

Abstract
Homologous recombination is an important biological process that facilitates genome rearrangement and repair of DNA double-strand breaks (DSBs). The induction of Epstein-Barr virus (EBV) lytic replication induces ataxia telangiectasia-mutated (ATM)-dependent DNA damage checkpoint signaling, leading to the clustering of phosphorylated ATM and Mre11/Rad50/Nbs1 (MRN) complexes to sites of viral genome synthesis in nuclei. Here we report that homologous recombinational repair (HRR) factors such as replication protein A (RPA), Rad51, and Rad52 as well as MRN complexes are recruited and loaded onto the newly synthesized viral genome in replication compartments. The 32-kDa subunit of RPA is extensively phosphorylated at sites in accordance with those with ATM. The hyperphosphorylation of RPA32 causes a change in RPA conformation, resulting in a switch from the catalysis of DNA replication to the participation in DNA repair. The levels of Rad51 and phosphorylated RPA were found to increase with the progression of viral productive replication, while that of Rad52 proved constant. Furthermore, biochemical fractionation revealed increases in levels of DNA-bound forms of these HRRs. Bromodeoxyuridine-labeled chromatin immunoprecipitation and PCR analyses confirmed the loading of RPA, Rad 51, Rad52, and Mre11 onto newly synthesized viral DNA, and terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling analysis demonstrated DSBs in the EBV replication compartments. HRR factors might be recruited to repair DSBs on the viral genome in viral replication compartments. RNA interference knockdown of RPA32 and Rad51 prevented viral DNA synthesis remarkably, suggesting that homologous recombination and/or repair of viral DNA genome might occur, coupled with DNA replication to facilitate viral genome synthesis.
Replication protein A (RPA), the eukaryotic single-stranded DNA (ssDNA)-binding protein, is a heterotrimeric complex composed of three tightly associated subunits of 70, 32, and 14 kDa (referred as to RPA70, RPA32, and RPA14, respectively) that is essential for DNA replication, recombination, and all major types of DNA repair (4). RPA participates in such diverse pathways through its ability to interact with DNA and numerous proteins involved in its processing. During DNA replication, RPA associates with ssDNA at forks and facilitates nascent-strand DNA synthesis by replicative DNA polymerases localized at replication foci during S phase. Under DNA-damaging conditions, RPA binds to ssDNA at damaged sites and interacts with repair and recombination components to process double-strand DNA breaks (DSBs) and other lesions (6, 14, 21, 32, 38, 41).
RPA undergoes both DNA damage-independent and -dependent phosphorylation on the N-terminal 33 residues of RPA32. Unstressed cell cycle-dependent phosphorylation occurs during the G1/S-phase transition and in M phase, primarily at the conserved cyclin-CDK phosphorylation sites of Ser-23 and Ser-29 in the N terminus of the RPA32 subunit (13, 15). In contrast, stress-induced hyperphosphorylation of RPA is much more extensive. Nine potential phosphorylation sites within the N-terminal domain of RPA32, Ser-4, Ser-8, Ser-11/Ser-12/Ser-13, Thr-21, Ser-23, Ser-29, and Ser-33, in response to DNA-damaging agents, have been suggested (33, 54). Although this region of RPA32 is not required for the ssDNA-binding activity of RPA (5, 22), a phosphorylation-induced subtle conformation change in RPA, resulting from altered intersubunit interactions, regulates the interaction of RPA with both interacting proteins and DNA (30). The hyperphosphorylated form of RPA32 is unable to localize to replication centers in normal cells, while binding to DNA damage foci is unaffected (46). Therefore, RPA phosphorylation following damage is thought to both prevent RPA from catalyzing DNA replication and potentially serve as a marker to recruit repair factors to sites of DNA damage. RPA localizes to nuclear foci where DNA repair is occurring after DNA damage and is essential for multiple DNA repair pathways, participating in damage recognition, excision, and resynthesis reactions (4, 56).
Mammalian cells can repair DSBs by homologous recombination (HR) or by nonhomologous end joining. HR is an accurate repair process, the first step of which is the resection of the 5′ ends of the DSB to generate 3′ ssDNA overhangs. This reaction is carried out by the Mre11/Rad50/Nbs1 (MRN) complex, which not only functions as a damage sensor upstream of ataxia telangiectasia-mutated (ATM)/ATM-Rad3-related (ATR) activation but also plays a role in DSB repair (4). RPA and members of the RAD52 epistasis group of gene products, such as Rad51, Rad52, and Rad54, bind to the resulting 3′ ssDNA strands and form a helical, nucleoprotein filament that facilitates the invasion of a damaged DNA strand into the homologous double-stranded DNA partner. The human Rad51 protein is a structural and functional homolog of the Escherichia coli RecA protein, which promotes homologous pairing and strand transfer reactions in vitro. Both Rad51 and Rad52 bind specifically to the terminal regions of tailed duplex DNA, the substrate thought to initiate recombination in vivo. Furthermore, nucleoprotein filaments of Rad51, formed on tailed DNA, catalyze strand invasion of homologous duplex DNA in a reaction that is stimulated by Rad52 and RPA (3).
Epstein-Barr virus (EBV) is a human herpesvirus that infects B lymphocytes, inducing their continuous proliferation. In B-lymphoblastoid cell lines, there is no production of virus particles, which is termed latent infection (52). Reactivation from latency is characterized by the expression of lytic genes, and one of the first detectable changes is the expression of the BZLF1 immediate-early gene product, which trans-activates viral promoters (16), leading to an ordered cascade of viral early and late gene expression. This lytic EBV DNA replication occurs in discrete sites in nuclei, called replication compartments, in which seven viral replication proteins are assembled (44). The viral genome is amplified several hundredfold by the viral replication machinery and is thought to generate highly branched replication intermediates through HR coupled with viral DNA replication (48). With the progression of lytic replication, the replication compartments become larger and appeared to fuse to form large globular structures that eventually filled the nucleus at late stages of infection (8, 45).
We previously isolated latently EBV-infected Tet-BZLF1/B95-8 cells in which the exogenous BZLF1 protein is conditionally expressed under the control of a tetracycline-regulated promoter, leading to a highly efficient induction of lytic replication (28). Using this system, we have demonstrated that the induction of the EBV lytic program results in the inhibition of replication of cellular DNA in spite of the replication of viral DNA (28) and elicits a cellular DNA damage response, with the activation of the ATM-Chk2-p53 DNA damage transduction pathway (29). The DNA damage sensor MRN complex and phosphorylated ATM are recruited and retained in viral replication compartments (29).
Here we report that RPA32 is extensively phosphorylated after EBV lytic replication is induced, with the phosphorylation sites in accordance with those for ATM. Phosphorylated RPA, Rad51, and Rad52, which are involved in HR repair (HRR), are recruited and retained in viral replication compartments as well as the MRN complex. Furthermore, DSBs could be demonstrated to occur during viral genome synthesis in the EBV replication compartments. HRR factors might be recruited to repair DSBs on the viral genome in viral replication compartments. RNA interference (RNAi) knockdown of RPA32 and Rad51 prevented viral DNA synthesis remarkably, suggesting that HR and/or repair of viral DNA genome might occur, coupled with DNA replication, to facilitate viral genome synthesis.
MATERIALS AND METHODS
Cells.
Tet-BZLF1/B95-8 cells, a marmoset B-cell line latently infected with EBV, and Tet-BZLF1/Akata cells, which are human EBV-positive Burkitt's lymphoma cells (27), were maintained in RPMI medium supplemented with 1 μg/ml of puromycin, 250 μg/ml of hygromycin B, and 10% tetracycline-free fetal calf serum. To induce lytic EBV replication, a tetracycline derivative, doxycycline, was added to the culture medium at a final concentration of 2 μg/ml. B95-8 cells were cultured in RPMI medium supplemented with 10% fetal calf serum. 293/EBV cells were prepared by transfection with EBV-Bac DNA (11) into HEK293 cells (kindly provided by H. J. Delecluse, German Cancer Research Center) by hygromycin selection (hygromycin B; 150 μg/ml). EBV-Bac was a gift from Wolfgang Hammerschmidt (Helmholtz Zentrum München-Haematologikum, Germany).
Protein preparation.
Cells were harvested at the indicated times posttreatment with doxycycline, washed with phosphate-buffered saline (PBS), and treated with lysis buffer (0.02% sodium dodecyl sulfate [SDS], 0.5% Triton X-100, 300 mM NaCl, 20 mM Tris-HCl [pH 7.6], 1 mM EDTA, 1 mM dithiothreitol) for 20 min on ice. Multiple protease inhibitors (protease inhibitor mixture for mammalian cell extracts, 25 μl/ml; Sigma), 200 μM sodium vanadate, and 20 mM sodium fluoride were added to the buffer. Samples were centrifuged at 18,000 × g for 10 min at 4°C, and clarified cell extracts were assayed for protein concentrations using a Bio-Rad protein assay kit.
Biochemical cellular fractionation.
If not stated otherwise, the buffer used for cell fractionation procedures was modified cytoskeleton buffer (mCSK) [10 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES) (pH 6.8), 100 mM NaCl, 300 mM sucrose, 1 mM MgCl2, 1 mM EGTA, 1 mM dithiothreitol] containing 0.1% Triton X-100 and 0.1 mM ATP. Where necessary, 200 μM sodium vanadate and 10 mM sodium fluoride were added as phosphatase inhibitors, along with multiple protease inhibitors (protease inhibitor mixture for mammalian cell extracts, 25 μl/ml; Sigma). Tet-BZLF1/B95-8 cells were washed with cold PBS and then resuspended at 1.5 × 107 cells/ml in mCSK buffer containing 0.5% Triton and 0.1 mM ATP. Cell extraction was carried out for 10 min on ice. Cell lysates were separated into a soluble fraction and a nuclear pellet by centrifugation for 1 min at 1,500 × g at 4°C. The pellet nuclei were washed once with mCSK buffer and resuspended in mCSK buffer containing 0.1% Triton, 1 mM ATP, and 4 mM MgCl2 at 1.5 × 107 nuclei/ml. The extracted nuclei were digested with 250 units/ml DNase I (Boehringer Mannheim) at 25°C for 30 min. The samples were then centrifuged (2,000 × g for 3 min at 4°C) to obtain the solubilized chromatin fraction and the remaining nonchromatin nuclear structures. Each sample was adjusted to the same volume by adding 2× SDS sample buffer and boiled, and aliquots corresponding to 1.8 × 104 cells per each lane were applied for SDS-polyacrylamide gel electrophoresis (PAGE).
Antibodies.
Primary monoclonal antibodies to RPA32, the BMRF1 protein, PCNA, Rad51, and 5-bromodeoxyuridine (BrdUrd) were purchased from Calbiochem (RPA2 Ab-2 for immunoprecipitation [IP] and Rad51 Ab-2), Chemicon (EBV BMRF1-R3), MBL (PCNA clone 5A10), BD Transduction Laboratories (Mre11 clone 18 for immunoblotting [IB] and Nbs1 clone 34), Upstate (Rad51 clone 3C10), and Abcam (BrdUrd clone 5llB), respectively. Primary rabbit polyclonal antibodies were purchased from Cell Signaling (Rad52), Bethyl (Rad52 A300-759A for IP and A300-760A for IB, RPA32 for IB, phospho-RPA32 Ser-4/Ser-8, and phospho-RPA32 Ser-33), Santa Cruz (Rad52 for immunofluorescence and MCM6), and Upstate (Mre11 for immunofluorescence and IP), respectively. Rabbit polyclonal antibodies to the BZLF1, BALF2, BMRF1, and MCM7 proteins were prepared as described previously (10, 19, 53). Highly cross-absorbed secondary reagents for dual-color detection (Alexa 488 and 594) and the Zenon mouse immunoglobulin G (IgG) labeling kit (Alexa 647) were obtained from Molecular Probes.
Immunofluorescence.
Cells were treated with 0.5% Triton X-100-mCSK buffer for 10 min on ice, followed by fixation with 70% methanol for 180 min at −20°C. The fixed cells were washed with PBS and blocked for 20 min in 10% normal goat serum in PBS. Staining with primary antibodies was performed overnight at 4°C in PBS-0.5% goat serum. Staining with a rabbit polyclonal antibody to BALF2 or a mouse monoclonal antibody to BMRF1 was carried out for 1 h at room temperature. Species-specific secondary antibodies were applied for 1 h at room temperature. Alexa 488 and Alexa 594, highly cross-absorbed secondary reagents, were purchased from Molecular Probes. For three-color staining, anti-BMRF1 antibodies were directly labeled with a Zenon tricolor mouse IgG1 labeling kit for deep red fluorescent imaging (purchased from Molecular Probes). Cells were incubated with Alexa Flour 647-labeled anti-BMRF1 antibodies for 50 min at room temperature and washed three times with PBS, followed by a second fixation with 4% formaldehyde solution in PBS for 15 min at room temperature. The slides were mounted in ProLong Gold antifade reagent with or without 4′,6′-diamidino-2-phenylindole (DAPI) (Molecular Probes) and analyzed by fluorescence confocal microscopy. Confocal fluorescence images were captured and processed using an LSM510 Meta microscope (Carl Zeiss MicroImaging, Inc.) with a plan-apochromat 63×/1.4-numerical-aperture oil immersion objective. All the primary antibodies were employed at 1:100 dilutions, and the secondary antibodies were employed at 1:500 dilutions. All washes after antibody incubation were performed with 0.05% Tween 20 in PBS at room temperature. The specificity of the second antibodies and reliability of discrimination with fluorescent microscopy filters were tested. When cells were stained singly for either antigen with inappropriate combinations of first and second antibodies, no fluorescence was observed, and also, no immunofluorescence was observed with alternate filters.
TUNEL assays.
Terminal deoxynucleotidyltransferase (TdT)-mediated dUTP nick end labeling (TUNEL) assays in which Bodipy FL-14 (green)-conjugated dUTPs were coupled to 3-OH ends of DSBs by TdT were conducted. Cells were treated with 0.5% Triton X-100-mCSK buffer for 10 min on ice, followed by fixation with 70% methanol for 180 min at −20°C, washing, and incubation at 37°C for 1 h in a buffer containing 22.5 U of terminal transferase (Sigma), 120 mM sodium cacodylate, 1 mM cobalt chloride, 30 mM Tris-HCl (pH 7.2), and 0.1 mM Bodipy FL-14-dUTP. Control reactions were conducted with the same buffer, but TdT was omitted. Cells were then treated with 1× TB buffer (300 mM NaCl and 30 mM sodium citrate) at room temperature for 15 min and rinsed with water. TUNEL-stained cells were incubated with 10% normal goat serum in PBS and then stained with a rabbit polyclonal antibody to RPA32 Ser-33 or Rad51 overnight and with a mouse monoclonal antibody to the BMRF1 protein for 1 h. Species-specific secondary antibodies were applied for 1 h at room temperature.
IP.
Tet-BZLF1/B95-8 cells were cultured in the presence of 2 μg/ml doxycycline and harvested at 24 h postinduction (p.i.). The nuclear extracts were prepared with a nuclear complex coimmunoprecipitation (co-IP) kit (Active Motif) according to the instructions of the manufacturer. Briefly, nuclear extracts were prepared by collection of cells in ice-cold PBS with phosphatase inhibitors, suspension in hypotonic buffer to swell cell membranes and make them fragile, and the addition of detergent to cause the leakage of cytoplasmic proteins into the supernatant. The nuclear fraction was collected, and the nuclear proteins were recovered in a low-salt buffer in the presence of protease inhibitor cocktail and phenylmethylsulfonyl fluoride. This was followed by addition of a proprietary enzymatic DNA-shearing cocktail. The enzymatic DNA-shearing steps make DNA digested into 200 to 1,500 bp in length. These were collected for IP reactions. The nuclear extracts (100 μg) were diluted with 1× high-co-IP buffer (Active Motif) with a high-stringency starting composition, and the aliquots were mixed with 2 μg each of control IgG, anti-BMRF1, anti-RPA, anti-phospho-RPA32 Ser-4/Ser-8, and anti-phospho-RPA32 Ser-33 antibodies for 180 min at 4°C. After the addition of 20 μl protein A-Sepharose beads, the mixtures were further incubated for 1 h at 4°C under conditions of gentle rocking. The beads were then washed with NET buffer (50 mM Tris-HCl [pH 7.6], 150 mM NaCl, 1 mM EDTA, 0.1% NP-40), and the immunoprecipitates were boiled in SDS sample buffer without β-mercaptoethanol. The supernatants were collected, mixed with β-mercaptoethanol, reboiled, and subjected to immunoblot analysis. For PCR analysis, immunoprecipitated beads were treated with 0.05% SDS and 0.5 mM EDTA for 10 min at 95°C, and DNA was then purified using a Chroma Spin-10 gel filtration column from Clontech Laboratories, Inc. The DNA was applied for PCR analysis using primers specific for the EBNA1 gene (5′-GTCATCATCATCCGGGTCTC-3′ and 5′-TTCGGGTTGGAACCTCCTTG-3′), the BZLF1 promoter (5′-TAGCCTCGAGGCCATGCATATTTCAACTGG-3′ and 5′-GCCAAGCTTCAAGGTGCAATGTTTAGTGAG-3′), and the BALF5 gene (5′-TGTACACGCACGAGAAATGCGCCGTCATT-3′ and 5′-ACTGCAAACTCCACGTC-3′); those primers were designed to generate around 250-bp products. The PCR products were then analyzed by agarose gel electrophoresis and visualized with ethidium bromide staining.
For the detection of BMRF1 proteins, anti-BMRF1 IgG (BMRF1-R3; Chemicon) was directly labeled with peroxidase using an NH2 peroxidase labeling kit (purchased from Dojindo Laboratories).
BrdUrd-labeled chromatin IP assays.
Chromatin IP assays were performed as follows. Tet-BZLF1/B95-8 cells (5 × 107 cells per sample) were cultured in 100-mm plates in the presence of 2 μg/ml doxycycline for 48 h. Newly synthesized DNA was labeled by incubation with 20 μM BrdUrd added directly to the incubation medium 1 h prior to harvesting. Nuclear extracts were prepared by using a nuclear complex co-IP kit (Active Motif) according to the instructions of the manufacturer. Nuclear extracts (100 μg) were diluted with 1× high-co-IP buffer (Active Motif), and aliquots were mixed with 2 μg each of control IgG, anti-BMRF1, anti-MCM7, and anti-phospho-RPA32 Ser33 antibodies for 180 min at 4°C. Immune complexes of proteins were precipitated with protein G-Sepharose 4 Fast Flow or protein A-Sepharose Fast Flow beads, immunoprecipitated samples were treated with 0.05% SDS and 0.5 mM EDTA for 10 min at 95°C, and DNA then was purified using a Chroma Spin-10 gel filtration column from Clontech. Precipitates were boiled and dot blotted onto Hybond N+ membranes (Amersham) and analyzed by Western blotting using an anti-BrdUrd antibody (llB5; Abcam).
Quantification of viral DNA synthesis during lytic replication.
293/EBV cells were transfected with short interfering RNA (siRNA) oligonucleotides specific for green fluorescent protein (GFP) (Qiagen), Rad51 (Santa Cruz), or RPA32 (Santa Cruz) by using a microporator (Digital Bio Technology) and cultured for 24 h; subsequently, EBV lytic replication was induced by the transfection of the BZLF1 protein expression vector (pcDNA-BZLF1; kindly provided by K. Kuzushima, Aichi Cancer Center Research Institute). Whole DNAs were purified from a total of 3.5 × 106 cells and quantified. Dot blot hybridization was performed, and quantification of the copy numbers of viral genomes per cell were determined as described previously (28).
Western blotting.
Western blots were performed using standard procedures (28). Protein samples were separated by SDS-PAGE and subsequently transferred onto polyvinylidene difluoride membranes (Amersham). The detection of target proteins was achieved with an enhanced chemiluminescence detection system (Amersham), and images were processed by Lumi Vision Pro 400EX (Aisin/Taitec, Inc.). Signal intensity was quantified with a Lumi Vision Analyzer 400. The system applied features a cooled charge-coupled-device camera with a 16-bit (65,535) gray-scale dynamic range.
RESULTS
Increases in levels of HR factors such as Rad51 and phosphorylated RPA32 upon induction of EBV productive replication.
We previously reported that the induction of productive EBV replication elicits the activation of ATM-dependent DNA damage responses and that MRN complexes are recruited to the viral replication compartments as DNA damage sensors, along with a series of viral replication proteins (29). It is known that upon DNA damage, the N terminus of RPA32 becomes hyperphosphorylated and localized to nuclear foci where DNA repair occurs (4). To investigate whether the induction of EBV lytic replication changes the phosphorylation state of RPA, the phosphorylation of Ser-4/Ser-8 and Ser-33 residues on RPA32 was examined in Tet-BZLF1/B95-8 cells before or after the induction of lytic replication. Cells were treated with or without doxycycline, harvested at the indicated times after induction of lytic replication, and applied for Western blot analysis with specific antibodies (Fig. 1). Detailed expression profiles of viral proteins after the induction of lytic replication with doxycycline were described previously (28), with the EBV BALF2 protein becoming detectable at 4 h p.i. (28) and reaching a plateau at 24 h p.i., as shown in Fig. 1. We found that slowly migrating RPA32 bands accumulated as lytic EBV infection proceeded (Fig. 1). Alkaline phosphatase treatment of lysates from lytic replication-induced cells changed the slower-migrating forms of RPA32 to the faster-migrating form (data not shown), confirming that the slower-migrating forms were hyperphosphorylated. This was not the case with B95-8 cells treated with doxycycline. Furthermore, using phospho-specific antibodies, it was established that the levels of RPA32 phosphorylated at Ser-4/Ser-8 and Ser-33 increased with the progression of EBV lytic replication. The anti-RPA32 Ser-33-specific antibody reacted with both faster-migrating and slowly migrating forms of RPA32, while the anti-RPA32 Ser-4/Ser-8 antibody reacted mainly with slowly migrating bands. Thus, it should be noted that the faster-migrating bands also include phosphorylated RPA32. Interestingly, levels of Rad51, a member of the Rad52 epistasis group of gene products involved in HR, increased as lytic replication proceeded, while those of Rad52 remained constant. The level of Mre11, a component of the MRN complex that plays an essential role in HR, were constant throughout the lytic infection, but an increase in levels of the Ser-343-phosphorylated form of Nbs1 was observed (data not shown), as we reported previously (29).
FIG. 1.

Phosphorylation of Ser-33 and Ser-4/Ser-8 residues of RPA2 and an increase in the expression level of Rad51 upon induction of EBV lytic replication. Tet-BZLF1/B95-8 and B95-8 (parent) cells were cultured in the presence of 2 μg/ml doxycycline and harvested at the indicated h p.i. Equal amounts of proteins for each sample (∼25 to 50 μg) were subjected to immunoblot analysis with the specific antibodies indicated on the left. Anti-PCNA antibodies were used to confirm equal protein loading.
Increases in levels of DNA-bound forms of Rad51 and RPA with progression of lytic infection.
We previously reported the subcellular dynamics of viral replication proteins such as the BZLF1, BALF2, BMRF1, and BALF5 proteins and human chromosomal DNA replication initiation proteins such as CDC6, MCM4, and MCM7 in lytic program-induced Tet-BZLF1/B95-8 cells using biochemical fractionation methods (8, 9, 18, 20). Cells were first treated with mCSK buffer containing the nonionic detergent Triton X-100 under relatively physiological conditions, extracting not only cytoplasmic but also nuclear proteins not tightly bound to nuclear structures. Detergent-extracted nuclei were digested with DNase I to remove the bulk of chromosomal and EBV DNA, releasing DNA-binding proteins into DNase I-solubilized supernatants. The proteins remaining in the pellets were shown to bind to nonchromatin nuclear structures (18, 20). As illustrated in Fig. 2, about two-thirds of the MCM6 protein was extracted with mCSK buffer, and most of the remainder was solubilized after DNase I treatment, which is indicative of chromatin binding. Core histones were resistant to the detergent treatment but were liberated by DNase I digestion (data not shown). In contrast, almost all of the CDC6 protein detected in the detergent-extracted nuclei was resistant to DNase I treatment, indicating an association with nonchromatin nuclear structures. Almost all the BZLF1 viral protein was in a DNA-bound form. These results were consistent with data from our previous reports (8-10), confirming the reproducibility of the fractionation procedure.
FIG. 2.

Biochemical analysis of the subcellular distribution of proteins involved in HRR. Lytic replication was induced in Tet-BZLF1/B95-8 cells with 2 μg/ml of doxycycline, and cells were harvested at the indicated h p.i. As described in Materials and Methods, whole-cell lysates (W), Triton X-100-extractable supernatants (S), and extracted nuclear fractions (P) were prepared. The extracted nuclei were further treated with DNase I to obtain DNase I-solubilized supernatants (S under DNase) and remaining insoluble nuclear pellets (P under DNase). These fractions were applied for immunoblotting analyses with the antibodies indicated on the right. Samples corresponding to 1.8 × 104 cells were applied to each lane.
The same fractions were then examined with anti-RPA32 antibody (Fig. 2). During latent infection, about 9/10 of RPA32 was detected in the extractable nucleoplasmic fraction, and the remaining RPA was sensitive to DNase I treatment. RPA32 exists mostly as a DNA-unbound form in the latent phase of EBV replication. After the induction of lytic replication, the levels of the DNA-bound form increased with time to attain about 70% of the total after 48 h p.i. Levels of slower-migrating and hyperphosphorylated forms of RPA32 increased, and these forms especially were found to bind to DNA (RPA32 at 48 h). Although the faster-migrating and slowly migrating bands of RPA32 included the protein phosphorylated at Ser-33, almost all the phosphorylated RPA at Ser-33 was in the DNA-unbound form before induction. In contrast, after induction, about 40% of the slowly migrating RPA32 phosphorylated at Ser-33 bound to DNA, while the faster-migrating RPA32 Ser-33 did not. Phosphorylated RPA32 at Ser-4/Ser-8 was at an undetectable level in the latent state. However, after the induction of lytic replication, the levels of the DNA-bound form increased with time to attain about 30% of the total after 48 h p.i. Thus, RPA is actively phosphorylated and becomes loaded onto DNA after the induction of lytic replication.
Mammalian Rad51 and Rad52 are key HRR proteins promoting the pairing and exchange of strands between homologous DNA molecules. Rad51 was barely detectable in the cytoplasm and nuclei of uninduced cells but was massively induced after induction. One-third of Rad51 was found in the chromatin-bound fraction. In contrast, the Rad52 protein was detected in the chromatin- and nuclear structure-bound fractions, and the levels of the DNase I-resistant and nuclear structure-associated form increased with the progression of lytic replication. Since the nonchromatin nuclear structure is a dynamic subnuclear compartment that supports the temporal and spatial organization and regulation of HRR processes, Rad52 would appear to be involved in such processes. On the other hand, half of Mre11 bound to DNA consistently before and after induction.
Assembly of RPA, Rad51, and Rad52 into viral replication compartments after induction of lytic replication.
Tet-BZLF1/B95-8 cells were treated with doxycycline to induce lytic replication (28), harvested, and treated with a buffer containing the nonionic detergent Triton X-100 under relatively physiological salt conditions, which extracts not only cytoplasmic but also nuclear proteins not tightly bound to nuclear structures. Triton X-100 treatment is known to disrupt nuclear envelopes, but the nuclear lamina remains intact, thus maintaining nuclear structures (25), and proteins remaining in extracted nuclei have been shown to indeed bind to chromatin or viral DNA/nuclear matrix material (10, 18, 20). We previously demonstrated that the BMRF1 protein displays a homogeneous, not-dot-like, distribution in viral replication compartments, while the BALF2 ssDNA-binding protein is distributed as distinct dots within the replication compartments (8). Also, the sites stained with anti-BMRF1 protein-specific antibodies completely coincided with the foci of viral DNA as judged by BrdUrd incorporation and fluorescent in situ hybridization analyses of confocal immunofluorescence analyses (8). Furthermore, the demonstration that a majority of the BrdUrd pulse-labeled DNA moved out of nucleus with time clarified that BrdUrd-labeled DNA is mostly viral and not cellular after the induction of lytic replication (10). Thus, BMRF1 and BALF2 protein staining was used as a marker for viral replication compartments.
To investigate whether HRR factors are recruited to EBV replication compartments, we examined the subnuclear localization of RPA32, its phosphorylated forms, Rad51, Rad52, and the MRN complex in lytic replication-induced Tet-BZLF1/B95-8 cells (Fig. 3). RPA is an abundant protein that is generally distributed throughout the nucleus. In S phase, it is found in aggregates that have been shown to colocalize with newly replicated DNA labeled with BrdUrd in nuclei (12), but phosphorylated RPA also localizes to nuclear foci where DNA repair is occurring after DNA damage (21). In most latently infected cells, RPA proved sensitive to detergent treatment (Fig. 2), although a portion of the cells probably in S phase was resistant to the detergent extraction, being observed as punctuate spots throughout nuclei (Fig. 3Ac). RPA32 phosphorylated at Ser-33 and Ser-4/Ser-8 was scarcely detectable in latently infected cells (Fig. 3Aa and b). Thus, RPA foci in the latent phase were mostly unphosphorylated. After the induction of lytic replication, RPA32 was found to become resistant to mild-detergent extraction and colocalized with the BALF2 ssDNA-binding protein as distinct spots within replication compartments (Fig. 3Bd). RPA32 phosphorylated at Ser-33 and Ser-4/Ser-8 was found in foci of aggregates within the BMRF1 protein-localized replication compartments (Fig. 3Bb and c) and also colocalized with Nbs1, a component of the MRN complex, within EBV lytic replication compartments (Fig. 3C). Such recruitments of phosphorylated RPA32 to the EBV lytic replication compartments were also found in lytic replication-induced Tet-BZLF1/Akata cell lines (Fig. 3D). In contrast, Rad51 and Rad52 were distributed throughout nuclei in the latent phase (Fig. 3Ad and e) and were detected within replication compartments after the induction of the lytic phase (Fig. 3Ba and f). These results clearly showed that HRR proteins such as phosphorylated RPA32, Rad51, Rad52, and the MRN complex are recruited and localize to sites of EBV genome DNA synthesis.
FIG. 3.

RPA, Rad51, Rad52, and MRN complexes are recruited to EBV replication compartments. (A) Subnuclear localization of RPA, phosphorylated RPA, Rad51, and Rad52 in latent Tet-BZLF1/B95-8 cells treated with 0.5% Triton X-100-mCSK buffer, fixed with methanol, and stained with the indicated combinations of antibodies as described in Materials and Methods. (a) DAPI (blue), anti-BMRF1 (green), and anti-RPA32 Ser-4/Ser-8 (red); (b) DAPI (blue), anti-BMRF1 (green), and anti-RPA32 Ser-33 (red); (c) anti-BALF2 (green) and anti-RPA32 (red); (d) anti-BALF2 (green) and anti-Rad51 (red); (e) anti-BMRF1 (green) and anti-Rad52 (red) antibodies. Panels labeled DAPI (blue) are DAPI-stained nuclei. (Right) Merged images. (B) Phosphorylated RPA32, Rad51, Rad52, and Mre11 are colocalized to EBV replication compartments. Tet-BZLF1/B95-8 cells were cultured in the presence of 2 μg of doxycycline/ml and harvested at 24 h p.i. Cells were treated with 0.5% Triton X-100-mCSK buffer, fixed with methanol, and stained with the indicated combinations of anti-BMRF1 (green), anti-BALF2 (green), anti-Rad52 (red), anti-RPA32 Ser-33 (red), anti-RPA32 Ser-4/Ser-8 (red), anti-RPA32 (red), anti-Mre11 (red), and anti-Rad51 (red) antibodies as described in Materials and Methods. Panels labeled DAPI (blue) are DAPI-stained nuclei. (Right) Merged images. (C) Phosphorylated RPA32 and NBS1 are colocalized to discrete nuclear foci in EBV lytic replication compartments. Tet-BZLF1/B95-8 cells were cultured in the presence of 2 μg of doxycycline/ml and harvested at 24 h p.i. Cells were treated with 0.5% Triton X-100-mCSK buffer, fixed with methanol, and stained with the indicated combinations of anti-NBS1 antibody (green), Alexa Fluor 647-labeled BMRF1 antibody (blue), and phospho-specific anti-RPA32 Ser-4/Ser-8 or anti-RPA32 Ser-33 antibody (red). (Right) Merged images. (D) Recruitment of phosphorylated RPA32 to the EBV replication compartment in Tet-BZLF1/Akata cell lines. Tet-BZLF1/Akata cells were cultured in the presence of 2 μg of doxycycline/ml and harvested at 24 h p.i. Cells were treated as described above (B) and stained with the indicated combinations of anti-BMRF1 (green), anti-RPA32 Ser-33 (red), and anti-RPA32 Ser-4/Ser-8 (red) antibodies as described in Materials and Methods. (Right) Merged images.
Anti-RPA32 Ser-33, -RPA32 Ser-4/-8, -Rad52, and -Mre11 antibodies immunoprecipitate the BMRF1 replication protein.
We next determined whether HRR factors are loaded onto the viral genome DNA during lytic replication. It has been predicted that the BMRF1 protein not only acts as a viral polymerase processivity factor but also binds to the entire viral genome synthesized during lytic replication (8). Since HRR proteins such as phosphorylated RPA32, Rad51, and Rad52 were recruited to viral replication compartments (Fig. 3), these proteins might be loaded onto the EBV genome. IP experiments were therefore performed with nuclear extracts from lytic replication-induced Tet-BZLF1/B95-8 cells according to the nuclear complex co-IP kit method (Active Motif). Cells were collected in the presence of phosphatase inhibitors, which limit further protein modifications, resuspended in hypotonic buffer, and treated with detergent to remove detergent-soluble proteins. Although the BMRF1 protein was expressed abundantly and localized throughout nuclei after induction, the detergent treatment stripped the DNA-unbound form from nuclei. The resultant nuclei retained the BMRF1 protein localized only in the replication compartments. Nuclear protein-DNA complexes were recovered from the resultant nuclei with a low-salt buffer, followed by the addition of an enzymatic shearing cocktail, and gentle DNA digestion allowed a gentle release of protein-DNA complexes that were 200 to 1,500 bp in length. Thus, the method appears to maintain protein-DNA complexes in nuclear compartments of the cell. As shown in Fig. 4, anti-Mre11, anti-Rad52, anti-RPA32 Ser-33, and anti-RPA32 Ser-4/Ser-8 antibodies could coimmunoprecipitate the BMRF1 protein, in contrast to normal rabbit and mouse IgGs (Fig. 4). However, the anti-BMRF1 protein-specific antibody could hardly immunoprecipitate the phosphorylated RPA32, Rad51, Rad52, and Mre11 proteins (data not shown), probably due to an excess amount of the viral genome-bound form of the BMRF1 protein compared with those for the HRR proteins. Co-IP might be mediated by viral genome DNA rather than by direct protein-protein interactions between the BMRF1 and HRR proteins. To ascertain whether the BMRF1 protein directly binds to HRR proteins or not, we performed IP using nuclear extracts treated with DNase I. As shown in Fig. 4 (bottom), interactions between the BMRF1 protein and phosphorylated RPA, Mre11, and Rad52 proteins were decreased after treatment with DNase I. These observations further support the idea that HRR factors are associated with the viral DNA genome synthesized during lytic infection.
FIG. 4.

Anti-RPA32 Ser-33, -RPA32 Ser-4/Ser-8, -Mre11, and -Rad52 antibodies immunoprecipitate the BMRF1 replication protein. Tet-BZLF1/B95-8 cells were cultured in the presence of 2 μg of doxycycline/ml and harvested at 24 h p.i. Nuclear extracts were prepared as described in Materials and Methods and incubated with nonspecific anti-rabbit or mouse IgGs as a control or with anti-RPA32 Ser-33-, anti-RPA32 Ser-4/Ser-8-, anti-BMRF1-, anti-Mre11-, or anti-Rad52-specific antibodies. Twenty percent of the nuclear extract used for IP reactions was loaded as an input control. Proteins from the immunoprecipitates were applied for SDS-PAGE, and the BMRF1 protein was detected by Western blotting with peroxidase-labeled anti-BMRF1 antibodies. The top and bottom show IP from nuclear extracts treated without and with DNase I, respectively. IB, immunoblot.
Phosphorylated RPA, Rad51, Rad52, and Mre11 are loaded onto newly synthesized viral DNA during EBV lytic replication.
In order to determine further whether HRR factors such as RPA, Rad51, and Rad52 are loaded onto newly synthesized viral DNA, BrdUrd-labeled chromatin IP analyses were performed. During EBV lytic infection, chromosomal DNA replication is arrested while viral DNA synthesis occurs (8, 28). Although the precise molecular mechanisms remain to be determined, one of the reasons is the inhibition of the DNA helicase activity of the MCM4-6-7 complex through its phosphorylation by EBVPK and/or CDK1/2 (8, 26). Also, as was previously demonstrated in pulse-chase experiments and fluorescence in situ hybridization analyses, almost all BrdUrd-incorporating DNA is newly synthesized viral DNA (10). Thus, lytic infection-induced Tet-BZLF1/B95-8 cells were pulse-labeled with BrdUrd for 1 h prior to harvesting at 24 h p.i. Nuclear extracts were prepared according to the above-mentioned nuclear complex co-IP kit method, and protein-DNA complexes were immunoprecipitated with each antibody from the samples using protein G-Sepharose beads. BrdUrd-labeled DNAs were blotted onto membranes and stained with anti-BrdUrd monoclonal antibodies and secondary horseradish peroxidase-labeled anti-mouse IgG.
As shown in Fig. 5, each specific antibody could immunoprecipitate the corresponding protein from the samples except for nonimmune mouse IgGs. The anti-BALF2- and -BMRF1-specific IgGs markedly immunoprecipitated BrdUrd-labeled DNA from the samples, demonstrating that these viral replication proteins that are resistant to detergent treatments bind to newly synthesized viral DNAs. Similarly, anti-phospho-RPA32 Ser-33-, anti-phospho-Ser-4/Ser-8-, anti-RPA32-, anti-Rad51-, anti-Rad52-, and anti-Mre11-specific antibodies also immunoprecipitated BrdUrd-labeled DNA, while nonimmune mouse control IgG did not, and only trace amounts were found with anti-MCM7-specific IgG (Fig. 5A). The detergent-resistant form of the MCM7 protein localizes outside of viral replication compartments in nuclei (26), indicating chromosomal DNA binding of the MCM complex. Our results verified that RPA32 including phosphorylated forms, Rad51, Rad52, and Mre11 are loaded onto the viral DNA genome synthesized after the induction of lytic infection.
FIG. 5.

HRR factors such as phosphorylated RPA32, Mre11, Rad 51, and Rad52 are loaded onto EBV genomes. (A) Association of newly synthesized DNA with RPA32 Ser-33, RPA32 Ser-4/Ser-8, Rad52, Rad51, BMRF1, Mre11, and RPA32. Lytic replication was induced in Tet-BZLF1/B95-8 cells with 2 μg/ml of doxycycline, and newly synthesized DNAs were labeled for 1 h with 15 μM BrdUrd at 24 h p.i. Nuclear extracts were prepared as described in Materials and Methods and subjected to IP with each of the anti-RPA32 Ser-33, anti-RPA32 Ser-4/Ser-8, anti-Rad52, anti-Rad51, anti-BMRF1, anti-Mre11, and anti-RPA32 antibodies. An anti-MCM7 antibody and nonimmune mouse IgG were also applied as negative controls. The proteins and DNAs were eluted from the beads using SDS and EDTA, and DNAs were then purified with Chroma Spin-10 gel filtration columns (Clontech Laboratories, Inc.) and dot blotted onto Hybond N+ membranes. The BrdUrd (BrdU)-incorporating DNA in the precipitate was immunostained with an anti-BrdUrd antibody, and the included proteins were subjected to immunoblot analysis with the specific antibodies indicated at the top. (B) IP of the EBV genome by anti-RPA32 Ser-33, anti-RPA32 Ser-4/Ser-8, anti-Rad52, anti-Mre11, anti-BALF2, anti-Rad51, and anti-BMRF1 antibodies. Tet-BZLF1/B95-8 cells were cultured in the presence of 2 μg of doxycycline/ml and harvested at 24 h p.i. Nuclear extracts were prepared as described in Materials and Methods and subjected to IP using anti-RPA32 Ser-33, anti-RPA32 Ser-4/Ser-8, anti-Rad52, anti-Rad51, anti-BMRF1, anti-Mre11, and anti-BALF2 protein-specific antibodies, respectively. Nonimmune mouse IgG and anti-MCM6 or anti-16E7 antibodies were applied as negative controls. The proteins and DNAs were eluted from the beads using SDS and EDTA, and DNAs were then purified through Chroma Spin-10 gel filtration columns and amplified by PCR using primers specific for the EBV EBNA1 gene, the BZLF1 promoter, and the BALF5 gene. The amount of sample used in the PCR of the immunoprecipitated lane represents 1.2% of the precipitated DNA. These experiments were repeated three times, and the PCRs were performed in triplicate.
To evaluate the assembly of HRR factors onto the EBV genome further, we repeated the IP, and coimmunoprecipitated DNA was analyzed by PCR amplification using primers specific for the open reading frames of the EBNA1 and BALF5 genes and the promoter region of the BZLF1 gene. As shown in Fig. 5B, these regions were amplified with DNAs immunoprecipitated using anti-phosphorylated RPA32, Rad52, Rad51, Mre11, and EBV lytic protein (BALF2 and BMRF1) antibodies. In contrast, no PCR products were detected with nonspecific mouse IgG, MCM6, and 16E7 (human papillomavirus) antibodies. These results further support that phosphorylated RPA32, Rad52, Mre11, and Rad51 are loaded onto EBV genomes during lytic infection.
DSBs are detected in viral replication compartments.
We then asked whether DSB accumulation was accompanied by viral DNA replication by using TUNEL (7). Ionizing radiation resulted in a higher number of DSBs in chromatin DNA throughout nuclei than that found with mock irradiation (Fig. 6). As shown in Fig. 6, TUNEL staining as fine distinct dots was positive in viral replication compartments identified by BMRF1 protein staining, whereas it was negative in latent-phase Tet-BZLF1/B95-8 cells. Phosphorylated RPA32 at Ser-33 and Rad51 were found to localize with foci of DSBs (Fig. 6), and the induction of EBV lytic replication resulted in positive TUNEL staining in viral replication compartments. The positive TUNEL staining was clearly not due to apoptosis since caspase activity was not induced (data not shown), and there were no signs of DSBs in chromatin-occupying areas located outside of the replication compartments.
FIG. 6.

Presence of DNA DSBs in the viral genome synthesized in the replication compartments. TUNEL staining, performed as described in Materials and Methods, was used to detect DSBs in viral replication compartments. Tet-BZLF1/B95-8 cells were cultured in the presence of 2 μg of doxycycline/ml and harvested at 24 h p.i. Cells were treated with 0.5% Triton X-100-mCSK buffer, fixed with methanol, and coimmunostained with anti-BMRF1 (blue) antibody and anti-RPA32 Ser-33 (red) or anti-Rad51 (red) antibodies and with fluorescein-dUTP in the presence of TdT (green) as described in Materials and Methods. (Right) Merged images. (IR) for a TUNEL-staining positive control, Tet-BZLF1/B95-8 cells were exposed to 10 Gy of gamma irradiation and harvested 4 h posttreatment. Cells were stained with fluorescein-dUTP in the presence of TdT.
RNAi knockdown of RPA32 and Rad51 prevented viral genome synthesis.
HRR factors assembled onto EBV genomes during EBV lytic genome synthesis, suggesting their involvement in viral genome synthesis. The effects of RNAi knockdown of Rad51 and RPA32 on viral genome synthesis were examined in lytic EBV-induced cells. The siRNA-dependent knockdown of Rad51 decreased the number of EBV genome copies to one-fourth of that of the GFP siRNA-treated control by 48 h p.i. RPA32 knockdown by siRNA also decreased levels of EBV genome synthesis more dramatically (Fig. 7). RPA32 is reported to accumulate at ssDNA tracts and to undergo hyperphosphorylation, signifying the activation of HR. We speculate that HRR factors might amplify EBV genome synthesis through HR.
FIG. 7.
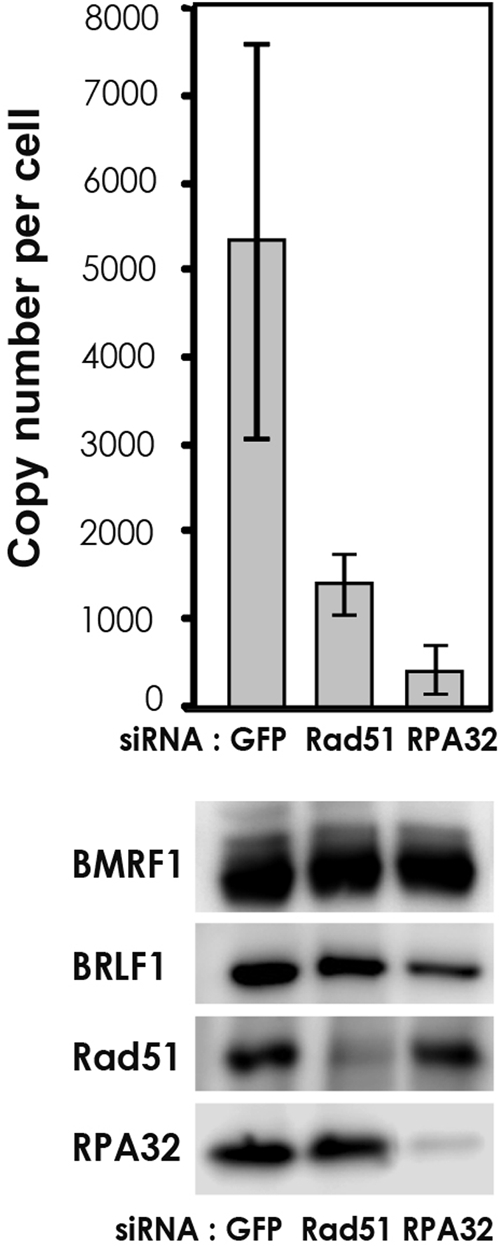
RNAi knockdown of RPA32 and Rad51 prevented viral genome synthesis. To examine the influence of Rad51 or RPA32 on EBV genome synthesis, 293/EBV cells were transfected with siRNA specific for Rad51, RPA32, or control GFP, respectively, and cultured for 24 h. Subsequently, cells were transfected with expression vector pBZLF1 and harvested 48 h after the induction of EBV lytic replication. (Top) Viral DNA synthesis was determined by slot blot assay as described in Materials and Methods. The means ± standard deviations of data from three independent experiments are shown. (Bottom) Cell lysates were analyzed by immunoblotting with antibodies to the indicated proteins.
DISCUSSION
We report here that RPA32 is extensively phosphorylated after the induction of EBV lytic replication, with the phosphorylation sites being in accordance with those for ATM. Phosphorylated RPA, Rad51, and Rad52, which are involved in HRR, are recruited and retained in viral replication compartments as well as the MRN complex. Furthermore, DSBs could be demonstrated to be generated in the EBV replication compartments. Thus, the MRN complex, hyperphosphorylated RPA, Rad51, and Rad52 might be recruited to repair DSBs on the viral genome in viral replication compartments. HRR is required to maintain viral genome integrity and to survive fatal DNA damage. Since the EBV genome is amplified to several hundred copies during lytic infection, host cellular HRR factors might help to increase the fidelity of viral replication. Furthermore, the depletion of RPA32 by siRNA treatment reduced viral DNA synthesis markedly, suggesting that HR and/or repair of the viral DNA genome might occur, coupled with EBV DNA replication. Thus, our findings offer fresh insight into the coordination of EBV genome synthesis with DNA damage-dependent induced host HRR factors and provide direct evidence that Rad51 and RPA32 are necessary for the completion of EBV lytic infection.
Similar to the cellular genome, the EBV genome contains repeated GC-rich recombinogenic sequences whose numbers vary remarkably in different virus isolates, even after only one passage of the same isolate (34, 36). During superinfection with two different EBV strains, hybrid viruses containing crossovers within the first internal repeats can be frequently isolated. Also, the formation of ends of EBV DNA, terminal repeats (TR) that are the sites of linearization and circularization, is not a simple cleavage-and-ligation reaction but an intricate process mediated by recombination. The total number of TR may be increased or decreased in linear virion DNA relative to their number in circular latent DNA (43, 55). Thus, TR and internal repeats in EBV are recombinogenic. In this study, we provide direct evidence that cellular HRR factors are loaded onto EBV genome DNA, in line with participation in viral genome synthesis in replication compartments.
The present study revealed that TUNEL-positive sites of DSBs were present in the viral replication compartments. An important issue raised is how DSBs are generated on EBV replicative intermediates. Although this remains to be confirmed, viral genome DNAs are presumably synthesized in a rolling-circle manner to produce head-to-tail concatemers that are subsequently cleaved into unit-length genomes, resulting in the generation of DSBs. Alternatively, there is a possibility that EBV-encoded DNase might generate DSBs. Direct visualization of herpes simplex virus type 1 (HSV-1) replication intermediates by electron microscopy showed ∼50% of molecules to consist of large tangled networks of DNA (2, 31). Also, Severini et al. previously isolated replication-intermediate HSV-1 DNA from the well of a pulsed-field gel and observed both Y-shaped arches and X-shaped junctions following restriction enzyme digestion (37). Thus, replicating DNA adopts a complex and most likely branched structure generated by recombination. One can speculate that HR is involved in the repair or editing of replication intermediates, and the observation of DSBs in EBV replication compartments suggests that the repair of such DSBs by HR would account for the prevalence of branched replication intermediates. Therefore, the EBV replication process might generate DSBs that will initiate HR.
As a cellular response to DNA damage, RPA32 can be hyperphosphorylated by members of the phosphatidylinositol 3-kinase family, including DNA-dependent protein kinase, ATM, and ATR. The phosphorylation occurs primarily within the N-terminal 33 residues. From mutational analyses of RPA32 phosphorylation sites, this prevents its recruitment to replication centers while having no effect on localization at sites of DNA damage (46). Current views of eukaryotic HRR are as follows (1, 35, 47). DSBs are processed to yield 3′ ssDNA overhangs, which are then protected by RPA containing phosphorylated RPA32. Processing of DSBs requires an MRN complex (24). Rad52 is implicated in displacing RPA to aid Rad51 in binding to ssDNA within a primary binding site, thus promoting nucleoprotein filament formation. Rad51 and Rad52 preferentially interact with hyperphosphorylated RPA32 (39, 40, 42). In concert with other factors, the active Rad51 nucleoprotein filament binds to a second double-stranded DNA substrate containing homology and exchanges DNA strands in an ATP-dependent manner, resulting in the formation of Holliday junctions and branch migration. Although DNA can be damaged as a result of failures during replication or exposure to damage-inducing agents, DSBs are the most plausible lesion for HRR and require a homologous sequence as a template. Thus, HR represents an error-free subpathway of damage tolerance, allowing the replicational bypass of lesions through a template switch. From the observations described here, we propose that HR occurs during EBV lytic genome replication with the aid of host cellular HRR factors. Although the siRNA depletion of Rad51 or RPA32 reduced newly viral DNA synthesis (Fig. 7), the detailed mechanism for the inhibition of EBV lytic replication is still unclear. Further studies of the functional interaction of the EBV lytic replication machinery with host HRR factors are needed to understand the EBV genome replication system.
We previously reported that postreplicative mismatch repair (MMR) factors are also recruited to viral replication compartments (10). MMR is a mutation avoidance system that eliminates base-base mismatches and small insertion/deletion mispairs accumulating in the genome during DNA replication. MMR proteins such as MSH2, MSH6, MSH3, and PCNA recognize these lesions, and the “wrong” bases are excised from newly synthesized strands of DNA, with a repair patch being synthesized by using the parental DNA strand as a template. RPA plays important roles in both excision and resynthesis reactions of MMR, with functions regulated by its phosphorylation state (23). Unphosphorylated RPA possesses a high level of DNA-binding affinity and preferentially stimulates mismatch-provoked excision, while phosphorylated RPA preferentially facilitates DNA resynthesis via reducing its DNA-binding ability, allowing displacement by DNA polymerases to make a DNA template available for nucleotide polymerization (23). Since the staining patterns differ between anti-phosphorylated RPA-specific and anti-RPA antibodies, it should be noted that both types of RPA are present in the EBV replication compartments. MSH2 was previously reported to be required for the relocalization of HRR factors such as Mre11 and Rad51 (17). MMR might connect with HRR through MSH2 or RPA in EBV replication compartments.
Wilkinson and Weller previously reported that RPA, Rad51, and NBS1 are recruited to HSV-1 replication compartments, although their steady-state levels remain unchanged throughout the course of infection (51). However, unlike the case of EBV reported here, there is no induction of hyperphosphorylation of RPA upon productive HSV-1 infection. Instead, endogenous hyperphosphorylated RPA is sequestered away from replication compartments into discrete nuclear foci, VICE domains that are enriched for cellular components involved in protein folding and degradation (49). The induction of hyperphosphorylation of RPA has been observed only in the presence of a viral polymerase inhibitor such as phosphonoacetic acid or acyclovir (50). The results are totally different from those upon EBV replicative infection, and they speculated that the unphosphorylated general population of RPA found within the HSV-1 replication compartments possibly acts at unperturbed viral forks or replication intermediates. Since neither the induction of RPA hyperphosphorylation nor the recruitment of phosphorylated RPA to HSV-1 replication compartments was observed, those authors concluded that these signaling molecules are excluded from sites that contain viral DNA. Furthermore, from the observation that RPA hyperphosphorylation does not occur in HSV-1-infected cells in the absence of phosphonoacetic acid, they presumed that the signal for this type of DNA damage is not transmitted under conditions in which viral DNA replication occurs and replication compartments form. Although the discrepancy remains unclear, our results clearly showed that hyperphosphorylated RPA32, Rad51, Rad52, and the MRN complex were colocalized within the EBV replication compartments and interacted with EBV genomic DNA, suggesting that EBV replication compartments contain HRR substrates. HRR is required to maintain viral genome integrity and to survive fatal DNA damage. Since the EBV genome is amplified to several hundred copies during lytic infection, host cellular HRR factors might help to increase the fidelity of viral replication.
Acknowledgments
We thank W. Hammerschmidt, H. J. Delecluse, T. Takahashi, and K. Kuzushima for invaluable materials and Yasuhiro Nishikawa for technical assistance.
This work was supported by grants-in-aid for scientific research on priority areas from the Ministry of Education, Science, Sports, Culture, and Technology of Japan (grants 19041078, 20012056, and 20390137 to T.T.). Y.S. and A.K. were supported by research fellowships from the Japanese Society for the Promotion of Science for Young Scientists.
Footnotes
Published ahead of print on 22 April 2009.
REFERENCES
- 1.Aylon, Y., B. Liefshitz, and M. Kupiec. 2004. The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J. 234868-4875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ben-Porat, T., A. S. Kaplan, B. Stehn, and A. S. Rubenstein. 1976. Concatemeric forms of intracellular herpesvirus DNA. Virology 69547-560. [DOI] [PubMed] [Google Scholar]
- 3.Benson, F. E., P. Baumann, and S. C. West. 1998. Synergistic actions of Rad51 and Rad52 in recombination and DNA repair. Nature 391401-404. [DOI] [PubMed] [Google Scholar]
- 4.Binz, S. K., A. M. Sheehan, and M. S. Wold. 2004. Replication protein A phosphorylation and the cellular response to DNA damage. DNA Repair (Amsterdam) 31015-1024. [DOI] [PubMed] [Google Scholar]
- 5.Bochkarev, A., and E. Bochkareva. 2004. From RPA to BRCA2: lessons from single-stranded DNA binding by the OB-fold. Curr. Opin. Struct. Biol. 1436-42. [DOI] [PubMed] [Google Scholar]
- 6.Braun, K. A., Y. Lao, Z. He, C. J. Ingles, and M. S. Wold. 1997. Role of protein-protein interactions in the function of replication protein A (RPA): RPA modulates the activity of DNA polymerase alpha by multiple mechanisms. Biochemistry 368443-8454. [DOI] [PubMed] [Google Scholar]
- 7.Costanzo, V., K. Robertson, M. Bibikova, E. Kim, D. Grieco, M. Gottesman, D. Carroll, and J. Gautier. 2001. Mre11 protein complex prevents double-strand break accumulation during chromosomal DNA replication. Mol. Cell 8137-147. [DOI] [PubMed] [Google Scholar]
- 8.Daikoku, T., A. Kudoh, M. Fujita, Y. Sugaya, H. Isomura, N. Shirata, and T. Tsurumi. 2005. Architecture of replication compartments formed during Epstein-Barr virus lytic replication. J. Virol. 793409-3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daikoku, T., A. Kudoh, M. Fujita, Y. Sugaya, H. Isomura, and T. Tsurumi. 2004. In vivo dynamics of EBNA1-oriP interaction during latent and lytic replication of Epstein-Barr virus. J. Biol. Chem. 27954817-54825. [DOI] [PubMed] [Google Scholar]
- 10.Daikoku, T., A. Kudoh, Y. Sugaya, S. Iwahori, N. Shirata, H. Isomura, and T. Tsurumi. 2006. Postreplicative mismatch repair factors are recruited to Epstein-Barr virus replication compartments. J. Biol. Chem. 28111422-11430. [DOI] [PubMed] [Google Scholar]
- 11.Delecluse, H. J., T. Hilsendegen, D. Pich, R. Zeidler, and W. Hammerschmidt. 1998. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc. Natl. Acad. Sci. USA 958245-8250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dimitrova, D. S., and D. M. Gilbert. 2000. Stability and nuclear distribution of mammalian replication protein A heterotrimeric complex. Exp. Cell Res. 254321-327. [DOI] [PubMed] [Google Scholar]
- 13.Din, S., S. J. Brill, M. P. Fairman, and B. Stillman. 1990. Cell-cycle-regulated phosphorylation of DNA replication factor A from human and yeast cells. Genes Dev. 4968-977. [DOI] [PubMed] [Google Scholar]
- 14.Dutta, A., J. M. Ruppert, J. C. Aster, and E. Winchester. 1993. Inhibition of DNA replication factor RPA by p53. Nature 36579-82. [DOI] [PubMed] [Google Scholar]
- 15.Dutta, A., and B. Stillman. 1992. cdc2 family kinases phosphorylate a human cell DNA replication factor, RPA, and activate DNA replication. EMBO J. 112189-2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Flemington, E. K., A. E. Goldfeld, and S. H. Speck. 1991. Efficient transcription of the Epstein-Barr virus immediate-early BZLF1 and BRLF1 genes requires protein synthesis. J. Virol. 657073-7077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franchitto, A., P. Pichierri, R. Piergentili, M. Crescenzi, M. Bignami, and F. Palitti. 2003. The mammalian mismatch repair protein MSH2 is required for correct MRE11 and RAD51 relocalization and for efficient cell cycle arrest induced by ionizing radiation in G2 phase. Oncogene 222110-2120. [DOI] [PubMed] [Google Scholar]
- 18.Fujita, M., Y. Ishimi, H. Nakamura, T. Kiyono, and T. Tsurumi. 2002. Nuclear organization of DNA replication initiation proteins in mammalian cells. J. Biol. Chem. 27710354-10361. [DOI] [PubMed] [Google Scholar]
- 19.Fujita, M., T. Kiyono, Y. Hayashi, and M. Ishibashi. 1996. Inhibition of S-phase entry of human fibroblasts by an antisense oligomer against hCDC47. Biochem. Biophys. Res. Commun. 219604-607. [DOI] [PubMed] [Google Scholar]
- 20.Fujita, M., C. Yamada, H. Goto, N. Yokoyama, K. Kuzushima, M. Inagaki, and T. Tsurumi. 1999. Cell cycle regulation of human CDC6 protein. Intracellular localization, interaction with the human mcm complex, and CDC2 kinase-mediated hyperphosphorylation. J. Biol. Chem. 27425927-25932. [DOI] [PubMed] [Google Scholar]
- 21.Golub, E. I., R. C. Gupta, T. Haaf, M. S. Wold, and C. M. Radding. 1998. Interaction of human rad51 recombination protein with single-stranded DNA binding protein, RPA. Nucleic Acids Res. 265388-5393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gomes, X. V., L. A. Henricksen, and M. S. Wold. 1996. Proteolytic mapping of human replication protein A: evidence for multiple structural domains and a conformational change upon interaction with single-stranded DNA. Biochemistry 355586-5595. [DOI] [PubMed] [Google Scholar]
- 23.Guo, S., Y. Zhang, F. Yuan, Y. Gao, L. Gu, I. Wong, and G. M. Li. 2006. Regulation of replication protein A functions in DNA mismatch repair by phosphorylation. J. Biol. Chem. 28121607-21616. [DOI] [PubMed] [Google Scholar]
- 24.Haber, J. E. 1998. The many interfaces of Mre11. Cell 95583-586. [DOI] [PubMed] [Google Scholar]
- 25.Hozak, P., A. B. Hassan, D. A. Jackson, and P. R. Cook. 1993. Visualization of replication factories attached to nucleoskeleton. Cell 73361-373. [DOI] [PubMed] [Google Scholar]
- 26.Kudoh, A., T. Daikoku, Y. Ishimi, Y. Kawaguchi, N. Shirata, S. Iwahori, H. Isomura, and T. Tsurumi. 2006. Phosphorylation of MCM4 at sites inactivating DNA helicase activity of the MCM4-MCM6-MCM7 complex during Epstein-Barr virus productive replication. J. Virol. 8010064-10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kudoh, A., T. Daikoku, Y. Sugaya, H. Isomura, M. Fujita, T. Kiyono, Y. Nishiyama, and T. Tsurumi. 2004. Inhibition of S-phase cyclin-dependent kinase activity blocks expression of Epstein-Barr virus immediate-early and early genes, preventing viral lytic replication. J. Virol. 78104-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kudoh, A., M. Fujita, T. Kiyono, K. Kuzushima, Y. Sugaya, S. Izuta, Y. Nishiyama, and T. Tsurumi. 2003. Reactivation of lytic replication from B cells latently infected with Epstein-Barr virus occurs with high S-phase cyclin-dependent kinase activity while inhibiting cellular DNA replication. J. Virol. 77851-861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kudoh, A., M. Fujita, L. Zhang, N. Shirata, T. Daikoku, Y. Sugaya, H. Isomura, Y. Nishiyama, and T. Tsurumi. 2005. Epstein-Barr virus lytic replication elicits ATM checkpoint signal transduction while providing an S-phase-like cellular environment. J. Biol. Chem. 2808156-8163. [DOI] [PubMed] [Google Scholar]
- 30.Liu, Y., M. Kvaratskhelia, S. Hess, Y. Qu, and Y. Zou. 2005. Modulation of replication protein A function by its hyperphosphorylation-induced conformational change involving DNA binding domain B. J. Biol. Chem. 28032775-32783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinez, R., R. T. Sarisky, P. C. Weber, and S. K. Weller. 1996. Herpes simplex virus type 1 alkaline nuclease is required for efficient processing of viral DNA replication intermediates. J. Virol. 702075-2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.New, J. H., T. Sugiyama, E. Zaitseva, and S. C. Kowalczykowski. 1998. Rad52 protein stimulates DNA strand exchange by Rad51 and replication protein A. Nature 391407-410. [DOI] [PubMed] [Google Scholar]
- 33.Nuss, J. E., S. M. Patrick, G. G. Oakley, G. M. Alter, J. G. Robison, K. Dixon, and J. J. Turchi. 2005. DNA damage induced hyperphosphorylation of replication protein A. 1. Identification of novel sites of phosphorylation in response to DNA damage. Biochemistry 448428-8437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raab-Traub, N., and K. Flynn. 1986. The structure of the termini of the Epstein-Barr virus as a marker of clonal cellular proliferation. Cell 47883-889. [DOI] [PubMed] [Google Scholar]
- 35.Rodrigue, A., M. Lafrance, M. C. Gauthier, D. McDonald, M. Hendzel, S. C. West, M. Jasin, and J. Y. Masson. 2006. Interplay between human DNA repair proteins at a unique double-strand break in vivo. EMBO J. 25222-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sato, H., T. Takimoto, S. Tanaka, J. Tanaka, and N. Raab-Traub. 1990. Concatameric replication of Epstein-Barr virus: structure of the termini in virus-producer and newly transformed cell lines. J. Virol. 645295-5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Severini, A., D. G. Scraba, and D. L. Tyrrell. 1996. Branched structures in the intracellular DNA of herpes simplex virus type 1. J. Virol. 703169-3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shao, R. G., C. X. Cao, H. Zhang, K. W. Kohn, M. S. Wold, and Y. Pommier. 1999. Replication-mediated DNA damage by camptothecin induces phosphorylation of RPA by DNA-dependent protein kinase and dissociates RPA:DNA-PK complexes. EMBO J. 181397-1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shinohara, A., H. Ogawa, and T. Ogawa. 1992. Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell 69457-470. [DOI] [PubMed] [Google Scholar]
- 40.Shinohara, A., M. Shinohara, T. Ohta, S. Matsuda, and T. Ogawa. 1998. Rad52 forms ring structures and co-operates with RPA in single-strand DNA annealing. Genes Cells 3145-156. [DOI] [PubMed] [Google Scholar]
- 41.Stigger, E., R. Drissi, and S. H. Lee. 1998. Functional analysis of human replication protein A in nucleotide excision repair. J. Biol. Chem. 2739337-9343. [DOI] [PubMed] [Google Scholar]
- 42.Sugiyama, T., and S. C. Kowalczykowski. 2002. Rad52 protein associates with replication protein A (RPA)-single-stranded DNA to accelerate Rad51-mediated displacement of RPA and presynaptic complex formation. J. Biol. Chem. 27731663-31672. [DOI] [PubMed] [Google Scholar]
- 43.Sun, R., T. A. Spain, S. F. Lin, and G. Miller. 1997. Sp1 binds to the precise locus of end processing within the terminal repeats of Epstein-Barr virus DNA. J. Virol. 716136-6143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takagi, S., K. Takada, and T. Sairenji. 1991. Formation of intranuclear replication compartments of Epstein-Barr virus with redistribution of BZLF1 and BMRF1 gene products. Virology 185309-315. [DOI] [PubMed] [Google Scholar]
- 45.Tsurumi, T., M. Fujita, and A. Kudoh. 2005. Latent and lytic Epstein-Barr virus replication strategies. Rev. Med. Virol. 153-15. [DOI] [PubMed] [Google Scholar]
- 46.Vassin, V. M., M. S. Wold, and J. A. Borowiec. 2004. Replication protein A (RPA) phosphorylation prevents RPA association with replication centers. Mol. Cell. Biol. 241930-1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ward, J. D., L. J. Barber, M. I. Petalcorin, J. Yanowitz, and S. J. Boulton. 2007. Replication blocking lesions present a unique substrate for homologous recombination. EMBO J. 263384-3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weber, P. C., M. D. Challberg, N. J. Nelson, M. Levine, and J. C. Glorioso. 1988. Inversion events in the HSV-1 genome are directly mediated by the viral DNA replication machinery and lack sequence specificity. Cell 54369-381. [DOI] [PubMed] [Google Scholar]
- 49.Wilkinson, D. E., and S. K. Weller. 2006. Herpes simplex virus type I disrupts the ATR-dependent DNA-damage response during lytic infection. J. Cell Sci. 1192695-2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wilkinson, D. E., and S. K. Weller. 2005. Inhibition of the herpes simplex virus type 1 DNA polymerase induces hyperphosphorylation of replication protein A and its accumulation at S-phase-specific sites of DNA damage during infection. J. Virol. 797162-7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wilkinson, D. E., and S. K. Weller. 2004. Recruitment of cellular recombination and repair proteins to sites of herpes simplex virus type 1 DNA replication is dependent on the composition of viral proteins within prereplicative sites and correlates with the induction of the DNA damage response. J. Virol. 784783-4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yates, J. L., and N. Guan. 1991. Epstein-Barr virus-derived plasmids replicate only once per cell cycle and are not amplified after entry into cells. J. Virol. 65483-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yokoyama, N., M. Hirata, K. Ohtsuka, Y. Nishiyama, K. Fujii, M. Fujita, K. Kuzushima, T. Kiyono, and T. Tsurumi. 2000. Co-expression of human chaperone Hsp70 and Hsdj or Hsp40 co-factor increases solubility of overexpressed target proteins in insect cells. Biochim. Biophys. Acta 1493119-124. [DOI] [PubMed] [Google Scholar]
- 54.Zernik-Kobak, M., K. Vasunia, M. Connelly, C. W. Anderson, and K. Dixon. 1997. Sites of UV-induced phosphorylation of the p34 subunit of replication protein A from HeLa cells. J. Biol. Chem. 27223896-23904. [DOI] [PubMed] [Google Scholar]
- 55.Zimmermann, J., and W. Hammerschmidt. 1995. Structure and role of the terminal repeats of Epstein-Barr virus in processing and packaging of virion DNA. J. Virol. 693147-3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zou, Y., Y. Liu, X. Wu, and S. M. Shell. 2006. Functions of human replication protein A (RPA): from DNA replication to DNA damage and stress responses. J. Cell. Physiol. 208267-273. [DOI] [PMC free article] [PubMed] [Google Scholar]