

Abstract
After penetrating the host cell, the herpesvirus capsid is transported to the nucleus along the microtubule network and docks to the nuclear pore complex before releasing the viral DNA into the nucleus. The viral and cellular interactions involved in the docking process are poorly characterized. However, the minor capsid protein pUL25 has recently been reported to be involved in viral DNA uncoating. Here we show that herpes simplex virus type 1 (HSV-1) capsids interact with the nucleoporin CAN/Nup214 in infected cells and that RNA silencing of CAN/Nup214 delays the onset of viral DNA replication in the nucleus. We also show that pUL25 interacts with CAN/Nup214 and another nucleoporin, hCG1, and binds to the pUL36 and pUL6 proteins, two other components of the herpesvirus particle that are known to be important for the initiation of infection and viral DNA release. These results identify CAN/Nup214 as being a nuclear receptor for the herpesvirus capsid and pUL25 as being an interface between incoming capsids and the nuclear pore complex and as being a triggering element for viral DNA release into the nucleus.
Many nucleus-replicating viruses have evolved different strategies for delivering their genomes into the nucleus of their host cell through the nuclear pores, which provide the only route of transit across the physical barrier of the nuclear envelope. These strategies depend mainly on the nature of the capsid, which acts both as a protective element for the genome and as a delivery agent (for reviews, see references 21 and 60).
Alphaherpesviruses are large, double-stranded DNA viruses. Their genomes are contained within a 125-nm-diameter capsid that is surrounded sequentially by a thick proteinaceous layer, called the tegument, and a lipid envelope. The herpes simplex virus type 1 (HSV-1) capsid structure has been extensively studied (66) and is a general model for other alphaherpesviruses. It has icosahedral symmetry with the major capsid protein VP5, forming hexamers and pentamers (termed hexons and pentons) at the faces and vertices, respectively, of the icosahedron. There are 150 hexons and 11 pentons per capsid. At one vertex, the penton is replaced by a portal, a structure common to tailed bacteriophages and herpesviruses, through which the viral DNA is encapsidated and released (7, 8). In HSV-1, the portal is a dodecamer of the UL6 gene product, pUL6 (38, 57).
The nuclear pore complex (NPC) is a multiprotein complex that selectively controls the passage of material through the nuclear envelope (for a review, see reference 28). The NPC has three structural components: the nuclear basket, the central framework, which is embedded in the nuclear envelope, and the cytoplasmic filaments. The diameter of the cytoplasmic face is ∼125 nm, whereas the central channel is ∼60 nm in diameter (3). Its component proteins, termed nucleoporins, perform various roles, being important both in forming a selective gate and in carrying out nucleocytoplasmic transport (41, 55). Several models have been proposed to explain the selectivity of the NPC, all of them involving the phenylalanine-glycine (FG) repeat domains that are present in some nucleoporins (32, 42, 46, 49).
In herpesviruses, transcription, DNA replication, assembly of new capsids, and DNA packaging all take place in the nuclei of infected cells. Infection of new cells is initiated when the virion envelope fuses with the plasma membrane, releasing the tegument and capsid into the cytoplasm. However, the capsid does not itself enter the nucleus but binds to the NPC, where the viral DNA is released and is transferred into the nucleus through the NPC (2, 39, 51, 52). Thus, the binding of the capsid to the NPC is necessary for the initiation of infection. However, the nature of this process and the viral and NPC proteins involved are poorly understood.
Studies have highlighted two herpesvirus structural proteins that are suspected to play roles in the targeting of capsids to the NPC and/or in viral DNA uncoating. The first is the tegument protein pUL36 (also termed VP1/2), the gene product of the UL36 open reading frame (ORF). Tegument proteins have been implicated in the transport of capsids (30, 62), and pUL36 has been shown to be necessary for this transport (31). Furthermore, an HSV-1 temperature-sensitive (ts) mutant of UL36, tsB7, has been described as being impaired in viral DNA release at nonpermissive temperatures (2), and a recent study has demonstrated that the proteolytic cleavage of pUL36 is required for viral DNA uncoating (24). A second HSV-1 ts mutant (ts1249) has a phenotype similar to that of tsB7 (44). The mutation is in the UL25 gene, which encodes a minor capsid protein, pUL25, that is needed to retain packaged viral DNA within the capsid (34, 53). pUL25 was previously suggested to bind as a heterodimer with another minor capsid protein, pUL17, around the capsid vertices (58) and is also known to interact with pUL36 (9).
In the present study, we identified an interaction between HSV-1 capsids and the nucleoporin CAN/Nup214. We further showed that the minor capsid protein pUL25 binds directly to both CAN/Nup214 and a second nucleoporin, hCG1. In addition, we demonstrated the binding of pUL25 to the portal protein pUL6 and the tegument protein pUL36, which both have key roles in viral DNA release (2, 24, 36). Thus, the pUL25 capsid protein appears to play a central role in linking the processes of nuclear pore binding and release of viral DNA.
MATERIALS AND METHODS
Cells and viruses.
African green monkey kidney (Vero), 293T, and HeLa cells were grown at 37°C in Dulbecco's modified Eagle medium (DMEM; Invitrogen) supplemented with 10% fetal calf serum. Baby hamster kidney (BHK) cells were grown in Glasgow modified Eagle medium (Invitrogen) supplemented with 10% newborn calf serum and tryptose phosphate broth.
Wild-type (WT) HSV-1 (strain 17+) and tsK/Luci (provided by C. Preston) were propagated on BHK cells infected at 0.01 PFU per cell, and virions were concentrated from supernatant medium by centrifugation at 15,000 × g for 2 h. To generate the tsK/Luci virus, the β-galactosidase coding region of pMJ102 (23) was replaced by the firefly luciferase coding sequences (with an additional in-frame, C-terminal fusion of chloramphenicol acetyltransferase coding sequences that is irrelevant to the experiments described here). This resulted in a plasmid in which the luciferase coding region was controlled by the HSV-1 immediate-early ICP0 promoter, with the entire construct embedded in the nonessential thymidine kinase (TK) coding region. This plasmid was recombined with genomic DNA of the HSV-1 ts mutant tsK (15, 43), and TK-negative mutants were isolated as described previously (23, 29). As the tsK virus has a ts lesion in the ICP4 protein, all experiments using this virus were performed at a permissive temperature (31°C).
The UL37 null mutant of HSV-1 (FRΔUL37) was propagated as described previously (47). vICP4CFP-VP26RFP was made by coinfecting Vero cells with vECFP-ICP4, which expresses the immediate-early protein ICP4 linked to enhanced cyan fluorescent protein (CFP) (19), and vUL35RFP1D1, which was made by fusing monomeric red fluorescent protein (RFP) (Clontech) to the N terminus of the small capsid protein VP26. Progeny virus was collected and serially diluted on fresh cells. Plaques exhibiting both CFP and RFP fluorescence were selected and purified through four rounds of plaque purification.
Antibodies.
Rabbit antibody PTNC raised against purified nuclear C capsids recognizes the capsid proteins VP23 and VP26 and the inner tegument protein pUL36 on Western blots. The following antibodies were used. Mouse monoclonal antibody (MAb) DM165 (30) and MAb 4846 (provided by A. Cross, MRC, Glasgow, United Kingdom) against the HSV-1 major capsid protein VP5 and glycoprotein D, respectively, were used. Rabbit antibody NRP14 against respiratory syncytial virus protein N (Nrsv) was provided by V. Cowton (35). Rabbit antibody Y11 and MAb F7 against a defined epitope (hemagglutinin [HA]) from influenza virus HA and rabbit antibody A14 against c-myc were obtained from Santa Cruz Biotechnology. MAb 9E10 against c-myc and MAb 414, which recognizes four nuclear pore proteins, were obtained from Sigma and Covance Research, respectively. Alexa Fluor 633-conjugated goat anti-mouse antibody and horseradish peroxidase-conjugated goat anti-mouse antibody were obtained from Molecular Probes and Sigma, respectively.
Plasmids.
Vector pCDNA3.1-myc was made by cloning the six-Myc tag sequence, isolated as a BamHI-EcoRI fragment from pCS2-myc (48) (provided by S. Benichou, Institut Cochin, Paris, France), between the BamHI and EcoRI restriction sites of pCDNA3.1 (Invitrogen). Vector pCDNA3.1-HA was made by cloning sequences encoding the HA tag (YPYDVPDYA) between the same restriction sites in pcDNA3.1. pCS2-myc-hCG1 was provided by S. Benichou. pCMV10-HA-UL32 and pECFP-ICP4 were provided by E. Palmer and R. Everett (MRC, Glasgow, United Kingdom), respectively. The HSV-1 UL25 gene was isolated from pCG-UL25 HSV-1 (provided by K. Kaelin, Université Paris) as an EcoRI-XhoI fragment and was subcloned into pCDNA3.1-HA to generate pCDNA3.1-HA UL25. pLexA-UL25 PrV was provided by H. Raux (CNRS, Gif-sur-Yvette, France). The other constructs used were generated by PCR and cloned into pCDNA3.1-myc as described in Table 1.
TABLE 1.
PCR primers used to generate DNA fragments used in interaction studiese
| Construct (residues) | Templatea | PCR primerf |
|---|---|---|
| myc-UL36 (2037-3164) | cos14 | CGCGAATTCGATGTGTCCCGAGGCGGCA* |
| CGATCTAGACTAGCCCAGTAACATGCGCAC | ||
| myc-UL36 (2037-2572) | cos14 | CGCGAATTCGATGTGTCCCGAGGCGGCA* |
| AATTCTAGACTACATGCGAGGGGGGACGGG | ||
| myc-UL36 (2037-2504) | cos14 | CGCGAATTCGATGTGTCCCGAGGCGGCA* |
| GGATCTAGACTACGGCCGCCCGAGGATAGC | ||
| myc-UL36 (2037-2353) | cos14 | CGCGAATTCGATGTGTCCCGAGGCGGCA* |
| CGCTCTAGACTAGGGCGCGCCCAGGAGCAC | ||
| myc-UL36 (2037-2200) | cos14 | CGCGAATTCGATGTGTCCCGAGGCGGCA* |
| GCCTCTAGACGGAGGGCCTCCGGCCG | ||
| myc-UL36 (2446-3164) | cos14 | GGAGAATTCCATGACCCCGGTCGCGGTG* |
| CGATCTAGACTAGCCCAGTAACATGCGCAC | ||
| myc-UL36 (2446-3104) | cos14 | GGAGAATTCCATGACCCCGGTCGCGGTG* |
| TGCTCTAGAGGGCGGGGGGCCGAATTG | ||
| myc-UL36 (2446-2986) | cos14 | GGAGAATTCCATGACCCCGGTCGCGGTG* |
| ATATCTAGAGGGTGCTACATGCCCG | ||
| myc-hNup133 | pLex12-hs133b | CGCGAATTCGGGTACCGGGTCCCGAAGG* |
| TAGCTCGAGTTATATTTGTCCCTGAACATA | ||
| myc-hNup50 | IMAGE clone 6463796 | GAAGCGGCCGCATGGCCAGTGAGGAAGTC |
| TTTTCTAGATCAGGCATCCTTTTTCTCC | ||
| myc-hNup54 | IMAGE clone 3931652 | CTTCTCGAGCATGGCCTTCAATTTTGGG* |
| TGTCTCGAGTCAACTAAAGACACCACC | ||
| myc-hCAN Ct | psRET-CAN Ctc | CTCGAATTCTGCAGCTGTCACAGCAGCT* |
| GACCTCGAGTCAGCTTCGCCAGCC | ||
| myc-UL6 HSV | pAC-UL6 HSV1d | GGCGAATTCGACCGCACCACGCTCGCG* |
| GGCTCTAGATCATCGTCGGCCGTC |
cos14 is described in reference 12.
Provided by V. Doye (4).
Provided by M. Fornerod (56).
Provided by F. Homa (34).
The PCR primers are listed in pairs, with the forward primer listed first and the reverse primer second. All PCR products were cloned into pCDNA3.1-myc, and the restriction enzyme sites used for cloning are underlined.
* indicates that these primers have a +1 nucleotide after the restriction site to put the gene in frame with the tag.
shRNA.
The use of lentiviruses expressing small hairpin RNA (shRNA) was described previously (18). Briefly, 293T cells were cotransfected with three plasmids: (i) pVSV-G, (ii) pCMVDR8.91 (provided by Didier Trono [http://tronolab.com/index.php]), and (iii) pLKO.1Puro-shLuc or pLKO.1Puro-shCAN. The cell supernatant containing recombinant lentivirus was harvested 3 days posttransfection and used to transduce HeLa cells in the presence of hexadimethrine bromide (5 μg/ml polybrene; Sigma). After overnight incubation, the cells were maintained in selective medium containing puromycin (2 μg/ml) for 2 to 3 days before being harvested for Western blot analysis or infected with HSV-1. Silencing of CAN/Nup214 was done by using the 19-nucleotides sequence GAATCACATCCGCATCAAA. Silencing efficiency was estimated by Western blotting using MAb 414 and quantified using ImageJ software (NIH [http://rsb.info.nih.gov/ij/]) to measure the ratios of Nup358, Nup214, and Nup153 relative to p62, which was used as a loading control. All silencing experiments were carried out in quadruplicate.
Yeast two-hybrid system.
The yeast two-hybrid system used in this study was described previously (45). Briefly, the pseudorabies virus (PrV) UL25 ORF was cloned into the EcoRI-BamHI sites of vector pLexA. Saccharomyces cerevisiae L40 cells were transformed using lithium acetate, and transformants were selected by their ability to grow on tryptophan-depleted agar plates. These transformants were then transformed with a library of nucleoporin ORFs cloned into a pGAD-based vector (provided by S. Benichou, Institut Cochin, Paris, France) as described previously (27). Double-transformed yeasts were selected on tryptophan- and leucine-depleted agar plates and assayed for β-galactosidase activity.
Plasmid transfection and superinfection.
Vero cells grown to 80% confluence on 60-mm plates were transfected with 1.0 μg of a single plasmid DNA or cotransfected with 0.5 μg each of two plasmid DNAs using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. The cells were incubated for 24 h in antibiotic-free DMEM complemented with 10% fetal calf serum and 2 mM glutamine before harvesting. If cells were to be superinfected with FRΔUL37, the transfection mix was removed after 3 h and replaced with fresh medium. After a further 3 h, the cells were infected with 5 PFU/cell, and incubation was continued for 15 h. If cells were to be superinfected with WT HSV-1, the transfection mix was removed after overnight incubation and replaced with fresh medium. The cells were infected with WT HSV-1 at 50 PFU/cell, and incubation was continued for 30 min.
Immunoprecipitation.
To prepare extracts for immunoprecipitation, transfected cells were lysed with cytoplasmic protein extraction buffer (CPEB) {50 mM Tris-HCl (pH 8), 150 mM NaCl, 5 mM EDTA, 0.5% IGEPAL [(octylphenoxyl)polyethoxyethano] 630} (Sigma) and protease inhibitors (complete, EDTA free; Roche) for 30 min on ice. Nuclei and cell debris were pelleted at 13,000 × g for 2 min. The supernatant was retained (cytoplasmic fraction), and the pellet was resuspended in nuclear protein extraction buffer (20 mM HEPES-KOH [pH 7.9], 420 mM KCl, 20% glycerol, 1 mM EDTA, 2 mM β-mercaptoethanol, and protease inhibitors) for 30 min on ice (33). The nuclear extracts were clarified at 13,000 × g for 2 min, and the supernatants from the nuclear and cytoplasmic fractions were pooled to constitute the whole-cell extract. Superinfected cells were lysed with CPEB only, and immunoprecipitation was performed on the cytoplasmic extract. All extracts were mixed with protein A for 30 min at 4°C and centrifuged at 13,000 × g for 2 min to eliminate nonspecific binding. A 30-μl sample of the supernatant, constituting 6% of the total extract, was removed before the addition of antibodies for 90 min at 4°C. The immune complexes were collected on protein A-Sepharose beads (Sigma) by incubation for 1 h at 4°C and washed three times in cold CPEB. Proteins were separated by 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and detected by Western blotting as described previously (47). The immunoprecipitate/cell extract ratio was estimated by quantifying the band of the immunoprecipitated protein and the corresponding band in the cell extract using ImageJ software.
Fluorescence microscopy.
For detecting autofluorescent proteins, cells were fixed with 4% paraformaldehyde (PFA), washed three times in phosphate-buffered saline, and mounted directly onto 2.5% 1,4-diazabicyclo[2,2,2]octane (Sigma) in Mowiol (Harco) containing 1 μg/ml 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI) (Sigma). For immunofluorescent detection, fixed cells were permeabilized with 0.1% Triton X-100 and incubated sequentially with MAb 4846 and Alexa Fluor 633-conjugated goat anti-mouse antibody (90 min for each incubation) before mounting. All samples were examined using a Zeiss LSM 510 Meta confocal microscope using a 63× oil immersion objective and in the multitrack recording mode. For studies of the association between gD and capsids, z stacks of whole cells were recorded and assembled for further analysis. Colocalizing signals were extracted from the original image using the histogram colocalization function of the LSM 510 software (version 4). The extracted images were analyzed using the counting and tracking function of the AutDeblur&AutoVisualize software (version 1.4.1; MediaCybernetics).
ICP4-CFP fluorescence in transfected silenced cells was measured 24 h after transfection by fixing the cells with 4% PFA and taking images of nine randomly chosen cells exhibiting CFP fluorescence per condition, using the same gain settings in every case. Quantification was carried out using the profile function of the LSM 510 software.
Luciferase assay.
Replicate monolayers of ∼106 HeLa cells were infected with 5 PFU/cell of tsK/Luci and incubated at 31°C for the times required. Luciferase assays were performed using the luciferase assay system (Promega) according to the manufacturer's instructions, and luciferase activity was assessed using a Glomax 20/20 luminometer (Promega).
Infectious-center experiments.
Cells infected with a control lentivirus expressing an shRNA directed against luciferase (shLuc cells) or with a lentivirus expressing a CAN/Nup214-specific shRNA (shCAN cells) were infected with 5 PFU/cell of vICP4CFP-VP26RFP for 1 h at 37°C. The viral inoculum was removed, and the cells were acid washed (0.14 M NaCl, 0.1 M glycine [pH 3]) to remove any residual input virus. The cells were then resuspended by trypsinization and serially diluted in DMEM. The dilutions were mixed with Vero cells in suspension and plated out into six-well dishes in DMEM containing 10% human serum. Once monolayers of adherent cells were formed (6 h later), the medium was replaced with methylcellulose. After incubation at 37°C for 48 h, the cells were stained with Giemsa (VWR), and plaques were counted.
RESULTS
Identification of capsid-binding nucleoporins.
Herpesvirus capsids accumulate in the vicinity of nuclear pores before releasing their DNA into the nucleus (39, 52), and it is likely that this process requires interactions between the capsid and proteins of the NPC. Therefore, we exogenously expressed a subset of nucleoporins to examine their potential roles in capsid binding. We selected a set of nucleoporins associated with different parts of the nuclear pore (cytoplasmic face, nucleoplasmic face, and pore membrane) and containing a variety of motifs (Table 2) and cloned them, adding an N-terminal myc tag for detection (Table 1). Because full-length myc-tagged CAN/Nup214 was poorly expressed, we cloned the C-terminal region of the protein, encompassing residues 1594 to 2090 and containing the FG repeat sequences, into pCDNA3.1-myc to produce myc-hCAN Ct. This C-terminal domain was previously shown to bind to the adenovirus capsid (56).
TABLE 2.
Interactions described in this study
| Nucleoporin | NPC localizationa | Motifb | Resultf
|
||
|---|---|---|---|---|---|
| HSV-1 capsid IPc | HSV-1 pUL25 IPc | PrV pUL25 yeastd | |||
| CAN/Nup214e | Cyto | FG, Gly, CC, BP, LZ, AH | + | + | + |
| hNup153 | Nuc | FG, Gly, ZF | NT | NT | − |
| hNup133 | Cyto + Nuc | BP, AHR | − | − | NT |
| hNup54 | Cyto + Nuc | FG, Gly, CC | − | − | NT |
| hNup50 | Nuc | FG, RBD | − | − | NT |
| hCG1 | Cyto | FG, ZF, CC | − | + | +g |
| POM121 | PM | FG, Gly, TM | NT | NT | − |
| Nup98 | Nuc | FG, Gly | NT | NT | − |
Cyto, cytoplasmic face; Nuc, nucleoplasmic face; PM, pore membrane (11).
FG, FG repeats; Gly, glycosylated; RBD, Ran-binding domain; ZF, zinc fingers; CC, coiled coil; LZ, leucine zipper; AH, amphipathic helix; BD, β-propeller; TM, transmembrane helix.
See Table 3 for details.
NT, not tested.
Only amino acids 1 to 380 (see Table 3 for details).
To examine interactions between capsids and nucleoporins, Vero cells were transfected with a plasmid encoding the myc-tagged nucleoporins. After 6 h, the cells were infected with 5 PFU/cell of a UL37 null virus, FRΔUL37, and incubated for a further 15 h. FRΔUL37 has a failure in virion assembly that results in the accumulation of unenveloped capsids in the cytoplasm of infected cells. Recently, it was reported that these capsids retain the ability to deliver their genomes into nuclei, showing that they contain all the functions necessary for interacting with the NPC (47). The accumulation of large numbers of cytoplasmic capsids that are capable of binding to the NPC was expected to enhance the detection of interacting partners. Thus, the cells were lysed, and the cytoplasmic capsids were immunoprecipitated using the antibody PTNC, raised against purified capsids. The immunoprecipitated pellet was analyzed by Western blotting using MAb 9E10 to detect the presence of the myc-tagged nucleoporins. As shown in Fig. 1A, the only protein identified by coimmunoprecipitation with capsids was the C-terminal region of CAN/Nup214 expressed from myc-hCAN Ct.
FIG. 1.

Interaction of HSV-1 capsids with nucleoporins. (A) Vero cells were transfected with plasmids expressing myc-tagged nucleoporins hNup133, hNup54, hNup50, hCAN Ct, and hCG1 for 6 h before being mock infected or infected with FRΔUL37. Fifteen hours after infection, cells were lysed, and capsids were immunoprecipitated using anti-capsid antibody PTNC. Cell extracts (bottom) and immune complexes (top and middle) were separated by SDS-PAGE and analyzed by Western blotting using anti-VP5 MAb DM165 to reveal the capsids (top) or anti-myc MAb 9E10 to reveal the nucleoporins (middle and bottom). The positions of the VP5 and myc-hCAN Ct bands and of protein size standards are indicated to the left and right, respectively. Ig indicates the immunoglobulin heavy chain. (B) Vero cells were transfected as described above (A) and incubated for 15 h before being mock infected (−) or infected (+) with 50 PFU/cell of WT HSV-1. Thirty minutes after infection, cells were lysed, and capsids were immunoprecipitated using PTNC antibody. Cell extracts (bottom) and immune complexes (top and middle) were separated by SDS-PAGE and analyzed as described above (A). The relative amount of protein present in the immunoprecipitate (IP) is indicated as a percentage of the amount present in the cell extract (CE) (IP:CE ratio).
To rule out the possibility that the nucleoporins were interacting only with newly synthesized proteins rather than assembled capsids in these FRΔUL37-infected cells and to determine whether a similar interaction pattern could be observed during the initial stages of infection, we examined the behavior of incoming capsids following virus infection. Vero cells expressing the selected nucleoporins were infected with 50 PFU/cell of WT HSV-1 and incubated at 37°C for 30 min before undergoing lysis and immunoprecipitation of capsids. Once again, the only protein that coimmunoprecipitated with capsids was myc-hCAN Ct, although the amount of immunoprecipitated capsids was considerably lower than that of the FRΔUL37 capsids (Fig. 1B).
Identification of pUL25-binding nucleoporins.
Any specific interaction between capsids and nuclear pore proteins would be expected to involve capsid proteins that have a role in pore binding and DNA release. Two potential candidates, pUL25 and pUL36, have been identified by studying ts mutants. pUL36 is a very large tegument protein that also appears to have several functions during virus assembly and infection (2). pUL25 is a minor capsid protein that is required for viral DNA release at the nuclear pore (44). The UL25 ORF from HSV-1 with an HA tag inserted at the N terminus was cloned under the control of the human cytomegalovirus immediate-early promoter. Vero cells expressing HA-pUL25 and the different myc-tagged nucleoporins were lysed 15 h after transfection, and coimmunoprecipitations were carried out with myc-specific antibody A14. The immunoprecipitated pellet was analyzed by Western blotting using anti-HA MAb F7 to detect the presence of pUL25 (Fig. 2). HA-pUL25 was detected only in samples containing the C terminus of CAN/Nup214 (hCAN Ct) and hCG1. The specificity of the interaction was confirmed using another, irrelevant, HA-tagged HSV-1 protein, pUL32, which did not interact with any of the nucleoporins tested. The reverse experiment showed that myc-CAN Ct could also be coimmunoprecipitated by pUL25 and not by pUL32 (data not shown).
FIG. 2.

Interaction of pUL25 with nucleoporins. Vero cells cotransfected with plasmids expressing myc-tagged nucleoporins, as described in the legend of Fig. 1, and either HA-pUL25 or HA-pUL32 were lysed 15 h after transfection. Following immunoprecipitation with anti-myc antibody A14, cell extracts (top) and immune complexes (middle and bottom) were separated by SDS-PAGE and analyzed by Western blotting using anti-HA MAb F7 to reveal the presence of HA-pUL25 (left top and left middle) and HA-pUL32 (right top and right middle). The presence of the nucleoporins in the immunoprecipitate was confirmed by stripping the blots and reprobing with anti-myc MAb 9E10 (bottom). The positions of the HA-pUL25 and HA-pUL32 bands and of protein size standards are indicated to the left and right, respectively. UP indicates an unknown protein in the cell extracts that cross-reacts with anti-HA MAb F7. The relative amount of protein present in the immunoprecipitate (IP) is indicated as a percentage of the amount present in the cell extract (CE) (IP:CE ratio).
To provide an independent confirmation of the interaction between pUL25 and the nucleoporins, we used the yeast two-hybrid system. However, as reported previously (26), we observed that the fusion of the LexA domain to the N terminus of the HSV-1 UL25 ORF resulted in an autoactivation of the β-galactosidase reporter gene, making it unsuitable for yeast two-hybrid analyses. Therefore, we used the UL25 gene from a related herpesvirus, PrV, which did not cause autoactivation. The pUL25 proteins from HSV-1 and PrV share a high level of amino acid identity (54%) and have a conserved functional role in virion formation (25). Thus, PrV pUL25 should provide a functional substitute for pUL25 of HSV-1 and would have the additional benefit of demonstrating whether the interaction with CAN Ct is conserved among alphaherpesviruses. LexA-PrV UL25 was screened for interactions against a set of full-length nucleoporins and specific subdomains that had previously been used to identify interactions with the human immunodeficiency virus type 1 (HIV-1) Rev and Vpr proteins (27). In addition, the C-terminal region of CAN/Nup 214 (hCAN Ct) (amino acids 1594 to 2090) used in the immunoprecipitation studies was cloned into pGAD. The results are summarized in Table 3. HIV-1 Rev, which interacts with FG repeat domains, was used as a positive control and indeed gave a positive β-galactosidase signal with all of the FG domain-containing nucleoporins. In contrast, with PrV pUL25, we detected a positive signal only with hCG1 at amino acids 1 to 380 and hCAN Ct (Table 3). In the latter case, the specificity of the interaction could not be tested since yeast cells transformed with hCAN Ct and the LexA control plasmid did not grow. Nevertheless, these results supported the evidence obtained from the coimmunoprecipitation experiments.
TABLE 3.
Yeast two-hybrid interactions between PrV UL25 and a selection of nucleoporin constructs
| Gene (residues)a | β-Gal activityb
|
||
|---|---|---|---|
| HIV-1 Revc | UL25 PrVc | LexA alone | |
| CAN/Nup214 (1-256) | − | − | − |
| CAN/Nup214 (586-1059) | − | − | − |
| CAN/Nup214 FG | + | − | − |
| hCAN Ct (CAN/Nup214 Ct) (1594-2090) | NT | + | 0 |
| POM121 (73-801) | − | − | − |
| POM121 FG | + | − | − |
| Nup98 FG | + | − | − |
| hCG1 Full length (1-423) | 0 | 0 | 0 |
| hCG1 (1-380) | + | + | − |
| hCG1 FG | + | − | − |
| hCG1 Nt | − | − | − |
| Nup153 full length | + | − | − |
| Nup153 Nt | − | − | − |
Cloned into vector pGAD, fusing the GAL4-activating domain to the N terminus of the indicated protein. These plasmids were described previously in reference 27, except for hCAN Ct, which contains the C-terminal region of CAN/Nup214, encoding amino acids 1594 to 2090, which was also present in the myc-hCAN Ct plasmid used in the immunoprecipitation studies (Fig. 1 and 2).
− indicates no interaction, + indicates an interaction, and 0 indicates poor yeast growth. NT, not tested.
Cloned into vector pLexA, fusing the LexA domain to the N terminus of the indicated protein.
In all three types of assays, capsid immunoprecipitation, pUL25 immunoprecipitation, and yeast two-hybrid analyses, the C-terminal region of CAN/Nup214 was detected as a binding partner, with hCG1 also being identified by the latter experiments (Table 2). Therefore, from these results, we conclude that HSV-1 capsids are able to interact with CAN/Nup214 (and possibly with hCG1) and that this interaction is likely to be mediated by pUL25.
Effect of depletion of CAN/Nup214 on early stages of infection.
In order to examine its role during HSV-1 infection, we used shRNA technology to deplete CAN/Nup214. To monitor the effects of this depletion, we constructed an HSV-1 recombinant virus, vICP4CFP-VP26RFP. This virus has monomeric RFP fused to the VP26 capsid protein and CFP fused to ICP4, an immediate-early HSV-1 protein that associates with virus DNA in the nucleus of infected cells. VP26-RFP could be used to monitor the fate of virus capsids, while ICP4-CFP fluorescence acted as a marker for virus DNA entry into the nucleus (17). Thus, since DNA entry into the nucleus is dependent on the interaction between capsids and the NPC triggering the release of the virus genomes, the efficiency of capsid docking could be monitored as a function of the progress of viral DNA replication. When HeLa cells were infected with this virus, several different patterns of fluorescence were observed at 3 h postinfection (Fig. 3A). These patterns were arbitrarily divided into five categories. Cells in category 0 were uninfected (RFP and CFP minus). Category I had RFP capsids in the cytoplasm but no detectable CFP signal. Category II had cells with small ICP4-CFP foci, indicating the presence of viral DNA in the nucleus. Category III had larger but still-discrete ICP4-CFP foci in the nucleus, indicating early DNA synthesis. In category IV, the foci had started to merge into patches, indicating more-extensive DNA replication, and VP26-RFP was present in some nuclei.
FIG. 3.

CAN/Nup214 depletion delays HSV-1 DNA replication. (A) Cells 3 h after infection with vICP4CFP-VP26RFP. Cells were classified into different categories based on the extent of viral DNA replication as determined by ICP4-CFP fluorescence (green). Category 0 comprised cells showing no fluorescence (not shown). The panels show typical examples of cells assigned to category I, with capsids (red) in the cytoplasm but no detectable ICP4-CFP signal. The location of the nucleus is revealed by DAPI staining (blue). Categories II to IV exhibit increasing amounts of ICP4-CFP nuclear fluorescence localized in small (II), large (III), and coalescing (IV) foci. In the latter case, nuclear capsid assembly compartments are visible as VP26RFP foci. Scale bars, 10 μm. (B) Efficiency of CAN/Nup214 silencing. HeLa cells infected with lentiviruses expressing shRNA directed against the luciferase gene (shLuc) or CAN/Nup214 (shCAN) were harvested at 72 h postinfection and analyzed by Western blotting using MAb 414, which recognizes the nucleoporins RanBP2/Nup358, CAN/Nup214, Nup153, and Nup62. Nup62 was used as a loading control, and the relative intensities of the other bands were determined in shCAN and shLuc cells as indicated on the right (shLuc values were set at 100%). (C and D) Representative images from shLuc (C) or shCAN (D) cells infected for 3 h with vICP4CFP-VP26RFP. The pattern of infection was estimated by assessing the proportion of cells belonging to each of the categories shown above (A) as determined from the level of ICP4-CFP expression. A total of 248 randomly selected cells was observed, and the relative number of cells in each category, expressed as a percentage of the total number of cells counted, is shown in the graphs on the right. Scale bars, 10 μm.
To analyze the effect of the CAN/Nup214 depletion, HeLa cells were infected with a lentivirus expressing a CAN/Nup214-specific shRNA (shCAN) or with a control lentivirus expressing an shRNA directed against luciferase (shLuc). Western blotting with MAb 414, which recognizes the nucleoporins RanBP2/Nup358, CAN/Nup214, Nup153, and p62 (14), confirmed the selective depletion of CAN/Nup214 (Fig. 3B). Thus, there was a reduction in CAN/Nup214 levels of more than 80% in shCAN-infected cells compared to shLuc cells, whereas the levels of the other nucleoporins were not affected. The p62 protein was used as a loading standard since it is a core protein of the NPC, which was previously reported to be unaffected by CAN/Nup214 depletion (5).
To determine the impact of the CAN/Nup214 depletion on early stages of HSV-1 infection, shLuc and shCAN cells were infected with vICP4CFP-VP26RFP, and the ICP4-CFP fluorescence patterns were assessed at 3 h postinfection according to the classification scheme described above. The pattern seen with the shLuc control cells shows the majority of cells (∼90%) falling into categories III and IV, indicating that viral DNA had entered the nuclei and was replicating (Fig. 3C). In contrast, cells silenced for CAN/Nup214 exhibited very little ICP4-CFP signal, with virtually all the cells falling into categories I and II (Fig. 3D). The large number of cells in category I (>60%) were at the stage where capsids are present in the cytoplasm, but no ICP4-CFP signal was detectable in the nuclei. The other 40% of the cells were placed into category II, as they displayed weak ICP4-CFP fluorescence. These results show that the depletion of CAN/Nup214 leads to a marked delay in viral DNA entering the nucleus but does not entirely prevent it, since some ICP4-CFP signal could still be detected.
To confirm the effect of the depletion of CAN/Nup214 on the delivery of the viral genome into the nucleus and to provide a quantitative assay, we used another HSV-1 recombinant virus, tsK/Luci (provided by C. Preston). This virus is a derivative of the tsK virus in which the luciferase coding region was controlled by the HSV-1 immediate-early ICP0 promoter. As shLuc cells would suppress luciferase expression, HeLa cells were infected with a lentivirus encoding an shRNA specific for the GFP gene (shGFP) to act as a control. shCAN- and shGFP-infected cells were infected for 2 h, 3 h, or 7 h with 5 PFU/cell of tsK/Luci at 31°C, and their luciferase activities were measured. As shown in Fig. 4, the luciferase levels in shGFP-infected cells was markedly higher than that in shCAN-infected cells, with ∼400-times-more activity at 2 h postinfection, ∼200-times-more activity at 3 h postinfection, and ∼15-times-more activity at 7 h postinfection. This shows that the depletion of CAN/Nup214 has a strong effect at early times postinfection, indicating that an early stage of the viral cycle is affected. Furthermore, the rapid increase in the level of luciferase activity after the initial delay suggests that later functions are not disrupted as a consequence of the CAN/Nup214 depletion. This is the type of behavior that would be expected if the CAN/Nup214 depletion was affecting capsid docking and DNA release, leading to a delay in the entry of the viral genome into the nucleus, but was not inhibiting later replication processes. Thus, these results support the role of CAN/Nup214 in viral DNA release.
FIG. 4.
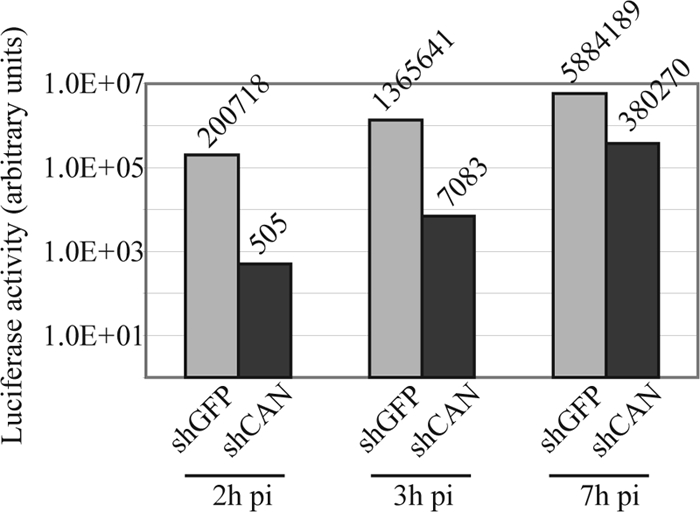
CAN/Nup214 depletion delays HSV-1 gene expression. HeLa cells infected with lentiviruses expressing shRNA directed against the GFP gene (shGFP) or CAN/Nup214 (shCAN) were infected with 5 PFU/cell of tsK/Luci. After incubation at 31°C for 2 h, 3 h, or 7 h, the cells were harvested and processed for luciferase assays. The luciferase activity is indicated on a logarithmic-scaled graph, with the numerical value indicated above each bar. pi, postinfection.
Depletion of CAN/Nup214 does not affect virus entry or ICP4 nuclear import.
Based on the CAN/Nup214 interaction studies described above, the likely explanation for the delay in the onset of replication is that the depletion of CAN/Nup214 inhibits or alters the binding of capsids to the NPC, which prevents the release of viral DNA. However, two other possibilities need to be considered.
The first possibility is that the cell's susceptibility to HSV-1 infection is reduced as a result of changes in cellular protein expression caused by reduced mRNA export consequent on the CAN/Nup214 depletion. This is unlikely, since it was previously reported that in mammalian cells, mRNA export is barely affected by the CAN/Nup214 depletion (22). However, we compared the efficiencies of virus entry in shLuc and shCAN cells by looking at the breakdown in colocalization between the envelope glycoprotein gD and capsids, which occurs when the virion envelope fuses with the plasma membrane and the capsids are released into the cytosol. For these experiments, shLuc or shCAN cells were infected with vICP4CFP-VP26RFP for 2 h in the presence of cycloheximide, a protein synthesis inhibitor. Capsids were recognized by VP26RFP fluorescence, while gD was identified using gD-specific MAb 4846. As a control, purified vICP4CFP-VP26RFP virions were applied directly onto a glass coverslip. The capsid (VP26RFP) signal colocalized with the gD signal (Fig. 5A) in 93% of the particles (Fig. 5C). Similarly, when membrane fusion was inhibited by adsorbing the virus onto cells for 1 h at 4°C, 88% of capsids were gD positive. In contrast, by 2 h postinfection, at 37°C, 80% of capsids in shLuc cells and 83% of capsids in shCAN cells were gD negative (Fig. 5B and C), thereby indicating that the CAN/Nup214 depletion does not affect the cell's susceptibility to infection.
FIG. 5.

Effect of CAN/Nup214 depletion on virus entry. (A) vICP4CFP-VP26RFP virions purified on 5-to-15% Ficoll gradients were spread onto collagen-coated glass coverslips, fixed with 4% PFA, and incubated sequentially with anti-gD antibody, MAb 4846, and Alexa Fluor 633-conjugated goat anti-mouse antibody. Capsids and gD were revealed by RFP fluorescence (red) and Alexa Fluor 633 fluorescence (pseudocolored in green), respectively. (B) shLuc (top and middle) and shCAN (bottom) cells (Fig. 3) were incubated with vICP4CFP-VP26RFP virions for 1 h at 4°C (top) or for 2 h at 37°C in the presence of 200 μM cycloheximide (middle and bottom). The cells were fixed, permeabilized, and stained for gD as described above (A). Deconvoluted pictures of z stacks are shown. Outlines of nuclei as determined by DAPI staining (not shown) are indicated (N). Scale bars, 10 μm. (C) RFP capsids on randomly chosen cells from the experiment shown above were counted. The presence or absence of gD was assessed by examining colocalization between RFP and Alexa Fluor 633 signals using the colocalization function of the LSM510 software. Results are shown as numbers of gD-positive (yellow) and gD-negative (red) capsids expressed as a percentage of the total number of capsids counted. A total of 1,102 particles were analyzed.
As a further demonstration that the depletion of CAN did not block the initial stages of virus infection, infectious-center assays were carried out. The numbers of infectious centers produced in shLuc and shCAN cells were comparable, with average titers of 4.9 × 105 PFU/ml in shLuc cells and 4.8 × 105 PFU/ml in shCAN cells (Table 4), thereby confirming that virus entry and replication are not affected by the depletion of CAN, as expected from the results obtained with the tsK/Luci virus (Fig. 4).
TABLE 4.
Titration of infectious centers in shLuc and shCAN cells infected with vICP4CFP-VP26RFP
| Cell type | Titer (PFU/ml) in expt:
|
||
|---|---|---|---|
| 1 | 2 | 3 | |
| shLuc | 4 × 105 | 4.9 × 105 | 5.8 × 105 |
| shCAN | 4 × 105 | 5.5 × 105 | 4.8 × 105 |
The second possible explanation for the delay in the appearance of the nuclear ICP4-CFP signal is that the depletion of CAN/Nup214 interferes either with ICP4-CFP production or with its nuclear import. This also appears to be unlikely, since earlier studies showed that the depletion of CAN/Nup214 did not affect nuclear protein import (22, 59). However, to examine the effects of the CAN/Nup214 depletion on the production and transport of ICP4, we carried out experiments that confirmed that the nuclear localization of ICP4-CFP expressed from a transfected plasmid was not affected in shCAN cells (Fig. 6A). Moreover, the fluorescence level of ICP4-CFP appeared to be unchanged in shCAN cells compared to that in shLuc cells, indicating no defect in the expression of the protein in the absence of CAN/Nup214 (Fig. 6B). Thus, we conclude that the delay in infection resulting from the depletion of CAN/Nup214 is compatible with its putative function as part of the NPC docking site for the capsid.
FIG. 6.
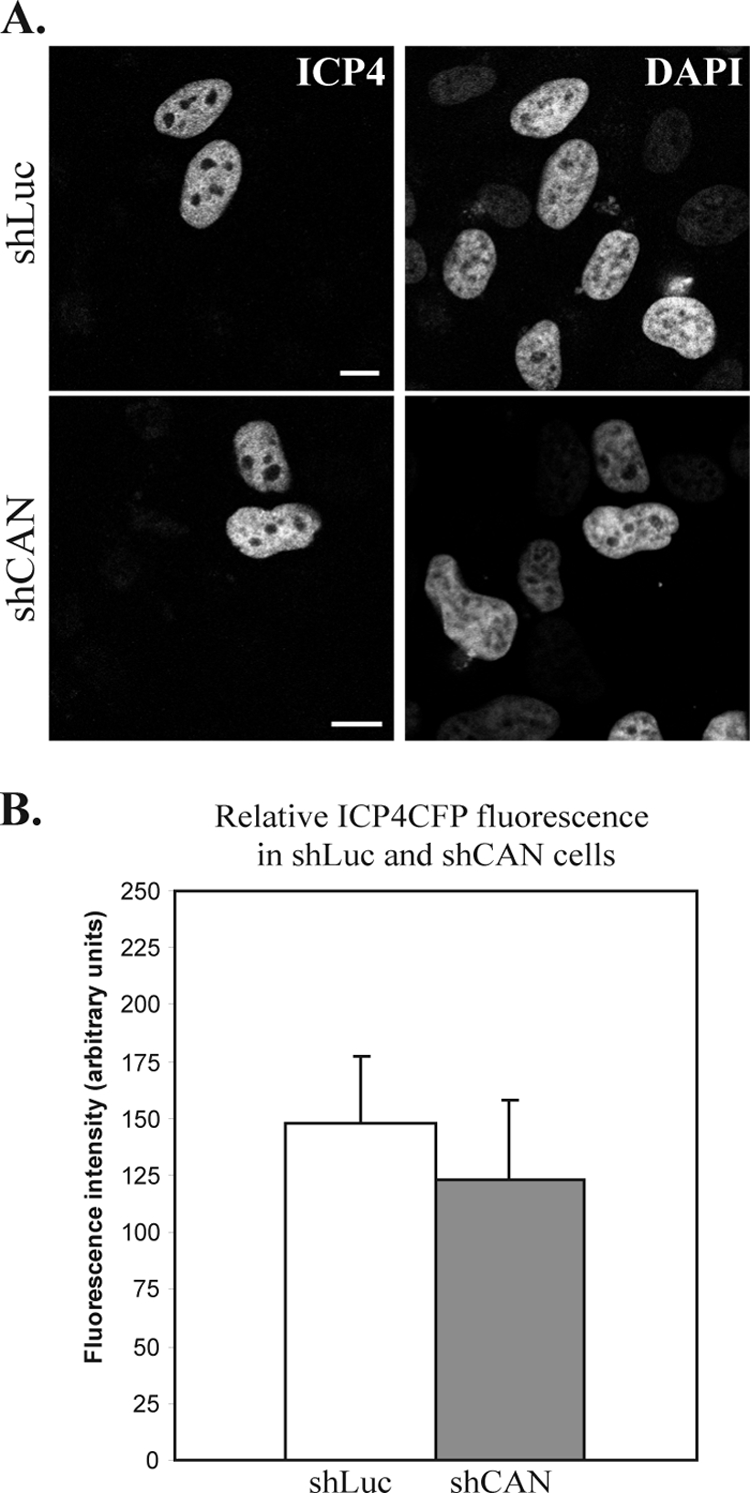
ICP4-CFP expression and localization in shLuc and shCAN cells. (A) shLuc or shCAN cells were transfected with a plasmid encoding ICP4-CFP. Twenty-four hours later, cells were fixed, and ICP4-CFP localization was assessed by direct CFP fluorescence. Nuclei were counterstained with DAPI. Scale bars, 5 μm. (B) The fluorescence level of nine randomly chosen CFP-positive cells per condition (shLuc or shCAN), imaged using the same acquisition parameters, was quantified on a scale from 0 (no fluorescence) to 250 (saturated fluorescence). The values obtained were averaged and compared on a graph.
pUL25 as an interface between capsids and the NPC: interaction with pUL6 and pUL36.
Binding to the NPC is thought to trigger viral DNA release from capsids. Since we have now demonstrated a potential role for pUL25 in the binding of the capsid to the NPC, we decided to investigate the relationship between pUL25 and two other proteins, pUL6 and pUL36, that are known to have a role in DNA release. The mechanism of release is very poorly understood, but comparison with tailed bacteriophages suggests that the DNA exits through the portal, which is a ring-shaped channel formed by a dodecamer of pUL6 proteins and is located at one vertex of the capsid (7, 8, 38). Also, as mentioned above, pUL36 is important for this process, as a ts mutant in UL36 makes particles that are blocked for DNA release at nonpermissive temperatures.
To investigate whether pUL25 interacts with pUL6, Vero cells were cotransfected with plasmids encoding HA-tagged pUL25 (HA-pUL25) and myc-tagged pUL6 (myc-pUL6). Immunoprecipitation was performed with HA antibody Y11 and resulted in the coprecipitation of myc-pUL6 when expressed in the presence of HA-pUL25 but not when expressed with the HA-tagged importin α1 (HA-Imp α1) control (Fig. 7A, lanes 1 and 4). That there was no nonspecific interaction between HA-pUL25 and the myc tag was shown by the failure of the myc-tagged dynamitin control (myc-Dyn) to coprecipitate with HA-pUL25 (Fig. 7A, lane 3). In addition, no myc-pUL6 band was detected when the immunoprecipitation was carried out using a control antibody, NRP14, directed against Nrsv (Fig. 7A, lane 2). In the inverse experiment, anti-myc antibody A14 pulled down HA-pUL25 only when coexpressed with myc-pUL6 and not with the unrelated myc-Dyn protein (Fig. 7B, lanes 1 and 3). As described above, there was no interaction between myc-pUL6 and HA-Imp α1 (Fig. 7B, lane 4), and NRP14 did not bring down HA-pUL25 (Fig. 7B, lane 2). Although it cannot be ascertained whether the interaction identified in our system involves the portal structure or monomers of pUL6, these results demonstrate that transiently expressed pUL25 and pUL6 can interact and suggest the possibility of an association between pUL25 and the portal in the virus particle.
FIG. 7.
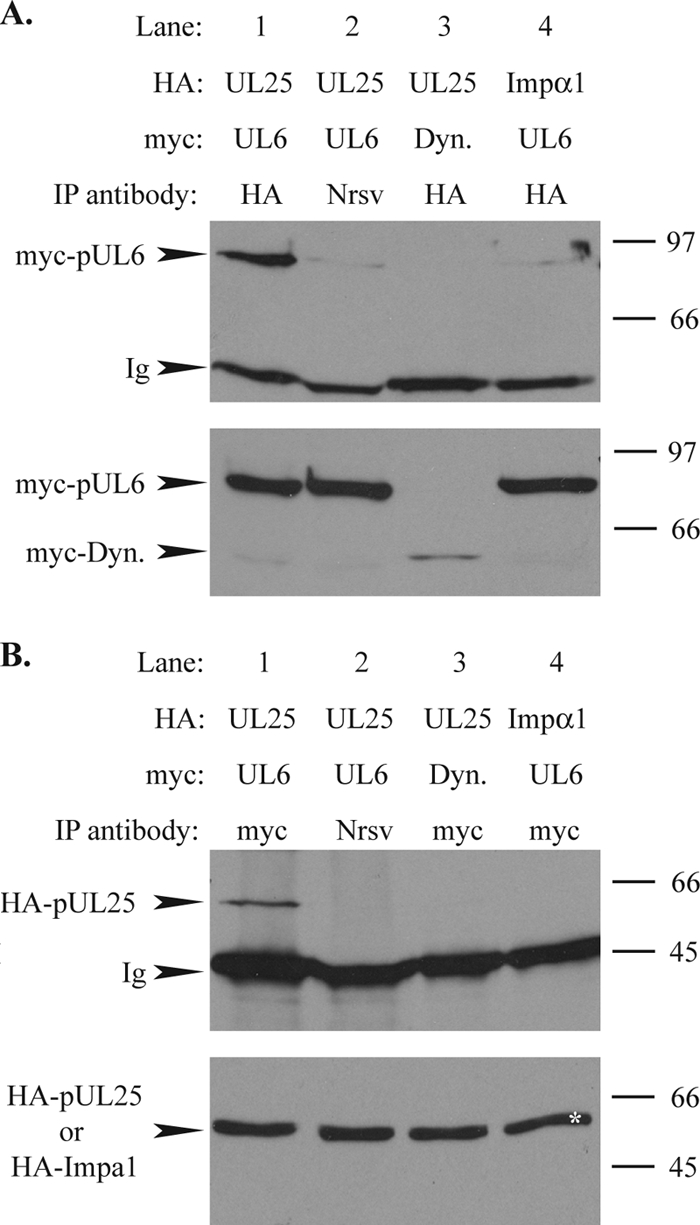
Interaction between pUL25 and pUL6. Vero cells were cotransfected with plasmids expressing HA-pUL25 (UL25) or HA-importin α1 (Impα1) and myc-pUL6 (UL6) or myc-dynamitin (Dyn.) and harvested 24 h after transfection. (A) Following immunoprecipitation with anti-HA antibody Y11 or an antibody against Nrsv, RPN14, cell extracts (bottom) and immune complexes (top) were separated by SDS-PAGE and analyzed by Western blotting using anti-myc MAb 9E10 to reveal the presence of myc-pUL6 and myc-dynamitin. The positions of the myc-pUL6 and myc-dynamitin (myc-Dyn) bands and of protein size standards are indicated to the left and right, respectively. Ig indicates the immunoglobulin heavy chain. (B) Extracts of the transfected cells described above (A) were immunoprecipitated (IP) with anti-myc antibody A14 (myc) or NRP14 (Nrsv). Cell extracts (bottom) and immune complexes (top) were separated by SDS-PAGE and analyzed by Western blotting using anti-HA MAb F7 to reveal the presence of HA-pUL25. The positions of the HA-pUL25 band and of protein size standards are indicated to the left and right, respectively. HA-importin α1, which is the same size as HA-pUL25, is marked with an asterisk in lane 4.
The tegument protein pUL36 has also been shown to be implicated in viral DNA uncoating (2, 24), and an interaction between pUL25 and the C terminus of pUL36 was reported previously for both PrV and HSV-1 (9). In order to confirm this interaction in HSV-1, plasmids expressing the last 1,127 or 719 amino acids of pUL36, each fused at its N terminus to a myc tag, were constructed (Table 1). Their products were designated myc-pUL36 2037 and myc-pUL36 2446, respectively, where the number specifies the position of the N-terminal pUL36 residue in the sequence of the full-length, 3,164-amino-acid protein. Both myc-pUL36 2037 and myc-pUL36 2446 were precipitated by HA antibody Y11 when coexpressed with HA-pUL25 (Fig. 8, lanes 1 and 3) but not when expressed on their own (Fig. 8, lanes 2 and 4). Similarly, carrying out the immunoprecipitation with myc antibody A14 resulted in the coimmunoprecipitation of HA-pUL25 when coexpressed with the myc-pUL36 proteins (Fig. 8, lanes 5 and 6) but not when it was expressed alone (Fig. 8, lane 7). As has been frequently observed with pUL36, both myc-pUL36 2037 and myc-pUL36 2446 gave rise to many breakdown products (Fig. 8, bottom, lanes 1 to 4). Interestingly, most of the myc-pUL36 2037 breakdown products coprecipitated with HA-pUL25, whereas those of myc-pUL36 2446 did not (Fig. 8, top, lanes 1 and 3). Since the myc tag is at the N terminus of each construct, all the fragments represent C-terminal truncations, suggesting that the deletion of the C terminus of myc-pUL36 2446 caused a loss of binding to pUL25, whereas the deletion of the same part of myc-pUL36 2037 did not. These observations suggested the existence of a second HA-pUL25-binding domain in myc-pUL36 2037 or, alternatively, a multimerization domain on pUL36 that could be indirectly involved in the interaction.
FIG. 8.

Interaction between pUL25 and pUL36. Vero cells were cotransfected with plasmids expressing HA-pUL25 and either myc-pUL36 2037 (encoding C-terminal amino acids 2037 to 3164 of pUL36) or myc-pUL36 2446 (encoding C-terminal amino acids 2446 to 3164) and harvested 24 h after transfection. Cell extracts were immunoprecipitated (IP) by anti-HA antibody Y11 (top left) or anti-myc antibody A14 (top right). Cell extracts (bottom) and immune complexes (top) were separated by SDS-PAGE and analyzed by Western blotting (WB) for the presence of myc-tagged pUL36 fragments (left) or HA-pUL25 (right), respectively. The positions of myc-pUL36 bands, HA-pUL25 bands, and protein size standards are indicated to the left, right, and center, respectively. The full-length myc-pUL36 2037 and myc-pUL36 2446 bands are marked with asterisks. Note that breakdown products of the myc-pUL36 fragments are present in both cell extracts and the HA-pUL25 and myc-pUL36 2037 coimmunoprecipitation sample (lane 1) but not in the HA-pUL25 and myc-pUL36 2446 coimmunoprecipitation sample (lane 3). Ig indicates the immunoglobulin heavy chain.
To verify this, we constructed several C-terminal truncations of myc-pUL36 2037 and myc-pUL36 2446 (Fig. 9A) and tested their interactions with HA-pUL25. In the case of myc-pUL36 2446, the deletion of the last 60 amino acids, which removes the previously mapped (9) capsid-binding domain (CBD/pUL25 BDI) (Fig. 9A), was sufficient to prevent the interaction with HA-pUL25 (Fig. 9C, lane 3104). However, with myc-pUL36 2037, the removal of CBD/pUL25 BD I did not block binding since truncations lacking up to 811 residues from the C terminus could still precipitate HA-pUL25 (Fig. 9B, lanes 2572, 2504, and 2353), although deletions lacking a further 153 amino acids could not (Fig. 9B, lane 2200). This surprising result confirms the observations shown in Fig. 8 and demonstrates the existence of a second, independent pUL25-binding domain (pUL25 BD II) (Fig. 9A) located between residues 2037 and 2353 of pUL36.
FIG. 9.

Mapping of pUL25-binding domains of pUL36. (A) Representation of the pUL36 fragment of amino acids 2037 to 3164. The arrowheads shown above mark the last amino acids encoded by the C-terminal truncations for myc-pUL36 2446, while those below are for myc-pUL36 2037. CBD/pUL25 BD I and pUL25 BD II indicate the pUL25-binding domains (pUL25 BD I is equivalent to the previously mapped capsid-binding domain) (9). (B and C) Interaction of HA-pUL25 with C-terminal truncations of myc-pUL36 2037 and myc-pUL36 2446. Vero cells were cotransfected with plasmids expressing HA-pUL25 and either full-length myc-pUL36 2037 or C-terminal truncations of myc-pUL36 2037 (B) or full-length myc-pUL36 2446 or C-terminal truncations of myc-pUL36 2446 (C), as specified above (A). Twenty-four hours after transfection, the cells were harvested, and extracts were immunoprecipitated (IP) with anti-myc antibody A14. Cell extracts (middle) and immune complexes (top) were separated by SDS-PAGE and analyzed for the presence of HA-pUL25 by Western blotting using anti-HA MAb F7. The immunoprecipitate blots were then stripped and reprobed with anti-myc MAb 9E10 to reveal the presence of the myc-pUL36 truncation products (bottom). The positions of the HA-pUL25 bands and of protein size standards are indicated to the left and right, respectively. Control extracts from cells coexpressing HA-pUL25 and myc-pUL6 were immunoprecipitated using the same procedure. The full-length myc-pUL36 truncation products and the myc-pUL6 band are marked with asterisks.
DISCUSSION
CAN/Nup214 is a nuclear receptor for the herpesvirus capsid.
Large nuclear-replicating DNA viruses face the problem of transmitting their genomes into the nucleus. To achieve this, herpesviruses have developed mechanisms for transporting the genome-containing capsid to the nuclear pore, which provides the route through the nuclear membrane. It has long been known that incoming capsids localize to the vicinity of nuclear pores before releasing their DNA. However, the nature of any interaction with the pore and the trigger for DNA release are largely unknown. In this study, we examined interactions between the HSV-1 capsid and selected components of the NPC. Since endogenous nucleoporins are sequestered predominantly in NPCs, we looked at interactions of capsids with uncomplexed proteins by overexpressing selected nucleoporins from plasmids. The set of nucleoporins examined included members from different parts of the NPC. Thus, CAN/Nup214 and hCG1 are associated with the cytoplasmic face; hNup153, hNup50, and hNup98 are components of the nucleoplasmic face; and hNup133 and hNup54 are located on both faces (Table 2). Many NPC proteins contain characteristic FG repeat sequences, which are known to be an important part of the selective gate formed by the pore (for a review, see reference 13). To examine whether they played a role in HSV-1 capsid binding, we included several FG repeat proteins (CAN/Nup214, hNup153, hNup54, hNup50, hCG1, hPOM121, and hNup98). Of the nucleoporins tested, only CAN/Nup214 and hCG1 were identified as being binding partners of pUL25, and only CAN/Nup214 coprecipitated with HSV-1 capsids. Although in our assays the nucleoporins were overexpressed and are unlikely to be incorporated into NPCs, it is interesting that both CAN/Nup214 and hCG1 contain FG repeats, and both are normally localized to the cytoplasmic face of the nuclear pore, where they would be accessible to incoming capsids. Although the precise role of the pUL25-CAN/Nup214 interaction in capsid docking and DNA release is unknown, the functional importance of CAN/Nup214 in these processes was confirmed by shRNA depletion experiments. These experiments showed that reducing the amount of the endogenous protein seriously inhibited the entry of the virus genome into the nucleus. Thus, these studies have identified a likely NPC receptor for HSV-1 capsids.
A recent study has suggested a role for pUL36 and the nucleoporins Nup358/RanBP2 and CAN/Nup214 in the attachment of capsids to the nucleus by syringe-loading and RNA silencing experiments (10). That report demonstrated that antibodies directed against Nup358/RanBP2 had an inhibitory effect on the number of incoming capsids associating with the nucleus, and a similar effect was observed when Nup358/RanBP2 levels were reduced by RNA silencing. Nup358/RanBP2 is a large nucleoporin that is a major component of the NPC cytoplasmic filaments (59, 61, 63, 64) and is therefore appropriately located to act as a receptor for the docking of herpesvirus capsids. A role for CAN/Nup214 in the attachment of capsids to the nucleus was also suggested by RNA silencing experiments, but targeting CAN/Nup214 with an antibody failed to inhibit the capsid-nucleus association. However, it should be noted that the antibody used in this case, QE5, also recognizes other nucleoporins (Nup153 and Nup62) (40) and therefore cannot be considered to be CAN/Nup214 specific. Our silencing data also suggest a role of CAN/Nup214 in nuclear pore binding and/or viral DNA release, in agreement with the results described previously by Copeland et al. (10). However, by using immunoprecipitation to examine capsid-nucleoporin interactions directly, we were able to identify CAN/Nup214 as being a likely docking target for incoming capsids. Interestingly, Nup358/RanBP2 and CAN/Nup214 interact with each other (5) and may therefore constitute a complex that is targeted by herpesvirus capsids.
It is interesting to compare our findings from interactions of HSV-1 capsids with the NPC with findings from studies of another nuclear-replicating virus. Adenovirus capsids are also transported to nuclear pores. However, unlike the case with herpesviruses, where the capsid retains its integrity and releases its genome through the specialized portal vertex, binding to the NPC triggers capsid disassembly in adenoviruses, resulting in viral DNA uncoating. Despite these very different strategies, it is interesting that adenovirus capsids also interact with CAN/Nup214 and that the same domain is involved in binding to both types of capsid (56). These data show that despite adopting different strategies for uncoating their viral DNA at the NPC, adenovirus and herpesvirus capsids target the same domain of the NPC for binding.
pUL25 acts as an interface between the incoming capsid and the NPC.
pUL25 was recently shown to have a role in viral DNA uncoating at the nuclear pore (44). It also has a well-characterized role in stabilizing newly packaged DNA within capsids (34, 53). The crystallographic structure of residues 134 to 580 of pUL25 has been solved and shows a compact, α-helix-rich central core with several solvent-exposed loops, which suggests an ability to interact with a number of different partners (6). Here we demonstrate that pUL25 is able to interact with at least four different proteins, the nucleoporins CAN/Nup214 and hCG1 and the viral proteins pUL6 and pUL36, all of which are likely to be involved in the release of viral DNA at nuclear pores.
The finding that pUL25 binds the C-terminal region of CAN/Nup214 suggests a direct role in capsid binding to the NPC. We also identified a second pUL25-binding nucleoporin, hCG1, although it did not bind to HSV-1 capsids in our assays. It is interesting that CAN/Nup214 and hCG1 share numerous properties. Thus, both are localized on the cytoplasmic side of the NPC (for a review, see reference 50), the yeast homologues of CAN/Nup214 and hCG1 have been shown to act as “modular duplicates” in the NPC architecture in terms of position and fold arrangements (1), and both were previously shown to be involved in the localization of other virus genomes into the nucleus. As mentioned above, adenovirus capsids bind to CAN/Nup214, while the HIV-1 Vpr protein binds hCG1 (27, 56). The pUL25-pUL36 interaction was already known, and a pUL25-binding domain was mapped to the last 60 residues of pUL36 (9). We have confirmed this interaction and identified a second interaction domain between amino acids 2037 and 2353 of pUL36. The independence of these two binding domains indicates that the binding of pUL36 to pUL25 can occur through different mechanisms. The interaction of pUL25 with the portal protein pUL6 has not been described previously but is also compatible with the role of pUL25 in viral DNA packaging and release.
From transport to viral DNA release.
Both pUL25 and pUL36 have a role in viral DNA uncoating (2, 44). Since the two proteins interact with each other, it is likely that they form a complex involved in DNA release. pUL36 is an inner tegument protein that is also involved in particle assembly and is essential for cytoplasmic envelopment. It has been demonstrated that pUL36 remains associated with incoming capsids until they reach the nucleus (20, 30). The inner tegument has been shown to promote capsid movement on microtubules (62), and pUL36 has been directly implicated in microtubule-based capsid transport (31). It is likely, therefore, that pUL36 contains the viral receptors for microtubule-linked motors. Altogether, these data strongly suggest a role for the inner tegument, and particularly pUL36, in the transport of capsids toward the nucleus.
pUL6, pUL25, and pUL36 are all associated with the capsid. pUL6 forms the portal that occupies a unique vertex in the capsid shell (7, 8, 38). pUL25 is a minor capsid protein, which preferentially associates with DNA-containing capsids (C capsids) (54). It localizes close to the capsid vertices (37) and has been reported to form part of the additional density seen associated with the vertices in reconstructions of DNA-containing C capsids (58). pUL36 has also been suggested to be on the vertices of capsids present within mature virions (65). The interaction between these two proteins is therefore consistent with their proposed locations on the capsid. In contrast, the interaction of pUL25 and pUL6 described here is not supported by their known spatial arrangements but is consistent with their functions in DNA packaging and release. This suggests the possibility that pUL25 occupies a second position on the capsid in close proximity to the portal.
Given the duality of binding sites of pUL36 for pUL25 and of pUL25 for the capsid (through the pentonal vertices and the portal), we hypothesize that there are two forms of the pUL36-pUL25 complex on the capsid, each with different roles. Thus, penton-associated pUL25-pUL36 would be involved in tegument assembly, envelopment, and egress (16, 47). However, we suggest that pUL25 is also likely to be present at the portal, probably together with pUL36, as a consequence of the interaction between pUL25 and pUL6. In this location, both pUL25 and pUL36 would be suitably positioned to interact with the NPC and trigger the release of the encapsidated DNA through the nuclear pore.
Acknowledgments
We thank Yves Gaudin and Hélène Raux for helpful advice and Valerie Preston and Duncan McGeoch for critical reading of the manuscript. We also thank David McNab and Marion McElwee for excellent technical assistance. We are very grateful to those listed throughout the text for the provision of reagents.
This work was funded by the MRC (United Kingdom), by the CNRS (France), and by a grant from the French Education and Research Ministry.
Footnotes
Published ahead of print on 22 April 2009.
REFERENCES
- 1.Alber, F., S. Dokudovskaya, L. M. Veenhoff, W. Zhang, J. Kipper, D. Devos, A. Suprapto, O. Karni-Schmidt, R. Williams, B. T. Chait, A. Sali, and M. P. Rout. 2007. The molecular architecture of the nuclear pore complex. Nature 450695-701. [DOI] [PubMed] [Google Scholar]
- 2.Batterson, W., D. Furlong, and B. Roizman. 1983. Molecular genetics of herpes simplex virus. VIII. Further characterization of a temperature-sensitive mutant defective in release of viral DNA and in other stages of the viral reproductive cycle. J. Virol. 45397-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beck, M., F. Forster, M. Ecke, J. M. Plitzko, F. Melchior, G. Gerisch, W. Baumeister, and O. Medalia. 2004. Nuclear pore complex structure and dynamics revealed by cryoelectron tomography. Science 3061387-1390. [DOI] [PubMed] [Google Scholar]
- 4.Belgareh, N., G. Rabut, S. W. Bai, M. van Overbeek, J. Beaudouin, N. Daigle, O. V. Zatsepina, F. Pasteau, V. Labas, M. Fromont-Racine, J. Ellenberg, and V. Doye. 2001. An evolutionarily conserved NPC subcomplex, which redistributes in part to kinetochores in mammalian cells. J. Cell Biol. 1541147-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bernad, R., H. van der Velde, M. Fornerod, and H. Pickersgill. 2004. Nup358/RanBP2 attaches to the nuclear pore complex via association with Nup88 and Nup214/CAN and plays a supporting role in CRM1-mediated nuclear protein export. Mol. Cell. Biol. 242373-2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bowman, B. R., R. L. Welschhans, H. Jayaram, N. D. Stow, V. G. Preston, and F. A. Quiocho. 2006. Structural characterization of the UL25 DNA-packaging protein from herpes simplex virus type 1. J. Virol. 802309-2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cardone, G., D. C. Winkler, B. L. Trus, N. Cheng, J. E. Heuser, W. W. Newcomb, J. C. Brown, and A. C. Steven. 2007. Visualization of the herpes simplex virus portal in situ by cryo-electron tomography. Virology 361426-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang, J. T., M. F. Schmid, F. J. Rixon, and W. Chiu. 2007. Electron cryotomography reveals the portal in the herpesvirus capsid. J. Virol. 812065-2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coller, K. E., J. I. Lee, A. Ueda, and G. A. Smith. 2007. The capsid and tegument of the alphaherpesviruses are linked by an interaction between the UL25 and VP1/2 proteins. J. Virol. 8111790-11797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Copeland, A. M., W. W. Newcomb, and J. C. Brown. 2009. Herpes simplex virus replication: roles of viral proteins and nucleoporins in capsid-nucleus attachment. J. Virol. 831660-1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cronshaw, J. M., A. N. Krutchinsky, W. Zhang, B. T. Chait, and M. J. Matunis. 2002. Proteomic analysis of the mammalian nuclear pore complex. J. Cell Biol. 158915-927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cunningham, C., and A. J. Davison. 1993. A cosmid-based system for constructing mutants of herpes simplex virus type 1. Virology 197116-124. [DOI] [PubMed] [Google Scholar]
- 13.D'Angelo, M. A., and M. W. Hetzer. 2008. Structure, dynamics and function of nuclear pore complexes. Trends Cell Biol. 18456-466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davis, L. I., and G. Blobel. 1987. Nuclear pore complex contains a family of glycoproteins that includes p62: glycosylation through a previously unidentified cellular pathway. Proc. Natl. Acad. Sci. USA 847552-7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davison, M. J., V. G. Preston, and D. J. McGeoch. 1984. Determination of the sequence alteration in the DNA of the herpes simplex virus type 1 temperature-sensitive mutant ts K. J. Gen. Virol. 65(Pt. 5)859-863. [DOI] [PubMed] [Google Scholar]
- 16.Desai, P. J. 2000. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J. Virol. 7411608-11618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Everett, R. D., and J. Murray. 2005. ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J. Virol. 795078-5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Everett, R. D., S. Rechter, P. Papior, N. Tavalai, T. Stamminger, and A. Orr. 2006. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol. 807995-8005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Everett, R. D., G. Sourvinos, and A. Orr. 2003. Recruitment of herpes simplex virus type 1 transcriptional regulatory protein ICP4 into foci juxtaposed to ND10 in live, infected cells. J. Virol. 773680-3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Granzow, H., B. G. Klupp, and T. C. Mettenleiter. 2005. Entry of pseudorabies virus: an immunogold-labeling study. J. Virol. 793200-3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greber, U. F., and A. Fassati. 2003. Nuclear import of viral DNA genomes. Traffic 4136-143. [DOI] [PubMed] [Google Scholar]
- 22.Hutten, S., and R. H. Kehlenbach. 2006. Nup214 is required for CRM1-dependent nuclear protein export in vivo. Mol. Cell. Biol. 266772-6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jamieson, D. R., L. H. Robinson, J. I. Daksis, M. J. Nicholl, and C. M. Preston. 1995. Quiescent viral genomes in human fibroblasts after infection with herpes simplex virus type 1 Vmw65 mutants. J. Gen. Virol. 76(Pt. 6)1417-1431. [DOI] [PubMed] [Google Scholar]
- 24.Jovasevic, V., L. Liang, and B. Roizman. 2008. Proteolytic cleavage of VP1-2 is required for release of herpes simplex virus 1 DNA into the nucleus. J. Virol. 823311-3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuhn, J., T. Leege, B. G. Klupp, H. Granzow, W. Fuchs, and T. C. Mettenleiter. 2008. Partial functional complementation of a pseudorabies virus UL25 deletion mutant by herpes simplex virus type 1 pUL25 indicates overlapping functions of alphaherpesvirus pUL25 proteins. J. Virol. 825725-5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee, J. H., V. Vittone, E. Diefenbach, A. L. Cunningham, and R. J. Diefenbach. 2008. Identification of structural protein-protein interactions of herpes simplex virus type 1. Virology 378347-354. [DOI] [PubMed] [Google Scholar]
- 27.Le Rouzic, E., A. Mousnier, C. Rustum, F. Stutz, E. Hallberg, C. Dargemont, and S. Benichou. 2002. Docking of HIV-1 Vpr to the nuclear envelope is mediated by the interaction with the nucleoporin hCG1. J. Biol. Chem. 27745091-45098. [DOI] [PubMed] [Google Scholar]
- 28.Lim, R. Y., and B. Fahrenkrog. 2006. The nuclear pore complex up close. Curr. Opin. Cell Biol. 18342-347. [DOI] [PubMed] [Google Scholar]
- 29.Lowenstein, P. R., E. E. Morrison, D. Bain, P. Hodge, C. M. Preston, P. Clissold, N. D. Stow, T. A. McKee, and M. G. Castro. 1994. Use of recombinant vectors derived from herpes simplex virus 1 mutant tsK for short-term expression of transgenes encoding cytoplasmic and membrane anchored proteins in postmitotic polarized cortical neurons and glial cells in vitro. Neuroscience 601059-1077. [DOI] [PubMed] [Google Scholar]
- 30.Luxton, G. W., S. Haverlock, K. E. Coller, S. E. Antinone, A. Pincetic, and G. A. Smith. 2005. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc. Natl. Acad. Sci. USA 1025832-5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luxton, G. W., J. I. Lee, S. Haverlock-Moyns, J. M. Schober, and G. A. Smith. 2006. The pseudorabies virus VP1/2 tegument protein is required for intracellular capsid transport. J. Virol. 80201-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Macara, I. G. 2001. Transport into and out of the nucleus. Microbiol. Mol. Biol. Rev. 65570-594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McBride, K. M., G. Banninger, C. McDonald, and N. C. Reich. 2002. Regulated nuclear import of the STAT1 transcription factor by direct binding of importin-alpha. EMBO J. 211754-1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McNab, A. R., P. Desai, S. Person, L. L. Roof, D. R. Thomsen, W. W. Newcomb, J. C. Brown, and F. L. Homa. 1998. The product of the herpes simplex virus type 1 UL25 gene is required for encapsidation but not for cleavage of replicated viral DNA. J. Virol. 721060-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murray, J., C. Loney, L. B. Murphy, S. Graham, and R. P. Yeo. 2001. Characterization of monoclonal antibodies raised against recombinant respiratory syncytial virus nucleocapsid (N) protein: identification of a region in the carboxy terminus of N involved in the interaction with P protein. Virology 289252-261. [DOI] [PubMed] [Google Scholar]
- 36.Newcomb, W. W., F. P. Booy, and J. C. Brown. 2007. Uncoating the herpes simplex virus genome. J. Mol. Biol. 370633-642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Newcomb, W. W., F. L. Homa, and J. C. Brown. 2006. Herpes simplex virus capsid structure: DNA packaging protein UL25 is located on the external surface of the capsid near the vertices. J. Virol. 806286-6294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Newcomb, W. W., R. M. Juhas, D. R. Thomsen, F. L. Homa, A. D. Burch, S. K. Weller, and J. C. Brown. 2001. The UL6 gene product forms the portal for entry of DNA into the herpes simplex virus capsid. J. Virol. 7510923-10932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ojala, P. M., B. Sodeik, M. W. Ebersold, U. Kutay, and A. Helenius. 2000. Herpes simplex virus type 1 entry into host cells: reconstitution of capsid binding and uncoating at the nuclear pore complex in vitro. Mol. Cell. Biol. 204922-4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pante, N., R. Bastos, I. McMorrow, B. Burke, and U. Aebi. 1994. Interactions and three-dimensional localization of a group of nuclear pore complex proteins. J. Cell Biol. 126603-617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patel, S. S., B. J. Belmont, J. M. Sante, and M. F. Rexach. 2007. Natively unfolded nucleoporins gate protein diffusion across the nuclear pore complex. Cell 12983-96. [DOI] [PubMed] [Google Scholar]
- 42.Peters, R. 2005. Translocation through the nuclear pore complex: selectivity and speed by reduction-of-dimensionality. Traffic 6421-427. [DOI] [PubMed] [Google Scholar]
- 43.Preston, C. M. 1979. Control of herpes simplex virus type 1 mRNA synthesis in cells infected with wild-type virus or the temperature-sensitive mutant tsK. J. Virol. 29275-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Preston, V. G., J. Murray, C. M. Preston, I. M. McDougall, and N. D. Stow. 2008. The UL25 gene product of herpes simplex virus type 1 is involved in uncoating of the viral genome. J. Virol. 826654-6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raux, H., A. Flamand, and D. Blondel. 2000. Interaction of the rabies virus P protein with the LC8 dynein light chain. J. Virol. 7410212-10216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ribbeck, K., and D. Gorlich. 2001. Kinetic analysis of translocation through nuclear pore complexes. EMBO J. 201320-1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roberts, A. P., F. Abaitua, P. O'Hare, D. McNab, F. J. Rixon, and D. Pasdeloup. 2009. Differing roles of inner tegument proteins pUL36 and pUL37 during entry of herpes simplex virus type 1. J. Virol. 83105-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roth, M. B., A. M. Zahler, and J. A. Stolk. 1991. A conserved family of nuclear phosphoproteins localized to sites of polymerase II transcription. J. Cell Biol. 115587-596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rout, M. P., J. D. Aitchison, M. O. Magnasco, and B. T. Chait. 2003. Virtual gating and nuclear transport: the hole picture. Trends Cell Biol. 13622-628. [DOI] [PubMed] [Google Scholar]
- 50.Schwartz, T. U. 2005. Modularity within the architecture of the nuclear pore complex. Curr. Opin. Struct. Biol. 15221-226. [DOI] [PubMed] [Google Scholar]
- 51.Shahin, V., W. Hafezi, H. Oberleithner, Y. Ludwig, B. Windoffer, H. Schillers, and J. E. Kuhn. 2006. The genome of HSV-1 translocates through the nuclear pore as a condensed rod-like structure. J. Cell Sci. 11923-30. [DOI] [PubMed] [Google Scholar]
- 52.Sodeik, B., M. W. Ebersold, and A. Helenius. 1997. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 1361007-1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stow, N. D. 2001. Packaging of genomic and amplicon DNA by the herpes simplex virus type 1 UL25-null mutant KUL25NS. J. Virol. 7510755-10765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thurlow, J. K., M. Murphy, N. D. Stow, and V. G. Preston. 2006. Herpes simplex virus type 1 DNA-packaging protein UL17 is required for efficient binding of UL25 to capsids. J. Virol. 802118-2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tran, E. J., and S. R. Wente. 2006. Dynamic nuclear pore complexes: life on the edge. Cell 1251041-1053. [DOI] [PubMed] [Google Scholar]
- 56.Trotman, L. C., N. Mosberger, M. Fornerod, R. P. Stidwill, and U. F. Greber. 2001. Import of adenovirus DNA involves the nuclear pore complex receptor CAN/Nup214 and histone H1. Nat. Cell Biol. 31092-1100. [DOI] [PubMed] [Google Scholar]
- 57.Trus, B. L., N. Cheng, W. W. Newcomb, F. L. Homa, J. C. Brown, and A. C. Steven. 2004. Structure and polymorphism of the UL6 portal protein of herpes simplex virus type 1. J. Virol. 7812668-12671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Trus, B. L., W. W. Newcomb, N. Cheng, G. Cardone, L. Marekov, F. L. Homa, J. C. Brown, and A. C. Steven. 2007. Allosteric signaling and a nuclear exit strategy: binding of UL25/UL17 heterodimers to DNA-filled HSV-1 capsids. Mol. Cell 26479-489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walther, T. C., H. S. Pickersgill, V. C. Cordes, M. W. Goldberg, T. D. Allen, I. W. Mattaj, and M. Fornerod. 2002. The cytoplasmic filaments of the nuclear pore complex are dispensable for selective nuclear protein import. J. Cell Biol. 15863-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Whittaker, G. R., M. Kann, and A. Helenius. 2000. Viral entry into the nucleus. Annu. Rev. Cell Dev. Biol. 16627-651. [DOI] [PubMed] [Google Scholar]
- 61.Wilken, N., J. L. Senecal, U. Scheer, and M. C. Dabauvalle. 1995. Localization of the Ran-GTP binding protein RanBP2 at the cytoplasmic side of the nuclear pore complex. Eur. J. Cell Biol. 68211-219. [PubMed] [Google Scholar]
- 62.Wolfstein, A., C. H. Nagel, K. Radtke, K. Dohner, V. J. Allan, and B. Sodeik. 2006. The inner tegument promotes herpes simplex virus capsid motility along microtubules in vitro. Traffic 7227-237. [DOI] [PubMed] [Google Scholar]
- 63.Wu, J., M. J. Matunis, D. Kraemer, G. Blobel, and E. Coutavas. 1995. Nup358, a cytoplasmically exposed nucleoporin with peptide repeats, Ran-GTP binding sites, zinc fingers, a cyclophilin A homologous domain, and a leucine-rich region. J. Biol. Chem. 27014209-14213. [DOI] [PubMed] [Google Scholar]
- 64.Yokoyama, N., N. Hayashi, T. Seki, N. Pante, T. Ohba, K. Nishii, K. Kuma, T. Hayashida, T. Miyata, U. Aebi, et al. 1995. A giant nucleopore protein that binds Ran/TC4. Nature 376184-188. [DOI] [PubMed] [Google Scholar]
- 65.Zhou, Z. H., D. H. Chen, J. Jakana, F. J. Rixon, and W. Chiu. 1999. Visualization of tegument-capsid interactions and DNA in intact herpes simplex virus type 1 virions. J. Virol. 733210-3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou, Z. H., M. Dougherty, J. Jakana, J. He, F. J. Rixon, and W. Chiu. 2000. Seeing the herpesvirus capsid at 8.5 Å. Science 288877-880. [DOI] [PubMed] [Google Scholar]