

Abstract
During microRNA (miRNA) biogenesis, one strand of a ∼21–22-nucleotide RNA duplex is preferentially selected for entry into a silencing complex. The other strand, known as the miRNA* species, has typically been assumed to be a carrier strand. Here we show that, although Drosophila melanogaster miRNA* species are less abundant than their partners, they are often present at physiologically relevant levels and can associate with Argonaute proteins. Comparative genomic analyses revealed that >40% of miRNA* sequences resist nucleotide divergence across Drosophilid evolution, and at least half of these well-conserved miRNA* species select for conserved 3′ untranslated region seed matches well above background noise. Finally, we validated the inhibitory activity of miRNA* species in both cultured cells and transgenic animals. These data broaden the reach of the miRNA regulatory network and suggest an important mechanism that diversifies miRNA function during evolution.
miRNAs are an abundant class of ∼21–22-nucleotide (nt) RNAs that typically function as post-transcriptional repressors of gene activity1,2. The biogenesis of animal miRNAs involves stepwise processing of precursor transcripts containing hairpin structures. Canonical primary miRNA transcripts are cleaved in the nucleus by the RNase III enzyme Drosha, releasing ∼60–80-nt pre-miRNA hairpins3. In addition, splicing and debranching of short hairpin introns termed ‘mirtrons’ can directly generate pre-miRNA—like hairpins4–6. In both cases, the hairpins are exported to the cytoplasm and cleaved by the RNase III enzyme Dicer, resulting in a ∼21-nt miRNA duplex7–10. Although both strands of miRNA duplexes are necessarily produced in equal amounts by transcription, their accumulation is asymmetric at steady state. The convention is to refer to the more abundant product of a pre-miRNA or mirtron hairpin as the miRNA and its rarer partner as a miRNA* species11.
The function of miRNA strands is evident from the preferential conservation of 7-nt sequences in target transcripts with Watson-Crick complementarity to positions 2–8 of mature miRNAs (the ‘seed’ region). Although other features influence target-site efficacy, miRNA seed matches are often necessary and sufficient for target regulation12–15 and are the basis of most genome-wide predictions of miRNA regulatory sites16–18. Such studies conclude that most animal genes are either actively regulated by one or more miRNAs or actively avoid the acquisition of miRNA binding sites17,19. The reach of the miRNA regulatory network may in fact be larger, depending on the extent to which additional miRNA genes remain to be discovered, the extent to which noncanonical target sites are functional, and the extent to which nonconserved sites are relevant in vivo20.
The nonrandom nature of miRNA strand selection was posited to reflect an active process that minimizes the population of silencing complexes with illegitimate miRNA* species. The mechanism of strand selection correlates with the relative free energies of the duplex ends, as the small RNA whose 5′ end inhabits the less stable end is preferentially maintained in the mature silencing complex21,22. Nevertheless, miRNA* species are necessarily present in the cell and have been detected in increasing numbers during large-scale small RNA sequencing efforts23–25. Although previous studies did not explicitly address their potential trans-regulatory function, it is difficult to imagine how miRNA* species might be entirely excluded from entering regulatory complexes. In this study, we combine experimental and computational methods to show that many D. melanogaster miRNA* species are bona fide trans-regulatory RNAs with demonstrable effects on endogenous regulatory effects. Furthermore, we show that the inherent ‘dual’ nature of miRNA hairpins has tangible consequences for miRNA gene evolution.
RESULTS
Some miRNA* species are relatively abundant in total RNA
Initial analysis of ∼4,000 D. melanogaster small RNA sequences yielded clones from 62 miRNA loci26. miRNA* species were cloned for nine loci; however, only one of these was cloned more than twice (miR-2a-2*, four clones). These and other early cloning efforts contributed to the prevailing view that miRNA* species are, by and large, rare RNAs. More recent analysis of > 1 million small RNA sequences that aligned to the D. melanogaster genome (GEO dataset GSE7448) not only identified new miRNA genes, but also yielded a nearly comprehensive set of cloned miRNA* species25. These data permitted detailed analyses of miRNA* biology.
Inspection of 316,927 miRNA and 28,465 miRNA* clones revealed that many miRNA* species are actually relatively abundant. Whereas 60 out of 134 miRNA genes showed ≥20:1 strand bias, 29 out of 134 miRNA genes showed ≤5:1 strand bias. Some of these ratios were uncertain owing to low numbers of reads; still, 16 genes in the ‘low’ strand bias set were confidently sampled by > 100 reads. The entire set of miRNA:miRNA* read counts and ratios, organized by gene and library of origin, is presented in Supplementary Table 1 online.
The absolute number of miRNA* clones recovered from abundantly expressed loci was greater than the miRNA clone counts from many lowly expressed loci. The interpretation of this is ambiguous, as seemingly ‘rare’ miRNAs cloned from a whole animal might be highly expressed in a specific cell type. However, analysis of S2 cells showed that many miRNA* species were more abundant than many miRNA species in this single cell type. For example, we recovered > 50 clones for seven miRNA* species in S2 cells (miR-276a*, bantam*, miR-34*, miR-2a-2* miR-282*, miR-996* and miR-306*), whereas 40 of the S2-expressed miRNAs had <50 clones.
We validated the steady-state accumulation of miRNA and miRNA* species using northern analysis of total D. melanogaster RNAs (Fig. 1 and Supplementary Fig. 1 online). We easily detected miRNA* species from members of miRNA families (mir-10, mir-276a and mir-281-1) and from unique miRNA genes (mir-306, mir-184 and mir-iab-4). Together with the cloning data, this suggested that many miRNA* species are present at levels that are conceivably biologically relevant.
Figure 1.

Both miRNA and miRNA* species can be detected in total RNA. RNA was analyzed from D. melanogaster cells at different stages: E, 0–24 h embryos; LP, 3rd instar larvae and mixed pupae; A, adult males and females; S, S2 cells. Each blot was sequentially hybridized, stripped and rehybridized to detect a miRNA* species, its partner miRNA species, then 2S rRNA (the exception is miR-10*, whose companion 2S blot is shown in Supplementary Fig. 1). Arrows indicate the mature 21–24 species and brackets indicate the pre-miRNA hairpin.
Preferential stability of highly conserved miRNA* species
The mere existence of miRNA* species in total RNA does not establish their function as regulatory RNAs. A trivial alternative interpretation is that certain discarded miRNA* strands are degraded more slowly than others. We addressed this by examining the ratio of miRNA: miRNA* reads across successive time points in D. melanogaster embryonic development. Indeed, the miRNA:miRNA* ratio of many loci became increasingly skewed as development proceeded. For example, the ratio of miR-286 to miR-286* increased from 1.6 to 4.8 to 51.6 across three consecutive stages of embryo development (Supplementary Table 2 online). This trend is consistent with the preferred stability of miRNA species and concomitant turnover of miRNA* species.
miR-286 derives from a cluster of eight miRNAs: mir-309, mir-3, mir-286, mir-4, mir-5, mir-6-1, mir-6-2 and mir-6-3 (Fig. 2a). Note that although the miRNA products of the mir-6 genes are identical, their respective miRNA* species are distinct. The isolation of 297 miR-6-1* clones, 373 miR-6-2* clones and 858 miR-6-3* clones provided evidence for fairly comparable processing of each of the three mir-6 genes (Supplementary Table 1). As was done in a previous analysis25, we divided the total miR-6 clone counts in each library by three to estimate the output of each individual mir-6 gene.
Figure 2.

Highly conserved miRNA* species accumulate to higher relative levels at steady state. (a) Evolution of the mir-309->mir-6 cluster. Both miRNA (green) and miRNA* (yellow) sequences of mir-4 and mir-5 are perfectly conserved across 12 Drosophilids. The other six miRNA* species in this cluster all accumulated divergence (red nucleotides); mir-6-1 is shown. (b) Temporal dynamics of miRNA:miRNA* ratios. Six genes show read ratios that rise progressively with embryo age (time after egg laying (AEL)). Only the genes with highly conserved miRNA* (mir-4 and mir-5) maintain a low miRNA:miRNA* ratio. (c) The miRNA:miRNA* ratios of all 26 miRNA loci that generate at least 50 clones in each of four embryonic time points. Loci whose miRNA* species is perfectly conserved in 11 or 12 Drosophilids are in blue; all other loci are in gray. The most highly conserved miRNA* species tend to be present at a more comparable level to their miRNA partners and are concentrated at the bottoms of these graphs.
All eight miRNA* species from this operon were abundant at the earliest time point (0–1h after egg laying), with six miRNA:miRNA* pairs cloned at a ratio of 4:1 or less. However, six loci showed miRNA:miRNA* ratios that rose rapidly with age (in 2–6-h, 6–10-h and 12–24-h embryos), usually exceeding 50:1 (Fig. 2b). The exceptions were mir-4 and mir-5, whose ratios rose to only 8:1 (mir-4) or actually decreased to a terminal ratio of 2.2:1 (mir-5). Notably, these were the same genes of the cluster whose miRNA* sequences were most highly conserved. In fact, both miRNA and miRNA* of mir-4 and mir-5 are perfectly conserved among 12 Drosophilids (Fig. 2a).
We asked whether these trends applied among miRNA genes more generally. For this purpose, we selected the 26 miRNA loci that produced at least 50 clones in each of four successive embryo time points (Supplementary Table 2), values that ensured that their miRNA:miRNA* ratios were quantitatively meaningful. These genes collectively showed miRNA:miRNA* ratios that rose sharply, were stable or even decreased with embryo age (Fig. 2c). Notably, the nine genes whose ratios remained lowest (bantam, mir-5, mir-92a, mir-2a-2, mir-4, mir-8, mir-996, mir-7 and mir-2b-2) were all genes whose miRNA* species were perfectly conserved among 11 or 12 Drosophilid species (Fig. 2c, ‘blue’ genes, and Supplementary Fig. 2a,b online). These observations generalize the correlation between the degree of nucleotide conservation of miRNA* species and their tendency to accumulate to higher levels at steady state.
miRNA* species populate AGO1 complexes
We proposed that the correlation between the evolutionary constraint of miRNA* strands and their expression level might reflect their usage as endogenous regulatory RNAs and sought evidence for this by asking whether any miRNA* species were physically associated with effector complexes. To do so, we immunoprecipitated endogenous Argonaute-1 (AGO1) and probed this fraction for endogenous miRNA and miRNA* species. These experiments detected miR-34:miR-34*, miR-184:miR-184* and miR-276:miR-276* in association with AGO1 in S2 cells, and miR-5:miR-5* and miR-10:miR-10* from 0–10-h embryos (Fig. 3). The fraction of miRNA* species that associated with AGO1, relative to their total cellular content, was in many cases comparable to that of their partner miRNA species. A notable exception was mir-184, for which much less of the miRNA* detected in total RNA was associated with AGO1 relative to miRNA. This provided compelling evidence that the immunoprecipitation assay reports on a small RNA population that is distinct from total RNAs, and probably reflects the active sorting of miRNAs and miRNA* species into regulatory complexes27,28. We take these data as evidence for active miRNA* sorting in both cultured cells and in the animal. At the same time, these data suggest that caution should be applied in ascribing function to small RNAs detected in total RNA because they contain species that are rejected from sorting complexes and/or await their degradation.
Figure 3.
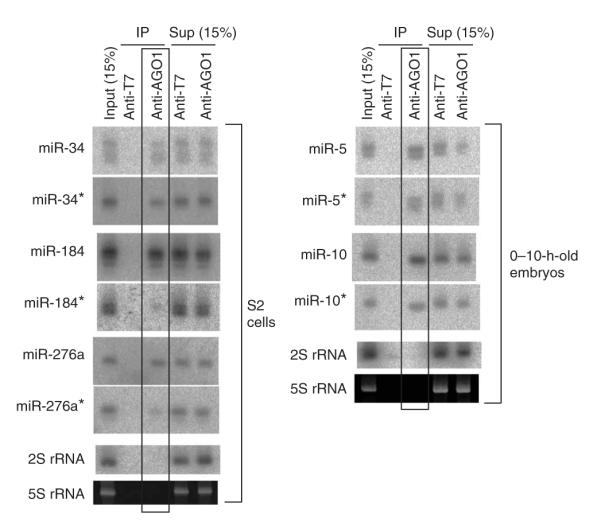
miRNA and miRNA* species can be co-immunoprecipitated with AGO1. Each blot contains input total RNA, the immunoprecipitate (IP) of mouse anti-T7 (as a control) and mouse anti-AGO1, and the supernatant (sup) of anti-T7 and anti-AGO1 incubations. IPs were performed from S2 cells and 0–10-h-old embryos, as indicated, and probed for the indicated small RNAs. In the cases of mir-34, mir-5 and mir-10, both miRNA and miRNA* were effectively coimmunoprecipitated with AGO1 but not T7 antibody; miR-184* and miR-276a* were weakly, but specifically, coimmunoprecipitated with AGO1.
Validation of the regulatory activity of miRNA* species
We next tested the regulatory potential of miRNA* species using assays previously used to validate miRNA targets. Active sorting of miRNA processing intermediates was reported to influence the type and/or level of target regulation in heterologous tests27. Nevertheless, we find that perfect target sites are usually more sensitive than imperfect target sites, even when the bulk of the small RNA partitions into AGO1 complexes. This is probably due to the much stronger cleavage activity of AGO2 relative to AGO1 (ref. 27).
We therefore designed artificial targets containing four tandem sequences antisense to miR-iab-4-5p or miR-iab-4-3p (the left- and right-arm products of mir-iab-4) downstream of the Renilla luciferase coding region in psiCHECK2, a vector that also contains a control firefly luciferase gene. We then examined their response to expression constructs for mir-iab-4 or mir-315. We earlier showed that this mir-315 construct is biologically active and strongly represses miR-315 target genes29 and, thus, represents an appropriate noncognate control. The miRNA and miRNA* sensors were strongly repressed (20-fold to 40-fold) by mir-iab-4, whereas these sensors showed no response to mir-315 (Fig. 4a). We and others have shown that the antisense strand of mir-iab-4 encodes a functionally distinct miRNA hairpin termed mir-iab-8 (refs. 30–33). We tested left-arm and right-arm sensors for this pre-miRNA and again observed strong and specific repression of both miR-iab-8-5p and miR-iab-8-3p sensors (Fig. 4a). Together, these results clearly demonstrate that miRNA* species can have regulatory capability.
Figure 4.

Sensor assays in cultured cells and transgenic animals validate the regulatory activity of miRNA* species. (a) Repression by miRNA* species (colored bars) from canonical precursors. Luciferase sensors bearing complementary sites to miR-iab-4-5p/miR-iab-3p and miR-iab-8-5p/miR-iab-8-3p were all repressed by cognate but not noncognate pre-miRNAs. Mean values and s.d. are shown. (b) Repression by miRNA* species from a mirtron precursor. Sensors for both miR-1010 and miR-1010* were repressed by mir-1010 but not by the unrelated mirtron mir-1003. (c,d) Repression of miRNA* targets in transgenic animals. Shown are the wing pouch regions of third instar imaginal discs that express the indicated DsRed-miRNA constructs and GFP sensors. Inhibition of both 5p and 3p sensors by mir-iab-4 (c) and mir-iab-8 (d) is reflected by the loss of GFP in DsRed+ cells.
Short intronic hairpins termed ‘mirtrons’ provide a secondary source of miRNA precursors that are independent of canonical nuclear miRNA processing4–6. Drosophila melanogaster mirtrons are strongly biased to yield right-arm products, possibly because their left-arm products begin with a G residue, which is rare among mature miRNAs. Still, we asked whether the left-arm products of a mirtron might also be functional. We assayed the response of sensors for miR-1010 and miR-1010* to ectopic mirtron expression constructs for mir-1010 and mir-1003. We observed strong repression of both sensors by mir-1010 but not by mir-1003 (Fig. 4b), indicating that miRNA* functionality extends to mirtron precursors as well.
Finally, we tested whether miRNA* species could regulate target genes in the animal using a repression assay in the D. melanogaster wing imaginal disc34. We recently used this assay to show that mir-iab-4 and mir-iab-8 could selectively repress tub-GFP sensor transgenes carrying perfect target sites for their respective left-arm products, miR-iab-4-5p and miR-iab-8-5p (ref. 33). We now prepared transgenic animals carrying sensors for the right-arm, ‘3p’ species and tested these in parallel with their partner, the left-arm, ‘5p’ sensors. We observed that ectopic mir-iab-4 could repress both its 5p sensor (Fig. 4c, above) and its 3p sensor (Fig. 4c, below), although the regulation of the 3p sensor was weaker. We also found that ectopic mir-iab-8 strongly inhibited both its 5p (Fig. 4d, above) and 3p (Fig. 4d, below) sensors. Overall, these data provide convincing evidence that miRNA* species are capable of repressing targets in both cultured cells and in the animal.
Patterns of miRNA* evolution are consistent with their regulatory potential
Our tests show that miRNA* species can populate regulatory complexes to guide target repression. Nevertheless, this could, in principle, be fortuitous. For example, a certain degree of imprecision in miRNA strand selection might be of neutral consequence and thus tolerated in vivo. However, this view is not consistent with the well-documented and adverse consequences of small interfering RNA (siRNA) off-targeting35. Rather, to avoid undesirable regulation of cellular transcripts, we proposed that many miRNA* species may have infiltrated endogenous regulatory networks during evolution. To obtain evidence for this model, we examined the patterns of miRNA gene conservation across 12 Drosophilid species. The conservation of miRNA sequences, with particular constraint on their seed regions, was previously taken to reflect their sequence-based, trans-regulatory activity16. We reasoned that the same logic might apply to miRNA* sequences and seeds.
In fact, the possibility of trans-acting activity for miRNA* species was hinted at by earlier computational efforts for miRNA gene finding36. We observed that miRNA* species diverge much more slowly than miRNA terminal loops, a property that strongly aids the identification of functional animal miRNA hairpins as ‘saddle’ structures36,37. The extent to which miRNA* strands are constrained in their primary nucleotide sequence is not adequately explained solely by pressures to maintain particular secondary structures, which would predict a higher frequency of compensatory mutations than is observed during evolution.
We systematically examined a set of 131 D. melanogaster canonical miRNA genes, almost all of which had cloned miRNA* species25. Of these, 31 miRNA* sequences were completely conserved among all 12 sequenced Drosophilids (Supplementary Fig. 2a), and another 23 miRNA* sequences were nearly perfectly conserved (Supplementary Fig. 2b), with up to 4 aggregate mismatches among all orthologs (that is, only 4 out of ∼260 bases). The fact that so many ( ∼40%) miRNA* sequences resist nucleotide divergence across a broad species range is inconsistent with the idea that miRNA* species are merely carrier strands whose only constraint is to maintain hairpin pairing to their miRNA partners. We also classified 11 additional genes as ‘highly conserved’, in that no more than two miRNA* nucleotide positions had diverged among 12 orthologs (Supplementary Fig. 2b). In total, 65 genes satisfied highly conserved (HC) criteria, or nearly half of all D. melanogaster miRNA loci. We divided the remaining miRNA gene alignments on the basis of their presence in non-Sophophoran Drosophilids (Drosophila virilis, Drosophila mojavensis and Drosophila grimshawi), the most distantly related sequenced species relative to D. melanogaster. There were 46 genes with non-Sophophoran orthologs (Supplementary Fig. 2c) and 20 genes that were restricted to the Sophophora (Supplementary Fig. 2d; we refer to these as the poorly conserved (PC) gene set).
We calculated the relative conservation of each 7-nt window along the orthologs of all miRNA strands using a scheme that was weighted according to evolutionary branch length (Methods). A previous survey of paralogous miRNA families revealed that positions 2–8 showed the highest constraint of all such 7-nt windows16. Our analysis differed in that we considered all orthologous miRNAs, which allowed us to evaluate many more gene comparisons and also to consider more recent evolutionary trends (as gene orthologs are much more recently diverged than gene paralogs).
Analysis of 131 miRNAs revealed two discernable evolutionary patterns. First, the 5′ and 3′ ends of miRNAs were more conserved than their central regions (Fig. 5a, dark green). This is consistent with the idea that there is general pressure to maintain the immediate sequence of Drosha and Dicer processing sites. Second, the 5′ ends of miRNAs were slightly more conserved than their 3′ ends. In particular, the miRNA seed window at positions 2–8 was most conserved (Fig. 5a, below, indicated by an asterisk on the dark green bar). These trends paralleled the results of paralog analysis16 and reflect the experimental demonstration that the sequence at the 5′ end of the miRNA is most crucial for target identification14,15,36. The 65 HC miRNA genes were nearly universally conserved along their miRNA strands and thus generated little in the way of evolutionary signal. Nevertheless, it was evident that the central region of even highly conserved miRNAs showed some measure of divergence, resulting in a slight dip in their aggregate conservation scores (Fig. 5a, below, light green; see also the closer view above).
Figure 5.

Bioinformatic evidence for the endogenous usage of both miRNAs and miRNA* species as regulatory RNAs. (a) miRNA-miRNA* sequence evolution. Above is a schematic of a typical miRNA hairpin, showing that the miRNA seed pairs to the 3′ end of the miRNA*, and vice versa. Analysis of all miRNAs (below, dark green) shows that miRNA termini are more conserved than their central regions, but that windows 1–7 and 2–8 are the most highly conserved. Similar trends apply to the subset of 65 highly conserved (HC) miRNAs and miRNA* species; the highest-scoring portion of the graph has been enlarged (middle). (b) The relative conservation of 3′ UTR complements to 7-nt windows across the 65 HC miRNA-miRNA* (above) and 20 poorly conserved (PC) miRNA/miRNA* (below), assessed between D. melanogaster and its distant relatives D. mojavensis and D. virilis. There is preferential conservation of heptamers complementary to positions 1–7 and 2–8 (and, to a lesser extent, 3–9) of both miRNAs and miRNA* species. (c) The proportion of 3′ UTR matches to miRNA 2–8 seeds that are conserved between D. melanogaster (Dm) and the increasingly divergent species D. simulans (Ds), D. yakuba (Dy), D. ananassae (Da), D. pseudoobscura (Dp), D. mojavensis (Dmo) and D. virilis (Dv). The signal-to-noise ratio (S/N) of HC miRNA and miRNA* seeds, but not of PC seeds, increases steadily with evolutionary distance. (d) Estimated number of conserved target sites for HC miRNAs-miRNA*s. miRNA and miRNA* seeds with similar hit frequencies as their controls in Dm (left) have more conserved matches than their controls in Dmo/Dv (right).
If the primary purpose of a miRNA* species is simply to promote accurate processing of its miRNA partner, then we might expect that miRNA* species should be more tightly constrained at their 3′ ends, which pair with the miRNA seed (Fig. 5a). On the contrary, systematic analysis of the 65 HC miRNA* species produced a profile that was notably analogous to miRNA strands. In particular, the 5′ and 3′ termini of miRNA* arms were more conserved than their central regions, but miRNA* 5′ ends were slightly more conserved than their 3′ ends (Fig. 5a, yellow). miRNA* conservation dropped off noticeably between the positions 2–8 and 3–9 windows, a feature that was suggestive of preferred seed constraint for miRNA* species. Therefore, although miRNA* species are less well-conserved than miRNA species, they show patterns of nucleotide divergence that are consistent with their selection for regulatory activity.
Selective conservation of miRNA* seed matches in target 3′ UTRs
The evolutionary rigidity of ∼50% of Drosophila miRNA* species was suggestive of their functional constraint. We sought to corroborate this by comparing the evolutionary behavior of miRNA and miRNA* seed matches. Watson-Crick complements to miRNA seeds, namely positions 2–8 from their 5′ ends, identify significantly more conserved matches in 3′ UTRs than do matched cohorts of shuffled seeds16,18,34. We asked whether this applied to miRNA* seeds as well. Because sequence randomization necessarily yields some motifs that are not representative of a true genome, we took care to create control heptamers that had the same nucleotide composition and the same hit frequency (±∼10%) in D. melanogaster 3′ UTRs as genuine miRNA heptamers (see Methods).
We first asked whether miRNA:miRNA* seed matches were more conserved than matches to all other heptamers along these small RNAs16. We used the pairwise conservation score (PCS) method to rank the relative conservation of D. melanogaster 3′ UTR sequences with that of divergent Drosophilids38. This score represents the log rank ratio between the number of seed matches in D. melanogaster and the species of comparison, for which positive values imply functional constraint. As expected, seed matches to 7-nt windows at the 5′ ends of D. melanogaster miRNAs were preferentially conserved in the highly diverged species D. mojavensis and D. virilis, with other 7-nt windows evolving neutrally (Fig. 5b, above, green). The highest-scoring windows were positions 2–8 and 1–7, consistent with their known role in determining miRNA target specificity. Analysis of the 65 highly conserved miRNA* species yielded a similar picture. Although the trends were more modest, heptamer matches to the 5′ ends of miRNA* species clearly showed preferential conservation (Fig. 5b, above, yellow).
For comparison, we analyzed the set of 20 D. melanogaster miRNAs (Supplementary Fig. 4d online) that lack orthologs or homologs outside of the Sophophoran subgenus. We designated these as PC miRNAs, although their conservation status is heterogeneous: some PC genes have orthologs in nine species. Neither PC miRNAs nor PC miRNA* species showed preferred conservation of 3′ UTR matches across any 7-nt window (Fig. 5b, below, light blue and pink), negative data that provided reassurance that our control sets were selected appropriately.
We next examined the numbers of seed matches to the 2–8 window that were conserved between species of increasing evolutionary distance from D. melanogaster. The fraction of conserved hits to functional miRNA seeds relative to controls increases with evolutionary distance, resulting in a rising signal-to-noise profile across speciation16,18. For the 65 HC miRNA genes, average miRNAs showed a ∼3:1 ratio in the most diverged species (Fig. 5c, green diamonds). On the other hand, the 20 PC miRNA seed matches showed no enrichment across evolution, so their values stayed flat at a ratio of ∼1.0 (Fig. 5c, blue triangles). The same was true for the PC miRNA* seeds (Fig. 5c, black circles).
In light of the PC miRNA:miRNA* data, the behavior of the 65 HC miRNA* species was noteworthy. Their values increased steadily to a terminal signal-to-noise of 1.48 to 1 in D. mojavensis and D. virilis (Fig. 5c, yellow squares). A caveat to this value is that some miRNA* species have the same seed as some miRNAs; notably, miR-5* shares the K box seed of miR-2/6/11/13/308, whereas miR-9a* shares the Brd box seed of miR-79. We consider it appropriate to include their contributions to the miRNA* target network because, at least in the case of mir-5, we presented biochemical evidence that its precursor actively loads appreciable amounts of miRNA* into AGO1 (Fig. 3). Nevertheless, even when discounting these miRNA* species to afford a more conservative interpretation, the remaining HC miRNA* still reached a signal-to-noise ratio of 1.38:1.
Another way to demonstrate that the HC miRNA* data was not dominated by a few genes was to consider the percentage of genes that select for targets. Of 65 HC miRNAs, 62 showed a positive PCS score in 3′ UTRs, and 54 out of 65 (83%) reached a P-value significance of 95% relative to their controls (see Supplementary Table 3 online for individual gene values). On the other hand, 40 out of 65 HC miRNA* species showed a positive PCS score in their 3′ UTRs, with 23 out of 65 (35%) with P > 95%. Therefore, the biological signal of targeting by HC miRNA* species was not dominated by a few genes. Only three PC miRNAs and two PC miRNA* achieved a P > 95% difference with their random controls (see Supplementary Table 4 online for individual gene values).
According to these measures based solely on conserved seed matches, highly conserved Drosophila miRNAs have at least conserved 30 targets above noise, whereas their corresponding miRNA* species have ∼10 targets above noise (Fig. 5d). Although these miRNA* target networks are smaller than those of average miRNAs (Fig. 5c), their significance can be measured in light of the fact that at least one-fifth of all miRNA* species can be confidently described as showing some endogenous 3′ UTR targets that are conserved among the Drosophilids, whereas one-sixth of D. melanogaster miRNAs (that is, the PC gene set) lack such conserved targets.
Experimental evidence for endogenous miRNA* activity
Our bioinformatic studies strongly support the idea that a significant fraction of miRNA* species contribute to 3′ UTR—mediated regulatory networks. In our final experimental tests, we wished to generate experimental evidence for the regulatory activity of an endogenous miRNA* and/or regulation of an endogenous miRNA* target.
We focused on mir-276a for tests of the former, as we recovered similar numbers of small RNAs from both its hairpin left arm(5p) and right arm(3p) from various samples. For example, there were 408 miR-276a-5p and 479 miR-276a-3p clones from S2 cells, and we could corroborate the steady-state accumulation of both strands using northern analysis (Fig. 1). We note that the mir-276a and mir-276b loci encode identical left-arm products, which might obscure their assignment to a particular locus. However, their right-arm (3p) products have unique sequences. As no miR-276b-3p clones were recovered from S2 cells, despite > 1,000 clones of this RNA in other libraries (Supplementary Table 1), we inferred that mir-276a is uniquely expressed by S2 cells. We verified this by performing quantitative reverse-transcription PCR (qPCR) for pri-mir-276a and pri-mir-276b, which provided evidence for a > 15:1 discrepancy in the level of their primary transcripts in S2 cells (Supplementary Fig. 3 online).
Using four-tandem site sensors for miR-276a or miR-276a* into psiCHECK2, we first assayed their response to ub-Gal4 and UAS-DsRed-mir-276a. We observed that either mir-276a or mir-276b induced 4-fold to 8-fold repression of both 5p and 3p sensors. Evidence for specificity of repression came with their insensitivity to a noncognate expression vector for mir-315. Thus, both mir-276 genes (which are both perfectly conserved on their left and right arms across Drosophilid evolution, Supplementary Fig. 4) produce functional small RNAs (Fig. 6a), consistent with our previous tests with mir-iab-4, mir-iab-8 and mir-1010 (Fig. 4).
Figure 6.

Endogenous relevance of miRNA*-mediated repression. (a) Ectopic mir-276a and mir-276b, but not DsRed or mir-315, specifically repress both miR-276a-5p and miR-276a-3p sensors. The experimental design is the same as for Figure 4a; mean values and s.d. are shown. (b) 2′O-methylated antisense oligos (ASO) against endogenous miR-276a-5p and miR-276a-3p specifically derepress cognate sensors. Data were collected and analyzed as in a. (c) Abrupt is an endogenous target of the miRNA* species, miR-iab-4-3p. Of three ‘2–8’ seed matches (highlighted yellow) near the start of the abrupt 3′ UTRs (denoted ab#1, ab#2 and ab#3) two are highly conserved among divergent Drosophilids and one has a t1A feature (red). (d) Endogenous Abrupt protein in a wing pouch carrying one copy of ptc-Gal4 and two copies of UAS-DsRed. Abrupt accumulates to a high level in the L5 wing vein primordium and a lower level in the L3 vein domain (arrows). (e) Wing pouch of an animal carrying two copies of ptc-Gal4 and one copy of UAS-DsRed-mir-iab-4. Abrupt protein is reduced in L3 (arrows). Genotypes d and e express roughly equivalent amounts of DsRed and control for the neutral effect of DsRed on Abrupt. (f) In ptc-Gal4, UAS-DsRed-mir-iab-4; tub-GFP-abrupt 3′ UTR wing imaginal discs, GFP is strongly suppressed in DsRed/miR+ cells.
We then analyzed the effect of depleting endogenous miR-276a-5p and miR-276a-3p using 2′O-methylated antisense oligonucleotides (ASO)39,40. As a control, we used a similarly sized ASO to miR-288. We observed that the miR-276a-5p sensor was specifically derepressed by its cognate ASO but not by miR-276a-3p or miR-288 ASO (Fig. 6b). Conversely, the miR-276a-3p sensor was specifically derepressed by its cognate ASO and was unaffected by noncognate ASO. These data provided evidence for the endogenous regulatory activity of both small RNAs derived from a single pre-miRNA hairpin.
To obtain experimental evidence for an endogenous 3′ UTR target of a miRNA* species, we returned to the Hox miRNA mir-iab-4. We showed that its miRNA* species miR-iab-4-3p is an active repressor in cultured cells and in the animal (Fig. 4), and its strict conservation suggested that it might have endogenous targets. Searches for miR-iab-4-3p seed matches revealed abrupt as a top candidate18. The abrupt 3′ UTR contains three seed matches that are Watson-Crick complements to positions 2–8 of miR-iab-4-3p (Fig. 6c). Two of these sites are perfectly conserved across 12 sequenced Drosophilid genomes (Supplementary Fig. 4) and are located close enough to each other to mediate synergism41,42. One of these sites also has a t1A feature, which increases site efficacy43. Finally, all three sites are located near the stop codon (Fig. 6c), an optimal location for miRNA target-site function41,44,45. For these reasons, abrupt is a compelling miRNA* target. We note that miR-iab-4-5p shows plausible matching to these sites; however, all of the sites are mispaired with position two of miR-iab-4-5p (Supplementary Fig. 4b), a disruption that has explicitly been shown to nearly eliminate target regulation in D. melanogaster14. We therefore consider miR-iab-4-5p unlikely to be functionally relevant to abrupt regulation.
We tested the ability of ectopic mir-iab-4 to repress Abrupt in the wing imaginal disc. Endogenous Abrupt protein is present at the highest level in the L5 wing primordium46 (Fig. 6d). Expression of ectopic mir-iab-4 using bx-Gal4 did not suppress L5 expression of Abrupt (data not shown), possibly because of the high target level, compensatory regulation and/or occluding factors bound to abrupt transcripts in this domain. However, when we examined discs that ectopically expressed mir-iab-4 using ptc-Gal4, which specifically overlaps a region of lower Abrupt expression in the L3 wing primoridium, we detected mild downregulation of endogenous Abrupt (Fig. 6e). To obtain clearer evidence for this regulatory relationship, we analyzed a tub-GFP-abrupt 3′ UTR transgenic sensor. The heterologous promoter excludes the possibility of compensation at the transcriptional level. These assays clearly revealed repression of the abrupt sensor in mir-iab-4—expressing cells (Fig. 6f), confirming it as a genuine miRNA* target. In summary, these experimental tests provide functional evidence for the regulatory activity of endogenous miRNA* species and targets.
DISCUSSION
miRNA hairpins produce two distinct regulatory RNAs
It has long been recognized that pre-miRNA hairpins necessarily produce a duplex composed of two potential small RNAs. Because one of the two strands usually accumulates to a higher level than its partner, it has been widely assumed that the poorly expressed products, termed miRNA* species, represent functionally irrelevant carrier strands. However, this idea is not consistent with the fact that most miRNA* sequences are substantially constrained during evolution (Supplementary Fig. 2), nor with the fact that several miRNA* species are cloned at high frequency relative to their miRNA partner (Fig. 2 and Supplementary Table 1).
High-throughput methods allowed us to collect comprehensive data on miRNA* sequences and quantitative data on miRNA:miRNA* ratios25. With cloned miRNA* species in hand, we found that a significant fraction of pre-miRNA hairpins produce a miRNA* species whose seed sequence identifies 3′ UTR target sites that are under demonstrable selective conservation. This indicates that miRNA* species are not only present in cells, as they must be, but that a significant proportion of them have acquired endogenous regulatory targets. In support of this, we found that miRNA* species can populate AGO1 complexes and obtained evidence for their ability to repress target transcripts. Therefore, although miRNA* are by no means equivalent to miRNA species, they nonetheless comprise a functionally relevant and substantial aspect of small RNA regulatory networks.
A concurrent computational study proposed that 11 miRNA* species in D. melanogaster show characteristics of regulatory miRNA species47. Notably, ten of these genes were independently scored in our bioinformatic surveys as HC miRNA*s that select for targets above noise at a significance level of 98% (Supplementary Table 3). We found that the eleventh gene (miR-959*) selects for targets at a significance of 94%, even though we designated it as a PC gene (Supplementary Table 4). mir-959 is actually the most highly conserved member of the PC gene set, and its miRNA* seed sequence is maintained precisely across nine Sophophoran orthologs (Supplementary Fig. 2d). Thus, some signature of its targeting capability may linger in the other Drosophilids. Our studies go further in demonstrating that 23 out of 131 miRNA genes select targets above noise at a significance level of 95%. Undoubtedly, this is an underestimate of the number of functional miRNA* species that are incorporated into endogenous regulatory networks, as we were able to validate the function of other miRNA* species that did not reach this significance level (that is, miR-iab-4-3p and miR-276a*; Figs. 4 and 6, and Supplementary Table 3). We infer from this that up to ∼50% of all D. melanogaster miRNA genes—that is, genes that show exceptional evolutionary constraint on their miRNA* species (Supplementary Fig. 2a,b)—may prove to have some endogenous targets.
Dual miRNA-miRNA* function of Hox miRNAs
Because of our historical interest in Hox miRNAs30,33, we dedicated particular effort to experimental tests of Hox miRNA* species. It was earlier speculated that the Hox miRNA locus mir-10 might have the capacity to repress independent targets via its left- and right-arm products22. Target predictions revealed miR-10-5p as a likely repressor of the Hox gene Abd-B25,48 and miR-10-3p as a regulator of the Hox gene Scr14. We support this idea with experimental evidence that both miR-10-5p and miR-10-3p are indeed loaded into AGO1 (Fig. 3). In a more extreme case, we and others showed that bidirectional transcription and processing of the Hox mir-iab-4/mir-iab-8 locus results in four different miRNAs31–33. We show that all four mir-iab-4/mir-iab-8 miRNAs have regulatory activity in the animal (Fig. 4), and three of these now have validated endogenous targets30,32,33 (Fig. 6). Thus, dual miRNA-miRNA* function is a shared feature of the different Hox miRNA genes.
In light of our computational and experimental studies on miRNA* functionality, additional examples of compelling miRNA* targets will undoubtedly come to be recognized. The Bartel laboratory maintains a web server that searches for conserved matches to user-defined sequences (http://www.targetscan.org/fly_10/seedmatch.html), and can be used to generate candidate target lists for miRNA* species. It is important to bear in mind that, although the endogenous miRNA* regulatory network is considerable, it is not as broad as the miRNA regulatory network. Therefore, predicted miRNA* target lists will have substantial background, with 2 out of 3 attributable to noise (Fig. 5). As with miRNAs, then, experimental tests are necessary to draw firm conclusions on the biological utility of individual computationally inferred target sites.
Dual-function hairpins may drive miRNA diversification
Our data suggest that all miRNA loci are, at least to some extent, dualfunction genes that produce distinct regulatory RNAs from both left- and right-hairpin arms (Fig. 7a). Of course, the principal mode is for one regulatory RNA to predominate over its partner, owing to preferential degradation of miRNA* species and/or preferential loading of miRNA strands into effector complexes. On the other hand, it would seem challenging, if not impossible, to exclude all miRNA* species from entering effector complexes. A considerable amount of siRNA off-targeting is attributable to the unintended regulatory activity of siRNA* passenger strands from nonasymmetrically loaded siRNA duplexes21,22. On this basis, we propose that the regulatory activity of miRNA* strands will not generally be neutral with respect to a cell. Instead, this must be accompanied by their functional incorporation into endogenous regulatory networks, just as with miRNA strands.
Figure 7.

The inherent dual-regulatory capability of miRNA hairpins may influence miRNA evolution. (a) Although bulk miRNA* species are degraded, miRNA hairpins are designed to load specific fractions of miRNA and miRNA* species into AGO complexes. As with miRNA strands, miRNA* functionality has been accompanied by their incorporation into endogenous regulatory networks. (b) The relative ratio of left-arm and right-arm products can differ among members of the same miRNA family. In some cases, the dominant ‘miRNA’ strand has switched between family members. Other miRNA families share a preferred strand, but the level of strand asymmetry can differ dramatically among family members. Perhaps these are miRNA loci in the midst of ‘switching’ their dominant arm.
For unique miRNA loci, the extent to which miRNA* species are transferred to AGO complexes may also depend on the degree to which the remaining amount of miRNA species is sufficient to serve the normal regulatory needs of an organism. In theory, diversion of hairpin output toward the miRNA* species might be better tolerated in cases of miRNA gene duplication, which could free a gene copy from normal constraints. In this scenario, we might expect to identify cases in which paralogous miRNA loci produce dominant small RNAs from opposite arms. We previously found this to be true for both the K box and Brd box gene families49. Further inspection of small RNA sequences revealed two other clear examples of families that show ‘miRNA arm switching’ (mir-310/311/312/313 and mir-276a/276b) and four other examples where families include highly asymmetric and more equivalent miRNA hairpin outputs, possibly representing loci that are in the process of switching arms (including mir-252/1002, mir-12/960, mir-279/286 and mir-285/995/998; Fig. 7b). Thus, the phenomenon of arm switching has a demonstrable impact on miRNA gene evolution.
In summary, the inhabitance of miRNA* species in AGO complexes, the demonstration of their regulatory activity and the detection of selective evolutionary pressures on miRNA* seeds and their complementary sequences in 3′ UTRs indicate that miRNA* function has measurable effects on gene regulatory networks in living animals, and during the course of species evolution. In addition, they have strong implications for the interpretation of ectopic expression of miRNAs, which in our experiments frequently result in the repression of both miRNA and miRNA* targets.
METHODS
Northern analysis
Total RNA was isolated from staged Drosophila samples or S2 cells using Trizol (Gibco). RNAs associated with AGO1 were isolated from 0–10-h embryos or from S2 cells as described4.
Quantitative reverse transcription PCR
We reverse-transcribed 2 μg DNA-free RNA with random primer and Superscript III (Invitrogen), and used 1 μl cDNA as a template for qPCR using SYBR Green (ABI). qPCR primers are listed in the Supplementary Methods.
Luciferase sensor assays
Single-site and four copy—site sensors for the various miRNAs were cloned into a modified version of psiCHECK2 with 5′ NotI and 3′ XhoI cloning sites. miRNA and mirtron expression constructs consisted of ∼400 nt of genomic sequence cloned into the 3′ UTR of UAS-DsRed34. Luciferase sensor assays and 2′Omethyl antisense oligonucleotide treatments were performed as previously described4. Fold repression was normalized to the effect of the miRNA construct on the empty psiCHECK sensor, and all data were pooled from two sets of quadruplicate transfections performed on independent batches of cells. Sensor primers are listed in the Supplementary Methods.
GFP sensor assay in transgenic flies
UAS-DsRed-mir-iab-4 (ref. 30) and UAS-DsRed-mir-iab-8 (ref. 33) were described previously. We inserted a 1.8-kb PCR product that including the entire abrupt 3′ UTR and the flanking downstream sequence (to ensure normal polyadenylation) was inserted into tub-GFP34 to generate the abrupt sensor. We prepared perfect mir-iab4/mir-iab8 GFP sensors by inserting oligonucleotides with pairs of complementary sites into the XbaI/XhoI sites of tub-GFP; sensor primers are listed in the Supplementary Methods. We selected GFP+ DsRed+ late third instar larvae from appropriate crosses to obtain animals bearing Gal4, UAS-DsRed-miRNA and tub-GFP transgenes. It was necessary to express ptc-Gal4 with UAS-DsRed-mir-iab-4, and dpp-Gal4 with UAS-DsRed-mir-iab-8, because of early lethality with reciprocal Gal4-UAS combinations. We used a standard immunostaining technique50 with rabbit anti-GFP (Molecular Probes, 1:1250) and goat anti—rabbit-Alexa 488 (Molecular Probes, 1:500). Abrupt staining was performed with a rabbit antibody51 (Stephen Crews, 1:500).
Drosophilid miRNA gene alignments
We retrieved 15-way multiz alignments for each miRNA precursor from the University of California, Santa Cruz (UCSC) Genome Browser Database (http://genome.ucsc.edu) using the Table Browser25. We extracted the 12-fly data from these files, made manual adjustments to the alignments as necessary, and color-coded the output so that mature miRNAs were in green, miRNA* species in yellow, inferred miRNA* species in blue and positions of divergence in red. The complete alignment data are reported in Supplementary Fig. 2.
Analysis of miRNA gene conservation
The conservation of individual miRNA bases was assessed by pairwise comparison of each position in each ortholog with D. melanogaster Dm2 as the reference. Matches were scored as 1, whereas mismatched or gapped nucleotides in the alignment were scored as 0. To capture the significance of evolutionary distance across the species, we weighted the score in each species using the following scheme: droSim1 = 0.1, droSec1 = 0.1, droYak2 = 0.25, droEre2 = 0.25, droAna3 = 0.3, dp4 = 0.7, droPer1 = 0.7, droWil1 = 0.9, droVir3 = 1, droMoj3 = 1, droGri2 = 1. For each 7-nt window across the miRNA-miRNA* sequence, we summed the seven scores and rescaled them from 0–100, with the maximum score reflecting perfect conservation of all 7 nts in the window across all 12 Drosophilids.
Generation of miRNA seed controls
To establish biologically appropriate controls, we first tabulated the occurrence of all (47 = 16,384) possible heptamers among annotated Dm 3′ UTRs. We then generated controls for each consecutive heptamer across all miRNA and miRNA* species by calculating the unique permutations of a given heptamer and selecting those with the closest frequency in Dm 3′ UTRs to the reference heptamer. Specifically, we generated sets of controls containing all permutations within ± x% of the experimental frequency where x = (1, 2, 3, 4, 5, 8, 10 or 15), choosing the lowest x whose corresponding set contained at least five controls. If no x met this criterion, we chose the lowest x that gave at least three controls. 91% of the control heptamers to miRNAs and 89% of the control heptamers to miRNA* species were within 4% tolerance for the number of hits to D. melanogaster 3′ UTRs.
Analysis of 3′ UTR seed match conservation
To study the conservation patterns of sequences complementary to miRNA-miRNA* sequences, we identified 3′ UTR heptamers complementary to 7-nt windows slid across a given small RNA. We then used a lookup table of ∼16,000 relative conservation values for different heptamers across different pairs of Drosophilids (that is, the PCS score as defined in ref. 38) and plotted the average values for various sets of miRNA or miRNA* seed matches and their corresponding control sets.
To specifically evaluate canonical miRNA target sites, we considered 3′ UTR seed matches to positions 2–8 of miRNAs and miRNA* species. Controls were generated for these seeds using our standard method. We then used a lookup table of the number of heptamer instances that were conserved between the following species pairs: dm-dm, dm-dy, dm-da, dm-dp, dm-dmo and dm-dv38 (that is, the AC score as defined in ref. 38). We calculated the average of these values across all genes in the data set, and took the ratio of experimental seed match values to control seed match values as the signal-to-noise value.
To measure the significance of target conservation, we followed previously described methods48 and calculated the following P-value based on the binomial distribution:
where p is the average conservation level of the control shuffled heptamers. That is,
where S simply denotes the fact that we are considering shuffled controls. Stated more simply, this value is the probability of obtaining the same or greater number of conserved sequences given the average conservation of the shuffled controls. Therefore, for small values, we conclude that the level of target conservation is significant.
Supplementary Material
ACKNOWLEDGMENTS
We thank B. Tam (University of California, Davis) for helping to construct transgenic sensors. We thank J.G. Ruby and D. Bartel (Howard Hughes Medical Institute and Whitehead Institute) for sharing their initial analysis of Drosophila miRNAs; S. Crews (University of North Carolina) for the Abrupt antibody; the University of California, Santa Cruz genome center, Agencourt, and the Baylor College of Medicine for Drosophila genome sequences, assemblies and alignments; and J. Major (Sloan-Kettering Institute) for software and technical support. K.O. was supported by a grant from the Charles Revson Foundation. E.C.L. was supported by grants from the Leukemia and Lymphoma Society, the Burroughs Wellcome Foundation, the V Foundation for Cancer Research, the Sidney Kimmel Cancer Foundation and the US National Institutes of Health (GM083300).
Footnotes
Reprints and permissions information is available online at http://npg.nature.com/ reprintsandpermissions
Note: Supplementary information is available on the Nature Structural & Molecular Biology website.
References
- 1.Lai EC. MicroRNAs: runts of the genome assert themselves. Curr. Biol. 2003;13:R925–R936. doi: 10.1016/j.cub.2003.11.017. [DOI] [PubMed] [Google Scholar]
- 2.Bushati N, Cohen SM. MicroRNA functions. Annu. Rev. Cell Dev. Biol. 2007;23:175–205. doi: 10.1146/annurev.cellbio.23.090506.123406. [DOI] [PubMed] [Google Scholar]
- 3.Lee Y, et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- 4.Okamura K, Hagen JW, Duan H, Tyler DM, Lai EC. The mirtron pathway generates microRNA-class regulatory RNAs in Drosophila. Cell. 2007;130:89–100. doi: 10.1016/j.cell.2007.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruby JG, Jan CH, Bartel DP. Intronic microRNA precursors that bypass Drosha processing. Nature. 2007;448:83–86. doi: 10.1038/nature05983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berezikov E, Chung W-J, Willis J, Cuppen E, Lai EC. Mammalian mirtron genes. Mol. Cell. 2007;28:328–336. doi: 10.1016/j.molcel.2007.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hutvagner G, et al. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science. 2001;293:834–838. doi: 10.1126/science.1062961. [DOI] [PubMed] [Google Scholar]
- 8.Grishok A, et al. Genes and mechanisms related to RNA interference regulate expression of the small temporal RNAs that control C. elegans developmental timing. Cell. 2001;106:23–34. doi: 10.1016/s0092-8674(01)00431-7. [DOI] [PubMed] [Google Scholar]
- 9.Ketting RF, et al. Dicer functions in RNA interference and in synthesis of small RNAs involved in developmental timing in C. elegans. Genes Dev. 2001;15:2654–2659. doi: 10.1101/gad.927801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knight SW, Bass BL. A role for the RNase III enzyme DCR-1 in RNA interference and germ line development in Caenorhabditis elegans. Science. 2001;293:2269–2271. doi: 10.1126/science.1062039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lau NC, Lim L, Weinstein E, Bartel DP. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science. 2001;294:858–862. doi: 10.1126/science.1065062. [DOI] [PubMed] [Google Scholar]
- 12.Lai EC, Tam B, Rubin GM. Pervasive regulation of Drosophila Notch target genes by GY-box-, Brd-box-, and K-box-class microRNAs. Genes Dev. 2005;19:1067–1080. doi: 10.1101/gad.1291905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lai EC. MicroRNAs are complementary to 3′ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat. Genet. 2002;30:363–364. doi: 10.1038/ng865. [DOI] [PubMed] [Google Scholar]
- 14.Brennecke J, Stark A, Russell RB, Cohen SM. Principles of microRNA-target recognition. PLoS Biol. 2005;3:e85. doi: 10.1371/journal.pbio.0030085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Doench JG, Sharp PA. Specificity of microRNA target selection in translational repression. Genes Dev. 2004;18:504–511. doi: 10.1101/gad.1184404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 17.Stark A, Brennecke J, Bushati N, Russell RB, Cohen SM. Animal microRNAs confer robustness to gene expression and have a significant impact on 3′UTR evolution. Cell. 2005;123:1133–1146. doi: 10.1016/j.cell.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 18.Grun D, Wang YL, Langenberger D, Gunsalus KC, Rajewsky N. MicroRNA target predictions across seven Drosophila species and comparison to mammalian targets. PLoS Comp. Biol. 2005;1:e13. doi: 10.1371/journal.pcbi.0010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farh KK, et al. The widespread impact of mammalian microRNAs on mRNA repression and evolution. Science. 2005;310:1817–1821. doi: 10.1126/science.1121158. [DOI] [PubMed] [Google Scholar]
- 20.Lai EC. Predicting and validating microRNA targets. Genome Biol. 2004;5:115. doi: 10.1186/gb-2004-5-9-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell. 2003;115:209–216. doi: 10.1016/s0092-8674(03)00801-8. [DOI] [PubMed] [Google Scholar]
- 22.Schwarz DS, et al. Asymmetry in the assembly of the RNAi enzyme complex. Cell. 2003;115:199–208. doi: 10.1016/s0092-8674(03)00759-1. [DOI] [PubMed] [Google Scholar]
- 23.Kloosterman WP, et al. Cloning and expression of new microRNAs from zebrafish. Nucleic Acids Res. 2006;34:2558–2569. doi: 10.1093/nar/gkl278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ruby JG, et al. Large-scale sequencing reveals 21U-RNAs and additional microRNAs and endogenous siRNAs in C. elegans. Cell. 2006;127:1193–1207. doi: 10.1016/j.cell.2006.10.040. [DOI] [PubMed] [Google Scholar]
- 25.Ruby JG, et al. Evolution, biogenesis, expression, and target predictions of a substantially expanded set of Drosophila microRNAs. Genome Res. 2007;17:1850–1864. doi: 10.1101/gr.6597907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aravin AA, et al. The small RNA profile during Drosophila melanogaster development. Dev. Cell. 2003;5:337–350. doi: 10.1016/s1534-5807(03)00228-4. [DOI] [PubMed] [Google Scholar]
- 27.Forstemann K, Horwich MD, Wee L, Tomari Y, Zamore PD. Drosophila microRNAs are sorted into functionally distinct Argonaute complexes after production by Dicer-1. Cell. 2007;130:287–297. doi: 10.1016/j.cell.2007.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tomari Y, Du T, Zamore PD. Sorting of Drosophila small silencing RNAs. Cell. 2007;130:299–308. doi: 10.1016/j.cell.2007.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Silver SJ, Hagen JW, Okamura K, Perrimon N, Lai EC. Functional screening identifies miR-315 as a potent activator of Wingless signaling. Proc. Natl. Acad. Sci. USA. 2007;104:18151–18156. doi: 10.1073/pnas.0706673104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ronshaugen M, Biemar F, Piel J, Levine M, Lai EC. The Drosophila microRNA iab-4 causes a dominant homeotic transformation of halteres to wings. Genes Dev. 2005;19:2947–2952. doi: 10.1101/gad.1372505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bender W. MicroRNAs in the Drosophila bithorax complex. Genes Dev. 2008;22:14–19. doi: 10.1101/gad.1614208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stark A, et al. A single Hox locus in Drosophila produces functional microRNAs from opposite DNA strands. Genes Dev. 2008;22:8–13. doi: 10.1101/gad.1613108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tyler DM, et al. Functionally distinct regulatory RNAs generated by bidirectional transcription and processing of microRNA loci. Genes Dev. 2008;22:26–36. doi: 10.1101/gad.1615208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stark A, Brennecke J, Russell RB, Cohen SM. Identification of Drosophila microRNA targets. PLoS Biol. 2003;1:e60. doi: 10.1371/journal.pbio.0000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jackson AL, et al. Expression profiling reveals off-target gene regulation by RNAi. Nat. Biotechnol. 2003;21:635–637. doi: 10.1038/nbt831. [DOI] [PubMed] [Google Scholar]
- 36.Lai EC, Tomancak P, Williams RW, Rubin GM. Computational identification of Drosophila microRNA genes. Genome Biol. 2003;4:R42. doi: 10.1186/gb-2003-4-7-r42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Berezikov E, et al. Phylogenetic shadowing and computational identification of human microRNA genes. Cell. 2005;120:21–24. doi: 10.1016/j.cell.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 38.Gu J, Fu H, Zhang X, Li Y. Identifications of conserved 7-mers in 3-UTRs and microRNAs in Drosophila. BMC Bioinformatics. 2007;8:432. doi: 10.1186/1471-2105-8-432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hutvagner G, Simard MJ, Mello CC, Zamore PD. Sequence-specific inhibition of small RNA function. PLoS Biol. 2004;2:e98. doi: 10.1371/journal.pbio.0020098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meister G, Landthaler M, Dorsett Y, Tuschl T. Sequence-specific inhibition of microRNA- and siRNA-induced RNA silencing. RNA. 2004;10:544–550. doi: 10.1261/rna.5235104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grimson A, et al. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell. 2007;27:91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nielsen CB, et al. Determinants of targeting by endogenous and exogenous micro-RNAs and siRNAs. RNA. 2007;13:1894–1910. doi: 10.1261/rna.768207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 44.Gaidatzis D, van Nimwegen E, Hausser J, Zavolan M. Inference of miRNA targets using evolutionary conservation and pathway analysis. BMC Bioinformatics. 2007;8:69. doi: 10.1186/1471-2105-8-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Majoros WH, Ohler U. Spatial preferences of microRNA targets in 3′ untranslated regions. BMC Genomics. 2007;8:152. doi: 10.1186/1471-2164-8-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cook O, Biehs B, Bier E. brinker and optomotor-blind act coordinately to initiate development of the L5 wing vein primordium in Drosophila. Development. 2004;131:2113–2124. doi: 10.1242/dev.01100. [DOI] [PubMed] [Google Scholar]
- 47.Stark A, et al. Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures. Nature. 2007;450:219–232. doi: 10.1038/nature06340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stark A, et al. Systematic discovery and characterization of fly microRNAs using 12 Drosophila genomes. Genome Res. 2007;17:1865–1879. doi: 10.1101/gr.6593807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lai EC, Wiel C, Rubin GM. Complementary miRNA pairs suggest a regulatory role for miRNA:miRNA duplexes. RNA. 2004;10:171–175. doi: 10.1261/rna.5191904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lai EC, Rubin GM. neuralized functions cell-autonomously to regulate a subset of Notch-dependent processes during adult Drosophila development. Dev. Biol. 2001;231:217–233. doi: 10.1006/dbio.2000.0124. [DOI] [PubMed] [Google Scholar]
- 51.Hu S, Fambrough D, Atashi JR, Goodman CS, Crews ST. The Drosophila abrupt gene encodes a BTB-zinc finger regulatory protein that controls the specificity of neuromuscular connections. Genes Dev. 1995;9:2936–2948. doi: 10.1101/gad.9.23.2936. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.