

Abstract
Among previously healthy children with severe influenza, the mechanisms leading to increased pathology are not understood. We hypothesized that children with severe influenza would have high levels of circulating cytokines. To examine this, we recruited patients with severe influenza and examined plasma cytokine levels as well as the ability of peripheral blood cells to respond to stimuli. Ten patients with severe influenza were enrolled during the 2005–2007 influenza seasons. We evaluated plasma cytokine levels, circulating NK cells, and responses to TLR ligands during the illness. We compared these patients with five patients with moderate influenza, six patients with respiratory syncytial virus (RSV), and 24 noninfected controls. Patients with influenza showed depressed responses to TLR ligands when compared with RSV patients and healthy controls (P<0.05). These normalized when retested during a convalescent phase. Plasma levels of IL-6, IL-12, and IFN-γ were elevated in influenza patients compared with controls (P<0.05). A compromised ability to produce TNF-α was reproduced by in vitro infection, and the magnitude of the effect correlated with the multiplicity of infection and induction of IFN regulatory factor 4 expression. Aberrant, systemic, innate responses to TLR ligands during influenza infection may be a consequence of specific viral attributes such as a high inoculum or rapid replication and may underlie the known susceptibility of influenza-infected patients to secondary bacterial infections.
Keywords: TLR, IRF4, IFN, NK cells, IL-6, NKp46
INTRODUCTION
Epidemics of influenza typically occur during the winter months and have been responsible for an average of 36,000 deaths per year in the United States from 1990 to 1999 [1, 2]. Among young children, hospitalization rates have ranged from 500/100,000 children for those with high-risk medical conditions to 100/100,000 children for those without high-risk medical conditions [3, 4]. Complications are more frequent in the elderly, young children, and patients with neuromuscular disease or immune compromise and can include respiratory failure, bacterial superinfection, myocarditis, encephalitis, and death. Approximately one-quarter of the deaths in children are a result of bacterial superinfection [5].
Host responses to influenza include early innate viral responses. Type I IFN production after engagement of TLRs or retinoic acid-inducible gene-I is critical for activation of cytotoxic T cells and initial restraint of viral replication and spread [6,7,8]. Some of the known, high-risk populations are presumed to have increased susceptibility to influenza on the basis of impaired cytotoxic T cell responses; however, severe influenza occurs in children who have no other evidence of immune compromise [9,10,11,12]. One patient has been identified with a TLR3 mutation, confirming the importance of this innate pathway [13].
There are also virus-specific determinants of severity, and this has been highlighted by recent work about the 1918 pandemic strain as well as the emerging H5N1 strains [14,15,16]. In both cases, pathogenicity appears to relate to altered innate responses and hypercytokinemia. Virus-specific attributes reduce early type I IFN production, and this is believed to lead to enhanced spread of virus and subsequent production of high levels of proinflammatory cytokines driven by the high viral load [17, 18]. Similar to what has been reported in children, bacterial superinfection was extremely common in the lungs of patients from the 1918 pandemic [19, 20].
We hypothesized that a process similar to that seen with highly pathogenic strains of influenza could underlie severe influenza arising in patients who were previously healthy. Limited data support this hypothesis. Serum cytokines are elevated in patients with severe influenza [21,22,23,24,25,26]. A study looking at healthy adult patients who were experimentally inoculated with the influenza A virus found that IL-6 and IFN-α levels in nasal lavage fluid peaked on Day 2 and correlated with viral titers, temperature, mucus production, and symptom scores, also suggesting that cytokine effects might underlie severe disease [27].
Taken together, these data support a hypothesis in which severity of influenza correlates with cytokine production. The current study investigated severe influenza in children, who are less likely to exhibit co-morbidities such as smoking or diabetes, which could alter host responses, and are less likely to have pre-existing immunity to influenza. Our data demonstrate significant levels of inflammatory cytokines in the plasma of patients with influenza. Elevated plasma levels of IL-12 and IFN-γ were seen specifically with influenza and not respiratory syncytial virus (RSV) infection. This was not unexpected based on previous published data; however, unexpectedly, culture of PBMCs revealed markedly compromised production of cytokines in response to TLR ligands. This phenomenon could be replicated partly by infection of PBMCs with influenza ex vivo and correlated with the multiplicity of infection (MOI) and induction of IFN regulatory factor 4 (IRF4) expression. Our data suggest that in the absence of known immune deficiency, the most likely contributor to severity of influenza is at the level of virus. In vivo, this could reflect the size of the inoculum or the ability of the virus to replicate efficiently. The inhibition of TLR responses could contribute to bacterial superinfection.
MATERIALS AND METHODS
Patient and control subjects
Fifteen patients with influenza (10 with severe influenza and five with moderate influenza), who presented in the 2005–2006 and 2006–2007 influenza seasons, were enrolled in this study. Influenza was detected by PCR in a Clinical Laboratory Improvement Amendments-certified laboratory. Severe influenza patients were defined as those patients admitted to the Intensive Care Unit (ICU) with encephalopathy or respiratory failure. Moderate influenza patients were defined as those admitted with fever or wheezing with an uncomplicated hospital course. Children with known co-morbid disorders were excluded (immune deficiency, neuromuscular disease, severe developmental delay, cardiac disease, bacterial superinfection). Twenty-four noninfected, control subjects were recruited from the outpatient department. As an additional control group, we compared influenza patients with five RSV patients admitted to the hospital with fever or wheezing. Three patients with severe influenza were restudied between 1 and 3 months after infection.
This study was approved by the Institutional Review Board at the Children’s Hospital of Philadelphia (PA, USA) for protection of human subjects. Informed consent was obtained from all participating patients and their parents or legal guardians.
Assays
Plasma was removed and frozen within 1 h of the blood draw. Plasma IL-1, IL-6, IL-10, IL-12, MIP-1α, TNF-α, and IFN-γ were evaluated by cytokine bead array using the Becton Dickinson cytometric bead array flex kit (BD Biosciences, San Jose, CA, USA). IL-18 was analyzed by ELISA using a kit from Biosource International, Inc. (Camarillo, CA, USA). IFN-α plasma concentrations were analyzed by a capture ELISA (for all types of IFN-α) from PBL Biomedical Laboratories (Piscataway, NJ, USA). All cytokines were measured as triplicates and averaged. Responses to TLR ligands were measured at 24 h as described previously on fresh PBMCs [28]. TNF-α and IFN-α were measured by ELISA at 6 or 24 h poststimulus. Statistical analyses were performed using t-tests. sd is shown in the figures.
CD3-FITC, NKp46-PE, and CD56-PeCy5 antibodies (BD Biosciences) were used, and CD3-negative cells anchored the NKp46 and CD56 analyses. Flow cytometry was performed using a four-color FACSCalibur (Becton Dickinson, San Diego, CA, USA).
The Affymetrix (Santa Clara, CA, USA) U133A 2.0 array was hybridized according to the manufacturer’s recommendations. RNA was prepared by sequential TRIzol (Invitrogen, Carlsbad, CA, USA) and RNeasy (Qiagen, Valencia, CA, USA) and complementary RNA (cRNA) prepared according standard protocols. Ingenuity (Redwood City, CA, USA) was used for network analysis of array data. Ingenuity is a program based on the largest curated database of mammalian biological interaction available. Focus or nodal genes are identified on the basis of connectivity to genes with altered expression from the expression arrays. Nodal genes need not themselves have perturbed expression. Networks of gene interactions are generated algorithmically based on their connectivity. Database for Annotation, Visualization and Integrated Discovery (DAVID) [29] was used to define functional categories of genes with altered expression. For Ingenuity and DAVID, severe influenza samples were compared with noninfected control samples.
In vitro infections
PBMCs from healthy adult controls were plated at 2 × 105/well. Parallel plates were infected with PR8 at a MOI of 0, 1, 5, and 10 (calculated from the hectare units). PR8 virus is a human H1N1 strain (A/PR/8/34; Charles River Laboratories, Wilmington, MA, USA). Twenty-four and 48 h later, the cells were stimulated with TLR ligands as above. Supernatants were harvested at 6 h, and ELISAs for IFN-α and TNF-α were performed. The Western blot used rabbit anti-IRF4 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and donkey anti-rabbit (GE Healthcare, Piscataway, NJ, USA).
RESULTS
Patient characteristics
The characteristics of the patients with severe influenza, moderate influenza, RSV, and the controls are summarized in Table 1. Nearly all patients in the influenza groups were infected with influenza A. Nine of the 10 patients in the severe group were in the ICU, and eight experienced respiratory failure. One patient with a pericardial effusion was managed predominantly on the Cardiology Unit. The majority of patients in the severe influenza group (seven of 10) received oseltamivir, and only one patient in the moderate group received oseltamivir. The severe influenza group had a slightly younger mean age and a slightly longer time from disease onset to participation in the study compared with the moderate influenza group.
Table 1.
Patient Characteristics
| Severe influenza | Moderate influenza | RSV | Healthy controls | ||
|---|---|---|---|---|---|
| # of Patients | 10 | 5 | 6 | 24 | |
| Mean age (range) | 3.4 (0.2–12.6 years) | 6.3 (3 months–12 years) | 2.2 (22 days–4 years) | 6.9 (0.5–19 years) | |
| Sex, M/F | 5/5 | 2/3 | 3/3 | 11/13 | |
| Influenza virus type A/B | 8/2 | 5/0 | N/A | N/A | |
| Days into illness on day of admission | 5 (1–14) | 2.6 (2–4) | 6 (2–14) | N/A | |
| Days into illness for blood draw | 10.8 (6–28) | 5.6 (4–8) | 8.5 (3–16) | N/A | |
| ICU admission | 9 | 0 | 2 | N/A | |
| Intubation | 7 | 0 | 1 | N/A | |
| Oscillator | 4 | 0 | 0 | N/A | |
| Encephalopathy | 3 | 0 | 0 | N/A | |
| Coagulapathy | 5 | 0 | 0 | N/A | |
| Oxygen | 8 | 0 | 3 | – | |
| Flu vaccinea | 3 | 1 | b | N/A | |
| Oseltamivir | 7 | 1 | b | – | |
| Hospitalized days | 22.7 (4–53 days) | 6 (2–12 days) | 7.5 (2–17 days) | N/A | |
| WBC | 10,470 (6500–25,200) | 16,666 (6800–26,000) | 13,867 (6700–27,300) | N/A | |
| ALC | 2356 (711–4032) | 2266.6 (408–5495) | 2802 (871–5733) | N/A |
During current season.
Historical information not obtained about RSV patients and healthy controls. N/A, Not applicable; WBC, white blood cells; ALC, absolute lymphocyte count.
Plasma cytokines
Plasma levels of TNF-α, MIP-1α, IL-1β, IL-18, and IFN-α were not increased in infected patients compared with controls (not shown). Plasma levels of IL-6 and IL-10 were increased comparably in patients infected with influenza and RSV compared with controls (Fig. 1). Plasma levels of IL-12 and IFN-γ were increased markedly in influenza patients compared with RSV patients and controls; however, there were no significant differences in patients with severe influenza compared with moderate influenza (Fig. 1). IFN-γ may be relevant for the hemophagocytic process that can accompany influenza in children [5].
Fig. 1.

Plasma cytokines were measured in fresh-frozen plasma from the defined patient populations. The means are shown in the graphs along with sd. Open stars, P < 0.01, compared with controls; closed stars, P < 0.05, compared with controls using a t-test.
Decreased TLR responses in influenza patients
To probe the status of innate responses in patients acutely infected with influenza, we used PBMCs cultured with TLR ligands and assayed for TNF-α and IFN-α production at 24 h. Generally, responses to TLR ligands were comparable in RSV patients and controls. However, TLR responses in influenza patients were consistently, significantly lower than controls (Fig. 2). The ability to produce TNF-α and IFN-α was affected. Samples obtained from patients after recovery from influenza were comparable with controls with respect to TNF-α production, suggesting that the defective TNF-α responses were a consequence of the infection, not a hard-wired defect. In contrast, the IFN-α responses were more mixed, and responses to poly(I:C) and CpG oligonucleotides exhibited defective responses, even after clinical recovery from infection.
Fig. 2.

TLR responses. PBMCs taken from patients in the defined groups were plated and stimulated with TLR ligands. Supernatants were harvested at 24 h and ELISAs performed. For TNF-α, the differences between the severe influenza and control groups were significant with a P < 0.05 (t-test) for palmitoyl-3-cysteine-serine-lysine-4 (Pam3CSK4; Pam), LPS, and zymosan (Zym). When all influenza patients were combined, the differences between all influenza patients and controls were significant for Pam3CSK4, LPS, flagellin (Flag), zymosan, and loxoribine (Lox). The differences between all influenza and RSV were significant with P < 0.05 (t-test) for Pam3CSK4, LPS, flagellin, zymosan, and loxoribine. For IFN-α, the differences between severe influenza and controls were significant with P < 0.01 for all stimuli (t-test). Poly, Polyinosinic:polycytidylic acid [poly(I:C)]; ODN, ODN2216; Unstim, unstimulated.
To determine whether cells could have altered signaling molecule expression, which could contribute to this phenotype, gene expression arrays were performed on a small number of samples from patients (Fig. 3). This study demonstrates a distinction between patients with severe and moderate influenza. This is consistent with what was seen in a murine study [30]. Network analysis with Ingenuity revealed that MAPK activation could potentially underlie multiple overexpressed genes. This MAPK activation has also been seen with H5N1 infections and is hypothesized to underlie the cytokine storm seen with H5N1 [31, 32]. Down-regulated expression of cytokines could be connected by multiple IFN nodes. To gain additional insight into the systemic consequences of influenza infection, DAVID was used to identify functional gene classes among up-regulated genes. Glycoproteins had the highest enrichment score followed by inflammatory response genes and chemokines. This corresponded to up-regulated cadherins, matrix proteins, and cytokine/chemokine families. Chemokines were up-regulated significantly, and they were largely chemokines acting on monocytes. The full gene list is available as Supplemental Data. The cytokines demonstrated to be abnormal in the serum or after TLR stimulation are comparable between severe influenza samples and controls in the array study, as cytokine mRNA levels are low in the absence of stimulation. Another notable group of genes also had RNA levels that were comparable between patients and controls—the TLR gene family. This suggests that TLR down-regulation by influenza is not responsible for the diminished responses.
Fig. 3.

Gene expression analysis of patients with severe influenza. PBMCs were prepared at the time of plasma cytokine analysis. RNA was isolated and cRNA prepared and labeled. (A) Results from the array were clustered. Patients with severe influenza have a markedly disordered gene expression. The convalescent sample (Conv.) and the moderate influenza (Mod. Inf.) sample are much more like the controls than the severe influenza group. In evaluating the overexpressed genes, many related to potential changes in the cellular composition of the PBMCs. (B) Using network analysis, the major affected pathways for up-regulated genes are ERK, p38, Akt, AP-1, NF-κB, and peroxisome proliferator-activated receptor γ. The major affected pathways for down-regulated genes are IFN-γ, IL-12, IFN-induced protein with tetratricopeptide repeat 2 (IFIT2), and cyclin-dependent kinase inhibitor 1A. FCERIA, FcεRIα; GBP1, guanylate-binding protein 1; Hsp70, heat shock protein 70; GPR37, G protein-coupled receptor 37; KLRC2, killer cell lectin-like receptor subfamily C, member 2; IFNG, IFN-γ; XAF1, X-linked inhibitor of apoptosis protein-associated factor 1; ST8SIA1, ST8 α-N-acetyl-neuraminide α-2,8-sialyltransferase 1; TBX21, T-box 21; LDL, low-density lipoprotein; GVIN1, GTPase, very large IFN-inducible 1 protein; MMP19, matrix metalloproteinase 19; EREG, epiregulin; SERPINB2, serpin peptidase inhibitor, clade B (OVA), member 2; C1QA, complement 1 QA; AREG, amphiregulin; RAB13, member RAS oncogene family; Pka, protein kinase A; Pdgf, platelet-derived growth factor; SYN1, Synapsin I; CES1, carboxylesterase 1; SPRY2, sprouty homolog 2; SPP1, secreted phosphoprotein 1; N-cor, nuclear receptor corepressor; EDNRB, endothelin receptor type B; GEM, glycolipid-enriched membrane; OLR1, oxidized LDL (lectin-like) receptor 1.
In vitro influenza infection
Our data suggested that innate responses during infection were compromised globally. We considered whether features related to the characteristics of the initial infection could compromise innate responses. PBMCs from healthy controls were infected with PR8 virus with different MOIs. Higher levels of infection led to decreased TNF-α production (Fig. 4). This effect was more marked at 48 h postinfection compared with 24 h postinfection. This could not be attributed to cell death, as viability was >95% in all cultures. The finding was most pronounced for bacterial mimic ligands. In fact, prior influenza actually improved TNF-α responses to poly(I:C), a viral mimic. IFN-α responses after viral infection were increased in this in vitro assay, contrary to what was seen ex vivo in Figure 2.
Fig. 4.

Infection of PBMC with influenza. PBMCs were prepared and infected with PR8 influenza at the indicated MOI. Twenty-four hours postinfection, TLR ligands were added. TNF-α and IFN-α were analyzed 6 h after stimulation. The data represent averages of two independent experiments with triplicate wells.
To examine potential mechanisms for reduced TLR responses, we considered the role of IRF4, which competitively inhibits IRF5 and decreases TLR responses [33]. This negative regulator is induced by IFN-α and could link the array results with the TLR results [34]. We evaluated PBMC infected with PR8 influenza at different MOIs and harvested at 24 h (Fig. 5). The Western blot clearly demonstrates a dose-response effect of influenza on IRF4 expression. When cells were purified from PBMC and examined as purified populations, we were not able to reproduce the effect of PR8 on TLR responses, although we did observe increased IRF4 expression in T cells (not shown). These data suggest that cell interactions may be required for the effect.
Fig. 5.
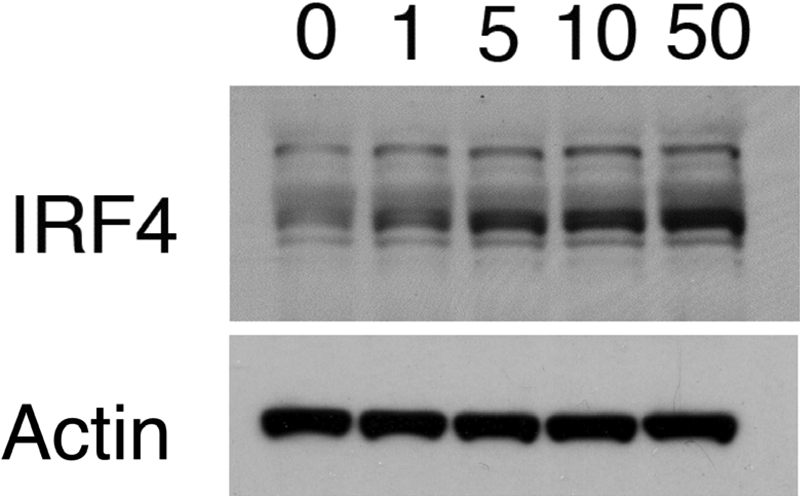
Western blot analysis of IRF4. PBMCs were prepared and infected with PR8 influenza at the indicated MOI. Twenty-four hours after infection, IRF4 was detected.
NKp46 expression
NK cell function is altered markedly by cytokine exposure, particularly type I IFN exposure [35]. We hypothesized that NK cells would be altered in severe influenza. In the setting of influenza, NK cells were found to be decreased in peripheral blood in influenza and RSV-infected patients (Fig. 6). The convalescent influenza patients had normal numbers of circulating NK cells, suggesting apoptosis or tissue migration during the acute infection.
Fig. 6.

NK cells were evaluated by flow cytometry. Peripheral blood NK cell counts were diminished in influenza and RSV. Total NK cells and CD56dim NK cells were diminished significantly in RSV and severe influenza patients with P < 0.05 (t-test).
NK cells were analyzed according to the level of CD56 expression, as low CD56 expression is associated with an increased capacity for cytotoxicity. The influenza patients and RSV patients had a decreased fraction of CD56dim NK cells compared with controls. NKp46 is a germline-encoded receptor for viral hemagglutinin (HA). Its expression was evaluated on NK cells and was found to be comparable across all patient groups. The percent of NKp46-positive cells in the NK cell subsets and the mean fluorescent intensity were comparable (data not shown).
DISCUSSION
The data from this study suggest that most cases of severe influenza occurring in previously healthy children are not associated with a specific innate immunologic deficit. Although it is clear that immune deficiencies, acquired and congenital, predispose to severe influenza, this study provides a framework for understanding patients who have no other signs of immune deficiency and develop severe influenza.
During the course of local spread of influenza, type I IFNs induce a set of genes whose products impair translation and viral replication [7, 36]. This study did not identify type I IFN in the plasma, possibly as a result of the timing of sample acquisition [16, 31]. In a setting where early containment is defective, the spread of virus leads to direct cytopathic effects [6, 7, 37]. Viral spread can then induce high levels of proinflammatory cytokines locally [38]. One interpretation of the data is that viral characteristics allow the virus to outrun early containment in some cases and drive increased expression of proinflammatory cytokines, which spill into the bloodstream. The MAPK, p38, may have a central role in this process. Circulating cells exposed to the pulmonary bed develop dysfunctional responses to TLR ligands, which can then contribute to increased mortality and morbidity. Supporting this hypothesis is a model of local spread of influenza, which demonstrates that small changes in infection characteristics lead to markedly altered cytokine production [39].
The plasma cytokines were elevated paradoxically in the face of diminished TLR responses. Among plasma cytokine levels evaluated, only IL-6 tracked with severity. IL-6 has been found previously to be elevated in patients with severe influenza and to correlate with severity early in infection [26, 27, 40]. The serum levels seen in our patients were comparable with those seen in a study of patients infected with H5N1 [16].
The finding of diminished responses to TLR ligands after ex vivo influenza infection has broad implications. In the murine lung, alveolar macrophages have been found to be desensitized to bacteria and flagellin after influenza infection [30]. The phenomenon identified in our study may be similar. The heightened susceptibility to Streptococcus pneumoniae seen with influenza and the frequency of bacterial superinfection leading to death could be a result of compromised TLR responses [19, 41, 42]. Recent studies of pediatric deaths from influenza suggest that bacterial superinfection is common, and a pathologic review of cases from the 1918 pandemic demonstrated that bacterial superinfection was the leading contributor to death [5, 19, 20]. Our study is unique in finding that the patients exhibited systemically diminished TLR responses during the course of a clinical infection. This implies that the effect of influenza on a subsequent infectious challenge is not just limited to the lung, which is surprising, as influenza is typically not found in the bloodstream. A sustained remodeling of innate responses could contribute to secondary infectious susceptibility but could also contribute to qualitatively altered responses. In mice, type I IFNs are critical for humoral responses to influenza, and therefore, the dysregulated TLR responses could also affect adaptive immunity [43].
One consequence of the dysregulated cytokine expression could be its effects on NK cells. IL-12, a cytokine that was identified as elevated in influenza specifically, has an important role in the generation of cytotoxicity [44, 45]. The HA moiety of influenza can specifically engage NKp46, also important for cytotoxicity activation [46,47,48]. The levels of NKp46 were comparable across all patient groups; however, activation through this receptor may have contributed to tissue migration or altered survival, which would account for the diminished NK cells.
Conclusion
This study is significant in the demonstration of altered, systemic TLR responses acutely with influenza. In vitro infection of PBMCs replicated this finding with respect to TNF-α responses but did not replicate the reduction in IFN-α production. PBMCs were used in an effort to provide biological cell–cell interactions, but in vitro analyses cannot replicate tissue migration or longer-term effects of cytokine exposure. High titers of influenza have been shown to induce dendritic cell apoptosis, which could explain the ex vivo findings [49]. In our model, the failure of early containment as a result of high viral load leads to increased expression of proinflammatory cytokines. These same cytokines appear to participate in the severe manifestations associated with H5N1 infection, although our network analysis did not appear to implicate similar signaling molecules [16]. Instead, the network analysis implicated type I IFNs and IRF4 in the deficient TLR responses. This is the first demonstrations of systemically altered TLR responses in humans and has implications for the care of patients with influenza. These data have limitations, however. This is one of the largest studies of pediatric patients with severe influenza; nevertheless, the sample size is small. Relatively few patients consented for follow-up studies. In spite of these limitations, the data are consistent in demonstrating elevated plasma cytokines coupled with compromised TLR responses in vivo and in vitro.
Supplementary Material
Acknowledgments
This work was supported by the Amanda Kanowitz Foundation and National Institutes of Health grant NO1-AI-50024. The contributions of the patients and families and the assistance of Marissa Kuba are gratefully acknowledged.
References
- Sabin A B. Mortality from pneumonia and risk conditions during influenza epidemics. High influenza morbidity during nonepidemic years. JAMA. 1977;237:2823–2828. [PubMed] [Google Scholar]
- Glezen W P. Considerations of the risk of influenza in children and indications for prophylaxis. Rev Infect Dis. 1980;2:408–420. doi: 10.1093/clinids/2.3.408. [DOI] [PubMed] [Google Scholar]
- Izurieta H S, Thompson W W, Kramarz P, Shay D K, Davis R L, DeStefano F, Black S, Shinefield H, Fukuda K. Influenza and the rates of hospitalization for respiratory disease among infants and young children. N Engl J Med. 2000;342:232–239. doi: 10.1056/NEJM200001273420402. [DOI] [PubMed] [Google Scholar]
- Ampofo K, Gesteland P H, Bender J, Mills M, Daly J, Samore M, Byington C, Pavia A T, Srivastava R. Epidemiology, complications, and cost of hospitalization in children with laboratory-confirmed influenza infection. Pediatrics. 2006;118:2409–2417. doi: 10.1542/peds.2006-1475. [DOI] [PubMed] [Google Scholar]
- Guarner J, Paddock C D, Shieh W J, Packard M M, Patel M, Montague J L, Uyeki T M, Bhat N, Balish A, Lindstrom S, Klimov A, Zaki S R. Histopathologic and immunohistochemical features of fatal influenza virus infection in children during the 2003–2004 season. Clin Infect Dis. 2006;43:132–140. doi: 10.1086/505122. [DOI] [PubMed] [Google Scholar]
- Garcia-Sastre A, Durbin R K, Zheng H, Palese P, Gertner R, Levy D E, Durbin J E. The role of interferon in influenza virus tissue tropism. J Virol. 1998;72:8550–8558. doi: 10.1128/jvi.72.11.8550-8558.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koerner I, Kochs G, Kalinke U, Weiss S, Staeheli P. Protective role of β interferon in host defense against influenza A virus. J Virol. 2007;81:2025–2030. doi: 10.1128/JVI.01718-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewell N A, Vaghefi N, Mertz S E, Akter P, Peebles R S, Jr, Bakaletz L O, Durbin R K, Flano E, Durbin J E. Differential type I interferon induction by respiratory syncytial virus and influenza A virus in vivo. J Virol. 2007;81:9790–9800. doi: 10.1128/JVI.00530-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans K D, Kline M W. Prolonged influenza A infection responsive to rimantadine therapy in a human immunodeficiency virus-infected child. Pediatr Infect Dis J. 1995;14:332–334. doi: 10.1097/00006454-199504000-00022. [DOI] [PubMed] [Google Scholar]
- Hayden F G. Prevention and treatment of influenza in immunocompromised patients. Am J Med. 1997;102:55–60. doi: 10.1016/s0002-9343(97)80013-7. [DOI] [PubMed] [Google Scholar]
- Maricich S M, Neul J L, Lotze T E, Cazacu A C, Uyeki T M, Demmler G J, Clark G D. Neurologic complications associated with influenza A in children during the 2003–2004 influenza season in Houston, Texas. Pediatrics. 2004;114:e626–e633. doi: 10.1542/peds.2004-0143. [DOI] [PubMed] [Google Scholar]
- Quach C, Piche-Walker L, Platt R, Moore D. Risk factors associated with severe influenza infections in childhood: implication for vaccine strategy. Pediatrics. 2003;112:e197–e201. doi: 10.1542/peds.112.3.e197. [DOI] [PubMed] [Google Scholar]
- Hidaka F, Matsuo S, Muta T, Takeshige K, Mizukami T, Nunoi H. A missense mutation of the Toll-like receptor 3 gene in a patient with influenza-associated encephalopathy. Clin Immunol. 2006;119:188–194. doi: 10.1016/j.clim.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Szretter K J, Gangappa S, Lu X, Smith C, Shieh W J, Zaki S R, Sambhara S, Tumpey T M, Katz J M. Role of host cytokine responses in the pathogenesis of avian H5N1 influenza viruses in mice. J Virol. 2007;81:2736–2744. doi: 10.1128/JVI.02336-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipatov A S, Andreansky S, Webby R J, Hulse D J, Rehg J E, Krauss S, Perez D R, Doherty P C, Webster R G, Sangster M Y. Pathogenesis of Hong Kong H5N1 influenza virus NS gene reassortants in mice: the role of cytokines and B- and T-cell responses. J Gen Virol. 2005;86:1121–1130. doi: 10.1099/vir.0.80663-0. [DOI] [PubMed] [Google Scholar]
- De Jong M D, Simmons C P, Thanh T T, Hien V M, Smith G J, Chau T N, Hoang D M, Chau N V, Khanh T H, Dong V C, Qui P T, Cam B V, Ha do Q, Guan Y, Peiris J S, Chinh N T, Hien T T, Farrar J. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med. 2006;12:1203–1207. doi: 10.1038/nm1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiss G K, Salvatore M, Tumpey T M, Carter V S, Wang X, Basler C F, Taubenberger J K, Bumgarner R E, Palese P, Katze M G, García-Sastre A. Cellular transcriptional profiling in influenza A virus-infected lung epithelial cells: the role of the nonstructural NS1 protein in the evasion of the host innate defense and its potential contribution to pandemic influenza. Proc Natl Acad Sci USA. 2002;99:10736–10741. doi: 10.1073/pnas.112338099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao P, Tian G, Li Y, Deng G, Jiang Y, Liu C, Liu W, Bu Z, Kawaoka Y, Chen H. A single-amino-acid substitution in the NS1 protein changes the pathogenicity of H5N1 avian influenza viruses in mice. J Virol. 2008;82:1146–1154. doi: 10.1128/JVI.01698-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morens D M, Taubenberger J K, Fauci A S. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J Infect Dis. 2008;198:962–970. doi: 10.1086/591708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenberger J K, Morens D M. The pathology of influenza virus infections. Annu Rev Pathol. 2008;3:499–522. doi: 10.1146/annurev.pathmechdis.3.121806.154316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togashi T, Matsuzono Y, Narita M, Morishima T. Influenza-associated acute encephalopathy in Japanese children in 1994–2002. Virus Res. 2004;103:75–78. doi: 10.1016/j.virusres.2004.02.016. [DOI] [PubMed] [Google Scholar]
- Kawada J, Kimura H, Ito Y, Hara S, Iriyama M, Yoshikawa T, Morishima T. Systemic cytokine responses in patients with influenza-associated encephalopathy. J Infect Dis. 2003;188:690–698. doi: 10.1086/377101. [DOI] [PubMed] [Google Scholar]
- Hosoya M, Nunoi H, Aoyama M, Kawasaki Y, Suzuki H. Cytochrome c and tumor necrosis factor-α values in serum and cerebrospinal fluid of patients with influenza-associated encephalopathy. Pediatr Infect Dis J. 2005;24:467–470. doi: 10.1097/01.inf.0000160995.07461.b8. [DOI] [PubMed] [Google Scholar]
- Nunoi H, Mercado M R, Mizukami T, Okajima K, Morishima T, Sakata H, Nakayama S, Mori S, Hayashi M, Mori H, Kagimoto S, Kanegasaki S, Watanabe K, Adachi N, Endo F. Apoptosis under hypercytokinemia is a possible pathogenesis in influenza-associated encephalopathy. Pediatr Int. 2005;47:175–179. doi: 10.1111/j.1442-200x.2005.02042.x. [DOI] [PubMed] [Google Scholar]
- Ichiyama T, Morishima T, Isumi H, Matsufuji H, Matsubara T, Furukawa S. Analysis of cytokine levels and NF-κB activation in peripheral blood mononuclear cells in influenza virus-associated encephalopathy. Cytokine. 2004;27:31–37. doi: 10.1016/j.cyto.2004.03.012. [DOI] [PubMed] [Google Scholar]
- Ichiyama T, Isumi H, Ozawa H, Matsubara T, Morishima T, Furukawa S. Cerebrospinal fluid and serum levels of cytokines and soluble tumor necrosis factor receptor in influenza virus-associated encephalopathy. Scand J Infect Dis. 2003;35:59–61. doi: 10.1080/0036554021000026986. [DOI] [PubMed] [Google Scholar]
- Kaiser L, Fritz R S, Straus S E, Gubareva L, Hayden F G. Symptom pathogenesis during acute influenza: interleukin-6 and other cytokine responses. J Med Virol. 2001;64:262–268. doi: 10.1002/jmv.1045. [DOI] [PubMed] [Google Scholar]
- Deering R P, Orange J S. Development of a clinical assay to evaluate Toll-like receptor function. Clin Vaccine Immunol. 2006;13:68–76. doi: 10.1128/CVI.13.1.68-76.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G, Jr, Sherman B T, Hosack D A, Yang J, Gao W, Lane H C, Lempicki R A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- Didierlaurent A, Goulding J, Patel S, Snelgrove R, Low L, Bebien M, Lawrence T, van Rijt L S, Lambrecht B N, Sirard J C, Hussell T. Sustained desensitization to bacterial Toll-like receptor ligands after resolution of respiratory influenza infection. J Exp Med. 2008;205:323–329. doi: 10.1084/jem.20070891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee N, Wong C K, Chan P K, Lun S W, Lui G, Wong B, Hui D S, Lam C W, Cockram C S, Choi K W, Yeung A C, Tang J W, Sung J J. Hypercytokinemia and hyperactivation of phospho-p38 mitogen-activated protein kinase in severe human influenza A virus infection. Clin Infect Dis. 2007;45:723–731. doi: 10.1086/520981. [DOI] [PubMed] [Google Scholar]
- Lee D C, Cheung C Y, Law A H, Mok C K, Peiris M, Lau A S. p38 Mitogen-activated protein kinase-dependent hyperinduction of tumor necrosis factor α expression in response to avian influenza virus H5N1. J Virol. 2005;79:10147–10154. doi: 10.1128/JVI.79.16.10147-10154.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negishi H, Ohba Y, Yanai H, Takaoka A, Honma K, Yui K, Matsuyama T, Taniguchi T, Honda K. Negative regulation of Toll-like-receptor signaling by IRF-4. Proc Natl Acad Sci USA. 2005;102:15989–15994. doi: 10.1073/pnas.0508327102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Hochhaus A, Konig-Merediz S A, Brendel C, Proba J, Hoppe G J, Wittig B, Ehninger G, Hehlmann R, Neubauer A. Expression of interferon regulatory factor 4 in chronic myeloid leukemia: correlation with response to interferon α therapy. J Clin Oncol. 2000;18:3331–3338. doi: 10.1200/JCO.2000.18.19.3331. [DOI] [PubMed] [Google Scholar]
- Malaponte G, Passero E, Leonardi S, Monte V, Lauria C, Mazzarino C, Sciotto A, Russo Mancuso G, Di Gregorio F, Musumeci S. Effect of α-interferon on natural killer cell activity and lymphocyte subsets in thalassemia patients with chronic hepatitis C. Acta Haematol. 1997;98:83–88. doi: 10.1159/000203603. [DOI] [PubMed] [Google Scholar]
- Gresser I, Tovey M G, Maury C, Bandu M T. Role of interferon in the pathogenesis of virus diseases in mice as demonstrated by the use of anti-interferon serum. II. Studies with herpes simplex, Moloney sarcoma, vesicular stomatitis, Newcastle disease, and influenza viruses. J Exp Med. 1976;144:1316–1323. doi: 10.1084/jem.144.5.1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wabuke-Bunoti M A, Bennink J R, Plotkin S A. Influenza virus-induced encephalopathy in mice: interferon production and natural killer cell activity during acute infection. J Virol. 1986;60:1062–1067. doi: 10.1128/jvi.60.3.1062-1067.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsukura S, Kokubu F, Noda H, Tokunaga H, Adachi M. Expression of IL-6, IL-8, and RANTES on human bronchial epithelial cells, NCI-H292, induced by influenza virus A. J Allergy Clin Immunol. 1996;98:1080–1087. doi: 10.1016/s0091-6749(96)80195-3. [DOI] [PubMed] [Google Scholar]
- Baccam P, Beauchemin C, Macken C A, Hayden F G, Perelson A S. Kinetics of influenza A virus infection in humans. J Virol. 2006;80:7590–7599. doi: 10.1128/JVI.01623-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Nomura F, Kawai T, Takeuchi O, Muhlradt P F, Takeda K, Akira S. Synergy and cross-tolerance between Toll-like receptor (TLR) 2- and TLR4-mediated signaling pathways. J Immunol. 2000;165:7096–7101. doi: 10.4049/jimmunol.165.12.7096. [DOI] [PubMed] [Google Scholar]
- Giebink G S, Berzins I K, Marker S C, Schiffman G. Experimental otitis media after nasal inoculation of Streptococcus pneumoniae and influenza A virus in chinchillas. Infect Immun. 1980;30:445–450. doi: 10.1128/iai.30.2.445-450.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerone P J, Ward T G, Chappell W A. Combined infections in mice with influenza virus and Diplococcus pneumoniae. Am J Hyg. 1957;66:331–341. doi: 10.1093/oxfordjournals.aje.a119906. [DOI] [PubMed] [Google Scholar]
- Heer A K, Shamshiev A, Donda A, Uematsu S, Akira S, Kopf M, Marsland B J. TLR signaling fine-tunes anti-influenza B cell responses without regulating effector T cell responses. J Immunol. 2007;178:2182–2191. doi: 10.4049/jimmunol.178.4.2182. [DOI] [PubMed] [Google Scholar]
- Perussia B, Chan S H, D'Andrea A, Tsuji K, Santoli D, Pospisil M, Young D, Wolf S F, Trinchieri G. Natural killer (NK) cell stimulatory factor or IL-12 has differential effects on the proliferation of TCR-α β+, TCR-γ δ+ T lymphocytes, and NK cells. J Immunol. 1992;149:3495–3502. [PubMed] [Google Scholar]
- Hirai N, Kato Y, Kobayashi K, Hattori N. Natural killer (NK) cell activity and its in vitro response to interferon-α(Le) in chronic liver diseases and hepatocellular carcinoma. Liver. 1986;6:212–220. doi: 10.1111/j.1600-0676.1986.tb01068.x. [DOI] [PubMed] [Google Scholar]
- Mandelboim O, Lieberman N, Lev M, Paul L, Arnon T I, Bushkin Y, Davis D M, Strominger J L, Yewdell J W, Porgador A. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature. 2001;409:1055–1060. doi: 10.1038/35059110. [DOI] [PubMed] [Google Scholar]
- Pessino A, Sivori S, Bottino C, Malaspina A, Morelli L, Moretta L, Biassoni R, Moretta A. Molecular cloning of NKp46: a novel member of the immunoglobulin superfamily involved in triggering of natural cytotoxicity. J Exp Med. 1998;188:953–960. doi: 10.1084/jem.188.5.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnon T I, Achdout H, Lieberman N, Gazit R, Gonen-Gross T, Katz G, Bar-Ilan A, Bloushtain N, Lev M, Joseph A, Kedar E, Porgador A, Mandelboim O. The mechanisms controlling the recognition of tumor- and virus-infected cells by NKp46. Blood. 2004;103:664–672. doi: 10.1182/blood-2003-05-1716. [DOI] [PubMed] [Google Scholar]
- Oh S, McCaffery J M, Eichelberger M C. Dose-dependent changes in influenza virus-infected dendritic cells result in increased allogeneic T-cell proliferation at low, but not high, doses of virus. J Virol. 2000;74:5460–5469. doi: 10.1128/jvi.74.12.5460-5469.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.