

Summary
TRPV1 (transient receptor potential vanilloid subfamily member 1) receptors have classically been defined as ligand-gated, non-selective cation channels that act as heat-, proton- and ligand-activated integrators of nociceptive stimuli in sensory neurons, and there has been great interest in TRPV1 as a novel therapeutic target for pain relief. TRPV1 receptors have also been identified in the brain, but their physiological role is poorly understood. Here we report for the first time that TRPV1 channel activation is necessary and sufficient to trigger long-term synaptic depression (LTD). Excitatory synapses onto hippocampal interneurons were depressed either by capsaicin, a potent TRPV1 activator, or by 12-(S)-HPETE, an endogenous eicosanoid released during synaptic stimulation, while neither compound affected excitatory synapses onto CA1 pyramidal cells. TRPV1 receptor antagonists also prevented the induction of interneuron LTD. Furthermore, in brain slices from transgenic mice lacking TRPV1 receptors, LTD was absent and neither capsaicin nor 12-(S)-HPETE elicited synaptic depression. Our results suggest that TRPV1 channel activation represents a novel mechanism capable of selectively modifying synapses onto hippocampal interneurons. Like other forms of synaptic plasticity, TRPV1-mediated LTD may have a role in long-term changes in the physiological and pathological behavior of neural circuits during learning and epileptic activity.
Introduction
The TRPV1 channel, also known as vanilloid receptor VR1, was cloned ten years ago and is a member of a large family of calcium-permeable non-selective cation channels (Caterina et al., 1997; Szallasi and Blumberg, 1999). TRPV1 receptors are gated by heat, low pH, or endogenous ligands termed ‘endovanilloids’ including anandamide, lipoxygenase derivatives of arachidonic acid, and long-chain, linear fatty acid dopamines such as N-arachidonyldopamine (NADA) (Caterina et al., 1997; Tominaga et al., 1998; Zygmunt et al., 1999; Hwang et al., 2000; Smart et al., 2000; Huang et al., 2002; Shin et al., 2002; De Petrocellis and Di Marzo, 2005; Matta et al., 2007). In the peripheral nervous system (PNS), TRPV1 receptors are activated by thermal and chemical stimuli, by capsaicin (8-methyl-N-vanillyl-6-nonenamide; the pungent ingredient of red hot chili peppers), and by the Euphorbia toxin, resiniferatoxin, causing pain, inflammation and hyperalgesia. Bipolar neurons with unmyelinated axons (C-fibres) and somata in dorsal root and trigeminal ganglia, as well as a subset of sensory neurons with thin myelinated axons (Aδ fibres) are capsaicin-sensitive (Holzer, 1988). The TRP family of proteins is currently under intense investigation in health and disease because these ion channels respond to a diverse range of stimuli and because of their widespread distribution in a number of organs and tissues. Currently, TRPV1 receptors are a novel therapeutic target in the PNS, and agonists and antagonists are being tested for the treatment of inflammatory and chronic neuropathic pain (Szallasi and Appendino, 2004; Steenland et al., 2006; Szallasi et al., 2006).
In contrast to the well established function of TRPV1 receptors in the PNS, their role in the central nervous system (CNS) is not well defined. The presence of TRPV1 receptors in the mammalian brain has been demonstrated using in situ hybridization and reverse transcription polymerase chain reaction (RT-PCR) (Sasamura et al., 1998; Mezey et al., 2000), immunochemical staining methods (Sanchez et al., 2001; Toth et al., 2005; Cristino et al., 2006) and [3H]resiniferatoxin autoradiography comparing wild-type and TRPV1 receptor knockout mice (Roberts et al., 2004). These studies indicate the presence of potentially functional TRPV1 receptors in brain regions including the thalamic and hypothalamic nuclei, the locus coeruleus, periaqueductal grey and cerebellum, cortical and limbic structures including the hippocampus, the caudate putamen and the substantia nigra pars compacta. Nonetheless, the functional significance of TRPV1 receptor expression in the brain remains elusive, although there is evidence that TRPV1 receptors in the CNS are involved in pain modulation and may serve as useful drug targets (Cui et al., 2006). TRPV1 receptor mRNA and protein are expressed in hippocampal neurons (Sasamura et al., 1998; Roberts et al., 2004; Toth et al., 2005; Cristino et al., 2006) including those of the human hippocampus (Mezey et al., 2000), and functional effects of these receptors have been shown using electrophysiological methods (Al-Hayani et al., 2001; Huang et al., 2002; Marsch et al., 2007). A recent study using mice lacking TRPV1 receptors suggests their involvement in anxiety-related behavior and two behavioral measures of hippocampal-dependent learning, conditioned and sensitized fear (Marsch et al., 2007). Moreover, hippocampal long-term potentiation (LTP) was attenuated in the CA1 region of brain slices from TRPV1 knockout mice, indicating alterations in synaptic circuit function in this brain region, although the mechanism remains unknown (Marsch et al., 2007).
TRPV1 receptors in the CNS are less likely than those in the PNS to be activated by heat or low pH, and therefore it has been suggested that other endogenous ligands of this ion channel, such as the endovanilloids mentioned above, are likely activators (Huang et al., 2002; Marinelli et al., 2003; Van Der Stelt and Di Marzo, 2004; De Petrocellis and Di Marzo, 2005; Marsch et al., 2007). Anandamide and NADA are also members of the endocannabinoid family, activating CB1 receptors as well (Zygmunt et al., 1999; Huang et al., 2002), and it remains unclear whether or not any of these ligands are responsible for the TRPV1-mediated physiological and pathological effects in and outside of the CNS (Van Der Stelt and Di Marzo, 2004).
Synaptic plasticity in the brain is a fundamental process underlying information storage and adaptation to external stimuli (Malenka and Bear, 2004), and the cellular mechanisms underlying synaptic plasticity are of great interest since manipulation of these mechanisms could be used to modify neural function. Plasticity of synapses onto GABAergic interneurons can modify the output of cortical circuits, since interneurons are essential in the precise control of firing of groups of principle cells as well as in network oscillations (Kullmann and Lamsa, 2007; Mann and Paulsen, 2007). Some years ago we demonstrated that following high-frequency afferent stimulation, excitatory synapses onto CA1 hippocampal interneurons exhibit long-term depression (LTD) (McMahon and Kauer, 1997). Here we report that TRPV1 channel activation is a novel cellular element required for this form of LTD.
Results
In rat brain slices, AMPA receptor-mediated excitatory postsynaptic currents (AMPAR EPSCs) were locally stimulated and recorded from hippocampal CA1 interneurons in stratum radiatum. Since NMDA receptor (NMDAR) activation is an essential component of many forms of synaptic plasticity, we first asked whether LTD at these synapses requires NMDARs. In the presence of D-AP5 (50 μM), high-frequency electrical stimulation (HFS) of glutamatergic afferents triggered robust depression of EPSCs onto interneurons, indicating that NMDARs are not necessary for LTD induction (Figure 1A, B; EPSC amplitudes 15–20 minutes post-HFS: 62.0 ± 5.3% of control values before HFS; P < 0.001, n = 26). These values are similar to those found previously in the absence of D-AP5 (McMahon and Kauer, 1997), and all subsequent experiments were carried out in the presence of the NMDAR antagonist. Stable LTD could be elicited even after 40 minutes in the whole-cell recording configuration (Figure 1B; EPSC amplitudes 15–20 minutes post-HFS: 52.0 ± 23.2% of control values before HFS; P < 0.05, n = 3). Hippocampal interneurons are a diverse group of cells, expressing different neuropeptides and with different axonal innervation patterns (Freund and Buzsaki, 1996; Parra et al., 1998). Nonetheless, synaptic depression followed HFS in the majority of interneurons (26/29 experiments), supporting previous findings that distinct interneuron classes in stratum radiatum can express this form of LTD (McMahon and Kauer, 1997).
Figure 1. LTD at excitatory synapses on interneurons is NMDAR-independent and is maintained by a decrease in presynaptic glutamate release.

A. A single experiment illustrating interneuron LTD. NMDARs were blocked throughout the experiment using 50 μM D-AP5. At the arrow, HFS was delivered to the afferent pathway. The dotted line in this and all other single examples is an approximation of the mean EPSC response before HFS. Right panel: average of 10 consecutive EPSCs taken just before (black) and 20 minutes after HFS (gray). Calibration: 100 pA, 10 msec.
B. Left panel; Averaged LTD experiments in the presence of 50 μM D-AP5 (n = 26). The dotted line in this and all other time course averages represents the mean normalized EPSC value before HFS. Right panel; Averaged LTD experiments in which LTD was not triggered until 40 minutes following break-in to the whole-cell configuration, showing that the ability to induce LTD does not “wash out” over this time period (n = 3). This and all other experiments in the paper were carried out in the presence of 50 μM D-AP5. Error bars in this and all figures indicate mean ± s.e.m.
C, D, E. Consistent with a presynaptic mechanism, 1/CV2 (squared mean EPSC amplitude divided by EPSC variance) decreased, the PPR (EPSC2/EPSC1) increased and the number of synaptic failures increased significantly during interneuron LTD (average synaptic failures pre-HFS: 39.6 ± 3.3%; average synaptic failures post-HFS: 98.5 ± 0.7%; P < 0.001, n = 6). The paired-pulse ratio and coefficient of variation were calculated for 5 minute epochs before and between 15–20 minutes after HFS (see methods), and control cells with LTD of at least 10% in response to HFS were included in the PPR and 1/CV2 analysis. Non-normalized values of 1/CV2 (C), PPR (D) and synaptic failures (E) from each interneuron are shown (open circles). The thick black line and filled circles indicate the mean value for all cells. Using non-normalized values, all points are significantly different from pre-LTD values (P < 0.05). Inset (D): Example traces of EPSCs taken just before (black) and after HFS-induced LTD (gray) are shown, with the latter scaled so that the first EPSCs are of the same size, illustrating the increased paired pulse ratio during LTD. Calibration: 100 pA, 10 msec. Inset (E): Example traces illustrating consecutive EPSCs evoked using minimal stimulation before and during LTD from one experiment showing EPSCs identified as synaptic failures (gray). Calibration: 25 pA, 25 msec. Stimulus artifacts have been truncated for clarity.
F. NMDAR-mediated EPSCs were evoked while holding the interneuron at +40 mV, in the absence of D-AP5, and including 10 μM 6,7-dinitroquinoxaline-2,3-dione (DNQX) in the bathing solution. At the arrow, HFS was delivered to the afferents with the interneuron in current-clamp mode. Inset: average of 10 EPSCs recorded just before (black) and at 20 minutes after HFS (gray). Calibration for inset: 300 pA, 20 msec.
G. Averaged experiments showing LTD of NMDAR EPSCs recorded by holding the interneuron at +40 mV in the presence of 10 μM DNQX (n = 7).
The most commonly observed mechanisms underlying synaptic depression are a decrease in presynaptic neurotransmitter release or a decrease in postsynaptic receptor number or responsiveness (Malenka and Bear, 2004). When synaptic plasticity results from a change in neurotransmitter release, this is generally accompanied by an altered coefficient of variation of the EPSCs (CV), and changes in the paired-pulse ratio (PPR) and synaptic failure rate (del Castillo and Katz, 1954; Malinow and Tsien, 1990; Manabe et al., 1992). Consistent with this interpretation, we observed a decrease in 1/CV2 and an increase in the PPR and number of synaptic failures during LTD (Figure 1C, D, E). If LTD at interneuron synapses results from a persistent decrease in presynaptic glutamate release, we would also predict depression of the NMDAR-mediated component as well as the AMPAR-mediated component of the EPSC (isolated in the experiments in Figure 1A, B). We therefore measured the isolated NMDAR-mediated EPSC at +40 mV and found that HFS delivered to the afferents elicited robust LTD of the NMDAR EPSC (Figure 1F, G; NMDAR EPSC amplitudes post-HFS: 64.2 ± 11.1% of control values before HFS; P < 0.001, n = 7). Taken together, these findings indicate that LTD is AMPAR- and NMDAR-independent and results from a persistent decrease in presynaptic glutamate release as monitored by both AMPA and NMDA receptors.
How might high-frequency activation of excitatory afferents trigger LTD at interneuron synapses? Neither NMDARs nor AMPARs are necessary for LTD (Figure 1), but group I metabotropic glutamate receptors (mGluRs) are expressed on these cells (Ferraguti et al., 2004) and will also be activated by the glutamate released during HFS. We found that LTD was entirely blocked in the presence of the selective mGluR1 antagonist, CPCCOEt (25–50 μM; Figure 2A; EPSC amplitudes 10–15 minutes post-HFS in CPCCOEt: 111.4 ± 17.0% of control values before HFS; P < 0.01 compared to control LTD, n = 9). Our findings thus far are reminiscent of other recent examples of LTD in which activation of postsynaptic group I mGluRs produces endocannabinoids (Maejima et al., 2001; Gerdeman et al., 2002; Robbe et al., 2002; Chevaleyre and Castillo, 2003, 2004; Ronesi et al., 2004; Kreitzer and Malenka, 2005; Takahashi and Castillo, 2006). Endocannabinoids can act as retrograde messengers, traveling across the synapse to activate presynaptic CB1 receptors, thereby reducing presynaptic neurotransmitter release (Llano et al., 1991; Pitler and Alger, 1992; Kreitzer and Regehr, 2001; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). To ask whether endocannabinoids might mediate LTD at interneuron synapses, we tested two CB1 receptor antagonists. SR141716A (rimonabant; 2–5 μM) effectively blocked LTD (Figure 2B; EPSC amplitudes 15–20 minutes post-HFS: 110.6 ± 19.3% of control values before HFS; P < 0.01 compared to control LTD, n = 10). However, the selective CB1 receptor antagonist, AM251 (2 μM), did not block LTD in any of the 8 interneurons tested (Figure 2C; EPSC amplitudes 15–20 minutes post-HFS: 48.6 ± 5.3% of control values before HFS, n = 8). To confirm that AM251 indeed blocks CB1 receptors under these experimental conditions, we found in separate experiments that AM251 (2 μM) blocked the synaptic depression of these synapses elicited by the CB1 receptor agonist, WIN 55,212–2 (1 μM) (Figure 2D, E; EPSC amplitudes after 10–15 minutes in WIN 55,212–2 alone: 66.1 ± 7.9% of pre-drug control values; P < 0.001, n = 14; EPSC amplitudes after 10–15 minutes in both WIN 55,212–2 and AM251: 101.7 ± 8.9% of pre-WIN 55,212–2 control values; P < 0.05 compared to WIN55,212–2 depression in the absence of AM251, n = 5). In addition, pre-treatment with WIN 55,212–2 (1 μM) for at least ten minutes did not prevent synaptic depression triggered by high-frequency synaptic stimulation (EPSC amplitudes 15–20 minutes post-HFS: 51.3 ± 7.5% of control values in WIN55,212–2 before HFS; P = 0.25 compared to control LTD, n = 12; data not shown). The block of LTD by SR141716A but not by AM251 was surprising, indicating that CB1 receptors are not necessary for this form of LTD and instead that SR141716A blocks LTD via a CB1 receptor-independent mechanism.
Figure 2. A group I mGluR antagonist and SR141716A block LTD, but AM251 does not.

A. Averaged data showing that when the group I mGluR antagonist, CPCCOEt (25–50 μM) was bath-applied for at least 10 minutes before HFS (arrow), LTD was blocked in all but one cell (n = 9). Inset: average of 10 EPSCs from an example neuron before (black) or 20 minutes after HFS (gray). Calibration for all insets: 100 pA, 10 msec.
B. Averaged data showing that SR141716A (2–5 μM) consistently blocked LTD (n = 10). Inset: 10 consecutive EPSCs from an example neuron were averaged before (black) or 20 minutes after HFS (gray).
C. The CB1 receptor antagonist, AM251 (2 μM) did not affect LTD (average of 8 experiments). Inset: average of 10 EPSCs taken from an example neuron just before (black) and at 20 minutes after HFS (gray).
D. The CB1 receptor agonist, WIN 55,212–2 (1 μM) was bath-applied and depresses synaptic transmission at excitatory synapses onto interneurons (n = 14). Inset: 10 consecutive EPSCs taken from an example neuron were averaged before (black) or 10 minutes after the addition of WIN 55,212–2 (gray).
E. The CB1 receptor antagonist, AM251 (2 μM), bath applied for at least 10 minutes prior to the addition of WIN 55,212–2 prevents synaptic depression (n = 5). Inset: average of 10 EPSCs taken from an example neuron just before (black) and at 10 minutes after the addition of WIN 55,212–2 in the continued presence of AM251 (gray).
SR141716A may antagonize not only CB1 receptors but also the TRP channel family member, TRPV1 (De Petrocellis et al., 2001). TRPV1 is found in hippocampal neurons (Hajos and Freund, 2002; Roberts et al., 2004; Toth et al., 2005; Cristino et al., 2006; Marsch et al., 2007) and we therefore first tested whether transient application of a TRPV1 agonist mimics LTD induction. The extremely selective TRPV1 agonist capsaicin (1 μM) significantly depressed excitatory synaptic currents in interneurons (Figure 3A, B; EPSC amplitudes after 10–15 minutes in capsaicin: 73.3 ± 4.6% of pre-drug control values; P < 0.001, n = 14). If capsaicin maximally activates signaling mechanisms in common with LTD, HFS following the synaptic depression elicited by capsaicin should not cause any further depression. As predicted, HFS after capsaicin exposure failed to produce further LTD (Figure 3A, B; EPSC amplitudes 15–20 minutes post-HFS: 112.5 ± 21.9% of control values in capsaicin before HFS; P < 0.05 compared to control LTD, n = 10). This result suggests that the processes underlying LTD induction are fully activated by treatment with capsaicin, again supporting the requirement for TRPV1 channels in LTD. Like synaptically-induced LTD, the synaptic depression elicited by capsaicin was accompanied by an increase in synaptic failures, supporting our hypothesis that LTD at interneuron synapses results from a persistent decrease in presynaptic glutamate release (Figure 3C; average synaptic failures pre-capsaicin application: 38.4 ± 3.5%; average synaptic failures in capsaicin: 73.5 ± 7.8%; P < 0.01, n = 5).
Figure 3. Capsaicin mimics interneuron LTD via TRPV1 receptors and TRPV1 receptor antagonists block LTD induction.

A. A single example illustrating that 12 minutes of bath-applied capsaicin (1 μM) depresses EPSC amplitudes so that no further depression is elicited following HFS (arrow). Right Inset: Top panel; averaged EPSCs taken just before (black) and after 10 minutes in capsaicin (gray). Lower panel; average of 10 EPSCs in capsaicin taken just before (black) and at 20 minutes after HFS (gray). Calibration for all insets: 100 pA, 10 msec.
B. Left panel: Twelve minutes of bath-applied capsaicin (1 μM) depresses EPSC amplitudes (average of 14 experiments). Right panel: After capsaicin (1 μM) caused a stable EPSC depression, HFS was delivered (arrow) but elicited no further depression (average of 10 experiments).
C. Capsaicin (1 μM) application is associated with an increase in synaptic failures using minimal stimulation (P< 0.01). Percent failures for 5 experiments are shown for the 10 minute baseline period just before capsaicin application and for the last 5 minutes in capsaicin. The thicker black line and filled circles represent the average of five experiments.
D. SR141716A (2 μM) prevents the synaptic depression by capsaicin (1 μM), as expected if SR141716A is blocking the capsaicin-sensitive receptors (average of 6 experiments). SR141716A was bath applied for at least 10 minutes before the application of capsaicin. Inset: averaged EPSCs from an example neuron in SR141716A before (black) and after 10 minutes in capsaicin (gray).
E. Interneuron LTD was blocked by the TRPV1 receptor antagonist capsazepine (10 μM), bath-applied prior to HFS (arrow) (average of 9 experiments). Inset: average of 10 EPSCs from an example neuron taken just before (black) and at 20 minutes after HFS (gray).
F. Bath-applied 5′-Iodoresiniferatoxin (100 nM), another TRPV1 receptor antagonist, also blocked LTD (average of 7 experiments). Inset: average of 10 EPSCs from an example neuron taken just before (black) and at 20 minutes after HFS (gray).
We reasoned that if SR141716A blocks LTD by an antagonist action at TRPV1 receptors on hippocampal neurons, then SR141716A should also prevent capsaicin-induced synaptic depression. After pretreatment with SR141716A (2 μM), capsaicin (1 μM) did not depress the synapses (Figure 3D; EPSC amplitudes after 10–15 minutes in capsaicin and in the presence of SR141716A: 102.8 ± 9.2% of pre-capsaicin control values; P < 0.01 compared to capsaicin depression in the absence of SR141716A, n = 6). This finding emphasizes that at this concentration SR141716A cannot be regarded as a selective CB1 receptor antagonist, but instead appears to antagonize capsaicin-sensitive receptors, presumably TRPV1 channels. If, as these data suggest, TRPV1 is necessary for LTD induction at interneuron synapses, then TRPV1 antagonists should interfere with LTD. Both capsazepine (10 μM) and 5′-Iodoresiniferatoxin (100 nM) potently blocked LTD when bath applied prior to HFS (Figure 3E, F; EPSC amplitudes 15–20 minutes post-HFS in capsazepine: 105.6 ± 8.6% of control values before HFS; P < 0.001 compared to control LTD, n = 9; in 5′-Iodoresiniferatoxin: 96.2 ± 13.6% of control values before HFS; P < 0.01 compared to control LTD, n = 7). These data support an essential role for TRPV1 receptors in LTD induction. Once LTD is initiated, TRPV1 channel activity is no longer necessary to maintain synaptic depression since following LTD induction, EPSC amplitudes were not restored to basal values by blocking TRPV1 receptors (10 μM capsazepine was added 10 minutes after HFS; EPSC amplitudes 10–15 minutes after adding capsazepine: 50.1 ± 6.1% of pre-HFS values, n = 6; data not shown).
The pharmacological data presented above are all consistent with an essential role for TRPV1 channels in the induction of LTD. To further test this hypothesis, we asked whether LTD could be elicited in transgenic mice lacking TRPV1 receptors (TRPV1−/−) (Caterina et al., 2000). LTD was markedly reduced in slices from TRPV1−/− mice, when compared to LTD in interleaved slices from wild-type control mice (Figure 4A, B; EPSC amplitudes 15–20 minutes post-HFS in TRPV1−/− mice: 95.8 ± 7.0% of control values before HFS; P < 0.001 compared to control LTD in wild-type mice, n = 9; in C57BL/6 wild-type mice: 52.1 ± 5.2% of control values before HFS, n = 15). While application of capsaicin (1 μM) to slices from wild-type mice elicited synaptic depression, this was not seen in slices from TRPV1−/− mice, confirming the lack of functional TRPV1 receptors (Figure 4C, D; EPSC amplitudes after 10–15 minutes in capsaicin in TRPV1−/− mice: 100.7 ± 6.6% of pre-drug control values; P < 0.01 compared to capsaicin response in wild-type mice, n = 8; EPSC amplitudes after 10–15 minutes in capsaicin in C57BL/6 wild-type mice: 50.5 ± 12.1% of pre-drug control values, n = 6). These data complement the pharmacological evidence, and strongly suggest that TRPV1 channels or TRPV1-containing heteromultimeric channels are signaling components required for interneuron LTD.
Figure 4. Slices from TRPV1−/− mice lack interneuron LTD and capsaicin-induced synaptic depression.

A. In hippocampal slices from TRPV1−/− mice, HFS does not elicit LTD. Left panel, single experiment. Inset: averaged EPSCs before and 15 minutes after HFS. Calibration for all figure insets: 100 pA, 10 msec. Right panel, averaged experiments from TRPV1−/− mice (n = 9 animals).
B. In slices from wild-type mice, HFS induces LTD. Left panel, single experiment. Inset: averaged EPSCs before and 15 minutes after HFS. Right panel, averaged experiments from wild-type mice (n = 15 animals). Experiments were interleaved with those from TRPV1−/− mice.
C. Capsaicin (1 μM) has no effect on interneuron synapses in slices from TRPV1−/− animals. Left panel, single experiment. Inset: averaged EPSCs before and after 10 minutes in capsaicin. 1 μM capsaicin was added as marked by the bar. Right panel, averaged experiments from slices from TRPV1−/− mice (n = 8 animals).
D. In slices from C57BL/6 wild-type mice, capsaicin (1 μM) elicits synaptic depression. Left panel, single experiment. Inset: averaged EPSCs before and after 10 minutes in capsaicin. Right panel, averaged experiments from slices from C57BL/6 wild-type mice (n = 6 animals).
How is LTD initiated by high-frequency synaptic stimulation? Our data are consistent with a model analogous to that of endocannabinoid-mediated LTD (Chevaleyre et al., 2006), in which activation of mGluR1 produces a lipid retrograde messenger capable of activating TRPV1 receptors located on presynaptic pyramidal cell terminals. Activation of group I mGluRs can produce both endocannabinoids and eicosanoid metabolites of arachidonic acid, and these endogenous messengers effectively activate TRPV1 receptors (Zygmunt et al., 1999; Hwang et al., 2000; Shin et al., 2002). The eicosanoid, 12-(S)-HPETE, is known to be liberated during electrical stimulation of hippocampal slices (Feinmark et al., 2003), and thus we asked whether or not this lipid messenger can mimic LTD at interneuron synapses. Application of 12-(S)-HPETE (100 nM) depressed excitatory synapses on interneurons (Figure 5A; EPSC amplitudes after 10–15 minutes in 12-(S)-HPETE: 40.6 ± 11.7% of pre-drug control values; P < 0.01, n = 8), and subsequent HFS did not produce further LTD (Figure 5B; EPSC amplitudes 15–20 minutes post-HFS: 98.7 ± 15.3% of control values in 12-(S)-HPETE before HFS; P < 0.05 compared to control LTD, n = 6). In addition, the synaptic depression as a result of 12-(S)-HPETE application, like that caused by HFS or capsaicin, was associated with an increase in synaptic failures (Figure 5C; average synaptic failures pre-12-(S)-HPETE application: 35.2 ± 1.9%; average synaptic failures in 12-(S)-HPETE: 89.7 ± 3.0%; P < 0.001, n = 6). 12-(S)-HPETE synthesis from arachidonic acid requires 12-lipoxygenase. To determine whether or not endogenously released 12-(S)-HPETE is responsible for triggering LTD following synaptic stimulation, we attempted to induce LTD using HFS in the presence of baicalein (500 nM), an inhibitor of 12-lipoxygenase. Synaptically-induced LTD was blocked in the presence of baicalein, and in fact in four of ten cells we observed potentiation (greater than 125% of control 20 minutes following the HFS)(Figure 5D; EPSC amplitudes 15–20 minutes post-HFS: 129.6 ± 20.3% of control values before HFS; P < 0.01 compared to control LTD, n = 10). Moreover, the depression caused by 12-(S)-HPETE was prevented by either capsazepine (10 μM) or SR141716A (2 μM) (Figure 5E, F; EPSC amplitudes after 10–15 minutes in 12-(S)-HPETE and in the presence of capsazepine: 103.0 ± 8.9% of pre-12-(S)-HPETE control values; P < 0.01 compared to 12-(S)-HPETE depression in the absence of capsazepine, n = 6; in 12-(S)-HPETE and in the presence of SR141716A: 106.9 ± 5.5% of pre-12-(S)-HPETE control values; P < 0.01 compared to 12-(S)-HPETE depression in the absence of SR141716A, n = 5). These observations demonstrate a similar pharmacological profile for 12-(S)-HPETE and synaptically-triggered LTD. Finally, we found that in slices from TRPV1−/−mice, 12-(S)-HPETE did not depress synaptic transmission at excitatory synapses on interneurons (Figure 5G, H; EPSC amplitudes after 10–15 minutes in 12-(S)-HPETE in TRPV1−/− mice: 109.0 ± 6.8% of pre-drug control values; P = 0.33, n = 6). To rule out any possible involvement of the retrograde messenger, nitric oxide (NO), we tested whether or not LTD was affected when nitric oxide synthase (NOS) was inhibited. When 200 μM L-NAME was bath applied 10 minutes prior to HFS, LTD appeared entirely normal, suggesting that NO does not have a role in this form of synaptic plasticity (EPSC amplitudes 15–20 minutes post-HFS: 46.3 ± 10.4% of control values before HFS, n = 5; data not shown). Together, our data strongly suggest that 12-(S)-HPETE acts at TRPV1 receptors to depress synaptic transmission at excitatory synapses onto interneurons, and that 12-(S)-HPETE liberated during HFS is essential for triggering LTD.
Figure 5. The endogenous TRPV1 receptor agonist 12-(S)-HPETE mimics LTD.

A. The endogenous TRPV1 receptor agonist 12-(S)-HPETE (100 nM) was bath applied for 15 minutes and depressed EPSC amplitudes (average of 8 experiments). Inset: average of 10 EPSCs taken from an example neuron just before (black) and at 10 minutes after 12-(S)-HPETE application (gray). Calibration for this and all insets: 100 pA, 10 msec.
B. Following the bath application of 12-(S)-HPETE for 15 minutes, resulting in a stable EPSC depression, HFS (arrow) failed to induce further LTD (average of 6 experiments). Inset: average of 10 EPSCs in 12-(S)-HPETE taken from an example neuron just before (black) and at 20 minutes after HFS (gray).
C. 12-(S)-HPETE (1nM) application is associated with an increase in synaptic failures using minimal stimulation (P< 0.001). Percent failures for 6 experiments are shown for the 10 minute baseline period just before 12-(S)-HPETE application and for the last 5 minutes in 12-(S)-HPETE. The thicker black line and filled circles represent the average of six experiments.
D. Bath–applied baicalein (500 nM), a 12-lipoxygenase inhibitor, blocked LTD induction (average of 10 experiments). Inset: averaged EPSCs taken from an example neuron before (black) or 20 minutes after HFS (gray) in the presence of baicalein.
E. The TRPV1 receptor antagonist capsazepine (10 μM) prevents the synaptic depression caused by 12-(S)-HPETE (100 nM), as expected if 12-(S)-HPETE acts as a TRPV1 receptor agonist (average of 6 experiments). Inset: averaged EPSCs taken from an example neuron in capsazepine before (black) and after 10 minutes in 12-(S)-HPETE (gray).
F. SR141716A (2 μM) prevents the synaptic depression resulting from the application of 12-(S)-HPETE (100 nM) (average of 5 experiments). Inset: averaged EPSCs taken from an example neuron in SR141716A before (black) and after 10 minutes in 12-(S)-HPETE (gray).
G. 12-(S)-HPETE (100 nM) has no effect on interneuron synapses in slices from TRPV1−/− animals. Left panel, single experiment. Inset: averaged EPSCs before and after 10 minutes in 12-(S)-HPETE. 12-(S)-HPETE was added as marked by the bar. Right panel, averaged experiments from slices from TRPV1−/− mice showing the lack of effect of 1 μM 12-(S)-HPETE (n = 6 animals).
Interneurons in stratum radiatum of hippocampal area CA1 receive their major excitatory synaptic inputs from CA3 pyramidal cells but can also receive recurrent collaterals from CA1 pyramidal cells (Freund and Buzsaki, 1996). We next tested whether or not field excitatory postsynaptic potentials (fEPSPs) from synapses between CA3 pyramidal cells and CA1 pyramidal cells also exhibit TRPV1-mediated synaptic depression. Surprisingly, 1 μM capsaicin, a concentration that significantly depressed excitatory synapses on interneurons (Figure 3A, B), did not depress synapses on CA1 pyramidal cells (Figure 6A; fEPSP slopes after 10–15 minutes in capsaicin: 102.8 ± 5.4% of pre-drug control values; P = 0.58, n = 5). Although 10 μM capsaicin depressed synaptic transmission at the CA3–CA1 synapse, as previously reported (Hajos and Freund, 2002), we found that this was often associated with a depression in the presynaptic fiber volley component of the field potential, suggesting a possible confounding effect on presynaptic excitability. Furthermore, we found that 100 nM 12-(S)-HPETE, a concentration that significantly depressed excitatory synapses on area CA1 interneurons (Figure 5A), did not depress synapses between CA3 and CA1 pyramidal cells (Figure 6B; fEPSP slopes after 10–15 minutes in 12-(S)-HPETE: 99.0 ± 3.5% of pre-drug control values; P = 0.83, n = 5). Our data strongly suggest that TRPV1 channel activation does not depress glutamate release at these CA3 excitatory synapses onto CA1 hippocampal pyramidal cells, but potently inhibits excitatory synapses on interneurons in area CA1 stratum radiatum.
Figure 6. Field potential recordings from synapses on CA1 pyramidal cells are unaffected by concentrations of capsaicin or 12-(S)-HPETE that depress synapses on CA1 interneurons.

A. Field potentials (fEPSPs) recorded at the excitatory synapses between CA3 and CA1 pyramidal cells are not affected by capsaicin (1 μM). Inset: averaged fEPSPs taken from a single experiment before (black) and after 15 minutes in capsaicin (gray). Calibration for the insets: 250 μV, 10 msec.
B. Field potentials (fEPSPs) recorded at the excitatory synapses between CA3 and CA1 pyramidal cells are not affected by 12-(S)-HPETE (100 nM). Inset: average of 10 fEPSPs taken from a single experiment before (black) and after 15 minutes in 12-(S)-HPETE (gray).
We next investigated the involvement of the recorded interneuron in the generation of LTD. We found that intracellular perfusion of recorded interneurons with either GDPβS(250 μM), to block G-protein signaling, or BAPTA, (25–40 mM) to chelate postsynapticCa2+, reduced interneuron LTD (Figure 7), a result that can be explained if lipid retrograde messengers required for LTD are largely produced by the interneuron. Furthermore, delivery of the 12-lipoxygenase inhibitor, baicalein (140 nM) into the postsynaptic neuron via the patch pipette also markedly attenuated LTD (Figure 7). Together, these data indicate that the 12-(S)-HPETE necessary for LTD induction is produced in the recorded interneuron, and that G-protein signaling and postsynaptic Ca2+ play an important role.
Figure 7. Intracellular blockade of G-protein signaling or the enzyme 12-lipoxygenase, or chelation of intracellular Ca2+ reduces the incidence of LTD triggered by HFS.

A. The amount of synaptic depression present 15–20 minutes following HFS is plotted for interneurons in four separate conditions: 1. control intracellular patch pipette solution (control LTD; open circles, n = 26), 2. patch pipette solution containing 250 μM GDPβS (filled circles, n = 10), 3. patch pipette solution containing 25–40 mM BAPTA (filled circles, n = 14), and 4. patch pipette solution containing 140 nM baicalein (filled circles, n = 12). Each circular symbol represents the LTD observed in one experiment. Interneurons were held in the whole-cell recording configuration for at least 15 minutes before delivering HFS. The mean for each population is indicated by the horizontal black bar within each set of points. The dotted line in this figure represents the mean normalized EPSC value before HFS. Although the average amount of LTD elicited with each intracellular drug is significantly different from control LTD induced using control intracellular patch pipette solution (* P < 0.05, ** P < 0.01), in each case there are a few cells that appear to undergo LTD (EPSC amplitudes 15–20 minutes post-HFS with intracellular GDPβS: 88.2 ± 10.6% of control values before HFS; P < 0.05 compared to control LTD, n = 10; 6 of 10 cells recorded from with intracellular GDPβS had LTD. EPSC amplitudes 15–20 minutes post-HFS with intracellular BAPTA: 90.5 ± 8.5% of control values before HFS; P < 0.01 compared to control LTD, n = 14, 6 of 14 cells recorded from with intracellular BAPTA had LTD. EPSC amplitudes 15–20 minutes post-HFS with intracellular baicalein: 110.8 ± 15.4% of control values before HFS; P < 0.05 compared to control LTD, n = 12; 4 of 12 cells recorded from with intracellular baicalein had LTD).
Where are the TRPV1 receptors located that must be activated during LTD? Capsaicin was bath applied to determine whether we could detect TRPV1-mediated inward currents in different types of hippocampal neurons. Following bath application of 3 μM capsaicin, inward currents were elicited in both CA3 and CA1 pyramidal cells (Figure 8A, black bars; peak capsaicin response in CA1 pyramidal cells: 160.1 ± 55.3 pA, n = 7; in CA3 pyramidal cells: 180.1 ± 48.0 pA, n = 5). In contrast, CA1 stratum radiatum interneurons consistently exhibited little or no response to 3 μM capsaicin (Figure 8A, black bar; peak capsaicin response in interneurons: 41.6 ± 16.2 pA; P < 0.05 compared to CA1 and CA3 pyramidal cell responses, n = 5). In interleaved control experiments, 10 μM capsazepine blocked the effects of 3 μM capsaicin application, indicating that the capsaicin-induced inward currents were caused by TRPV1 receptor activation (Figure 8A). These results demonstrate the presence of functional TRPV1 receptors on pyramidal cell bodies as well as on some interneurons. The TRPV1 responses on interneurons were variable at best, suggesting that TRPV1 receptors on interneurons themselves do not play a significant role in LTD. To examine this directly, we delivered the TRPV1 receptor antagonist capsazepine (2 μM) into the recorded interneuron where it can inhibit the channel from the inside (Jordt and Julius, 2002). We found intracellular capsazepine to be ineffective at blocking either synaptically induced LTD or capsaicin-triggered depression (Figure 8B, C; EPSC amplitudes 15–20 minutes post-HFS with intracellular capsazepine: 47.5 ± 10.3% of control values before HFS; P = 0.20 compared to control LTD observed using control intracellular patch pipette solution, n = 7; EPSC amplitudes after 10–15 minutes in 1 μM capsaicin and in the presence of intracellular capsazepine: 46.8 ± 10.3% of pre-capsaicin control values; P < 0.05 compared to capsaicin depression in the absence of intracellular capsazepine, n = 6). These data indicate that TRPV1 receptors on interneurons are not necessary for LTD, and are instead consistent with a model in which the TRPV1 receptors responsible for interneuron LTD may be located on the nerve terminals of pyramidal cells (Figure 8D).
Figure 8. Functional TRPV1 receptors are found on pyramidal cells and interneurons, but the TRPV1 receptor necessary for LTD is not located on the recorded interneuron.
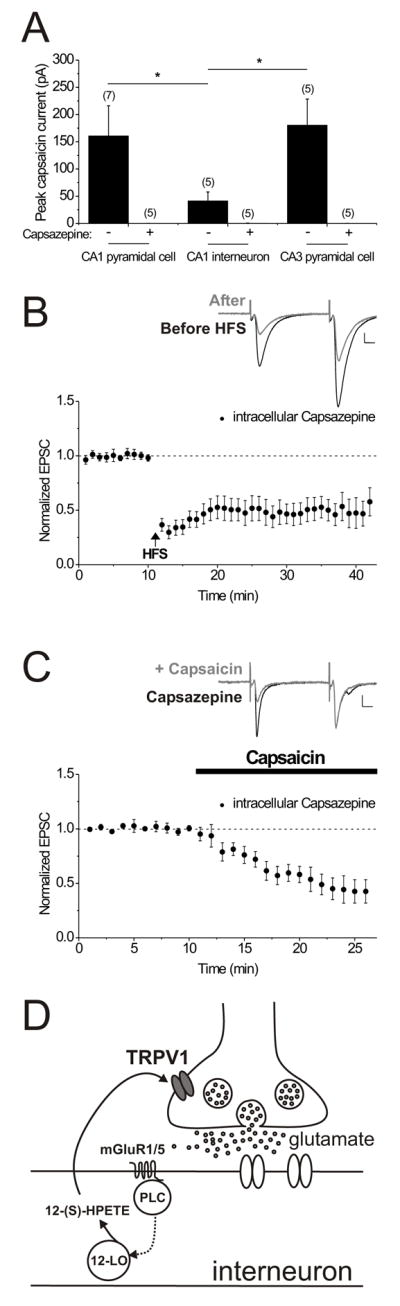
A. CA1 and CA3 pyramidal cells exhibit a greater peak inward current response to capsaicin (3 μM) when compared to CA1 interneurons. The holding current in three different hippocampal neuron classes was monitored while capsaicin was bath-applied. The peak (mean ± s.e.m.) capsaicin response (black bars) was significantly reduced in the presence of the TRPV1 receptor antagonist, capsazepine (10 μM; bars marked ‘+’) in CA1 and CA3 pyramidal cells. Peak capsaicin response in the presence of capsazepine: in CA1 pyramidal cells: 0.1 ± 0.1 pA; P < 0.05 compared to that without capsazepine, n = 5; in CA1 interneurons: 0.4 ± 0.2 pA; P = 0.06 compared to that without capsazepine, n = 5; and in CA3 pyramidal cells: 0.1 ± 0.1 pA; P < 0.05 compared to that in the absence of capsazepine, n = 5). In separate experiments, 1 μM capsaicin elicited a small and variable response in pyramidal cells but essentially no response in interneurons (1 μM capsaicin response in interneurons: 0.7 ± 0.3 pA; P = 0.78 compared to pre-drug control values, n = 5, data not shown).
B. Intracellular capsazepine does not block LTD. Average of 7 experiments with capsazepine (2 μM) included in the intracellular patch pipette solution. After at least 15 minutes, HFS was delivered (at the arrow). Inset: average of 10 EPSCs taken from an example neuron just before (black) and at 20 minutes after HFS (gray). Calibration for all insets: 100 pA, 10 msec.
C. Intracellular capsazepine does not prevent capsaicin-induced synaptic depression. Average of 6 experiments with capsazepine (2 μM) included in the intracellular patch pipette solution. After at least 15 minutes, capsaicin (1 μM) was bath applied to the slice (bar). Inset: average of 10 EPSCs taken from an example neuron just before (black) and after 10 minutes in capsaicin (gray).
D. Possible scheme to account for the induction of LTD at excitatory synapses onto CA1 interneurons. Glutamate release during synaptic stimulation activates mGluR1/5 receptors, leading to the activation of phospholipase C. Arachidonic acid is converted to 12-(S)-HPETE by a pathway requiring 12-lipoxygenase. 12-(S)-HPETE then activates TRPV1 receptors on presynaptic excitatory nerve terminals. Glutamate release is persistently altered, perhaps by a Ca2+-activated signaling cascade.
Discussion
A rapidly growing body of evidence suggests a functional role for the TRPV channel family in brain function (Marinelli et al., 2003; Lipski et al., 2006; Marinelli et al., 2007; Marsch et al., 2007; Shibasaki et al., 2007). In this study we show for the first time that TRPV1 receptors are necessary and sufficient for a novel form of long-term depression at excitatory synapses. The broad distribution of TRPV1 receptors in the brain suggests that these receptors could play a similar role in synaptic plasticity throughout the CNS. TRPV1 receptors may even contribute to some examples of previously reported endocannabinoid-mediated LTD, since anandamide can activate TRPV1 in addition to CB1 receptors.
We also report that in the hippocampus at least, SR141716A appears to be insufficiently selective to distinguish CB1 from TRPV1 receptors. In our study, SR141716A blocked LTD, in addition to responses to capsaicin and to 12-(S)-HPETE, whereas the very similar CB1 receptor antagonist, AM251, was ineffective. SR141716A has been shown to attenuate responses to capsaicin in other systems as well, particularly at concentrations above 1 μM (Zygmunt et al., 1999; De Petrocellis et al., 2001). A pharmacological profile similar to what we have observed was reported for the vasorelaxation of small mesenteric blood vessels that was mediated by an endothelial receptor in response to NADA, also blocked by SR141716A but not AM251 (O’Sullivan et al., 2004). Our findings may also relate to previous reports of a vanilloid receptor-like response at hippocampal excitatory synapses (Al-Hayani et al., 2001; Hajos and Freund, 2002). SR141716A (also known as rimonabant or Acomplia) is in wide clinical use outside the United States as an anti-obesity aid (Tucci et al., 2006; Padwal and Majumdar, 2007). A large percentage of patients stop taking this drug as a result of psychiatric side-effects, and our findings suggest the possibility that some of the central effects of rimonabant result from the antagonism of TRPV1 receptors as well as CB1 receptors (Pegorini et al., 2006).
TRPV1 receptors are expressed in hippocampal neurons (Mezey et al., 2000; Szabo et al., 2002; Toth et al., 2005; Cristino et al., 2006) and may be activated in several different ways, including by lipoxygenase derivatives that can be released as a result of group 1 mGluR activation, as we have shown here (Hwang et al., 2000; Sohn et al., 2007). 12-(S)-HPETE is known to be released during field stimulation of hippocampal slices (Feinmark et al., 2003), and our data indicate that 12-(S)-HPETE production is necessary and sufficient for LTD at excitatory interneuron synapses. Our previous study showed that LTD was triggered simultaneously at both activated and non-activated synapses on interneurons, indicating that the LTD is not synapse-specific or activity-dependent (McMahon and Kauer, 1997). The heterosynaptic nature of interneuron LTD may be accounted for by the local spread of 12-(S)-HPETE from interneurons activated during HFS. The most likely source of this eicosanoid is the recorded interneuron itself, based on our data using internally-perfused drugs; when applied intracellularly to the interneuron the Ca2+ chelator, BAPTA, the G-protein inhibitor, GDPβS, and the 12-lipoxygenase inhibitor, baicalein, all reduced the number of interneurons exhibiting LTD, suggesting that a Ca2+-sensitive process, a GPCR-mediated process and 12-lipoxygenase generation within the interneuron are necessary for LTD. If pyramidal cells, whose processes surround stratum radiatum interneurons, were a significant source of 12-(S)-HPETE following HFS, drugs delivered intracellularly to the recorded interneuron should not block LTD. Instead, in most experiments the intracellularly delivered drugs blocked LTD (Figure 7). However in some interneurons even with BAPTA, GDPβS or baicalein present, LTD of normal magnitude was induced, suggesting that the necessary signaling molecules can also arise elsewhere; we favor the idea that in some cases neighboring interneurons may release sufficient 12-(S)-HPETE to depress synapses, even when postsynaptic processes are blocked in the recorded cell. It is alternatively possible that perhaps the heterogeneity of hippocampal interneurons could also account for these data (Freund and Buzsaki, 1996; Parra et al., 1998).
The simplest model to account for our results is that synaptic stimulation releases glutamate that activates group 1 mGluRs producing 12-(S)-HPETE, which may act as a retrograde messenger (Feinmark et al., 2003). 12-(S)-HPETE in turn may open TRPV1 channels on the presynaptic glutamatergic terminals of CA1 and/or CA3 pyramidal cells that synapse onto interneurons (Figure 8D). How might activation of a Ca2+-permeable ion channel lead to persistent synaptic depression? Calcium entry through TRPV1 channels on glutamatergic terminals could initiate a signaling cascade responsible for the persistent downregulation of glutamate release observed during LTD. In dorsal root ganglion neurons, TRPV1 channel opening triggers calcineurin activation, which then rapidly depresses multiple voltage-gated calcium channels (Wu et al., 2005, 2006). Moreover, presynaptic NMDARs are required for spike-timing dependent LTD in neocortical neurons (Sjostrom et al., 2003) and Ca2+ arising from presynaptic activity is required for LTD at striatal synapses (Singla et al., 2007), suggesting that presynaptic Ca2+ signals are required to initiate these forms of LTD as well. However, in both of these examples, co-active CB1 receptors are also required for LTD, whereas CB1 receptors are not required for LTD at excitatory synapses onto hippocampal interneurons, since AM251 was ineffective in blocking this form of LTD. The model we present is the simplest to account for all of our data; however, while we report here that functional TRPV1 receptors are present on CA3 and CA1 pyramidal cell bodies, TRPV1 receptors are also expressed in glial cell populations (Doly et al., 2004; Kim et al., 2006), so it remains possible that an alternative, more complex signaling pathway is involved.
TRPV1 was first identified as a heat-sensitive ion channel in peripheral sensory neurons (Caterina et al., 1997). The temperature threshold of 43°C for TRPV1 channels (Caterina et al., 1997) is normally outside the brain’s physiological range, but the sensitivity of the channel to heat and other activating stimuli can be modulated by endogenous lipids and by the phosphorylation state of the channel (Vellani et al., 2001; Benham et al., 2003). It is therefore conceivable that during fever TRPV1 channels in the hippocampus may be activated, producing LTD at interneuron synapses. Depression of these synapses is expected to increase the excitability of innervated pyramidal cells. In this regard, it is intriguing that the in vivo treatment of animals with SR141716A after the induction of febrile seizures reduced hyperexcitability in hippocampal area CA1 and prevented the emergence of long-term limbic hyperexcitability (Chen et al., 2007). Our data suggest that the blockade of TRPV1 receptors could contribute to the anticonvulsant effect of SR141716A. The selective depression of excitatory synapses on interneurons but not on CA1 pyramidal cells that we report suggests that TRPV1 receptors are differentially distributed on hippocampal excitatory afferents and offers the potential to target hippocampal inhibitory circuits selectively through TRPV1 receptors.
Recently there has been great interest in therapeutic agents targeting TRPV1 receptors for several disorders, most notably inflammatory and neuropathic pain (Szallasi and Appendino, 2004; Steenland et al., 2006; Szallasi et al., 2006). Although drugs binding to peripheral TRPV1 receptors exert analgesic effects on their own, there is also evidence that TRPV1 receptors in the CNS are involved in pain modulation and may serve as useful drug targets (Cui et al., 2006). Our results as well as others (Marsch et al., 2007) indicate that drugs that bind to CNS TRPV1 receptors are likely to influence more than just pain-related functions. The human hippocampus expresses relatively high levels of TRPV1 mRNA (Mezey et al., 2000), suggesting that effects such as those reported here in rodent brain may occur in humans as well. Further work will help to ascertain whether hippocampal TRPV1 receptors could provide novel drug targets for neurological disorders.
Experimental Procedures
Preparation of brain slices
The basic methods have been detailed previously (McMahon and Kauer, 1997). Sprague-Dawley rats (15–22 days old) were used in the majority of experiments. In addition, we used TRPV1−/− mice (Caterina et al., 2000) and wild-type C57BL/6 mice aged between 15 and 21 days (Jackson Laboratory). The TRPV1−/− mice we used have been backcrossed at least 10 times onto a C57BL/6 background and were obtained from homozygous breeding pairs. Control mice were therefore not littermates but were age-matched, wild-type C57BL/6 animals received from the same supplier in the same shipment. All animal protocols were approved by the Brown University Institutional Animal Care and Use Committee. For mouse experiments, only one brain slice per mouse was used for each experiment, so that reported ‘n’ numbers represent the number of animals. Animals were anaesthetized using halothane or isoflurane and quickly decapitated. The brain was rapidly removed and 300 μm thick coronal slices prepared and stored for at least one hour submerged on a net in artificial cerebrospinal fluid (ACSF) containing in mM: 119 NaCl, 26 NaHCO3, 2.5 KCl, 1.0 NaH2PO4, 2.5 CaCl2, 1.3 MgSO4 and 11 dextrose, saturated with 95% O2/5% CO2 (pH 7.4). Slices were then transferred to a submerged recording chamber and bathed in oxygenated ACSF (28–32 °C) containing elevated divalent cations to reduce epileptiform activity (4 mM CaCl2 and 4 mM MgCl2, replacing MgSO4). A surgical cut was made between the CA3 and CA1 regions. The storage of slices submerged on a net rather than in an interface chamber on filter paper may be important in maintaining slice health and improving the likelihood of observing LTD.
Electrophysiological recordings from interneurons
Slices were continuously perfused with ACSF warmed to 28–32 °C at a flow rate of 1–2 ml/min. Picrotoxin (100 μM) and D-AP5 (50 μM) were added to block GABAA receptor- and NMDAR-mediated synaptic transmission. Whole-cell patch clamp recordings were made from interneurons identified visually in the CA1 stratum radiatum of the hippocampus. No specific cell morphology was targeted, although we did not record from cells with the “giant cell” morphology as these have been reported to be glutamatergic interneurons (Gulyas et al., 1998). Patch pipettes were filled with internal recording solution containing in mM: 117 cesium gluconate, 2.8 NaCl, 5 MgCl2, 20 HEPES, 2 ATP-Na+, 0.3 GTP-Na+ and 0.6 EGTA. In some experiments 2 μM capsazepine, 140 nM baicalein, or 250 μM GDPβS were also included in the intracellular patch pipette solution. In experiments with BAPTA-containing patch electrodes, EGTA was omitted from the intracellular solution and 25 or 40 mM BAPTA replaced a corresponding amount of cesium gluconate. EPSCs were stimulated at 0.1 Hz (100 μsec) using a bipolar stainless steel stimulating electrode placed in stratum radiatum at least 200 μm from the recorded cell. CA1 interneurons were voltage clamped at −65 mV (not corrected for the liquid junction potential, of ~10 mV), and EPSCs were evoked by paired pulses with an interval of 50 msec (stimulus intensity typically 50–400 μA). In early experiments, we measured rectification ratios of EPSCs evoked at +40 mV/−60 mV in the presence of 50 μM D-AP5, measured at the time of peak inward synaptic current seen at −70 mV (Lei and McBain, 2004). Rectification ratios did not correlate with the incidence of LTD: interneurons with no LTD, 0.63 ± 0.19, n = 3, range 0.25–0.86; interneurons with transient LTD, 0.47 ± 0.05, n = 4, range 0.42–0.52; interneurons with persistent LTD, 0.58 ± 0.11, n = 9, range 0.11–1.28.
High-frequency stimulation was used to induce LTD (HFS; two 1 sec trains at 100 Hz, inter-train interval 20 sec, at 1.5 times test current intensity) with the neuron held in current-clamp mode, so that the HFS trains were delivered with the membrane potential free to vary. Receptor antagonists were added directly to the ACSF at known concentrations for at least 10 minutes prior to HFS. Control experiments were interleaved with those experiments using receptor antagonists or involving slices from TRPV1−/− mice. The cell input resistance and series resistance were monitored throughout each experiment; cells were discarded if these values changed by more than 10% during the experiment. EPSCs were amplified using an AxoClamp 2B amplifier (Axon instruments) and Brownlee Precision Model 410 post-amplifier (AutoMate Scientific), low-pass filtered at 3 kHz and digitally sampled to a PC at 30 kHz using an analogue to digital interface (National Instruments).
Field EPSP recordings
Extracellular field potential recordings were made from synapses between CA3 and CA1 pyramidal cells in hippocampal slices prepared from rats as previously described (McMahon and Kauer, 1997). Briefly, 400 μm thick coronal slices were cut using a vibratome and individual slices were stored for at least one hour submerged on a net in ACSF. Slices were then transferred to a submersion chamber and held between two nylon nets. The chamber was constantly perfused with high divalent ACSF including 100 μM picrotoxin, oxygenated and warmed to 29–31°C at a flow rate of ~2–3 ml/min. A bipolar stainless steel stimulating electrode placed in stratum radiatum was used to stimulate CA1 field potentials, while a recording electrode filled with 2M NaCl was positioned about 500 μm from the stimulating electrode in stratum radiatum. Stimuli (intensity typically 50–200 μA, 100 μsec duration) were delivered at 0.1 Hz and the current intensity was adjusted to elicit a fEPSP of 0.5 mV at the start of each experiment. fEPSPs were amplified using an AxoPatch 1D amplifier (Axon instruments) and Brownlee Precision Model 410 post-amplifier (AutoMate Scientific), low-pass filtered at 1–2 kHz and digitally sampled to a PC at 10–20 kHz using an analogue to digital interface (National Instruments). Capsaicin (1 μM) or 12-(S)-HPETE (100 nM) were added directly to the ACSF bathing solution after at least a 15 minute baseline period of consistent fEPSPs.
Analysis
The maximal initial slope of fEPSPs was calculated using a LabVIEW-based program (National Instruments). The peak amplitude of each EPSC was measured by comparing a 10 msec time period immediately prior to the stimulus with the peak of the EPSC using this program as well. Occasionally polysynaptic responses were evoked, and in these cases, only the initial monosynaptic event was measured. To positively identify LTD, EPSCs measured every 10 seconds were averaged in 1 minute intervals. EPSC amplitude values were normalized to control pre-HFS EPSC amplitude values (baseline period of at least 5 minutes prior to HFS) and subjected to analysis of variance (ANOVA) repeated measures analysis with a post-hoc Dunnett’s test (GraphPad Prism, Version 4). A significant decrease (P < 0.05) in EPSC amplitude in 5 minute periods following HFS that persisted more than 10 minutes post-HFS, indicated that LTD had been induced. EPSC amplitude values 15 to 20 minutes post-HFS were compared between control LTD experiments and those carried out either in transgenic TRPV1−/− mice, or in the presence of drug using a t-test (unpaired, two-tailed, with Welch’s correction if the variances between the groups were unequal). To calculate the effects of capsaicin, 12-(S)-HPETE or WIN 55,212–2 application on basal excitatory glutamatergic transmission, normalized EPSC amplitudes or fEPSP slopes were averaged in the final 5 minutes of drug application and compared with EPSCs/EPSPs 5 minutes prior to drug application. In addition, to measure capsaicin’s effects on holding current, the peak change in holding current was measured during bath application of 3 μM capsaicin. The n-values reported refer to the number of slices. All combined data are expressed as mean ± the standard error of the mean (s.e.m.). All results reported in this study were significant to at least P < 0.05.
Paired-pulse ratios (PPR; EPSC2/EPSC1) and coefficient of variation (1/CV2) were calculated within 5 minute epochs of 30 EPSCs each, starting 5 minutes immediately before HFS or drug addition. The PPR was calculated by dividing the mean of all 30 EPSC2 amplitudes by the mean of all 30 corresponding EPSC1 amplitudes within each epoch. 1/CV2 was determined by dividing the squared mean amplitude of 30 EPSCs within 5 minute epochs by the variance of these EPSC amplitudes. Experiments in which the EPSC was depressed by more than 10% in response to HFS were included in the PPR and 1/CV2 analysis. Given that in some of the experiments the synaptic depression following HFS returned to baseline values after 15 to 20 minutes, we are most confident of the PPR and 1/CV2 data over the 20 minute time period immediately following HFS. For statistical analysis of significance of the changes in non-normalized values of 1/CV2 and PPR, we used distribution-free, non-parametric inferential statistics (Wilcoxon Matched-Pairs Signed-Ranks Test) to assess these values obtained from the same cell before and after HFS with a significance level of P < 0.05. Non-parametric statistics were used since the response values did not meet assumptions of normality and homogeneity of variance.
For synaptic failure analysis, EPSCs were evoked using minimal stimulation intensities that resulted in at least 20% failures of synaptic transmission. The number of failures for each experiment was determined by eye for the baseline period of at least 10 minutes; the largest amplitude value associated with a failure was then defined as the threshold value for individual failures in that experiment. This analysis necessarily groups both failures of transmitter release and transmission failures. Failures reported in the figures were assessed as the percentage of failures occurring during a 10 minute control baseline period, for the 15–20 minute time period post-HFS (Figure 1E) or for the 10–15 minute time period following the application of capsaicin or 12-(S)-HPETE (Figure 3C and 5C).
Materials
SR141716A was generously provided by NIDA. 12-(S)-HPETE [12-(S)-Hydroperoxyeicosa-5Z, 8Z, 10E, 14Z-tetraenoic acid] was purchased from Biomol International and BAPTA [1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid] was purchased from Calbiochem. AM251, baicalein, capsaicin, capsazepine, CPCCOEt [7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylate ethyl ester], D-AP5 [D(−)-2-amino-5-phosphonovaleric acid], 5′-Iodoresiniferatoxin, L-NAME and WIN 55,212–2 mesylate were obtained from Tocris Bioscience. All other chemicals were purchased from Sigma-Aldrich. AM251, baicalein, capsaicin, capsazepine, CPCCOEt, 5′-Iodoresiniferatoxin, SR141716A and WIN 55,212–2 mesylate were dissolved in DMSO and then diluted at least 1:1000 to the final concentration in ACSF, or for baicalein and capsazepine, at least 1:5000 to the final concentration in the intracellular patch pipette solution. Control experiments showed that 0.1% DMSO did not block LTD (EPSC amplitudes post-HFS: 67.7 ± 17.8% of baseline values, n = 3; not significantly different from control LTD).
Acknowledgments
The authors thank Drs. Barry Connors and Robert Malenka as well as members of our lab for helpful discussions and reading of the manuscript, Dr. Kevin Gormley at NIDA for providing SR141716A, and Jeannette Downing-Park for technical assistance. This work was supported by National Institutes of Health grants DA11289, NS050570 (JK) and NS049779 (JE).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Al-Hayani A, Wease KN, Ross RA, Pertwee RG, Davies SN. The endogenous cannabinoid anandamide activates vanilloid receptors in the rat hippocampal slice. Neuropharmacol. 2001;41:1000–1005. doi: 10.1016/s0028-3908(01)00145-9. [DOI] [PubMed] [Google Scholar]
- Benham CD, Gunthorpe MJ, Davis JB. TRPV channels as temperature sensors. Cell Calcium. 2003;33:479–487. doi: 10.1016/s0143-4160(03)00063-0. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- Chen K, Neu A, Howard AL, Foldy C, Echegoyen J, Hilgenberg L, Smith M, Mackie K, Soltesz I. Prevention of plasticity of endocannabinoid signaling inhibits persistent limbic hyperexcitability caused by developmental seizures. J Neurosci. 2007;27:46–58. doi: 10.1523/JNEUROSCI.3966-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron. 2003;38:461–472. doi: 10.1016/s0896-6273(03)00235-6. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Endocannabinoid-mediated metaplasticity in the hippocampus. Neuron. 2004;43:871–881. doi: 10.1016/j.neuron.2004.08.036. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci. 2006;29:37–76. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- Cristino L, de Petrocellis L, Pryce G, Baker D, Guglielmotti V, Di Marzo V. Immunohistochemical localization of cannabinoid type 1 and vanilloid transient receptor potential vanilloid type 1 receptors in the mouse brain. Neurosci. 2006;139:1405–1415. doi: 10.1016/j.neuroscience.2006.02.074. [DOI] [PubMed] [Google Scholar]
- Cui M, Honore P, Zhong C, Gauvin D, Mikusa J, Hernandez G, Chandran P, Gomtsyan A, Brown B, Bayburt EK, et al. TRPV1 receptors in the CNS play a key role in broad-spectrum analgesia of TRPV1 antagonists. J Neurosci. 2006;26:9385–9393. doi: 10.1523/JNEUROSCI.1246-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Petrocellis L, Bisogno T, Maccarrone M, Davis JB, Finazzi-Agro A, Di Marzo V. The activity of anandamide at vanilloid VR1 receptors requires facilitated transport across the cell membrane and is limited by intracellular metabolism. J Biol Chem. 2001;276:12856–12863. doi: 10.1074/jbc.M008555200. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Di Marzo V. Lipids as regulators of the activity of transient receptor potential type V1 (TRPV1) channels. Life Sci. 2005;77:1651–1666. doi: 10.1016/j.lfs.2005.05.021. [DOI] [PubMed] [Google Scholar]
- del Castillo J, Katz B. Quantal components of the end-plate potential. J Physiol. 1954;124:560–573. doi: 10.1113/jphysiol.1954.sp005129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doly S, Fischer J, Salio C, Conrath M. The vanilloid receptor-1 is expressed in rat spinal dorsal horn astrocytes. Neurosci Lett. 2004;357:123–126. doi: 10.1016/j.neulet.2003.12.051. [DOI] [PubMed] [Google Scholar]
- Feinmark SJ, Begum R, Tsvetkov E, Goussakov I, Funk CD, Siegelbaum SA, Bolshakov VY. 12-lipoxygenase metabolites of arachidonic acid mediate metabotropic glutamate receptor-dependent long-term depression at hippocampal CA3-CA1 synapses. J Neurosci. 2003;23:11427–11435. doi: 10.1523/JNEUROSCI.23-36-11427.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraguti F, Cobden P, Pollard M, Cope D, Shigemoto R, Watanabe M, Somogyi P. Immunolocalization of metabotropic glutamate receptor 1alpha (mGluR1alpha) in distinct classes of interneuron in the CA1 region of the rat hippocampus. Hippocampus. 2004;14:193–215. doi: 10.1002/hipo.10163. [DOI] [PubMed] [Google Scholar]
- Freund TF, Buzsaki G. Interneurons of the hippocampus. Hippocampus. 1996;6:347–470. doi: 10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci. 2002;5:446–451. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- Gulyas AI, Toth K, McBain CJ, Freund TF. Stratum radiatum giant cells: a type of principal cell in the rat hippocampus. Eur J Neurosci. 1998;10:3813–3822. doi: 10.1046/j.1460-9568.1998.00402.x. [DOI] [PubMed] [Google Scholar]
- Hajos N, Freund TF. Pharmacological separation of cannabinoid sensitive receptors on hippocampal excitatory and inhibitory fibers. Neuropharmacol. 2002;43:503–510. doi: 10.1016/s0028-3908(02)00157-0. [DOI] [PubMed] [Google Scholar]
- Holzer P. Local effector functions of capsaicin-sensitive sensory nerve endings: involvement of tachykinins, calcitonin gene-related peptide and other neuropeptides. Neurosci. 1988;24:739–768. doi: 10.1016/0306-4522(88)90064-4. [DOI] [PubMed] [Google Scholar]
- Huang SM, Bisogno T, Trevisani M, Al-Hayani A, De Petrocellis L, Fezza F, Tognetto M, Petros TJ, Krey JF, Chu CJ, et al. An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc Natl Acad Sci U S A. 2002;99:8400–8405. doi: 10.1073/pnas.122196999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SW, Cho H, Kwak J, Lee SY, Kang CJ, Jung J, Cho S, Min KH, Suh YG, Kim D, Oh U. Direct activation of capsaicin receptors by products of lipoxygenases: endogenous capsaicin-like substances. Proc Natl Acad Sci U S A. 2000;97:6155–6160. doi: 10.1073/pnas.97.11.6155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordt SE, Julius D. Molecular basis for species-specific sensitivity to “hot” chili peppers. Cell. 2002;108:421–430. doi: 10.1016/s0092-8674(02)00637-2. [DOI] [PubMed] [Google Scholar]
- Kim SR, Kim SU, Oh U, Jin BK. Transient receptor potential vanilloid subtype 1 mediates microglial cell death in vivo and in vitro via Ca2+-mediated mitochondrial damage and cytochrome c release. J Immunol. 2006;177:4322–4329. doi: 10.4049/jimmunol.177.7.4322. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001;29:717–727. doi: 10.1016/s0896-6273(01)00246-x. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci. 2005;25:10537–10545. doi: 10.1523/JNEUROSCI.2959-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM, Lamsa KP. Long-term synaptic plasticity in hippocampal interneurons. Nat Rev Neurosci. 2007;8:687–699. doi: 10.1038/nrn2207. [DOI] [PubMed] [Google Scholar]
- Lei S, McBain CJ. Two Loci of expression for long-term depression at hippocampal mossy fiber-interneuron synapses. J Neurosci. 2004;24:2112–2121. doi: 10.1523/JNEUROSCI.4645-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipski J, Park TI, Li D, Lee SC, Trevarton AJ, Chung KK, Freestone PS, Bai JZ. Involvement of TRP-like channels in the acute ischemic response of hippocampal CA1 neurons in brain slices. Brain Res. 2006;1077:187–199. doi: 10.1016/j.brainres.2006.01.016. [DOI] [PubMed] [Google Scholar]
- Llano I, Leresche N, Marty A. Calcium entry increases the sensitivity of cerebellar Purkinje cells to applied GABA and decreases inhibitory synaptic currents. Neuron. 1991;6:565–574. doi: 10.1016/0896-6273(91)90059-9. [DOI] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M. Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron. 2001;31:463–475. doi: 10.1016/s0896-6273(01)00375-0. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malinow R, Tsien RW. Presynaptic enhancement shown by whole-cell recordings of long-term potentiation in hippocampal slices. Nature. 1990;346:177–180. doi: 10.1038/346177a0. [DOI] [PubMed] [Google Scholar]
- Manabe T, Renner P, Nicoll RA. Postsynaptic contribution to long-term potentiation revealed by the analysis of miniature synaptic currents. Nature. 1992;355:50–55. doi: 10.1038/355050a0. [DOI] [PubMed] [Google Scholar]
- Mann EO, Paulsen O. Role of GABAergic inhibition in hippocampal network oscillations. Trends Neurosci. 2007;30:343–349. doi: 10.1016/j.tins.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Marinelli S, Di Marzo V, Berretta N, Matias I, Maccarrone M, Bernardi G, Mercuri NB. Presynaptic facilitation of glutamatergic synapses to dopaminergic neurons of the rat substantia nigra by endogenous stimulation of vanilloid receptors. J Neurosci. 2003;23:3136–3144. doi: 10.1523/JNEUROSCI.23-08-03136.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli S, Di Marzo V, Florenzano F, Fezza F, Viscomi MT, van der Stelt M, Bernardi G, Molinari M, Maccarrone M, Mercuri NB. N-arachidonoyl-dopamine tunes synaptic transmission onto dopaminergic neurons by activating both cannabinoid and vanilloid receptors. Neuropsychopharmacol. 2007;32:298–308. doi: 10.1038/sj.npp.1301118. [DOI] [PubMed] [Google Scholar]
- Marsch R, Foeller E, Rammes G, Bunck M, Kossl M, Holsboer F, Zieglgansberger W, Landgraf R, Lutz B, Wotjak CT. Reduced anxiety, conditioned fear, and hippocampal long-term potentiation in transient receptor potential vanilloid type 1 receptor-deficient mice. J Neurosci. 2007;27:832–839. doi: 10.1523/JNEUROSCI.3303-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta JA, Miyares RL, Ahern GP. TRPV1 is a novel target for omega-3 polyunsaturated fatty acids. J Physiol. 2007;578:397–411. doi: 10.1113/jphysiol.2006.121988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon LL, Kauer JA. Hippocampal interneurons express a novel form of synaptic plasticity. Neuron. 1997;18:295–305. doi: 10.1016/s0896-6273(00)80269-x. [DOI] [PubMed] [Google Scholar]
- Mezey E, Toth ZE, Cortright DN, Arzubi MK, Krause JE, Elde R, Guo A, Blumberg PM, Szallasi A. Distribution of mRNA for vanilloid receptor subtype 1 (VR1), and VR1-like immunoreactivity, in the central nervous system of the rat and human. Proc Natl Acad Sci U S A. 2000;97:3655–3660. doi: 10.1073/pnas.060496197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan SE, Kendall DA, Randall MD. Characterisation of the vasorelaxant properties of the novel endocannabinoid N-arachidonoyl-dopamine (NADA) Br J Pharmacol. 2004;141:803–812. doi: 10.1038/sj.bjp.0705643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron. 2001;29:729–738. doi: 10.1016/s0896-6273(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Padwal RS, Majumdar SR. Drug treatments for obesity: orlistat, sibutramine, and rimonabant. Lancet. 2007;369:71–77. doi: 10.1016/S0140-6736(07)60033-6. [DOI] [PubMed] [Google Scholar]
- Parra P, Gulyas AI, Miles R. How many subtypes of inhibitory cells in the hippocampus? Neuron. 1998;20:983–993. doi: 10.1016/s0896-6273(00)80479-1. [DOI] [PubMed] [Google Scholar]
- Pegorini S, Zani A, Braida D, Guerini-Rocco C, Sala M. Vanilloid VR1 receptor is involved in rimonabant-induced neuroprotection. Br J Pharmacol. 2006;147:552–559. doi: 10.1038/sj.bjp.0706656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitler TA, Alger BE. Postsynaptic spike firing reduces synaptic GABAA responses in hippocampal pyramidal cells. J Neurosci. 1992;12:4122–4132. doi: 10.1523/JNEUROSCI.12-10-04122.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ. Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci U S A. 2002;99:8384–8388. doi: 10.1073/pnas.122149199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts JC, Davis JB, Benham CD. [3H]Resiniferatoxin autoradiography in the CNS of wild-type and TRPV1 null mice defines TRPV1 (VR-1) protein distribution. Brain Res. 2004;995:176–183. doi: 10.1016/j.brainres.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Ronesi J, Gerdeman GL, Lovinger DM. Disruption of endocannabinoid release and striatal long-term depression by postsynaptic blockade of endocannabinoid membrane transport. J Neurosci. 2004;24:1673–1679. doi: 10.1523/JNEUROSCI.5214-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez JF, Krause JE, Cortright DN. The distribution and regulation of vanilloid receptor VR1 and VR1 5′ splice variant RNA expression in rat. Neurosci. 2001;107:373–381. doi: 10.1016/s0306-4522(01)00373-6. [DOI] [PubMed] [Google Scholar]
- Sasamura T, Sasaki M, Tohda C, Kuraishi Y. Existence of capsaicin-sensitive glutamatergic terminals in rat hypothalamus. Neuroreport. 1998;9:2045–2048. doi: 10.1097/00001756-199806220-00025. [DOI] [PubMed] [Google Scholar]
- Shibasaki K, Suzuki M, Mizuno A, Tominaga M. Effects of body temperature on neural activity in the hippocampus: regulation of resting membrane potentials by transient receptor potential vanilloid 4. J Neurosci. 2007;27:1566–1575. doi: 10.1523/JNEUROSCI.4284-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, Cho H, Hwang SW, Jung J, Shin CY, Lee SY, Kim SH, Lee MG, Choi YH, Kim J, et al. Bradykinin-12-lipoxygenase-VR1 signaling pathway for inflammatory hyperalgesia. Proc Natl Acad Sci U S A. 2002;99:10150–10155. doi: 10.1073/pnas.152002699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singla S, Kreitzer AC, Malenka RC. Mechanisms for synapse specificity during striatal long-term depression. J Neurosci. 2007;27:5260–5264. doi: 10.1523/JNEUROSCI.0018-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjostrom PJ, Turrigiano GG, Nelson SB. Neocortical LTD via coincident activation of presynaptic NMDA and cannabinoid receptors. Neuron. 2003;39:641–654. doi: 10.1016/s0896-6273(03)00476-8. [DOI] [PubMed] [Google Scholar]
- Smart D, Gunthorpe MJ, Jerman JC, Nasir S, Gray J, Muir AI, Chambers JK, Randall AD, Davis JB. The endogenous lipid anandamide is a full agonist at the human vanilloid receptor (hVR1) Br J Pharmacol. 2000;129:227–230. doi: 10.1038/sj.bjp.0703050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn JW, Lee D, Cho H, Lim W, Shin HS, Lee SH, Ho WK. Receptor-specific inhibition of GABAB-activated K+ currents by muscarinic and metabotropic glutamate receptors in immature rat hippocampus. J Physiol. 2007;580:411–422. doi: 10.1113/jphysiol.2006.125914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenland HW, Ko SW, Wu LJ, Zhuo M. Hot receptors in the brain. Mol Pain. 2006;2:34. doi: 10.1186/1744-8069-2-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo T, Biro T, Gonzalez AF, Palkovits M, Blumberg PM. Pharmacological characterization of vanilloid receptor located in the brain. Brain Res Mol Brain Res. 2002;98:51–57. doi: 10.1016/s0169-328x(01)00313-8. [DOI] [PubMed] [Google Scholar]
- Szallasi A, Blumberg PM. Vanilloid (Capsaicin) receptors and mechanisms. Pharmacol Rev. 1999;51:159–212. [PubMed] [Google Scholar]
- Szallasi A, Appendino G. Vanilloid receptor TRPV1 antagonists as the next generation of painkillers. Are we putting the cart before the horse? J Med Chem. 2004;47:2717–2723. doi: 10.1021/jm030560j. [DOI] [PubMed] [Google Scholar]
- Szallasi A, Cruz F, Geppetti P. TRPV1: a therapeutic target for novel analgesic drugs? Trends Mol Med. 2006;12:545–554. doi: 10.1016/j.molmed.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Takahashi KA, Castillo PE. The CB1 cannabinoid receptor mediates glutamatergic synaptic suppression in the hippocampus. Neurosci. 2006;139:795–802. doi: 10.1016/j.neuroscience.2006.01.024. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 1998;21:531–543. doi: 10.1016/s0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- Toth A, Boczan J, Kedei N, Lizanecz E, Bagi Z, Papp Z, Edes I, Csiba L, Blumberg PM. Expression and distribution of vanilloid receptor 1 (TRPV1) in the adult rat brain. Brain Res Mol Brain Res. 2005;135:162–168. doi: 10.1016/j.molbrainres.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Tucci SA, Halford JC, Harrold JA, Kirkham TC. Therapeutic potential of targeting the endocannabinoids: implications for the treatment of obesity, metabolic syndrome, drug abuse and smoking cessation. Curr Med Chem. 2006;13:2669–2680. doi: 10.2174/092986706778201512. [DOI] [PubMed] [Google Scholar]
- Van Der Stelt M, Di Marzo V. Endovanilloids. Putative endogenous ligands of transient receptor potential vanilloid 1 channels. Eur J Biochem. 2004;271:1827–1834. doi: 10.1111/j.1432-1033.2004.04081.x. [DOI] [PubMed] [Google Scholar]
- Vellani V, Mapplebeck S, Moriondo A, Davis JB, McNaughton PA. Protein kinase C activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamide. J Physiol. 2001;534:813–825. doi: 10.1111/j.1469-7793.2001.00813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- Wu ZZ, Chen SR, Pan HL. Transient receptor potential vanilloid type 1 activation down-regulates voltage-gated calcium channels through calcium-dependent calcineurin in sensory neurons. J Biol Chem. 2005;280:18142–18151. doi: 10.1074/jbc.M501229200. [DOI] [PubMed] [Google Scholar]
- Wu ZZ, Chen SR, Pan HL. Signaling mechanisms of down-regulation of voltage-activated Ca2+ channels by transient receptor potential vanilloid type 1 stimulation with olvanil in primary sensory neurons. Neurosci. 2006;141:407–419. doi: 10.1016/j.neuroscience.2006.03.023. [DOI] [PubMed] [Google Scholar]
- Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sorgard M, Di Marzo V, Julius D, Hogestatt ED. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]