

Abstract
Herpes simplex virus (HSV) glycoproteins gB, gD, and gH/gL are necessary and sufficient for virus entry into cells. Structural features of gB are similar to those of vesicular stomatitis virus G and baculovirus gp64, and together they define the new class III group of fusion proteins. Previously, we used mutagenesis to show that three hydrophobic residues (W174, Y179, and A261) within the putative gB fusion loops are integral to gB function. Here we expanded our analysis, using site-directed mutagenesis of each residue in both gB fusion loops. Mutation of most of the nonpolar or hydrophobic amino acids (W174, F175, G176, Y179, and A261) had severe effects on gB function in cell-cell fusion and null virus complementation assays. Of the six charged amino acids, mutation of H263 or R264 also negatively affected gB function. To further analyze the mutants, we cloned the ectodomains of the W174R, Y179S, H263A, and R264A mutants into a baculovirus expression system and compared them with the wild-type (WT) form, gB730t. As shown previously, gB730t blocks virus entry into cells, suggesting that gB730t competes with virion gB for a cell receptor. All four mutant proteins retained this function, implying that fusion loop activity is separate from gB-receptor binding. However, unlike WT gB730t, the mutant proteins displayed reduced binding to cells and were either impaired or unable to bind naked, cholesterol-enriched liposomes, suggesting that it may be gB-lipid binding that is disrupted by the mutations. Furthermore, monoclonal antibodies with epitopes proximal to the fusion loops abrogated gB-liposome binding. Taken together, our data suggest that gB associates with lipid membranes via a fusion domain of key hydrophobic and hydrophilic residues and that this domain associates with lipid membranes during fusion.
Herpes simplex virus (HSV) entry into cells requires four viral envelope glycoproteins (gB, gD, and the heterodimer gH/gL) as well as a cell surface gD receptor (reviewed in references 31, 42, 43, and 49). When gD binds its receptor, it undergoes conformational changes that are essential to activate the fusion machinery, gB and gH/gL. In addition to being essential for virus entry, both gH/gL and gB play important roles in primary fusion events that occur during egress of the capsid from the nuclei of infected cells (22). gB and gH/gL constitute the core fusion machinery of all members of the Herpesviridae.
The mechanisms by which gB and gH/gL function individually and in concert during fusion are topics of intense investigations. Peptides based on predicted heptad repeats in gH block virus entry and have the ability to bind and disrupt model membranes (24, 26, 27). In addition, gH/gL can achieve hemifusion of adjacent cells in the absence of other herpesvirus proteins (50). These studies imply that gH/gL has fusogenic properties. Previously, we showed that both virion gB and soluble wild-type (WT) gB (gB730t), but not gD or gH/gL, bind to cells and associate with lipid rafts (10). Like gH/gL, several synthetic gB peptides induced the fusion of large unilamellar vesicles and inhibited herpesvirus infection (23, 24). Thus, it appears that both gB and gH/gL may be fusion proteins, a theory strengthened by data showing that either gB or gH/gL is sufficient for membrane fusion during nuclear egress (22). Additionally, gB730t blocks virus entry into cells deficient in heparan sulfate proteoglycans (HSPGs), suggesting that it competes with virion gB for an obligate cell surface receptor (9). A recent study suggested that paired immunoglobulin-like type 2 receptor alpha (PILRα) may serve this role for at least some cell types (47).
The crystal structure of gB is now known for both HSV type 1 (HSV-1) (32) and Epstein-Barr virus (EBV) (6). Interestingly, gB is structurally related to two other viral fusion proteins, the vesicular stomatitis virus (VSV) G protein (45) and the baculovirus gp64 protein (34). VSV G, gB, and most recently, baculovirus gp64 were placed into a newly formed group of fusion proteins, the class III proteins. Class III fusion proteins have similar individual domain structures and contain a central three-stranded coiled coil reminiscent of the class I proteins. Whereas class I proteins have an N-terminal fusion peptide, class III proteins have internal bipartite fusion loops within domain I (shown in Fig. 1A for gB) which are similar to the single fusion loop of class II fusion proteins. However, the class II fusion loop is composed entirely of hydrophobic amino acids, whereas the fusion loops of gB have both hydrophobic and charged residues (32, 34, 45). Unlike G or gp64, which are the sole fusion proteins for their respective viruses, gB requires gH/gL to function in fusion and entry.
FIG. 1.

HSV gB hydrophobic ridge is surrounded by charged residues on the surface of the molecule. A ribbon diagram of the HSV protomer (A) and molecular surface representation of the trimer (B) are shown. In each, one protomer is colored by secondary structure succession, using blue (domain I), green (domain II), yellow (domain III), orange (domain IV), and red (domain V). The box in panel A shows the primary amino acid sequences of the fusion loops. The box in panel B shows the base of the gB trimer, rotated 90°. For the boxes in both panels A and B, highlighted hydrophobic residues are colored in blue and charged residues are shown in red. All structural figures were generated, in part, using PyMOL Molecular Graphics System software.
In our previous study, we used site-directed mutagenesis to show that three hydrophobic amino acids within the gB loops (W174, Y179, and A261) are essential for gB function (29). Similar studies of VSV G, gp64, and EBV gB support the notion that hydrophobic amino acids of both fusion loops are critical for fusion (34, 44, 51) and together constitute a fusion domain. Recently, bimolecular complementation was used to show that gB and gH/gL interact with each other concomitantly with fusion and that this interaction is triggered by binding of gD to its cellular receptor (3, 4). Thus, gB may function cooperatively with gH/gL, yet each may have some fusogenic potential on its own.
The goal of the experiments reported here was twofold. First, we wanted to complete our mutagenic analysis of all of the residues in the two putative fusion loops of HSV gB. Our data show that the two fusion loops constitute a structural “subdomain” wherein key hydrophobic amino acids form a ridge that is supported on both sides by charged residues. We hypothesize that two charged residues on one side of the ridge enhance the ability of the hydrophobic residues to interact with target membranes and to function in fusion.
Our second goal was to assess the effects of mutations in the fusion loops on the function of gB in cell binding, blocking of entry, and insertion into lipid membranes. Therefore, we constructed recombinant baculoviruses, with each carrying the gene for a truncated version (residues 31 to 730) of one of four mutant forms of gB (W174R, Y179S, H263A, and R264A). We found that the mutant proteins were able to efficiently block virus entry, suggesting that the fusion loops do not participate in protein-receptor binding. However, all four mutant proteins were impaired in cell binding compared to WT gB730t. Whereas WT gB730t associated with liposomes in a flotation assay, soluble truncated forms of HSV gD and gH/gL did not, consistent with our previous finding that gB730t associates with lipid rafts on cell surfaces (8). In contrast to WT gB730t, the gB mutant proteins were either impaired or unable to bind liposomes. Our data suggest that gB has an intrinsic ability to associate with a target membrane via its fusion domain.
MATERIALS AND METHODS
Cells and viruses.
Mouse fibroblast L cells and derived mutant Gro2C cells (gifts of F. Tufaro) (7) were grown in Dulbecco's modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS). African green monkey kidney (Vero) cells were grown in DMEM with 5% FBS. Mouse B78H1 melanoma cells engineered to express the gD receptor nectin-1 (C10 cells) were grown in DMEM supplemented with 5% FBS and 500 μg/ml of G418 (36). Chinese hamster ovary K1 (CHO-K1) cells were grown in Ham's F-12 medium containing 10% FBS. CHO-HVEM12 cells, expressing the HSV receptor HVEM (52), were grown in F-12 medium containing 10% FBS and 250 μg/ml G418. HSV-1 KOS/tk12, which carries the lacZ gene under the control of the ICP4 promoter (54), was purified on sucrose gradients as described previously (28). CHO-K1, CHO-HVEM12, and C10 cells and HSV-1 KOS/tk12 were kindly provided by P. G. Spear. Propagation of the gB-null virus K082 (gift of S. Person) on VB38 cells (gift of D. C. Johnson) was done as previously described (12, 22).
Construction of gB mutants.
A QuikChange site-directed mutagenesis kit (Stratagene Cloning Systems, La Jolla, CA) was used to generate full-length mutant gB constructs as described previously (19). Primers designed to mutate individual gB residues were used to amplify the gB gene of plasmid pPEP98 (41) by PCR. The mutations were confirmed by sequencing of the entire gB gene. Plasmids encoding the gB substitutions were named as follows: gB-F175K, pBH839; gB-G176K, pBH807; gB-H177A, pBH812; gB-R178A, pBH784; gB-R258A, pBH792; gB-E260A, pBH876; gB-F262D, pBH874; gB-H263A, pBH809; gB-R264A, pBH786; and gB-Y265R, pBH828. We also studied the following gB mutant constructs first reported by Hannah et al. (29): gB-W174Y (pBH730), gB-W174R (pBH739), gB-W174K (pBH776), gB-Y179S (pBH777), gB-Y179K (pBH877), gB-V259R (pBH738), gB-A261W (pBH750), gB-A261D (pBH732), and gB-F262L (pBH733).
Truncated versions of gB (residues 31 to 730) carrying the amino acid substitutions Y179S, H263A, W174R, and R264A were generated by changing the codon at residue 730 of pBH777, pBH809, pBH739, and pBH786, respectively, into a stop codon that also created a BclI restriction site. These gB mutant sequences were then subcloned into pFB686, a baculovirus expression vector that expresses gB730t, by NotI/NheI double digestion and subsequent ligation. gB730t comprises amino acids 31 to 730 (numbered starting at the first methionine) of the gB ectodomain; the native gB signal sequence (residues 1 to 30) is replaced with the melittin signal sequence (10). Mutant gB730t proteins were encoded by plasmids pBH861 (Y179S), pBH868 (H263A), pBH890 (W174R), and pBH873 (R264A). The truncation mutant gB670t was constructed by PstI digestion of pFB679 (8) and ligation of the insert into pCW289 (10), resulting in plasmid pFB688. Recombinant baculoviruses were generated as previously described (48).
Production and purification of HSV glycoproteins.
Soluble gD306t was purified from baculovirus-infected insect cells (Sf9) as previously described (46, 48). Soluble gH1t/gL1 was purified from a stably transfected L-cell line as described by Peng et al. (40). The complex contains gH1 truncated at residue 792 and full-length gL1. To make the soluble gH2t/gL2 complex, we used the FastBac Dual system (Invitrogen) to construct a single baculovirus recombinant that expressed both gH2t (truncated just before the transmembrane region at residue 803) and full-length gL2. The sequence for gL2 was PCR amplified from pWF318 (13) by use of a primer (5′-GGGTTTATACGGTACCTCTAGACTCG), which encodes a KpnI restriction site and a primer complementary to the region 5′ of the pcDNA3.1 multiple cloning site. The gL fragment was digested with NheI-KpnI and ligated into vector pFastBac Dual to generate the plasmid pTC604. Next, plasmid pCW333 (containing the sequence for gH2 residues 1 to 803) (13) was digested with EcoRI, and the resulting small fragment was ligated into pTC604, generating the gH2t/gL2-expressing plasmid pTC605. Unlike our other soluble glycoproteins, gH2t/gL2 retains its native signal sequence. A six-His tag on the C terminus of gH2t facilitated purification of the complex by use of Ni-nitrilotriacetic acid resin and elution with imidazole (14). Both WT and mutant forms of soluble gB730t and gB670t were purified from baculovirus-infected Sf9 cells by use of a DL16 immunosorbent column as described earlier (10).
Antibodies.
Polyclonal antibodies (PAbs) used in this study were as follows: rabbit (R) sera R68, R69 (both against gB), and R8 (gD) were raised against proteins purified from infected cells (33), whereas R137 and R176 were prepared against purified gH1t/gL1 and gH2t/gL2, respectively (13, 40). gB-specific monoclonal antibodies (MAbs) used in this study were as follows. C226 was provided by Becton Dickinson (8). MAbs numbered SS10 to SS145, DL16, and DL21 were prepared and characterized previously (8, 9). MAbs DL16, DL21, SS55, and SS145 recognize discontinuous (conformation-dependent) epitopes, SS106 and SS121 recognize continuous (linear) epitopes, and C226, SS10, SS68, SS69, SS120, and SS144 recognize pseudo-continuous epitopes, as defined by Bender et al. (8). The anti c-myc MAb 9E10 (21) was used as a negative control for immunoprecipitation.
Western blotting and immunoprecipitation.
Purified proteins from baculovirus-infected cells were mixed with an equal volume of polyacrylamide gel electrophoresis (PAGE) sample buffer containing either no reducing agent and 0.2% sodium dodecyl sulfate (SDS) (“native” conditions) or 200 mM dithiothreitol and 2% SDS (“denaturing” conditions) (8, 17). Proteins from denatured samples were also boiled for 5 min at 100°C before electrophoresis. Proteins were resolved by SDS-PAGE and transferred to nitrocellulose for Western blotting. Anti-gB MAbs were used to immunoprecipitate full-length gB from total cell lysates of transfected L cells. Cell extracts were diluted in lysis buffer (10 mM Tris, pH 8, 150 mM NaCl, 10 mM EDTA, 1% NP-40, 0.5% deoxycholic acid) and incubated with 5 μg of either DL16, SS55, SS145, or anti-c-myc (negative control) immunoglobulin G (IgG) overnight at 4°C. Immunoprecipitation of purified, truncated gB was performed as described above, with the exception that proteins were diluted in IP binding buffer (10 mM Tris, pH 8.0, 100 mM NaCl, 0.01% NP-40, 0.05% bovine serum albumin) for the antibody incubation step. Proteins were precipitated with protein A-agarose beads (Gibco BRL) for 2 h at 4°C. Beads were washed extensively with cold phosphate-buffered saline (PBS), and the proteins were eluted with PAGE sample buffer, boiled, and electrophoresed. After transfer to nitrocellulose, the blots were probed with PAb R69.
CELISA.
To detect gB cell surface expression, we used a modification of a cell-based enzyme-linked immunosorbent assay (CELISA) (25, 37). CHO-K1 cells growing in 96-well plates were transfected with T7 polymerase and with gD, gH, gL, and gB plasmids by using 40 ng of each plasmid/well and 0.5 μl of Lipofectamine 2000 (Invitrogen), both diluted in Opti-MEM1 (Gibco). Cells were exposed to the DNA-Lipofectamine 2000 mixture for 5 h, after which the mixture was replaced with growth medium. Cells were grown overnight at 37°C, fixed in 3% paraformaldehyde, and rinsed with PBS containing Ca2+ and Mg2+. Cells were incubated for 1 h with PAb R69 diluted in 3% bovine serum albumin-PBS and then incubated for 30 min with goat anti-rabbit antibodies coupled to horseradish peroxidase, all at room temperature. Cells were rinsed with 20 mM citrate buffer (pH 4.5), 2,2′-azino-di(3-ethylbenzthiazoline) sulfonic acid peroxidase substrate (Moss, Inc.) was added, and the absorbance at 405 nm was recorded using a microtiter plate reader. The absorbance reading of cells transfected with the empty vector pCAGGS/MCS was subtracted, and data were normalized to WT gB. To measure the binding of soluble glycoproteins to the cell surface, cells in 96-well plates were incubated for 1 h at 4°C with serial dilutions of soluble glycoproteins diluted in PBS. After being washed with ice-cold PBS, cells were fixed with 3% paraformaldehyde, and the remainder of the assay was performed as described above.
Quantitative fusion assay.
To detect cell-cell fusion, we used a modified version of a previously described luciferase reporter gene activation assay (39, 41). Effector cells (4 × 104 CHO-K1 cells per well) growing in 96-well plates were transfected with plasmids encoding T7 RNA polymerase (pCAGT7), gD (pPEP99), gH (pPEP100), gL (pPEP101), and either wild-type gB (pPEP98), empty vector (pCAGGS/MCS), or one of the mutant gBs. Forty nanograms of each plasmid and 0.5 μl of Lipofectamine 2000/well (Invitrogen), both diluted in Opti-MEM1 (Gibco), were added to cells in each well. Transfections were performed in triplicate. To prepare receptor-bearing target cells, CHO-HVEM12 cells growing in six-well plates were transfected with 10 μl of Lipofectamine 2000 containing 4 μg/well of a plasmid carrying the firefly luciferase gene under the control of the T7 promoter (pT7EMCLuc). Cells were exposed to the DNA-Lipofectamine 2000 mixture for 5 h at 37°C, after which the transfection mixtures were replaced with fresh medium and incubated for 1 h at 37°C. Target cells were trypsinized, and 4 × 104 cells were added to each well of effector cells and incubated at 37°C for 18 h. Cells were washed with PBS, lysed with 30 μl/well of 1× reporter lysis buffer (luciferase assay system; Promega), and then frozen at −80°C for at least 2 h. Finally, the extracts were thawed, and 25 μl was mixed with 100 μl of luciferase substrate (Promega). Light output was measured with a Luminoskan Ascent instrument (Thermo Labsystems). Plasmids pCAGGS/MCS, pT7EMCLuc, pCAGT7, pPEP98, pPEP99, pPEP100, and pPEP101 were gifts of P. G. Spear (39, 41).
Complementation assay.
The complementation assay was performed essentially as described for complementation of gD-negative virus (16, 18, 38). Vero cells in six-well plates were first transfected (using the Lipofectamine 2000 protocol) with 1.0 μg of plasmid expressing either WT gB or mutant gB or with an empty vector control plasmid. Transfected cells were infected with a gB-negative mutant, HSV-1(KOS)K082 (12), that had been grown in the gB-complementing cell line VB38 (22). Virus titrations were performed on VB38 cells.
Inhibition of viral entry by soluble gB730t and mutants.
Soluble gB730t was used to block HSV entry into cells (9). Briefly, cells were seeded in 96-well plates and incubated overnight at 37°C. Serial dilutions of soluble glycoproteins diluted in DMEM containing 5% FBS and 30 mM HEPES were allowed to bind to cells at 4°C for 30 min, and then HSV-1 KOS/tk12 was added at a multiplicity of infection (MOI) of 10. The cell-virus mixture was incubated at 4°C for 30 min to allow virus attachment, and then the temperature was shifted to 37°C to allow synchronous infection. Cells were lysed 6 h later by the addition of PBS containing 1% NP-40. β-Galactosidase activity was determined by the addition of substrate (chlorophenol red-β-d-galactopyranoside; Roche) to an aliquot of each extract, and absorbance was measured at 570 nm, using a microtiter plate reader. The background (extracts of uninfected cells) was subtracted, and values were expressed as % of control (infected cells with no protein added).
Liposome flotation assay.
Conditions for liposome flotation experiments were adapted from previously described methods (20, 35). Liposomes were purchased from Encapsula Nanosciences (Nashville, TN) at a size of 400 nm, containing either a 1.7:1 molar ratio of soy-phosphatidylcholine to cholesterol (PC/C) or phosphatidylcholine only (PC). Liposomes were stored at 4°C and used for up to 1 month per the manufacturer's instructions. Purified soluble proteins (1 μg), liposomes (25 μg), and 15 μl of 200 mM sodium citrate were combined, and the final reaction volume was adjusted to 50 μl with PBS. Alternatively, purified proteins were incubated with MAbs (10 μg) on ice for 1 h prior to the addition of liposomes. Protein-liposome mixtures were then incubated at 37°C for 1 h. To eliminate unwanted electrostatic protein-lipid associations, mixtures were incubated with 1 M KCl for 15 min at 37°C (20) before being loaded onto a sucrose gradient. Mixtures were adjusted to 40% sucrose in a final volume of 500 μl and overlaid with 4 ml of 25% sucrose-PBS and 500 μl of 5% sucrose-PBS. The gradients were centrifuged for 3 h in a Beckman SW55Ti rotor at 246,000 × g and 4°C. Seven equal fractions (approximately 700 μl each) were collected, starting from the top of the gradient. For dot blots, 200 μl of each fraction was spotted onto a nitrocellulose filter by use of a vacuum manifold (Schleicher and Schuell). Western blots were performed using 25 μl/well of each fraction that had been subjected to SDS-PAGE. Blots were then probed using PAbs raised against purified gB (R68 and R69), gH1/gL1 (R137), gH2/gL2 (R176), or gD (R8), incubated with horseradish peroxidase-conjugated goat anti-rabbit antibodies, and visualized using enhanced chemiluminescence (Amersham). When anti-gB MAbs were included in the assay, blots were probed with biotin-conjugated R68 and then incubated with streptavidin-horseradish peroxidase conjugate (GE Healthcare) to avoid background caused by potential antibody cross-reactivity.
RESULTS
Site-directed mutagenesis of amino acids in the two “fusion loops” of gB. (i) Residues that were mutated.
HSV-1 gB is a 904-amino-acid protein whose ectodomain consists of the first 773 amino acids. The crystal structure of a truncated form of HSV gB (residues 31 to 730) revealed that it is a large trimeric spike (32). The structural model of gB has been divided into five domains (Fig. 1A). The two putative fusion loops are at the bottom of domain I. Of their 15 amino acids (Fig. 1A), 6 are charged and 8 are hydrophobic or uncharged. We previously mutated four of the hydrophobic residues and found that three are critical for gB function in cell fusion (W174, Y179, and A261) (29). The fourth, F262, was mutated to either leucine (F262L) or aspartic acid (F262D). Due to a sequencing error, the F262D change was incorrectly reported (29, 30). We have now made the proper mutant, and this report examines the bona fide F262D mutation. In this study, we report on the contributions of the other residues that make up the fusion loops in gB structural domain I.
Interestingly, the charged amino acids of the fusion loops surround key hydrophobic residues, i.e., those shown previously to be critical for fusion (Fig. 1B). We hypothesized that most of the uncharged residues would likely share properties with the first set we examined. The potential role of the charged residues was less clear, but their proximity to the other amino acids suggested that one or more might may also play an important role in gB structure and function.
To test these ideas, we prepared single amino acid substitutions of all of the remaining residues and tested their biochemical and functional properties as previously reported for W174, Y179, and A261. The three additional hydrophobic residues (F175, G176, and Y265) were changed to charged amino acids, and the six charged residues (H177, R178, R258, E260, H263, and R264) were each mutated to alanine (Table 1).
TABLE 1.
Properties of full-length gB fusion loop mutantsa
| Mutant | Surface expression (% WT) | Reactivity with MAb
|
Fusion (% WT) | Complementation (% WT) | ||
|---|---|---|---|---|---|---|
| DL16 | SS145 | SS55 | ||||
| WT | 100 | + | + | + | 100 | 100 |
| W174Yb | 123 | + | + | + | 46 | 5 |
| W174Kb | 55 | − | − | − | 0 | ND |
| W174Rb | 146 | + | + | + | 9 | 0 |
| F175K | 110 | + | + | + | 26 | 0 |
| G176K | 92 | + | + | + | 0 | 0 |
| Y179Sb | 142 | + | + | + | 0 | 0 |
| Y179Kb | 100 | + | + | + | 0 | 0 |
| V259Rb | 22 | − | − | − | 4 | ND |
| A261Wb | 108 | + | + | + | 18 | 0 |
| A261Db | 125 | + | + | + | 9 | 0 |
| F262Lb | 105 | + | + | + | 68 | 27 |
| F262D | 30 | + | + | + | 0 | 0 |
| Y265R | 82 | ND | ND | ND | 93 | 44 |
| H177A | 84 | + | + | + | 63 | 46 |
| R178A | 68 | ND | ND | ND | 83 | 30 |
| R258A | 91 | ND | ND | ND | 81 | 72 |
| E260A | 85 | ND | ND | ND | 74 | 72 |
| H263A | 95 | + | + | + | 51 | 12 |
| R264A | 64 | + | + | + | 31 | 19 |
Data shown are averages for at least three independent experiments. Mutants selected for cloning into baculovirus in order to express and purify soluble proteins are shown in bold. ND, not determined.
Data are for mutants reported in our previous study (27).
(ii) Examination of mutant gB proteins with MAbs and for cell surface expression.
To test the new set of mutant proteins for proper folding, we used three MAbs specific to different conformational epitopes (8, 29). SS55 has strong virus-neutralizing activity and recognizes an epitope that includes structural domains I and V (Fig. 1A). Thus, we predicted that its binding to gB may be affected by local structural changes caused by the mutations. SS145 recognizes an epitope in domain IV, the crown of gB (Fig. 1A). We used the trimer-specific MAb DL16 to test the mutants for proper oligomerization. An anti-c-myc MAb was used as a negative control. All new mutants were immunoprecipitated by DL16, SS55, and SS145, and none were immunoprecipitated by the control antibody (Table 1). These data indicate that the mutated proteins retained proper folding of epitopes in several domains. By CELISA, all mutants (except the F262D mutant) were expressed on the surfaces of transfected cells at WT or nearly WT levels (Fig. 2A; Table 1). Although the F262D mutant was expressed on the cell surface at only 30% of WT gB levels, the previously characterized F262L mutant was expressed at 100% of the WT level (29). Taken together with our previous data (Table 1), mutation of each of the residues in both loops (with the exception of V259) yielded properly folded proteins expressed on the cell surface.
FIG. 2.

Characterization of gB fusion loop mutants. (A) Protein surface expression detected by CELISA. Transfected CHO-K1 cells were fixed with 3% paraformaldehyde and then incubated with the anti-gB PAb R69 and goat anti-mouse-horseradish peroxidase. Cells transfected with empty vector DNA were used as a negative control, and this value was subtracted from the other experimental samples. Percent WT was calculated as follows: (sample absorbance/WT absorbance) × 100. (B) Quantitative cell-cell fusion assay. Target CHOK1 cells (expressing the luciferase protein and the HSV receptor HVEM) were cocultivated with effector CHO cells (expressing T7 polymerase, gD, gH, and gL plus either WT gB, mutant gB, or empty vector DNA) and tested for light production 20 h later. Percent WT was calculated as follows: (relative light units of test sample/relative light units of WT) × 100. (C) gB-null virus complementation. Vero cells were transfected with plasmids encoding WT or mutant gB and then infected with gB-null HSV that had been complemented phenotypically with WT gB to allow for entry. Cell lysates containing progeny virions complemented with gB were harvested and assayed for virus entry into gB-expressing VB38 cells. After 2 days of incubation at 37°C, cells were stained and plaques were counted. Percent WT was calculated as follows: (sample titer/WT titer) × 100. For each of the assays (CELISA and fusion and complementation assays), the data are shown as averages for at least three experiments. Standard deviations are shown as lines above each bar. In each graph, gray bars indicate hydrophobic residues, white bars indicate hydrophilic/charged residues, and black bars indicate the WT control. Mutants that were selected for expression as soluble proteins are indicated with gray boxes.
(iii) Testing of mutants for function with a cell-cell fusion assay.
Effector CHO-K1 cells were transfected with plasmids encoding T7 RNA polymerase, gD, gH, gL, and either WT gB or one of the gB mutants. Target cells that stably express the gD receptor HVEM (CHO-HVEM12) were transfected with a plasmid carrying the open reading frame for luciferase under the control of the T7 promoter. Target and effector cells were cocultivated for 18 h and assayed for luciferase activity as a measure of cell-cell fusion. Effector cells that substituted empty vector for WT gB were used as a negative control.
We previously (29) showed that the W174R, Y179S, Y179K, A261D, and A261W mutants were nonfunctional in the fusion assay and that the W174Y mutant was impaired (Table 1). Among the new mutants, both the G176K and F262D mutants were nonfunctional in fusion (Fig. 2B), although the low surface expression of the F262D mutant (30% that of the WT) probably indicates more of a transport/folding problem. The F175K and R264A mutants were impaired, functioning at levels of 26% and 31% of WT gB fusion, respectively. A third mutant, the H263A mutant, retained slightly more activity and functioned at 51% of the WT level. Thus, two additional uncharged (or hydrophobic) and two charged residues in the fusion loops are important for gB function. The remaining mutants, representing four charged residues (H177, R178, R258, and E260) as well as one hydrophobic amino acid (Y265), functioned between 60% and 100% of the WT level of fusion. We suggest that these five residues play limited individual roles in the function of gB in fusion. To summarize our findings, mutation of nonpolar or hydrophobic amino acids W174, F175, G176, Y179, and A261 as well as of two polar residues, H263 and R264, had the greatest effect on the cell-cell fusion activity of gB. Mutagenesis studies of the fusion loops of gp64 (34), VSV G (51), and EBV gB (5) revealed similar phenotypes for hydrophobic residues in those class III proteins.
(iv) Complementation of HSV gB null virus with fusion loop mutants.
HSV glycoprotein-mediated cell-cell fusion is used as a surrogate for virus-cell fusion. To test the function of all of the mutants in the context of the virus, we studied them in a null virus complementation assay. We omitted the W174K and V259R mutants because they were misfolded but included the F262D mutant because it did exhibit some cell surface expression (Table 1). To prepare virus for this assay, HSV-1(K082) (11), a strain lacking the open reading frame for gB, was propagated in VB38 cells (22). VB38 cells were engineered to express full-length WT gB under the control of its own HSV promoter (22). Vero cells were transiently transfected with plasmids for either WT gB or one of the mutant gB constructs and then infected 24 h later with complemented HSV-1(K082). Progeny virions were harvested at 24 h postinfection. The infectivity of these particles, i.e., their ability to enter cells, was determined by plaque assay on VB38 cells and represents HSV-1(K082) complemented by plasmid-derived gB. The titer of each stock of complemented virus on VB38 cells therefore measures the ability of the gB mutants to function in entry.
We considered a mutant to be severely impaired in the ability to function in virus entry when the titer on VB38 cells was 20% or less of that for HSV-1(K082) complemented with WT gB. Six mutants (F262L, Y265R, H177A, R178A, R258A, and E260A) were above 20% of the WT level of complementation (Fig. 2C). Ten mutants (W174Y, W174R, F175K, G176K, Y179S, A261W, A261D, F262D, H263A, and R264A) were severely impaired or nonfunctional. The data for the two functional assays were in good agreement, i.e., mutants that were impaired for cell-cell fusion were also impaired in complementation (Table 1). Interestingly, three of these important residues (F175, H263, and R264) cluster together and border the hydrophobic ridge (Fig. 1B) that was previously shown to be critical for gB function (29).
Cloning and expression of the ectodomains of four gB mutants as baculovirus recombinants.
Having identified important residues in the two fusion loops of gB, we wanted to know more about their properties as proteins to better understand the functional mechanism(s) interrupted by the mutation. Therefore, we chose four mutants (W174R, Y179S, H263A, and R264A) to clone into a baculovirus expression vector, using codons for residues 31 to 730 (the mature gB ectodomain) of each mutant. The recombinant viruses were used to infect Sf9 cells for protein production and purification. As done before for the crystallization of WT gB730t (32), each protein was purified by affinity chromatography using the trimer-specific anti-gB MAb DL16 and examined by SDS-PAGE and Western blotting (Fig. 3A). Under “native” (nonreducing and nonboiling) conditions, most of each protein migrated slowly (200 kDa), and we interpret this form to be the trimer (T) (8, 32). A smaller (∼100 kDa) band, interpreted to be the monomer (M), was also observed by both silver staining and Western blotting with the anti-gB PAb R69. Interestingly, we noted some minor differences between the W174R, Y179S, and H263A mutants and WT gB in the relative amounts of the slower-migrating forms. Under denaturing conditions, each protein migrated more rapidly, and all were located at the same position on the gel, at about 80 to 85 kDa (Fig. 3A).
FIG. 3.

Truncated forms of gB mutant proteins are expressed and folded correctly. Four gB mutants (Y179S, W174R, H263A, and R264A) were cloned and expressed in a baculovirus expression system as secreted forms truncated after amino acid 730. (A) Purified proteins were extracted and separated by SDS-PAGE under “native” (N) or denaturing (D) conditions (as described in Materials and Methods) and then visualized either by silver staining or by detection with the PAb R69 via Western blotting. (B) The gB mutant proteins were immunoprecipitated using MAbs to conformational epitopes (DL16, SS55, and SS145). The anti-myc MAb was used as a negative control. PAb R69 was used for detection of gB via Western blotting, and construct designations are indicated to the side of each blot. Molecular size markers are indicated to the side of each blot or gel, in kilodaltons. T, trimer; M, monomer.
The purified glycoproteins were tested for proper folding by immunoprecipitation, using MAbs DL16, SS55, and SS145 (Fig. 3B). The W174R, Y179S, and H263A mutants reacted with each of these MAbs and were not recognized by the anti-c-myc control antibody. Thus, these three mutant glycoproteins are antigenically correct for three distinct epitopes (8). In contrast, the R264A mutant reacted with MAbs DL16 and SS145 as well as WT gB did, but its reactivity with SS55 was reduced compared with the background reactivity of the control antibody for c-myc. In further tests, the R264A mutant was readily immunoprecipitated with MAbs C226 and SS10 (data not shown), neutralizing MAbs that react with pseudo-continuous epitopes (8). These results indicate that while at least four epitopes remain intact in the soluble R264A mutant (those for C226, DL16, SS10, and SS145), the SS55 epitope may have been altered specifically by the mutation at residue 264. In previous studies, this MAb mapped to structural domains I (which includes R264) and II (8).
Mutant gB proteins block virus entry but are impaired in binding to cell surfaces.
We previously showed that HSV gB binds to cell surfaces independently of HSPGs and blocks virus entry (9). These observations provided evidence for a cellular entry receptor for gB, and recent studies suggested that PILRα may serve this function in some cell types (47). It is possible that domain I of gB binds a proteinaceous cell surface receptor and that fusion loop mutants are unable to function in fusion because they fail to recognize this receptor. If this is true, then the soluble fusion loop mutants should be impaired in the ability to block virus entry into cells. We tested this by using Gro2C cells because they lack HSPGs on the cell surface, thus limiting the binding of gB cell surface proteoglycans. Although Gro2C cells contain chondroitin sulfate, previous work has shown that it is not directly involved in gB attachment to cells (7). To test our hypothesis, decreasing equimolar concentrations of soluble WT gB730t or mutant gB protein were added to Gro2C cells for 30 min at 4°C. Next, HSV-1 KOS/tk12 (which carries the lacZ gene under the control of the IC4 promoter) was added for 30 min at 4°C, and then the virus-cell mixture was shifted to 37°C for 6 h. Virus entry was quantified by β-galactosidase expression. The results for gB blocking were essentially the same as those previously reported for soluble WT gB (9). gB W174R and R264A mutant proteins blocked virus entry as well as WT gB did (50% inhibition at 1 μM gB). Y179S and H263A mutant proteins also blocked virus entry but were not as efficient as WT gB; for these mutants, a concentration of approximately 2 μM gB was needed to achieve 50% inhibition (Fig. 4A). These data suggest that the fusion loops are not critical for binding of gB to a cellular receptor.
FIG. 4.

Fusion loop mutant proteins block entry like WT gB does, but they are impaired in binding to Gro2C cells. (A) Soluble WT and mutant gB proteins inhibit HSV entry into Gro2C cells. Decreasing equimolar concentrations of gB proteins were incubated with cells in an ELISA plate for 30 min at 4°C. HSV was then added to cells and allowed to attach for 30 min, after which the temperature was raised to 37°C and incubation continued for 6 h. Cells were lysed, and β-galactosidase activity was assayed. Error bars show standard deviations. (B) WT gB730t and four mutant gB proteins were tested for binding to Gro2C cells by CELISA in parallel to the experiment shown in panel A. Decreasing equimolar concentrations of gB were added to cells in an ELISA plate for 1 h at 4°C. Wells were washed with PBS and fixed with 3% paraformaldehyde. Cell-bound gB was detected using the PAb R69.
Because the mutant proteins blocked virus entry, we fully expected them to be able to bind to cell surfaces as efficiently as WT gB730t did; however, this was not the case. Increasing equimolar concentrations of WT or mutant gB were added to monolayers of Gro2C cells for 1 h at 4°C. CELISA was used to detect gB binding to the cell surface (Fig. 4B). Unexpectedly, all of the mutant proteins were impaired (albeit to different levels) in the ability to bind to Gro2C cells. At a 5 μM concentration, the W174R mutant bound cells at 62%, the R264A mutant bound at 51%, the H263A mutant bound at 31%, and the Y179S mutant bound at 22% of WT gB730t levels (Fig. 4B; Table 2). These results suggest that gB binds to two different non-HSPG moieties on the Gro2C cell surface. In support of this theory, the two mutants that were the most impaired in entry were the same ones that were most impaired for cell binding. One possible binding partner is the putative cellular receptor involved in entry. We propose that the second binding partner involves lipids and that the effect of the mutations in the fusion loops is most pronounced in studies of gB binding to cells.
TABLE 2.
Properties of soluble, purified gB fusion loop mutant proteins
gB, but not gD or gH/gL, binds to liposomes.
Our laboratory previously showed that HSV virions bind to liposomes when incubated in the presence of a soluble form of the gD receptor, HVEM, at pH 5.0 (55). Second, gB730t, but not soluble forms of gD or gC, can bind to cholesterol-rich lipid rafts (10). Together, these data suggest that HSV may interact with lipids through gB. In support of this hypothesis, mutations in the fusion loops of two class II fusion proteins from flavivirus (2) and Semliki Forest virus (35) impair the ability of these proteins to associate with liposomes. Therefore, we theorized that if gB inserts into target membranes via the putative fusion loops, gB730t should associate and float with liposomes, while the soluble forms of the fusion loop mutants should be impaired in this association.
To test this hypothesis, purified soluble gB, gD, and gH/gL were each incubated with two different compositions of liposomes, i.e., PC only or PC/C, at 37°C for 1 h. We then added KCl to eliminate any nonspecific electrostatic interactions between the proteins and liposomes (20). Each protein-liposome mixture was then adjusted to 40% sucrose, layered beneath a sucrose step gradient (35), and centrifuged for 3 h to allow the liposomes and any associated proteins to float to the top of the gradient. Under these conditions, proteins that fail to bind liposomes should remain at the bottom of the gradient. To verify our experimental conditions, we used liposomes containing a fluorescent dye and found that the majority of liposomes were in the first (top) fraction (data not shown).
Seven equal fractions were collected, starting from the top of each gradient, and were analyzed by immunodot blotting, using a PAb specific for each viral protein (Fig. 5A). Of the four glycoproteins essential for HSV entry, only gB730t bound and cofloated with liposomes (Fig. 5A, fraction 1). When liposomes were omitted from the reaction mixture, gB730t did not float (Fig. 5A, last lane). More gB was detected at the top of the gradient when the liposomes contained cholesterol (PC/C) than when the liposomes contained PC only. Western blot analysis of the gradient fractions showed that the majority of gB was at the bottom of the gradient for PC-only liposomes but at the top for cholesterol-containing PC/C liposomes (Fig. 5B). Thus, gB binds to cholesterol-enriched liposomes in a specific and nonsuperficial fashion, i.e., in the presence of high salt. These results are consistent with previous studies showing the specific association of gB730t with cholesterol-rich rafts and may help to explain the cholesterol dependence of HSV infection (10). Unlike the binding of HSV virions to liposomes (55), the association of gB with liposomes did not require low pH. The presence or absence of cholesterol in the liposomes had no effect on the distribution of gD and gH/gL in the gradient, since neither protein cofloated with either type of liposome. In fact, we found that neither truncated forms of gH1/gL1 nor gH2/gL2 cofloated with liposomes and, as such, remained at the bottom of the gradient (Fig. 5A, fraction 7).
FIG. 5.

Binding of soluble HSV glycoproteins to liposomes. Purified soluble glycoproteins were incubated with (PC/C and PC) or without (no lip) liposomes for 1 h at 37°C. Samples were then adjusted to 1 M KCl, incubated for an additional 15 min, and then layered beneath a discontinuous 5 to 40% sucrose gradient. (A) Gradients were centrifuged for 3 h, fractionated, and analyzed by dot blotting. Three separate dot blots are shown, separated by black lines, and the top and bottom fractions of the gradients are indicated. Fraction numbers are shown to the right. An arrow indicates the direction of flotation, from the bottom of the gradient (B) to the top (T). Blots were probed with either the anti-gB PAb R69, anti-gH1/gL1 PAb R137, anti-gH2/gL2 PAb R176, or anti-gD PAb R8. Two separate film exposures are shown for gB to illustrate the difference between protein association with PC/C- and PC-only-containing liposomes. (B) Fractions 1, 4, and 7 (representing the top, middle, and bottom of each gradient) were also analyzed by Western blotting and probed with the PAb R69. Molecular size markers are indicated to the left, in kilodaltons. PC, phosphatidylcholine; PC/C, phosphatidylcholine-cholesterol.
gB proteins with mutations in the fusion loops are impaired in liposome binding.
We hypothesized that if gB inserts into target membranes via the putative fusion loops, then soluble forms of the gB fusion loop mutants should be impaired in liposome binding. Indeed, three of the four mutants (W174R, Y179S, and R264A) were unable to associate and cofloat with liposomes (Fig. 6). These data correlate with the failure of each mutant's full-length counterpart to participate in cell-cell fusion (Table 1). Interestingly, a portion of the H263A mutant protein was detected in the top fraction by dot blot analysis (Fig. 6A). However, the bulk of the protein sample remained in the bottom fraction, as best demonstrated by Western blotting (Fig. 6B). The full-length version of this mutant protein retained function in the cell-cell fusion assay (51% of the WT level) (Fig. 2B), so it follows that the soluble form might retain some ability to bind lipids.
FIG. 6.
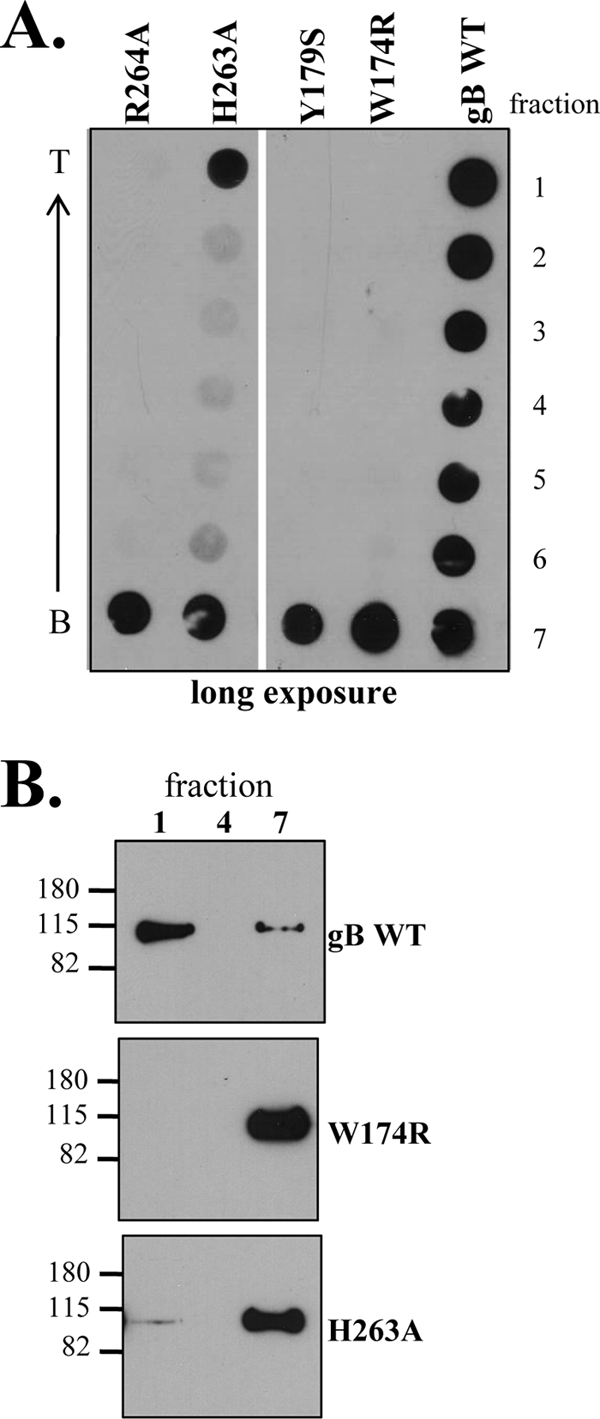
Mutations in the putative gB fusion loops impair liposome binding. The flotation assay was performed as described in the legend to Fig. 5, using liposomes containing PC/C. Both the dot blot (A) and Western blot (B) were probed with the PAb R69. Molecular size markers are indicated to the left of the Western blot, in kilodaltons.
gB-liposome binding can be blocked by MAbs.
We next questioned whether or not MAbs whose epitopes have been mapped in close proximity to the fusion loops would block gB-liposome binding. The putative gB fusion loops are located in structural domain I and are part of functional region 1 (FR1), which includes domains I and V and forms the base of the gB trimer (Fig. 7A) (8, 32). Bender et al. (8) epitope mapped a panel of MAbs to gB, and we chose a MAb series that represented each FR and the domains that comprise them. Among the 11 MAbs tested here, SS55, SS120, and SS144 effectively blocked the association of gB with liposomes (Fig. 7B and C; Table 3). These three MAbs have epitopes in FR1 (SS55 epitope in structural domain I and SS120 and SS144 epitopes in domain V) (Fig. 7A). However, SS106 and SS121, two other MAbs with epitopes in FR1, did not block binding of gB to liposomes (Table 3). These results suggest that only a subdomain of FR1 is involved in liposome association. MAbs tested with epitopes in FR2 and FR3 (C226, SS10, SS68, SS69, and DL21) were also unable to prevent gB-liposome binding. Thus, the MAb blocking data support the data for gB mutants, i.e., that liposome association of gB occurs via FR1, which contains the fusion loops. Furthermore, we could find no correlation between MAbs that were previously shown to neutralize virus infection with ones that block binding of gB to liposomes (Table 3).
FIG. 7.

gB-liposome association is inhibited by anti-gB MAbs. (A) Ribbon diagram of HSV protomer, colored by secondary structure succession as described in the legend to Fig. 1A. Functional regions (FR) of gB are outlined in black and are indicated along with representative MAbs that helped to define these regions. Soluble gB730t was incubated with MAbs on ice for 1 h prior to the addition of PC/C liposomes. The remainder of the flotation assay was done as described in the legend to Fig. 5. Both the dot blot (B) and Western blot (C) were probed with R68-biotin to eliminate potential cross-reactivity problems between the MAbs and secondary antibodies typically used for detection. Molecular size markers are indicated to the left of the Western blot, in kilodaltons. (D) Deletion of domain V inhibits gB-liposome binding. The dot blot was performed as described in the legend to Fig. 5 and probed with the PAb R69.
TABLE 3.
Properties of MAbs tested in functional assaysd
| MAb | Functional regiona | Epitope residuesa | Structural domaina,b | Epitope typea | Neutralizes virusa | Blocks gB-cell bindingc | Blocks gB-liposome binding |
|---|---|---|---|---|---|---|---|
| SS55 | 1 | 98-472 | I, II | Discontinuous | + | + | + |
| SS106 | 1 | 697-725 | V | Continuous | + | ND | − |
| SS120 | 1 | 697-725 | V | Pseudo-continuous | +e | ND | + |
| SS121 | 1 | 697-725 | V | Continuous | − | ND | − |
| SS144 | 1 | 697-725 | V | Pseudo-continuous | + | ND | + |
| C226 | 2 | 234-472 | II | Pseudo-continuous | + | − | − |
| SS10 | 3 | 640-670 | IV | Pseudo-continuous | + | + | − |
| SS68 | 3 | ND | ND | Pseudo-continuous | + | ND | − |
| SS69 | 3 | ND | ND | Pseudo-continuous | + | ND | − |
| DL21 | ND | ND | ND | Discontinuous | − | − | − |
To further localize the liposome binding activity of gB, we used the truncation mutant gB670t, which removes the entire gB structural domain V and was previously characterized by Bender et al. (8). Since two MAbs that map to domain V (SS120 and SS144) blocked gB-liposome association, we postulated that liposome association not only might involve the fusion loops but also might be influenced by residues 670 to 730. Indeed, when tested in the flotation assay, gB670t was unable to cofloat with liposomes (Fig. 7D). However, the loss of liposome association could also be because gB670t is no longer trimeric (6); it may be that cooperativity among protomers is important for liposome binding.
DISCUSSION
The crystal structure of gB730t shows that gB has the characteristic features of a class III fusion protein. Its two putative fusion loops, found at the base of the protein, are analogous to those of VSV G and gp64, two other members of this class. Our previous and present mutagenesis studies support this concept, as single amino acid mutations of many of the hydrophobic residues in both loops had profound effects on the ability of gB to function in fusion and gB-null virus complementation assays. Two charged residues, H263 and R264, are also important for gB function and inspection of the crystal structure shows that these residues support a ridge formed by key hydrophobic and nonpolar amino acids. In a surface representation model (Fig. 1B), the loops come together to form a scaffold. Although the hydrophobic residues of HSV gB fusion loops do not appear to be able to insert deeply into a target membrane (32), our liposome studies show that the protein does have the ability to accomplish this feat. We propose that the hydrophilic residues on either side of the hydrophobic ridge help to stabilize insertion of gB into cholesterol-enriched membranes. In support of this hypothesis, the gB mutant protein R264A was unable to associate with liposomes and H263A was impaired in this activity. Thus, we propose that during virus entry the hydrophobic ridge of the gB fusion structure inserts into the target membrane and that charged residues in the loops, primarily H263 and R264, interact with phospholipid head groups but do not penetrate the hydrophobic core. We also propose that this interaction likely guides and stabilizes insertion of the ridge into the hydrophobic core of the bilayer of the cell.
Completion of mutagenesis studies.
Only a few of the mutants in the fusion loops showed defects in transport to the cell surface, and none showed major defects (as full-length proteins) when examined for specific conformationally dependent epitopes. This overall structural integrity allowed us to examine the effects of the individual mutations on protein function without the need to invoke changes to overall protein conformation. When considered as a complete set, the majority of impaired proteins had mutations of hydrophobic residues. A similar story emerges for the other class III fusion proteins, baculovirus gp64 and VSV G, supporting the concept that these hydrophobic loops do indeed function as fusion loops and form a tripartite fusion structure functionally homologous to the class II fusion loops (31, 42). In gp64, for example, F153 is part of an exposed hydrophobic patch that when mutated to aspartic acid or threonine causes the protein to lose fusion activity (34). The adverse effect of the mutations of gB H263 and R264 was more of a surprise, although something analogous was seen for gp64. In that case, fusion activity was abolished by mutations of a serine and glutamic acid, but not by a change of a histidine in the vicinity of the fusion loops (34).
When we examined our set of proteins with two different functional assays, we found a more profound impairment of mutants in null virus complementation than in cell-cell fusion. However, both assays were in agreement over which mutants displayed an altered phenotype compared to WT gB. This observation could be due, at least in part, to differences in the way the two assays are set up and in what each assay measures. The fusion assay relies on transfection and overexpression of the mutant proteins; high levels of expression might mask some of the impairment that is more obvious in the complementation assays. In the latter case, the amount of mutant gB is governed by how much is in each virion and likely results in presentation of less gB to the target cell than the fusion assay.
Properties of recombinant baculovirus proteins.
In selecting mutants to express in the baculovirus system, we did so according to their hydrophobicity, charge and phenotype in the functional assays. Three of the mutant proteins (W174R, Y179S, and H263A) had different relative amounts of the slower migrating protein forms compared to WT gB (Fig. 3A). Although our data for the full-length versions of these proteins suggested that no major changes to protein structure had occurred (Table 1) (29), changes to the fusion loops could have a more profound effect on the structure of the ectodomain, especially since the changes were spatially close to the truncation site (residue 730). In the full-length protein, any subsequent effect that a change to the fusion loops might have on the structure of domain I might be compensated for by the presence of the downstream hydrophobic amino acids (residues 730 to 773), to the transmembrane anchor, or to insertion of gB into a lipid bilayer. It is interesting that two of the three soluble mutant proteins which show differences by native electrophoresis (H263A and Y179S) were less effective at blocking HSV entry and were also the most impaired in binding to Gro2C cells (Fig. 4; Table 2).
gB binds to two different non-HSPG moieties on the cell surface.
The existence of a cellular receptor(s) for gB has been hypothesized (9), and indeed, one such possible protein, PILRα, has been characterized (47). The observation that soluble gB blocks virus entry into cells lacking HSPG suggests that gB binds a cell receptor as an essential step in the entry process (9). Since the soluble forms of the fusion loop mutants also blocked entry (W174R, R264A at WT levels; Y179S, H263A at lower levels) (Fig. 4A), we hypothesize that these mutations do not interfere with binding to this putative entry receptor. However, since the soluble fusion loop mutant proteins do not bind to cells with the same efficiency as WT gB, we conclude that there is a second non-HSPG moeity to which gB binds that the fusion loop mutants are defective in binding. This binding partner is most likely lipids, as our data showed that these mutations interfere with gB-liposome binding in vitro. Fusion loop mutants within the baculovirus fusion protein gp64 also exhibited decreased binding to cells (56).
Wild-type gB-cell binding is blocked by MAbs SS10 (FR3, structural domain IV), SS55, and SS118 (FR1, structural domains I and II) (8, 9). Interestingly, SS55 and SS118 have epitopes in the same FR as the fusion loops (FR1) (8). These data suggest that both non-HSPG moieties can be blocked by MAbs: MAbs that map to FR3 block receptor-gB association while MAbs that map to FR1, which contain the fusion loops, block the association of gB with lipids. Antibodies directed against the N terminus of gp64, which encompasses the fusion loops, also showed strong neutralizing activity and inhibited binding of baculovirus to Sf9 cells (34). Our present work strengthens the concept of individual functional regions in HSV gB.
gB associates with cholesterol-containing liposomes.
HSV virions bind to liposomes under certain conditions (55). We had hypothesized that HSV may interact with lipids through gB. Here, we found that gB binds to cholesterol-enriched liposomes in a specific and nonsuperficial fashion, i.e., in the presence of high salt. A study by Gianni et al. (26) found that gB did not associate with PC-only liposomes, which fits with our data as we found that the majority of soluble gB remained at the bottom of the gradient when incubated with PC-only liposomes (Fig. 5). Unfortunately, they did not report experiments using PC/C liposomes. Our results are consistent with previous studies showing the specific association of gB730t with cholesterol-rich rafts and may also help to explain the cholesterol-dependence of HSV infection (10). We had hypothesized that cholesterol-containing lipid rafts might contain the putative gB entry receptor (10). Here we showed that gB can associate with cholesterol-containing liposomes, suggesting that the cholesterol dependence of gB is not necessarily because of a protein receptor in rafts, but because cholesterol itself enhances insertion of the fusion loops. In Semliki Forest virus fusion protein E1, a class II fusion protein that inserts preferentially into membranes enriched in cholesterol and sphingolipid (1), a point mutation in the loop adjacent to the fusion loop confers increased cholesterol independence (15, 53). It is theorized that this loop regulates a cholesterol-dependent interaction of the E1 fusion loop with target membranes.
The binding of gB to liposomes is specific and involves the fusion loops.
We hypothesized that if gB inserts into a target membrane via its fusion loops, then mutation of the fusion loops should impair liposome association. Soluble fusion loop mutants W174R, Y179S, and R264A were unable to bind to liposomes, and these proteins remained at the bottom of the gradient in our flotation assay (Fig. 6A). The H263A mutant retained some liposome binding capability, but the majority of protein did not cofloat with liposomes but rather was found at the bottom of the gradient (Fig. 6B). The data for the H263A mutant were not completely surprising, as it also retaining some cell-cell fusion activity (50% levels of WT gB fusion). The liposome flotation data constitute powerful evidence that gB interacts with liposomes via its fusion loops and are the first demonstration of insertion of class III fusion loops into target membranes.
Does gB associate specifically with liposomes? Our MAb data suggest that the answer is yes. Coflotation of gB with liposomes was inhibited by a subset of gB-specific MAbs, in particular those MAbs with epitopes that map to FR1. This region is comprised of structural domain I and the αF helix of structural domain V (8). Since structural domain I contains the fusion loops, it is logical that neutralizing MAbs to this domain interfere with virus-cell fusion. Within structural domain V, it is possible that the αF helix works by maintaining domain I in close contact with the gB core and in this conformation keeps the fusion loops hidden. MAbs that map to αF may alter this conformation, thereby accounting for how these MAbs neutralize virus.
The specificity of gB-liposome binding was also demonstrated by the observation that neither gD nor gH/gL cofloated with liposomes, regardless of the presence or absence of cholesterol. Our finding correlates with data showing that neither gD nor gH/gL associates with lipid rafts during HSV entry (10). However, our data for gH/gL and liposomes are in contradiction to the observation of Gianni et al. (26) that soluble gH1/gL1 floated with liposomes. The reason for this discrepancy is not clear, although one possible explanation is that what the authors saw was a nonspecific electrostatic association of gH/gL with liposomes, as they did not include an incubation with high salt in their protocol.
Why do the conditions for gB-liposome binding differ from those needed for HSV-liposome binding?
HSV particles associate with liposomes when incubated with the ectodomain of HVEM, one of the gD receptors, at 37°C and low pH (55). It was theorized that the combination of HVEM-gD binding, mildly acidic pH and a physiological temperature provided coactivation signals for the fusion process, resulting in virion-liposome binding in vitro. However, gB-liposome binding does not require low pH (Fig. 5) nor physiological temperature (data not shown). It is possible that insertion of gB730t into target membranes is due to exposure of the fusion loops by deletion of the two hydrophobic regions (residues 731 to 773) upstream of the transmembrane domain. Furthermore, compared with the pre- and postfusion forms of VSV G (45), it is likely that gB730t is in a “triggered” (postfusion) form. By definition, virion gB is in the pre-fusion form, and we posit that additional activation signals such as low pH and receptor binding are required to expose the fusion loops for membrane insertion.
Binding an entire virion to lipids could also require more than just the intrinsic lipid binding ability provided by gB. In addition to gB, gD, a gD receptor, and gH/gL are all required to achieve membrane fusion and it is known that gD triggers gB and gH/gL to interact with each other during this process (3, 4). We suggest that the ability of gB730t, but not mutant forms with changes to the fusion loops, reflects a functional interaction between full-length gB and membranes during fusion. The fact the same mutants that fail in cell-cell fusion assays also fail in null-virus complementation supports this concept.
Our studies also suggest that soluble gH/gL cannot associate on its own with liposomes or with cells. Perhaps insertion of gB into target membranes coupled with its interaction with gH/gL brings the entire fusion machinery together to carry out fusion. The order of this process is not known but it will be interesting to see if mutations that affect gB-lipid interaction impact the association of gB-gH/gL, or gB with its putative receptor (9, 47), during virus entry.
Acknowledgments
Funding for this project was through NIH grants AI-076231 (R.J.E.), AI-05645 (R.J.E.), and AI-18289 (G.H.C.) from the National Institute of Allergy and Infectious Diseases. B.P.H. was supported by training grant AI-076234.
We are grateful to D. C. Johnson, S. Person, P. G. Spear, and F. Tufaro for providing reagents. We thank P. Bates and R. Doms for their help in optimizing the liposome assay, I. Baribaud for her suggestions regarding gH/gL purification, and D. Atanasiu and M. Ponce-de-Leon for testing the gB MAbs in a neutralization assay. We also thank E. E. Heldwein and all of our lab colleagues for helpful advice.
Footnotes
Published ahead of print on 15 April 2009.
REFERENCES
- 1.Ahn, A., D. L. Gibbons, and M. Kielian. 2002. The fusion peptide of Semliki Forest virus associates with sterol-rich membrane domains. J. Virol. 763267-3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allison, S. L., J. Schalich, K. Stiasny, C. W. Mandl, and F. X. Heinz. 2001. Mutational evidence for an internal fusion peptide in flavivirus envelope protein E. J. Virol. 754268-4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Atanasiu, D., J. C. Whitbeck, T. M. Cairns, B. Reilly, G. H. Cohen, and R. J. Eisenberg. 2007. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc. Natl. Acad. Sci. USA 10418718-18723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Avitabile, E., C. Forghieri, and G. Campadelli-Fiume. 2007. Complexes between herpes simplex virus glycoproteins gD, gB, and gH detected in cells by complementation of split enhanced green fluorescent protein. J. Virol. 8111532-11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Backovic, M., T. S. Jardetzky, and R. Longnecker. 2007. Hydrophobic residues that form putative fusion loops of Epstein-Barr virus glycoprotein B are critical for fusion activity. J. Virol. 819596-9600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Backovic, M., R. Longnecker, and T. S. Jardetzky. 2009. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc. Natl. Acad. Sci. USA 1062880-2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Banfield, B. W., Y. Leduc, L. Esford, K. Schubert, and F. Tufaro. 1995. Sequential isolation of proteoglycan synthesis mutants by using herpes simplex virus as a selective agent: evidence for a proteoglycan-independent virus entry pathway. J. Virol. 693290-3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bender, F. C., S. Saswati, E. E. Heldwein, M. Ponce de Leon, E. Bilman, H. Lou, J. C. Whitbeck, R. J. Eisenberg, and G. H. Cohen. 2007. Antigenic and mutational analyses of herpes simplex virus glycoprotein B reveal four functional regions. J. Virol. 813827-3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bender, F. C., J. C. Whitbeck, H. Lou, G. H. Cohen, and R. J. Eisenberg. 2005. Herpes simplex virus glycoprotein B binds to cell surfaces independently of heparan sulfate and blocks virus entry. J. Virol. 7911588-11597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bender, F. C., J. C. Whitbeck, M. Ponce de Leon, H. Lou, R. J. Eisenberg, and G. H. Cohen. 2003. Specific association of glycoprotein B with lipid rafts during herpes simplex virus entry. J. Virol. 779542-9552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cai, W. H., B. Gu, and S. Person. 1988. Role of glycoprotein B of herpes simplex virus type 1 in viral entry and cell fusion. J. Virol. 622596-2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cai, W. Z., S. Person, S. C. Warner, J. H. Zhou, and N. A. DeLuca. 1987. Linker-insertion nonsense and restriction-site deletion mutations of the gB glycoprotein gene of herpes simplex virus type 1. J. Virol. 61714-721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cairns, T. M., D. J. Landsburg, J. C. Whitbeck, R. J. Eisenberg, and G. H. Cohen. 2005. Contribution of cysteine residues to the structure and function of herpes simplex virus gH/gL. Virology 332550-562. [DOI] [PubMed] [Google Scholar]
- 14.Cairns, T. M., M. S. Shaner, Y. Zuo, M. Ponce-de-Leon, I. Baribaud, R. J. Eisenberg, G. H. Cohen, and J. C. Whitbeck. 2006. Epitope mapping of herpes simplex virus type 2 gH/gL defines distinct antigenic sites, including some associated with biological function. J. Virol. 802596-2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chatterjee, P. K., C. H. Eng, and M. Kielian. 2002. Novel mutations that control the sphingolipid and cholesterol dependence of the Semliki Forest virus fusion protein. J. Virol. 7612712-12722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiang, H. Y., G. H. Cohen, and R. J. Eisenberg. 1994. Identification of functional regions of herpes simplex virus glycoprotein gD by using linker-insertion mutagenesis. J. Virol. 682529-2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohen, G. H., V. J. Isola, J. Kuhns, P. W. Berman, and R. J. Eisenberg. 1986. Localization of discontinuous epitopes of herpes simplex virus glycoprotein D: use of a nondenaturing (“native” gel) system of polyacrylamide gel electrophoresis coupled with Western blotting. J. Virol. 60157-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Connolly, S. A., D. J. Landsburg, A. Carfi, D. C. Wiley, G. H. Cohen, and R. J. Eisenberg. 2003. Structure-based mutagenesis of herpes simplex virus glycoprotein D defines three critical regions at the gD-HveA/HVEM binding interface. J. Virol. 778127-8140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Connolly, S. A., D. J. Landsburg, A. Carfi, D. C. Wiley, R. J. Eisenberg, and G. H. Cohen. 2002. Structure-based analysis of the herpes simplex virus glycoprotein D binding site present on herpesvirus entry mediator HveA (HVEM). J. Virol. 7610894-10904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doms, R. W., A. Helenius, and J. White. 1985. Membrane fusion activity of the influenza virus hemagglutinin. The low pH-induced conformational change. J. Biol. Chem. 2602973-2981. [PubMed] [Google Scholar]
- 21.Evan, G. I., G. K. Lewis, G. Ramsay, and J. M. Bishop. 1985. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol. Cell. Biol. 53610-3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farnsworth, A., T. W. Wisner, M. Webb, R. Roller, G. Cohen, R. Eisenberg, and D. C. Johnson. 2007. Herpes simplex virus glycoproteins gB and gH function in fusion between the virion envelope and the outer nuclear membrane. Proc. Natl. Acad. Sci. 10410187-10192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galdiero, S., M. Vitiello, M. D'Isanto, A. Falanga, M. Cantisani, H. Browne, C. Pedone, and M. Galdiero. 2008. The identification and characterization of fusogenic domains in herpes virus glycoprotein B molecules. Chembiochem 9758-767. [DOI] [PubMed] [Google Scholar]
- 24.Galdiero, S., M. Vitiello, M. D'Isanto, A. Falanga, C. Collins, K. Raieta, C. Pedone, H. Browne, and M. Galdiero. 2006. Analysis of synthetic peptides from heptad-repeat domains of herpes simplex virus type 1 glycoproteins H and B. J. Gen. Virol. 871085-1097. [DOI] [PubMed] [Google Scholar]
- 25.Geraghty, R. J., C. R. Jogger, and P. G. Spear. 2000. Cellular expression of alphaherpesvirus gD interferes with entry of homologous and heterologous alphaherpesviruses by blocking access to a shared gD receptor. Virology 268147-158. [DOI] [PubMed] [Google Scholar]
- 26.Gianni, T., R. Fato, C. Bergamini, G. Lenaz, and G. Campadelli-Fiume. 2006. Hydrophobic alpha-helices 1 and 2 of herpes simplex virus gH interact with lipids, and their mimetic peptides enhance virus infection and fusion. J. Virol. 808190-8198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gianni, T., A. Piccoli, C. Bertucci, and G. Campadelli-Fiume. 2006. Heptad repeat 2 in herpes simplex virus 1 gH interacts with heptad repeat 1 and is critical for virus entry and fusion. J. Virol. 802216-2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Handler, C. G., R. J. Eisenberg, and G. H. Cohen. 1996. Oligomeric structure of glycoproteins in herpes simplex virus type 1. J. Virol. 706067-6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hannah, B. P., E. E. Heldwein, F. C. Bender, G. H. Cohen, and R. J. Eisenberg. 2007. Mutational evidence of internal fusion loops in herpes simplex virus glycoprotein B. J. Virol. 814858-4865. (Erratum, 82:7249, 2008.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harrison, S. C. 2005. Mechanism of membrane fusion by viral envelope proteins. Adv. Virus Res. 64231-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heldwein, E. E., and C. Krummenacher. 2008. Entry of herpesviruses into mammalian cells. Cell. Mol. Life Sci. 651653-1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heldwein, E. E., H. Lou, F. C. Bender, G. H. Cohen, R. J. Eisenberg, and S. C. Harrison. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313217-220. [DOI] [PubMed] [Google Scholar]
- 33.Isola, V. J., R. J. Eisenberg, G. R. Siebert, C. J. Heilman, W. C. Wilcox, and G. H. Cohen. 1989. Fine mapping of antigenic site II of herpes simplex virus glycoprotein D. J. Virol. 632325-2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kadlec, J., S. Loureiro, N. G. Abrescia, D. I. Stuart, and I. M. Jones. 2008. The postfusion structure of baculovirus gp64 supports a unified view of viral fusion machines. Nat. Struct. Mol. Biol. 151024-1030. [DOI] [PubMed] [Google Scholar]
- 35.Kielian, M., M. R. Klimjack, S. Ghosh, and W. A. Duffus. 1996. Mechanisms of mutations inhibiting fusion and infection by Semliki Forest virus. J. Cell Biol. 134863-872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller, C. G., C. Krummenacher, R. J. Eisenberg, G. H. Cohen, and N. W. Fraser. 2001. Development of a syngenic murine B16 cell line-derived melanoma susceptible to destruction by neuroattenuated HSV-1. Mol. Ther. 3160-168. [DOI] [PubMed] [Google Scholar]
- 37.Milne, R. S., S. A. Connolly, C. Krummenacher, R. J. Eisenberg, and G. H. Cohen. 2001. Porcine HveC, a member of the highly conserved HveC/nectin 1 family, is a functional alphaherpesvirus receptor. Virology 281315-328. [DOI] [PubMed] [Google Scholar]
- 38.Muggeridge, M. I., T. T. Wu, D. C. Johnson, J. C. Glorioso, R. J. Eisenberg, and G. H. Cohen. 1990. Antigenic and functional analysis of a neutralization site of HSV-1 glycoprotein D. Virology 174375-387. [DOI] [PubMed] [Google Scholar]
- 39.Okuma, K., M. Nakamura, S. Nakano, Y. Niho, and Y. Matsuura. 1999. Host range of human T-cell leukemia virus type I analyzed by a cell fusion-dependent reporter gene activation assay. Virology 254235-244. [DOI] [PubMed] [Google Scholar]
- 40.Peng, T., M. Ponce-de-Leon, H. Jiang, G. Dubin, J. M. Lubinski, R. J. Eisenberg, and G. H. Cohen. 1998. The gH-gL complex of herpes simplex virus (HSV) stimulates neutralizing antibody and protects mice against HSV type 1 challenge. J. Virol. 7265-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pertel, P. E., A. Fridberg, M. L. Parish, and P. G. Spear. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279313-324. [DOI] [PubMed] [Google Scholar]
- 42.Reske, A., G. Pollara, C. Krummenacher, B. M. Chain, and D. R. Katz. 2007. Understanding HSV-1 entry glycoproteins. Rev. Med. Virol. 17205-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rey, F. A. 2006. Molecular gymnastics at the herpesvirus surface. EMBO Rep. 71000-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roche, S., A. A. Albertini, J. Lepault, S. Bressanelli, and Y. Gaudin. 2008. Structures of vesicular stomatitis virus glycoprotein: membrane fusion revisited. Cell. Mol. Life Sci. 651716-1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roche, S., S. Bressanelli, F. A. Rey, and Y. Gaudin. 2006. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 313187-191. [DOI] [PubMed] [Google Scholar]
- 46.Rux, A. H., S. H. Willis, A. V. Nicola, W. Hou, C. Peng, H. Lou, G. H. Cohen, and R. J. Eisenberg. 1998. Functional region IV of glycoprotein D from herpes simplex virus modulates glycoprotein binding to the herpesvirus entry mediator. J. Virol. 727091-7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Satoh, T., J. Arii, T. Suenaga, J. Wang, A. Kogure, J. Uehori, N. Arase, I. Shiratori, S. Tanaka, Y. Kawaguchi, P. G. Spear, L. L. Lanier, and H. Arase. 2008. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 132935-944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sisk, W. P., J. D. Bradley, R. J. Leipold, A. M. Stoltzfus, M. Ponce de Leon, M. Hilf, C. Peng, G. H. Cohen, and R. J. Eisenberg. 1994. High-level expression and purification of secreted forms of herpes simplex virus type 1 glycoprotein gD synthesized by baculovirus-infected insect cells. J. Virol. 68766-775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Spear, P. G., and R. Longnecker. 2003. Herpesvirus entry: an update. J. Virol. 7710179-10185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Subramanian, R. P., and R. J. Geraghty. 2007. Herpes simplex virus type 1 mediates fusion through a hemifusion intermediate by sequential activity of glycoproteins D, H, L, and B. Proc. Natl. Acad. Sci. USA 1042903-2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun, X., S. Belouzard, and G. R. Whittaker. 2008. Molecular architecture of the bipartite fusion loops of vesicular stomatitis virus glycoprotein G, a class III viral fusion protein. J. Biol. Chem. 2836418-6427. [DOI] [PubMed] [Google Scholar]
- 52.Terry-Allison, T., R. I. Montgomery, J. C. Whitbeck, R. Xu, G. H. Cohen, R. J. Eisenberg, and P. G. Spear. 1998. HveA (herpesvirus entry mediator A), a coreceptor for herpes simplex virus entry, also participates in virus-induced cell fusion. J. Virol. 725802-5810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vashishtha, M., T. Phalen, M. T. Marquardt, J. S. Ryu, A. C. Ng, and M. Kielian. 1998. A single point mutation controls the cholesterol dependence of Semliki Forest virus entry and exit. J. Cell Biol. 14091-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Warner, M. S., R. J. Geraghty, W. M. Martinez, R. I. Montgomery, J. C. Whitbeck, R. Xu, R. J. Eisenberg, G. H. Cohen, and P. G. Spear. 1998. A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2, and pseudorabies virus. Virology 246179-189. [DOI] [PubMed] [Google Scholar]
- 55.Whitbeck, J. C., Y. Zuo, R. S. Milne, G. H. Cohen, and R. J. Eisenberg. 2006. Stable association of herpes simplex virus with target membranes is triggered by low pH in the presence of the gD receptor, HVEM. J. Virol. 803773-3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou, J., and G. W. Blissard. 2008. Identification of a gp64 subdomain involved in receptor binding by budded virions of the baculovirus Autographica californica multicapsid nucleopolyhedrovirus. J. Virol. 824449-4460. [DOI] [PMC free article] [PubMed] [Google Scholar]