

Abstract
Laboratories working with closely related viruses need simple and cost-effective ways to rapidly validate viral stocks, detect contamination and measure the abundance of viral RNA species. Using RT-PCR and specific primers an approach for the specific detection of rhinovirus type 14 (RV14) or poliovirus type 1 (PV1) is presented. It is demonstrated that viral sequences can be amplified directly from viral stocks or from infected cells. In addition, the utility of this protocol for the detection of low levels of contaminating PV1 in RV14 stocks is shown. Further, using quantitative real-time PCR It is shown that this approach can be used for the quantitative analysis of viral RNA and replication kinetics in infected cells. This method should be useful for laboratories working with PV and RV14 and could be adapted easily for use by laboratories working with other rhinovirus and enterovirus serotypes.
Keywords: real-time PCR, Rhinovirus 14, Poliovirus type 1, contamination
1. Introduction
Rhinovirus (RV) and poliovirus (PV) are positive stranded RNA viruses that belong to the family picornaviridae. Rhinovirus and poliovirus exhibit extensive sequence similarity (Callahan et al., 1985; Stanway et al., 1984) and in fact the International Committee on Taxonomy of Viruses have approved recently the reclassification of RV into the enterovirus (EV) genus; the same genus as poliovirus. In addition to their sequence similarities, RV and PV grow well on many of the same cell lines and induce similar cytopathic effects on susceptible cell lines making it hard to distinguish these viruses without further testing. This close similarity, along with the more rapid replication of PV has resulted in several reports in the literature of cross-contamination of RV stocks with PV (Davies et al., 2003; Peng et al., 2007; Savolainen and Hovi, 2003). Inadvertent contamination with PV can result in a significant loss of time, effort and money and create uncertainty in the scientific community regarding the validity of results. In addition, inadvertent contamination makes compiling a complete inventory of PV stocks, as recommended by the World Health Organization as part of the Polio Eradication campaign much more difficult.
Current methods used for the identification of PV and RV rely on differential acid lability and neutralization assays with type specific antisera (Couch and Atmar, 1999; Schnurr, 1999). While effective, these methods are laborious, time consuming and, in the case of neutralization require sera that may be in limited supply. More rapid and sensitive detection methods based on PCR have been reported. While effective, most of these approaches utilize primers that will amplify both RV and EV sequences (Andreoletti et al., 2000; Bruce et al., 1990; Hyypia et al., 1989; Johnston et al., 1993; Kammerer et al., 1994; Santti et al., 1997; Torgersen et al., 1989). Distinguishing, between EV and RV then requires additional steps such as hybridization with type-specific oligonucleotides (Andreoletti et al., 2000; Freymuth et al., 1997; Halonen et al., 1995; Lonnrot et al., 1999; Olive et al., 1990; Santti et al., 1997), nested PCR (Andeweg et al., 1999; Ireland et al., 1993; Mori and Clewley, 1994), restriction digestion (Kammerer et al., 1994; Torgersen et al., 1989) or gel electrophoresis (Atmar and Georghiou, 1993; Mori and Clewley, 1994). More recently, real-time PCR assays for the detection of both RV and EV have been described, however, all of these assays rely on oligonucleotide probes tagged with expensive fluorescent molecules to distinguish RVs from EVs (Costa et al., 2008; Dagher et al., 2004; Deffernez et al., 2004; Kares et al., 2004; Lu et al., 2008; Nijhuis et al., 2002; Petitjean et al., 2006; Scheltinga et al., 2005; Verstrepen et al., 2002; Wright et al., 2007).
For laboratories working with only a limited number of RV or EV serotypes a simple, rapid and relatively inexpensive method for distinguishing specific viral types would be very useful. Such an approach utilizing RT-PCR for distinguishing poliovirus type 1 (PV1) and rhinovirus type 14 (RV14) is described. This method is shown to be suitable for the rapid and accurate identification of PV1 and RV14 sequences from infected cells or directly from viral stocks. Using this approach, very small amounts of contaminating virus can be detected easily and quickly in virus stocks. Finally, it is shown that this method can be adapted for use with quantitative real-time PCR and SYBR green dye for quantitation of both virion-associated RNA in stocks and viral RNA levels in infected cells.
2. Materials and methods
2.1. Cell culture and viruses
HeLa cells were grown in monolayer in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2mM L-glutamine and penicillin-streptomycin at 37°C in 5% CO2. A549 cells, a human lung alveolar epithelial cell line, was purchased from ATCC and cultured in F-12 K medium (Invitrogen) supplemented with 10% FBS and Penicillin-Streptomycin as described above. Rhinovirus type 14 strain 1059 was obtained from ATCC (VR-284) and the specificity of the virus was confirmed by RT-PCR and RV14-specific antisera from ATCC (VR-284AS/RB). RV14 viral stocks were prepared as described previously (Gustin and Sarnow, 2002). Poliovirus type 1 Mahoney (PV1) stocks were prepared by infecting sub-confluent HeLa monolayers at a multiplicity of infection (MOI) of 10. Virus was adsorbed for 30 min at 37°C in CPBS (phosphate-buffered saline (PBS) containing 1mM MgCl2 and 1mM CaCl2). Following adsorption, residual virus was removed and DMEM with 10% FBS, 2mM L-glutamine and penicillin-streptomycin was added. At 7 hours post infection (hpi), cells were collected and washed in CPBS by centrifugation. Cell pellets were resuspended in CPBS and subjected to three freeze and thaw cycles followed by centrifugation at 10,000 × g for five minutes at 4°C. The supernatant was taken and stored at -70 °C. Sub-confluent HeLa/A549 cells were either mock-infected or infected with PV1/RV14 at an MOI of 50. Virus was adsorbed for 30 min at 32°C (RV14) or 37°C (PV1) in CPBS. Following adsorption, residual virus was removed and medium with 10% FBS was added.
2.2. Plasmids
A plasmid harboring the full length infectious cDNA of PV1 was described previously (Sarnow, 1989). A Plasmid with the full length RV14 genome was constructed by cloning full length RV14 cDNA (Gen Bank accession no. K02121). Plasmids harboring genomes of other picornaviruses were kindly provided by different scientists. Poliovirus type 2 (MEF strain) and poliovirus type 3 (Leon strain) (Dr. Eckard Wimmer); Poliovirus type 2 (Lansing strain, 39-2600 nucleotides) and poliovirus type 3 (Sabin strain, 1-2604 nucleotides) (Dr. Vincent Racaniello); Coxsackievirus B3 (Dr. Ralph Feuer); Human rhinovirus type (HRV) 16 and HRV1A (Dr. William Jackson).
2.3. Primer sequences
The primer sequences designed for PCR amplification of RV14 and PV1 are shown in Figure 1. RV14 primers corresponded to nt positions 299-318 in the 5′ UTR (forward) and nt positions 744-763 in VP4 (reverse). The PV1 conventional PCR primers corresponded to nt positions 631-650 in the 5′ UTR (forward) and nt positions 1410-1420 in ????? (reverse). The PV1 nested/real-time primers corresponded to nt positions 710-730 in the 5′ UTR (forward) and nt positions 862-880 in ????? (reverse). The primers for β-actin were described previously (Kotla et al., 2008). The specificity of primers was confirmed by sequencing of amplified products.
Figure 1.

RV14 and PV1 primers. HRV14 (Gen Bank accession no. K02121) and PV1 (Gen Bank accession no. V01149) genome sequences were obtained from NCBI nucleotide database and were aligned using the ClustalW sequence alignment program from EMBL website (http://www.ebi.ac.uk/Tools/clustalw2/index.html). Arrows represent forward and reverse primer sequences. Asterisk (*) indicates identical nucleotide bases.
2.4. Conventional PCR
Conventional PCR was conducted using the Advantage 2 Polymerase mix (Clontech; 639201). Unless otherwise indicated the following PCR cycling conditions were used for amplification: Initial denaturation at 95°C for 1′; 25 cycles of 95°C for 30″, 56°C for 1′, 68°C for 1′; and a final extension at 70°C for 10′. The PCR products were visualized by electrophoresis on ethidium bromide stained agarose gels. For assessing the cross-reactivity of RV14 and PV1 primers with other picornaviruses 0.25 ng of the plasmid DNA was used in the PCR.
2.5. RNase treatment, RNA isolation and cDNA synthesis
Virion-associated RNA was isolated by first treating 6 μl of virus stock with 10.5 units of RNase at 37°C for 30 minutes to remove unpackaged RNA (Promega; M426A). Virion RNA was then isolated using the Qiagen RNeasy mini kit (Qiagen; 74104) with the volumes of the buffers reduced to half at each step in the extraction protocol. Isolated virion-associated RNA was reverse transcribed to cDNA using the Superscript III First-Strand Synthesis System for RT-PCR (Invitrogen; 18080-051). Free viral RNA in stocks was detected by using 6 μl of virus stock directly in a cDNA synthesis reaction. In the case of RNAs extracted from infected cells, the standard Qiagen RNeasy mini kit protocol was followed and one microgram of RNA was reverse transcribed to cDNA.
2.6. Quantitative real-time PCR (qRT-PCR)
Either 0.2 or 0.4 percent of the total cDNA volume was used in a SYBR green PCR assay (New England Biolabs; F-410l). PCR was performed on a 7500 Fast Real-Time PCR System (Applied Biosystems). The following PCR cycling conditions were used for amplifying: first stage, 50°C for 2′; second stage 95°C for 10′; third stage, 35 cycles of 94°C for 10″, 56°C for 30″ and 72°C for 30″; fourth stage, dissociation. The cycle threshold (Ct) for each gene was determined by setting the Ct line at the center of the logarithmic phase of amplification for that particular amplicon.
2.7. Detection of viral contamination
RV14 virus stock was spiked with serial dilutions of PV1 stock ranging from 104 to 10-1 plaque forming units (pfu). Total RNA was extracted from the spiked samples using the Qiagen RNeasy mini kit. Fifty percent of the RNA was used for cDNA synthesis and 40% of the resulting cDNA was used for primary amplification by the conventional PCR method. For the nested PCR amplification 16% of the PCR product from the primary PCR was used.
3. Results
3.1. Identification of RV14 and PV1 by PCR
It was observed that the antisera raised against the virion proteins of PV1 cross-reacted with RV14 by immunoblot analysis (data not shown). Examination of the amino acid sequence similarity between RV14 and PV1 using the ClustalW sequence alignment program revealed nearly 53% sequence identity and 70% sequence similarity between these viruses (data not shown). Thus, the cross reactivity is likely due to the high level of sequence homology shared by PV1 and RV14. To circumvent this problem and to develop a simple and rapid method for the detection of PV1 and RV14 primers were designed to unique regions of these viruses for use in PCR analysis. To identify regions of PV1 and RV14 suitable for primer design the complete genome sequences were aligned using the ClustalW sequence alignment program. The alignment results revealed approximately 59% identity between RV14 and PV1 at the nucleotide level. Based on this alignment, primers were designed that were predicted to bind specifically to either the PV1 or RV14 sequences (Figure 1). For PV1 two primers sets were designed; One set was used for conventional PCR and the other set was used for nested PCR in conjunction with this first primer set or for real-time PCR. To examine the specificity of these primers conventional PCR was undertaken using plasmids harboring the genomes of RV14 and PV1 along with other representative members of the picornavirus family. Figure 2A demonstrates that the RV14 primers can amplify readily the appropriate size amplicon from the RV14 template but not from any of the three poliovirus types or coxsackievirus type B3 or even from the other rhinovirus templates examined. Similarly, the PV1 primers produce amplified product in the presence of PV1 template but not from any of the rhinovirus templates, the coxsackievirus B3 or even the more closely related poliovirus type 2 or 3 templates tested (Figure 2B and 2C). To confirm that these primers can be used for the detection of viral RNA from infected cells, RT-PCR was performed on RNA isolated from cells that had been infected with RV or PV1. RT-PCR analysis on the RNA extracted from infected cells indicated that the RV14 and PV1 primers could indeed detect RV14 and PV1, respectively (Figure 2D). These results indicate that the primers designed for RV14 and PV1 are specific for their respective templates and can be used for the detection of viral RNA in vivo.
Figure 2.
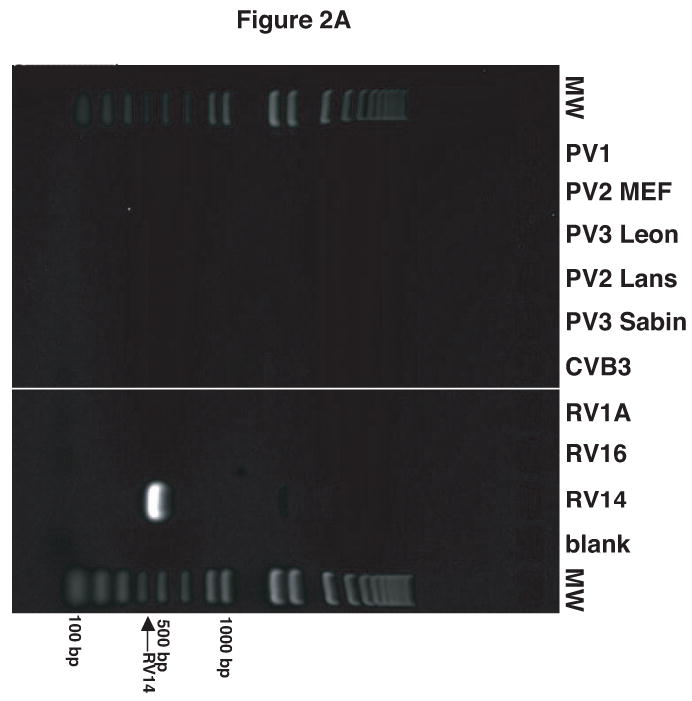
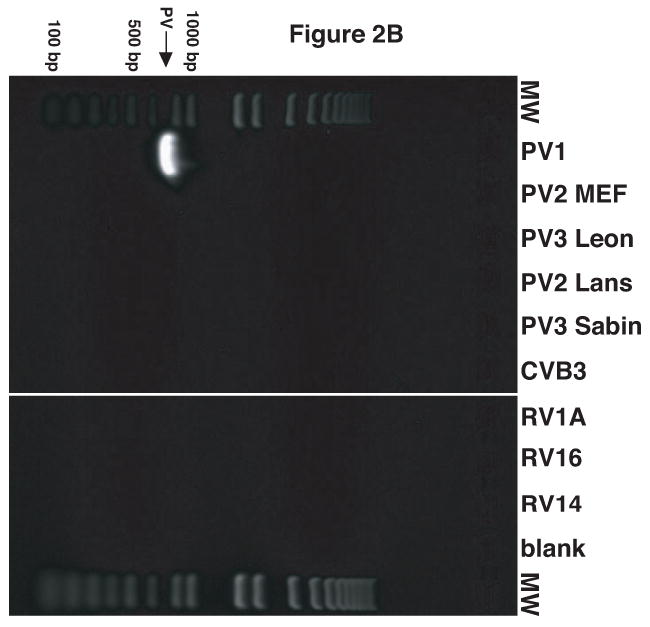
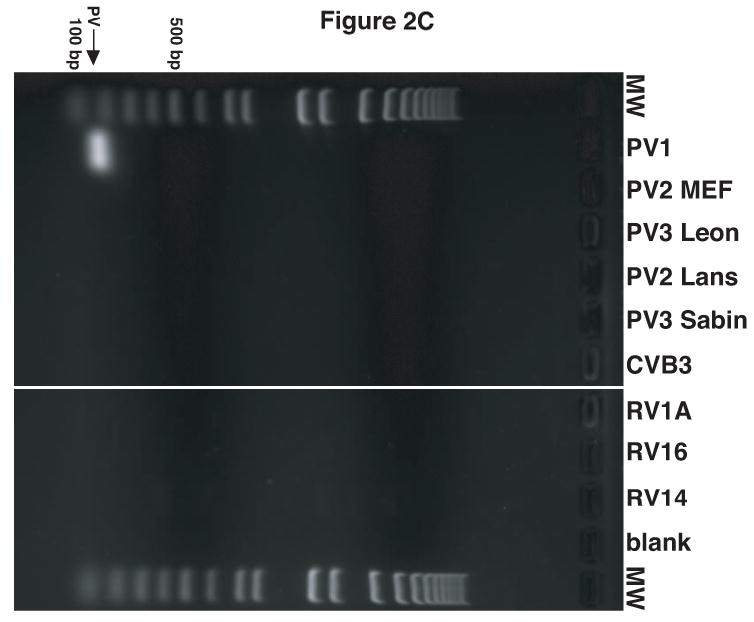
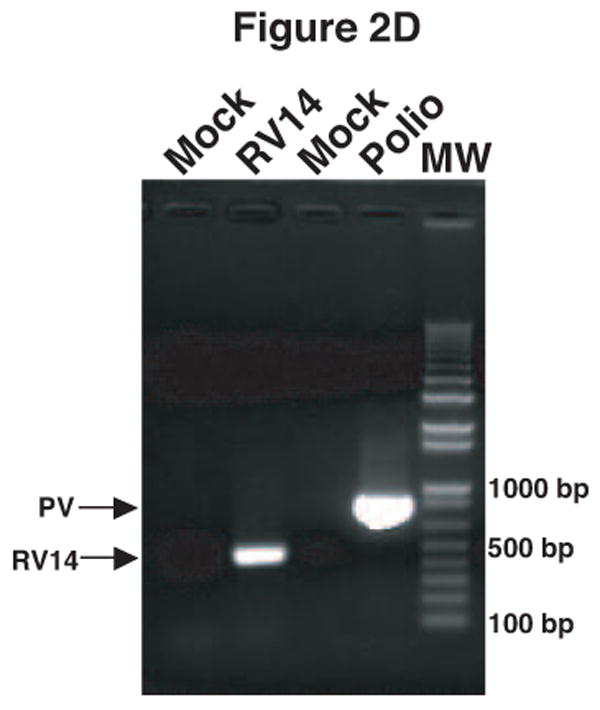
Validation of RV14 and PV1 primers. Conventional PCR was conducted using RV14 primers (A), PV1 primers (B) and PV1 nested PCR primers (C) and DNA templates harboring the sequences from the indicated viruses; Poliovirus type 1 (PV1), Poliovirus type 2 MEF-1 strain (PV2 MEF), Poliovirus type 3 Leon strain (PV3 Leon), Poliovirus type 2 Lansing strain (PV2 Lans), Poliovirus type 3 Sabin strain (PV3 Sabin), Coxsackievirus B3 (CVB3) and human rhinovirus types 1A, 16 and 14 (HRV1A, HRV16 and HRV14. Ethidium bromide stained agarose gels show the location of the 465 bp rhinovirus (RV14) (A) and 798 and 171 bp poliovirus (PV1) (B and C, respectively) specific amplicons. Blank indicates no template was added to the PCR reaction. MW; molecular weight markers. D. Identification of RV14 and PV1 from infected cells. Total RNAs extracted from uninfected, RV14 or PV-infected cells were reverse transcribed into cDNA and analyzed by PCR with primers for both RV14 and PV1. Labeling and analysis is as described above.
3.2. Detection of RV14 and PV1 directly from viral stocks
The results above indicated that the RV14 and PV1 primers could be used in a conventional PCR to detect viral RNA isolated directly from infected cells. However, to reduce the amount of time and effort needed to confirm the identity of viral stocks it would be helpful to eliminate the need for infection and the culturing of virus. Consequently, the ability to amplify viral sequences directly from viral stocks was examined. To improve sensitivity and allow for quantitative analysis of viral sequences quantitative real-time PCR (qRT-PCR) was utilized. This necessitated the design of a new set of PV1 primers that amplify a 171 bp fragment as this size amplicon is more suitable for real-time PCR analysis (Figure 1). The specificity of this primer set was confirmed by showing that it amplified the appropriate sized amplicon from a PV1 template, but not from other picornavirus templates (Figure 2C) and by sequencing (data not shown).
The viral stocks used in this laboratory contain a mixture of free viral RNA consisting of single stranded, replicative-intermediate and replicative form RNA that is not associated with virions and RNA that is packaged into virions. To measure free viral RNA viral stocks were used directly for cDNA synthesis, without subjecting them to denaturing conditions that might disrupt virions. Virion-associated RNA was measured by first treating viral stocks with RNase to remove free RNA, followed by standard RNA isolation and cDNA synthesis. Analysis of PV1 stocks by qRT-PCR indicated that in the absence of RNase treatment and denaturing conditions free viral RNA could easily be measured (Figure 3. Free RNA; Ct-26.2). If samples were treated with RNase before cDNA synthesis very little viral RNA was detected indicating that RNase treatment was able to degrade essentially all the free viral RNA and that virion RNA was not released during processing (Figure 3, RNase Treated). However, if RNA was extracted under denaturing conditions from RNAse-treated samples a strong signal was seen that corresponded to the virion-associated RNA (Figure 3. Virion RNA; Ct-17.1). A similar analysis using the RV14 primers was able to detect both free viral RNA and virion-associated RNA in RV14 stocks (data not shown). These results indicate that this method is able to measure directly free and virion-associated RNA in viral stocks.
Figure 3.
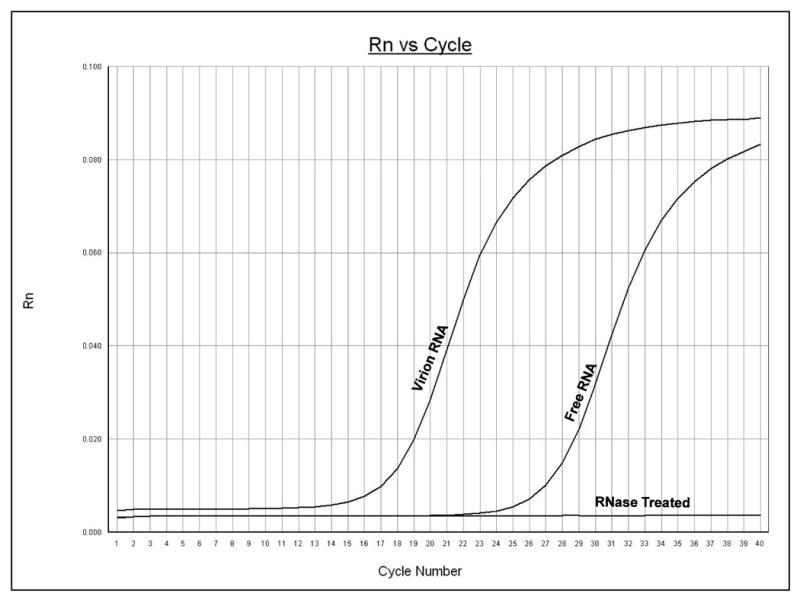
Detection of RNA directly from viral stocks. Poliovirus stock samples were analyzed by qRT-PCR using poliovirus primers. The y-axis represents fluorescence and the x-axis represents cycle number. The increase in fluorescence at a particular cycle number represents the cycle threshold (Ct) for that sample, which indicates the detection of nucleic acids. The PCR was carried out for 40 cycles. Graph shows amplification from a representative well. Free RNA represents the signal detected when viral stocks were used directly for cDNA synthesis, i.e., without disrupting virions. Virion RNA represents the signal detected when stocks were first digested with RNase to remove free RNA followed by purification under denaturing conditions to release virion-associated RNA. RNase Treated represents the signal observed when stocks were treated with RNase and then used directly for cDNA synthesis.
3.3. Detection of PV1 contamination in RV stocks
To determine if this method could be used as a rapid assay to detect PV1 contamination in viral stocks, RV14 stocks were spiked with various amounts of PV1. Total RNA was then extracted from these spiked samples, reverse transcribed and analyzed by PCR. The results from this primary PCR indicated that as little as 104 pfu of PV1 contamination could be detected (Figure 4A, lane 1). To increase further the limit of detection nested PCR using the primary PCR product as template was examined. The results indicate that nested PCR increased the detection limit to as little as 1 pfu of PV1 in RV14 stocks (Figure 4B, lane 5). Thus, this method appears suitable for rapid detection of very low amounts of contaminating PV1 in RV14 stocks.
Figure 4.
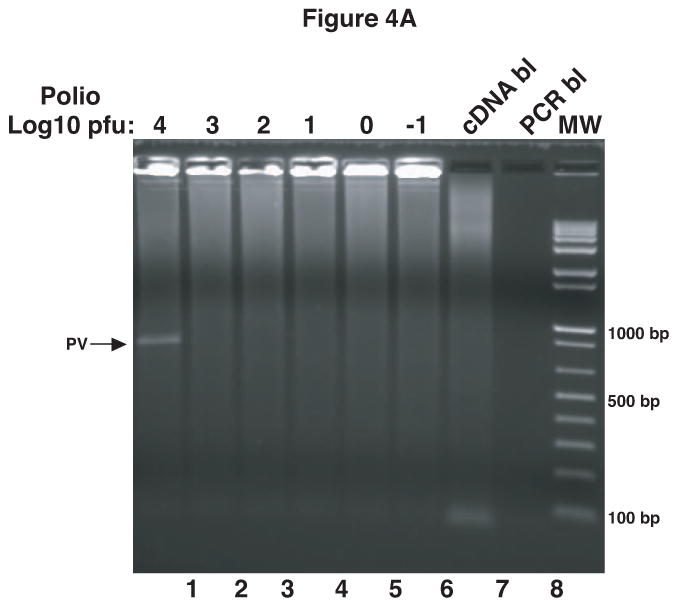
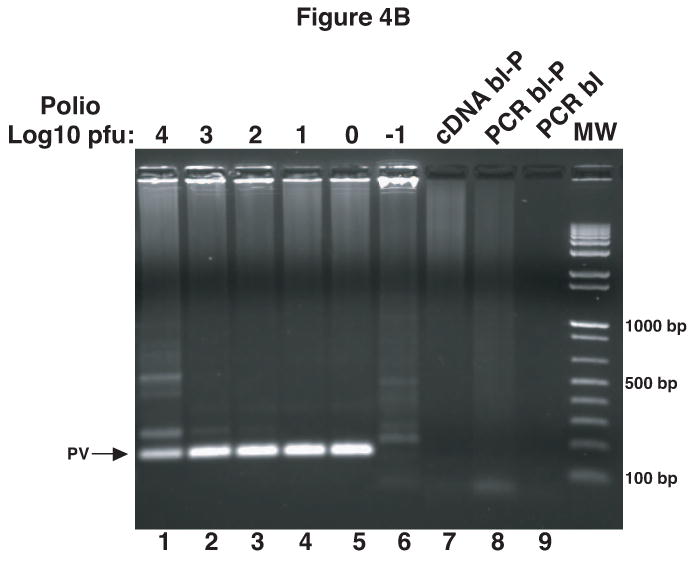
Detection of PV1 contamination in RV14 stocks. RV14 stocks were spiked with104 to 10-1 pfu of PV1 stock solution. RNA was extracted from these spiked samples followed by cDNA synthesis and primary PCR (A). Portions of primary PCR reactions were then used as template in a nested PCR reaction (B). Ethidium bromide stained agarose gels show the 798 bp and 171 bp PCR products that arise from primary PCR (A) and nested PCR (B), respectively. The PCR was carried out for 40 cycles and in the case of nested PCR the extension time was reduced to 30″. cDNA bl: cDNA blank-negative control for cDNA synthesis with no RNA template; PCR bl: PCR blank-negative control for PCR reaction with no cDNA template; PCR bl-P and cDNA bl-P indicate the blanks carried over from the primary PCR reaction and used in nested PCR.
3.4. Quantitative analysis of viral RNA in infected cells
Methods for measuring PV1 and RV viral RNA levels in infected cells have relied on Northern blotting, RNase protection assays or pulse labeling with 3H-uridine in the presence of actinomycin D. These methods are quite effective, but they are laborious and often require the use of radioactive materials, which increase further the cost and add to the time spent by requiring compliance with radiation safety guidelines. To provide a faster, more economical method for measuring viral RNA levels, the RV14 and PV1 primers were used in a quantitative real-time PCR (qRT-PCR) assay using SYBR green to determine viral replication kinetics at different times post infection. Initially, qRT-PCR was conducted using serial dilutions of plasmids harboring complete viral genomes to generate a standard curve for quantitation. Figure 5A shows that the PV1 primers can quantify effectively serial dilutions of the plasmid with the expected average Ct difference of 3.32 between the 10 fold dilutions (3.32=Log2 (10)). Analysis of RNA from PV1-infected cells using this primer set revealed that PV1 RNA levels increased from 1-5 hours post infection (hpi) after which RNA levels plateaued (Figure 5B). These results are in good agreement with results from earlier studies showing that the majority of viral RNA synthesis occurs after 3 hpi (reviewed in (Johnson and Sarnow, 1995). Similarly, it was shown that the RV14 primers can be used to accurately measure viral RNA synthesis in RV14-infected cells (Kotla et al., 2008). Thus the use of these primers in a qRT-PCR format represents a rapid and effective way to quantitate the accumulation of viral RNA in infected cells.
Figure 5.
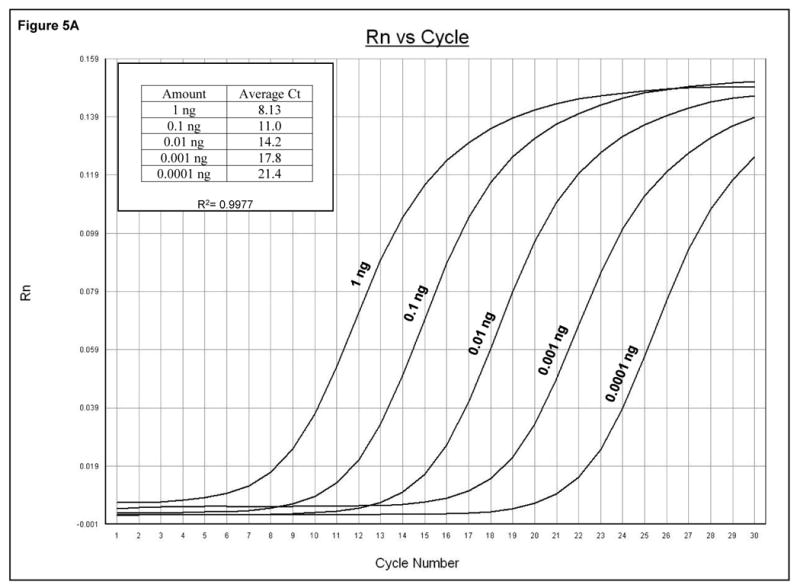
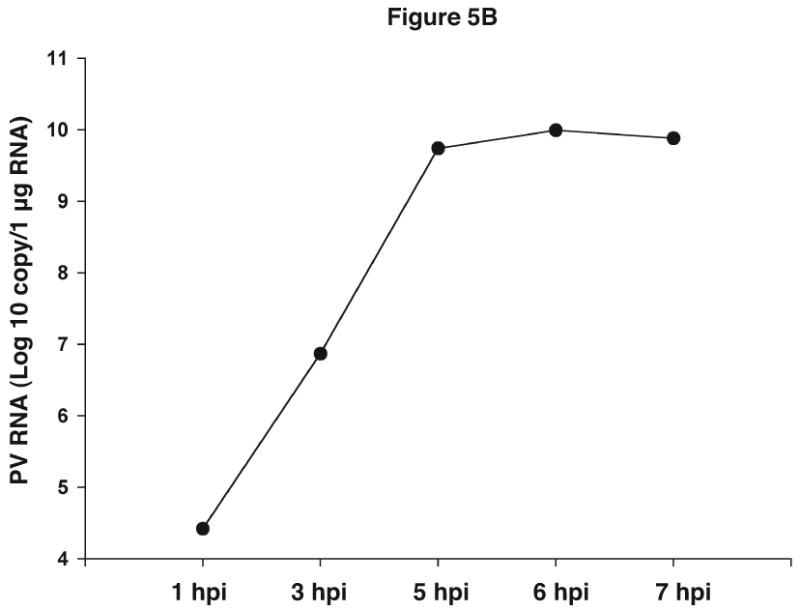
Quantitative analysis of viral sequences. A. qRT-PCR was conducted on 10-fold serial dilutions of a plasmid harboring the complete PV1 genome using the PV1 primer set that amplifies a 171 bp product. The y-axis represents fluorescence and the x-axis represents cycle number. The PCR was carried out for 30 cycles. Graph shows amplification from a representative well. The amount of plasmid DNA and the corresponding average Ct from triplicate samples are indicated in the upper left. R2 value obtained from the standard curve generated by plotting Ct values against plasmid copy number is shown. B. Total RNAs were extracted from uninfected or PV1-infected cells at an MOI of 10 at the indicated times post-infection and analyzed by qRT-PCR with primers for both PV1 and β-actin mRNAs. PV1 RNA copy number was determined from the standard curve obtained in A. The amount of PV1 RNA was normalized to the amount of β-actin mRNA using 1 hpi as reference. Values on the y-axis are expressed after log10 transformation. The data represents viral RNA levels from a representative experiment. hpi: hours post infection.
4. Discussion
The existence of several reports in the current literature documenting contamination of RV stocks with PV suggest that this may be a more common problem than is appreciated currently (Davies et al., 2003; Peng et al., 2007; Savolainen and Hovi, 2003). In this study an approach is described that allows for the rapid identification of PV1 type 1 and RV14 sequences in cell lysates and viral stocks. For laboratories working with these closely related viruses, these methods provide an inexpensive way to confirm the identity of viral stocks, detect contamination and measure viral RNA synthesis in infected cells. Using available sequence data and alignment algorithms it should be possible to design primers that are specific for other PVs, RVs or other enteroviruses as needed. Thus, these methods may prove useful for laboratories working with other members of the picornaviridae family of viruses and perhaps viruses in other families.
The design of species-specific primer sets for RV14 and PV1 eliminates the need for expensive TaqMan probes or time consuming Southern or dot blot analysis to confirm the identity of viral sequences. In addition, the use of SYBR green detection obviates the need to design a specific third primer for the target sequence. Of course, with SYBR green detection it is not possible to distinguish on-target from off-target amplifications without further analysis. Hence, for each primer set it is necessary to analyze amplified products by agarose gel electrophoresis and melting curve analysis to confirm that the fluorescent signal is due to the presence of the desired amplicon.
For the detection of PV1 contamination in RV14 stocks a nested-PCR approach was used. Obviously one limitation of a nested PCR is the increased chance of contamination that occurs with increased manipulation. The increased sensitivity of this assay also increases the problems associated with very low levels of contamination. For this reason it is essential to include a control in the nested PCR that uses as template the blank from the primary PCR reaction (as shown in Figure 4B, PCR bl-P). While a one step real-time PCR assay has the advantage of reduced likelihood of contamination, in our hands real-time assays were not as sensitive as a conventional nested-PCR reaction (S. K., S.C.M. and K.E.G unpublished). Consequently if the goal is optimum sensitivity, as it would be for detecting low levels of contamination, nested PCR with appropriate controls is superior. For general detection or quantitation of viral RNA in stocks (Figure 3) or cell cultures (Figure 5B) real-time PCR is the desired method.
Previous studies have shown that the guanidinium isothiocyanate based method of extraction of viral RNA and DNA from the cerebrospinal fluids was superior over other methods of purification (Casas et al., 1995). In this study it was found that viral RNA can be quickly and easily extracted from as little as 5 μl of crude viral stock using guanidine isothiocyanate lysis buffer and the Quiagen RNeasy Mini kit. In addition, by including an RNase treatment of the viral stocks prior to exposure to the guanidine isothiocyanate lysis buffer virion-associated RNA can be isolated efficiently and quantitated (Figure 3). By plotting the amount of virion-associated RNA on a standard curve the copy number of packaged viral genomes can be calculated. If it is assumed that viral genome number is equivalent to particle number then the particle to pfu ratio can be determined if the titer of the stock is known. Such an analysis of PV1 and RV14 stocks in the laboratory revealed a particle/pfu ratio of approximately 20 and 129, respectively. As the particle to pfu ratio for most picornaviruses is in the range of 30 to 1000 the results are in good agreement with previous reports (Racaniello, 2007).
In this study it was shown that RT-PCR in conjunction with appropriate primers can be used to distinguish PV1 from RV14 and to detect very low amounts of contaminating PV1 in RV14 stocks. Using this approach the addition of stock corresponding to as little as 1 pfu of PV1 was easily detected in RV14 stocks. Based on the finding that free RNA represents a small fraction of the total RNA present in viral stocks (Figure 3) and the measured particle/pfu ratio of 20, this indicates that contamination with as little as 20 viral genomes can be detected readily using this approach. It should be pointed out that this represents only 2×10-7 μl of a typical PV stock with a titer of 5×109 pfu/ml. Such a minute volume of contamination is quite possible from aerosols, equipment or reagents in a laboratory and thus this method would allow rapid detection of such contamination. Furthermore this method could be extended easily for the parallel detection of multiple viral contaminants using the appropriate primer sets and the limit of detection can likely be improved further by the use of molecular beacons (O'Shea and Cane, 2004).
Acknowledgments
The authors would like to thank Dr. Madhusudhan Reddy Papasani for helpful discussions and Tamara Olenyik for timely ordering of the material needed for this study. We would also like to thank Eckard Wimmer, Vincent Racaniello, William Jackson and Ralph Feuer for providing plasmids. This work was supported by grants from the NIH (AI059467 and AI064432), the American Cancer Society (RSG109705) and the National Center for Research Resources COBRE (P20 RR15587) and INBRE (P20 RR016454) Programs to K.E.G.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andeweg AC, Bestebroer TM, Huybreghs M, Kimman TG, de Jong JC. Improved detection of rhinoviruses in clinical samples by using a newly developed nested reverse transcription-PCR assay. J Clin Microbiol. 1999;37:524–30. doi: 10.1128/jcm.37.3.524-530.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreoletti L, Lesay M, Deschildre A, Lambert V, Dewilde A, Wattre P. Differential detection of rhinoviruses and enteroviruses RNA sequences associated with classical immunofluorescence assay detection of respiratory virus antigens in nasopharyngeal swabs from infants with bronchiolitis. J Med Virol. 2000;61:341–6. doi: 10.1002/1096-9071(200007)61:3<341::AID-JMV10>3.0.CO;2-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atmar RL, Georghiou PR. Classification of respiratory tract picornavirus isolates as enteroviruses or rhinoviruses by using reverse transcription-polymerase chain reaction. J Clin Microbiol. 1993;31:2544–6. doi: 10.1128/jcm.31.9.2544-2546.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce CB, Gama RE, Hughes PJ, Stanway G. A novel method of typing rhinoviruses using the product of a polymerase chain reaction. Arch Virol. 1990;113:83–7. doi: 10.1007/BF01318355. [DOI] [PubMed] [Google Scholar]
- Callahan PL, Mizutani S, Colonno RJ. Molecular cloning and complete sequence determination of RNA genome of human rhinovirus type 14. Proc Natl Acad Sci U S A. 1985;82:732–6. doi: 10.1073/pnas.82.3.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casas I, Powell L, Klapper PE, Cleator GM. New method for the extraction of viral RNA and DNA from cerebrospinal fluid for use in the polymerase chain reaction assay. J Virol Methods. 1995;53:25–36. doi: 10.1016/0166-0934(94)00173-e. [DOI] [PubMed] [Google Scholar]
- Costa AM, Lamb D, Garland SM, Tabrizi SN. Evaluation of LightCycler as a platform for nucleic acid sequence-based amplification (NASBA) in real-time detection of enteroviruses. Curr Microbiol. 2008;56:80–3. doi: 10.1007/s00284-007-9043-2. [DOI] [PubMed] [Google Scholar]
- Couch RB, Atmar RL. Rhinoviruses. In: Lennette EH, Smith TF, editors. Laboratory Diagnosis of Viral Infections. Marcel Dekker, Inc.; New York: 1999. pp. 787–802. [Google Scholar]
- Dagher H, Donninger H, Hutchinson P, Ghildyal R, Bardin P. Rhinovirus detection: comparison of real-time and conventional PCR. J Virol Methods. 2004;117:113–21. doi: 10.1016/j.jviromet.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Davies M, Bruce C, Bewley K, Outlaw M, Mioulet V, Lloyd G, Clegg C. Poliovirus type 1 in working stocks of typed human rhinoviruses. Lancet. 2003;361:1187–8. doi: 10.1016/S0140-6736(03)12919-4. [DOI] [PubMed] [Google Scholar]
- Deffernez C, Wunderli W, Thomas Y, Yerly S, Perrin L, Kaiser L. Amplicon sequencing and improved detection of human rhinovirus in respiratory samples. J Clin Microbiol. 2004;42:3212–8. doi: 10.1128/JCM.42.7.3212-3218.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freymuth F, Vabret A, Galateau-Salle F, Ferey J, Eugene G, Petitjean J, Gennetay E, Brouard J, Jokik M, Duhamel JF, Guillois B. Detection of respiratory syncytial virus, parainfluenzavirus 3, adenovirus and rhinovirus sequences in respiratory tract of infants by polymerase chain reaction and hybridization. Clin Diagn Virol. 1997;8:31–40. doi: 10.1016/s0928-0197(97)00060-3. [DOI] [PubMed] [Google Scholar]
- Gustin KE, Sarnow P. Inhibition of nuclear import and alteration of nuclear pore complex composition by rhinovirus. J Virol. 2002;76:8787–96. doi: 10.1128/JVI.76.17.8787-8796.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halonen P, Rocha E, Hierholzer J, Holloway B, Hyypia T, Hurskainen P, Pallansch M. Detection of enteroviruses and rhinoviruses in clinical specimens by PCR and liquid-phase hybridization. J Clin Microbiol. 1995;33:648–53. doi: 10.1128/jcm.33.3.648-653.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyypia T, Auvinen P, Maaronen M. Polymerase chain reaction for human picornaviruses. J Gen Virol. 1989;70(Pt 12):3261–8. doi: 10.1099/0022-1317-70-12-3261. [DOI] [PubMed] [Google Scholar]
- Ireland DC, Kent J, Nicholson KG. Improved detection of rhinoviruses in nasal and throat swabs by seminested RT-PCR. J Med Virol. 1993;40:96–101. doi: 10.1002/jmv.1890400204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KL, Sarnow P. Viral RNA synthesis. In: Rotbart HA, editor. Human Enterovirus Infections. ASM Press; Washington, D.C.: 1995. pp. 95–112. [Google Scholar]
- Johnston SL, Sanderson G, Pattemore PK, Smith S, Bardin PG, Bruce CB, Lambden PR, Tyrrell DA, Holgate ST. Use of polymerase chain reaction for diagnosis of picornavirus infection in subjects with and without respiratory symptoms. J Clin Microbiol. 1993;31:111–7. doi: 10.1128/jcm.31.1.111-117.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammerer U, Kunkel B, Korn K. Nested PCR for specific detection and rapid identification of human picornaviruses. J Clin Microbiol. 1994;32:285–91. doi: 10.1128/jcm.32.2.285-291.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kares S, Lonnrot M, Vuorinen P, Oikarinen S, Taurianen S, Hyoty H. Real-time PCR for rapid diagnosis of entero- and rhinovirus infections using LightCycler. J Clin Virol. 2004;29:99–104. doi: 10.1016/s1386-6532(03)00093-3. [DOI] [PubMed] [Google Scholar]
- Kotla S, Peng T, Bumgarner RE, Gustin KE. Attenuation of the type I interferon response in cells infected with human rhinovirus. Virology. 2008;374:399–410. doi: 10.1016/j.virol.2008.01.022. [DOI] [PubMed] [Google Scholar]
- Lonnrot M, Sjoroos M, Salminen K, Maaronen M, Hyypia T, Hyoty H. Diagnosis of enterovirus and rhinovirus infections by RT-PCR and time-resolved fluorometry with lanthanide chelate labeled probes. J Med Virol. 1999;59:378–84. [PubMed] [Google Scholar]
- Lu X, Holloway B, Dare RK, Kuypers J, Yagi S, Williams JV, Hall CB, Erdman DD. Real-time reverse transcription-PCR assay for comprehensive detection of human rhinoviruses. J Clin Microbiol. 2008;46:533–9. doi: 10.1128/JCM.01739-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori J, Clewley JP. Polymerase chain reaction and sequencing for typing rhinovirus RNA. J Med Virol. 1994;44:323–9. doi: 10.1002/jmv.1890440403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijhuis M, van Maarseveen N, Schuurman R, Verkuijlen S, de Vos M, Hendriksen K, van Loon AM. Rapid and sensitive routine detection of all members of the genus enterovirus in different clinical specimens by real-time PCR. J Clin Microbiol. 2002;40:3666–70. doi: 10.1128/JCM.40.10.3666-3670.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Shea MK, Cane PA. Development of a highly sensitive semi-quantitative real-time PCR and molecular beacon probe assay for the detection of respiratory syncytial virus. J Virol Methods. 2004;118:101–10. doi: 10.1016/j.jviromet.2004.01.022. [DOI] [PubMed] [Google Scholar]
- Olive DM, Al-Mufti S, Al-Mulla W, Khan MA, Pasca A, Stanway G, Al-Nakib W. Detection and differentiation of picornaviruses in clinical samples following genomic amplification. J Gen Virol. 1990;71(Pt 9):2141–7. doi: 10.1099/0022-1317-71-9-2141. [DOI] [PubMed] [Google Scholar]
- Peng T, Kotla S, Bumgarner RE, Gustin KE. Human rhinovirus attenuates the type I interferon response by disrupting activation of interferon regulatory factor 3. J Virol. 2007;81:6161. doi: 10.1128/JVI.00433-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitjean J, Vabret A, Dina J, Gouarin S, Freymuth F. Development and evaluation of a real-time RT-PCR assay on the LightCycler for the rapid detection of enterovirus in cerebrospinal fluid specimens. J Clin Virol. 2006;35:278–84. doi: 10.1016/j.jcv.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Racaniello V. Picornaviridae: The Viruses and Their Replication. In: K DM, et al., editors. Fields Virology. Lippincott Williams and Wilkins; Philadelphia, PA: 2007. p. 795. [Google Scholar]
- Santti J, Hyypia T, Halonen P. Comparison of PCR primer pairs in the detection of human rhinoviruses in nasopharyngeal aspirates. J Virol Methods. 1997;66:139–47. doi: 10.1016/s0166-0934(97)00049-9. [DOI] [PubMed] [Google Scholar]
- Sarnow P. Role of 3′-end sequences in infectivity of poliovirus transcripts made in vitro. J Virol. 1989;63:467–70. doi: 10.1128/jvi.63.1.467-470.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savolainen C, Hovi T. Caveat: poliovirus may be hiding under other labels. Lancet. 2003;361:1145–6. doi: 10.1016/S0140-6736(03)12965-0. [DOI] [PubMed] [Google Scholar]
- Scheltinga SA, Templeton KE, Beersma MF, Claas EC. Diagnosis of human metapneumovirus and rhinovirus in patients with respiratory tract infections by an internally controlled multiplex real-time RNA PCR. J Clin Virol. 2005;33:306–11. doi: 10.1016/j.jcv.2004.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnurr DP. Enteroviruses. In: Lennette EH, Smith TF, editors. Laboratory Diagnosis of Viral Infections. Marcel Dekker, Inc.; New York: 1999. pp. 373–383. [Google Scholar]
- Stanway G, Hughes PJ, Mountford RC, Minor PD, Almond JW. The complete nucleotide sequence of a common cold virus: human rhinovirus 14. Nucleic Acids Res. 1984;12:7859–75. doi: 10.1093/nar/12.20.7859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torgersen H, Skern T, Blaas D. Typing of human rhinoviruses based on sequence variations in the 5′ non-coding region. J Gen Virol. 1989;70(Pt 11):3111–6. doi: 10.1099/0022-1317-70-11-3111. [DOI] [PubMed] [Google Scholar]
- Verstrepen WA, Bruynseels P, Mertens AH. Evaluation of a rapid real-time RT-PCR assay for detection of enterovirus RNA in cerebrospinal fluid specimens. J Clin Virol. 2002;25 1:S39–43. doi: 10.1016/s1386-6532(02)00032-x. [DOI] [PubMed] [Google Scholar]
- Wright PF, Deatly AM, Karron RA, Belshe RB, Shi JR, Gruber WC, Zhu Y, Randolph VB. Comparison of results of detection of rhinovirus by PCR and viral culture in human nasal wash specimens from subjects with and without clinical symptoms of respiratory illness. J Clin Microbiol. 2007;45:2126–9. doi: 10.1128/JCM.02553-06. [DOI] [PMC free article] [PubMed] [Google Scholar]