

Abstract
Rap1 is a small GTPase that modulates adhesion of T cells by regulating inside-out signaling through LFA-1. The bulk of Rap1 is expressed in a GDP-bound state on intracellular vesicles. Exocytosis of these vesicles delivers Rap1 to the plasma membrane, where it becomes activated. We report here that phospholipase D1 (PLD1) is expressed on the same vesicular compartment in T cells as Rap1 and is translocated to the plasma membrane along with Rap1. Moreover, PLD activity is required for both translocation and activation of Rap1. Increased T-cell adhesion in response to stimulation of the antigen receptor depended on PLD1. C3G, a Rap1 guanine nucleotide exchange factor located in the cytosol of resting cells, translocated to the plasma membranes of stimulated T cells. Our data support a model whereby PLD1 regulates Rap1 activity by controlling exocytosis of a stored, vesicular pool of Rap1 that can be activated by C3G upon delivery to the plasma membrane.
Regulated adhesion of lymphocytes is required for immune function. The β2 integrin lymphocyte function-associated antigen 1 (LFA-1) mediates lymphocyte adhesion to endothelium, antigen-presenting cells, and virally infected target cells (14). These cell-cell adhesions enable lymphocyte trafficking in and out of lymphoid organs, T-cell activation, and cytotoxicity, respectively (2, 34). Thus, the regulation of LFA-1 adhesiveness is central to adaptive immunity.
LFA-1 is a bidirectional receptor in that it mediates both outside-in and inside-out signaling (30). Outside-in signaling is analogous to signaling by conventional receptors and is defined as stimulation of intracellular signaling pathways as a consequence of ligation of LFA-1 with any of its extracellular ligands, such as intracellular adhesion molecule 1 (ICAM-1). Inside-out signaling refers to intracellular signaling events that result in a higher-affinity state of the ectodomain of LFA-1 for its cognate ligands. Regulatory events that mediate inside-out signaling converge on the cytoplasmic tails of the LFA-1 α and β chains, which transduce signals to their ectodomains (14). Signaling molecules implicated in inside-out signaling through LFA-1 include talin, Vav1, PKD1, several adaptor proteins (SLP-76, ADAP, and SKAP-55), the Ras family GTPase Rap1, and two of its effectors, RAPL and RIAM (26). How these proteins interact to activate LFA-1 remains poorly understood.
Rap1 is a member of the Ras family of GTPases and has been implicated in growth control, protein trafficking, polarity, and cell-cell adhesion (6). The ability of activated Rap1 to promote LFA-1-mediated lymphocyte adhesion is well established (33). The physiologic relevance of this pathway is highlighted by leukocyte adhesion deficiency type III (LAD III), where immunocompromised patients have a congenital defect in GTP loading of Rap1 in leukocytes (24). LFA-1 is a plasma membrane protein, consistent with its role in cell-cell adhesion, which by definition is a cell surface phenomenon. Paradoxically, the bulk of Rap1 is expressed on intracellular vesicles. We have characterized these vesicles as recycling endosomes and have shown that the intracellular pool of Rap1 can be mobilized by exocytosis to augment the expression of Rap1 at the plasma membranes of lymphocytes, leading to increased adhesion (5). We used a fluorescent probe of activated Rap1 in live cells to show that only the pool of Rap1 at the plasma membrane becomes GTP bound upon lymphocyte activation. Thus, it appears that delivery of Rap1 via vesicular transport to the plasma membrane and activation of the GTPase on that compartment are linked. Among the signaling enzymes known to regulate vesicular trafficking is phospholipase D (PLD). Whereas PLD type 2 (PLD2) is expressed at the plasma membranes of lymphocytes, PLD1 is expressed on intracellular vesicles (29). We now show that PLD1 resides on the same vesicles as Rap1, is delivered along with Rap1 to the plasma membranes of stimulated T cells, and is required for Rap1 activation and T-cell adhesion.
MATERIALS AND METHODS
General reagents.
RPMI medium, Dulbecco's modified Eagle's medium, 5-carboxyfluorescein, and Opti-MEM I were purchased from Invitrogen Corporation/Molecular Probes (Carlsbad, CA). Primary and tertiary butanol was purchased from Sigma-Aldrich (St. Louis, MO).
Cell culture, transfection, and stimulation.
Jurkat T cells were obtained from the American Type Culture Collection (Manassas, VA). Cells were maintained in 5% CO2 at 37°C in RPMI 1640 medium supplemented with 10% fetal bovine serum and 100 U/ml penicillin G and streptomycin. Transfection of Jurkat cells was performed with DMRIE-C (Invitrogen, Carlsbad, CA), and cells were examined 24 to 48 h later. COS-1 and HeLa cells were maintained in 5% CO2 at 37°C in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum. Transfection of COS-1 and HeLa cells was performed with SuperFect (Qiagen, Valencia, CA) according to the manufacturer's instructions, and cells were examined the following day. Jurkat T cells were serum starved at 37°C for 2 to 6 h, followed by incubation with 5 μg/ml of mouse anti-human CD3 antibodies (Ancell, Bayport, MN).
Mice.
Wild-type C57BL/6 mice were bred under pathogen-free conditions. Primary CD4+ T lymphocytes were isolated from spleens of 6- to 8-week-old female mice as previously described (29) and were kept in supplemented RPMI 1640 medium.
DNA constructs.
GFP-Rap1WT, GFP-Rap1V12, GFP-Rap1N17, pcDNA3.1-Rap1WT, pcDNA3.1-Rap1V12, pcDNA3.1-Rap1N17, and YFP-RalGDS RBD constructs were described previously (5). GFP-TC10 was also described earlier (28). To make RFP-PLD1, the coding sequence of PLD1 was amplified by PCR and cloned in frame into the mammalian expression vector mRFP-C1. Plasmids were verified by bidirectional sequencing. GFP-PLD1, PLD1 short hairpin RNA (shRNA) (RFP/H1-PLD1), and PLD2 shRNA (RFP/H1-PLD2) were previously described (37). GFP-C3G was a gift from Philip Stork (The Vollum Institute, Portland, OR). A Rap1-Kras tail fusion construct was made by an overlapping PCR designed to replace the last 19 codons of Rap1a with those of Kras4B (primer sequences are available upon request).
Antibodies.
Mouse anti-human CD3 (Ancell, Bayport, MN) was used for T-cell-receptor (TCR)-dependent activation. Anti-PLD1 antibody (3832) was purchased from Cell Signaling Technology, Inc. (Danvers, MA). Anti-PLD2 antibody was a gift from Yoshiko Banno (Gifu International Institute of Biotechnology, Gifu, Japan). Anti-Rap1 antibody (610195) and anti-RhoGDI antibody (clone 16) were purchased from BD Biosciences (San Jose, CA). Anti-C3G (C-19) and anti-Erk (K-23) antibodies were purchased from Santa Cruz Biotechnologies (Santa Cruz, CA). Monoclonal anti-pan-Ras antibody (Ras10) was purchased from Calbiochem (San Diego, CA).
Microscopy.
Live cells were plated in 35-mm dishes containing a no. 0 glass coverslip over a 15-mm cutout (MatTek, Ashland, MA). Cells were maintained at 37°C, using a PDMI-2 microincubator (Harvard Apparatus, Holliston, MA). Individual cells were imaged continuously, before and after the addition of stimuli, for periods of up to 30 min. Images were acquired with an inverted Zeiss 510 laser scanning confocal microscope (Carl Zeiss Microimaging, Inc., Thornwood, NY) and processed with Adobe Photoshop CS2. For quantification of GFP-Rap1 upregulation at the plasma membrane, line scans of the edge of the cell were obtained before and 5 min after stimulation with anti-CD3, fluorescence intensity was plotted as a function of distance along this line, and the integrated areas under the curves representing plasma membrane fluorescence were compared.
Rap activation assays.
Detection of activated Rap1 was performed as described previously (12). Immunoblots were developed using 125I-protein A and quantified by a phosphorimager.
Subcellular fractionation.
Jurkat T cells (3.5 × 108) grown in supplemented RMPI medium were washed three times in ice-cold phosphate-buffered saline. Cells were resuspended in relaxation buffer (10 mM HEPES, 100 mM KCl, 3 mM NaCl, 3.5 mM MgCl2, 0.1 mM phenylmethylsulfonyl fluoride, and protease inhibitors) and subsequently equilibrated with N2 at 350 lb/in2 for 20 min at 4°C in a nitrogen bomb (Parr Instrument Company, Moline, IL). Dropwise release from the bomb resulted in disruption of the cells by cavitation. The cavitated sample was centrifuged at 500 × g for 10 min to remove unbroken cells and nuclei. The postnuclear supernatant was loaded on top of a discontinuous sucrose density gradient (20 to 60% [vol/vol]) and separated by centrifugation at 133,000 × g for 120 min at 4°C. Fractions were collected from the top of the gradient. To detect Rap1, proteins were precipitated with ice-cold 10% trichloroacetic acid, resuspended in sodium dodecyl sulfate sample buffer, and loaded into 14% acrylamide gels. To detect PLD1, fractions were immunoprecipitated with anti-PLD1 antibodies and analyzed by 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. For subcellular localization of endogenous Rap1, Ras, and Erk and for C3G translocation assays, cytosolic (S175) and membrane (P175) fractions were generated from postnuclear supernatants of nitrogen cavitation samples by centrifugation at 175,000 × g for 45 min at 4°C.
Adhesion assay.
Jurkat T-cell adhesion to ICAM-1-coated multiwell plates was performed as described previously (5). Recombinant ICAM-1 was produced as described previously (19). Cells were either transfected with green fluorescent protein (GFP)-tagged constructs (as indicated) or labeled with 5-carboxyfluorescein. Cells (1.5 × 105) were plated for 20 min before removal of nonadherent cells by serial washes. Adherent cells were detected with a plate reader at 525 nm (DTX 800 multimode detector; Beckman Coulter, Fullerton, CA).
RESULTS
Rap1 and PLD1 colocalize on cytoplasmic vesicles.
Both PLD1 (10) and Rap1 (5) have been reported to reside on cytoplasmic vesicles. To determine if these two signaling molecules are expressed on the same class of vesicle, we tagged PLD1 with monomeric red fluorescent protein (mRFP) and Rap1 with GFP and coexpressed the fusion proteins in Jurkat T cells. As we previously reported (5), GFP-Rap1 was expressed on both the plasma membrane and intracellular vesicles that coalesced in a paranuclear region consistent with recycling endosomes. mRFP-PLD1 was observed on the same vesicles (Fig. 1A). Colocalization of GFP-Rap1 and mRFP-PLD1 was also observed in fibroblasts and epithelial cells (Fig. 1B). To test whether the pool of vesicles decorated by Rap1 and PLD1 represented a distinct class of endocytic vesicles, we coexpressed mRFP-PLD1 with GFP-TC10, a Rho family GTPase expressed on Glut4-containing vesicles (39). Jurkat cell vesicles decorated with these two fusion proteins were entirely distinct (Fig. 1A), indicating that PLD1 decorates only a subset of endocytic vesicles that also carry Rap1.
FIG. 1.

Rap1 colocalizes with PLD1 on cytoplasmic vesicles of resting cells and on the plasma membranes of activated T cells. (A) GFP-Rap1 or GFP-TC10 (vesicular GTPase control) was coexpressed with RFP-PLD1 in Jurkat T cells, which were imaged alive with a laser scanning confocal microscope. (B) GFP-Rap1 and RFP-PLD1 were expressed in COS-1 and HeLa cells and imaged live with a laser scanning confocal microscope. Colocalization is evident on both the plasma membrane and cytoplasmic vesicles. The inset shows a high-magnification view of vesicles. (C) Endogenous Rap1 and PLD1 in Jurkat T cells were analyzed for vesicle association by nitrogen cavitation, sucrose density centrifugation, and immunoblotting. Both molecules were expressed on vesicles that have the same buoyant density profile. (D) The cytosolic (S175) and membrane (P175) fractions of the postnuclear supernatant of Jurkat cell cavitation samples were analyzed by immunoprecipitation and immunobloting (Rap1 and Ras) or by immunoblotting only (Erk), as indicated. (E) The subpopulation of serum-starved Jurkat T cells expressing neither GFP-Rap1 nor mRFP-PLD1 at the plasma membrane (20% of total) was imaged before and after stimulation with an anti-CD3 antibody. Bars, 5 μm (A and E) and 10 μm (B).
Antibodies to PLD1 and Rap1 did not support immunofluorescence analysis of endogenous proteins. We therefore turned to subcellular fractionation to analyze the localization of endogenous Rap1 and PLD1. Jurkat T cells were disrupted by nitrogen cavitation to preserve the integrity of cytoplasmic vesicles, and the postnuclear supernatant was subjected to equilibrium centrifugation through discontinuous sucrose gradients. Fractions were analyzed by immunoblotting for PLD1 and Rap1. The distributions of the two proteins were found to overlap (Fig. 1C). These data support the observation that Rap1 and PLD1 have affinity for the same subpopulation of endocytic vesicles.
Since small GTPases are peripheral membrane proteins, their translocation from one membrane compartment to another can be either via vesicular transport, through the fluid phase of the cytosol, or both. Whereas farnesylated Ras can cycle off membranes and into the fluid phase without a chaperone (16, 36), geranylgeranylated Rho proteins require RhoGDI as a cytosolic chaperone (27, 28). To determine if geranylgeranylated Rap1, without a known cytosolic chaperone, can be found in the fluid phase, we analyzed subcellular fractions of Jurkat T cells generated by nitrogen cavitation. Whereas a significant portion of endogenous Ras proteins could readily be detected in the cytosol (S175; 175,000 × g supernatant), along with most of the cellular Erk, endogenous Rap1 was found exclusively in the membrane fraction (P175) (Fig. 1D). These data demonstrate that the subcellular transport of Rap1 does not involve a cytosolic phase and is therefore entirely vesicular.
We have previously shown that stimulation of the antigen receptor on T cells rapidly increases the expression of Rap1 at the plasma membrane via exocytosis of Rap1-laden vesicles (5). To determine if PLD1 is translocated to the plasma membrane in parallel with Rap1, we studied Jurkat T cells coexpressing GFP-Rap1 and mRFP-PLD1 before and after stimulation. Indeed, stimulation of the TCR induced translocation of mRFP-PLD1 to the plasma membrane (Fig. 1E). Whereas all transfected Jurkat cells grown in serum revealed GFP-Rap1 on both the plasma membrane and cytoplasmic vesicles (Fig. 1A), two populations of GFP-Rap1-expressing cells were observed after serum starvation. Eighty percent of cells showed some GFP-Rap1 at the plasma membrane, but 20% of cells revealed GFP-Rap1 only on intracellular vesicles that were also marked by mRFP-PLD1 (Fig. 1E). Upon TCR stimulation of serum-starved cells expressing GFP-Rap1 and mRFP-PLD1 only on vesicles, both molecules translocated to the plasma membrane (Fig. 1E). For the population of cells that retained plasma membrane-associated GFP-Rap1 after serum starvation, GFP-Rap1 upregulation on this compartment was confirmed by quantitative analysis of line scans of confocal micrographs and revealed that TCR stimulation for 10 min increased GFP-Rap1 expression on the plasma membrane to 131% ± 3% of control levels (n = 5; P < 0.0001). These data suggest that Rap1 and PLD1 are transported to the plasma membrane in tandem via exocytosis of the vesicles upon which both proteins reside.
PLD1 controls Rap1 trafficking.
PLD activity has been implicated in vesicular trafficking (7). We therefore explored the dependence on PLD activity of GFP-Rap1 translocation in response to TCR stimulation. Whereas 95% ± 3% of Jurkat cells transfected with GFP-Rap1 and stimulated with anti-CD3 antibodies manifested plasma membrane fluorescence, this number dropped to 60% ± 5% (P < 0.005) for cells treated with n-butanol (Fig. 2A). t-Butanol was inactive, indicating that the effect of the alcohol was mediated by inhibition of PLD.
FIG. 2.

Rap1 expression on the plasma membrane depends on PLD1. (A) Inhibition of PLDs with n-butanol affects the plasma membrane (PM) localization of GFP-Rap1 but not GFP-Rap1V12. (B) n-butanol inhibits anti-CD3-stimulated translocation of both GFP-Rap1 and RFP-PLD1 from cytoplasmic vesicles to the plasma membrane. (C) Endogenous PLD1 and PLD2 in HeLa cells, detected by immunoblotting before and 72 h after expression of the indicated shRNA. (D) PLD1 knockdown in Jurkat cells, shown by cytofluorimetry of cells expressing GFP-PLD1 (x axis) and the indicated shRNA, which also directs expression of RFP from an internal ribosome entry site (y axis). (E) Silencing PLD1 but not PLD2 expression with shRNA inhibits plasma membrane localization of GFP-Rap1. Images show representative cells (bars, 5 μm), and the bar graph shows means ± standard errors of the means (SEM) (n = 3) for the percentage of cells with plasma membrane expression of each fluorescent protein.
Interestingly, unlike wild-type GFP-Rap1, which can undergo GTP-GDP exchange, the plasma membrane localization of constitutively active GFP-Rap1V12 was insensitive to n-butanol. This suggests that either delivery of Rap1-laden vesicles to the plasma membrane is PLD independent when Rap1 is activated or the removal of Rap1 from the plasma membrane by endocytosis requires GAP-induced GTP hydrolysis that is blocked by the G12V substitution. The absence of plasma membrane expression of GFP-Rap1N17 (not shown), a dominant-negative mutant that is either GDP bound or nucleotide free, supports the latter possibility. These data indicate that upregulation of wild-type Rap1 at the plasma membrane requires both PLD activity and the ability of Rap1 to cycle between the GDP- and GTP-bound states.
If PLD1 and Rap1 reside on the same cytoplasmic vesicles, the effects of n-butanol on GFP-Rap1 translocation should parallel the effects on mRFP-PLD1 translocation. Indeed, n-butanol but not t-butanol inhibited plasma membrane expression of mRFP-PLD1 (Fig. 2B), indicating that like the case for Rap1, PLD1 delivery to the plasma membrane required the catalytic activity of the enzyme. These data suggest that the lipase activity of PLD serves to promote the exocytic events that deliver both the lipase and Rap1 to the plasma membrane.
To confirm the dependence of Rap1 translocation on PLD activity, we silenced the two PLD isoforms with shRNA (Fig. 2C and D). Whereas knockdown of PLD2 had little effect on the expression of GFP-Rap1 on the plasma membranes of Jurkat cells, silencing of PLD1 inhibited plasma membrane expression by 43% ± 3% (Fig. 2E) (P < 0.01). Thus, expression of Rap1 at the plasma membrane depends on PLD1.
PLD lipase activity is required for Rap1 activation on the plasma membrane.
Because GTP loading of Rap1 and its delivery to the plasma membrane appeared to be linked, we studied the requirement of PLD activity for Rap1 activation. n-Butanol but not t-butanol inhibited TCR-stimulated GTP exchange on Rap1, as measured in cell lysates with a glutathione S-transferase (GST)-RalGDS-RBD pulldown assay (Fig. 3A). YFP-RalGDS-RBD can be used as a biosensor for activated Rap1 in live cells (5). Using this probe, we confirmed that although wild-type Rap1 is expressed on both the plasma membrane and intracellular vesicles (Fig. 1), the only pool that can recruit the probe is on the plasma membrane (Fig. 3B, left panel), indicating that the intracellular pool remains GDP bound. Rap1V12 is constitutively GTP bound and, like wild-type Rap1, is distributed on both the plasma membrane and the endomembrane (Fig. 2A). Rap1V12 therefore served as a control in this analysis and demonstrated that the probe has access to Rap1 decorating intracellular vesicles if the Rap1 is GTP bound (Fig. 3B, center panel). As expected, Rap1N17 recruited the probe to no membrane compartment (Fig. 3B, right panel). Using this live-cell assay, we determined that n-butanol but not t-butanol inhibited Rap1 activation at the plasma membrane (Fig. 3C and D).
FIG. 3.

Activation of Rap1 requires PLD activity. (A) Jurkat T cells were serum starved for 2 h and treated with or without t-butanol or n-butanol for 15 min prior to stimulation with anti-CD3 antibodies for 10 min, as indicated. Cell lysates were immunoblotted for total Rap1 and GTP-Rap1 (GST-RalGDS-RBD pulldown assay). (B) Jurkat T cells expressing YFP-RalGDS-RBD, a probe for GTP-bound Rap1, and the indicated Rap1 construct were imaged live growing in serum. (C) Serum-starved Jurkat T cells expressing YFP-RalGDS-RBD and Rap1 were treated with or without t-butanol or n-butanol and imaged live before and after stimulation with anti-CD3 antibodies. Bars, 5 μm. (D) Quantification of the results shown in panel C, plotted as means± SEM (n = 4).
Rap1-dependent lymphocyte adhesion requires PLD1.
Rap1 controls lymphocyte adhesion by regulating inside-out signaling to the integrin LFA-1 (30). Whereas Jurkat T cells expressing wild-type Rap1 remained responsive to TCR stimulation of increased adhesion to ICAM-1, cells expressing constitutively active Rap1V12 were relatively sticky but unresponsive to TCR cross-linking (Fig. 4A). This result confirms that the constitutively active mutant of Rap1 does not require upstream signals for full activity. Cells expressing Rap1N17 were less adhesive and were unresponsive to TCR stimulation (Fig. 4A), consistent with the dominant-negative properties of this mutant. These results suggest that the Rap1 pathway mediates the full effect of TCR signaling for increased adhesion. Although basal adhesion to ICAM-1 was not affected by n-butanol, TCR-stimulated Jurkat cell adhesion was inhibited 56% (P < 0.01) after treatment with n-butanol, whereas t-butanol had no significant effect (Fig. 4B). n-Butanol versus t-butanol had the same differential effect on primary murine T cells (Fig. 4C), demonstrating that the dependence on PLD activity was not specific to Jurkat cells. Silencing the gene for PLD1 had an effect on lymphocyte adhesion to ICAM-1 similar to that of n-butanol, but knockdown of PLD2 had no significant effect (Fig. 4D). Thus, TCR-stimulated T-cell adhesion to an LFA-1 ligand required PLD1, indicating that inside-out signaling through Rap1 requires this lipase.
FIG. 4.

PLD activity is required for TCR-stimulated lymphocyte adhesion to ICAM-1. (A) Jurkat T cells transfected with the indicated Rap1 construct tagged with GFP were plated on ICAM-1 and stimulated with or without anti-CD3 antibodies, and adherent, transduced cells were measured with a fluorescent plate reader. Untransfected Jurkat cells (B) or CD4+ splenocytes from C57BL/6 mice (C) were labeled with 5-carboxyfluorescein, treated with or without t-butanol or n-butanol, and stimulated with or without anti-CD3 antibodies, and adhesion to ICAM-1 was measured as in panel A. (D) Jurkat cells labeled with 5-carboxyfluorescein and transfected with the indicated shRNA were stimulated with or without anti-CD3 antibodies, and adhesion to ICAM-1 was measured as in panel A. Results are shown as means ± SEM (n = 4), except in panel C, where the results of an experiment representative of two are shown.
C3G translocates from the cytosol to the plasma membrane and regulates adhesion in a PLD-dependent fashion.
The results discussed above suggest that PLD1 controls the delivery of Rap1 to the plasma membrane via exocytosis of storage vesicles and that translocation to the cell surface is associated with GTP-GDP exchange on Rap1. This model requires a plasma membrane-associated guanine nuclear exchange factor (GEF). The best-characterized exchange factor for Rap1 is C3G (17, 38). We found that C3G is expressed at significantly higher levels in Jurkat T cells than in fibroblasts (not shown), suggesting a prominent role in T-cell signaling. GFP-tagged C3G gave a cytosolic pattern in Jurkat T cells that did not change with TCR stimulation (not shown). In contrast, following endogenous C3G by subcellular fractionation revealed translocation. Whereas in resting T cells virtually all of the endogenous C3G was found in the cytosol, in cells stimulated by cross-linking of the TCR 10% of the total pool translocated to membranes (Fig. 5). RhoGDI, a cytoplasmic protein, did not appear in the membrane fraction under the same conditions (Fig. 5). The pool of membrane-associated C3G was too small to analyze by sucrose gradient centrifugation, such that the identity of the light membranes with which C3G associated could not be determined. Nevertheless, the result is consistent with translocation of C3G to the plasma membranes of T cells, as previously shown with PC12 cells (38).
FIG. 5.
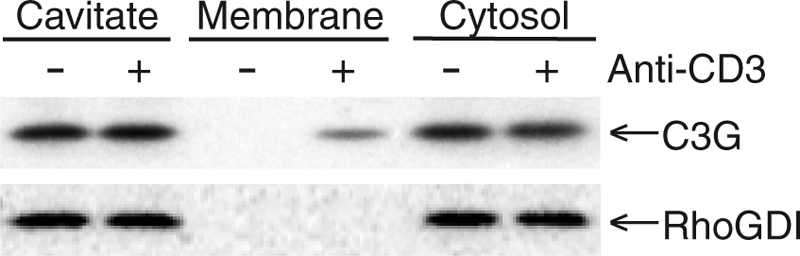
TCR activation stimulates translocation of C3G from cytosol to membranes of Jurkat T cells. Serum-starved Jurkat T cells were stimulated with or without anti-CD3 antibodies and disrupted by nitrogen cavitation. The cavitated samples were separated into total membranes (P175) and cytosol (S175) and analyzed for C3G and RhoGDI by immunoblotting. Results shown are representative of three independent experiments.
To confirm a functional role for C3G translocated to the plasma membrane, we studied T-cell adhesion to ICAM-1. Overexpression of C3G in Jurkat T cells increased the level of adhesion to ICAM-1 stimulated by TCR engagement (Fig. 6A). The increased adhesion was sensitive to n-butanol but not t-butanol (Fig. 6A), suggesting that the activity of C3G requires PLD1-mediated upregulation of Rap1 at the plasma membrane. This was confirmed using shRNA. Silencing of PLD1 but not PLD2 diminished C3G-dependent, TCR-stimulated increased adhesion (Fig. 6B). If PLD1 regulates Rap1-dependent adhesion by controlling delivery of Rap1 to the plasma membrane, where it can encounter C3G, then Rap1V12, which requires neither C3G nor any other exchange factor for full activity, should be insensitive to inhibition by PLD. Indeed, the proadhesive effect of Rap1V12 was independent of PLD activity (Fig. 6C).
FIG. 6.

C3G-dependent TCR-stimulated T-cell adhesion to ICAM-1 requires PLD1. (A) Jurkat T cells transfected with GFP-C3G or GFP vector and treated with or without t-butanol or n-butanol were assayed for adhesion to ICAM-1 as described in the legend to Fig. 5, before and after stimulation with anti-CD3 antibodies. (B) Jurkat T cells were transfected with GFP-C3G and the indicated shRNA, and the adhesion of transduced, fluorescent cells to ICAM-1 was determined. (C) Jurkat T cells transfected with GFP-Rap1V12 and treated with or without t-butanol or n-butanol were stimulated or not with anti-CD3 antibodies, and adhesion to ICAM-1 was measured. Results are shown as means ± SEM (n = 3).
PLD does not regulate adhesion when Rap1 is delivered directly to the plasma membrane by nonvesicular transport.
To test further the idea that PLD1 regulates adhesion by controlling the trafficking of Rap1, we constructed a Rap1/Kras fusion protein in which the membrane-targeting C terminus of Rap1 was replaced with the C terminus of Kras (Rap1-Ktail). We predicted that this chimera would traffic directly to the plasma membrane, like Kras (9), in a fashion independent of PLD1-regulated exocytic vesicles. Jurkat T cells expressing Rap1-Ktail displayed an enhanced adhesion response to TCR stimulation (Fig. 7), indicating that artificially targeted Rap1 was biologically active in inside-out signaling to LFA-1. Importantly, this effect was insensitive to n-butanol, demonstrating that PLD1 regulates the activity of Rap1 at the level of protein trafficking rather than distal signaling to LFA-1.
FIG. 7.

Rap1 targeted directly to the plasma membrane with the Kras tail regulates T-cell adhesion in a PLD-independent fashion. Jurkat T cells were transfected with GFP vector or GFP-Rap1-Ktail, as indicated, and adhesion to ICAM-1 was measured with or without stimulation with anti-CD3 antibodies and with or without pretreatment with either t- or n-butanol. Results are shown as means ± SEM (n = 3).
DISCUSSION
Our data support the idea that the trafficking of Rap1 and PLD1 is linked and depends on the enzymatic activity of the lipase. Mature Rap1 possesses a potent membrane-targeting motif at the C terminus, consisting of a 20-carbon geranylgeranyl group and an adjacent polybasic region. Geranylgeranylated Rho family proteins traffic through the cytosol by virtue of association with a chaperone, RhoGDI (28), but no analogous chaperone has been described for Rap1. This suggests that Rap1 is constitutively associated with membranes. Indeed, whereas we readily observed farnesylated Ras proteins in Jurkat cell cytosol, we detected no Rap1 in this fraction. Therefore, although Rap1 is a peripheral membrane protein that, in principle, could be transported between membrane compartments by modulation of its affinity for the membranes, the evidence presented here argues against such fluid-phase trafficking. We concluded that the level of expression of Rap1 at the plasma membrane is acutely modulated by the delivery of the GTPase to the cell surface via vesicular transport (5). Exocytosis of recycling endosomes upregulates Rap1 expression at the cell surface, and endocytosis has the opposite effect.
PLDs are signaling enzymes best known for their ability to generate phosphatidic acid (PA), an important lipid second messenger (15). Among the cellular processes regulated by PLDs and their lipid products is vesicular trafficking, including endoplasmic reticulum-to-Golgi transport (4), post-Golgi secretion (8), and endocytosis (35). In lymphocytes, PLD has been found to regulate the exocytosis of CTLA-4-containing vesicles (25). Our results add exocytosis of recycling endosomes laden with Rap1 to this list.
In addition to affecting vesicular transport, PA and diacylglycerol (DAG) produced from PA can regulate GEFs that act on Ras (29, 40). Thus, although the effects of PLD inhibition on exocytosis were unambiguous, PLD inhibition might also affect one or more GEFs that act on Rap1. To explore this possibility, we sent Rap1 to the plasma membrane by using the Kras C-terminal targeting motif, which delivers proteins to the plasma membrane independent of vesicular transport. The Rap1-Ktail chimera was biologically active in our adhesion assay, but the activity was independent of PLD, providing strong support for PLD operating at the level of vesicular trafficking rather than GTP-GDP exchange.
Three classes of GEFs activate Rap1 in leukocytes. C3G is regulated through adaptor proteins that bind phosphotyrosine (20). Epac1 is regulated by cyclic AMP (cAMP) (13, 22). CalDAG-GEFI (a splice variant of RasGRP2) is regulated by DAG and calcium (23). The physiological significance of the last of these observations was highlighted by a recent report that revealed that the genetic basis for two cases of LAD III could be attributed to a splice junction defect in the CalDAG-GEFI gene (31). Because much of the PA produced by PLDs is converted by phosphatidic acid phosphatase into DAG, in addition to controlling the upregulation of Rap1 at the plasma membrane, PLD may also regulate its activation through CalDAG-GEFI. However, CalDAG-GEFI is activated more effectively by chemokines than by TCR signaling (31), and TCR stimulation alone does not induce translocation of this GEF to the plasma membrane (32). Moreover, CalDAG-GEFI is highly expressed in the murine brain (23) and in myeloid cells but is undetectable in thymocytes (11), and mice deficient in CalDAG-GEFI have normal lymphopoiesis but severely impaired neutrophil and platelet function (3), similar to the phenotype of patients with LAD III (31). Thus, although overexpression of CalDAG-GEFI in Jurkat T cells promotes Rap1 activation and adhesion (21), it is not likely to represent the physiologically relevant Rap1 GEF downstream of the TCR.
Epac1 is also not likely to play a physiologic role in stimulating T-cell adhesion, since elevated cAMP inhibits T-cell activation, is associated with anergy, and results in decreased GTP loading of Rap1 (18). In contrast, C3G is an attractive candidate for the GEF that transmits the signal from the TCR to Rap1. This exchange factor and its signaling partners Cbl and CrkL are highly expressed in thymocytes (1). Indeed, we found that C3G is much more highly expressed in Jurkat T cells than in fibroblasts. Moreover, we found that TCR stimulation led to the translocation of C3G to the membranes of Jurkat cells. The relatively small fraction of endogenous C3G that could be driven to the membrane from the cytosol in response to TCR stimulation is consistent with that observed for many Ras family GEFs, e.g., SOS. This may explain why we failed to observe translocation of GFP-C3G, even when we co-overexpressed Crk and CrkL. The high background fluorescence of the untranslocated pool did not permit resolution of the small portion that associates with the membrane. Our observation that C3G overexpression markedly increased TCR-stimulated T-cell adhesion strongly supports a role for C3G in Rap1-mediated inside-out signaling to LFA-1. The inhibition by n-butanol of this augmented adhesion is consistent with a requirement for PLD for the full effect of C3G. This suggests a model whereby Rap1 activation at the plasma membrane is the consequence of two independent translocation events. Rap1 is delivered to the plasma membrane by exocytosis concomitant with the delivery of its GEF, C3G, to the same compartment by fluid-phase translocation from the cytosol as a consequence of phosphotyrosine-binding adaptor proteins (Fig. 8).
FIG. 8.

Model of Rap1 upregulation and activation at the plasma membrane. In resting cells, Rap1 is stored on a pool of intracellular, endocytic vesicles in an inactive, GDP-bound state. PLD1 is expressed on the same pool of vesicles. T-cell activation leads to PLD-dependent exocytosis of these vesicles, which results in increased expression of Rap1 at the plasma membrane. TCR signaling induces concomitant translocation of C3G in a complex with an SH2/SH3 adapter, such as Crk, from the cytosol to the plasma membrane such that both the GTPase and its cognate GEF meet only at the plasma membrane, coupling upregulation to activation and leading to increased T-cell adhesion.
The spatial separation of small GTPases and their GEFs is a common theme in the biology of these regulatory molecules. In some systems, such as growth factor stimulation of Ras, the GTPase is constitutively present at the membrane and activation involves translocation only of the GEF. In other systems, such as the activation of Rac1 by Vav1, both GTPase and GEF translocate to the membrane from the aqueous cytosol, affording a higher order of regulation and therefore more stringent control. Our data suggest that the control of Rap1 in T cells fits the second pattern. However, unlike Rho proteins that translocate from cytosolic chaperones, Rap1 is delivered by vesicular transport, and this process is controlled in part by PLD1. PLD antagonists, in particular those specific for PLD1, may therefore prove to be immunosuppressive.
Acknowledgments
This work was supported by grants GM055279 and CA116034 from the National Institutes of Health, the Arthritis National Research Foundation, and the New York State Department of Health.
Footnotes
Published ahead of print on 30 March 2009.
REFERENCES
- 1.Amsen, D., A. Kruisbeek, J. L. Bos, and K. Reedquist. 2000. Activation of the Ras-related GTPase Rap1 by thymocyte TCR engagement and during selection. Eur. J. Immunol. 302832-2841. [DOI] [PubMed] [Google Scholar]
- 2.Anikeeva, N., K. Somersalo, T. N. Sims, V. K. Thomas, M. L. Dustin, and Y. Sykulev. 2005. Distinct role of lymphocyte function-associated antigen-1 in mediating effective cytolytic activity by cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. USA 1026437-6442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bergmeier, W., T. Goerge, H. W. Wang, J. R. Crittenden, A. C. Baldwin, S. M. Cifuni, D. E. Housman, A. M. Graybiel, and D. D. Wagner. 2007. Mice lacking the signaling molecule CalDAG-GEFI represent a model for leukocyte adhesion deficiency type III. J. Clin. Investig. 1171699-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bi, K., M. G. Roth, and N. T. Ktistakis. 1997. Phosphatidic acid formation by phospholipase D is required for transport from the endoplasmic reticulum to the Golgi complex. Curr. Biol. 7301-307. [DOI] [PubMed] [Google Scholar]
- 5.Bivona, T. G., H. H. Wiener, I. M. Ahearn, J. Silletti, V. K. Chiu, and M. R. Philips. 2004. Rap1 up-regulation and activation on plasma membrane regulates T cell adhesion. J. Cell Biol. 164461-470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bos, J. L., J. de Rooij, and K. A. Reedquist. 2001. Rap1 signalling: adhering to new models. Nat. Rev. Mol. Cell Biol. 2369-377. [DOI] [PubMed] [Google Scholar]
- 7.Cazzolli, R., A. N. Shemon, M. Q. Fang, and W. E. Hughes. 2006. Phospholipid signalling through phospholipase D and phosphatidic acid. IUBMB Life 58457-461. [DOI] [PubMed] [Google Scholar]
- 8.Chen, Y. G., A. Siddhanta, C. D. Austin, S. M. Hammond, T. C. Sung, M. A. Frohman, A. J. Morris, and D. Shields. 1997. Phospholipase D stimulates release of nascent secretory vesicles from the trans-Golgi network. J. Cell Biol. 138495-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choy, E., V. K. Chiu, J. Silletti, M. Feoktistov, T. Morimoto, D. Michaelson, I. E. Ivanov, and M. R. Philips. 1999. Endomembrane trafficking of ras: the CAAX motif targets proteins to the ER and Golgi. Cell 9869-80. [DOI] [PubMed] [Google Scholar]
- 10.Corrotte, M., S. Chasserot-Golaz, P. Huang, G. Du, N. T. Ktistakis, M. A. Frohman, N. Vitale, M. F. Bader, and N. J. Grant. 2006. Dynamics and function of phospholipase D and phosphatidic acid during phagocytosis. Traffic 7365-377. [DOI] [PubMed] [Google Scholar]
- 11.Crittenden, J. R., W. Bergmeier, Y. Zhang, C. L. Piffath, Y. Liang, D. D. Wagner, D. E. Housman, and A. M. Graybiel. 2004. CalDAG-GEFI integrates signaling for platelet aggregation and thrombus formation. Nat. Med. 10982-986. [DOI] [PubMed] [Google Scholar]
- 12.de Rooij, J., and J. L. Bos. 1997. Minimal Ras-binding domain of Raf1 can be used as an activation-specific probe for Ras. Oncogene 14623-625. [DOI] [PubMed] [Google Scholar]
- 13.de Rooij, J., F. J. Zwartkruis, M. H. Verheijen, R. H. Cool, S. M. Nijman, A. Wittinghofer, and J. L. Bos. 1998. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396474-477. [DOI] [PubMed] [Google Scholar]
- 14.Dustin, M. L., T. G. Bivona, and M. R. Philips. 2004. Membranes as messengers in T cell adhesion signaling. Nat. Immunol. 5363-372. [DOI] [PubMed] [Google Scholar]
- 15.Exton, J. H. 1999. Regulation of phospholipase D. Biochim. Biophys. Acta 1439121-133. [DOI] [PubMed] [Google Scholar]
- 16.Goodwin, J. S., K. R. Drake, C. Rogers, L. Wright, J. Lippincott-Schwartz, M. R. Philips, and A. K. Kenworthy. 2005. Depalmitoylated Ras traffics to and from the Golgi complex via a nonvesicular pathway. J. Cell Biol. 170261-272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gotoh, T., S. Hattori, S. Nakamura, H. Kitayama, M. Noda, Y. Takai, K. Kaibuchi, H. Matsui, O. Hatase, H. Takahashi, et al. 1995. Identification of Rap1 as a target for the Crk SH3 domain-binding guanine nucleotide-releasing factor C3G. Mol. Cell. Biol. 156746-6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grader-Beck, T., A. A. van Puijenbroek, L. M. Nadler, and V. A. Boussiotis. 2003. cAMP inhibits both Ras and Rap1 activation in primary human T lymphocytes, but only Ras inhibition correlates with blockade of cell cycle progression. Blood 101998-1006. [DOI] [PubMed] [Google Scholar]
- 19.Grakoui, A., S. K. Bromley, C. Sumen, M. M. Davis, A. S. Shaw, P. M. Allen, and M. L. Dustin. 1999. The immunological synapse: a molecular machine controlling T cell activation. Science 285221-227. [DOI] [PubMed] [Google Scholar]
- 20.Ichiba, T., Y. Kuraishi, O. Sakai, S. Nagata, J. Groffen, T. Kurata, S. Hattori, and M. Matsuda. 1997. Enhancement of guanine-nucleotide exchange activity of C3G for Rap1 by the expression of Crk, CrkL, and Grb2. J. Biol. Chem. 27222215-22220. [DOI] [PubMed] [Google Scholar]
- 21.Katagiri, K., M. Shimonaka, and T. Kinashi. 2004. Rap1-mediated lymphocyte function-associated antigen-1 activation by the T cell antigen receptor is dependent on phospholipase C-gamma1. J. Biol. Chem. 27911875-11881. [DOI] [PubMed] [Google Scholar]
- 22.Kawasaki, H., G. M. Springett, N. Mochizuki, S. Toki, M. Nakaya, M. Matsuda, D. E. Housman, and A. M. Graybiel. 1998. A family of cAMP-binding proteins that directly activate Rap1. Science 2822275-2279. [DOI] [PubMed] [Google Scholar]
- 23.Kawasaki, H., G. M. Springett, S. Toki, J. J. Canales, P. Harlan, J. P. Blumenstiel, E. J. Chen, I. A. Bany, N. Mochizuki, A. Ashbacher, M. Matsuda, D. E. Housman, and A. M. Graybiel. 1998. A Rap guanine nucleotide exchange factor enriched highly in the basal ganglia. Proc. Natl. Acad. Sci. USA 9513278-13283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kinashi, T., M. Aker, M. Sokolovsky-Eisenberg, V. Grabovsky, C. Tanaka, R. Shamri, S. Feigelson, A. Etzioni, and R. Alon. 2004. LAD-III, a leukocyte adhesion deficiency syndrome associated with defective Rap1 activation and impaired stabilization of integrin bonds. Blood 1031033-1036. [DOI] [PubMed] [Google Scholar]
- 25.Mead, K. I., Y. Zheng, C. N. Manzotti, L. C. Perry, M. K. Liu, F. Burke, D. J. Powner, M. J. Wakelam, and D. M. Sansom. 2005. Exocytosis of CTLA-4 is dependent on phospholipase D and ADP ribosylation factor-1 and stimulated during activation of regulatory T cells. J. Immunol. 1744803-4811. [DOI] [PubMed] [Google Scholar]
- 26.Menasche, G., S. Kliche, N. Bezman, and B. Schraven. 2007. Regulation of T-cell antigen receptor-mediated inside-out signaling by cytosolic adapter proteins and Rap1 effector molecules. Immunol. Rev. 21882-91. [DOI] [PubMed] [Google Scholar]
- 27.Michaelson, D., W. Ali, V. K. Chiu, M. Bergo, J. Silletti, L. Wright, S. G. Young, and M. Philips. 2005. Postprenylation CAAX processing is required for proper localization of Ras but not Rho GTPases. Mol. Biol. Cell 161606-1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Michaelson, D., J. Silletti, G. Murphy, P. D'Eustachio, M. Rush, and M. R. Philips. 2001. Differential localization of Rho GTPases in live cells. Regulation by hypervariable regions and RhoGDI binding. J. Cell Biol. 152111-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mor, A., G. Campi, G. Du, Y. Zheng, D. A. Foster, M. L. Dustin, and M. R. Philips. 2007. The lymphocyte function-associated antigen-1 receptor costimulates plasma membrane Ras via phospholipase D2. Nat. Cell Biol. 9713-719. [DOI] [PubMed] [Google Scholar]
- 30.Mor, A., M. L. Dustin, and M. R. Philips. 2007. Small GTPases and LFA-1 reciprocally modulate adhesion and signaling. Immunol. Rev. 218114-125. [DOI] [PubMed] [Google Scholar]
- 31.Pasvolsky, R., S. W. Feigelson, S. S. Kilic, A. J. Simon, G. Tal-Lapidot, V. Grabovsky, J. R. Crittenden, N. Amariglio, M. Safran, A. M. Graybiel, G. Rechavi, S. Ben-Dor, A. Etzioni, and R. Alon. 2007. A LAD-III syndrome is associated with defective expression of the Rap-1 activator CalDAG-GEFI in lymphocytes, neutrophils, and platelets. J. Exp. Med. 2041571-1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pérez de Castro, I., T. Bivona, M. Philips, and A. Pellicer. 2004. Ras activation in Jurkat T cells following low-grade stimulation of the T-cell receptor is specific to N-Ras and occurs only on the Golgi. Mol. Cell. Biol. 243485-3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reedquist, K. A., E. Ross, E. A. Koop, R. M. Wolthuis, F. J. Zwartkruis, Y. van Kooyk, M. Salmon, C. D. Buckley, and J. L. Bos. 2000. The small GTPase, Rap1, mediates CD31-induced integrin adhesion. J. Cell Biol. 1481151-1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rose, D. M., R. Alon, and M. H. Ginsberg. 2007. Integrin modulation and signaling in leukocyte adhesion and migration. Immunol. Rev. 218126-134. [DOI] [PubMed] [Google Scholar]
- 35.Shen, Y., L. Xu, and D. A. Foster. 2001. Role for phospholipase D in receptor-mediated endocytosis. Mol. Cell. Biol. 21595-602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Silvius, J. R., P. Bhagatji, R. Leventis, and D. Terrone. 2006. K-ras4B and prenylated proteins lacking “second signals” associate dynamically with cellular membranes. Mol. Biol. Cell 17192-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Su, W., P. Chardin, M. Yamazaki, Y. Kanaho, and G. Du. 2006. RhoA-mediated phospholipase D1 signaling is not required for the formation of stress fibers and focal adhesions. Cell. Signal. 18469-478. [DOI] [PubMed] [Google Scholar]
- 38.Wang, Z., T. J. Dillon, V. Pokala, S. Mishra, K. Labudda, B. Hunter, and P. J. Stork. 2006. Rap1-mediated activation of extracellular signal-regulated kinases by cyclic AMP is dependent on the mode of Rap1 activation. Mol. Cell. Biol. 262130-2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Watson, R. T., and J. E. Pessin. 2001. Subcellular compartmentalization and trafficking of the insulin-responsive glucose transporter, GLUT4. Exp. Cell Res. 27175-83. [DOI] [PubMed] [Google Scholar]
- 40.Zhao, C., G. Du, K. Skowronek, M. A. Frohman, and D. Bar-Sagi. 2007. Phospholipase D2-generated phosphatidic acid couples EGFR stimulation to Ras activation by Sos. Nat. Cell Biol. 9706-712. [DOI] [PubMed] [Google Scholar]