

Abstract
The respiratory syncytial virus (RSV) M2-1 protein is an essential cofactor of the viral RNA polymerase complex and functions as a transcriptional processivity and antitermination factor. M2-1, which exists in a phosphorylated or unphosphorylated form in infected cells, is an RNA-binding protein that also interacts with some of the other components of the viral polymerase complex. It contains a CCCH motif, a putative zinc-binding domain that is essential for M2-1 function, at the N terminus. To gain insight into its structural organization, M2-1 was produced as a recombinant protein in Escherichia coli and purified to >95% homogeneity by using a glutathione S-transferase (GST) tag. The GST-M2-1 fusion proteins were copurified with bacterial RNA, which could be eliminated by a high-salt wash. Circular dichroism analysis showed that M2-1 is largely α-helical. Chemical cross-linking, dynamic light scattering, sedimentation velocity, and electron microscopy analyses led to the conclusion that M2-1 forms a 5.4S tetramer of 89 kDa and ∼7.6 nm in diameter at micromolar concentrations. By using a series of deletion mutants, the oligomerization domain of M2-1 was mapped to a putative α-helix consisting of amino acid residues 32 to 63. When tested in an RSV minigenome replicon system using a luciferase gene as a reporter, an M2-1 deletion mutant lacking this region showed a significant reduction in RNA transcription compared to wild-type M2-1, indicating that M2-1 oligomerization is essential for the activity of the protein. We also show that the region encompassing amino acid residues 59 to 178 binds to P and RNA in a competitive manner that is independent of the phosphorylation status of M2-1.
Human respiratory syncytial virus (HRSV) is the leading cause of severe respiratory tract infections in newborn children worldwide (5). It infects close to 100% of infants within the first 2 years of life and is the main cause of bronchiolitis. Bovine RSV (BRSV) is very similar to its human counterpart and is a major cause of respiratory diseases in calves, resulting in substantial economic losses to the cattle industry worldwide (25). RSV belongs to the order Mononegavirales, family Paramyxoviridae, genus Pneumovirus (5). The RSV genome consists of a ∼15-kb single-stranded nonsegmented negative-sense RNA molecule encoding 11 proteins, of which 4, N, P, M2-1, and L (the main component of the RNA-dependent RNA polymerase, RdRp), are associated with the viral genome, forming the holonucleocapsid complex. N, P, and L are sufficient to mediate viral replication in a reconstituted replication and transcription system (14, 35). In contrast to replication, RSV transcription requires the M2-1 protein, which is encoded by the first of the two open reading frames (ORFs) in the M2 cistron (6).
M2-1 is a basic protein of 194 amino acids that is found in all known pneumoviruses. It was first described as a structural membrane phosphoprotein (7, 19). More recent studies have shown that it functions as an intragenic transcription antitermination factor allowing the synthesis of complete mRNA (6, 11) and that it inhibits termination at gene end signals (11, 15). In infected cells, M2-1 is associated with cytoplasmic inclusion bodies that contain P and N proteins and most likely the L protein (13). P-M2-1 and N-M2-1 interactions have been described previously (1, 13). The P-M2-1 interaction is abolished by the phosphorylation of P on residue Thr108, resulting in the loss of the transcriptional antitermination activity of M2-1 on the viral RNA polymerase (1). An interaction between M2-1 and RSV M protein has also been described previously (23). However, the domains on M2-1 responsible for the interactions with P and M have not been defined.
M2-1 demonstrates RNA-binding activity (3, 8), but its binding specificity remains unclear. Specific binding to a short (∼90-nucleotide) RNA containing the 42-nucleotide, positive-sense antigenomic leader sequence has been reported previously (8). However, this specificity was not apparent when longer RNAs (700 nucleotides or more) were tested (8). In a separate study, Cartee and Wertz observed that M2-1 interacts predominantly with viral mRNA and that the M2-1-N interaction is mediated by RNA (3). An RNA-binding domain on M2-1 was mapped to amino acid residues 59 to 85 (8). In RSV-infected cells, M2-1 exists largely in an unphosphorylated form but a minor phosphorylated form is also present. The phosphorylated amino acid residues have been identified as Thr56 and Ser58 (8) and Ser58 and Ser61 (3) by different groups. M2-1 phosphorylation is important for the function of the protein but not for interaction with N or RNA (3). A CCCH (CX7CX5CX3H) motif, a putative zinc finger (ZnF), is found at the N terminus of RSV M2-1 protein and is conserved in M2-1 proteins from all known pneumoviruses (16). This motif has been shown previously to be essential for the M2-1 antitermination function but also for the interactions with RNA (3) and N (16, 31). Within the CCCH motif, along with the cysteine and histidine residues, other residues including Leu16 and Asn17 are critical for M2-1 function (36). For unknown reasons, the disruption of the CCCH motif by mutagenesis prevents M2-1 phosphorylation (16). The exact role of this putative ZnF is not known, and direct RNA binding to this domain has not been reported for any members of the Pneumovirus and Metapneumovirus genera. It is assumed that the correct conformation of the M2-1 putative ZnF is essential for the proper folding of the entire protein and the maintenance of the functional integrity of RSV M2-1 (16). A similar motif (CX8CX4CX3H) is present in filovirus VP30 proteins, and it has been shown previously that Ebola virus (EBOV) VP30 is a dimer (18) that binds zinc in vitro (26). Interestingly, EBOV VP30, like RSV M2-1, is a transcriptional antitermination factor that also binds RNA (20). Like M2-1, the EBOV VP30 CCCH motif does not bind RNA directly but is essential for VP30 RNA-binding activity (20). The first CCCH motif was first identified in Nup475 (10), which is a zinc-binding cellular protein (33) that binds to AU-rich elements present on some mRNAs (34).
The functionality of RSV M2-1 has been studied using transient transfections and reverse-genetics approaches, but little structural information is available. In this study, we expressed and purified M2-1 and demonstrated that the protein is a tetramer. We mapped the sequences necessary to maintain M2-1 as an oligomer to residues 32 to 63. The deletion of this region abolished M2-1 activity when the protein was tested in an RSV minigenome replicon system. The secondary structure of recombinant M2-1 consists mainly of α-helices, as determined by circular dichroism (CD) analysis, and M2-1 binds RNA and P in a competitive manner in vitro through an α-helical domain comprising residues 53 to 177.
MATERIALS AND METHODS
Plasmid construction.
Randomly primed cDNA synthesis from total cytoplasmic RNA isolated from HEp-2 cells infected with the RSV strain Long (4) was performed with SuperScript II reverse transcriptase (Gibco BRL). The cDNA carrying the M2-1 ORF was amplified by PCR with high-fidelity PfuTurbo polymerase (5 U; Stratagene) and 100 ng of the following primers: M2-1Bam(+), 5′-GAGGGATCCATGTCACGAAGGAATCCTTGCA-3′, and M2-1(−), 5′-GTCAGGTAGTATCATTATTTTTGGC-3′. PCR-amplified DNA was digested by BamHI and inserted at the BamHI-SmaI sites in the pGEX-4T3 vector (Pharmacia) to engineer the pGEX-M2-1 plasmid. This plasmid was designed to produce a protein that contained glutathione S-transferase (GST) fused at the amino terminus of M2-1, with GST and M2-1 separated by a thrombin cleavage site. Thrombin cleavage of the fusion protein resulted in the addition of two amino acids, a glycine and a serine, at the amino terminus of M2-1. The M2-1 ORF was subsequently subcloned into the pET-28a(+) vector (Novagen) at the BamHI-XhoI sites to make a pET-M2-1 plasmid that encodes M2-1 containing an amino-terminal poly-His tag. N-terminal deletion mutants and internal M2-1 domains were obtained by PCR using Pfu DNA polymerase from Stratagene (primer sequences are available upon request), and the PCR products were subcloned into pGEX-4T3 at the BamHI-SmaI sites. C-terminal deletion mutants were obtained using the QuikChange site-directed mutagenesis kit (Stratagene) to introduce stop codons in the M2-1 sequence (primer sequences are available upon request). We used the following designations for M2-1 mutant proteins: M2-1[53-177] consists of residues 53 to 177, M2-1Δ31N has an amino-terminal deletion of the first 31 residues of M2-1, M2-1Δ35-58 contains an internal deletion of residues 35 to 58, and M2-1Δ33C carries a C-terminal deletion of residues 33 to 194.
Plasmids expressing the HRSV proteins N, P, M2-1, and L, designated pN, pP, pM2-1, and pL, have been described previously (17, 35). The sequence encoding M2-1Δ35-58 was inserted at the BamHI-HindIII sites in pM2-1 in place of the M2-1 gene. To generate the M-luciferase (M-Luc) subgenomic replicon, the firefly Luc gene was inserted at the KpnI-XhoI restriction sites in the pM/SH vector in place of the SH coding region (17), yielding pM/Luc. The integrity of all constructs was assessed by DNA sequencing.
Cell transfections and luciferase assays.
BHK-21 cells constitutively expressing the T7 RNA polymerase (clone BSRT7/5) (2) were grown overnight to 90% confluence in six-well dishes containing Dulbecco modified essential medium (BioWhittaker) supplemented with 10% fetal calf serum. The cells were transfected using Lipofectamine 2000 (Invitrogen) with a plasmid mixture containing 1 μg of pM/Luc, 1 μg of pN, 1 μg of pP, 0.5 μg of pL, and 0.25 μg of pM2-1 or the M2-1Δ35-58 expression plasmid, pM2-1Δ35-58, as well as 0.25 μg of pRSV-β-Gal (Promega) to normalize transfection efficiencies. A DNA mixture omitting pM2-1 served as a negative control. Transfections were done in triplicate. Cells were harvested at 24 h posttransfection and lysed in luciferase lysis buffer (30 mM Tris, pH 7.9, 10 mM MgCl2, 1 mM dithiothreitol [DTT], 1% [vol/vol] Triton X-100, and 15% [vol/vol] glycerol), and luciferase activities were evaluated twice for each cell lysate with an Anthos Lucy 3 luminometer (Bio Advance).
Expression and purification of recombinant proteins.
Escherichia coli BL21(DE3) (Novagen) cells transformed with the pGEX-M2-1-derived plasmids or pGEX-P (4) were grown at 37°C for 8 h in 1 liter of Luria-Bertani (LB) medium containing 100 μg/ml ampicillin. Cells transformed with pET-M2-1 were grown in LB medium containing kanamycin (50 μg/ml). The same volume of LB medium was then added, and protein expression was induced by adding 80 μg/ml isopropyl-β-d-thiogalactopyranoside (IPTG) and 50 μM ZnSO4 to the medium. The bacteria were incubated for a further 15 h at 28°C and harvested by centrifugation. Bacterial pellets were resuspended in 100 ml of standard low-salt lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM DTT, 2% Triton X-100, 10 mM MgSO4, 1 mM CaCl2, 1 mg/ml lysozyme) or high-salt lysis buffer (50 mM Tris-HCl, pH 7.4, 1 M NaCl, 1 mM DTT, 2% Triton X-100, 10 mM MgSO4, 1 mM CaCl2, 1 mg/ml lysozyme) supplemented with a complete protease inhibitor cocktail (Roche, Mannheim, Germany), and the suspensions were incubated for 1 h on ice, sonicated, and centrifuged twice at 4°C for 30 min at 10,000 × g. Glutathione-Sepharose 4B beads (GE Healthcare) were added to the clarified supernatants (1 ml of beads per 2 liters of induced bacterial culture), and the mixtures were incubated at 4°C for 15 h. The beads were washed three times with lysis buffers and then stored at 4°C in a volume of lysis buffer equal to the bead volume. To isolate GST-free M2-1, beads containing bound GST-M2-1 were extensively washed with 1× phosphate-buffered saline (PBS)-1 mM DTT, resuspended in a volume of PBS-1 mM DTT equal to the bead volume, and incubated with biotinylated thrombin (Novagen) for 16 h at 20°C. Thrombin was then removed using the thrombin cleavage capture kit according to the instructions of the manufacturer (Novagen).
DLS.
Dynamic light scattering (DLS) was performed at 20°C with a Malvern Zetasizer Nano series instrument. Purified M2-1-derived proteins at 1 mg/ml in PBS were centrifuged at 100,000 × g for 10 min and analyzed in plastic cuvettes. Measurements were made at a fixed angle of 173° by using an incident laser beam with a wavelength of 633 nm. Eleven measurements were made, with an acquisition time of 10 s for each measurement. Hydrodynamic diameters (DH) were calculated using the DLS software provided by the instrument manufacturer.
Cross-linking analysis.
GST-free M2-1 was centrifuged for 10 min at 100,000 × g. Samples containing ∼1 μg of proteins in 10 μl of PBS supplemented with 1 mM DTT were incubated for 1 h at 4°C with increasing amounts of ethylene glycol disuccinate (EGS). Reactions were stopped by the addition of glycine to a final concentration of 50 mM, the cross-linked products were analyzed by sodium dodecyl sulfate (SDS)-10% polyacrylamide gel electrophoresis (10% PAGE), and the proteins were visualized by Coomassie blue staining.
Absorbance-based analytical ultracentrifugation.
Sedimentation velocity studies of RNA-free M2-1 (45 μM) were performed at a wavelength of 280 nm using a Beckman Optima XL-A70 centrifuge equipped with an An-60 Ti rotor at 15°C. Double-sector centrifuge cells with quartz windows were loaded with 380 μl of the sample and 400 μl of the reference compound (1× PBS-1 mM DTT). Data from sedimentation at 45,000 rpm (147,280 × g) were collected at 5-min intervals (giving a total of 55 scans). Sedimentation velocity data were fitted to a c(s) continuous size distribution model using SEDFIT (29; www.analyticalultracentrifugation.com) and Svedberg (27) software. The solvent density and viscosity at 15°C were determined to be 1.00796 g/ml and 1.1652 cP, respectively. The M2-1 partial specific volume was calculated as 0.7312 ml/g.
Electron microscopy.
GST-free M2-1 in a mixture of 10 mM Tris-HCl, pH 7.4, and 150 mM NaCl was applied to an airglow-discharged carbon-coated grid, and the grid was stained with a 2% (wt/vol) uranyl acetate aqueous solution. Grids were observed with a CM12 electron microscope (Philips) operated at 80 kV. Micrographs were recorded at a nominal magnification of ×35,000 on a Kodak SO163 electron plate and developed in Kodak D-19 for 12 min. The scanned micrographs were viewed with Photoshop (Adobe Systems).
Mass spectrometry.
Bands were cut from the gels and washed in 50% (vol/vol) acetonitrile-50 mM ammonium bicarbonate, pH 8.0. Tryptic digestion was performed with 0.1 μg of modified trypsin (sequencing grade; Promega) in 50 mM bicarbonate, pH 8.0, in a thermomixer (Eppendorf) for 18 h at 37°C and 500 rpm. A volume of 0.5 μl from the supernatant was spotted directly onto a matrix-assisted laser desorption ionization (MALDI) plate. The sample was allowed to dry at room temperature before the addition of 0.5 μl of a matrix solution, 5 mg/ml α-cyano-4-hydroxycinnamic acid in 50% (vol/vol) acetonitrile-0.05% (wt/vol) trifluoroacetic acid. Mass spectra were acquired on a Voyager-DE-STR MALDI-time of flight (TOF) mass spectrometer (Applied Biosystems, Framingham, MA) equipped with a nitrogen laser (Laser Science, Franklin, MA) emitting at a wavelength of 337 nm. The accelerating voltage used was 20 kV. All spectra were recorded in the positive reflector mode with a delayed extraction of 130 ns and a 62% grid voltage. MALDI-TOF was performed using internal calibration with two peptides from trypsin autolysis with (M + H)+ values of 2,211.1040 and 842.5090.
Analysis of the presence of nucleic acids in GST-M2-1 complexes.
GST-M2-1 complexes bound to glutathione-Sepharose beads were resuspended in PBS to an estimated concentration of 1 mg/ml as determined by the Bradford method. For analysis of the presence of nucleic acids in the GST-M2-1 complexes, samples were separated into two equal parts. For protein analysis, beads were boiled for 10 min in 1× Laemmli buffer before being loaded onto a 12% polyacrylamide gel. For nucleic acid analysis, beads were boiled for 5 min in 1× PBS and pelleted by centrifugation at 2,000 rpm for 1 min and the supernatant was subjected to Tris-borate-EDTA-2% agarose gel electrophoresis, stained with EtBr, and visualized under UV light. The nature of the nucleic acids present in the complexes was determined by incubating supernatants with RQ1 DNase (0.1 U/μl; Promega) or RNase A (0.1 mg/ml) for 30 min at 37°C.
Subsequently, to remove nucleic acids from GST-M2-1 complexes, beads were washed twice with 1 M NaCl in 10 mM Tris, pH 7.4, and then washed twice with PBS.
CD spectroscopy.
GST-free M2-1 and M2-1 deletion mutants in PBS-1 mM DTT were dialyzed against a mixture 150 mM NaF, and 5 mM MOPS, pH 7.5, at 4°C. The molar absorption coefficient (ɛ) for M2-1 was determined to be 13,200 M−1 cm−1 by using the Expasy ProtParam tool (http://www.expasy.ch/tools/protparam.html), and protein concentrations were determined by absorption at 280 nm. Samples were analyzed with a Jasco J-810 spectropolarimeter at 20°C using 1-mm-thick quartz cells. Protein samples (200 μl) were loaded into the cuvettes, and spectra between 170 and 280 nm were recorded. The baseline was determined with the same buffer without protein and subtracted from the protein spectra. The results are expressed as degrees per square centimeter per decamole. The proportions of secondary structures were determined by the self-consistent method (30) using the Dicroprot program (9).
In vitro transcription of RNAs.
RNAs were synthesized by in vitro transcription using a T7 Ribomax large-scale RNA production system (Promega). Transcription was performed using the pBluescript SK(−) plasmid (Stratagene) digested by BamHI, the pCDNA3 plasmid (Invitrogen) digested by SmaI, and pET-P (4) digested by XhoI, resulting in the synthesis of RNAs of 81, 1,213, and 778 bases, respectively. The RSV leader region was amplified by reverse transcription-PCR using RNA extracted from RSV-infected HEp-2 cells as described previously (4) and the following primers: for the transcript designated PCR-A, primer T7Leader+ (containing a T7 promoter), 5′-CGCCAAGCTTAATACGACTCACTATAGGGACGCGAAAAAATGCGTACAACAAACTTGCGTA A-3′, and primer NS1−, 5′-GCGGCGCTCGAGTTATGGATTAAGATCAAATCCAAG-3′; for the PCR-B transcript, primer T7Leader+ and primer Leader−, 5′-TAAGTGGTACTTATCAAATTC-3′; and for the PCR-C transcript, primer T7Leader− (containing a T7 promoter), 5′-GCGGCGTTAATACGACTCACTATAGGGTAAGTGGTACTTATCAAATTC-3′, and primer Leader+, 5′-ACGCGAAAAAATGCGTACAACAAACTTGC-3′. Purified amplified DNA was used for in vitro transcription, resulting in the synthesis of a plus-polarity transcript of 533 bases containing the RSV leader sequence plus three extra G residues at the 5′ end and the NS1 coding region (PCR-A), a plus-polarity transcript of 80 bases containing the RSV leader sequence plus three extra G residues at the 5′ end (PCR-B), and a minus-polarity transcript of 80 bases containing the RSV leader region and three extra G residues at the 5′ end (PCR-C). After RNA synthesis, DNA was digested by RQ1 DNase (Promega).
In vitro phosphorylation.
Samples of 100 μl of a protein-bead suspension containing GST-M2-1 at a concentration of ∼70 μM were washed once with 10 volumes of 1× casein kinase I (CKI) reaction buffer (50 mM Tris-HCl [pH 7.5], 10 mM MgCl2, 5 mM DTT) supplemented with 200 μM ATP and 0.1 mCi/ml [γ-32P]ATP (Amersham) as a tracer and resuspended in an equal volume of 1× CKI buffer. M2-1 phosphorylation by CKI was carried out by incubating the mixture for 1 h at 30°C after the addition of 500 U of CKI (New England Biolabs). The reactions were analyzed by SDS-PAGE and autoradiography.
Mapping of RNA- and P-binding domains by pulldown assays.
The pulldown of RNA by GST-M2-1 was performed by incubating 100 μg of GST-M2-1 complexes bound to beads with 100 μg of RNA. Beads were washed three times with PBS. Half of each sample was boiled in PBS and analyzed for the presence of nucleic acids, and the other half was used for SDS-PAGE protein analysis. For mapping of the RNA- and P-binding domains on M2-1, a volume of 10 μl of the 50% bead-PBS slurry, containing the different GST-M2-1-derived fusion proteins at concentrations of ∼100 μM, was incubated with a twofold molar excess of either P or RNA for 1 h at 20°C with agitation. Beads were washed extensively with PBS, boiled with 10 μl of Laemmli buffer, and analyzed by SDS-PAGE. The pulldown of RNA by GST-M2-1 was analyzed as described above.
RNA/P competitions.
Bead samples containing 200 μg of GST-M2-1 complexes (4 nmol) were incubated for 1 h at 20°C with 600 μg of P (22 nmol) with or without RNA (100 μg) in a total volume of 300 μl in 1× PBS supplemented with 1 mM DTT. Beads were washed extensively with PBS, and the bead samples were separated into two equal parts and analyzed as described above. The recombinant RSV P protein was obtained by biotinylated-thrombin cleavage of GST-P complexes purified from E. coli and further removal of biotinylated-thrombin as described previously (4).
Bioinformatic analysis.
The M2-1 sequence used in this study was from the HRSV Long strain, ATCC VR-26, with NCBI accession number AAX23995. Hydrophobic cluster analysis was carried out using the DRAWHCA program (12). Secondary structure predictions were performed with PSI-PRED (21).
RESULTS
Expression and purification of recombinant HRSV M2-1.
The RSV M2-1 protein has been poorly characterized at the biochemical level, due to difficulties met by different groups when attempting to purify this protein in sufficient amounts (8, 24). To explore the structure of the protein, M2-1 cDNA was amplified by reverse transcription-PCR from RSV-infected HEp-2 cells and cloned into the pGEX-4T3 expression vector. The GST-M2-1 fusion protein was purified from bacterial lysates by glutathione-Sepharose 4B bead affinity chromatography. Since M2-1 can bind to long RNA with no sequence specificity (8), the presence of nucleic acids in the purified protein samples was investigated. Purified GST-M2-1 migrated as a single band with an apparent mass of ∼45 kDa, as analyzed by SDS-PAGE and Coomassie blue staining (Fig. 1B). GST-M2-1 was treated with RQ1 DNase, RNase A, or both and run on a 1% agarose gel stained with ethidium bromide (EtBr) to visualize any associated nucleic acids. A band with RNase but not DNase sensitivity was observed (Fig. 1A). Washing of GST-M2-1, captured on glutathione-Sepharose beads, with 1 M NaCl eliminated the RNA from the isolated complexes (Fig. 1A). No nucleic acid associated with GST without the M2-1 fusion was observed (data not shown).
FIG. 1.

Purification of M2-1 from E. coli and analysis of the presence of nucleic acids. (A and B) Agarose gel electrophoresis analysis of nucleic acids present in the GST-M2-1 complexes (A) and SDS-PAGE analysis of GST-M2-1 complexes from the same samples (B). GST-M2-1 fusion proteins from standard bacterial lysates containing 150 mM NaCl were purified by using glutathione-Sepharose beads, resuspended in an equal volume of PBS, and then treated with RNase A, RQ1 DNase, or both or washed with 1 M NaCl, as indicated. Lane M, molecular mass markers. (C) SDS-PAGE analysis of M2-1 purified from standard bacterial lysates (lanes 2, 5, and 7) or high-salt lysates (lanes 3, 6, and 8). Proteins were cleaved by thrombin, and the pellets (lanes 5 and 6) or soluble fractions (lanes 7 and 8) were analyzed in parallel with uncleaved GST-M2-1 adsorbed on glutathione-Sepharose beads (lanes 2 and 3; 1 μl of beads per lane). Lanes 1 and 4, molecular mass markers. (D) UV spectra of tRNA and M2-1 purified from standard (150 mM NaCl) or high-salt (1 M NaCl) bacterial lysates, as indicated.
We modified our purification protocol for GST-M2-1 by adding NaCl to the bacterial lysates to a final concentration of 1 M before adding glutathione-Sepharose beads. The final yield of GST-M2-1 was higher when we used high-salt conditions instead of the standard salt concentration (150 mM NaCl), with corresponding yields of approximately 30 and 15 mg of protein liter−1 of bacterial culture, respectively. In both cases, the M2-1 proteins were recovered in the supernatants after thrombin cleavage and migrated as a single band of about 25 kDa (Fig. 1C, lanes 7 and 8); GST was retained with the beads (Fig. 1C, lanes 5 and 6). The purities of proteins and nucleic acids can be estimated by using the optical density spectrum and by calculating the ratios of absorption coefficients at 260 nm (A260) and 280 nm (A280); pure nucleic acid samples would have an A260/A280 ratio of 2.0, while the ratio for pure protein would be 0.57. As shown in Fig. 1D, M2-1 had a spectrum typical of pure proteins when washed with 1 M NaCl, while M2-1 purified from low-salt extracts had a spectrum resembling that for RNA. A260 and A280 values for both unwashed M2-1 and M2-1 washed with 1 M NaCl were measured. The calculated A260/A280 ratios were approximately 2 for unwashed M2-1 and approximately 0.6 for M2-1 washed with a high-salt solution. These values are characteristic of the presence and absence of RNA, respectively. These results showed that bacterial RNA was present in the purified recombinant M2-1 protein sample and that RNA-M2-1 interactions can be disrupted in the presence of high-salt solutions. Subsequent experiments all used RNA-free complexes washed with high-salt solutions.
Determination of the M2-1 secondary structure in solution by CD spectroscopy.
To study the secondary structure of M2-1, recombinant M2-1 was analyzed by far-UV CD spectroscopy. The far-UV CD spectrum of M2-1 at neutral pH is typical of a structured protein, with a predominant α-helical content, as seen by the positive ellipticity between 185 and 200 nm and by two minima at 208 and 222 nm (Fig. 2). The proportions of secondary structures were determined by the self-consistent method (30) using the Dicroprot program (9). The estimations were 38% α-helix structure, 12% β-sheet structure, 40% random coils, and 10% turns, confirming that M2-1 is largely α-helical (Table 1).
FIG. 2.
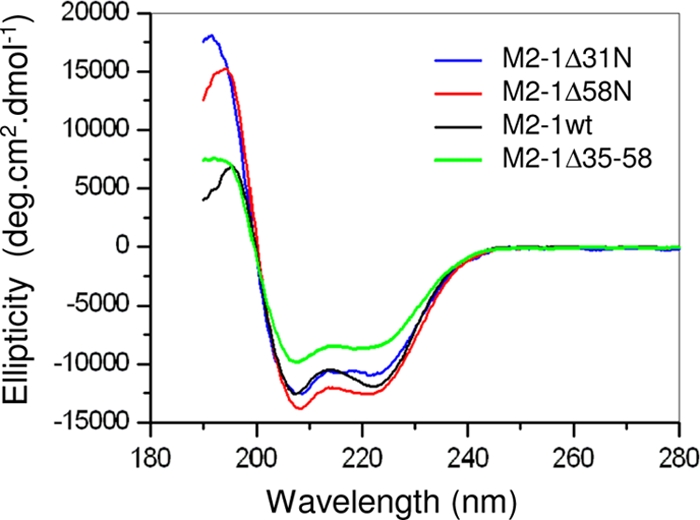
Far-UV CD spectra of wild-type (wt) M2-1 and the deletion mutants M2-1Δ31N, M2-1Δ58N, and M2-1Δ35-58. The results are expressed as degrees per square centimeter per decamole (deg.cm2.dmol−1).
TABLE 1.
Estimation of proportions of secondary structuresa present in M2-1 and deletion mutants
| Protein | % Comprising:
|
|||
|---|---|---|---|---|
| α-Helix structure | β-Sheet structure | Turns | Random structure | |
| M2-1 wild type | 38 | 12 | 10 | 40 |
| M2-1Δ31N | 49 | 7 | 8 | 35 |
| M2-1Δ58N | 45 | 6 | 13 | 37 |
| M2-1Δ35-58 | 28 | 17 | 12 | 43 |
The proportions of secondary structures were determined using the Dicroprot program.
Quaternary structure of M2-1.
The homogeneity of M2-1 in terms of quaternary structure was then investigated by DLS. Figure 3A shows the volume-based particle size distribution histogram for RNA-free M2-1 (solid line in Fig. 3A) in PBS. M2-1 appeared as a single peak with a polydispersity index of 0.33, indicating that the M2-1 sample was highly monodisperse, consisting of a single protein species with an average diameter ± standard deviation of 7.25 ± 0.5 nm. From this value, one can derive the corresponding hydrodynamic volume (VH ≈ 195,000 Å3). According to Uversky (32), this volume corresponds to a globular protein of approximately 800 residues, suggesting that M2-1 (194 residues) may be either a partially structured monomer or an oligomer, possibly a tetramer.
FIG. 3.

M2-1 forms oligomers in solution. (A) DLS analysis. Purified M2-1 (solid line) and M2-1Δ35-58 (dashed line) in filtered PBS at 20°C were subjected to DLS with a Malverne Zetasizer Nano series instrument. The results are presented as volume-based particle size distributions. M2-1 is shown to exist as a monomodal, monodisperse protein with a DH of approximately 7.25 nm. M2-1Δ35-58 is also monodisperse, with a DH of ∼5 nm. d. nm, diameter in nanometers. (B) M2-1 cross-linking. Samples of 2 μg of purified recombinant M2-1 free of RNA were subjected to cross-linking with increasing concentrations of EGS as indicated below each lane. The reaction products were resolved on an SDS-4 to 12% gradient PAGE gel and stained with Coomassie blue. The molecular mass markers are indicated. 1X, monomers; 2X, dimers; 3X, trimers; 4X, tetramers.
To confirm that M2-1 exists as an oligomer, we employed chemical cross-linking. GST-free M2-1 protein, which has an apparent mass of ∼22 or 25 kDa on an SDS-4 to 12% polyacrylamide gradient gel, depending on the electrophoresis conditions used (Fig. 3B), was cross-linked by increasing concentrations of EGS. Bands migrating with apparent masses of ∼50, ∼75, and ∼100 kDa were apparent when EGS concentrations increased. These masses were compatible with M2-1 dimers, trimers, and tetramers, respectively (indicated by arrows in Fig. 3B). Since cross-linked products with more units than tetramers were not observed, we conclude that M2-1 is likely to be a tetramer.
We next carried out analytical ultracentrifugation to accurately determine the protein oligomeric state and further assess the size and shape of M2-1. Figure 4 shows sedimentation boundaries determined at 5-min intervals by using a loading concentration of 1 mg/ml. Analysis of the sedimentation velocity profiles using a c(s) distribution model and the Lamm equation (in SEDFIT software) gave excellent fits (Fig. 4A) and yielded the sedimentation coefficient distributions shown in Fig. 4B, which revealed the presence of a species with an s value of 4.6, corresponding to a standardized sedimentation coefficient (s20,w) of 5.4. This value is compatible with the behavior of an 89-kDa globular protein with a viscosity coefficient (frictional ratio [f/f0]) of 1.3 and a hydrodynamic radius of 3.83 nm. Alternatively, this value represents a species with the features of an elongated molecule with a larger mass, but not monomeric, dimeric, or trimeric forms of M2-1, whatever the viscosity coefficient. However, with an oligomer bigger than a tetramer such as a pentamer or hexamer, the molecular complex should be more elongated, with a higher f/f0 ratio and a larger hydrodynamic radius, to conserve an s20,w value of 5.4, which was not consistent with the DLS observations. Thus, these results were consistent with DLS and cross-linking data and confirmed the tetrameric nature of M2-1 under our experimental conditions.
FIG. 4.

Analytical ultracentrifugation of M2-1. (A) Sedimentation velocity analysis of RNA-free M2-1 in PBS. The positions of the moving boundaries shown were recorded at intervals of 5 min by spectrometric scanning at 280 nm. The continuous lines are best fits of the experimental data (points) analyzed by the Lamm equation using SEDFIT. The rotor speed was 45,000 rpm, and the temperature was 15°C. OD, optical density. (B) The calculated c(s) distribution was plotted as a function of the sedimentation coefficient and reveals the presence of a major species with an s value of 4.6 (at 15°C in 1× PBS-1 mM DTT) that corresponds to an s20,w value of 5.4 (at 20°C in H2O).
To obtain information about the shape of M2-1, the purified protein was analyzed by negative-stain electron microscopy (Fig. 5). Despite the limitations of this technique for small molecules, grids prepared with M2-1 revealed the presence of small, uniform objects of about 10 ± 2 nm in diameter with a cogwheel-like appearance. These observations were again in agreement with DLS and analytical ultracentrifugation findings, supporting the hypothesis that M2-1 is an oligomer.
FIG. 5.

Electron microscopy image of M2-1. Original magnification, ×35,000. Bar, 50 nm.
Mapping of the M2-1 oligomerization domain.
To map the M2-1 oligomerization domain, GST-M2-1 (from the pGEX-M2-1 plasmid) was coexpressed with His-M2-1 (from the pET-M2-1 plasmid) in bacteria by using a double-antibiotic selection protocol. GST-M2-1 was purified from bacteria and analyzed by SDS-PAGE. The M2-1-M2-1 interaction was revealed by the ability of His-M2-1 to copurify with GST-M2-1. As shown in Fig. 6B, two polypeptides with apparent masses of ∼45 and ∼27 kDa, corresponding to GST-M2-1 and His-M2-1, respectively, were observed. The identities of GST-M2-1 and His-M2-1 were confirmed by MALDI-TOF mass spectrometry, showing that M2-1 forms stable oligomers. To map the M2-1 regions necessary for homo-oligomerization, a series of GST-M2-1 deletion mutants (Fig. 6A) were coexpressed with His-M2-1 in E. coli and purified as described above. As shown in Fig. 6B, the N-terminal deletion mutant M2-1Δ31N and the C-terminal deletion mutants M2-1Δ60C and M2-1Δ71C, with deletions of residues 60 to 194 and 71 to 194, respectively, retained the ability to associate with His-M2-1. On the contrary, no band corresponding to His-M2-1 was visible for either GST or the M2-1Δ58N and M2-1Δ85N constructs, with N-terminal deletions through residues 58 and 85, respectively (Fig. 6B). These results indicate that the M2-1 oligomerization domain is situated between residues 32 and 58 or at least that this region contributes to the interaction. To confirm this prediction, M2-1 residues 35 to 58 were deleted (generating mutant M2-1Δ35-58). As shown in Fig. 6B, this mutant was unable to interact with His-M2-1. Together, these results strongly indicate that a region situated between residues 35 and 58 of M2-1 is necessary for M2-1-M2-1 intermolecular interactions and is an essential part of the oligomerization domain.
FIG. 6.

Analysis of M2-1 oligomerization and mapping of the M2-1 oligomerization domain. (A) Schematic representation of the deletion mutants of the GST-M2-1 fusion protein that were used to map the M2-1 oligomerization domain by coexpression with M2-1 in bacteria and sequence of the putative M2-1 ZnF motif. The column to the right summarizes the interactions between GST-M2-1 mutants and His-M2-1 analyzed as depicted in panel B. (B) Coomassie blue-stained SDS-12% polyacrylamide gel with purified GST-M2-1 deletion mutants expressed alone (−) or coexpressed with M2-1 (+) in E. coli. The arrowhead points to the His-M2-1 band. Lane M, molecular mass markers. (C) M2-1Δ35-58 cross-linking. Two micrograms of purified M2-1Δ35-58 free of RNA was subjected to cross-linking and analyzed as described in the legend to Fig. 3. The upper band corresponds to M2-1Δ35-58, and the faster-migrating band indicated by a star results from an N-terminal cleavage of the M2-1Δ35-58 protein.
To confirm that the region between residues 35 and 58 of M2-1 is necessary for oligomerization, we used chemical cross-linking. No cross-linked products were detected by treating M2-1Δ35-58 with EGS (Fig. 6C), confirming that the region is necessary for M2-1 oligomerization. M2-1Δ35-58 was partially cleaved at the N terminus by thrombin (Fig. 6C, with the faster-migrating band indicated by a star), as determined by MALDI-TOF analysis (data not shown). When analyzed by DLS, this mutant exhibited a monodisperse profile with a DH of approximately 5 nm (Fig. 3A, dashed line). The derived VH (≈65,000 Å3) corresponds to a globular protein of ∼270 residues. According to Uversky (32), these results suggest that M2-1Δ35-58 (169 amino acid residues) is a monomer with either an extended shape or a partially unfolded (extended-coil) region.
The secondary structures of the M2-1Δ31N, M2-1Δ58N, and M2-1Δ35-58 deletion mutants were evaluated by CD spectroscopy. As shown in Fig. 2, the three deletion mutants showed spectra similar to that of full-length M2-1 and typical of α-helical proteins, indicating that the secondary structure was not adversely affected by the deletions. Interestingly, the M2-1Δ35-58 mutant had a lower α-helix content than full-length M2-1, indicating that the deleted oligomerization region was probably folded as an α-helix (Table 1). Together, these results indicate that the deletion mutants used in these experiments were correctly folded and that their inability to oligomerize was not due to denaturation.
Effect of deletion of the M2-1 oligomerization domain on RSV transcription.
The M2-1 protein has been characterized as a transcription antiterminator that increases the processivity of the transcriptase and decreases the efficiency of transcription termination at gene junctions (11, 17). To determine whether the domain we identified as being responsible for M2-1 oligomerization was important for M2-1 to function as a transcription antiterminator, the sequence encoding the M2-1Δ35-58 mutant was subcloned into the pGEM3 vector in place of the sequence encoding M2-1. The effect of the deletion of residues 35 to 58 on M2-1 function as an antiterminator was assayed using a dicistronic subgenomic replicon, pM/Luc, which contained the authentic M-SH gene junction and the Luc reporter gene downstream of the gene start sequence present in this gene junction. BHK-21 BSRT7/5 cells expressing T7 RNA polymerase were cotransfected with the pM/Luc plasmid and pRSV-β-Gal, pL, pP, pN, and pM2-1. Luciferase activities were determined and normalized based on β-galactosidase (β-Gal) expression. As shown in Table 2, the expression of the luciferase gene in this system was absolutely dependent on the presence of M2-1. The deletion of the M2-1 oligomerization domain (in M2-1Δ35-58) reduced the luciferase minigenome activity to near background levels. Thus, the deletion that prevented M2-1 oligomerization in vitro resulted in the loss of the transcription antiterminator activity of M2-1, suggesting that this activity depends on the tetrameric nature of M2-1.
TABLE 2.
Effect of deletion of the M2-1 oligomerization domain on luciferase minigenome activity
| System content | Luc/β-Gal ratioa |
|---|---|
| M2-1 | 0.425 ± 0.066 |
| M2-1Δ35-58 | 0.040 ± 0.015 |
| No M2-1 | 0.065 ± 0.060 |
| No L | 0.037 ± 0.015 |
Each luciferase minigenome activity value was normalized based on β-Gal expression and is the average ± the standard deviation of results from three separate experiments performed in duplicate.
Mapping P and RNA interaction domains on M2-1.
It has been reported previously that the RNA-binding domain of M2-1 is situated between residues 59 and 85 (8). However, the P-binding domain of M2-1 remains undefined. To determine whether RNA-free GST-M2-1 was still competent to bind RNA, 100-μg samples of GST-M2-1 complexes bound to glutathione-Sepharose beads were incubated with 100-μg samples of different RNAs in PBS, and the beads were washed extensively with PBS and analyzed by agarose gel electrophoresis and SDS-PAGE. As shown in Fig. 7A, GST-M2-1 was able to pull down RNA containing either plasmid (pcDNA3) sequences, RSV plus-polarity sequences (P and leader-NS1 sequences), and short yeast tRNA (which has an average size of 100 nucleotides) with similar efficiencies.
FIG. 7.

The domain of residues 59 to 177 is sufficient for binding to P and RNA in a competitive manner. (A) Binding of different RNAs to GST-M2-1. GST-M2-1 complexes bound to glutathione-Sepharose beads (100 μg) were incubated with 100 μg of RNA transcribed from pCDNA3 (lane 1), pET-P (lane 2), or a leader-NS1 sequence (lane 3) or 100 μg of yeast tRNA (lane 4) for 1 h at room temperature. After extensive washing, GST-M2-1-RNA complexes were analyzed by agarose gel electrophoresis by double staining with EtBr (top) and amido black (bottom) to reveal RNAs and proteins, respectively. (B) Schematic representation of the deletion mutants of the GST-M2-1 fusion protein that were used in the pulldown assays. (C and D) Mapping of the RNA (C)- and P (D)-binding domains on M2-1 by in vitro pulldown assays using deletion mutants of the GST-M2-1 fusion protein, tRNA, and purified recombinant RSV P protein. After extensive washing, the sample of GST-M2-1-RNA complexes was separated into two equal parts and analyzed by agarose gel electrophoresis and EtBr staining (C, bottom) and SDS-PAGE and Coomassie blue staining (C, top) to reveal RNA and proteins, respectively. Lane M, molecular size markers; +, M2-1 was inoculated with P in vitro. (E) Evidence for competition between RNA and P for binding to M2-1. Glutathione-Sepharose beads containing RNA-free GST-M2-1 (lane 1) were incubated with tRNA alone (lane 2), P alone (lane 3), or P in the presence of yeast tRNA (lane 4), an 81-nucleotide RNA transcribed from pBluescript plasmid (lane 5), or an 80-nucleotide RNA containing the HRSV leader sequence in the plus (lane 6) or minus (lane 7) polarity. After further incubation for 1 h at 20°C, the reaction products were washed extensively and separated into two parts for analysis to detect the presence of both P (top) and RNA (bottom) bound to M2-1, as described above. The GST-M2-1 and P protein bands are indicated by arrowheads.
A pulldown assay was then performed to assess whether GST-M2-1 could interact with purified recombinant P protein in the absence of additional viral or cellular proteins. A twofold molar excess of P was added to 100 μg of GST-M2-1 immobilized on glutathione-Sepharose beads. After extensive washing, proteins were analyzed by SDS-PAGE and Coomassie blue staining. As shown in Fig. 7D, GST-M2-1 was able to efficiently interact with P.
To map the RNA- and P-binding regions on M2-1, a further set of N-terminal and C-terminal deletions throughout the 194-amino-acid sequence of the M2-1 protein fused to GST were made in the pGEX-4T3 vector (Fig. 7B). N-terminal deletions were tolerated until they extended past residue 86 (Fig. 7C and D), after which proteins were insoluble (data not shown). For C-terminal mutations, deletions that extended past residue 177 were also not tolerated. The central region of residues 53 to 177 could be efficiently produced and purified (Fig. 7C and D). These observations suggest that deletions within the domain of residues 86 to 177 result in M2-1 misfolding, rendering the protein insoluble. A GST pulldown assay using either tRNA or recombinant P protein was then performed with the purified mutants listed in Fig. 7C and D. P and RNA binding was observed with M2-1Δ58N and M2-1Δ53-177, but not with M2-1Δ152N, which has an N-terminal deletion through residue 152. The M2-1Δ85N mutant bound both P and RNA with reduced efficiencies. These results demonstrate that the RNA- and P-binding domains of M2-1 are both situated between residues 59 and 153, thus excluding the putative ZnF, the oligomerization domain, and the C-terminal region.
We investigated whether RNA and P could compete for binding to M2-1. Nucleic acid-free GST-M2-1 fixed on glutathione-Sepharose beads was incubated with P in the presence or absence of tRNA. After extensive washing, the pulled-down products were analyzed by agarose gel electrophoresis and SDS-PAGE to detect the presence of RNA and P, respectively. As shown in Fig. 7E, the presence of tRNA prevented the loading of P, demonstrating that the interactions of RNA and P with M2-1 were mutually exclusive. The same results were obtained with shorter (80-nucleotide) RNAs that contained HRSV leader sequences with plus or minus polarity or were transcribed from the pBluescript plasmid linearized by BamHI digestion and were unrelated to RSV sequences (Fig. 7E). These results showed that under our experimental conditions, M2-1 bound RNA, including short (80-nucleotide) sequences, with no sequence specificity and that this binding competed with the P-M2-1 interaction.
Effects of M2-1 phosphorylation on oligomerization and binding to P and RNA.
In eukaryotic cells, M2-1 is phosphorylated on serine residues 58 and 61 (3) or on threonine 56 and serine 58 (8). However, bacterially expressed M2-1 is not phosphorylated. As shown above, unphosphorylated M2-1 binds P efficiently, confirming earlier observations (24). However, phosphorylation may potentially result in the loss of M2-1 oligomerization, affecting binding to either P or RNA, which may constitute a switch for binding to either moiety. RNA-free GST-M2-1 bound to glutathione-Sepharose beads was in vitro phosphorylated by CKI as described previously (3), washed, and cleaved by thrombin, and the soluble fraction was analyzed by SDS-PAGE. M2-1 phosphorylation was demonstrated using [γ-32P]ATP (Fig. 8A), and SDS-PAGE revealed that phosphorylated M2-1 migrated more slowly than untreated M2-1 (Fig. 8A and D). A pulldown assay with P (Fig. 8B) or tRNA (Fig. 8C and D) showed no differences between the activities of unphosphorylated and phosphorylated M2-1 proteins in their abilities to pull down P or tRNA.
FIG. 8.

In vitro M2-1 phosphorylation has no visible effect on binding to P or RNA or on oligomerization. (A) In vitro phosphorylation of M2-1. GST-M2-1 was in vitro phosphorylated (lane 1) or not (lane 2) by CKI in the presence of [γ-32P]ATP32, cleaved by thrombin, resolved by SDS-PAGE, stained with Coomassie blue (lanes 1 and 2), and exposed by autoradiography (lanes 3 and 4). +, with. (B) Unphosphorylated GST-M2-1 (lanes 1 and 3) and phosphorylated GST-M2-1 (lanes 2 and 4) were incubated with P (lane 5) and washed, and the presence of P pulled down by GST-M2-1 was revealed by SDS-PAGE after thrombin cleavage. (C and D) Unphosphorylated GST-M2-1 (lanes 3 and 4) and phosphorylated GST-M2-1 (lanes 5 and 6) were incubated with tRNA and resolved by agarose gel electrophoresis. The agarose gel was stained with EtBr (C) and amido black (D). Lanes: 1, GST; 2, GST and tRNA; 3, GST-M2-1; 4, GST-M2-1 and tRNA; 5, phosphorylated GST-M2-1; 6, phosphorylated GST-M2-1 and tRNA; and 7, tRNA. (E) Phosphorylated M2-1 cross-linking. In vitro-phosphorylated M2-1 was cross-linked with EGS as indicated in the legend to Fig. 6.
To determine whether phosphorylation could affect M2-1 oligomerization, phosphorylated GST-M2-1 was cleaved by thrombin and the soluble phosphorylated M2-1 was used for chemical cross-linking with EGS. As shown in Fig. 8E, phosphorylated M2-1 was efficiently cross-linked by EGS, with the same pattern as that for unphosphorylated M2-1 (see above and Fig. 3B).
Finally, M2-1 was also efficiently phosphorylated when it was incubated with either RNA or P before in vitro phosphorylation, indicating that interaction with P or RNA did not prevent its phosphorylation (data not shown).
Taken together, these results indicate that M2-1 phosphorylation does not affect its oligomeric status or its binding with either RNA or P, at least in vitro.
DISCUSSION
In the present study, we expressed and purified recombinant M2-1 protein for preliminary structural studies. We observed that nucleic acids copurified with M2-1 could be removed using strong ionic wash conditions. This intrinsic RNA-binding activity has been observed previously with a recombinant M2-1 protein that was renatured after SDS-PAGE treatment (8). This property may partly explain the difficulties that were encountered by different groups when attempting to purify RSV M2-1 in sufficient amounts to allow biochemical and structural studies (3, 8, 19, 24).
The secondary structure of M2-1 was analyzed by CD spectroscopy, and the measured spectra showed that M2-1 adopts a highly structured α-helical conformation in solution, which is in agreement with computed secondary structure predictions that most of the protein (residues 33 to 170) adopts a helix bundle architecture (Fig. 9B).
FIG. 9.

Organization of HRSV M2-1. (A) Sequence alignment of HRSV, BRSV, mouse pneumovirus (MPV), human metapneumovirus (HMPV), and avian metapneumovirus (AMPV) M2-1 proteins by ClustalW. Acidic residues are in red, basic residues are in blue, hydrophobic residues are in green, and glycines are in boldface. Asterisks and colons indicate identities and homologies, respectively. (B) Hydrophobic cluster analysis plot for HRSV M2-1. Structured regions are characterized by a high number of hydrophobic clusters, while unstructured regions have low numbers or are devoid of hydrophobic clusters. Secondary structure predictions were performed with PSI-PRED (21) and are shown below the plot.
The quaternary structure of M2-1 was investigated with several complementary techniques. DLS analysis showed that recombinant M2-1 was highly homogenous in solution, consisting of a single protein species with a DH of approximately 7.3 nm and a VH of ≈195,000 Å3. Considering the frictional ratio generated from ultracentrifugation studies, we calculated that M2-1 is a globular 89-kDa complex with a DH of 7.66 nm, which is consistent with findings from DLS studies. According to Uversky (32), the theoretical VH expected for a native, globular protein of the size of M2-1 would be about 45,000 Å3. The experimentally measured VH may correspond to either a partially or completely unfolded state for M2-1 or an oligomeric molecule, since both cases would result in an increased VH compared to the theoretical value. Since M2-1 was not intrinsically disordered when analyzed by CD spectroscopy, the latter scenario is more likely. The experimentally observed VH (≈195,000 Å3) obtained in this study indicates that M2-1 is a globular protein of approximately 800 residues, suggesting that M2-1 is tetrameric. These data were supported by observations from electron microscopy showing that the recombinant M2-1 protein is a small object of about 10 nm in diameter, which was in agreement with the DLS measurements. Chemical cross-linking also indicated that M2-1 is a tetramer. These are the first direct findings that M2-1 is oligomeric, though previous experiments using UV cross-linking suggested that M2-1 may form dimers in solution (8).
The coexpression of GST-M2-1 and His-M2-1 in bacteria and further purification by GST affinity chromatography showed that M2-1 can also form oligomers in bacteria, despite the occurrence of affinity tags. We used this coexpression strategy to map the M2-1 oligomerization domain, or at least part of it, to amino acids 35 to 58. Notably, this region is well conserved among pneumoviruses (Fig. 9A). The CD spectrum of M2-1Δ35-58 indicates that this mutant has a lower α-helix content than full-length M2-1. The region of amino acids 35 to 58 was predicted by the PSI-PRED program to fold as an α-helix (Fig. 9B), and this putative α-helix was characterized by regularly spaced hydrophobic and charged residues, in particular, Arg38, Arg45, Lys48, and Lys52, that should be found on the same face of the helix. The end of this region comprised a series of three acidic residues, each separated by 3 residues that would also be found on the same side of the putative α-helix. Thus, although this region was not predicted to be a coiled-coil domain by bioinformatic tools (data not shown), it has the characteristics of an amphipathic α-helix. Interestingly, this region contains two of the predicted phosphorylation sites of M2-1 (3). The in vitro phosphorylation of M2-1 did not inhibit self-interaction, as determined by EGS cross-linking, discounting a role, at least in vitro, for phosphorylation in affecting oligomerization. To evaluate whether the M2-1 transcription antiterminator activity requires M2-1 oligomerization, we used a luciferase reporter RSV minigenome. The deletion of the region responsible for M2-1 tetramerization in vitro abolished the transcription antiterminator activity of the protein. These results indicate that, while interaction with P or RNA does not, the positive effect of M2-1 on viral transcription depends on its oligomeric status, for reasons that remain to be clarified.
It has been shown previously by several groups that the HRSV M2-1 protein can bind to P and RNA in vitro. Cuesta et al. observed that RNA binding by M2-1 is relatively promiscuous for long RNA but specific for short (≤100-nucleotide) RNA containing the leader sequence (8). Using recombinant proteins, Mason et al. discovered that M2-1 can also interact directly with P in vitro (24). Using GST pulldown and band shift assays, we confirmed that our recombinant M2-1 protein was also able to bind P and RNA but with no sequence specificity. However, since we used a molar excess of RNA, we cannot exclude the possibility that some sequence specificity does exist for M2-1 at lower concentrations of RNA. We observed that recombinant P and RNA were efficiently pulled down by GST-M2-1 (Fig. 7), and a band shift was clearly observed when tRNA was added to M2-1 (Fig. 8C and D). RNA was retarded upon native agarose gel electrophoresis, when M2-1 migration was accelerated, probably due to the neutralization of M2-1 positive charges by RNA.
By using a series of N- and C-terminal deletions, the RNA- and P-binding domains of M2-1 were identified. It was noted during these studies that deletions within the region of amino acids 86 to 173 were not tolerated, since they resulted in the production of insoluble GST fusion proteins. These results suggest that these deletion mutant forms of M2-1 were misfolded and that the correct folding of the central region of M2-1 (residues 86 to 173) depends on the entire core sequence. Previous results, obtained using an M2-1 mutant with cysteine 7 converted to a serine (C7S), indicated that the CCCH motif is important for the adoption of the correct conformation and for RNA binding (3). However, CD analysis of M2-1Δ31N, M2-1Δ58N, and M2-1Δ35-58 mutants, which all lack the putative ZnF domain and which we purified efficiently, did not reveal any change in the global, α-helical folds of the molecules. It is possible that the presence of GST at the N termini aids the folding of these molecules and/or protects them from degradation. By using GST pulldowns, the region of residues 59 to 177 of M2-1 was shown to bind to both tRNA and P protein in vitro. Furthermore, we found that yeast tRNA and P protein bind to M2-1 in a competitive manner, the presence of RNA preventing M2-1-P interactions. The deletion of amino acids 59 to 86 slightly affected RNA and P binding, possibly because of the conformational modification of the domain of residues 59 to 177 as a result of the truncation. Because RNA and P are both negatively charged molecules, they may compete for the same positively charged domain on M2-1. These results suggest that either a conformational change on M2-1 occurs after M2-1 binding to RNA, affecting the P-binding site, or P and RNA compete for the same site on M2-1, with M2-1 possibly having higher affinity for RNA. Cuesta et al. reported that M2-1 is an RNA-binding protein that preferentially binds to short RNAs (<100 nucleotides) containing plus-polarity (antigenomic) RSV leader sequences but that can also bind to long RNA with no apparent sequence specificity (8). On the contrary, Cartee and Wertz reported that M2-1 interacts predominantly with RSV mRNAs that do not contain a leader sequence (3). Thus, although affinity for specific viral sequences is suggested by these data, our results support the suggestion that M2-1 has intrinsic RNA-binding activity with no sequence specificity, at least in vitro.
In a previous study, the RNA-binding domain of M2-1 was mapped to residues 59 to 85 (8). Nonspecific RNA-binding domains are expected to contain basic residues that will interact with phosphate groups of RNA. The region of positions 59 to 85 contains only one basic residue (Arg68). Examination of the sequence of M2-1 shows that a cluster of basic residues is present in the region of positions 135 to 165. This positively charged region is a good candidate for being part of a domain responsible for binding to negatively charged molecules.
A study by Mason et al. (24) and the experiments reported here demonstrated the interaction between P and M2-1 in vitro by using recombinant proteins. However, it is not clear whether this interaction really occurs within cells. Using coimmunoprecipitation, Asenjo et al. found that the P-M2-1 interaction could be detected in cells cotransfected with plasmids encoding P and M2-1 (1). On the contrary, other groups did not detect this interaction in cells cotransfected with M2-1 and P expression plasmids (13, 24) but did detect it in cells expressing N, P, and M2-1 (13). Furthermore, the interaction between M2-1 and N proteins was probably mediated by RNA (3). Indeed, it is possible that the M2-1-P interaction observed in vitro does not occur in transfected or infected cells or is only transient and masked by M2-1-RNA interactions and that the colocalization of P, N, and M2-1 in infected cells is due only to the binding of M2-1 to viral RNA.
The role of M2-1 phosphorylation remains unclear. The mutation of amino acid residues Ser58 and Ser61, the targets for phosphorylation, results in a decrease in the efficiency of M2-1 transcriptional antitermination activity (3). In RSV-infected cells, unphosphorylated M2-1 is the predominant form of the protein (16, 22, 28). This is also the case when cells are cotransfected to express M2-1 and P (1). However, when cells are transfected to express M2-1 without P, the phosphorylated form of the protein predominates, suggesting that the interaction between M2-1 and P modulates M2-1 phosphorylation. The unphosphorylated recombinant M2-1 purified from bacteria was able to bind to RNA and to P in our in vitro assays. We did not observe any change in the RNA- or P-binding activity of M2-1 after phosphorylation by CKI. These results were in agreement with previously reported data that indicated that M2-1 phosphorylation is necessary for the transcription antitermination function but not for RNA or P binding (1, 3, 8, 24). Residues Ser58 and Ser61 are situated at the end of the oligomerization domain of M2-1, and their phosphorylation may affect the oligomeric status of M2-1. Because M2-1 was ∼100% phosphorylated by CKI in vitro, these residues must be accessible to CKI within tetramers and thus are probably situated at the surface of the molecule; no disturbance of M2-1 tetramerization by chemical cross-linking was observed. Furthermore, the presence of P or RNA did not affect the in vitro phosphorylation of M2-1, indicating that Ser58 and Ser61 within the complexes were still accessible to CKI.
In conclusion, we have shown that HRSV M2-1 forms tetramers in solution and can be divided into four distinct parts: a putative ZnF (amino acid residues 1 to 32), the function of which remains unknown; a domain responsible for M2-1 oligomerization, situated between amino acid residues His33 and Gly62; a globular domain composed of α-helices, encompassing amino acid residues 63 to 170, capable of binding P and RNA with no sequence specificity; and a predicted unstructured C-terminal region (amino acid residues 171 to 194) with a low degree of conservation.
Acknowledgments
We thank G. Wertz for providing the minigenome system, Human Rezaei for help in CD spectroscopy and critical reading of the manuscript, Anny Slama-Schwok for help in DLS, Sonia Longhi, Jean Lepault, Yves Gaudin, and Noël Tordo for helpful discussions, José A. Melero for advice and provision of anti-M2-1 monoclonal antibodies, and Wendy Brand-Williams for critical reading of the manuscript. We are grateful to Karine Madiona for ultracentrifugation experiments and to Ronald Melki for advice. The ultracentrifugation work has benefited from the LEBS facilities of the IMAGIF Structural Biology and Proteomic Unit of the Gif campus (www.imagif.cnrs.fr).
Footnotes
Published ahead of print on 22 April 2009.
REFERENCES
- 1.Asenjo, A., E. Calvo, and N. Villanueva. 2006. Phosphorylation of human respiratory syncytial virus P protein at threonine 108 controls its interaction with the M2-1 protein in the viral RNA polymerase complex. J. Gen. Virol. 873637-3642. [DOI] [PubMed] [Google Scholar]
- 2.Buchholz, U. J., S. Finke, and K. K. Conzelmann. 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 73251-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cartee, T. L., and G. W. Wertz. 2001. Respiratory syncytial virus M2-1 protein requires phosphorylation for efficient function and binds viral RNA during infection. J. Virol. 7512188-12197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Castagne, N., A. Barbier, J. Bernard, H. Rezaei, J. C. Huet, C. Henry, B. Da Costa, and J. F. Eleouet. 2004. Biochemical characterization of the respiratory syncytial virus P-P and P-N protein complexes and localization of the P protein oligomerization domain. J. Gen. Virol. 851643-1653. [DOI] [PubMed] [Google Scholar]
- 5.Collins, P. L., and J. E. Crowe. 2007. Respiratory syncytial virus and metapneumovirus, p. 1601-1646. In D. M. Knipe and P. M. Howley (ed.), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 6.Collins, P. L., M. G. Hill, E. Camargo, H. Grosfeld, R. M. Chanock, and B. R. Murphy. 1995. Production of infectious human respiratory syncytial virus from cloned cDNA confirms an essential role for the transcription elongation factor from the 5′ proximal open reading frame of the M2 mRNA in gene expression and provides a capability for vaccine development. Proc. Natl. Acad. Sci. USA 9211563-11567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collins, P. L., and G. W. Wertz. 1985. The envelope-associated 22K protein of human respiratory syncytial virus: nucleotide sequence of the mRNA and a related polytranscript. J. Virol. 5465-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cuesta, I., X. Geng, A. Asenjo, and N. Villanueva. 2000. Structural phosphoprotein M2-1 of the human respiratory syncytial virus is an RNA binding protein. J. Virol. 749858-9867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deleage, G., and C. Geourjon. 1993. An interactive graphic program for calculating the secondary structure content of proteins from circular dichroism spectrum. Comput. Appl. Biosci. 9197-199. [DOI] [PubMed] [Google Scholar]
- 10.DuBois, R. N., M. W. McLane, K. Ryder, L. F. Lau, and D. Nathans. 1990. A growth factor-inducible nuclear protein with a novel cysteine/histidine repetitive sequence. J. Biol. Chem. 26519185-19191. [PubMed] [Google Scholar]
- 11.Fearns, R., and P. L. Collins. 1999. Role of the M2-1 transcription antitermination protein of respiratory syncytial virus in sequential transcription. J. Virol. 735852-5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gaboriaud, C., V. Bissery, T. Benchetrit, and J. P. Mornon. 1987. Hydrophobic cluster analysis: an efficient new way to compare and analyse amino acid sequences. FEBS Lett. 224149-155. [DOI] [PubMed] [Google Scholar]
- 13.Garcia, J., B. Garcia-Barreno, A. Vivo, and J. A. Melero. 1993. Cytoplasmic inclusions of respiratory syncytial virus-infected cells: formation of inclusion bodies in transfected cells that coexpress the nucleoprotein, the phosphoprotein, and the 22K protein. Virology 195243-247. [DOI] [PubMed] [Google Scholar]
- 14.Grosfeld, H., M. G. Hill, and P. L. Collins. 1995. RNA replication by respiratory syncytial virus (RSV) is directed by the N, P, and L proteins; transcription also occurs under these conditions but requires RSV superinfection for efficient synthesis of full-length mRNA. J. Virol. 695677-5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hardy, R. W., S. B. Harmon, and G. W. Wertz. 1999. Diverse gene junctions of respiratory syncytial virus modulate the efficiency of transcription termination and respond differently to M2-mediated antitermination. J. Virol. 73170-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hardy, R. W., and G. W. Wertz. 2000. The Cys3-His1 motif of the respiratory syncytial virus M2-1 protein is essential for protein function. J. Virol. 745880-5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hardy, R. W., and G. W. Wertz. 1998. The product of the respiratory syncytial virus M2 gene ORF1 enhances readthrough of intergenic junctions during viral transcription. J. Virol. 72520-526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hartlieb, B., T. Muziol, W. Weissenhorn, and S. Becker. 2007. Crystal structure of the C-terminal domain of Ebola virus VP30 reveals a role in transcription and nucleocapsid association. Proc. Natl. Acad. Sci. USA 104624-629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang, Y. T., P. L. Collins, and G. W. Wertz. 1985. Characterization of the 10 proteins of human respiratory syncytial virus: identification of a fourth envelope-associated protein. Virus Res. 2157-173. [DOI] [PubMed] [Google Scholar]
- 20.John, S. P., T. Wang, S. Steffen, S. Longhi, C. S. Schmaljohn, and C. B. Jonsson. 2007. Ebola virus VP30 is an RNA binding protein. J. Virol. 818967-8976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones, D. T. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292195-202. [DOI] [PubMed] [Google Scholar]
- 22.Lambert, D. M., J. Hambor, M. Diebold, and B. Galinski. 1988. Kinetics of synthesis and phosphorylation of respiratory syncytial virus polypeptides. J. Gen. Virol. 69313-323. [DOI] [PubMed] [Google Scholar]
- 23.Li, D., D. A. Jans, P. G. Bardin, J. Meanger, J. Mills, and R. Ghildyal. 2008. Association of respiratory syncytial virus M protein with viral nucleocapsids is mediated by the M2-1 protein. J. Virol. 828863-8870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mason, S. W., E. Aberg, C. Lawetz, R. DeLong, P. Whitehead, and M. Liuzzi. 2003. Interaction between human respiratory syncytial virus (RSV) M2-1 and P proteins is required for reconstitution of M2-1-dependent RSV minigenome activity. J. Virol. 7710670-10676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meyer, G., M. Deplanche, and F. Schelcher. 2008. Human and bovine respiratory syncytial virus vaccine research and development. Comp. Immunol. Microbiol. Infect. Dis. 31191-225. [DOI] [PubMed] [Google Scholar]
- 26.Modrof, J., S. Becker, and E. Muhlberger. 2003. Ebola virus transcription activator VP30 is a zinc-binding protein. J. Virol. 773334-3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Philo, J. S. 1997. An improved function for fitting sedimentation velocity data for low-molecular-weight solutes. Biophys. J. 72435-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Routledge, E. G., M. M. Willcocks, L. Morgan, A. C. Samson, R. Scott, and G. L. Toms. 1987. Heterogeneity of the respiratory syncytial virus 22K protein revealed by Western blotting with monoclonal antibodies. J. Gen. Virol. 681209-1215. [DOI] [PubMed] [Google Scholar]
- 29.Schuck, P. 2000. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 781606-1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sreerama, N., and R. W. Woody. 1994. Poly(pro)II helices in globular proteins: identification and circular dichroic analysis. Biochemistry 3310022-10025. [DOI] [PubMed] [Google Scholar]
- 31.Tang, R. S., N. Nguyen, X. Cheng, and H. Jin. 2001. Requirement of cysteines and length of the human respiratory syncytial virus M2-1 protein for protein function and virus viability. J. Virol. 7511328-11335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uversky, V. N. 2002. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 11739-756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Worthington, M. T., B. T. Amann, D. Nathans, and J. M. Berg. 1996. Metal binding properties and secondary structure of the zinc-binding domain of Nup475. Proc. Natl. Acad. Sci. USA 9313754-13759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Worthington, M. T., J. W. Pelo, M. A. Sachedina, J. L. Applegate, K. O. Arseneau, and T. T. Pizarro. 2002. RNA binding properties of the AU-rich element-binding recombinant Nup475/TIS11/tristetraprolin protein. J. Biol. Chem. 27748558-48564. [DOI] [PubMed] [Google Scholar]
- 35.Yu, Q., R. W. Hardy, and G. W. Wertz. 1995. Functional cDNA clones of the human respiratory syncytial (RS) virus N, P, and L proteins support replication of RS virus genomic RNA analogs and define minimal trans-acting requirements for RNA replication. J. Virol. 692412-2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou, H., X. Cheng, and H. Jin. 2003. Identification of amino acids that are critical to the processivity function of respiratory syncytial virus M2-1 protein. J. Virol. 775046-5053. [DOI] [PMC free article] [PubMed] [Google Scholar]