

Abstract
Although replicons have been used to demonstrate hepatitis C virus (HCV) inhibition by alpha interferon (IFN-α), the detailed inhibition kinetics required to mathematically model HCV RNA decline have been lacking. Therefore, we measured genotype 1b subgenomic replicon (sg1b) RNA levels under various IFN-α concentrations to assess the inhibition kinetics of intracellular HCV RNA. During nine days of IFN-α treatment, sg1b RNA decreased in a biphasic, dose-dependent manner. Using frequent measurements to dissect these phases during IFN-α treatments of 100 and 250 U/ml revealed that the first-phase sg1b RNA decline began ∼12 h posttreatment, continued for 2 to 4 days, and then exhibited a distinct flat or slower second phase. Based on these data, we developed a mathematical model of IFN-α-induced intracellular sg1b RNA decline, and we show that the mechanism(s) mediating IFN-α inhibition of HCV acts primarily by reducing sg1b RNA amplification, with an additional effect on HCV RNA stability/degradation detectable at a dose of 250 U/ml IFN-α. While the extremely slow or flat second phase of viral RNA inhibition observed in vitro, in which there is little or no cell death, supports the in vivo modeling prediction that the more profound second-phase decline observed in IFN-α-treated patients reflects immune-mediated death/loss of productively infected cells, the second-phase decline in viral RNA with a dose of 250 U/ml IFN-α suggests that a further inhibition of intracellular HCV RNA levels may contribute as well. As such, dissection of HCV IFN-α inhibition kinetics in vitro has brought us closer to understanding the mechanism(s) by which IFN-α may be inhibiting HCV in vivo.
Hepatitis C virus (HCV) is remarkably efficient at establishing persistent infection, affecting approximately 200 million persons worldwide of whom 20% to 30% will likely progress to cirrhosis (17). However, the current treatment option of alpha interferon (IFN-α) in combination with ribavirin is ineffective in eliminating the virus in a large proportion of chronic hepatitis C patients (8, 13). Hence, there is a compelling need to better understand HCV infection and the dynamics of HCV inhibition achieved under antiviral therapy in order to optimize the use of IFN-α and ribavirin as well as new agents that are in development.
Over the last decade, clinical studies that included frequent serum sampling during treatment with IFN-α have afforded a detailed description of the extracellular viral RNA inhibition kinetics achieved in patients and thus have provided insights into HCV infection dynamics and treatment outcomes (7, 22). Specifically, it has been shown that during chronic HCV infection in the absence of therapy, the level of serum HCV RNA does not vary significantly (<0.5 log) on time scales of weeks to months (18). However, when chronically infected patients are treated with IFN-α alone or IFN-α plus ribavirin, HCV RNA typically declines in a biphasic pattern. The first rapid-phase decline, which begins 8 to 10 h after IFN-α administration, lasts ∼1 to 2 days in which HCV RNA, on average, falls 1 to 2 logs in genotype 1-infected patients (16, 20). Subsequently, a slower second phase of HCV RNA decline ensues.
Modeling the kinetics of HCV RNA decline observed during IFN-α treatment of patients suggested that the initial rapid phase of HCV RNA decline was due to a decrease in progeny virus production from infected cells (22) while the slower final phase was attributed to the net loss of productively infected cells (i.e., the cells capable of producing virus), which has generally been thought to be immune system mediated (3, 16, 24). Consistent with this hypothesis, it was recently reported that pegylated-IFN-α-2a-based treatment of HCV genotype-1b-infected severe combined immunodeficient mice transplanted with human hepatocytes resulted in a flat second phase of HCV RNA decline (5, 9).
Importantly, the subgenomic HCV replicon cell culture system has also become an important tool in investigating HCV replication and antiviral treatment (1). Previous studies have already shown that IFN-α reduces HCV replicon RNA levels in cells in a dose-dependent manner during the first days of treatment (i.e., before the first split of the culture) and can, with prolonged treatment and cell passaging, entirely eliminate HCV RNA from the cells (2, 11). However, it is not known if the elimination (i.e., “curing”) of the viral RNA from the host cells is mediated solely by IFN-α or if it is dependent upon the combination of IFN-α and the repeated splitting of the culture that by default occurs during this type of experiment. Theoretically, if the eradication of viral RNA was caused solely by IFN-α, then in the presence of IFN-α treatment one would expect to observe evidence of a continual decline in viral RNA in the absence of culture manipulations such as cell passaging. To try and specifically determine the kinetics of HCV RNA IFN-α-mediated inhibition achieved when cell culture passaging is eliminated from the experiment, several groups have designed “long-term” replicon culture assays in which subconfluent replicon cells are cultured and maintained in the presence or absence of IFN-α without splitting for 9 days (11, 12, 25). However, in these studies HCV genotype 1b subgenomic replicon (sg1b) RNA levels were measured only approximately every 3 days, which is insufficient for kinetic analysis and mathematical modeling.
In order to more thoroughly characterize the kinetics of HCV sg1b RNA inhibition during IFN-α treatment, we monitored with frequent sampling the level of intracellular sg1b RNA in long-term replicon cell cultures in the presence of various IFN-α concentrations. Interestingly, we observed a biphasic HCV RNA inhibition pattern in which a rapid first phase of sg1b RNA decline begins at ∼12 h posttreatment, followed by a slower or flat second phase of decline. To further elucidate the nature of the biphasic HCV sg1b RNA inhibition observed, we developed simple mathematical models and used those models to investigate the process (e.g., lowered production or enhanced degradation) by which sg1b RNA reduction is mediated in the presence of IFN-α. This and other in vitro mathematical modeling efforts should prove useful for dissecting the detailed mechanism(s) by which IFN-α inhibits HCV in infected patients.
MATERIALS AND METHODS
HCV subgenomic replicon cells and interferons.
The clone B HCV sg1b Huh7 cells were obtained from the NIH AIDS Research and Reference Reagent Program and have been previously described (2). The cells were cultured in complete Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 100 units/ml penicillin, 100 mg/ml streptomycin, 2 mM l-glutamine, and 500 μg/ml Geneticin (Invitrogen). Recombinant human IFN-α-2a (PBL Biomedical Laboratories, New Brunswick, NJ) was resuspended to a concentration of 1 U/μl in complete DMEM supplemented with 10% fetal bovine serum, aliquoted into single-use tubes, and stored at −80°C.
IFN-α HCV replicon inhibition assay.
To ensure that the cells would not reach confluence and require splitting during our 9-day IFN-α treatment experiment, sg1b cells were seeded at low density (∼5 × 104 cells/well) in six-well tissue-culture dishes in complete DMEM. One day later, cells were mock treated or treated with IFN-α at 100, 250, 500, and 1,000 U/ml concentrations in a total volume of 2 ml of complete DMEM without Geneticin. Fresh vehicle control or IFN-α containing medium was added to the cultures every 12 h for the entire duration of the experiment to ensure continuous IFN signaling. At the indicated times (see the figures), medium was harvested from triplicate wells for cytotoxicity analysis, and RNA was isolated from cells in 500 μl of 1× Nucleic Acid Purification Lysis Solution (Applied Biosystems, Foster City, CA) for real-time reverse transcription-quantitative PCR analysis (RT-qPCR).
Cytotoxicity assay.
The release of lactate dehydrogenase (LDH) from cells into the culture medium occurs when cell membrane integrity is compromised, and thus this release can be used to assess cytotoxicity. Hence, LDH levels in the medium of IFN-α-treated cultures were determined using a CytoTox-ONE Homogeneous Membrane Integrity Assay (Promega, Madison, WI), according to the manufacturer's instructions. Detection of the fluorescent resorufin product was assessed using an excitation wavelength of 560 nm and an emission wavelength of 590 nm (FLUOstar Optima fluorometer; BMG Labtech, Durham, NC). A complete cell lysis sample was run as a positive control, and medium alone was run as a negative control.
RNA analysis.
Total cellular RNA was isolated using a 1× Nucleic Acid Purification Lysis Solution (Applied Biosystems) and purified using an ABI Prism 6100 Nucleic Acid PrepStation (Applied Biosystems), as per the manufacturer's instructions. One microgram of purified RNA was used for cDNA synthesis using the TaqMan RT reagents (Applied Biosystems), followed by SYBR Green RT-qPCR using an Applied Biosystems 7300 real-time thermocycler (Applied Biosystems). Thermal cycling consisted of an initial denaturation step for 10 min at 95°C, followed by 40 cycles of denaturation (15 s at 95°C) and annealing/extension (1 min at 60°C). HCV, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), IFN-stimulated gene 15 (ISG15), and ISG56 transcript levels were determined relative to a standard curve comprised of serial dilutions of plasmid containing the relevant cDNAs. The PCR primers used to detect genotype 1b HCV were 5′-CGACACTCCACCATAGATCACT-3′ (sense) and 5′-GAGGCTGCACGACACTCATACT-3′ (antisense); for human GAPDH (NMX002046), the primers were 5′-GAAGGTGAAGGTCGGAGTC-3′ (sense) and 5′-GAAGATGGTGATGGGATTTC-3′ (antisense); for human ISG15 (NM_005101), the primers were 5′-CAGCGAACTCATCTTTGCCAGTA-3′ (sense) and 5′-CCAGCATCTTCACCGTCAGG-3′ (antisense); and for ISG56 (NM_001001887), the primers were 5′-GGGCAGACTGGCAGAAGC-3′ (sense) and 5′-TATAGCGGAAGGGATTTGAAAGC-3′ (antisense).
Indirect immunofluorescence.
At 5, 6, 7, and 8 days posttreatment, one four-chamber slide containing mock-treated and 100-U/ml and 250-U/ml IFN-α-treated sg1b cell cultures was fixed with 4% paraformaldehyde (Sigma), and intracellular staining was performed as previously described (10, 28). Briefly, the mouse monoclonal anti-HCV NS5A antibody E9-10 (a gift from Charlie Rice) was used at a dilution of 1:500, followed by incubation with a 1:1,000 dilution of an Alexa 555-conjugated goat anti-mouse immunoglobulin G antibody (Molecular Probes, Carlsbad, CA) for 1 h at room temperature. Cell nuclei were stained by Hoechst dye. Chambers were removed, and stained cells were coverslipped and mounted in Prolong antifade mounting medium (Molecular Probes) and visualized on a Zeiss Axiovert LSM 510. Images were generated using LSM Image Browser software and exported to Adobe Photoshop (Adobe Systems Inc., San Jose, CA).
Statistical analysis.
To compare mean sg1b RNA levels at each time point between IFN-α treatments, we used the general linear model univariate analysis method. All multiple comparisons were adjusted using a Bonferoni correction, and in all cases, a P value of ≤ 0.05 was considered significant (SPSS software, version 15, Chicago, IL). To determine whether sg1b RNA levels differ between IFN-α treatment doses over time, we used Friedman's test, the nonparametric analogue of the two-way analysis of variance (S-PLUS, version 7.0.2; Seattle, WA). HCV sg1b RNA decline slopes were measured by linear regression (SPSS, version 15; Chicago, IL). Nonlinear least-squares regression using a Levenberg-Marquardt algorithm was used to fit the solutions of mathematical models to the HCV RNA decline data.
RESULTS
Increasing concentrations of IFN-α are not toxic but inhibit replicon cell growth.
It has been reported that the growth state of the host cell can have detectable effects on the replication rate of HCV replicons (14, 23, 27). Because we wanted to ensure that our HCV IFN-α inhibition experiments were performed under conditions that limited any nonspecific effects IFN-α might have on the host cell, we initially monitored the effect that increasing IFN-α doses (i.e., 100, 250, 500, and 1,000 U/ml) had on Huh7 clone B cell viability and growth (Fig. 1). Mimicking the 9-day IFN-α inhibition assay described in Materials and Methods, replicon cells were plated at a low density (5 × 104 cells/well) in six-well tissue culture dishes and 1 day later were mock treated or treated every 12 h with IFN-α at various doses for 2, 3, 4, 6, 8, or 9 days. As shown in Fig. 1, cell growth was similar in all cultures regardless of the IFN-α dose until day 6 posttreatment. At this time, however, cells treated with 1,000 U/ml IFN-α stopped dividing and remained at a cell confluence of 60% (Fig. 1). Likewise, the expansion of cells treated with 500 U/ml IFN-α slowed between days 6 to 8 and then stopped by day 8 when the cells reached 70% confluence of (Fig. 1). In contrast, cells treated with 100 and 250 U/ml IFN-α expanded at a rate equivalent to mock-treated control cultures (Fig. 1) until at least day 8 (Fig. 1).
FIG. 1.

HCV replicon cell growth during treatment with various concentrations of IFN-α. HCV replicon cells were incubated with various concentrations of IFN-α for 2, 3, 4, 6, 8, or 9 days. Fresh IFN-α was added to the medium every 12 h. Cells were trypsinized and counted at indicated time points posttreatment. Cell number, expressed as relative confluence of the culture, was assessed as an indicator of relative cell growth rates.
To determine if any of these IFN-α treatment doses were toxic, we measured the release of LDH into the culture medium. The results indicated that none of the IFN-α concentrations tested (i.e., 100 to 1,000 U/ml) were toxic as LDH concentrations measured from treated cultures were comparable to the background LDH concentrations measured in the control cultures (data not shown).
IFN-α reduces intracellular subgenomic HCV RNA levels in a biphasic-like manner.
To analyze the effect of IFN-α on HCV sg1b RNA levels over time during this 9-day treatment, parallel replicon cell cultures were treated with the same four IFN-α concentrations tested above, i.e., 100, 250, 500, and 1,000 U/ml, and sg1b RNA was measured in triplicate samples by RT-qPCR analysis after 2, 3, 4, 6, 8, and 9 days of treatment (Fig. 2). Interestingly, when treated with 100 U/ml IFN-α, sg1b RNA levels fell in a biphasic manner that consisted of a rapid first-phase RNA decline (that lasted until about day 4), followed by an extremely slow or flat second phase (Fig. 2 and Table 1). The rapid first-phase decline appeared shorter (lasting only until day 2) in cultures treated with 250 U/ml, 500 U/ml, or 1,000 U/ml IFN-α (Fig. 2) and then was followed by a slower decline; however, since growth of the replicon cells treated with 500 U/ml or 1,000 U/ml IFN-α was compromised (i.e., slowed or stopped) by day 6 of treatment, the antiviral effect of IFN-α on HCV RNA levels cannot be accurately assessed after day 6 under these treatment conditions (i.e., some reduction in HCV RNA levels could be due to the decrease in cell growth). In Table 1 the sg1b RNA reduction rates (per day) and decline ratios from baseline for each IFN-α treatment are shown, indicating that the effect of IFN-α concentration on sg1b RNA levels was dose dependent (P < 0.01) (Fig. 2).
FIG. 2.

Subgenomic HCV RNA kinetics during IFN-α treatment is biphasic and dose dependent. HCV replicon cells were incubated with various concentrations of IFN-α and harvested from triplicate wells at the indicated time points. Detailed quantification analysis is provided in Table 1. Asterisks indicate time points at which sg1b RNA levels were significantly different (P < 0.01) among the various IFN-α concentrations using a one-way analysis of variance test and posthoc (Bonferroni) test. The size of the symbols is larger than the standard error of the mean.
TABLE 1.
Kinetics of sg1b HCV RNA decline under various IFN-α concentrations
| Sampling method and IFN-α concn (U/ml) | Rapid-phase kineticsa
|
Slow-phase kineticsa
|
||
|---|---|---|---|---|
| Mean sg1b RNA log10 reduction from baseline (V0/Vrapid) | sg1b RNA decline rate per day | Mean sg1b RNA log10 reduction from baseline (V0/Vend) | sg1b RNA decline rate per day | |
| Nonfrequente | ||||
| Control | 0.15 | 0.26 ± 0.11 | 0.02 | (0.14) ± 0.03 |
| 100 | 1.59 | 1.34 ± 0.15d | 1.81 | 0.1 ± 0.02d |
| 250 | 1.16 | 2.51 ± 0.22d | 2.29 | 0.33 ± 0.03d |
| 500c | 1.27 | 2.62 ± 0.04d | 2.51 | 0.70 ± 0.04d |
| 1,000c | 1.48 | 2.85 ± 0.01d | 2.88 | 0.79 ± 0.03d |
| Frequentb | ||||
| Control | 0.05 | 0.03 ± 0.02 | 0.05 | 0.03 ± 0.02 |
| 100 | 1.49 | 0.79 ± 0.02d | 1.53 | 0.05 ± 0.03 |
| 250 | 1.48 | 1.47 ± 0.07d | 2.47 | 0.33 ± 0.04d |
V0, sg1b level at baseline; Vrapid, sg1b level at day 2 (with 250, 500, and 1,000 U/ml IFN-α) or at day 4 (with 0 U/ml and 100 U/ml IFN-α); Vend, mean sg1b RNA levels at day 9 (with 0, 100, and 250 U/ml IFN) or day 6 (with 500 and 1,000 U/ml IFN-α). Parentheses are used to indicate an sg1b RNA increase.
Results from the frequent sampling of 100- and 250-U/ml IFN-α experiments shown in Fig. 3C. Because the first-phase decline was determined to last until 4.5 and 2.5 days in these experiments, respectively, the Vrapid and sg1b rapid decay rates were calculated through days 4.5 and 2.5, while the sg1b RNA decline rate during the slower phase was calculated from day 4.5 and 2.5, respectively, to day 8, i.e., Vend is 8.
Calculated until day 6 due to inhibition of cell growth from day 6 (Fig. 1).
sg1b decline rate is significantly different from 0 (P < 0.01).
Results from the nonfrequent sampling of 100- to 1,000-U/ml IFN-α experiments shown in Fig. 2.
Detailed sg1b RNA inhibition kinetics during 100- and 250-U/ml IFN-α treatment.
Because doses of 500 U/ml and 1,000 U/ml of IFN affect cell proliferation, which is known to negatively impact HCV RNA replication levels in cell culture (14, 23, 27), it may not be appropriate to analyze the kinetics of sg1b RNA decline under these conditions. However, to further characterize and dissect sg1b RNA IFN-α inhibition kinetics for the purposes of mathematical modeling, we repeated the inhibition assay at doses of 100 and 250 U/ml IFN-α but with more frequent sampling. Specifically, we harvested RNA from mock- and IFN-α-treated sg1b cells every 2 h during the first day, every 4 h during the second day, and then every 4 to 12 h until day 8 (Fig. 3).
FIG. 3.

Kinetic analysis and modeling of the in vitro data for doses of 100 and 250 U/ml of IFN-α. HCV replicon cells were incubated with 100 or 250 U/ml of IFN-α, and triplicate RNA samples were collected for real-time qPCR analysis every 2 h from 0 to 24 h, every 4 h from 24 to 60 h, and then every 12 h until day 8. (A) RT-qPCR analysis of ISG56 mRNA levels in mock-treated cell (circles) and in cells treated with 100 U/ml (squares) and 250 U/ml (triangles) IFN-α throughout the 8-day experiment. (B) RT-qPCR analysis of sg1b HCV RNA levels in mock-treated cells (circles) and in cells treated with 100 U/ml (squares) and 250 U/ml (triangles) IFN-α during the first 24 h of treatment. Open arrow, sg1b RNA levels in IFN-α-treated cells significantly (P = 0.05) lower than in control cells; filled arrow, sg1b RNA levels in 250 U/ml IFN-α-treated cells significantly (P = 0.05) lower than in 100 U/ml IFN-α-treated cells. (C) Fit of model equations to sg1b RNA decline data. Solid line denotes the best fit of equations 2 and 4 or equation 6 with g of ∼0 to HCV RNA levels obtained with 100 U/ml IFN-α. Dashed line indicates the best fit of equation 6 to the measured 250-U/ml IFN-α sg1b RNA levels. The parameter values that generated the best-fit theoretical curves (solid and dashed lines) are shown in Table 2. The size of the squares represents the largest standard error of the mean of sg1b RNA. Vertical lines represent standard error of the mean.
To confirm constitutive, dose-dependent IFN signaling throughout the 8-day experiment, cellular expression levels of ISGs, ISG56 (Fig. 3A) and ISG15 (data not shown), were monitored. In Fig. 3A we show that ISG56 mRNA levels in IFN-α-treated cells is ∼1.5 logs higher than in the mock-treated cells by 6 h posttreatment and that this level is maintained throughout the experiment. Notably, by 12 h posttreatment ISG56 levels were constitutively higher in cells treated with 250 U/ml IFN-α than in cells treated with 100 U/ml IFN-α, directly demonstrating the dose responsiveness of the cells to the concentrations of IFN-α. A similar expression pattern was observed for ISG15 (data not shown).
Reminiscent of the delayed response observed in infected patients after the initiation of IFN-α therapy (16), during the first ∼12 h of IFN-α treatment the levels of HCV sg1b RNA in both the 100- and 250-U/ml IFN-α-treated cultures were similar to the level measured in the mock-treated control cultures (Fig. 3B). After this delay, the first-phase decline of sg1b RNA in the 100-U/ml IFN-α-treated cultures occurred at a rate of 0.79 ± 0.02 day−1 ending at approximately day 4.5. This first-phase decline of 1.5 log10 represents a 97% reduction in HCV sg1b RNA; hence, we define 100 U/ml IFN-α as having an in vitro efficacy of 97%. After this rapid 97% drop, a flat second phase was observed (characterized by a “decline” rate of 0.04 ± 0.07 day−1, which is not significantly different from 0 [P = 0.6]), resulting in sg1b RNA levels that remained at ∼1.5 log10 below baseline until day 8 (Fig. 3C and Table 1). In contrast, in the 250-U/ml cultures, a first-phase 1.5 log10 decline of sg1b RNA occurred at a rate of 1.47 ± 0.07 day−1, ending at approximately day 2.5. This decline rate is significantly (P < 0.001) higher than with 100 U/ml IFN-α, which can be noticed from 18 h postinitiation of treatment (Fig. 3B). After this rapid 97% drop, a slower decline ensued, resulting in sg1b RNA levels reaching 2.47 log10 below baseline by day 8 (Fig. 3C and Table 1).
Notably, in both the 100- and 250-U/ml IFN-α-treated cultures, the majority of cells exhibited significantly reduced HCV NS5A protein levels by day 6, i.e., during the second phase of sg1b RNA decline (Fig. 4). However, it is interesting that a small percentage of cells (∼2%) in the IFN-α-treated cultures did retain slightly higher NS5A levels more comparable to the level observed in untreated cells (Fig. 4). Because these NS5A-positive cells did not disappear during treatment before the cells became confluent at day 8, it remains to be determined if HCV inhibition was randomly proceeding more slowly in these cells or if they represent a specific subset of cells that are more resistant to the effects of IFN-α. In either case, in the context of these experiments, the HCV RNA present in these cells does contribute to the number of HCV RNA copies present in the cultures throughout the experiment. Therefore, in the following sections we developed simple average or so-called mean-field models and fit them to the experimental data to provide insights into the viral RNA dynamics during IFN-α treatment.
FIG. 4.

HCV NS5A levels during second-phase IFN-α inhibition of sg1b RNA. At 5, 6, 7, and 8 days posttreatment, mock-treated cells (A) and sg1b cell cultures treated with 100 U/ml (B) and 250 U/ml (C) IFN-α were fixed and stained for HCV NS5A. Cell nuclei were stained by Hoechst dye. Shown are representative ×20 images taken at day 6 posttreatment. HCV NS5A is shown in red (Alexa 555), and nuclei are shown in blue (Hoechst). Bar, 50 μm.
Modeling sg1b RNA inhibition kinetics during 100-U/ml IFN-α treatment.
In order to model the effect of IFN-α on sg1b RNA, we initially focused on the 100-U/ml IFN-α inhibition data and assumed that in the absence of IFN-α, replicon cells produce sg1b RNA at a constant rate, α, and degrade sg1b RNA by a first-order process with rate constant μ. Notably, because replicon RNA is normalized to the total cellular RNA, reduction of sg1b RNA copy number on a per-cell basis due to cell division can be neglected, assuming that total cellular RNA and sg1b RNA are diluted to the same extent as replicon cells divide.
In order to account for the reduction of sg1b RNA levels during IFN-α therapy, we tested two alternative hypotheses. First, we assumed some process that supports sg1b RNA production after a time (t) delay, t0, is affected by IFN-α, resulting in the rate of sg1b RNA production changing from α to (1 − ɛin)α, where ɛin (0 ≤ ɛin ≤ 1) is the effectiveness of IFN-α in reducing intracellular sg1b RNA production. Thus, if R is the intracellular concentration of sg1b RNA, this model can be described by the equation
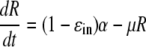 |
(1) |
with the solution
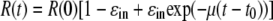 |
(2) |
where t0 is a parameter introduced to account for the delay from the time of IFN-α administration until sg1b RNA decline is observed, R(0) is the amount of intracellular sg1b RNA at the start of therapy, i.e., at t = 0, and we have assumed that at the start of therapy α = μR(0), i.e., replicon production has reached steady state. We make this assumption because the level of sg1b RNA is constant in control cells and because the level in IFN-α-treated cells is also approximately constant for the first 12 h after IFN-α administration (Fig. 3B). Fitting the natural logarithm of R(t), given by equation 2 to the natural logarithm of the experimental data using nonlinear least squares regression, we find that there is good agreement between the model and the data (Fig. 3C), with the best-fit parameter values being a ɛin of 0.972, μ of 0.98 day−1, and t0 of 9.5 h (Table 2). Hence, under the assumption that only the sg1b RNA production rate is being altered during IFN-α treatment, parameter estimates suggest that 100 U/ml of IFN can decrease sg1b RNA production by 97.2% and that at steady state sg1b RNA is degraded by a first-order process with a rate constant of ∼0.98 day−1, which corresponds to an sg1b RNA half-life (t1/2) of (ln 2)/0.98, or 0.71 days, i.e., 17 h (95% confidence interval [CI], 16 to 19 h). This t1/2 estimate is somewhat shorter but corresponds reasonably well with the 19 h (95% CI, 18 to 21) we calculated by linear regression during 4 days of treatment of the sg1b cells with the HCV NS5B inhibitor, NM107, which specifically blocks HCV RNA production (N. Baretto et al., unpublished data). Likewise, the 9.5-h estimated value of t0 calculated by equation 2 corresponds well with the empirically observed delay in the sg1b RNA decline we observed after IFN-α is added to the culture medium.
TABLE 2.
Parameter estimates obtained by fitting models to the dataa
| Modelb | IFN-α concn (U/ml) | R(0) (106 copies/μg RNA) | t0 (h) | ɛin | μ (per day) | g (per day) |
|---|---|---|---|---|---|---|
| Eq. 2 | 100 | 2.06 (1.81-2.35) | 9.5 (5.1-13.8) | 0.972 (0.966-0.988) | 0.98 (0.88-1.07) | NAc |
| Eq. 4 | 100 | 2.06 (1.81-2.35) | 9.5 (5.1-13.8) | 0.972 (0.966-0.978) | 0.027 (0.02-0.04) | NA |
| Eq. 6 | 100 | 2.06 (1.81-2.35) | 9.5 (3.6-15.4) | 0.972 (0.966-0.978) | 0.98 (0.85-1.11) | 3 × 10−8 (0-0.001)d |
| Eq. 6 | 250 | 2.29 (1.74-3.03) | 7.8 (3.1-12.4) | 0.927 (0.883-0.970) | 2.32 (1.57-3.07) | 0.33 (0.217-0.448) |
Best fits to the frequent sg1b data with 100 and 250 U/ml IFN-α are shown in Fig. 3C. Values in parentheses are 95% CIs. R(0), baseline sg1b RNA concentration; t0, delay until sg1b RNA starts to decline; ɛin, IFN-α effectiveness in blocking sg1b RNA production; μ, sg1b RNA degradation; g, slowing of sg1b RNA production.
Eq, equation.
The lower 95% CI (g = 0) is consistent with a flat second phase.
Alternatively, we examined the hypothesis that instead of reducing RNA production, IFN-α might increase replicon clearance/degradation. To describe this scenario, the following alternative form of the model is used:
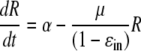 |
(3) |
where 1/(1 − ɛin) is the factor by which clearance is increased. Since ɛin is <1, this factor is greater than 1. If one again assumes the system is at steady state before IFN-α treatment, the solution of this model is
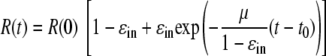 |
(4) |
Note that this equation is the same as equation 2 except that μ in equation 2 is replaced by the expression μ̂ = μ/(1 − ɛin) in equation 4. However, if we use equation 4 to fit the experimental data, we obtain the parameter estimates of a ɛin of 0.972, μ of 0.027 day−1, and t0 of 9.5 h (Table 2). This smaller estimate for the natural rate of loss of replicons, μ, occurs because we have assumed that in the presence of IFN-α the loss rate increases by the factor 1/(1 − ɛin), i.e., ∼30-fold. Thus, although this model gives exactly the same fit to the data as the one shown in Fig. 3C, it predicts that the intracellular t1/2 of replicon RNA in the absence of IFN-α is (ln 2)/0.027, or 26 days.
Given the usual rapid turnover of RNAs in cells and the sg1b t1/2 of 19 h measured in the presence of HCV NS5B inhibitor (Baretto et al., unpublished), we can therefore reject the latter hypothesis in favor of the former, which states that IFN-α is acts by reducing the level of HCV RNA production. While it is still possible that IFN-α not only reduces sg1b RNA production but also has a smaller effect on increasing sg1b RNA clearance, this analysis supports the conclusion that inhibition of HCV RNA synthesis is the primary effect observed during treatment with 100 U/ml IFN-α.
Modeling sg1b RNA inhibition kinetics during 250-U/ml IFN-α treatment.
Examining equation 1 suggests that, rather than having a flat second phase, a decline of sg1b RNA during treatment with 250 U/ml IFN would occur if the intrinsic rate of replicon production, α, in addition to being initially inhibited by a factor of 1 − ɛin, also decreases with time on therapy, say due to the net destruction of negative-strand RNA or the gradual loss of some other components of the replication complex. The continued reduction of such replication components would be expected to result in a further slowing of HCV sg1b RNA synthesis after the completion of the first phase, resulting in a decrease in sg1b RNA levels in the second phase. To incorporate this concept into the model, we assume that (1 − ɛin)α, rather than being a constant, is a function that decreases with time, for example, (1 −ɛin)α0 exp(−gt), where g is a constant and t is time posttreatment. Then equation 1 would become
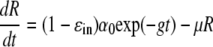 |
(5) |
with the solution
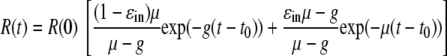 |
(6) |
where we have assumed a delay, t0, before IFN-α has any effect and that replicon production is at steady state at the start of therapy, i.e., α0 = μR(0). Importantly, comparing equation 6 to our previous simple model, equation 2, shows that when g is 0, the two equations are identical, and the same flat second phase predicted in Fig. 3C (solid line) is obtained. However, as intended, for any g that is >0, the flat second phase disappears, and we instead obtain a decline in the second phase (Fig. 3C). Fitting the natural logarithm of R(t), given by equation 6 to the natural logarithm of the 250-U/ml IFN-α experimental data (Fig. 3C) using nonlinear least squares regression, we find that there is good agreement between the model and the data, with the best-fit parameter values being a ɛin of 0.927, μ of 2.32 day−1, g of 0.33 day−1, and t0 of 7.8 h (Table 2). Interestingly, while the estimated degrees of inhibition of the sg1b RNA production rate under 100 and 250 U/ml IFN-α, ɛin, have overlapping 95% CIs (Table 2), the sg1b RNA degradation rate, μ, is approximately twofold higher in the presence of 250 U/ml.
DISCUSSION
In this study we investigated the kinetics of HCV sg1b RNA decline in the presence of IFN-α. Our results indicate that after a ∼12 h delay, IFN-α treatment leads to a biphasic decline in sg1b RNA in Huh7 HCV replicon cells. Under 100- and 250-U/ml IFN-α treatment, this biphasic pattern is characterized by a first-phase rapid decline in which sg1b RNA is reduced by ∼97% from baseline, followed by a flat or slow second-phase decline.
Using mathematical models to interpret the kinetics of sg1b RNA decline under IFN-α treatment and to deduce the nature of the mechanism(s) that mediate the observed decline, we conclude that IFN-α acts by profoundly reducing the production of sg1b RNA. Specifically, a model based on the hypothesis that IFN-α reduces replicon production fits the data obtained with 100 U/ml of IFN-α with reasonable biological parameters (Fig. 3C and Table 2). If only production of sg1b RNA is affected, as assumed in these model fits, this would correspond to 100 U/ml of IFN-α blocking sg1b production with ∼97% effectiveness, with an estimated sg1b RNA intracellular t1/2 of ∼17 h. While using a model based on the hypothesis that 100 U/ml IFN-α increases the rate of sg1b degradation could also fit the observed decline kinetics, it resulted in an estimate of the replicon t1/2 of about 26 days. Since we along with others (19; also Baretto et al., unpublished) have shown that intracellular HCV RNAs typically have t1/2 values on the order of 11 to 19 h in the presence of specific inhibitors against HCV replication, we believe this rules out the possibility that the main mode of IFN-α action at 100 U/ml is to increase sg1b RNA degradation.
Higher doses of IFN-α (e.g., 250 U/ml) (Fig. 3C) increased the sg1b RNA degradation rate (Table 1), accelerated the first-phase drop (from ∼4.5 to ∼2.5 days), and led to a more readily detectable second-phase decline in the sg1b RNA level. To model the observed sg1b RNA kinetics, we included the concept that in addition to sg1b RNA being initially inhibited by the factor of 1 − ɛin, the gradual loss of negative-strand RNA or some other components of the preexisting replication complexes would logically result in a further time-dependent inhibition of HCV sg1b RNA (equation 6). The resulting equation 6 illustrates that it is theoretically possible that intracellular RNA levels could continue to decline in the second phase (Fig. 3C) rather than reaching a lower plateau, as was observed during 100 U/ml of IFN-α treatment. Fitting equation 6 to experimental data, we predict that while the effectiveness of 250 U/ml IFN-α in blocking sg1b RNA production during the first phase is comparable to that of the 100-U/ml IFN-α dose, it results in more than a twofold increase in the sg1b degradation rate than under 100 U/ml IFN-α along with further inhibition of HCV sg1b RNA production with 250 U/ml IFN-α during the second phase (Table 2).
More complex modeling of replicon inhibition dynamics also supports the notion that IFN-α does to some extent enhance HCV RNA decay (H. Dahari. B. Sainz, K. Marsh, A. S. Perelson, and S. L. Uprichard, presented at the 15th International Symposium on HCV and Related Viruses, San Antonio, TX, 5 to 9 October, 2008). Briefly, expanding upon the mathematical model we previously developed to describe the virus-host dynamics of subgenomic HCV replication in vitro from transfection to steady state (6), we added the potential of IFN-α to act at multiple levels (e.g., blocking HCV protein production, blocking sg1b RNA synthesis, or enhancing sg1b RNA degradation). By running model simulations of a large spectrum of possible IFN-α modes of action and comparing the model predictions to the kinetics of sg1b RNA decline observed empirically (Fig. 3C), we found that a combination of slowing sg1b protein production and sg1b RNA synthesis fits the 100-U/ml IFN-α data well, and with an additional enhancement of the sg1b RNA degradation rate the model also agrees well with the 250-U/ml IFN-α data (H. Dahari et al., unpublished data). Consistent with this possibility, recent publications by Pedersen et al. (21) and Taylor et al. (26) report that intracellular viral RNA can be degraded by IFN system-induced cellular microRNAs and by RNA editing that acts on double-stranded RNA.
The model given by equation 6 gives rise to a biphasic sg1b RNA decline under therapy. Examining the decline obtained with 250 U/ml IFN-α closely (Fig. 3C), one could envision that a model with a triphasic decline could also fit the data, with an initial rapid phase followed by a plateau (from day 2.5 to 5) that is then followed by a further decline. This pattern could be accommodated within the context of our model by assuming that the destruction of components necessary for HCV RNA replication begins only after some delay, e.g., 5 days in the above example.
Of interest, the kinetics of intracellular sg1b decline during IFN-α treatment in vitro is in several ways reminiscent of the HCV RNA inhibition kinetics seen in IFN-α-treated patients, and thus a comparison of the inhibition patterns may provide insights into the nature of the biphasic inhibition observed in vivo. First, during both in vitro and in vivo IFN-α treatment, evidence of viral RNA decline is not observed until several hours after initiation of treatment. While the delay observed in patients was previously speculated to be due to the pharmacokinetics of IFN and/or the time it takes for the drug to reach its maximum serum concentration (16), the fact that a similar delay is also observed in vitro suggests that this delay may rather reflect the time needed for intracellular signaling events to impact HCV RNA levels.
Likewise, in both cases (i.e., IFN-α-treated replicon cells and IFN-α-treated patients), a biphasic decline pattern is observed, with viral RNA levels declining as much as 99% from baseline during the first phase. To explain the rapid first-phase decline in HCV RNA observed in patients treated with IFN-α, we previously had assumed that IFN-α reduces viral production by a factor of 1 − ɛ, where ɛ (0 ≤ ɛ ≤ 1) was the effectiveness of therapy. Thus, we assumed that while patients were on therapy, infected hepatocytes produced lower levels of virus (in contrast to enhancing virus removal from the serum). Using the in vitro replicon system, we have been able to directly monitor intracellular HCV RNA levels and confirm that in IFN-α-treated replicon cells, intracellular sg1b RNA levels have a rapid first-phase fall and attain a new rate of production consistent with a decrease by the factor of 1 −ɛin, where ɛin is the effectiveness of therapy in reducing intracellular viral RNA production, suggesting that the effectiveness of IFN-α therapy measured in patients, ɛ, may be directly related to ɛin.
While the pattern of HCV inhibition (i.e., biphasic) during IFN-α treatment in the replicon system and chronic HCV patients have many similarities, the slope and length of the first phase HCV RNA decline in replicon cells are slower, continuing ∼2 to 3 days longer with 100 U/ml IFN-α and ∼1 to 2 days longer with 250 U/ml IFN-α than seen in vivo (16). This raises questions as to whether the effect of IFN-α in reducing intracellular HCV RNA is more potent in vivo than in vitro or if additional factors contribute to and enhance the rate of extracellular viral clearance observed in patient serum during the first phase of inhibition. Of course, practically speaking, the slope of first-phase decline in patient serum would be expected to be governed not only by the reduction of intracellular de novo HCV production but also by virion clearance from the serum. Hence, the serum clearance rate in patients probably does reflect a variety of additional processes, such an antibody-mediated opsonization, complement-mediated lysis, and phagocytosis, and therefore would be expected to occur faster in patients than in vitro. Additionally, whether other factors, such as paracrine signaling (which is absent in Huh7 cell cultures), contribute to an amplification of the initial IFN-α effect and thus impact first- and/or second-phase inhibition kinetics remains to be determined.
It is known that different HCV genotypes exhibit differential sensitivity to IFN-α treatment and produce distinct liver pathologies (4, 15, 18). We used the HCV genotype-1b clone B replicon for our studies as it is a commonly used HCV replicon, which we previously used to mathematically model subgenomic HCV replication from transfection to steady state (6); however, similarly detailed modeling of other HCV genotypes and comparison of their IFN-α inhibition kinetics might reveal the basis of the distinct IFN-α responsiveness exhibited by the different HCV genotypes in patients. Likewise, comparing the inhibition kinetics of HCV under treatment with different IFNs (e.g., IFN-α-2a, IFN-α-2b, IFN-γ, and IFN-λ) might provide insight into the distinct activities of these related antiviral factors. One caveat to this approach, of course, is that for the moment HCV genotype 1 replication is restricted to a single human hepatoma cell line, Huh7, which is known to be defective in some aspects of IFN-α signaling. Nonetheless, here we have shown the value of comparing HCV inhibition kinetics in vitro with that observed in patients as this seems to provide a means of dissecting the more complex IFN-α effects observed in vivo.
In summary, we provide here a detailed description of sg1b RNA inhibition kinetics during IFN-α treatment and show that a comparison of both the similarities and differences in HCV IFN-α inhibition kinetics observed in vitro to the kinetics observed in vivo can help us understand the mechanism(s) by which IFN-α may be inhibiting HCV in vivo. Specifically, we found that sg1b RNA drops in a biphasic manner after an initial ∼12-h delay, similar to the HCV inhibition pattern observed in HCV-infected patients during IFN-α treatment. By creating mathematical models to test various hypotheses regarding the primary mechanism by which IFN-α reduces intracellular sglb RNA levels, we provide evidence that IFN-α acts mainly by reducing the rate of viral RNA production, with an additional effect on HCV RNA stability/degradation detectable at the 250-U/ml IFN-α dose. The extremely slow or flat second phase of viral RNA inhibition observed in the sglb RNA levels in vitro during 100-U/ml IFN-α treatment supports previous modeling predictions that the more profound second-phase decline observed in IFN-α-treated patients may reflect immune-mediated death/loss of productively infected cells. However, we show via mathematical modeling that more effective inhibition of HCV RNA can theoretically be achieved at higher doses of IFN-α such as 250 U/ml, where it appears that over long enough periods of time all intracellular RNA would be eliminated. By analogy, we hypothesize that it should be possible to completely eliminate serum HCV in patients in the absence of cell killing if higher IFN-α potency can be achieved.
Acknowledgments
This work was supported by NIH Public Health service grant AI070827 (S.L.U.) and the University of Illinois Chicago Council to Support Gastrointestinal and Liver Disease (S.L.U. and H.D.). Additionally, portions of this work were done under the auspices of the U.S. Department of Energy under contract DE-AC52-06NA25396 and supported by NIH Public Health service grants AI28433 and RR06555 (A.S.P.).
The clone B HCV sg1b Huh7 cell line from Charles M. Rice (via Apath, LLC) was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. We thank Thomas Layden for initiating this collaboration and Peter Corcoran for excellent technical assistance.
The authors have no conflicts of interest to disclose.
Footnotes
Published ahead of print on 15 April 2009.
REFERENCES
- 1.Bartenschlager, R. 2005. The hepatitis C virus replicon system: from basic research to clinical application. J. Hepatol. 43210-216. [DOI] [PubMed] [Google Scholar]
- 2.Blight, K. J., A. A. Kolykhalov, and C. M. Rice. 2000. Efficient initiation of HCV RNA replication in cell culture. Science 2901972-1974. [DOI] [PubMed] [Google Scholar]
- 3.Bowen, D. G., and C. M. Walker. 2005. Adaptive immune responses in acute and chronic hepatitis C virus infection. Nature 436946-952. [DOI] [PubMed] [Google Scholar]
- 4.Dahari, H., J. E. Layden-Almer, A. S. Perelson, and T. J. Layden. 2008. Hepatitis C viral kinetics in special populations. Curr. Hepat. Rep. 797-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dahari, H., and A. S. Perelson. 2007. Hepatitis C virus kinetics in chimeric mice during antiviral therapy. Hepatology 462048-2049. [DOI] [PubMed] [Google Scholar]
- 6.Dahari, H., R. M. Ribeiro, C. M. Rice, and A. S. Perelson. 2007. Mathematical modeling of subgenomic hepatitis C virus replication in Huh-7 cells. J. Virol. 81750-760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dahari, H., E. Shudo, R. M. Ribeiro, and A. S. Perelson. 2008. Mathematical modeling of HCV RNA kinetics. In H. Tang (ed.), Hepatitis C protocols, 2nd ed. Humana Press, Totowa, NJ.
- 8.Fried, M. W., M. L. Shiffman, K. R. Reddy, C. Smith, G. Marinos, F. L. Goncales, Jr., D. Haussinger, M. Diago, G. Carosi, D. Dhumeaux, A. Craxi, A. Lin, J. Hoffman, and J. Yu. 2002. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N. Engl. J. Med. 347975-982. [DOI] [PubMed] [Google Scholar]
- 9.Inoue, K., T. Umehara, U. T. Ruegg, F. Yasui, T. Watanabe, H. Yasuda, J. M. Dumont, P. Scalfaro, M. Yoshiba, and M. Kohara. 2007. Evaluation of a cyclophilin inhibitor in hepatitis C virus-infected chimeric mice in vivo. Hepatology 45921-928. [DOI] [PubMed] [Google Scholar]
- 10.Kapadia, S. B., A. Brideau-Andersen, and F. V. Chisari. 2003. Interference of hepatitis C virus RNA replication by short interfering RNAs. Proc. Natl. Acad. Sci. USA 1002014-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin, K., A. D. Kwong, and C. Lin. 2004. Combination of a hepatitis C virus NS3-NS4A protease inhibitor and alpha interferon synergistically inhibits viral RNA replication and facilitates viral RNA clearance in replicon cells. Antimicrob. Agents Chemother. 484784-4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin, K., R. B. Perni, A. D. Kwong, and C. Lin. 2006. VX-950, a novel hepatitis C virus (HCV) NS3-4A protease inhibitor, exhibits potent antiviral activities in HCV replicon cells. Antimicrob. Agents Chemother. 501813-1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manns, M. P., J. G. McHutchison, S. C. Gordon, V. K. Rustgi, M. Shiffman, R. Reindollar, Z. D. Goodman, K. Koury, M. Ling, and J. K. Albrecht. 2001. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: a randomised trial. Lancet 358958-965. [DOI] [PubMed] [Google Scholar]
- 14.Nelson, H. B., and H. Tang. 2006. Effect of cell growth on hepatitis C virus (HCV) replication and a mechanism of cell confluence-based inhibition of HCV RNA and protein expression. J. Virol. 801181-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neumann, A. U., N. P. Lam, H. Dahari, M. Davidian, T. E. Wiley, B. P. Mika, A. S. Perelson, and T. J. Layden. 2000. Differences in viral dynamics between genotypes 1 and 2 of hepatitis C virus. J. Infect. Dis. 18228-35. [DOI] [PubMed] [Google Scholar]
- 16.Neumann, A. U., N. P. Lam, H. Dahari, D. R. Gretch, T. E. Wiley, T. J. Layden, and A. S. Perelson. 1998. Hepatitis C viral dynamics in vivo and the antiviral efficacy of interferon-alpha therapy. Science 282103-107. [DOI] [PubMed] [Google Scholar]
- 17.National Institutes of Health. 2002. National Institutes of Health Consensus Development Conference statement: management of hepatitis C: 2002. Hepatology 36(Suppl. 1)S3-S20. [DOI] [PubMed] [Google Scholar]
- 18.Nguyen, T. T., A. Sedghi-Vaziri, L. B. Wilkes, T. Mondala, P. J. Pockros, K. L. Lindsay, and J. G. McHutchison. 1996. Fluctuations in viral load (HCV RNA) are relatively insignificant in untreated patients with chronic HCV infection. J. Viral Hepat. 375-78. [DOI] [PubMed] [Google Scholar]
- 19.Pause, A., G. Kukolj, M. Bailey, M. Brault, F. Do, T. Halmos, L. Lagace, R. Maurice, M. Marquis, G. McKercher, C. Pellerin, L. Pilote, D. Thibeault, and D. Lamarre. 2003. An NS3 serine protease inhibitor abrogates replication of subgenomic hepatitis C virus RNA. J. Biol. Chem. 27820374-20380. [DOI] [PubMed] [Google Scholar]
- 20.Pawlotsky, J. M., H. Dahari, A. U. Neumann, C. Hezode, G. Germanidis, I. Lonjon, L. Castera, and D. Dhumeaux. 2004. Antiviral action of ribavirin in chronic hepatitis C. Gastroenterology 126703-714. [DOI] [PubMed] [Google Scholar]
- 21.Pedersen, I. M., G. Cheng, S. Wieland, S. Volinia, C. M. Croce, F. V. Chisari, and M. David. 2007. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature 449919-922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perelson, A. S., E. Herrmann, F. Micol, and S. Zeuzem. 2005. New kinetic models for the hepatitis C virus. Hepatology 42749-754. [DOI] [PubMed] [Google Scholar]
- 23.Pietschmann, T., V. Lohmann, G. Rutter, K. Kurpanek, and R. Bartenschlager. 2001. Characterization of cell lines carrying self-replicating hepatitis C virus RNAs. J. Virol. 751252-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pilli, M., A. Zerbini, A. Penna, A. Orlandini, E. Lukasiewicz, J. M. Pawlotsky, S. Zeuzem, S. W. Schalm, M. von Wagner, G. Germanidis, Y. Lurie, J. I. Esteban, B. L. Haagmans, C. Hezode, M. Lagging, F. Negro, Y. Homburger, A. U. Neumann, C. Ferrari, and G. Missale. 2007. HCV-specific T-cell response in relation to viral kinetics and treatment outcome (DITTO-HCV project). Gastroenterology 1331132-1143. [DOI] [PubMed] [Google Scholar]
- 25.Simmen, K., O. Lenz, T. Lin, P. Fanning, H. Raoisson, H. de kock, G. Klooster, A. Rosenquist, M. Edlund, M. Nilsson, L. Vrang, and B. Samuelsson. 2007. In vitro activity and preclinical pharmacokinetics of the HCV protease inhibitor, TMC435350. Hepatology 461390. [Google Scholar]
- 26.Taylor, D. R., M. Puig, M. E. Darnell, K. Mihalik, and S. M. Feinstone. 2005. New antiviral pathway that mediates hepatitis C virus replicon Interferon sensitivity through ADAR1. J. Virol. 796291-6298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Windisch, M. P., M. Frese, A. Kaul, M. Trippler, V. Lohmann, and R. Bartenschlager. 2005. Dissecting the interferon-induced inhibition of hepatitis C virus replication by using a novel host cell line. J. Virol. 7913778-13793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhong, J., P. Gastaminza, G. Cheng, S. Kapadia, T. Kato, D. R. Burton, S. F. Wieland, S. L. Uprichard, T. Wakita, and F. V. Chisari. 2005. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 1029294-9299. [DOI] [PMC free article] [PubMed] [Google Scholar]