

Abstract
Cross-regulation of RUNX1 expression by RUNX3 plays a critical role in regulating proliferation of human B cells infected with Epstein-Barr virus (EBV). When EBV infection induces RUNX3, the consequent reduction in RUNX1 levels is required for the ensuing cell proliferation because forced expression of RUNX1 in an EBV lymphoblastoid cell line prevented cell proliferation. The TEL-RUNX1 fusion gene from acute B-lymphocytic leukemia retains almost all of the RUNX1 sequence but does not prevent B-cell proliferation in the same assay. B-cell maturation antigen (BCMA) was found to be induced by conditionally expressed RUNX3 in a lymphoma cell line. Chromatin immunoprecipitation assays confirmed that RUNX3 binds to the RUNX1 promoter in a lymphoblastoid cell line and a Burkitt's lymphoma cell line. The TLE binding VWRPY sequence from the C terminus of RUNX3 was found to be required for repression of the RUNX1 P1 promoter in a B-lymphoma cell line. The mechanism of repression in B-cell lines most likely involves recruitment of corepressor TLE3 or TLE4 to the RUNX1 promoter. The results demonstrate the importance of RUNX3-mediated repression of RUNX1 for EBV-driven B-cell proliferation and identify functional differences between human RUNX family proteins.
Latent infection of resting human B cells with Epstein-Barr virus (EBV) drives proliferation and immortalization of the cells, giving rise to lymphoblastoid cell lines (LCLs). During latent infection, the viral transcription factor EBNA2 induces viral and cell gene expression to cause the cell proliferation. We previously showed that RUNX3 is a direct target gene of EBNA2 in LCLs and that induction of RUNX3 is required for proliferation of human B-LCLs made by infection of primary human B cells with EBV (33, 34).
Although RUNX3 probably regulates many cell genes, we demonstrated that one effect of RUNX3 in EBV-transformed LCLs is downregulation of RUNX1 expression by a mechanism that involves the RUNX3 binding sites in the promoter of RUNX1 (34). Normal peripheral human B cells contain RUNX1 and are in a resting, nonproliferative state. Infection by EBV or stimulation of B cells with PMA causes induction of RUNX3 and results in a severe decrease in RUNX1 expression (34). However, it was not clear from that work whether the reduction in RUNX1 levels is required for LCL proliferation, particularly in view of the fact that latency I Burkitt's lymphoma (BL) cell lines (which lack EBNA2) normally express RUNX1 while proliferating in culture (33).
The RUNX family of transcriptional regulators plays key roles in B-cell development and maturation (28, 37), and RUNX1 is frequently translocated in acute lymphocytic leukemia and acute myeloid leukemia (AML). Several different translocation partners are known, including TEL1 (in about 20% of childhood acute lymphocytic leukemia cases). RUNX proteins all form heterodimers with the CBF-β protein and can then act as transcription factors, inducing or repressing genes, depending on the context. Human RUNX proteins all share the N-terminal Runt domain (which mediates CBF-β association and sequence-specific DNA binding), a transactivation domain, and several regions through which gene repression has also been shown to occur. One region associated with repression is the C-terminal sequence VWRPY, which has been shown to recruit TLE corepressor proteins that mediate repression of gene expression (19, 21, 24, 36). Members of the TLE family of corepressors have been shown to be involved in a wide variety of key regulatory events in B-cell biology.
Studies of transgenic mice with a conditional knockout of RUNX1 showed that loss of RUNX1 activity blocks B-cell differentiation at an early stage, reflecting the key role played by the RUNX family in normal B-cell development (9, 18). RUNX3 is induced by transforming growth factor β1 (31) and plays an important role in transforming growth factor β-induced B-cell antibody class switching (37). However, despite strong data demonstrating the importance of the RUNX family in key checkpoints during B-cell development, the precise roles of RUNX gene expression during these events, particularly in human cells, have yet to be fully clarified. The RUNX dominant-negative oncogenic fusion gene CBF-β-SMMHC has been shown to cause a block to S-phase entry in the pro-B-cell line Ba/F3 (5), but this cell cycle block was overcome by forced expression of c-MYC, indicating that c-MYC functions downstream of the RUNX proteins in those cells. Since BL cells characteristically have a translocated c-MYC allele that is expressed in a deregulated fashion, this can overcome the effects of RUNX1 and RUNX3 on cell proliferation, and BL cell lines thus provide an opportunity to investigate the mechanism of RUNX gene cross-regulation in B-cell lines without the complication of effects on cell proliferation.
In this paper, we demonstrate that downregulation of RUNX1 by RUNX3 is required for EBV-driven LCL growth, as demonstrated by the severe inhibition of LCL growth caused by expression of RUNX1 in an LCL line. We also use a B-lymphoma cell line to show that repression of the RUNX1 gene requires the RUNX3 VWRPY sequence and that RUNX3 is specifically bound to the RUNX1 promoter in the B-lymphoma cell line and in EBV LCL cells where repression occurs. The TLE3 and -4 genes are the TLE genes expressed in LCLs, and most likely, TLE3 acts as a corepressor in the cross-repression phenomenon. Finally, the B-cell survival protein BCMA (B-cell maturation antigen) is induced in BL cells when RUNX3 expression increases and RUNX1 levels decrease, suggesting at least one relevant target gene which may play a role in RUNX regulation of B-cell development.
MATERIALS AND METHODS
Plasmid construction.
pMEP4-RUNX3ΔVWRPY and pCEP4-RUNX3ΔVWRPY were constructed by amplifying the RUNX3 cDNA sequence lacking the VWRPY sequence and ligating into the KpnI and NotI sites of the constitutive pCEP4 and cadmium-inducible pMEP4 vectors (Invitrogen). The pCEP4ER root vector was constructed by amplifying the estrogen receptor (ER)-binding domain from a RUNX1-ER fusion plasmid kindly provided by Alan Friedmann (4) and translationally silent mutation of an internal KpnI site. RUNX3 and RUNX3ΔVWRPY were then cloned between the KpnI and NotI sites of this vector. The pEBtetD-RUNX1c construct was made by subcloning RUNX1c cDNA between the KpnI and NotI sites of pEBtetD (2) (from Dirk Gründemann). RUNX1 fusion cDNA TEL-RUNX1 (from Tony Ford) was subcloned into pEBtetD and pCEP4 by using KpnI and NotI sites. The BCMA promoter (459 bp) was amplified from DG75 BL cell line genomic DNA by using the primer sequences TTGATGCTGTGGGCTTGTCTGC (forward) and CGCTGACATGTTAGAGGAGG (reverse) and cloned into pCR-BluntII-TOPO (Invitrogen). The BCMA promoter was then excised and ligated between the KpnI and XhoI sites of pGL3-Basic (Promega).
Cell lines.
DG75 (3) and Akata 31 (20) are EBV-negative BL cell lines. IB4 is an EBV-immortalized LCL generated by infection of cord blood lymphocytes (35). The other B-cell lines were as described previously (33). The cell lines were maintained in RPMI 1640 medium (Gibco-BRL) supplemented with 10 to 20% (vol/vol) heat-inactivated fetal calf serum (FCS), glutamine, and antibiotics. Akata cell lines stably transfected with pMEP4-RUNX3 were maintained in RPMI 1640 supplemented with 10% FCS, glutamine, antibiotics, and 300 μg/ml hygromycin. HEK293 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% FCS, glutamine, and antibiotics.
Immunoblotting and antibodies.
Radioimmunoprecipitation assay (RIPA) lysates were prepared and quantitated, and immunoblotting was performed as described previously (8). Proteins were fractionated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes. After being blocked with 10% milk powder in phosphate-buffered saline (PBS)-0.05% Tween 20, membranes were probed with the following antibodies: 1/40 dilution of rabbit polyclonal antibodies (Oncogene research products), anti-AML-1 (RUNX1) RHD (Ab-2), anti-AML-2 (RUNX3) (Ab-1), 1/1,000 of TLE3 (M-201; Santa Cruz), 1/1,000 of ER (MC-20; Santa Cruz), 1/100 of BCMA (Abcam), or 1/1,000 of PCNA or a 1/5,000 dilution of mouse monoclonal anti-β-actin (AC-15; Sigma). Secondary antibodies were horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin (Sigma) and horseradish peroxidase-conjugated sheep anti-mouse immunoglobulin (Sigma). Bound immunocomplexes were detected by enhanced chemiluminescence (GE Healthcare).
ChIP.
The chromatin immunoprecipitation (ChIP) assay was performed using an EZ ChIP kit (Upstate) in accordance with the kit protocol. DNA was extracted from treated DG75 cells or IB4 cells, and sonication was performed. For sonication, the use of two sets of 15-s bursts with a Sonicator ultrasonic processor (Heat systems) at setting 5 was found to be optimal for generation of 100- to 500-bp fragments capable of amplifying the RUNX1P1 promoter in control samples. A total of 10 μg of anti-ER antibody was used for immunoprecipitation. A 252-bp fragment spanning the RUNX1 P1 promoter and part of the first exon was amplified by PCR using the primer sequences AAGGAAGGGCATTGCTCAGA and ACCCTGTGGTTTGCATTCAG.
Reverse transcription-PCR (RT-PCR).
Total RNA was extracted from cell lines by using TRI reagent (Sigma), and cDNA was generated using a Postscript first-strand cDNA synthesis kit (New England Biolabs). Target sequences were amplified by PCR amplification using primers TLE1 (forward, ATCTTTCTCATGGCCACGG; reverse, TCTGCGTTTATCTGTGCC), TLE2 (forward, AGCTCCAGCCGCTGTCCC; reverse, ATCACTCTTGTCTTCGTCG), TLE3 (forward, TCTCCCATGCCACACACG; reverse, TTCTCTCTCTCTGTGATCG), and TLE4 (forward, ACATGGACATGGTCTCCC; reverse, TTGGTGATCATTGTCATGG).
Microarrays and RPA.
For microarray analysis, RNA from cell lines was reversed transcribed using an oligo(dT) primer and the resulting probes were applied to Sanger Institute microarrays representing 5,300 genes from the human genome, as described previously (33). For the RNase protection assay (RPA) probe, the DNA sequence spanning the transcription start of the BCMA gene was amplified by PCR of DG75 genomic DNA with the primer sequences AGCTGCTTTGAGTGCTACGG and GGACAAAAAGCTGTATCGGC. The PCR product was cloned into pCR-II-TOPO and sequenced and the plasmid linearized at the 5′ end of the PCR product. One microgram of linearized plasmid was used to generate 32P-labeled antisense RNA probes with a MAXIscript SP6/T7 in vitro transcription kit, and RPAs were carried out using an Ambion RPA III kit. Cell RNA (10 μg) extracted using RNAzol B was hybridized overnight at 42°C with 50,000 cpm of probe. An equivalent amount of yeast RNA was included in a hybridization reaction mixture as a negative control. After hybridization, single-strand RNA was digested with an RNase A/T1 mixture for 30 min at 37°C, and protected fragments were precipitated, fractionated on an 8 M urea polyacrylamide gel, and analyzed with a phosphorimager.
Luciferase assays.
For transient expression in BL and LCL lines, exponentially growing cells (8 ×106) were electroporated at 250 mV and 960 μF in 0.4-cm cuvettes (Bio-Rad). Each transfection mixture contained 0.5 μg pCMV-β-galactosidase and 1 μg reporter construct. The total amount of DNA transfected was approximately 10 μg and was normalized by addition of empty vector (pCEP4). Following electroporation, cells were resuspended in 10 ml conditioned medium and incubated for 48 h. Cell pellets were harvested and lysed in 60 μl luciferase reporter lysis buffer (Promega). In total, 20 μl of lysate was analyzed for luciferase activity and an additional 20 μl assayed for β-galactosidase activity using chlorophenol red-β-d-galactopyranoside as a substrate.
Transient transfection of HeLa cells was performed in six-well dishes with cells at approximately 80% confluence. A total of 3 μg plasmid DNA was transfected into cells by using Lipofectamine (Invitrogen); cells were harvested in sample buffer and assayed for protein expression 24 h later.
Stable transfected cell lines.
Stable B-cell lines were generated using the Amaxa electroporation system (Amaxa Biosystems) with exponentially growing cells (8 ×106), 10 μg plasmid, and an Amaxa Nucleofector I electroporator. Cells were recovered in conditioned medium, and hygromycin selection was administered on the following day. Cultures were fed with fresh medium containing hygromycin every 3 or 4 days until stable outgrowth was obtained.
Confocal microscopy.
HEK293 cells were cultured on poly-l-lysine (Sigma)-coated coverslips in six-well dishes at approximately 50% density and were transfected with 3 μg plasmid DNA. Cells were fixed using 4% paraformaldehyde and permeabilized with 0.05% Triton X-100 in PBS. Primary anti-ER antibody was diluted 1/500 in blocking buffer (PBS-5% bovine serum albumin-0.05% Triton X-100), and samples were incubated overnight at 4°C with shaking. The coverslips were washed three times with blocking buffer, and fluorescein isothiocyanate-conjugated anti-rabbit secondary antibody (Sigma) was applied at a 1/1,000 dilution in blocking buffer. The samples were again washed in triplicate, and the coverslips were mounted in MOWIOL and visualized using a confocal microscope.
RESULTS
RUNX1c expression inhibits proliferation of EBV-infected LCL cells.
We previously reported that the EBV latent transcription factor EBNA2 induces RUNX3 expression upon infection of B cells (33). This process was shown to be essential for LCL proliferation and was associated with a severe repression of RUNX1 expression. However, it was previously unclear whether the repression of RUNX1 was incidental or is required for proliferation of these EBV infected cells. To investigate the functional significance of RUNX1, a transient electroporation system was developed to test the effect of RUNX1c on the growth of LCLs. The pCEP4 vector was used to constitutively express RUNX1c or, as controls, RUNX3 or TEL-RUNX1 in IB4 LCL cells. The transfected cells were selected with hygromycin, and the cultures were monitored for cell number after 2 weeks. Whereas cells electroporated with empty vector, RUNX3, or TEL-RUNX1 grew out normally, cells transfected with the RUNX1c expression plasmid did not grow out (Fig. 1A). Even though the TEL-RUNX1 oncogene fusion contains almost all of the RUNX1c sequence (Fig. 1B), TEL-RUNX1 did not prevent outgrowth; in fact, it consistently gave slightly stronger growth than the vector control (Fig. 1A). Protein expression of TEL-RUNX1 in the transfected IB4 cells was confirmed by Western blotting (Fig. 1C), and RUNX1 expression was also confirmed in the early stages of selection of the RUNX1 cells (Fig. 1C) before the cells died. Transfection of the RUNX3 expression vector did not noticeably change the high endogenous level of RUNX3 in the IB4 LCL (Fig. 1C). To ensure that the RUNX1 result shown in Fig. 1A was not due to interference of the transfected RUNX1 with hygromycin resistance encoded by the plasmid, we also checked whether the plasmids could be stably transfected under hygromycin selection and express their protein products in a cell line unaffected by RUNX expression (HeLa cells). All the constructs allowed outgrowth of hygromycin-resistant HeLa cells, and RUNX protein expression from the plasmids was confirmed by Western blotting (Fig. 1D). These data confirm that RUNX1c expression is incompatible with the growth of EBV-infected B cells and also confirm that the TEL-RUNX1 fusion has lost that property.
FIG. 1.

Constitutive RUNX1c expression prevents outgrowth of LCLs. (A) IB4 LCL cells were electroporated with pCEP4, pCEP4-RUNX3, pCEP4-RUNX1, and PCEP4-TEL-RUNX1. Hygromycin selection (150 μg/ml) was added the following day, and live cells were counted after 2 weeks of selection. (B) Protein sequence relationship of RUNX1c and TEL-RUNX1. The locations of the Runt homology domain (RHD) and the VWRPY sequence are shown. The fusion joins amino acids 1 to 336 of TEL1 (ETV6) to amino acids 20 to 480 of RUNX1c. (C) RIPA extracts (50 μg protein) from extracts of IB4 cells transfected as described for panel A were analyzed by Western blotting 7 days after transfection with antibodies that detect RUNX3, RUNX1, or TEL-RUNX1 as indicated. β-Actin was also blotted as a loading control. (D) HeLa cells were transfected with empty-vector pCEP4, pCEP4-RUNX1, pCEP4-TEL-RUNX1, or pCEP4-RUNX3. After 24 h, cells were selected with 100 μg/ml hygromycin for 2 weeks. These cells were lysed in sample buffer and fractionated by SDS-PAGE. Western blotting was performed, probing for RUNX1 or RUNX3. αRUNX-1, anti-RUNX-1.
To provide an additional experimental approach to investigate the effects of RUNX1, we also used a doxycycline-inducible expression system for controlled expression of RUNX1c in an LCL. Using an episomal plasmid, pEBtetD (2), stable cell lines in which expression of RUNX1c could be controlled by doxycycline were derived from the IB4 LCL (Fig. 2A). Expression of RUNX1c had a strongly inhibitory effect on the growth of these cell cultures compared to that of IB4 cells containing the pEBtetD empty vector (Fig. 2A). The level of RUNX1 expression upon 24 h of induction was similar to that present normally in Akata 31 BL cells (Fig. 2B), so the growth inhibition was not due to a nonphysiological expression level of RUNX1. As a control, a similar doxycycline-inducible cell line conditionally expressing TEL1-RUNX1 was established. Again, induction of TEL-RUNX1 did not prevent LCL proliferation (Fig. 2C). Expression of TEL-RUNX1 protein induced by doxycycline was confirmed (Fig. 2D).
FIG. 2.
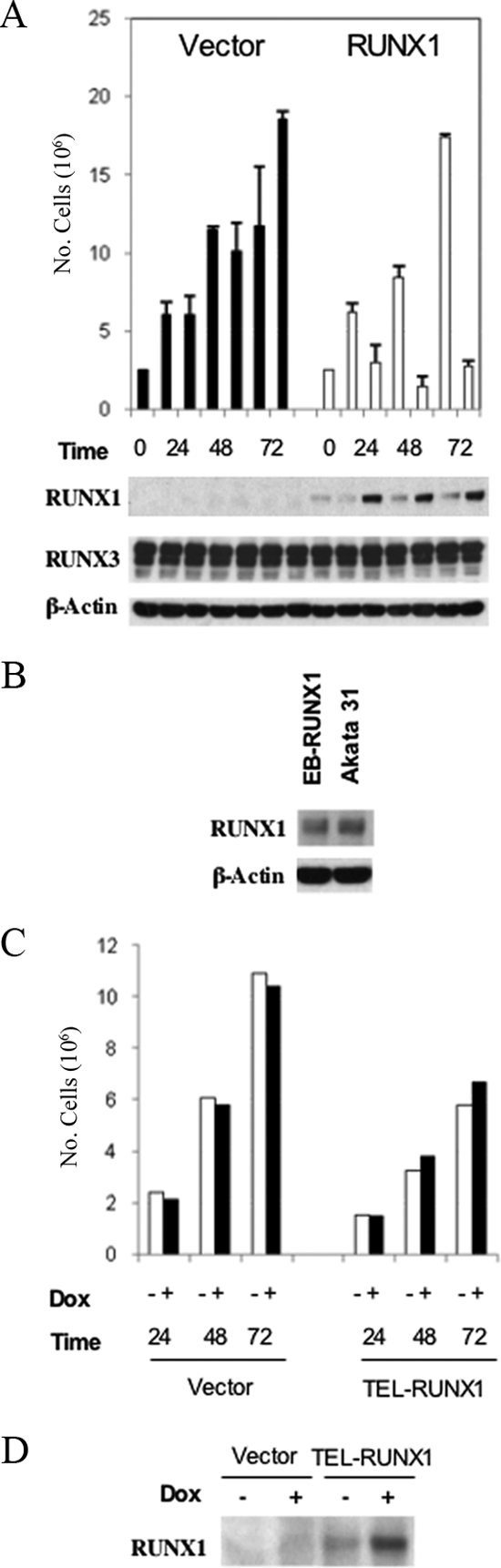
Doxycycline-inducible RUNX1c expression reduces growth in EBV-infected LCL cells. (A) IB4 LCLs were stably transfected with a doxycycline-inducible empty-vector construct (pEBtetD; filled bars) or one inducibly expressing RUNX1c (pEBtetD-RUNX1c; open bars). Cells were cultured in duplicate with and without 5 μg/ml doxycycline and were harvested at the indicated times. Cells were counted (counts are shown in the top panel as numbers of cells), and then RIPA extracts were prepared and 50 μg protein was analyzed by Western blotting for RUNX1, RUNX3, and β-actin. (B) The pEBtetD-RUNX1c cells plus doxycycline (track labeled EB-RUNX1) were coelectrophoresed at the 24-h time point with 50 μg of Akata 31 BL cell line RIPA extract, and samples were analyzed by Western blotting for RUNX1 expression. (C) IB4 LCL cells were stably transfected with empty-vector pEBtetD (vector) or doxycycline (Dox)-inducible pEBtetD-TEL-RUNX1. IB4:pEBtetD empty-vector or IB4:pEBtetD-TEL-RUNX1 cells were incubated with or without 5 μg/ml doxycycline for the indicated times and counted. (D) The IB4:pEBtetD-TEL-RUNX1 stably transfected cells or vector control cells were incubated with or without 5 μg/ml doxycycline for 24 h, and RIPA extracts were probed for TEL-RUNX1 expression.
RUNX3 can induce the survival factor BCMA (B-cell maturation antigen).
Although the RUNX genes presumably regulate some aspects of gene expression to modulate B-cell growth or survival, there is very little information about the identity of the target genes of RUNX in B cells. Technical limitations prevented us from studying this in LCLs, but we succeeded in performing gene expression profiling on an Akata 31 cell line stably transfected with CdCl2-inducible pMEP4-RUNX3 in which cross-repression of RUNX1 occurs (34). Two replica cell lines (33) of pMEP4 control and pMEP4-RUNX3 cells were incubated with and without CdCl2, and the mRNA was used to screen microarrays representing mRNAs from the human genome. One of the candidate target genes identified on the microarrays was the B-cell maturation factor BCMA, which has been shown to be critical for survival of late-stage differentiated B cells and is also induced after B-cell activation (13). The induction of BCMA RNA in these cells by RUNX3 was confirmed using an RPA (Fig. 3A).
FIG. 3.
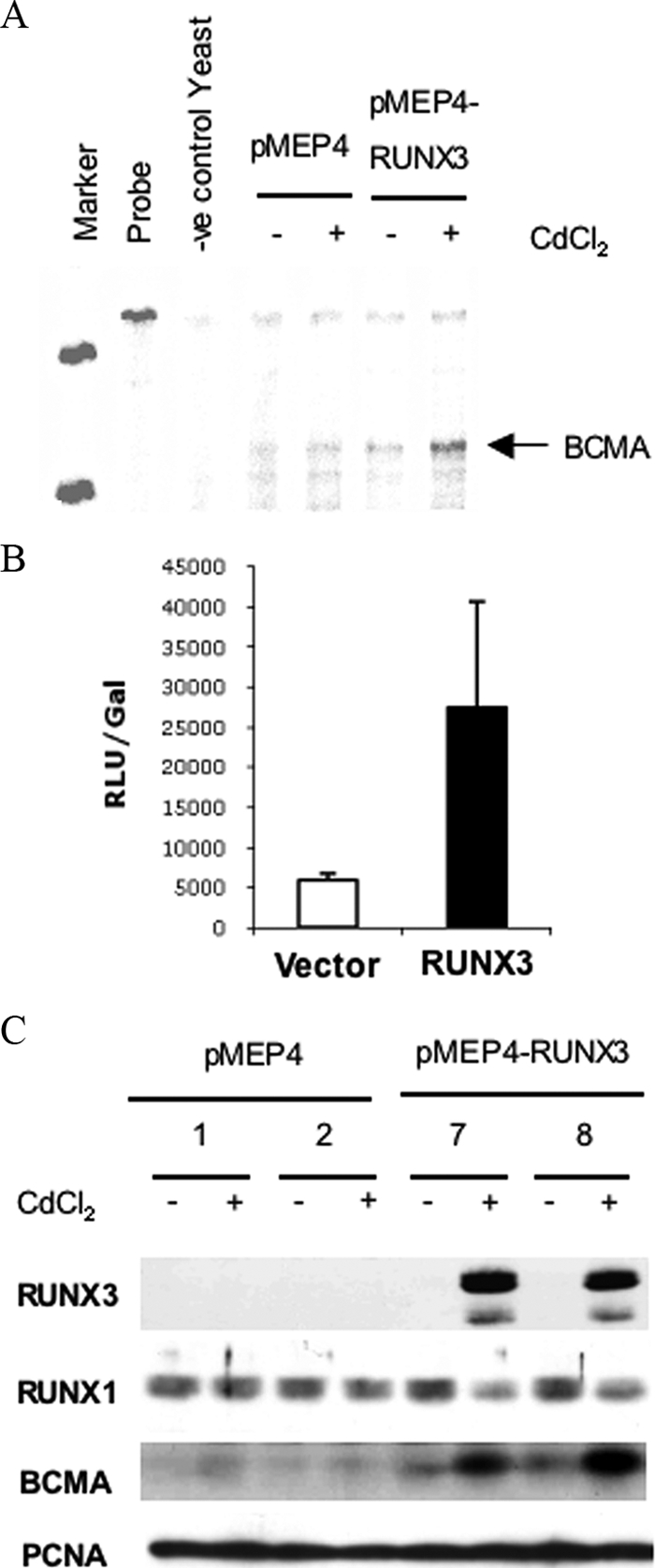
RUNX3 induces the survival factor BCMA (B-cell maturation antigen). (A) Akata31 pMEP4 and pMEP-RUNX3 cells were cultured for 12 h with or without 5 μM CdCl2. Total cell RNA (10 μg) from the cells was analyzed by RPA for BCMA RNA. M, P, and Y indicate size markers, undigested probe, and yeast negative control, respectively. (B) A 48-h transient transfection assay of DG75 cells with a BCMA promoter luciferase reporter construct (pGL3-BCMA) cotransfected with pCEP4 or pCEP-RUNX3. Results are expressed as means ± standard deviations of results from three transfections. Relative luciferase units (RLU) were adjusted for transfection efficiency according to a cotransfected β-galactosidase reporter plasmid. (C) Two isolates of Akata 31-pMEP4 cells (lanes 1 and 2) and two isolates of Akata 31-pMEP4-RUNX3 cells (lanes 7 and 8) were incubated for 12 h with or without 5 μM CdCl2. RIPA extracts were prepared, and 50 μg protein was separated by SDS-PAGE. Western blotting was performed, probing for RUNX3, RUNX1, BCMA, and PCNA (loading control).
We also tested whether RUNX3 could activate the BCMA promoter. A fragment of the BCMA promoter consisting of positions −343 to +115 relative to the transcription start site was cloned upstream of a luciferase reporter. This BCMA promoter fragment contains a single RUNX consensus sequence (TGTGGT) at position −189. DG75 cells were electroporated with the pGL3-pBCMA construct with or without a constitutive RUNX3 expression plasmid. In this system, RUNX3 caused a fourfold increase in reporter activity (Fig. 3B). BCMA induction by RUNX3 was also demonstrated at the protein level using the two separate isolates of Akata 31-MEP4-RUNX3 cells. When these cells were treated with CdCl2 for 12 h, BCMA protein was clearly induced (Fig. 3C). These data confirm that BCMA can be a target of RUNX3 in a BL cell line, but BCMA was expressed only at a low level in the EREB2.5 LCL (data not shown), and BCMA in those cells could not be investigated further at the protein level with the available antibodies.
Repression of the RUNX1 P1 promoter by RUNX3 requires the VWPRY sequence.
Having demonstrated that RUNX1 repression is critical for LCL proliferation, we further analyzed the mechanism of the repression. Our previous work showed that repression involves the ability of RUNX3 to bind to dual RUNX consensus sites in the P1 promoter. This had been demonstrated indirectly using gel shift and luciferase assays (34), but the mechanism of RUNX3 repression remained unclear.
One mechanism of RUNX transcriptional repression occurs by interaction with the Groucho/TLE family of transcriptional repressors through the C-terminal VWRPY sequence conserved in all RUNX family members. To test whether the repression of the RUNX1 gene involves this mechanism, we generated an Akata 31 BL cell line with CdCl2-inducible RUNX3 lacking the VWRPY sequence (ΔVWRPY). Expression of wild-type RUNX3 in this system caused a decrease in RUNX1c levels, as previously reported (34), but RUNX3 ΔVWRPY expression was unable to mediate this effect (Fig. 4A), showing that the VWRPY sequence is required.
FIG. 4.

Repression of the P1 promoter by RUNX3 requires the VWPRY sequence. (A) Akata 31 stable cell lines containing vector control pMEP4, pMEP4-RUNX3, or pMEP4-ΔVWRPY under the control of the metallothionein promoter were induced by the addition of CdCl2 at 5 μM for the times indicated. RIPA extracts were prepared, and 50 μg protein was analyzed by SDS-PAGE. Western blotting was performed, probing for RUNX3, RUNX1, and β-actin as a loading control. (B) Forty-eight-hour transient transfection assay of DG75 cells with a RUNX1c promoter luciferase reporter construct (pGL3-RUNX1P1) cotransfected with the pCEP4 empty-vector or RUNX3 expression plasmid (pCEP-RUNX3 or pCEP4-ΔVWRPY). Results are expressed as means ± standard deviations of results from three transfections. Relative luciferase units (RLU) were adjusted for transfection efficiency using a cotransfected β-galactosidase (Gal) reporter plasmid. (C) HEK293 cells were transfected with the pCEP4, RUNX3 wild-type, or ΔVWRPY expression plasmid and extracts analyzed by Western blotting, probing for RUNX3 or β-actin as a loading control.
RUNX3 was also previously shown (34) to repress the activity of a RUNX1 luciferase reporter construct containing a fragment of the RUNX1 P1 promoter spanning positions −151 to +100. We therefore examined the effect of ΔVWRPY expression on the activity of this promoter in a transfection assay. Although RUNX3 partially repressed RUNX1 P1 activity, ΔVWRPY did not have this effect in DG75 BL cells (Fig. 4B), again indicating a role for the VWRPY sequence in the mechanism of repression. Transient transfection of HEK293 cells with the constitutive RUNX3 and ΔVWRPY expression constructs confirmed the equal levels of expression of these proteins from the expression plasmids (Fig. 4C).
These data indicate that repression of the RUNX1 P1 promoter activity and the resulting reduction of RUNX1c protein levels in EBV-infected B cells require the RUNX3 VWRPY sequence. The CdCl2-inducible system effectively demonstrated the RUNX3 repression of RUNX1 within 24 h of induction, but cadmium toxicity to the cells was evident when BL cell lines were incubated for longer time periods (data not shown).
RUNX3-ER and ΔVWRPY-ER both translocate to the nucleus and bind to the RUNX1 P1 promoter.
To study the VWRPY-dependent repression phenomenon in a nontoxic system and provide a system in which ChIP assays could be used to test the association of RUNX3 with the RUNX1 promoter, we also generated ER/estrogen-binding domain constructs expressing ER fusion proteins of RUNX3 and ΔVWRPY. In the absence of β-estradiol, RUNX activity is inhibited by retention of the ER fusion protein in the cytoplasm. Addition of β-estradiol removes this inhibitory effect and allows appropriate protein localization to occur. This approach has previously been used for examining the biological effects of RUNX family members in mammalian cells (4). Transient transfection of the expression plasmids for the ER fusion proteins RUNX3-ER and ΔVWRPY-ER into HEK293 cells demonstrated that these proteins were expressed at equal levels and were detectable with the use of both anti-RUNX3 and anti-ER antibodies (Fig. 5A).
FIG. 5.

RUNX3-ER and ΔVWRPY-ER both translocate to the nucleus and bind to the RUNX1 P1 promoter. (A) HEK293 cells were transfected with plasmid pCEP4-RUNX3-ER or pCEP4-ΔVWRPY-ER, and cells were lysed in sample buffer and fractionated by SDS-PAGE. Western blotting was performed, probing for RUNX3 or ER. (B) HEK293 cells were transfected with the pCEP4-RUNX3-ER or pCEP4-ΔVWRPY-ER expression plasmid, and after 24 h, 5 μM β-estradiol (Est) was added. Twenty-four hours after β-estradiol addition, cells were fixed and probed with an anti-ER antibody followed by fluorescein isothiocyanate (FITC)-conjugated anti-rabbit secondary antibody. The locations of the fusion proteins were assessed by confocal microscopy. (C) DG75 cells stably transfected with pCEP4-RUNX3-ER or pCEP4-ΔVWRPY-ER were incubated with and without 5 μM β-estradiol for 24 h. RIPA extracts were prepared, and 50 μg protein was analyzed by Western blotting, probing for the ER component of the RUNX-ER fusion protein. (D) DG75 pCEP4-RUNX3-ER or pCEP4-ΔVWRPY-ER stable cells were incubated with 5 μM β-estradiol for 24 h. A ChIP assay was performed on extracts from these cells by using the ER antibody, a nonspecific negative-control antibody (NSA), and an RNA polymerase II (Pol II) positive control. PCR amplification using primers for the RUNX1 P1 promoter (R1P1) or the GAPDH promoter was performed on the immunoprecipitations, and products were fractionated by agarose gel electrophoresis. (E) Extracts of IB4 cells or IB4 cells stably expressing RUNX3-ER (as in Fig. 2), treated with estrogen, were analyzed by Western blotting with an ER antibody for RUNX3-ER expression. (F) The estrogen-treated IB4 cells expressing RUNX3-ER were tested in the ChIP assay, using either an ER antibody (RUNX3-ER), a nonspecific control antibody (NSA), or an RNA Pol II antibody. PCR was for the RUNX1 P1 promoter or the GAPDH promoter. (G) DG75 pCEP4-RUNX3-ER or pCEP4-ΔVWRPY-ER cells were incubated with 5 μM β-estradiol, RIPA extracts were prepared, and 50 μg protein was separated by SDS-PAGE. Western blotting was performed, probing for ER, RUNX1, and β-actin. (H) DG75 pCEP4-RUNX3-ER or pCEP4-ΔVWRPY-ER cells were incubated with 5 μM β-estradiol, harvested, and counted at the indicated times. Results are expressed as average total numbers of cells in a 10-cm dish.
To validate the system, we examined the effect of β-estradiol on localization of RUNX3-ER and ΔVWRPY-ER by using confocal microscopy. Both RUNX3-ER and ΔVWRPY-ER localized in the cytoplasm in the absence of β-estradiol (Fig. 5B, upper panels). Addition of β-estradiol resulted in normal localization of the RUNX3 fusion proteins to the nucleus, with characteristic nucleolar exclusion (Fig. 5B, lower panels). These constructs were then used to generate DG75 cell lines stably expressing the RUNX3-ER fusion proteins. After selection, these lines were shown to express RUNX3-ER and ΔVWRPY-ER protein (Fig. 5C); addition of β-estradiol also caused a slight stabilization of the ER fusion proteins, as has been previously reported for other ER fusion proteins in these cells (23). To determine if the VWRPY-dependent repression phenomenon also occurred with the use of this system, we examined the effect of β-estradiol on the RUNX1 levels. Again, activated RUNX3-ER caused decreases in RUNX1 levels, but ΔVWRPY-ER did not do this (Fig. 5G). As expected, in the BL cell line, neither fusion protein had any effect on the growth of DG75 cells when activated (Fig. 5H).
The presence of the ER tag in the RUNX3-ER fusion proteins allowed the use of a high-affinity ER antibody to test binding of RUNX3-ER to the RUNX1 P1 promoter in a ChIP assay. Cells incubated with β-estradiol were processed using the ChIP assay, and the RUNX1 P1 DNA fragment was found to be specifically immunoprecipitated with the anti-ER antibody (Fig. 5D). Both RUNX3-ER fusion proteins bound the RUNX1 P1 promoter equally in the presence of β-estradiol but did not bind to the GAPDH control fragment. The negative-control antibody did not associate with either the RUNX1 P1 promoter or the GAPDH control promoter, whereas the positive-control anti-RNA polymerase II antibody precipitated both GAPDH and RUNX1 P1, confirming that the binding of the RUNX-ER fusion proteins to the RUNX1P1 promoter is specific (Fig. 5D). Binding of the RUNX3-ER fusion protein to the RUNX1 promoter in an LCL background was also confirmed by transfecting the same RUNX3-ER expression plasmid into IB4 cells, checking the expression of the RUNX3-ER protein (Fig. 5E), and performing the same ChIP assay in the presence of β-estradiol (Fig. 5F).
These data demonstrate that, in the presence of β-estradiol, ER fusion proteins of RUNX3 and ΔVWRPY behave similarly to RUNX3 in terms of localization to the nucleus and that both bind equally to the RUNX1 P1 promoter. Both RUNX3 and RUNX3-ER require the VWRPY sequence to repress expression of RUNX1, but repression of RUNX1 did not affect proliferation of DG75 BL cells.
TLE3 and TLE4 are present in B-cell lines to act as corepressors.
We tested which TLE family members might be present to associate with RUNX3 to mediate the repression of RUNX1. Primers were designed for all four TLE family members, and RT-PCR was performed on RNA from LCL and BL cell lines, where the repression phenomenon has previously been demonstrated to occur (34). Although Namalwa, Jijoye, AM29, AM59, and EREB2.5 cells were found to express some TLE1, it was not detected in most of the lines with reciprocal RUNX3 and RUNX1 expression, making it unlikely to be responsible for the repression phenomenon (Fig. 6A). TLE2 was not detected in any of the lines tested (data not shown). Conversely, TLE3 and TLE4 RNAs were detected in all the LCL and BL lines tested (Fig. 6A). We examined the protein levels of TLE3 and TLE4. All the lines tested contained detectable TLE3 (Fig. 6B), but we were unable to detect protein levels of TLE4 by using the commercial TLE4 antibody (not shown). It therefore seems most likely that TLE3 is the main corepressor that is available in these cell lines to associate with RUNX3 and mediate the repression of RUNX1 by RUNX3.
FIG. 6.

TLE3 and TLE4 are present in B-cell lines to act as corepressors. (A) RNA from a panel of LCL and BL cell lines was used for RT-PCR assays for the presence of TLE1 to -4 and GAPDH RNA. Products were fractionated on a 1% agarose gel stained with ethidium bromide. (B) RIPA extracts (50 μg protein) from a panel of LCL and BL cell lines were analyzed by Western blotting, probing for TLE3 and β-actin.
DISCUSSION
The critical importance of removing RUNX1 from activated, proliferating B cells was demonstrated by controlled expression of RUNX1c. Expression of RUNX1 in the EBV-infected LCL line IB4 caused a substantial decrease in cell proliferation, suggesting that its expression is not tolerated in these cells after activation or in the presence of RUNX3. The expression of RUNX1 did not affect the levels of RUNX3 in these cells, showing that cross-repression is not responsible for this effect. The lack of cross-repression in this situation may be because only the P1 promoters of RUNX genes contain the RUNX binding sites for repression and, in LCLs, RUNX3 is transcribed from the P2 promoter. This facility for selective cross-repression may be part of the reason for the evolution of the two promoters for each RUNX gene. Since TEL-RUNX1 occurs in the proliferating cells of pediatric B lymphocytic leukemias, it is perhaps not surprising that its expression is compatible with growth of the IB4 B-LCL, and it may now be possible to use deletion analysis and chimeras to investigate how the TEL fusion protein overcomes the growth-inhibitory effect associated with normal RUNX1.
We used ER fusion proteins to show that the repression of RUNX1 depends on the VWRPY sequence at the C terminus of RUNX3. The homogeneity of cells in the ER fusion system allowed us to demonstrate that both wild-type and ΔVWRPY RUNX3 behaved the same in terms of localization and binding to the RUNX1 P1 promoter, indicating that incorrect cell location of the mutant RUNX3 is not the reason for lack of repressive function in the absence of the VWRPY sequence. The RUNX-ER fusion proteins also made it possible to perform ChIP assays to show that RUNX3-ER is bound to the RUNX1 promoter in LCL cells where repression occurs. Although we made considerable attempts (not shown) to further define the interactions by confirming that removal of the VWRPY sequence from RUNX3 reduced endogenous association between RUNX3-ER and TLE3 by immunoprecipitation, the available antibodies were not sufficiently specific to achieve consistent results for this experiment, a limitation also noted by others (11).
The differences in the activities of RUNX3 and RUNX1 in LCLs can be understood not solely in terms of the presence or absence of these transcriptional regulators but by their effect on downstream target genes within these cells. We have begun to investigate this in a BL cell line by examining genes affected when RUNX3 is expressed and RUNX1 becomes repressed. Our initial findings show that RUNX3 can cause increased expression of the APRIL receptor BCMA. This target might be important to EBV-driven proliferation in B cells or to the survival and emergence of lymphoma or leukemia cells. In fact, EBV LMP1 has been shown to induce the BAFF and APRIL ligands for BCMA (16). APRIL and BAFF have a critical role in mediating survival of B cells in a wide range of differentiation states (7), and BCMA is particularly important in memory and plasma B cells (13, 27, 38). BCMA has also been shown to be induced after B-cell activation, although its role at this stage is currently unknown (14). Current models for the EBV life cycle emphasize the role of the virus in aiding survival of infected B cells in the lymph nodes, where further differentiation can lead to the emergence of long-lived memory cells, in which the virus may persist indefinitely (1, 32). EBV is associated with about 30% of cases of Hodgkin's lymphoma (HL) (29), and HL cells express BCMA, which generates survival signals upon APRIL stimulation (10). APRIL activity has been reported to correlate with B-cell-lymphoma aggressiveness (30), and the expression of another BCMA ligand, BLyS, correlates with disease activity and patient outcome in non-HL cases (26). APRIL is also associated with survival of chronic lymphocytic leukemia cells (15), and APRIL-rich niches promote tumor cell proliferation (17). The induction of BCMA by RUNX3 could therefore play a role in EBV-associated cancers.
We propose that RUNX3-mediated repression of RUNX1 in LCLs and BL cell lines occurs mainly through recruitment of TLE3 or TLE4. These two family members were detected at the level of RNA in all of the cell lines where cross-repression was previously shown to occur. In an analysis of murine hemopoietic cells, the orthologue of TLE4, Grg4, was detected only in B cells, but the level of this protein was shown to decrease on B-cell activation (22, 25). We readily detected TLE3 protein in our cell lines, but we were unable to detect TLE4 protein by using commercial antibodies. This may be due to technical issues related to the antibody or may indicate that TLE3 is the predominant TLE protein in EBV-infected B cells. Interestingly, the E2A-HLF fusion oncogene drives expression of the murine TLE orthologues Grg1, -2, -4, and -6 in FL5.12 murine pro-B cells, and this effect is associated with a downregulation of RUNX1 (12). It would therefore be interesting to determine whether those cells express RUNX3, which might cause the RUNX downregulation by recruiting the TLE proteins.
In conclusion, in this study we demonstrate the importance of RUNX3-mediated repression of RUNX1 for EBV-driven B-cell proliferation and the mechanism of this repression. Very recent data (6) from a transgenic mouse model also support a role for RUNX1 in controlling B-cell proliferation. In addition to the significance for B-cell transformation by EBV, our system may be useful for identifying functional differences between different RUNX family members that, in other systems, have seemed to exhibit largely redundant activities.
Acknowledgments
We thank Tony Ford, Dirk Gründemann, and Alan Friedmann for kindly providing plasmids.
Funding for this project was provided by the Leukemia Research Fund and the Ludwig Institute for Cancer Research.
Footnotes
Published ahead of print on 29 April 2009.
REFERENCES
- 1.Al Tabaa, Y., E. Tuaillon, K. Bollore, V. Foulongne, G. Petitjean, J. M. Seigneurin, C. Duperray, C. Desgranges, and J. P. Vendrell. 2009. Functional Epstein-Barr virus reservoir in plasma cells derived from infected peripheral blood memory B cells. Blood 113604-611. [DOI] [PubMed] [Google Scholar]
- 2.Bach, M., S. Grigat, B. Pawlik, C. Fork, O. Utermohlen, S. Pal, D. Banczyk, A. Lazar, E. Schomig, and D. Grundemann. 2007. Fast set-up of doxycycline-inducible protein expression in human cell lines with a single plasmid based on Epstein-Barr virus replication and the simple tetracycline repressor. FEBS J. 274783-790. [DOI] [PubMed] [Google Scholar]
- 3.Ben-Bassat, H., N. Goldblum, S. Mitrani, T. Goldblum, J. M. Yoffey, M. M. Cohen, Z. Bentwich, B. Ramot, E. Klein, and G. Klein. 1977. Establishment in continuous culture of a new type of lymphocyte from a “Burkitt like” malignant lymphoma (line D.G.-75). Int. J. Cancer 1927-33. [DOI] [PubMed] [Google Scholar]
- 4.Bernardin, F., and A. D. Friedman. 2002. AML1 stimulates G1 to S progression via its transactivation domain. Oncogene 213247-3252. [DOI] [PubMed] [Google Scholar]
- 5.Bernardin, F., Y. Yang, C. I. Civin, and A. D. Friedman. 2002. c-Myc overcomes cell cycle inhibition by CBFbeta-SMMHC, a myeloid leukemia oncoprotein. Cancer Biol. Ther. 1492-496. [DOI] [PubMed] [Google Scholar]
- 6.Blyth, K., N. Slater, L. Hanlon, M. Bell, N. Mackay, M. Stewart, J. C. Neil, and E. R. Cameron. 2009. Runx1 promotes B-cell survival and lymphoma development. Blood Cells Mol. Dis. [Epub ahead of print.] doi: 10.1016/j.bcmd.2009.01.013. [DOI] [PubMed]
- 7.Bossen, C., and P. Schneider. 2006. BAFF, APRIL and their receptors: structure, function and signaling. Semin. Immunol. 18263-275. [DOI] [PubMed] [Google Scholar]
- 8.Brimmell, M., R. Mendiola, J. Mangion, and G. Packham. 1998. BAX frameshift mutations in cell lines derived from human haemopoetic malignancies are associated with resistance to apoptosis and microsatellite instability. Oncogene 161803-1812. [DOI] [PubMed] [Google Scholar]
- 9.Castilla, L. H. 2008. C/EBPalpha in leukemogenesis: a matter of being in the right place with the right signals. Cancer Cell 13289-291. [DOI] [PubMed] [Google Scholar]
- 10.Chiu, A., W. Xu, B. He, S. R. Dillon, J. A. Gross, E. Sievers, X. Qiao, P. Santini, E. Hyjek, J. W. Lee, E. Cesarman, A. Chadburn, D. M. Knowles, and A. Cerutti. 2007. Hodgkin lymphoma cells express TACI and BCMA receptors and generate survival and proliferation signals in response to BAFF and APRIL. Blood 109729-739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cinnamon, E., and Z. Paroush. 2008. Context-dependent regulation of Groucho/TLE-mediated repression. Curr. Opin. Genet. Dev. 18435-440. [DOI] [PubMed] [Google Scholar]
- 12.Dang, J., T. Inukai, H. Kurosawa, K. Goi, T. Inaba, N. T. Lenny, J. R. Downing, S. Stifani, and A. T. Look. 2001. The E2A-HLF oncoprotein activates Groucho-related genes and suppresses Runx1. Mol. Cell. Biol. 215935-5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Darce, J. R., B. K. Arendt, S. K. Chang, and D. F. Jelinek. 2007. Divergent effects of BAFF on human memory B cell differentiation into Ig-secreting cells. J. Immunol. 1785612-5622. [DOI] [PubMed] [Google Scholar]
- 14.Darce, J. R., B. K. Arendt, X. Wu, and D. F. Jelinek. 2007. Regulated expression of BAFF-binding receptors during human B cell differentiation. J. Immunol. 1797276-7286. [DOI] [PubMed] [Google Scholar]
- 15.Endo, T., M. Nishio, T. Enzler, H. B. Cottam, T. Fukuda, D. F. James, M. Karin, and T. J. Kipps. 2007. BAFF and APRIL support chronic lymphocytic leukemia B-cell survival through activation of the canonical NF-kappaB pathway. Blood 109703-710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He, B., N. Raab-Traub, P. Casali, and A. Cerutti. 2003. EBV-encoded latent membrane protein 1 cooperates with BAFF/BLyS and APRIL to induce T cell-independent Ig heavy chain class switching. J. Immunol. 1715215-5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hendriks, J., L. Planelles, J. de Jong-Odding, G. Hardenberg, S. T. Pals, M. Hahne, M. Spaargaren, and J. P. Medema. 2005. Heparan sulfate proteoglycan binding promotes APRIL-induced tumor cell proliferation. Cell Death Differ. 12637-648. [DOI] [PubMed] [Google Scholar]
- 18.Ichikawa, M., T. Asai, T. Saito, G. Yamamoto, S. Seo, I. Yamazaki, T. Yamagata, K. Mitani, S. Chiba, H. Hirai, S. Ogawa, and M. Kurokawa. 2004. AML-1 is required for megakaryocytic maturation and lymphocytic differentiation, but not for maintenance of hematopoietic stem cells in adult hematopoiesis. Nat. Med. 10299-304. [DOI] [PubMed] [Google Scholar]
- 19.Imai, Y., M. Kurokawa, K. Tanaka, A. D. Friedman, S. Ogawa, K. Mitani, Y. Yazaki, and H. Hirai. 1998. TLE, the human homolog of Groucho, interacts with AML1 and acts as a repressor of AML1-induced transactivation. Biochem. Biophys. Res. Commun. 252582-589. [DOI] [PubMed] [Google Scholar]
- 20.Jenkins, P., U. Binné, and P. Farrell. 2000. Histone acetylation and reactivation of Epstein-Barr virus from latency. J. Virol. 74710-720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levanon, D., R. E. Goldstein, Y. Bernstein, H. Tang, D. Goldenberg, S. Stifani, Z. Paroush, and Y. Groner. 1998. Transcriptional repression by AML1 and LEF-1 is mediated by the TLE/Groucho corepressors. Proc. Natl. Acad. Sci. USA 9511590-11595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Linderson, Y., D. Eberhard, S. Malin, A. Johansson, M. Busslinger, and S. Pettersson. 2004. Corecruitment of the Grg4 repressor by PU.1 is critical for Pax5-mediated repression of B-cell-specific genes. EMBO Rep. 5291-296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lucchesi, W., G. Brady, O. Dittrich-Breiholz, M. Kracht, R. Russ, and P. J. Farrell. 2008. Differential gene regulation by Epstein-Barr virus type 1 and type 2 EBNA2. J. Virol. 827456-7466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McLarren, K. W., F. M. Theriault, and S. Stifani. 2001. Association with the nuclear matrix and interaction with Groucho and RUNX proteins regulate the transcription repression activity of the basic helix loop helix factor Hes1. J. Biol. Chem. 2761578-1584. [DOI] [PubMed] [Google Scholar]
- 25.Milili, M., L. Gauthier, J. Veran, M. G. Mattei, and C. Schiff. 2002. A new Groucho TLE4 protein may regulate the repressive activity of Pax5 in human B lymphocytes. Immunology 106447-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Novak, A. J., D. M. Grote, M. Stenson, S. C. Ziesmer, T. E. Witzig, T. M. Habermann, B. Harder, K. M. Ristow, R. J. Bram, D. F. Jelinek, J. A. Gross, and S. M. Ansell. 2004. Expression of BLyS and its receptors in B-cell non-Hodgkin lymphoma: correlation with disease activity and patient outcome. Blood 1042247-2253. [DOI] [PubMed] [Google Scholar]
- 27.O'Connor, B. P., V. S. Raman, L. D. Erickson, W. J. Cook, L. K. Weaver, C. Ahonen, L. L. Lin, G. T. Mantchev, R. J. Bram, and R. J. Noelle. 2004. BCMA is essential for the survival of long-lived bone marrow plasma cells. J. Exp. Med. 19991-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pabst, T., and B. U. Mueller. 2007. Transcriptional dysregulation during myeloid transformation in AML. Oncogene 266829-6837. [DOI] [PubMed] [Google Scholar]
- 29.Rickinson, A. B., and E. Kieff. 2007. Epstein-Barr virus, p. 2680-2700. In D. Knipe and P. M. Howley (ed.), Fields virology, 5th ed., vol. 2. Lippincott-Raven, Philadelphia, PA. [Google Scholar]
- 30.Schwaller, J., P. Schneider, P. Mhawech-Fauceglia, T. McKee, S. Myit, T. Matthes, J. Tschopp, O. Donze, F. A. Le Gal, and B. Huard. 2007. Neutrophil-derived APRIL concentrated in tumor lesions by proteoglycans correlates with human B-cell lymphoma aggressiveness. Blood 109331-338. [DOI] [PubMed] [Google Scholar]
- 31.Shi, M. J., and J. Stavnezer. 1998. CBF alpha3 (AML2) is induced by TGF-beta1 to bind and activate the mouse germline Ig alpha promoter. J. Immunol. 1616751-6760. [PubMed] [Google Scholar]
- 32.Souza, T. A., B. D. Stollar, J. L. Sullivan, K. Luzuriaga, and D. A. Thorley-Lawson. 2005. Peripheral B cells latently infected with Epstein-Barr virus display molecular hallmarks of classical antigen-selected memory B cells. Proc. Natl. Acad. Sci. USA 10218093-18098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spender, L. C., G. H. Cornish, A. Sullivan, and P. J. Farrell. 2002. Expression of transcription factor AML-2 (RUNX3, CBFα-3) is induced by Epstein-Barr virus EBNA-2 and correlates with the B-cell activation phenotype. J. Virol. 764919-4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spender, L. C., H. J. Whiteman, C. E. Karstegl, and P. J. Farrell. 2005. Transcriptional cross-regulation of RUNX1 by RUNX3 in human B cells. Oncogene 241873-1881. [DOI] [PubMed] [Google Scholar]
- 35.van Santen, V., A. Cheung, M. Hummel, and E. Kieff. 1983. RNA encoded by the IR1-U2 region of Epstein-Barr virus DNA in latently infected, growth-transformed cells. J. Virol. 46424-433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang, W., Y. G. Wang, A. M. Reginato, D. J. Glotzer, N. Fukai, S. Plotkina, G. Karsenty, and B. R. Olsen. 2004. Groucho homologue Grg5 interacts with the transcription factor Runx2-Cbfa1 and modulates its activity during postnatal growth in mice. Dev. Biol. 270364-381. [DOI] [PubMed] [Google Scholar]
- 37.Whiteman, H. J., and P. J. Farrell. 2006. RUNX expression and function in human B cells. Crit. Rev. Eukaryot. Gene Expr. 1631-44. [DOI] [PubMed] [Google Scholar]
- 38.Zhang, X., C. S. Park, S. O. Yoon, L. Li, Y. M. Hsu, C. Ambrose, and Y. S. Choi. 2005. BAFF supports human B cell differentiation in the lymphoid follicles through distinct receptors. Int. Immunol. 17779-788. [DOI] [PubMed] [Google Scholar]