mutants such as SSA_1003 and SSA_2235, which produced weak signals in the blot from rabbit 2 but not from rabbit 1 (Fig. 2) or other rabbits tested (data not shown). In this regard, our results can be considered a validation of STM when a stringent technique for assessing mutants (53) is used.
We found strains in both this study (the lspA mutant) and the previous study (strain 16-29) (53) that never produced strong signals in any output blot and produced CI values of ∼0.6. One simple explanation for these results is that the STM analysis was highly sensitive to slight reductions in competitiveness. The SSA_0588 mutant provides another explanation. In the presence of reduced oxygen—our standard culture condition—this mutant grew to an OD of about a third less than the ODs of most of the other mutant strains in broth culture; yet, when spread on agar plates, this mutant produced tiny colonies that were barely discernible. The STM technique compares inoculum cells derived directly from liquid culture to output cells taken from agar plates. Therefore, any strain like the SSA_0588 mutant that exhibited poor plating efficiency would be expected to appear avirulent by STM. The CI assay employs plating for enumeration of both the inoculum and output bacteria, so inefficiencies in plating would be expected to cancel out, resulting in no net effect on the CI value. In any event, the combined results suggest that the mutant pool was thoroughly tested, with the CI assay serving as a complementary approach to STM analysis. It therefore seems unlikely that any truly avirulent mutant escaped detection.
Given the thoroughness of the screen, it was surprising to find only three lipoprotein mutants with significant reductions in competitiveness and only one, ssaB, with a survival defect that was much greater in vivo than in vitro. At least two previous random STM studies in S. pneumoniae identified multiple lipoprotein mutants with reduced virulence (26, 41). There are several explanations that could account for the smaller number of lipoprotein mutants identified in our study. First, the larger number of lipoproteins identified in S. sanguinis compared to that identified in S. pneumoniae (59 versus ∼40, considering only LipoP predictions) might suggest a greater degree of functional redundancy among the S. sanguinis lipoproteins. Such redundancy could allow for replacement of the function of a mutant lipoprotein gene by a second, related lipoprotein gene. It is indeed likely that functional redundancy of lipoproteins occurs in S. sanguinis. SSA_1340 has as its best match in GenBank, SSA_1990, and both are annotated as having Zn-binding activity, along with SSA_0138 (Table 3). Although SSA_1129, -1581, and -1742 are not close homologs, they are all annotated as potential transporters of iron. Three ABC-type iron transport systems have been found in S. pneumoniae. Double or triple mutants, in which two or three of these systems are disabled, display more-profound defects in virulence and metal uptake than single mutants, in which any one of the three systems is disabled (5). Nevertheless, the lipoprotein components of two of these systems were identified as virulence factors in one of these STM studies (26). Similarly, SK36 has five lipoproteins with homology to oligopeptide transporters (Table 1). Homologs of these lipoproteins, named AmiA, AliA, and AliB, have been shown to be partially redundant in S. pneumoniae (1). Yet, an amiA mutant was identified in one STM study (26) and an aliB mutant in another (41). Thus, proteins with partially redundant functions have been identified in previous STM studies.
Another possibility is that our bioinformatic strategy failed to identify some lipoproteins that are important for virulence. In this regard, SSA_0384 and SSA_1116, which were excluded as lipoproteins because they had no apparent homologs in GenBank at the time they were examined, currently have two homologs each that possess features of lipoproteins. Thus, these two proteins may also be lipoproteins. SSA_1792 and SSA_2139 were also excluded from the list of putative lipoproteins identified by the LipoP program and are questionable. These paralogous proteins belong to the OxaI/YidC family, which are highly conserved integral membrane proteins involved in protein secretion (14). Our analysis of sequences in GenBank from the order Lactobacillales identified homologous proteins both with and without a cysteine residue that could potentially serve as an acylation site. More generally, since most studies involving lipoproteins have not tested the accuracy of lipoprotein prediction algorithms or have done so indirectly (3), it is not possible to identify all lipoproteins with certainty in S. sanguinis or any other bacterium. Nevertheless, it is unlikely that we missed more than a small number of lipoprotein genes in our mutant screen, and we are unaware of any S. sanguinis ortholog of a known lipoprotein virulence factor that was not identified and mutagenized in our study. It is also possible that some of the insertions did not completely eliminate activity. While we cannot rule out this possibility entirely, it seems unlikely to account for our results, given the locations of the insertions and our creation of additional allelic exchange mutants.
Perhaps the simplest explanation for our finding fewer avirulent mutants is the disease being modeled. An STM study examining Staphylococcus aureus in mouse abscess, bacteremia, and wound models found that less than 10% of mutants that were attenuated in any single model were attenuated in all three (10). It is possible that fewer bacterial genes are required for survival in the relatively protected environment of the vegetation than in many other environments. In this regard, it should also be emphasized that this study employed a model of early infective endocarditis, with vegetations collected at approximately 20 h postinfection. Streptococcal infective endocarditis is usually a subacute disease, often progressing for weeks before diagnosis or treatment (48). Thus, mutations having little effect during the first 20 h might produce marked effects during persistent infections. Previous studies in the rabbit model indicate that bacteria become embedded within the vegetation within 30 min and then become protected from phagocytosis (16). Thus, any protein not required for the earliest stages of disease development is not likely to represent an attractive therapeutic target. Thus, it is significant that so few lipoproteins are required for early disease.
The minor effect of the other mutations examined in this study on virulence is in sharp contrast to the dramatic reduction in virulence caused by the ssaB mutation. Initial sequencing of the ssaB gene (22) and subsequent analysis of the entire operon (72) suggest that the ssaACB operon encodes an ABC-type transport system that is likely to transport Mn2+ and possibly Zn2+or Fe2+ based on homology to other systems (20, 46, 49, 50, 52). Mutation of the lipoprotein component in two of these systems, FimA of S. parasanguinis (7) and SloC of S. mutans (38), was shown to reduce virulence for endocarditis, although the extent of virulence reduction was not quantified. Similarly, psaA mutants have been shown to be severely attenuated in multiple infection models (33, 44, 46). Although adhesin functions have been suggested for a number of these lipoproteins (4, 7, 23), studies of S. mutans (52) and S. pneumoniae (33, 44, 46) have demonstrated that mutation of nonlipoprotein transport components within the lipoprotein-containing operon also results in virulence reduction. This suggests that the metal uptake function of these lipoproteins is paramount to any other virulence function they may possess. Moreover, Marra et al. (44) reported that mutation of psaA, psaB, or psaC abrogated murine intraperitoneal chamber proliferation, unless the mutants were provided with added MnSO4. The combined results suggest that metal transport by SsaB homologs is required not only for virulence but for mere survival in the mammalian host.
Another result that was somewhat surprising was that the loss of a single lipoprotein, SsaB, produced a far greater defect in virulence than the loss of either lipoprotein-processing enzyme Lgt or LspA. According to the model established in E. coli, prelipoproteins are secreted via the Sec pathway and then acylated at the conserved cysteine, which allows for cleavage of the signal peptide by LspA, leaving the mature lipoprotein anchored in the cell membrane by its lipid tail (59). An lgt mutant would be expected to be deficient in the acylation of all cellular lipoproteins. Experiments in which acylation is detected by the addition of labeled palmitate to the growth medium have borne out this expectation in many gram-positive bacteria (3, 25, 27, 47, 54), and we confirmed that this is also true in S. sanguinis (Fig. 4A). Contrary to the second part of this model, however, it has recently been shown in S. agalactiae (27) and Listeria monocytogenes (3) that nonacylated prolipoproteins produced in lgt mutants are subject to cleavage by LspA. The result is the enhanced release of cleaved, nonacylated proteins from the cell surface (3, 27). Although we did not examine culture supernatants for the presence of SsaB, Fig. 4B suggests a sharp reduction in cell-associated SsaB in the lgt mutant, with the remaining SsaB present in an uncleaved, nonacylated form. This is consistent with the results of Henneke et al. (27) and with a model suggesting partial cleavage and release of nonacylated proteins, with retention of a small amount of uncleaved protein via the signal peptide. It is interesting that the reduction in surface-localized SsaB levels observed in the lgt mutant is not sufficient to seriously compromise SsaB function.
Virulence phenotypes associated with lgt mutation in other streptococci range from profound attenuation in a mouse respiratory tract infection model by S. pneumoniae (54), to intermediate attenuation in a number of infection studies by Streptococcus equi (25), to enhancement of lethality in a mouse model of neonatal sepsis by S. agalactiae (27). The S. equi study produced a result similar to ours, in that lgt mutation reduced virulence less than mutation of the gene for a single lipoprotein, PrtM (25). Interestingly, S. sanguinis has a PrtM ortholog, SSA_0753, and mutation of its gene by insertion or allelic exchange had no marked effect on virulence in our study.
Recent studies have examined the effects of mutating the signal peptidase gene (designated lsp) on the structure and/or function of SsaB orthologs. Henneke et al. (27) determined that ScaA was localized mostly to the cytoplasmic membrane in wild-type and lsp mutant strains, whereas it was secreted into the supernatant in large amounts in lgt and lgt lsp double mutants. Microsequencing confirmed that secreted ScaA was cleaved at the normal site in the lgt mutant and suggested that it was cleaved by signal peptidase I in the lsp mutant. Processing of the SsaB ortholog MtuA in Streptococcus uberis was examined in an lsp mutant (13). Western blot analysis revealed either one or two bands in the lsp mutant that were larger than MtuA derived from wild-type cells. The latter finding is similar to that observed in our study (Fig. 4B). The authors mutagenized the eep gene, encoding an ortholog of an Enterococcus faecalis peptidase that is believed to process sex pheromones derived from the signal peptides of lipoproteins (9). The eep lsp double mutant produced only one larger MtuA band, corresponding to full-length MtuA, suggesting that Eep cleaves some MtuA molecules in an lsp mutant. An Eep ortholog encoded by SSA_2070 could potentially serve the same role in S. sanguinis, accounting for the smaller SsaB band observed in the lspA mutant (Fig. 4B). Finally, Khandavilli et al. (36) described the effect of the lsp mutation on PsaA structure and function and on virulence in S. pneumoniae. The lsp mutant possessed phenotypes typical of a psaA mutant and exhibited reduced competitiveness in disease models. Although individual lipoprotein mutants were not tested, the reduced competitiveness phenotype appeared less severe than that expected for psaA mutants (44, 46).
The overall picture that emerges from the lgt and lsp mutant analyses described above in combination with our own is that there is far less of an effect on growth or virulence than would be expected if all lipoprotein function were lost. This suggests that many lipoproteins retain at least partial activity in the absence of acylation or Lsp-mediated cleavage. This could explain the presence of proteins such as SSA_0499 in S. sanguinis, which does not possess an N-terminal cysteine for acylation but is encoded within an apparent ABC transport operon and has extensive homology to the substrate-binding component of ABC transport systems, including SSA_0493.
In conclusion, our systematic analysis of lipoproteins in S. sanguinis has identified a single lipoprotein, SsaB, that is critical for early endocarditis virulence. Although our data suggest that interference with acylation or signal peptide cleavage has little effect on its function, SsaB appears to be a singularly promising target for new inhibitors or for its renewed examination as a vaccine candidate (39, 70).
Supplementary Material
Acknowledgments
We thank Nicai Zollar for technical assistance and Lauren Senty Turner for useful discussions.
This work was supported by National Institutes of Health grants R01AI47841 and K02AI05490 (T.K.) and R01DE18138 (P.X.).
Footnotes
Published ahead of print on 24 April 2009.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Alloing, G., P. de Philip, and J. P. Claverys. 1994. Three highly homologous membrane-bound lipoproteins participate in oligopeptide transport by the Ami system of the gram-positive Streptococcus pneumoniae. J. Mol. Biol. 24144-58. [DOI] [PubMed] [Google Scholar]
- 2.Andersen, R. N., R. D. Lunsford, and P. E. Kolenbrander. 1997. Determination of the transcript size and start site of the putative sca operon of Streptococcus gordonii ATCC 51656 (formerly strain PK488), p. 657-660. In T. Horaud, A. Bouvet, R. Leclercq, H. de Montclos, and M. Sicard (ed.), Streptococci and the host, vol. 418. Plenum Press, New York, NY. [DOI] [PubMed] [Google Scholar]
- 3.Baumgartner, M., U. Karst, B. Gerstel, M. Loessner, J. Wehland, and L. Jansch. 2007. Inactivation of Lgt allows systematic characterization of lipoproteins from Listeria monocytogenes. J. Bacteriol. 189313-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berry, A. M., and J. C. Paton. 1996. Sequence heterogeneity of PsaA, a 37-kilodalton putative adhesin essential for virulence of Streptococcus pneumoniae. Infect. Immun. 645255-5262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown, J. S., S. M. Gilliland, J. Ruiz-Albert, and D. W. Holden. 2002. Characterization of Pit, a Streptococcus pneumoniae iron uptake ABC transporter. Infect. Immun. 704389-4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown, J. S., A. D. Ogunniyi, M. C. Woodrow, D. W. Holden, and J. C. Paton. 2001. Immunization with components of two iron uptake ABC transporters protects mice against systemic Streptococcus pneumoniae infection. Infect. Immun. 696702-6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burnette-Curley, D., V. Wells, H. Viscount, C. L. Munro, J. C. Fenno, P. Fives-Taylor, and F. L. Macrina. 1995. FimA, a major virulence factor associated with Streptococcus parasanguis endocarditis. Infect. Immun. 634669-4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carlsson, J. 1965. Zooglea-forming streptococci, resembling Streptococcus sanguis, isolated from dental plaque in man. Odontol. Revy 16348-358. [PubMed] [Google Scholar]
- 9.Chandler, J. R., and G. M. Dunny. 2008. Characterization of the sequence specificity determinants required for processing and control of sex pheromone by the intramembrane protease Eep and the plasmid-encoded protein PrgY. J. Bacteriol. 1901172-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coulter, S. N., W. R. Schwan, E. Y. Ng, M. H. Langhorne, H. D. Ritchie, S. Westbrock-Wadman, W. O. Hufnagle, K. R. Folger, A. S. Bayer, and C. K. Stover. 1998. Staphylococcus aureus genetic loci impacting growth and survival in multiple infection environments. Mol. Microbiol. 30393-404. [DOI] [PubMed] [Google Scholar]
- 11.Das, S., J. C. Noe, S. Paik, and T. Kitten. 2005. An improved arbitrary primed PCR method for rapid characterization of transposon insertion sites. J. Microbiol. Methods 6389-94. [DOI] [PubMed] [Google Scholar]
- 12.de Greeff, A., A. Hamilton, I. C. Sutcliffe, H. Buys, L. van Alphen, and H. E. Smith. 2003. Lipoprotein signal peptidase of Streptococcus suis serotype 2. Microbiology 1491399-1407. [DOI] [PubMed] [Google Scholar]
- 13.Denham, E. L., P. N. Ward, and J. A. Leigh. 2008. Lipoprotein signal peptides are processed by Lsp and Eep of Streptococcus uberis. J. Bacteriol. 1904641-4647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong, Y., S. R. Palmer, A. Hasona, S. Nagamori, H. R. Kaback, R. E. Dalbey, and L. J. Brady. 2008. Functional overlap but lack of complete cross-complementation of Streptococcus mutans and Escherichia coli YidC orthologs. J. Bacteriol. 1902458-2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Douglas, C. W., J. Heath, K. K. Hampton, and F. E. Preston. 1993. Identity of viridans streptococci isolated from cases of infective endocarditis. J. Med. Microbiol. 39179-182. [DOI] [PubMed] [Google Scholar]
- 16.Durack, D. T. 1975. Experimental bacterial endocarditis. IV. Structure and evolution of very early lesions. J. Pathol. 11581-89. [DOI] [PubMed] [Google Scholar]
- 17.Durack, D. T., P. B. Beeson, and R. G. Petersdorf. 1973. Experimental bacterial endocarditis. 3. Production and progress of the disease in rabbits. Br. J. Exp. Pathol. 54142-151. [PMC free article] [PubMed] [Google Scholar]
- 18.Dyson, C., R. A. Barnes, and G. A. J. Harrison. 1999. Infective endocarditis: an epidemiological review of 128 episodes. J. Infect. 3887-93. [DOI] [PubMed] [Google Scholar]
- 19.Eddy, S. R. 1998. Profile hidden Markov models. Bioinformatics 14755-763. [DOI] [PubMed] [Google Scholar]
- 20.Fenno, J. C., A. Shaikh, G. Spatafora, and P. Fives-Taylor. 1995. The fimA locus of Streptococcus parasanguis encodes an ATP-binding membrane transport system. Mol. Microbiol. 15849-863. [DOI] [PubMed] [Google Scholar]
- 21.Gan, K., S. D. Gupta, K. Sankaran, M. B. Schmid, and H. C. Wu. 1993. Isolation and characterization of a temperature-sensitive mutant of Salmonella typhimurium defective in prolipoprotein modification. J. Biol. Chem. 26816544-16550. [PubMed] [Google Scholar]
- 22.Ganeshkumar, N., P. M. Hannam, P. E. Kolenbrander, and B. C. McBride. 1991. Nucleotide sequence of a gene coding for a saliva-binding protein (SsaB) from Streptococcus sanguis 12 and possible role of the protein in coaggregation with actinomyces. Infect. Immun. 591093-1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ganeshkumar, N., M. Song, and B. C. McBride. 1988. Cloning of a Streptococcus sanguis adhesin which mediates binding to saliva-coated hydroxyapatite. Infect. Immun. 561150-1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ge, X., T. Kitten, Z. Chen, S. P. Lee, C. L. Munro, and P. Xu. 2008. Identification of Streptococcus sanguinis genes required for biofilm formation and examination of their role in endocarditis virulence. Infect. Immun. 762551-2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamilton, A., C. Robinson, I. C. Sutcliffe, J. Slater, D. J. Maskell, N. Davis-Poynter, K. Smith, A. Waller, and D. J. Harrington. 2006. Mutation of the maturase lipoprotein attenuates the virulence of Streptococcus equi to a greater extent than does loss of general lipoprotein lipidation. Infect. Immun. 746907-6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hava, D. L., and A. Camilli. 2002. Large-scale identification of serotype 4 Streptococcus pneumoniae virulence factors. Mol. Microbiol. 451389-1406. [PMC free article] [PubMed] [Google Scholar]
- 27.Henneke, P., S. Dramsi, G. Mancuso, K. Chraibi, E. Pellegrini, C. Theilacker, J. Hubner, S. Santos-Sierra, G. Teti, D. T. Golenbock, C. Poyart, and P. Trieu-Cuot. 2008. Lipoproteins are critical TLR2 activating toxins in group B streptococcal sepsis. J. Immunol. 1806149-6158. [DOI] [PubMed] [Google Scholar]
- 28.Hensel, M. 1998. Whole genome scan for habitat-specific genes by signature-tagged mutagenesis. Electrophoresis 19608-612. [DOI] [PubMed] [Google Scholar]
- 29.Hermans, P. W. M., P. V. Adrian, C. Albert, S. Estevao, T. Hoogenboezem, I. H. T. Luijendijk, T. Kamphausen, and S. Hammerschmidt. 2006. The streptococcal lipoprotein rotamase A (SlrA) is a functional peptidyl-prolyl isomerase involved in pneumococcal colonization. J. Biol. Chem. 281968-976. [DOI] [PubMed] [Google Scholar]
- 30.Horton, R. M. 1995. PCR-mediated recombination and mutagenesis. SOEing together tailor-made genes. Mol. Biotechnol. 393-99. [DOI] [PubMed] [Google Scholar]
- 31.Jakubovics, N. S., A. W. Smith, and H. F. Jenkinson. 2000. Expression of the virulence-related Sca (Mn2+) permease in Streptococcus gordonii is regulated by a diphtheria toxin metallorepressor-like protein ScaR. Mol. Microbiol. 38140-153. [DOI] [PubMed] [Google Scholar]
- 32.Jenkinson, H. F. 1994. Cell surface protein receptors in oral streptococci. FEMS Microbiol. Lett. 121133-140. [DOI] [PubMed] [Google Scholar]
- 33.Johnston, J. W., L. E. Myers, M. M. Ochs, W. H. Benjamin, Jr., D. E. Briles, and S. K. Hollingshead. 2004. Lipoprotein PsaA in virulence of Streptococcus pneumoniae: surface accessibility and role in protection from superoxide. Infect. Immun. 725858-5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones, A. L., K. M. Knoll, and C. E. Rubens. 2000. Identification of Streptococcus agalactiae virulence genes in the neonatal rat sepsis model using signature-tagged mutagenesis. Mol. Microbiol. 371444-1455. [DOI] [PubMed] [Google Scholar]
- 35.Juncker, A. S., H. Willenbrock, G. von Heijne, S. Brunak, H. Nielsen, and A. Krogh. 2003. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 121652-1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khandavilli, S., K. A. Homer, J. Yuste, S. Basavanna, T. Mitchell, and J. S. Brown. 2008. Maturation of Streptococcus pneumoniae lipoproteins by a type II signal peptidase is required for ABC transporter function and full virulence. Mol. Microbiol. 67541-557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kilian, M., L. Mikkelsen, and J. Henrichsen. 1989. Taxonomic study of viridans streptococci: description of Streptococcus gordonii sp. nov. and emended descriptions of Streptococcus sanguis (White and Niven 1946), Streptococcus oralis (Bridge and Sneath 1982), and Streptococcus mitis (Andrewes and Horder 1906). Int. J. Syst. Bacteriol. 39471-484. [Google Scholar]
- 38.Kitten, T., C. L. Munro, S. M. Michalek, and F. L. Macrina. 2000. Genetic characterization of a Streptococcus mutans LraI family operon and role in virulence. Infect. Immun. 684441-4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kitten, T., C. L. Munro, A. Wang, and F. L. Macrina. 2002. Vaccination with FimA from Streptococcus parasanguis protects rats from endocarditis caused by other viridans streptococci. Infect. Immun. 70422-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lai, S. H., W. M. Philbrick, and H. C. Wu. 1980. Acyl moieties in phospholipids are the precursors for the fatty acids in murein lipoprotein of Escherichia coli. J. Biol. Chem. 2555384-5387. [PubMed] [Google Scholar]
- 41.Lau, G. W., S. Haataja, M. Lonetto, S. E. Kensit, A. Marra, A. P. Bryant, D. McDevitt, D. A. Morrison, and D. W. Holden. 2001. A functional genomic analysis of type 3 Streptococcus pneumoniae virulence. Mol. Microbiol. 40555-571. [DOI] [PubMed] [Google Scholar]
- 42.Lei, B., M. Liu, G. L. Chesney, and J. M. Musser. 2004. Identification of new candidate vaccine antigens made by Streptococcus pyogenes: purification and characterization of 16 putative extracellular lipoproteins. J. Infect. Dis. 18979-89. [DOI] [PubMed] [Google Scholar]
- 43.Macrina, F. L., J. A. Tobian, K. R. Jones, R. P. Evans, and D. B. Clewell. 1982. A cloning vector able to replicate in Escherichia coli and Streptococcus sanguis. Gene 19345-353. [DOI] [PubMed] [Google Scholar]
- 44.Marra, A., S. Lawson, J. S. Asundi, D. Brigham, and A. E. Hromockyj. 2002. In vivo characterization of the psa genes from Streptococcus pneumoniae in multiple models of infection. Microbiology 1481483-1491. [DOI] [PubMed] [Google Scholar]
- 45.Martin, B., M. Prudhomme, G. Alloing, C. Granadel, and J. P. Claverys. 2000. Cross-regulation of competence pheromone production and export in the early control of transformation in Streptococcus pneumoniae. Mol. Microbiol. 38867-878. [DOI] [PubMed] [Google Scholar]
- 46.McAllister, L. J., H.-J. Tseng, A. D. Ogunniyi, M. P. Jennings, A. G. McEwan, and J. C. Paton. 2004. Molecular analysis of the psa permease complex of Streptococcus pneumoniae. Mol. Microbiol. 53889-901. [DOI] [PubMed] [Google Scholar]
- 47.McNab, R., and H. F. Jenkinson. 1998. Lipoproteins and other cell-surface associated proteins in streptococci. Methods Cell Sci. 20209-216. [Google Scholar]
- 48.Moreillon, P., and Y. A. Que. 2004. Infective endocarditis. Lancet 363139-149. [DOI] [PubMed] [Google Scholar]
- 49.Novak, R., J. S. Braun, E. Charpentier, and E. Tuomanen. 1998. Penicillin tolerance genes of Streptococcus pneumoniae: the ABC-type manganese permease complex Psa. Mol. Microbiol. 291285-1296. [DOI] [PubMed] [Google Scholar]
- 50.Oetjen, J., P. Fives-Taylor, and E. H. Froeliger. 2002. The divergently transcribed Streptococcus parasanguis virulence-associated fimA operon encoding an Mn2+-responsive metal transporter and pepO encoding a zinc metallopeptidase are not coordinately regulated. Infect. Immun. 705706-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Overweg, K., A. Kerr, M. Sluijter, M. H. Jackson, T. J. Mitchell, A. P. de Jong, R. de Groot, and P. W. Hermans. 2000. The putative proteinase maturation protein A of Streptococcus pneumoniae is a conserved surface protein with potential to elicit protective immune responses. Infect. Immun. 684180-4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paik, S., A. Brown, C. L. Munro, C. N. Cornelissen, and T. Kitten. 2003. The sloABCR operon of Streptococcus mutans encodes an Mn and Fe transport system required for endocarditis virulence and its Mn-dependent repressor. J. Bacteriol. 1855967-5975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paik, S., L. Senty, S. Das, J. C. Noe, C. L. Munro, and T. Kitten. 2005. Identification of virulence determinants for endocarditis in Streptococcus sanguinis by signature-tagged mutagenesis. Infect. Immun. 736064-6074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Petit, C. M., J. R. Brown, K. Ingraham, A. P. Bryant, and D. J. Holmes. 2001. Lipid modification of prelipoproteins is dispensable for growth in vitro but essential for virulence in Streptococcus pneumoniae. FEMS Microbiol. Lett. 200229-233. [DOI] [PubMed] [Google Scholar]
- 55.Polissi, A., A. Pontiggia, G. Feger, M. Altieri, H. Mottl, L. Ferrari, and D. Simon. 1998. Large-scale identification of virulence genes from Streptococcus pneumoniae. Infect. Immun. 665620-5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rice, P., I. Longden, and A. Bleasby. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16276-277. [DOI] [PubMed] [Google Scholar]
- 57.Roberts, G. J. 1999. Dentists are innocent! “Everyday” bacteremia is the real culprit: a review and assessment of the evidence that dental surgical procedures are a principal cause of bacterial endocarditis in children. Pediatr. Cardiol. 20317-325. [DOI] [PubMed] [Google Scholar]
- 58.Sampson, J. S., S. P. O'Connor, A. R. Stinson, J. A. Tharpe, and H. Russell. 1994. Cloning and nucleotide sequence analysis of psaA, the Streptococcus pneumoniae gene encoding a 37-kilodalton protein homologous to previously reported Streptococcus sp. adhesins. Infect. Immun. 62319-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sankaran, K., and H. C. Wu. 1994. Lipid modification of bacterial prolipoprotein. Transfer of diacylglyceryl moiety from phosphatidylglycerol. J. Biol. Chem. 26919701-19706. [PubMed] [Google Scholar]
- 60.Smith, A. J., P. N. Ward, T. R. Field, C. L. Jones, R. A. Lincoln, and J. A. Leigh. 2003. MtuA, a lipoprotein receptor antigen from Streptococcus uberis, is responsible for acquisition of manganese during growth in milk and is essential for infection of the lactating bovine mammary gland. Infect. Immun. 714842-4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Song, J. H., and K. S. Ko. 2008. Detection of essential genes in Streptococcus pneumoniae using bioinformatics and allelic replacement mutagenesis. Methods Mol. Biol. 416401-408. [DOI] [PubMed] [Google Scholar]
- 62.Sonnhammer, E. L. L., S. R. Eddy, E. Birney, A. Bateman, and R. Durbin. 1998. Pfam: multiple sequence alignments and HMM-profiles of protein domains. Nucleic Acids Res. 26320-322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Spellerberg, B., E. Rozdzinski, S. Martin, J. Weber-Heynemann, N. Schnitzler, R. Lutticken, and A. Podbielski. 1999. Lmb, a protein with similarities to the LraI adhesin family, mediates attachment of Streptococcus agalactiae to human laminin. Infect. Immun. 67871-878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sung, C. K., H. Li, J. P. Claverys, and D. A. Morrison. 2001. An rpsL cassette, Janus, for gene replacement through negative selection in Streptococcus pneumoniae. Appl. Environ. Microbiol. 675190-5196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sutcliffe, I. C., and D. J. Harrington. 2002. Pattern searches for the identification of putative lipoprotein genes in Gram-positive bacterial genomes. Microbiology 1482065-2077. [DOI] [PubMed] [Google Scholar]
- 66.Sutcliffe, I. C., and R. R. Russell. 1995. Lipoproteins of gram-positive bacteria. J. Bacteriol. 1771123-1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tatusov, R. L., D. A. Natale, I. V. Garkavtsev, T. A. Tatusova, U. T. Shankavaram, B. S. Rao, B. Kiryutin, M. Y. Galperin, N. D. Fedorova, and E. V. Koonin. 2001. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2922-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thanassi, J. A., S. L. Hartman-Neumann, T. J. Dougherty, B. A. Dougherty, and M. J. Pucci. 2002. Identification of 113 conserved essential genes using a high-throughput gene disruption system in Streptococcus pneumoniae. Nucleic Acids Res. 303152-3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68a.Turner, L. S., S. Das, T. Kanamoto, C. L. Munro, and T. Kitten. Development of genetic tools for in vivo virulence analysis of Streptococcus sanguinis. Microbiology, in press. [DOI] [PMC free article] [PubMed]
- 69.Unsworth, K. E., and D. W. Holden. 2000. Identification and analysis of bacterial virulence genes in vivo. Philos. Trans. R. Soc. Lond. B 355613-622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Viscount, H. B., C. L. Munro, D. Burnette-Curley, D. L. Peterson, and F. L. Macrina. 1997. Immunization with FimA protects against Streptococcus parasanguis endocarditis in rats. Infect. Immun. 65994-1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wilson, W., K. A. Taubert, M. Gewitz, P. B. Lockhart, L. M. Baddour, M. Levison, A. Bolger, C. H. Cabell, M. Takahashi, R. S. Baltimore, J. W. Newburger, B. L. Strom, L. Y. Tani, M. Gerber, R. O. Bonow, T. Pallasch, S. T. Shulman, A. H. Rowley, J. C. Burns, P. Ferrieri, T. Gardner, D. Goff, and D. T. Durack. 2007. Prevention of infective endocarditis: guidelines from the American Heart Association: a guideline from the American Heart Association Rheumatic Fever, Endocarditis and Kawasaki Disease Committee, Council on Cardiovascular Disease in the Young, and the Council on Clinical Cardiology, Council on Cardiovascular Surgery and Anesthesia, and the Quality of Care and Outcomes Research Interdisciplinary Working Group. Circulation 1161736-1754. [DOI] [PubMed] [Google Scholar]
- 72.Xu, P., J. M. Alves, T. Kitten, A. Brown, Z. Chen, L. S. Ozaki, P. Manque, X. Ge, M. G. Serrano, D. Puiu, S. Hendricks, Y. Wang, M. D. Chaplin, D. Akan, S. Paik, D. L. Peterson, F. L. Macrina, and G. A. Buck. 2007. Genome of the opportunistic pathogen Streptococcus sanguinis. J. Bacteriol. 1893166-3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yamagata, H., N. Taguchi, K. Daishima, and S. Mizushima. 1983. Genetic characterization of a gene for prolipoprotein signal peptidase in Escherichia coli. Mol. Gen. Genet. 19210-14. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.