

Abstract
Epstein-Barr virus (EBV) membrane glycoprotein 42 (gp42) is required for viral entry into B lymphocytes through binding to human leukocyte antigen (HLA) class II on the B-cell surface. EBV gp42 plays multiple roles during infection, including acting as a coreceptor for viral entry into B cells, binding to EBV glycoprotein H (gH) and gL during the process of membrane fusion, and blocking T-cell recognition of HLA class II-peptide complexes through steric hindrance. EBV gp42 occurs in two forms in infected cells, a full-length membrane-bound form and a soluble form generated by proteolytic cleavage that is secreted from infected cells due to loss of the N-terminal transmembrane domain. Both the full-length and the secreted gp42 forms bind to gH/gL and HLA class II, and the functional significance of gp42 cleavage is currently unclear. We found that in a virus-free cell-cell fusion assay, enhanced secretion of gp42 promoted fusion with B lymphocytes, and mutation of the site of gp42 cleavage inhibited membrane fusion activity. The site of gp42 cleavage was found to be physically distinct from the residues of gp42 necessary for binding to gH/gL. These results suggest that cleavage and secretion of gp42 are necessary for the process of membrane fusion with B lymphocytes, providing the first indicated functional difference between full-length and cleaved, secreted gp42.
Epstein-Barr virus (EBV) is a large DNA virus belonging to the human gammaherpesvirus subfamily. EBV is orally transmitted through saliva and persists for the lifetime of its human host, establishing a latency reservoir in B lymphocytes with intermittent viral reactivation (1, 27). More than 90% of the world's adult population is infected with EBV, although in healthy individuals, viral reactivation from latency is quickly controlled by the immune system. During primary infection and viral reactivation from latency, EBV infects epithelial cells as well as B lymphocytes (27). Primary infection with EBV can lead to development of infectious mononucleosis, and EBV has also been strongly associated with a number of human malignancies of epithelial and B-cell origin, including Burkitt's lymphoma and nasopharyngeal carcinoma (4, 9, 10, 33, 36).
EBV encodes a number of membrane glycoproteins important in a variety of viral processes, including entry of the virus into target host cells and virus-induced cell-cell fusion. The membrane glycoproteins necessary for fusion with both epithelial and B cells are glycoprotein B (gB), gH, and gL, and together, they form the core virus fusion machinery (7, 20, 24, 29). In addition to these glycoproteins, glycoprotein 42 (gp42) has been shown to play an essential role in membrane fusion with B cells (7, 18, 20). Attachment of EBV virions to B cells occurs through binding of the main envelope protein gp350/220 to CD21 (also known as complement receptor type 2) (5, 23, 34). This interaction enhances the efficiency of EBV infection of B cells but is not required for viral entry (12, 30). Antibodies to gp350/220 inhibit EBV infection of B cells but enhance infection of epithelial cells, possibly by facilitating the access of other viral glycoproteins to the epithelial cell membrane (35). Virus-cell membrane fusion is subsequently triggered by binding of gp42 to human leukocyte antigen (HLA) class II on the B-cell surface (6, 8, 11, 17, 31). Interestingly, gp42 appears to function as a switch of cellular tropism between epithelial and B cells. The presence of gp42 in the viral envelope is necessary for infection of B lymphocytes, and virions that are low in gp42 are better able to infect HLA class II-negative epithelial cells (3). Aside from its role in membrane fusion, gp42 plays a significant role in evasion of the host immune system. Gp42 binds to HLA class II-peptide complexes in infected cells, sterically hindering T-cell recognition of the complex by the T-cell receptor (25). This inhibition may allow EBV to delay detection by the host immune system.
Two different mature forms of gp42 are produced by EBV-positive B lymphocytes in the lytic cycle (26). The first form is a full-length type II membrane protein, and the second is a truncated soluble form (s-gp42) (26). s-gp42 is generated by posttranslational cleavage (most likely mediated by a cellular protease resident in the endoplasmic reticulum) and is secreted (26). Both forms of gp42 associate with HLA class II intracellularly, and both inhibit HLA class II-restricted antigen presentation to T cells (26). Both forms of gp42 produced by EBV-positive B cells in the lytic cycle were found to be present in gH-gL-gp42 complexes, indicating that s-gp42 retains the ability to bind gH/gL (26). The physiological significance of s-gp42 is currently unclear, but this form has been suggested to function in infection and immune evasion, blocking EBV entry receptors on lytically infected B cells to prevent reinfection and neutralizing gp42-specific antibodies following its secretion from infected cells (26).
Both forms of gp42 have been examined for their functions in mediating evasion from T-cell immunity through binding to HLA class II complexes (26), but the functions of the two forms of the protein in membrane fusion are unknown. To examine how each form of gp42 functions during membrane fusion, we have assayed the effect of gp42 cleavage site mutation on this process. Also, to distinguish residues important for gp42 cleavage from those necessary for association with gH/gL, we have constructed a number of fully secreted gp42 truncation mutants and examined their interaction with gH/gL and their ability to mediate fusion. Mutation or deletion of the gp42 cleavage site inhibited or eliminated cleavage of the protein, which had a direct effect on gp42 function in membrane fusion. An assay of N-terminal truncations of gp42 indicated that the region of gp42 necessary for cleavage was physically distinct from the region of gp42 necessary for association with gH/gL. We show that membrane association of gp42 has an inhibitory effect on gp42 function in membrane fusion and that increased secretion of gp42 stimulates membrane fusion in vitro. Cleavage of gp42 may be necessary for EBV gp42 to assume a functional position, interaction, or conformation for participation in membrane fusion.
MATERIALS AND METHODS
Cells and antibodies.
Mammalian cells were grown in 25-cm2 or 75-cm2 cell culture flasks (Corning) in medium supplemented with 10% FetalPlex animal serum complex (Gemini Bio-Products) and 1% penicillin-streptomycin (BioWhittaker). Mammalian B cells were Daudi B lymphocytes that are EBV positive, express HLA class II and CD21 (American Type Culture Collection), and are modified to stably express T7 RNA polymerase under selection of G418 (700 μg/ml) in RPMI 1640 medium (BioWhittaker) (28). Chinese hamster ovary K1 (CHO-K1) cells were kindly provided by Nanette Susmarski and were grown in complete Ham's F-12 medium (BioWhittaker). Versene (1 mM EDTA in phosphate-buffered saline [PBS]) or trypsin-Versene (BioWhittaker) was used to detach adherent cells. Polyclonal anti-gp42 antibody serum (PB1114) was used as previously described (21, 28). Monoclonal antibody 3H3 (anti-gp42) was obtained as previously described (14). Monoclonal antibody E1D1 (anti-gH/gL) was a gift kindly provided by L. Hutt-Fletcher (Louisiana State University Health Sciences Center, Shreveport, LA) (32).
Mutants.
Gp42 mutant Δ37-41 was kindly supplied by Amanda Lowery and was cloned as described previously (13). Mutant gp42-EK was generated using a previously described double-arm PCR approach (13). Mutant primers were generated to incorporate a 5-residue substitution at residues 37 to 41, replacing the wild-type gp42 sequence with the enterokinase cleavage site (VAAAA to DDDDK) as well as a silent restriction enzyme-cutting site. In the initial PCR, the mutant primers were paired with primers flanking the EBV gp42 gene in the pCAGGS.mcs vector (P1 and P2 [13]), each matched with its complementary directional mutant primer on a wild-type EBV gp42 plasmid template. PCR products were confirmed by gel electrophoresis and then used as templates with the flanking primers in the second PCR to generate full-length mutant gp42, which was confirmed by gel electrophoresis, cut with EcoRI and BglII, and ligated for 4 h at 25°C with vector that had been digested under the same conditions.
Gp42 truncation mutants Δ36, Δ41, Δ46, and Δ51 were cloned through a different PCR approach. The signal sequence of EBV gB (residues 1 to 22) was amplified from wild-type EBV gB in the pSG5 vector by using a forward primer, gBssAmp_Forward (CGACTCACTATAGGGCGAATTCG), and mutant-specific reverse primers that included the first 18 bases of the gp42 sequence after the end of the gB signal sequence in each mutant. These reverse primers were gBssAmpΔ36_Reverse (GATGGCCGCGGCTGCCACCGCACCGAGACGGCACGC), gBssAmpΔ41_Reverse (GGTTTGGGTACCCAGGTGATCGCACCGAGACGGCACGC), gBssAmpΔ46_Reverse (GACCTCTACATTTGGTTTCGCACCGAGACGGCACGC), and gBssAmpΔ51_Reverse (GGAGGATCCACCGGCCAGACCGCACCGAGACGGCACGC). The initial PCRs with these primers resulted in amplification of the gB signal sequence and the first 18 bases of the gp42 sequence in each gp42 truncation mutant. The products of each reaction were purified using a Qiagen PCR purification kit, and the products were each used as the forward primer in a second set of PCRs. These reactions amplified both the gB signal sequence and the wild-type gp42 sequence included in each gp42 truncation mutant, and wild-type gp42 in the pCAGGS vector was used as the template in these reactions. The reverse primer in each reaction was s-gp42TotalAmpReverse (CGAGATCTTTAGCTATTTGATCTTTGACTGACACATAAACATGG). PCR products were confirmed by gel electrophoresis, cut with EcoRI and BglII, and ligated for 4 h at 25°C with vector that had been digested under the same conditions. All ligated products were transformed and selected on ampicillin plates. Colonies were picked and grown overnight to generate minipreparations, which were digested to confirm the introduction of the mutation. Minipreparations were sequenced, and positive clones were then grown in large quantities, isolated using an EndoFree Plasmid maxikit (Qiagen), and sequenced again.
Transfection. (i) Fusion assay.
CHO-K1 cells were transfected in Opti-MEM I medium (Gibco) by a uniform protocol using Lipofectamine 2000 (Invitrogen), as previously described (28). Cells were plated in a six-well format, and after 24 h, each well received 5 μl of Lipofectamine 2000 and various combinations of expression vectors in the following amounts: 0.5 μg for gH, 0.5 μg for gL, 0.5 μg for gB, 0.8 μg for luciferase, 0.5 μg for green fluorescent protein, and 1.5 μg for the pCAGGS vector control. In the case of gp42-transfected CHO-K1 cells, green fluorescent protein and pCAGGS plasmids were replaced with 2 μg of wild-type or mutant gp42.
(ii) Expression and assay of binding of s-gp42 to HLA class II.
Four micrograms of pCAGGS vector, wild-type gp42 vector, or mutant gp42 vector was transfected as described above via Lipofectamine 2000 into each well in a six-well-plate format.
(iii) Immunoprecipitation.
Wild-type or mutant gp42, EBV gH, and EBV gL (1.33 μg each) was transfected as described above via Lipofectamine 2000 into each well in a six-well-plate format. For the condition with gH/gL alone, 1.33 μg of empty vector was transfected in place of gp42. For the condition with gp42 alone, 2.66 μg of empty vector was transfected in place of gH and gL.
Western blotting.
CHO-K1 cells were transfected as stated above. The medium was changed 6 h later to complete Ham's F-12 medium, and cells and culture supernatants were collected at 48 h posttransfection. Cells were detached with Versene, washed in PBS, and lysed using a 1% Triton X-100 buffer. Culture supernatants were collected prior to cell detachment and were spun down to pellet-detached cells. Supernatants and lysates were run on Bio-Rad Criterion gels in sodium dodecyl sulfate (SDS) sample buffer at 100 V for 100 min. Proteins were transferred to Immobilon-P membranes in transfer buffer at 100 V for 90 min with cooling. Blots were blocked in Tris-buffered saline with Tween 20 with 5% milk for 1 h at room temperature or overnight at 4°C and then incubated for 1 h at room temperature with a rabbit polyclonal anti-gp42 antibody (PB1114) diluted 1:2,000 in blocking solution. Blots were washed, and a horseradish peroxidase (HRP)-conjugated anti-rabbit immunoglobulin G (IgG; Cell Signaling) was applied for 45 min at room temperature. The blots were washed again and then mixed in equal volumes of enhanced chemiluminescence solutions and exposed to hyperfilm (Amersham Biosciences).
Following exposure, the blots were rotated with stripping buffer containing 100 mM beta-mercaptoethanol, 62.5 mM Tris-HCl (pH 6.8), and 2% SDS at 55°C for 1 h. They were washed twice in Tris-buffered saline with Tween 20 and blocked in Tris-buffered saline with Tween 20 with 5% milk for 1 h at room temperature or overnight at 4°C. The blots were then incubated for 1 h at room temperature with a mouse monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) antibody (Abcam) diluted 1:10,000 in blocking solution. The blots were washed, and an HRP-conjugated anti-mouse IgG (Cell Signaling) was applied for 45 min at room temperature. The blots were washed again and then mixed in equal volumes of enhanced chemiluminescence solutions and exposed to hyperfilm (Amersham Biosciences).
Fusion assay.
CHO-K1 cells were transiently transfected as described above. At 24 h posttransfection, the cells were detached with Versene and 2.5 × 105 cells/well in 0.5 ml were transferred to a 24-well format and overlaid with equal numbers of Daudi B target cells in 0.5 ml. Cells were counted using a Beckman Coulter Z1 particle counter. Equal numbers of CHO-K1 cells plated and Daudi cells overlaid were used in each experiment, and the total volume was 1 ml per well. Twenty-four hours after overlay, cells were washed with PBS and lysed with 100 μl passive lysis buffer (Promega) per well. Luciferase activity was quantified by transferring duplicate 20-μl aliquots of cell lysate to a 96-well opaque plate with clear well bottoms (Wallac), and luminescence was measured with a Perkin-Elmer Victor plate reader immediately after addition of 100 μl/well of luciferase assay reagents (Promega).
Cell enzyme-linked immunosorbent assay (CELISA).
CHO-K1 cells were transfected as described above via Lipofectamine 2000 with 1.3 μg of wild-type gp42 or mutant gp42 and either 1.3 μg each of EBV gH and gL or 2.6 μg of pCAGGS vector. After 6 h, the medium was changed, and at 24 h posttransfection, cells were detached with Versene and transferred to Corning 96-well plates, using 3 wells per sample. After incubation for 24 h at 37°C, cells were washed with PBS-ABC (PBS with 0.89 g of CaCl2 and 0.89 g of MgCl2·H2O per 8 liters), incubated for 30 min at room temperature with a mouse monoclonal anti-gp42 antibody (3H3), diluted 1:2,000 in PBS-BSA (PBS-ABC with 3% bovine serum albumin), washed again, and then fixed for 10 min in PBS with 2% formaldehyde and 0.2% glutaraldehyde. Cells were washed with PBS-BSA, incubated with a biotinylated anti-mouse IgG (Sigma) at 1:500 for 30 min, washed again, and then incubated with a streptavidin-HRP antibody (1:20,000) (Amersham) for 30 min, all at room temperature. Following a final wash with PBS-ABC with 0.1% Tween 20, cells were mixed with a peroxide substrate (BioFX Laboratories) and read with a Victor plate reader at 370 nm for 0.1 s.
Immunoprecipitation.
CHO cells were transiently transfected as described above. The medium was changed to complete Ham's F-12 medium at 6 h posttransfection, and at 48 h posttransfection, cells were detached with Versene, counted using a Beckman Coulter Z1 particle counter, washed in ice-cold PBS, and lysed with 1% Triton X-100 lysis buffer at a concentration of 10 million cells/ml. Fifty-microliter aliquots of each lysate were added to 2× SDS sample buffer, and the remainder of each lysate was split into three equal parts. One-third of each lysate was rotated with 25 μl of agarose beads cross-linked to protein G (Amersham Biosciences) alone, one-third was rotated with 1 μl of anti-gp42 monoclonal antibody 3H3 and 25 μl of agarose beads cross-linked to protein G, and one-third was rotated with 1 μl of anti-gH/gL monoclonal antibody E1D1 and 25 μl of agarose beads cross-linked to protein G. All samples rotated for at least 3 h at 4°C. Samples were washed a minimum of three times with ice-cold PBS, and 50 μl of 2× SDS sample buffer was added to each sample. Lysates and immunoprecipitations were run on Bio-Rad 12.5% Criterion gels in SDS sample buffer as described above and were transferred and blocked as described above. The lysates were blotted with anti-GAPDH monoclonal antibody (Abcam), and the immunoprecipitations were blotted with PB1114 as described above.
Binding of s-gp42 to HLA class II-expressing cells.
CHO-K1 cells were transfected as described above. The medium was changed 6 h later to complete Ham's F-12 medium, and culture supernatants were collected at 48 h posttransfection as described above. Supernatants were run on Bio-Rad 10% Criterion gels in SDS sample buffer, transferred, blocked, and blotted with gp42-specific antibody PB1114 as described above. After measurement of the relative levels of s-gp42 in CHO-K1 cell supernatants by Western blotting followed by densitometry (ImageJ), supernatants were diluted in complete Ham's F-12 medium to normalize protein concentrations between samples. Y107A, a previously described gp42 mutant known to not bind HLA class II, was utilized as a negative control (28). Normalized supernatants were incubated with 3.0 × 106 Daudi cells while rotating at 4°C for 20 min. Following this incubation, cells were washed three times with ice-cold PBS and each population was lysed with 300 μl 1% Triton X-100 buffer. Fifty-microliter aliquots of each lysate were added to 2× SDS sample buffer, and the remainder of each lysate was rotated with 1 μl of gp42-specific monoclonal antibody 3H3 and 25 μl of agarose beads cross-linked to protein G (Amersham Biosciences) for at least 3 h at 4°C. Samples were washed a minimum of three times with ice-cold PBS, and 50 μl of 2× SDS sample buffer was added to each sample. The cell lysates and immunoprecipitations were run on Bio-Rad 10% Criterion gels in SDS sample buffer, transferred, and blocked as described above. The immunoprecipitations were blotted with PB1114, and the lysates were blotted with anti-GAPDH monoclonal antibody (Abcam) as described above.
RESULTS
Mutants of the gp42 N-terminal site of cleavage.
EBV gp42 is a type II membrane protein with a number of identified functional domains (Fig. 1A). Within the N-terminal region is the site of gp42 cleavage, indicated to occur between residues 40 to 42, although the exact residues necessary for cleavage have not been identified (26). A signal sequence cleavage site is predicted to occur between residues 33 and 34 of gp42, but cleavage at this site is unlikely, since N-terminal sequencing of purified s-gp42 from culture supernatants of cells stably expressing gp42 indicates that cleavage occurs near amino acids 40 to 42 (26), and deletion of amino acids 32 to 36 from gp42 does not inhibit secretion of gp42 from transfected cells (13).
FIG. 1.
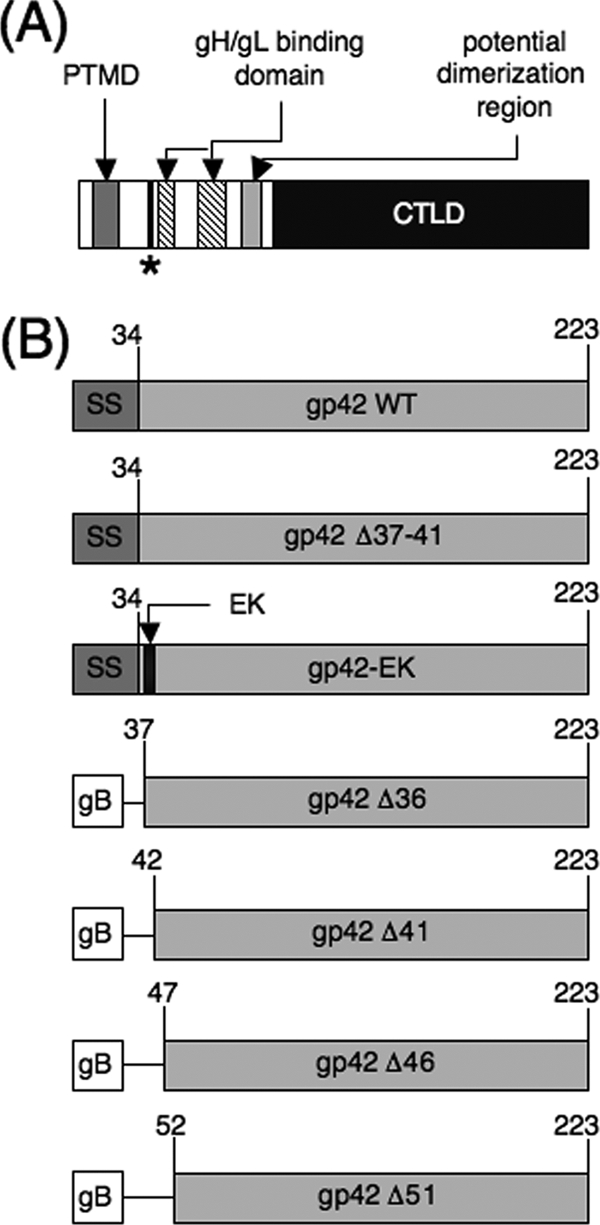
Schematic representation of EBV gp42 and gp42 mutants. (A) Wild-type gp42 including the cleavage site and the surrounding sequence. The relative locations of other known functional domains are also indicated. PTMD is the putative transmembrane domain, predicted to span residues 9 to 22. The site of gp42 cleavage has been narrowed to residues 40 to 42 and is indicated by an asterisk. The core gH/gL binding region is indicated to span residues 47 to 56 and 67 to 81, approximately. CTLD is the C-terminal C-type lectin domain, which includes the hydrophobic pocket and HLA class II-binding region. (B) gp42 mutants described in this study. SS indicates the gp42 signal sequence, which has been predicted to span residues 1 to 34. The boxes labeled gB indicate that the signal sequence of gp42 has been replaced with the signal sequence of EBV gB (residues 1 to 22). Lines connecting boxes indicate deleted regions in the gp42 mutants. The black box labeled EK indicates the replacement of residues 37 to 41 of gp42 (VAAAA) with an enterokinase cleavage site (DDDDK). WT, wild type.
A previously described gp42 deletion mutant, gp42 Δ37-41 (Fig. 1B), does not function in fusion, although it is expressed and trafficked to the surfaces of transfected cells similarly to wild-type gp42 (13). Unlike wild-type gp42, this mutant is not secreted from transfected cells (13). The Δ37-41 deletion spans residues potentially involved in gp42 cleavage (26). To further examine whether cleavage and secretion of gp42 are linked to the ability of the protein to mediate fusion, gp42-EK, a new mutant in which an enterokinase recognition sequence (DDDDK) was substituted for residues 37 to 41 of wild-type gp42 (VAAAA), was constructed (Fig. 1B).
Whole-cell expression and secretion of gp42 cleavage mutants.
Although inducible cleavage of gp42-EK was not observed by cotransfection with a construct expressing the catalytic subunit of bovine enterokinase (16) or by addition of recombinant enterokinase (Novagen) to cell culture supernatants (data not shown), we found that replacement of the wild-type gp42 sequence with the enterokinase cleavage site disrupted normal cleavage of gp42 (Fig. 2). When transfected into CHO-K1 cells, gp42-EK was largely retained within the cells, with a noticeable increase in this protein in transfected whole-cell lysate compared to the level for wild-type gp42 (Fig. 2A). Conversely, there was a marked decrease in secretion of gp42-EK from transfected CHO-K1 cells into the culture supernatant compared to the level for wild-type gp42 (Fig. 2B). Similar to previous observations (13), gp42 Δ37-41 was expressed well in transfected CHO-K1 cells but little to no Δ37-41 was secreted. The presence of multiple bands in lanes containing cell lysate from gp42-transfected cells is likely due to the facts that both forms of gp42 are heavily glycosylated (26) and that both immature and mature forms of the protein were detected. The increase in cell association and the decrease in secretion observed with gp42-EK indicated that substitution of residues 37 to 41 of gp42 disrupted but did not completely eliminate wild-type cleavage of gp42. Despite decreased secretion with both gp42 cleavage mutants, the cell surface expression levels of gp42-EK and gp42 Δ37-41 were similar to that for wild-type gp42 in transfected CHO-K1 cells, as shown by CELISA (see Fig. 5A).
FIG. 2.

Cellular expression and secretion of gp42 mutants Δ37-41 and gp42-EK. CHO-K1 cells were transiently transfected to express wild-type (WT) or mutant gp42. Transfected cells were lysed, and lysates (A) and culture supernatants (B) were analyzed by SDS-polyacrylamide gel electrophoresis (PAGE) and Western blotting with rabbit polyclonal anti-gp42 antibody PB1114. Size markers in kDa are noted to the left of the blots. Lysates and supernatants were also blotted with GAPDH as a loading control (A) and as a control for cell lysis (B).
FIG. 5.

Gp42 mutant cell surface expression and binding to gH/gL. Surface localization of gp42 mutants was measured by CELISA in the absence (A) and presence (B) of cotransfected gH/gL, using monoclonal 3H3 antibody, secondary biotinylated anti-mouse IgG antibody, tertiary streptavidin-HRP, and TMB substrate. Color development was measured by absorbance at 370 nm. Data shown are averages of results from three independent experiments, with cell surface expression normalized to wild-type levels, which were set to 100%. Error bars represent standard deviations for the normalized values.
Function of gp42 cleavage mutants in membrane fusion.
To examine the effect of mutation of gp42 residues 37 to 41 on membrane fusion, gp42 Δ37-41 and gp42-EK were tested in a virus-free cell-cell fusion assay (2, 14, 19). CHO-K1 cells were transiently transfected with gB, gH, gL, and wild-type or mutant gp42 and overlaid with Daudi B cells, and mutants were assayed for their ability to mediate membrane fusion. As previously described (13), gp42 Δ37-41 was unable to mediate cell fusion. The substitution mutant gp42-EK, however, showed ∼35% membrane fusion activity compared to the level for wild-type gp42 (Fig. 3). This decrease in the ability of gp42-EK to mediate membrane fusion closely correlated with the reduction in secretion from the wild-type level seen with this mutant (Fig. 2B). This correlation is also seen with gp42 Δ37-41, as this mutant appears to be at most only negligibly secreted (Fig. 2B) and has no membrane fusion activity (Fig. 3). These data suggest that the amount of gp42 cleavage may be directly linked to the ability of the protein to mediate membrane fusion and that cleavage of the protein is necessary for productive membrane fusion to occur.
FIG. 3.

Effect of gp42 cleavage site mutation on membrane fusion with B cells. Relative luciferase activity was measured in a cell fusion assay using CHO-K1 cells transfected with gB, gH, gL, and wild-type or mutant gp42 overlaid with B cells (DaudiT7). The data shown were averaged from a minimum of five independent experiments, and luciferase activity was normalized to the wild-type levels, which were set to 100%. Error bars represent standard deviations for the normalized values.
Construction of gp42 N-terminal truncation mutants.
The described mutants demonstrate that residues 37 to 41 of gp42 are important for membrane fusion, but an alternative explanation for the observed membrane fusion defects is that these residues are important for binding of gH/gL to gp42. A deletion study of the N terminus of gp42 indicated that residues 37 to 41 are not essential for binding to gH/gL (13), but the possibility remains that deletion or substitution of these amino acids may destabilize binding of gp42 to gH/gL, an association that is necessary for membrane fusion to occur. To determine whether residues 37 to 41 of gp42 are required for wild-type binding of gp42 to gH/gL, four N-terminal truncations of gp42 were constructed (Fig. 1B). These truncations were designed with progressive deletions of the gp42 N terminus, and all were deleted for the entire putative gp42 signal sequence, including the gp42 transmembrane domain. The entire gp42 transmembrane domain was removed to ensure 100% secretion of the truncation mutants, allowing the residues necessary for gH/gL binding to be further defined independent of gp42 cleavage and secretion. To ensure that the mutants were similar to wild-type gp42 in trafficking and cell surface localization, the EBV gB signal sequence, which does not contain a transmembrane domain, was substituted for the gp42 signal sequence.
Whole-cell expression and secretion of gp42 truncation mutants.
All four of the gp42 truncation mutants were expressed at levels similar to those for wild-type gp42 in cell culture, and all four appeared to be entirely secreted. Western blotting of lysates with gp42-specific polyclonal antibody PB1114 demonstrated that all the truncation mutants were expressed (Fig. 4A). Unsurprisingly, the molecular masses of the gp42 truncation mutants were slightly lower than those of wild-type gp42 and the predominantly membrane-bound gp42 cleavage mutants, due to loss of mature full-length gp42, which can be visualized just below the 50-kDa marker in Fig. 4A in the wild-type gp42 lane. All of the truncation mutants were secreted into cell culture supernatants similarly to wild-type gp42 (Fig. 4B). Following Western blotting, both the cell lysate and the culture supernatant blots were stripped and reprobed with anti-GAPDH monoclonal antibody as a loading control (Fig. 4A) and as a control for cell lysis (Fig. 4B). No GAPDH was visible in any of the culture supernatant lanes, indicating that cell lysis does not significantly contribute to the protein present in the culture supernatants (Fig. 4B). The data summarized in Fig. 4 indicate that the gp42 truncation mutants are expressed similarly to wild-type gp42 and are fully secreted from transfected CHO-K1 cells.
FIG. 4.
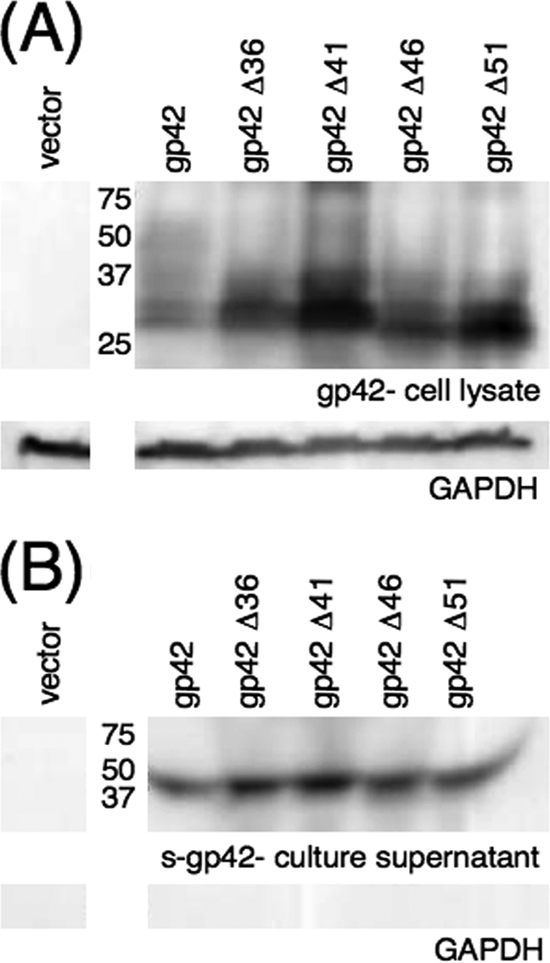
Cellular expression and secretion of gp42 truncation mutants. CHO-K1 cells were transiently transfected to express wild-type or mutant gp42. Transfected cells were lysed, and lysates (A) and culture supernatants (B) were analyzed by SDS-PAGE and Western blotting with rabbit polyclonal anti-gp42 antibody PB1114. Size markers in kDa are noted to the left of the blots. Lysates and supernatants were also blotted with GAPDH as a loading control (A) and as a control for cell lysis (B).
Cell surface expression of gp42 mutants.
As an alternative approach for determining the secretion levels of the gp42 truncation mutants from transfected cells and for assaying the binding of the mutants to gH/gL on the surfaces of transfected cells, the truncation mutants were examined by CELISA in the presence and absence of cotransfected gH/gL (Fig. 5). When transfected alone into CHO-K1 cells, none of the truncation mutants were detected on the transfected cell surface, unlike wild-type gp42, which was abundant (Fig. 5A). These data indicate that all of the gp42 truncation mutants are fully secreted and do not bind nonspecifically to the surfaces of transfected cells. In the presence of cotransfected gH/gL, truncation mutants Δ36, Δ41, and Δ46 were detected on the cell surface at levels similar to or higher than those for wild-type gp42, while mutant Δ51 was not detected (Fig. 5B). These data indicate that secreted truncation mutants Δ36, Δ41, and Δ46 were able to bind specifically to gH/gL at the surfaces of transfected cells but that mutant Δ51 lacked the wild-type ability to bind to gH/gL. The presence of cotransfected gH/gL had no effect on cell surface expression of gp42-EK or gp42 Δ37-41 (Fig. 5B).
Binding of gp42 mutants to gH/gL.
Binding of the gp42 truncation mutants to gH/gL was also assayed by immunoprecipitation to further understand the residues of gp42 important for this interaction. Immunoprecipitation with anti-gH/gL monoclonal antibody E1D1 followed by Western blotting with anti-gp42 polyclonal antibody PB1114 was performed to assay gH/gL binding with gp42 (Fig. 6B). Identical aliquots of each lysate were simultaneously immunoprecipitated with anti-gp42 monoclonal antibody 3H3, followed by Western blotting with anti-gp42 polyclonal antibody PB1114, to ensure the presence of gp42 in non-gH/gL transfected samples (Fig. 6A). Lysates were also subjected to Western blotting with anti-GAPDH monoclonal antibody as a loading control (Fig. 6A). The slight variations in the sizes of the gp42 truncation mutant products in Fig. 6A compared to the levels for Fig. 4A are likely due to the binding of mature secreted gp42 to cotransfected gH/gL. In Fig. 4A, this mature form of the protein is fully secreted from transfected cells, since gH/gL is not present.
FIG. 6.
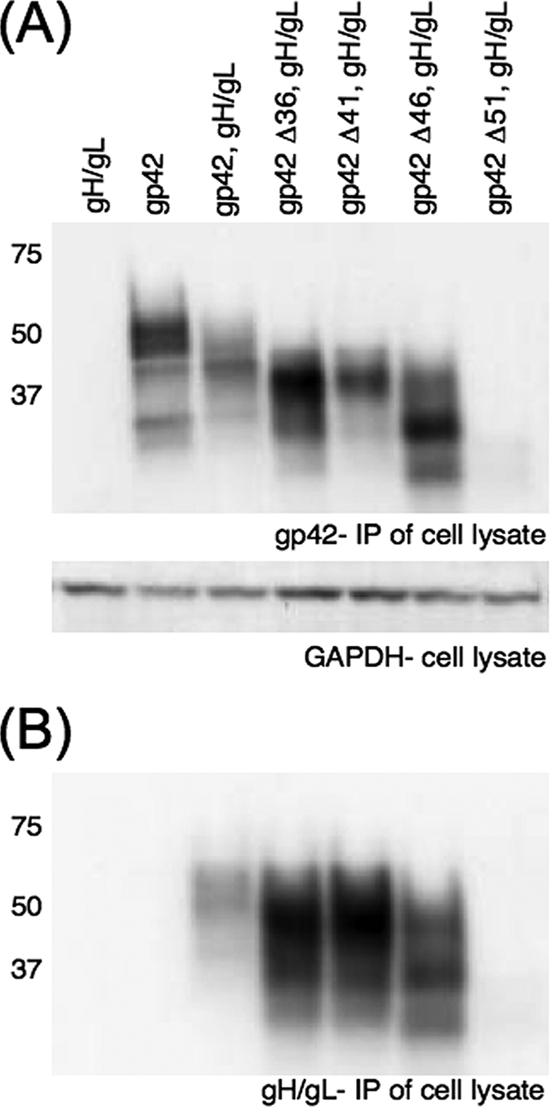
Binding of cotransfected secreted gp42 mutants and gH/gL in immunoprecipitation. (A and B) SDS-PAGE and Western blotting of wild-type and mutant gp42 coexpressed in CHO-K1 cells with EBV gH/gL, using rabbit polyclonal PB1114 anti-gp42 antibody, revealed gp42 levels in immunoprecipitations (IP) using either 3H3 anti-gp42 antibody or E1D1 anti-gHgL antibody, as labeled. Samples were from the same transfection and were simultaneously immunoprecipitated and run in nonadjacent lanes. Lysate from each transfection was blotted with anti-GAPDH antibody as a loading control. Size markers in kDa are noted to the left of the blots.
In agreement with the CELISA data, gp42 mutants Δ36, Δ41, and Δ46 bound to gH/gL, while mutant Δ51 did not (Fig. 6B). Wild-type gp42 alone was not immunoprecipitated nonspecifically with gH/gL-specific antibody (Fig. 6B). Gp42 Δ51 was fully secreted from transfected cells, with only immature, partially glycosylated forms of the protein immunoprecipitated with anti-gp42 monoclonal antibody 3H3 (Fig. 6A). Since all four of the truncation mutants were found to be fully secreted, the lack of detection of mature gp42 Δ51 with anti-gp42 antibody indicates that this mutant was unable to bind to gH/gL on the transfected-cell surface and was fully secreted into the culture medium. No gp42 Δ51 was seen after immunoprecipitation with anti-gH/gL antibody, further indicating that the mutant is missing residues necessary for binding to gH/gL (Fig. 6B).
Binding of gp42 truncation mutants to HLA class II-expressing B cells.
While the site of binding of gH/gL to gp42 is proximal to the putative site of gp42 cleavage (13, 26), the region of gp42 involved in binding to HLA class II is distal to the cleavage site in the C-terminal C-type lectin domain (22, 31). However, binding of gp42 to HLA class II is necessary for B-cell membrane fusion to occur (17), and mutation of gp42 could potentially disrupt the wild-type ability to bind HLA class II on the B-cell surface. To determine if residues proximal to the putative gp42 cleavage site are necessary for binding of gp42 to HLA class II, each of the gp42 truncation mutants was assayed for the ability to bind HLA class II on the B-cell surface (Fig. 7). Similar to previous studies examining binding of s-gp42 to HLA class II (28), the Daudi human B-cell line, known to express high levels of HLA class II, was utilized. Previous studies have shown that binding of gp42 to target B cells is dependent on HLA class II expression (17, 31). In this assay, supernatants of transfected CHO-K1 cells were collected and s-gp42 protein concentration was visualized by Western blotting with gp42-specific polyclonal antibody PB1114 (Fig. 7A). The relative concentrations of s-gp42 were normalized between samples through dilution with complete Ham's F-12 medium so that similar levels of s-gp42 were contained in all samples. Normalized supernatants were then rotated with Daudi cells at 4°C to allow binding of gp42 to HLA class II on the B-cell surface. Following rotation, Daudi cells were washed and lysed, and lysates were immunoprecipitated with anti-gp42 monoclonal antibody 3H3. Only s-gp42 that was able to bind HLA class II would be detected when immunoprecipitated. Immunoprecipitated gp42 was detected by Western blotting with anti-gp42 polyclonal antibody PB1114 (Fig. 7B). Lysates were also analyzed by Western blotting with anti-GAPDH monoclonal antibody as a loading control (Fig. 7B).
FIG. 7.

Immunoprecipitation analysis of binding of secreted gp42 mutants with HLA class II-expressing Daudi B cells. (A) Supernatants were collected from transfected CHO-K1 cell populations expressing wild-type or mutant gp42. Supernatants and untreated Daudi cell lysate were analyzed by SDS-PAGE and Western blotting with rabbit polyclonal PB1114 anti-gp42 antibody, and supernatants were normalized to approximate equal gp42 protein concentrations. (B) Daudi B lymphocytes expressing HLA class II were incubated with the normalized gp42-containing supernatants. The cells were then washed and lysed, and bound gp42 was immunoprecipitated with mouse monoclonal anti-gp42 antibody 3H3. The immunoprecipitations (IP) were analyzed by SDS-PAGE and Western blotting with rabbit polyclonal PB1114 anti-gp42 antibody. Cell lysate aliquots were saved and analyzed by SDS-PAGE and Western blotting with mouse monoclonal anti-GAPDH antibody as a loading control. Size markers in kDa are noted to the left of the blots.
Controls are shown in the first lanes of Fig. 7A. No protein was detected in the untreated Daudi cell lysate lane, indicating that there was no nonspecific binding of anti-gp42 polyclonal antibody to cellular protein. Only a nonspecific band was seen in the complete Ham's F-12 medium lane and the lane containing culture supernatant from empty-vector-transfected CHO-K1 cells. This nonspecific band was present in all the Ham's F-12 medium-containing supernatant lanes. Abundant wild-type or mutant s-gp42 was present in all the culture supernatants from g42-transfected CHO-K1 cells (Fig. 7A). After immunoprecipitation of Daudi cell lysates, gp42 was detected in the wild-type gp42 lane and each gp42 truncation mutant lane but not in the Ham's F-12, empty vector, or gp42 Y107A lane (Fig. 7B). Y107A is a previously described gp42 mutant known to not bind HLA class II (28) and was included as an additional negative control. These results indicate that loss of up to 51 amino acids from the N terminus of gp42 does not disrupt binding of gp42 to HLA class II.
Function of gp42 truncation mutants in membrane fusion.
To examine the effect of mutation of the gp42 N-terminal truncation mutants on membrane fusion, the truncations were tested in the above-described virus-free cell-cell fusion assay (Fig. 8). Truncation mutants Δ36 and Δ41 showed membrane fusion activities moderately higher than that of wild-type gp42, but these increases were statistically significant by Student's t test for the Δ41 mutant only (P < 0.01). Truncation mutant Δ46 showed a slight decrease in membrane fusion activity, although this decrease was not found to be statistically significant. Unsurprisingly, truncation mutant Δ51, which was shown to be unable to bind to gH/gL, showed no membrane fusion activity. These data indicate that deletion of up to the first 46 amino acids of the N terminus of gp42 has no significant inhibitory effect on wild-type expression, secretion, binding to gH/gL, binding to HLA class II, or ability to mediate membrane fusion with B cells.
FIG. 8.

Effect of N-terminal truncation on membrane fusion with B cells. Relative luciferase activity was measured in a cell fusion assay using CHO-K1 cells transfected with gB, gH, gL, and wild-type or mutant gp42 and overlaid with Daudi B cells. The data shown were averaged from a minimum of five independent experiments, and luciferase activity was normalized to wild-type levels, which were set to 100%. Error bars represent standard deviations for the normalized values.
DISCUSSION
In this study, we investigated the functional significance of gp42 cleavage and found that gp42 cleavage mutants lost the wild-type ability to mediate B-cell membrane fusion. Substitution or deletion of residues 37 to 41 of gp42 did not significantly alter normal gp42 expression. However, replacement of the wild-type sequence (VAAAA) with the cleavage site of enterokinase (DDDDK) or deletion of the residues inhibited or eliminated wild-type secretion of gp42, respectively. Experiments examining these gp42 cleavage mutants, Δ37-41 and gp42-EK, for their ability to mediate membrane fusion with B cells in a virus-free cell-cell fusion assay found that their function in membrane fusion closely correlated with the secretion of each mutant from transfected CHO-K1 cells. Deletion mutant Δ37-41 was negligibly secreted and did not mediate membrane fusion with B cells. Substitution mutant gp42-EK showed impaired secretion (approximately one-third that of wild-type gp42), and its ability to mediate fusion with B cells was ∼35% that of wild-type gp42. These data indicate that some or all of residues 37 to 41 of gp42 are necessary for wild-type cleavage and secretion and suggest that cleavage and secretion may be directly linked to gp42 function in membrane fusion.
The proximity of the gp42 cleavage site to the gH/gL binding region of gp42 was a concern, since the observed loss of the ability of the gp42 cleavage mutants to mediate membrane fusion could possibly be explained by disruption of binding of gp42 to gH/gL. However, we found through examination of the four truncation mutants created to address this possibility that up to 46 amino acids could be deleted from the N terminus of gp42 with no decrease in wild-type ability to bind gH/gL. Creation of gp42 mutants that were fully secreted allowed us to examine residues necessary for binding to gH/gL independent of gp42 cleavage and secretion, and we found that the gp42 cleavage site was physically distinct from the residues of gp42 necessary for binding to gH/gL.
An assay of gp42 truncation mutant binding to HLA class II by incubation of s-gp42 with Daudi B cells expressing HLA class II followed by immunoprecipitation of cell lysates with gp42-specific antibody determined that all of the truncation mutants retained the ability to bind to HLA class II. These data support the findings of a previous study examining gp42 truncations that found that deletion of 58 residues or more from the N terminus of gp42 does not disrupt binding of gp42 to HLA class II (31). These results indicated that loss of HLA class II binding was not responsible for any defect in ability to mediate membrane fusion observed with the gp42 cleavage site mutants or the N-terminal truncation mutants.
Truncation of the N terminus of gp42 did not have a deleterious effect on the ability of mutants Δ36, Δ41, and Δ46 to mediate membrane fusion with B cells. Interestingly, mutant Δ41, which begins at the predicted start of wild-type s-gp42, showed a significant increase in ability to mediate membrane fusion over wild-type gp42 (P < 0.01 by Student's t test). This increase may be due to the increased secretion of this mutant over that of wild-type gp42. Truncation mutant Δ51 was unable to mediate fusion with B cells, a defect that can be attributed to its inability to bind to gH/gL, an interaction necessary for virus-induced membrane fusion to occur. These data indicate that truncation to residue 46 of the gp42 N terminus does not negatively affect the ability of gp42 to mediate membrane fusion with B cells.
The site of gp42 cleavage was found to be physically distinct from the residues of gp42 necessary for binding to gH/gL, indicating that mutation of the gp42 cleavage site does not disrupt gH/gL binding. A likely explanation for the defect in ability to mediate membrane fusion observed with gp42 cleavage mutants Δ37-41 and gp42-EK is that wild-type cleavage and subsequent secretion of gp42 promote membrane fusion. Cleavage of the protein may allow it to assume a favorable position or conformation during membrane fusion, possibly by promoting binding of gp42 to gH/gL. Tethering of gp42 to the cell membrane by the N-terminal transmembrane domain may inhibit protein flexibility, blocking the ability to mediate membrane fusion observed with secreted gp42. It is tempting to speculate that cleavage of gp42 promotes stronger binding to gH/gL, since truncation mutant Δ41, which begins at the predicted site of gp42 cleavage, shows increased binding to gH/gL at the surfaces of transfected cells. Mutant Δ41 is able to mediate significantly greater cell-cell fusion than wild-type gp42 in our virus-free cell-cell fusion assay. However, further experiments are necessary to determine the mechanism behind the greater membrane fusion activity observed with the gp42 Δ41 mutant.
We cannot dismiss the possibility that the membrane fusion defect observed with the gp42 cleavage mutants is due to residues 37 to 41 being essential for full-length gp42 to maintain a conformation that promotes membrane fusion. Deletion or mutation of these residues may inhibit this conformation, causing the observed defect. This explanation seems less likely, however, since no cellular or viral factors are known to bind gp42 at this location and structures of both native and HLA class-II-bound gp42 show that this region of the N terminus is disordered, indicating that it is flexible and not bound to another region of the protein (15, 22). A greater understanding of the role of the gp42 N terminus in B-cell membrane fusion awaits a crystal structure of the N-terminal region of gp42 in complex with gH/gL.
Currently, the physiological significance of the alternate forms of gp42, secreted and full-length, is unclear. It has previously been suggested that s-gp42 functions in immune evasion, potentially binding EBV entry receptors on lytic B cells and preventing reinfection, blocking antiviral humoral immunity by neutralizing gp42-specific antibodies, and inhibiting HLA class II-restricted T-cell recognition of EBV-producing B cells (26). In the current study, we have found that cleaved, secreted gp42 may exclusively mediate membrane fusion with B cells but that full-length membrane-bound gp42 may be inactive in this process. Further understanding of the functions of the differentially processed forms of gp42, secreted and full-length, requires experiments examining in greater depth the role of each form of the protein in mediating both membrane fusion and immune evasion. This study suggests that cleavage and secretion of gp42 may be necessary for membrane fusion with B cells, potentially identifying a new target for therapeutics against EBV entry and spread.
Acknowledgments
We thank Lindsey Hutt-Fletcher for her generous gift of the E1D1 antibody cell line. We thank the members of the Longnecker, Jardetzky, and Spear laboratories for help and support.
This research was supported by grant AI076183 from the National Institute of Allergy and Infectious Diseases and grant CA117794 from the National Cancer Institute to R.L. and T.S.J. J.S. was supported by predoctoral training grant T32 AI060523 from the National Institute of Allergy and Infectious Diseases.
Footnotes
Published ahead of print on 15 April 2009.
REFERENCES
- 1.Amon, W., and P. J. Farrell. 2005. Reactivation of Epstein-Barr virus from latency. Rev. Med. Virol. 15149-156. [DOI] [PubMed] [Google Scholar]
- 2.Backovic, M., T. S. Jardetzky, and R. Longnecker. 2007. Hydrophobic residues that form putative fusion loops of Epstein-Barr virus glycoprotein B are critical for fusion activity. J. Virol. 819596-9600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borza, C. M., and L. M. Hutt-Fletcher. 2002. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat. Med. 8594-599. [DOI] [PubMed] [Google Scholar]
- 4.Burkitt, D. 1962. A tumour syndrome affecting children in tropical Africa. Postgrad. Med. J. 3871-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fingeroth, J. D., M. E. Diamond, D. R. Sage, J. Hayman, and J. L. Yates. 1999. CD21-Dependent infection of an epithelial cell line, 293, by Epstein-Barr virus. J. Virol. 732115-2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haan, K. M., W. W. Kwok, R. Longnecker, and P. Speck. 2000. Epstein-Barr virus entry utilizing HLA-DP or HLA-DQ as a coreceptor. J. Virol. 742451-2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haan, K. M., S. K. Lee, and R. Longnecker. 2001. Different functional domains in the cytoplasmic tail of glycoprotein B are involved in Epstein-Barr virus-induced membrane fusion. Virology 290106-114. [DOI] [PubMed] [Google Scholar]
- 8.Haan, K. M., and R. Longnecker. 2000. Coreceptor restriction within the HLA-DQ locus for Epstein-Barr virus infection. Proc. Natl. Acad. Sci. USA 979252-9257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Henle, W., and G. Henle. 1970. Evidence for a relation of Epstein-Barr virus to Burkitt's lymphoma and nasopharyngeal carcinoma. Bibl. Haematol. 1970706-713. [DOI] [PubMed] [Google Scholar]
- 10.Henle, W., and G. Henle. 1969. The relation between the Epstein-Barr virus and infectious mononucleosis, Burkitt's lymphoma and cancer of the postnasal space. East Afr. Med. J. 46402-406. [PubMed] [Google Scholar]
- 11.Hutt-Fletcher, L. M. 2007. Epstein-Barr virus entry. J. Virol. 817825-7832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Janz, A., M. Oezel, C. Kurzeder, J. Mautner, D. Pich, M. Kost, W. Hammerschmidt, and H. J. Delecluse. 2000. Infectious Epstein-Barr virus lacking major glycoprotein BLLF1 (gp350/220) demonstrates the existence of additional viral ligands. J. Virol. 7410142-10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kirschner, A. N., A. S. Lowrey, R. Longnecker, and T. S. Jardetzky. 2007. Binding-site interactions between Epstein-Barr virus fusion proteins gp42 and gH/gL reveal a peptide that inhibits both epithelial and B-cell membrane fusion. J. Virol. 819216-9229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kirschner, A. N., J. Omerovic, B. Popov, R. Longnecker, and T. S. Jardetzky. 2006. Soluble Epstein-Barr virus glycoproteins gH, gL, and gp42 form a 1:1:1 stable complex that acts like soluble gp42 in B-cell fusion but not in epithelial cell fusion. J. Virol. 809444-9454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirschner, A. N., J. Sorem, R. Longnecker, and T. S. Jardetzky. 2009. Structure of Epstein-Barr virus glycoprotein 42 suggests a mechanism for triggering receptor-activated virus entry. Structure 17223-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.LaVallie, E. R., A. Rehemtulla, L. A. Racie, E. A. DiBlasio, C. Ferenz, K. L. Grant, A. Light, and J. M. McCoy. 1993. Cloning and functional expression of a cDNA encoding the catalytic subunit of bovine enterokinase. J. Biol. Chem. 26823311-23317. [PubMed] [Google Scholar]
- 17.Li, Q., M. K. Spriggs, S. Kovats, S. M. Turk, M. R. Comeau, B. Nepom, and L. M. Hutt-Fletcher. 1997. Epstein-Barr virus uses HLA class II as a cofactor for infection of B lymphocytes. J. Virol. 714657-4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li, Q., S. M. Turk, and L. M. Hutt-Fletcher. 1995. The Epstein-Barr virus (EBV) BZLF2 gene product associates with the gH and gL homologs of EBV and carries an epitope critical to infection of B cells but not of epithelial cells. J. Virol. 693987-3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McShane, M. P., and R. Longnecker. 2005. Analysis of fusion using a virus-free cell fusion assay. Methods Mol. Biol. 292187-196. [DOI] [PubMed] [Google Scholar]
- 20.McShane, M. P., and R. Longnecker. 2004. Cell-surface expression of a mutated Epstein-Barr virus glycoprotein B allows fusion independent of other viral proteins. Proc. Natl. Acad. Sci. USA 10117474-17479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McShane, M. P., M. M. Mullen, K. M. Haan, T. S. Jardetzky, and R. Longnecker. 2003. Mutational analysis of the HLA class II interaction with Epstein-Barr virus glycoprotein 42. J. Virol. 777655-7662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mullen, M. M., K. M. Haan, R. Longnecker, and T. S. Jardetzky. 2002. Structure of the Epstein-Barr virus gp42 protein bound to the MHC class II receptor HLA-DR1. Mol. Cell 9375-385. [DOI] [PubMed] [Google Scholar]
- 23.Nemerow, G. R., R. A. Houghten, M. D. Moore, and N. R. Cooper. 1989. Identification of an epitope in the major envelope protein of Epstein-Barr virus that mediates viral binding to the B lymphocyte EBV receptor (CR2). Cell 56369-377. [DOI] [PubMed] [Google Scholar]
- 24.Pereira, L. 1994. Function of glycoprotein B homologues of the family Herpesviridae. Infect. Agents Dis. 39-28. [PubMed] [Google Scholar]
- 25.Ressing, M. E., D. van Leeuwen, F. A. Verreck, R. Gomez, B. Heemskerk, M. Toebes, M. M. Mullen, T. S. Jardetzky, R. Longnecker, M. W. Schilham, T. H. Ottenhoff, J. Neefjes, T. N. Schumacher, L. M. Hutt-Fletcher, and E. J. Wiertz. 2003. Interference with T cell receptor-HLA-DR interactions by Epstein-Barr virus gp42 results in reduced T helper cell recognition. Proc. Natl. Acad. Sci. USA 10011583-11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ressing, M. E., D. van Leeuwen, F. A. Verreck, S. Keating, R. Gomez, K. L. Franken, T. H. Ottenhoff, M. Spriggs, T. N. Schumacher, L. M. Hutt-Fletcher, M. Rowe, and E. J. Wiertz. 2005. Epstein-Barr virus gp42 is posttranslationally modified to produce soluble gp42 that mediates HLA class II immune evasion. J. Virol. 79841-852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rickinson, A. B., and E. Kieff. 2007. Epstein-Barr virus, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
- 28.Silva, A. L., J. Omerovic, T. S. Jardetzky, and R. Longnecker. 2004. Mutational analyses of Epstein-Barr virus glycoprotein 42 reveal functional domains not involved in receptor binding but required for membrane fusion. J. Virol. 785946-5956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spear, P. G., and R. Longnecker. 2003. Herpesvirus entry: an update. J. Virol. 7710179-10185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Speck, P., and R. Longnecker. 1999. Epstein-Barr virus (EBV) infection visualized by EGFP expression demonstrates dependence on known mediators of EBV entry. Arch. Virol. 1441123-1137. [DOI] [PubMed] [Google Scholar]
- 31.Spriggs, M. K., R. J. Armitage, M. R. Comeau, L. Strockbine, T. Farrah, B. Macduff, D. Ulrich, M. R. Alderson, J. Mullberg, and J. I. Cohen. 1996. The extracellular domain of the Epstein-Barr virus BZLF2 protein binds the HLA-DR beta chain and inhibits antigen presentation. J. Virol. 705557-5563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strnad, B. C., T. Schuster, R. Klein, R. F. Hopkins III, T. Witmer, R. H. Neubauer, and H. Rabin. 1982. Production and characterization of monoclonal antibodies against the Epstein-Barr virus membrane antigen. J. Virol. 41258-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Takada, K. 2001. Role of Epstein-Barr virus in Burkitt's lymphoma. Curr. Top. Microbiol. Immunol. 258141-151. [DOI] [PubMed] [Google Scholar]
- 34.Tanner, J., J. Weis, D. Fearon, Y. Whang, and E. Kieff. 1987. Epstein-Barr virus gp350/220 binding to the B lymphocyte C3d receptor mediates adsorption, capping, and endocytosis. Cell 50203-213. [DOI] [PubMed] [Google Scholar]
- 35.Turk, S. M., R. Jiang, L. S. Chesnokova, and L. M. Hutt-Fletcher. 2006. Antibodies to gp350/220 enhance the ability of Epstein-Barr virus to infect epithelial cells. J. Virol. 809628-9633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei, W. I., and J. S. Sham. 2005. Nasopharyngeal carcinoma. Lancet 3652041-2054. [DOI] [PubMed] [Google Scholar]