

Abstract
In the present study, we report that ubiquitin-mediated degradation of dMyc, the Drosophila homologue of the human c-myc proto-oncogene, is regulated in vitro and in vivo by members of the casein kinase 1 (CK1) family and by glycogen synthase kinase 3β (GSK3β). Using Drosophila S2 cells, we demonstrate that CK1α promotes dMyc ubiquitination and degradation with a mechanism similar to the one mediated by GSK3β in vertebrates. Mutation of ck1α or -ɛ or sgg/gsk3β in Drosophila wing imaginal discs results in the accumulation of dMyc protein, suggesting a physiological role for these kinases in vivo. Analysis of the dMyc amino acid sequence reveals the presence of conserved domains containing potential phosphorylation sites for mitogen kinases, GSK3β, and members of the CK1 family. We demonstrate that mutations of specific residues within these phosphorylation domains regulate dMyc protein stability and confer resistance to degradation by CK1α and GSK3β kinases. Expression of the dMyc mutants in the compound eye of the adult fly results in a visible defect that is attributed to the effect of dMyc on growth, cell death, and inhibition of ommatidial differentiation.
Myc is a member of a family of transcription factors that plays a pivotal role in the regulation of proliferation, growth and apoptosis. Myc expression is regulated by multiple signaling events acting at transcriptional, translational, and posttranslational levels (6, 50). Phosphorylation is one of the most relevant posttranslational mechanisms by which Myc stability is regulated. Activation of the Ras extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK), c-Jun kinase (JNK), and cyclin-dependent kinase 1 (CDK1) (36, 44) rapidly stabilizes c-Myc protein by phosphorylation on serine 62 (46). This event primes Myc for phosphorylation by glycogen synthase kinase 3β (GSK3β) on threonine 58 (45). Dephosphorylation of serine 62 by the phosphatase PP2A/B56α allows the ubiquitin ligase SCF (for Skip1-Cullin-F box) Fbw7 (SCFFbw7) to bind c-Myc on threonine 58, hence promoting its degradation by the proteasome pathway (54). In addition, the extent to which c-Myc exerts its effects is modulated by the SCF-SKP2 ubiquitin ligase, which enhances c-Myc transcriptional activity before triggering proteasomal degradation (25, 51). Coordination between phosphorylation and ubiquitination is a conserved feature in mammals and Drosophila, since mutations in archipelago (ago), encoding a ubiquitin ligase that is the Drosophila homologue of the mammalian Fbw7 ubiquitin ligase, result in dMyc protein accumulation (32), as does phosphorylation of dMyc by activated Ras (41).
In addition to phosphorylation at its N terminus, c-Myc protein stability is regulated by phosphorylation in a PEST domain (amino acids 253 to 266) located near the conserved acidic box (AB). Deletion of the c-Myc PEST domain enhances c-Myc protein stability (16).
Although the Drosophila Myc protein (dMyc) is only 26% identical to human c-Myc over its length, the homology increases up to 57% in critical functional regions such as the AB and the PEST domain (13). Moreover, dMyc and c-Myc proteins share similar functions; human c-Myc can rescue lethal dmyc mutations to viability (5), and dMyc is able to partially substitute for the proliferation defect of c-myc-deficient mouse embryo fibroblasts (49).
Analysis of the dMyc protein reveals the presence of evolutionarily conserved phosphorylation sites that correspond to optimal amino acid consensus sequences for phosphorylation by members of the casein kinase 1 (CK1) family and by the GSK3β kinases. GSK3β phosphorylation requires a priming event (10) that for some proteins, such as β-catenin/Armadillo or Cubitus interruptus (Ci), is carried out by members of the CK1 family (21, 28, 40, 53).
In the present study, we report that members of the CK1 kinases act as novel components of the process regulating dMyc protein stability through a mechanism that favors its ubiquitination and degradation, similar to the action of GSK3β. In vivo, decreasing levels of ck1α or -ɛ, or sgg/gsk3β in wing imaginal disc cells favor dMyc protein accumulation, preferentially in the presumptive hinge and notum territories of the disc. Through site-directed mutagenesis, we identified novel phosphorylation domains that are responsible for dMyc degradation induced by the action of members of the CK1 and GSK3β family of kinases. Expression of these dMyc phosphorylation mutants in vivo in the compound eyes of the adult fly (4) results in strong defects, which, at the cellular level, shows that expression of a proteasome-resistant form of dMyc inhibits ommatidial differentiation and increases cell death during the development of the eye imaginal discs.
MATERIALS AND METHODS
Fly husbandry and strains.
Fly cultures and crosses were maintained on standard fly medium at 25°C. UAS-DCO and FRT82B dco3/TM6B stocks were a gift from M. Noll and E. Frei, UAS-RNAi-CK1α (where RNAi-CK1α denotes the CK1α gene modified by RNA interference [RNAi]) stock was a gift from D. Kalderon, and yw hs-Flp; Act5C-FRT y+FRT-Gal4 stock was a gift from A. Yamamoto. UAS-HA-dMyc stock was described in reference 4; UAS-Sgg stock was a gift from M. Milán. We generated the dmycP0tub FRT-(dmyc cDNA)-FRT-Gal4-ey-FLP/Y line (hereafter called ey>dmycP0) that constitutively expresses the flippase (FLP-ase) under the control of the eyeless promoter (ey-FLP), allowing the dmyc cDNA construct to be flipped out, rendering the eye mutant for dmycP0. This line also expresses the Gal4 reporter. We used the UAS/Gal4 system to express UAS transgene in the eyeless domain. Line ey>yw carries the same construct as in ey>dmycP0 but in the yw background.
Cell culture and transfection of S2 cells.
Drosophila Schneider S2 cells and the S2-HA-dMyc cell line (4) were grown at 25°C using Schneider medium (Gibco) supplemented with 10% heat-inactivated fetal calf serum and 100 IU of penicillin-streptomycin (Gibco). S2 cells were transfected by using Cellfectin reagent (Invitrogen) as described in reference 3. The S2-tub-Gal4 line was generated in the present study by stable transfection of the tubulin Gal4 plasmids. The tubulin Gal4 construct was cotransfected with the pcP4 plasmid encoding the α-amanitin resistance gene, and cells were selected using 5 μg of α-amanitin/ml for 3 weeks (Sigma) (3).
Western blotting and chemical treatments.
Cells were lysed in 50 mM HEPES buffer (pH 7.5) containing 0.5% Triton X-100, 150 mM NaCl, 1 mM EDTA, and protease and phosphatase inhibitors. Lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, subjected to Western blot analysis, and visualized by using enhanced chemiluminescence (Amersham Pharmacia Biotech). For the ubiquitination analysis, N-ethylmaleimide, an inhibitor of deubiquitinating enzymes, was added to the lysis buffer, and immunoprecipitation was performed with rat anti-HA antibody bound to protein G-Sepharose (CL-4B; Amersham Pharmacia Biotech). We used the antibodies anti-HA (Roche), antiubiquitin (FK2-BioMol), antiactin and antivinculin (Sigma), and anti-dMyc (41). Chemicals were used at final concentrations of 50 μM for MG132, 10 mM for okadaic acid (OA), 10 μM for cycloheximide (CHX; Sigma), and 50 mM for LiCl and 10 μM CK1-7 (U.S. Biologicals).
Clonal analysis.
Clones mutant for dco3 were induced by mitotic recombination using the FLP-FRT method (52). yw hs-Flp; FRT82 Ubi-GFP/TM6B flies were crossed to yw; FRT82B dco3/TM6B flies, and clones were induced by a 20-min heat shock at 37°C at 65 ± 2 h after egg laying (AEL). sgg/gsk3β mutant clones were generated by mitotic recombination as described previously (21). For ectopic expression of UAS-DCO, UAS-Sgg, and UAS-RNAi-ck1α, Flp-out clones were induced using the yw hs-Flp; Act5C>FRTy+FRT>Gal4; UAS-GFP line and larvae were heat shocked at 37°C for 8 min at 65 ± 2 h AEL.
Immunofluorescence analysis of imaginal discs and antibody staining.
Discs were fixed in 4% paraformaldehyde in phosphate-buffered saline, permeabilized using 0.5% Triton X-100, and then blocked in 5% bovine serum albumin in phosphate-buffered saline. The antibodies used were anti-dMyc (1:5 dilution) (42), rat anti-ELAV (1:1,000 dilution; Developmental Study Hybridoma Bank), and rabbit anti-active caspase-3 (1:200 dilution; Invitrogen). The secondary antibodies, anti-mouse antibody-Alexa-555 and anti-rat antibody-fluorescein isothiocyanate (Invitrogen), were used at a 1:200 dilution in 5% bovine serum albumin, together with DAPI (4′,6-diamidino-2-phenylindole) at 1 μg/ml. After a washing step, discs were mounted in Vectashield and analyzed under a Leica microscope. The images were processed by using Open Lab and Adobe Photoshop software.
Site-directed mutagenesis.
Site-directed mutagenesis was carried out by using the QuikChange mutagenesis kit (Stratagene). Primer sequences are available in the supplemental material.
Determination of cell number and cell size in adult eyes.
Flies were reared under identical growth conditions and age matched (3-day-old males). To determine the ommatidial number, ommatidia were counted from scanning electron micrographs of eyes from different animals. From the same photographs, the sizes of the ommatidia were calculated by measuring the area of 20 ommatidia located in the center of the eye using Adobe Photoshop.
RESULTS
CK1 and GSK3β activity induces dMyc protein degradation.
Analysis of the dMyc amino acid sequence reveals the presence of conserved domains (Fig. 1A and Table 1) containing consensus sites for phosphorylation by members of the CK1 (S/T-XX-S/T) family and by GSK3β (S/T-XXX-S/T) (37). In vitro kinase assays demonstrated that dMyc and c-Myc proteins were directly phosphorylated by CK1α and GSK3β kinases (see Fig. S1 in the supplemental material), suggesting that Myc protein is a substrate for these kinases. In order to analyze whether CK1α and GSK3β affect dMyc protein stability, Drosophila S2 cells were treated with specific inhibitors of these kinases, and the dMyc protein levels were analyzed by Western blotting. As shown in Fig. 1B, treatment of the cells with either CK1-7, an inhibitor of the CK1 kinases, or LiCl, a specific inhibitor of GSK3β, led to the accumulation of dMyc protein, suggesting that both CK1 kinases and GSK3β act in the pathway of dMyc protein degradation. Since it was reported that GSK3β induces c-Myc degradation through the proteasomal pathway (45), we blocked proteasome activity with the drug MG132 and verified the effects on dMyc protein levels. As shown in Fig. 1C, treatment of the cells with MG132 resulted in the accumulation of two forms of immunoreactive dMyc protein (arrowhead, compare lane 1 to lane 2). Treatment of cells with the kinase inhibitor LiCl or CK1-7, in the presence of MG132, blocked the appearance of the slower-migrating dMyc band (compare lane 1 with lane 5 to lane 6), suggesting that the upper band of the doublet represents a hyperphosphorylated form of dMyc. The tumor suppressor protein phosphatase type 2A (PP2A) favors c-Myc degradation by dephosphorylating c-Myc at serine 62 (54). Since electrophoretic mobility shift has been used as an indication of the phosphorylation status of a protein, we used OA to inhibit PP2A and determine whether treatment with OA changes the dMyc electrophoretic mobility. As shown in Fig. 1C, OA caused an upward electrophoretic mobility shift of dMyc protein (compare lanes 1 and 3), indirectly suggesting that inhibition of PP2A affects dMyc dephosphorylation. In order to test whether the upward electrophoretic shift observed in the presence of OA was due to phosphorylation of dMyc exclusively by GSK3β, we treated cells with LiCl and OA together. LiCl only partially decreased the dMyc mobility shift induced by OA (compare lane 3 to lane 4), suggesting that another kinase, insensitive to LiCl, is responsible for the residual accumulation of the hyperphosphorylated form of dMyc (lane 4).
FIG. 1.

Inhibition of GSK3β and CK1 kinases increases dMyc protein level in S2 cells. (A) Schematic representation of dMyc protein with the amino acid sequences of dMyc phosphorylation mutants and homologous sequences in c-Myc. The amino acids changed by site-directed mutagenesis are underlined. (B) Treatment of S2 cells with LiCl and CK1-7 kinase inhibitors stabilizes endogenous dMyc protein. Cells were treated with the indicated inhibitors for 4 h; dMyc was immunoprecipitated from the cell extracts by using anti-dMyc antiserum, and its expression level was analyzed by Western blotting with anti-dMyc antiserum. The position of immunoglobulins is marked on the left. Total lysates were blotted with antiactin for control loading. (C) S2 cells were treated with MG132 (proteasome inhibitor), OA (an inhibitor of PP2A), LiCl (an inhibitor of GSK3β), and CK1-7 (an inhibitor of CK1s). Endogenous dMyc protein was analyzed in total lysates by Western blotting with anti-dMyc antibodies; actin was used as a loading control. The arrowhead on the left represents a dMyc doublet visible in cells treated with MG132. The arrows on the right point to dMyc forms of different electrophoretic mobilities. Molecular mass markers are shown on the right.
TABLE 1.
Conserved phosphorylation domains in Myc proteins from D. melanogaster and humans
| Mutant | Domain (aa) or mutationa | Sequence homologyb | % Identityc | Stability (min)d |
|---|---|---|---|---|
| dMyc-PI | D. melanogaster mutation | A---A-A | 50 | <30 |
| D. melanogaster (201-207) | SGELSGS | |||
| Human (67-73) | SGLCSPS | |||
| dMyc-PII | D. melanogaster mutation | A---A-A | 43 | 60 |
| D. melanogaster (324-330) | SPPTTGS | |||
| Human (58-64) | TPPLSPS | |||
| dMyc-PV | D. melanogaster mutation | A-A-A-- | 75 | 180 |
| D. melanogaster (405-411) | TPSDSDE | |||
| Human (247-253) | TSSDSEE | |||
| AB | D. melanogaster mutation | Q---N-NQQ-N--- | 54 | 180 |
| D. melanogaster (404-417) | ETPSDSDEEIDVVS | |||
| Human (253-266) | EEEQEDEEEIDVVS |
aa, amino acids.
Amino acid substitutions in D. melanogaster Myc protein are represented in boldface. Conserved substituted amino acids in the human Myc sequence are underlined. The residues in dMyc-PII and dMyc-PV that are putative substrates for GSK3β are in italics, and those that are putative substrates for CK1α are double underlined.
That is, the percentage of identical amino acids between D. melanogaster and human Myc protein sequences, calculated using DNA-Strider 1.3.
The stability of dMyc mutants was compared to that of dMyc-WT, which, in our biochemical experiments, was estimated to be 30 min (Fig. 6).
CK1α and GSK3β induce dMyc ubiquitination and degradation through the proteasome pathway.
We then analyzed whether expression of CK1α, GSK3β, or their kinase-dead mutant forms (KD) would affect dMyc protein levels in S2 cells. HA-tagged kinases cloned under the control of the copper-inducible pMT promoter (53) were transfected into S2 cells, and their expression was induced by using CuSO4. dMyc protein level was analyzed in lysates by Western blotting. Figure 2A shows that the dMyc protein levels were considerably reduced in the presence of CK1α or GSK3β kinases. Conversely, expression of the kinase-dead mutants caused the opposite effect and stabilized the dMyc protein. CK1ɛ, another member of the CK1 family of kinases, which sometimes acts as a substitute to CK1α in controlling protein degradation (53), was shown to have an effect similar to that of CK1α or GSK3β on reducing endogenous dMyc protein stability (see Fig. S2 in the supplemental material). This regulation was not exerted at the transcriptional level since dmyc mRNA did not significantly change in the presence of the kinases or their inactive forms (see Fig. S3 in the supplemental material). To further assess the contribution of these kinases to dMyc protein turnover, we used the protein synthesis inhibitor CHX to measure the dMyc half-life in the presence of CK1α and GSK3β or of their kinase-dead forms. S2 cells were treated with CHX, and the level of dMyc protein was analyzed at the indicated time points by Western blotting. The intensity of the respective bands was quantified and is reported in the graph shown in Fig. 2B. CK1α or GSK3β activity decreased the endogenous dMyc half-life from 35 min to ∼15 min. Conversely, expression of the kinase-dead forms of GSK3β or CK1α increased dMyc half-life to 80 and 75 min, respectively. These experiments were repeated at least three times, with similar results.
FIG. 2.

CK1α and GSK3β kinases induce ubiquitin-dependent degradation of dMyc. (A) CK1α and GSK3β regulate endogenous dMyc protein stability. S2 cells were transfected with the indicated HA-tagged kinase mutants and protein expression was induced by using copper sulfate. Their levels of expression were quantified by immunoblotting with anti-HA. Endogenous dMyc protein was quantified by using anti-dMyc antiserum. Actin was used as loading control. Molecular markers are indicated on the right. (B) Half-life of dMyc in the presence of CK1α and GSK3β or their KD mutants. The graph represents the quantification of endogenous dMyc levels after the addition of CHX. Cells were lysed at the indicated time points after CHX treatment, and dMyc expression was visualized by Western blotting with anti-dMyc antibodies. The intensity of the dMyc bands was quantified in pixels from the scanned X-ray film by using Adobe Photoshop. The background was calculated from an equivalent area in each lane and subtracted from the value for dMyc in the respective lanes. Time zero was set at 100. The data are plotted as percentages over the control for each point. These experiments were repeated three times. (C) GSK3β and CK1α kinases mediate dMyc ubiquitination. The stable cell line S2-HA-dMyc (3) was transfected with the various kinases, and expression was induced by using copper ions (3). In the upper panel, HA-dMyc was immunoprecipitated from cell extracts using anti-HA antiserum, and its ubiquitinated forms were visualized using antiubiquitin antiserum. In the lower panel, the expression of HA-dMyc and HA-kinases was visualized using anti-HA antiserum. MG132 was added to the medium at heat shock to avoid Myc degradation.
Since it was demonstrated that phosphorylation of c-Myc by GSK3β allows Myc ubiquitination by the ligase Fbw7, we tested whether the presence of GSK3β or CK1α kinase would also promote dMyc protein ubiquitination. We performed these experiments with the cell line S2-HA-dMyc, which expresses dMyc under the control of the inducible hsp70 promoter (4) and in the presence of MG132 to avoid rapid dMyc degradation. S2-HA-dMyc cells were transfected with the various wild-type kinases or the inactive mutants, and their expression was induced with CuSO4. Subsequently, a 1-h heat shock was applied to induce expression of HA-dMyc. Cells were lysed, dMyc was immunoprecipitated with anti-dMyc monoclonal antibodies, and ubiquitination levels were analyzed by Western blotting. Expression of CK1α or GSK3β kinases favored dMyc ubiquitination, whereas expression of the kinase-dead mutants blocked this process. Taken together, our data indicate that members of the CK1 family are novel components of the Myc degradation pathways, which, similar to GSK3β, regulate Myc protein stability through the ubiquitin-proteasome signaling.
GSK3β, CK1α, and CK1ɛ control dMyc protein expression in vivo.
To assess a possible role of GSKβ and CK1 kinases in the control of dMyc protein expression in vivo, we analyzed whether reduction or ectopic expression of these kinases affects dMyc protein levels in the clones of cells within the wing imaginal discs of Drosophila third-instar larvae. At this stage of development, dMyc is expressed within all cells of the presumptive notum, hinge, and wing pouch territories (Fig. 3B and 4A to A‴), except for a stripe of cells located along the dorsal-ventral boundary, an area also known as the zone of nonproliferative cells (ZNC), where it is transcriptionally repressed by Wg signaling (Fig. 3) (12, 19, 22).
FIG. 3.
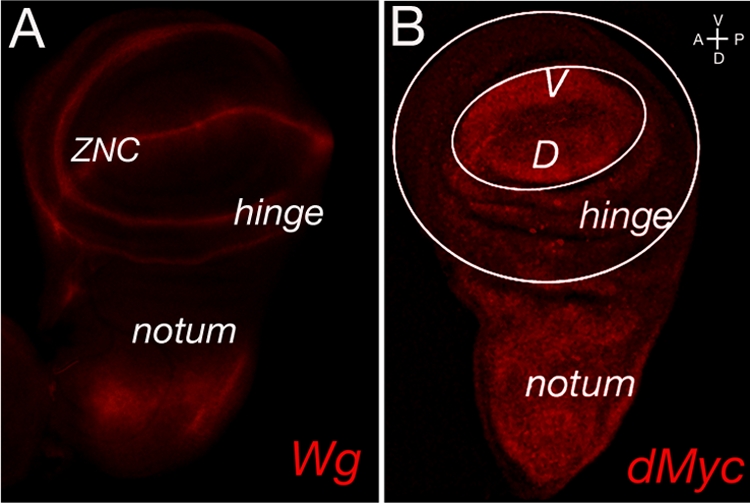
Wg and dMyc protein expression in late third-instar wing imaginal discs. (A) Subdivision of the Drosophila late third-instar wing imaginal disc relative to the Wg expression pattern (red). Two rings of Wg expression mark the presumptive hinge territory; the wing pouch primordium lies within the inner ring, while the notum is formed from tissue outside of the outer ring. Furthermore, Wg is expressed in a thin stripe of cells straddling the boundary between dorsal (D) and ventral (V) compartments, subdividing the prospective wing pouch into dorsal and ventral halves. (B) Late third-instar wing imaginal disc stained for dMyc protein (red). A white circle roughly outlines the boundary between prospective wing pouch and hinge, while a larger white circle distinguishes the prospective hinge area from the prospective notal area. dMyc protein is highly expressed within all cells of the presumptive notum and wing pouch domains except for a stripe of cells extending along the dorsal-ventral boundary. This area of reduced dMyc protein expression corresponds to the ZNC, where dmyc has been showed to be transcriptionally repressed by peak Wg activity associated with the dorsal-ventral boundary. Discs are oriented ventral up and anterior to the left.
FIG. 4.

dMyc is upregulated in sgg/gskβ mutant cells. sgg mutant clones from the third-instar wing imaginal disc were marked by the expression of GFP (D and E) or the protein Ci155, which is expressed autonomously in the anterior compartment of the disc and is upregulated in sgg mutant cells (A′ to C′, D‴, and E‴). Immunostaining of for endogenous dMyc (panels A to C in green and panels D′ and E′ in red) was performed using anti-dMyc antisera. Endogenous dMyc levels are higher in the wing pouch and notum but reduced along the ZNC by a peak activation of the Wg signaling pathway that has been shown to repress dmyc transcription (A). Consistent with this effect, the sgg mutant clones generated within this specific region of the disc are not associated with increased levels of dMyc protein (B, B′, C, and C′, arrowhead). However, accumulation of the dMyc protein is observed in sgg mutant clones located within the presumptive notum and hinge regions (B, C, D′, and E′, arrows). Higher dMyc protein levels are also observed in sgg clones located within the posterior compartment, marked by the absence of Ci (C and E′, asterisk). Discs are oriented ventral up and anterior to the left.
To reduce gene function in clones of cells, we used either mutant alleles (for GSKβ and CK1ɛ) or overexpression of RNAi transgenes (for CK1α) (40). To study ectopic kinase function, we used a combination of the UAS/Gal4 and Flp-out techniques (7, 15, 55) to create clones ectopically expressing a given kinase. The same method was used to ectopically express a given RNAi transgene. All overexpression clones were marked by coexpression of green fluorescent protein (GFP), and all types of clones were monitored for dMyc protein levels by using immunostaining with anti-dMyc antisera (41).
In Drosophila, GSK3β is encoded by the gene shaggy (sgg) (also called zeste-white3 [zw3] and referred to here as sgg). sgg acts in vivo as an inhibitory component of the Wnt/Wingless (Wg) signaling pathway, which regulates a wide variety of developmental processes, ranging from pattern formation to cell fate determination and proliferation (8).
sgg mutant clones were recognized either by GFP expression (Fig. 4D and E) or by increased levels of full-length Ci (Ci155), a component of the Hedgehog (Hh) signaling pathway that is known to accumulate autonomously and at high levels within sgg mutant clones generated within the anterior compartment (Fig. 4A′, B′, C′, D‴, and E‴) (21). As shown in the figure, accumulation of dMyc protein is observed in sgg mutant clones located within the presumptive notum and hinge regions (Fig. 4B, C, D′, and E′, arrows), confirming that Sgg/GSK3β activity is required to regulate dMyc protein expression in vivo. Moreover, our data exclude a role for Hh signaling in the regulation of dMyc expression, since higher dMyc protein levels are also observed in sgg clones located within the posterior compartment, where Hh signaling is not operating due to the absence of Ci (Fig. 4C, C″, and E′, asterisk) (31). Within the wing pouch, peak activation of the Wg signaling pathway has been shown to repress dmyc transcription (12, 19, 22). Consistent with this, we find that sgg mutant clones generated within this specific region of the disc are not associated with increased levels of dMyc protein (Fig. 4B, B′, C, and C′, arrowhead).
To understand how overexpression of Sgg affects dMyc protein stability in vivo, we measured the size of clones of cells that ectopically express UAS-Sgg and compared it to the size of control wild-type clones that only express UAS-GFP (see Fig. S4 in the supplemental material; Table 1). UAS-Sgg clones were much smaller than control wild-type clones, suggesting that they experience a growth disadvantage. This effect can be explained if overexpression of Sgg reduces dMyc protein levels, thereby causing the cells to be eliminated by “cell competition.” Cell competition is a phenomenon arising when two populations of cells with different growth properties are apposed (34). Cells expressing lower dMyc levels grow poorly and are outcompeted and eventually eliminated by cells expressing wild-type dMyc levels (48) (11, 35). Consistent with this effect, we found that coexpression of dMyc and Sgg partially rescues the growth disadvantage of clones overexpressing Sgg alone (see Fig. S4 in the supplemental material; Table 1). Nevertheless, these UAS-Sgg; UAS-dMyc clones were smaller in size than control wild-type or UAS-dMyc clones, further suggesting that overexpression of Sgg reduces dMyc protein expression.
Next, we analyzed how CK1s regulate dMyc expression in vivo. CK1s are believed to modulate the Wingless signaling cascade (26); however, their effect on this pathway has been subject to controversy.
CK1α acts as a negative regulator of Wnt/Wg signaling by phosphorylating β-catenin/Armadillo, thus priming it for phosphorylation by GSK3β and ensuing degradation (1).
CK1ɛ, on the other hand, has been identified as a positive regulator of Wnt/Wg signaling in mammals and Xenopus since it destabilizes the β-catenin degradation complex through the phosphorylation of Dishevelled (9, 14, 38).
Zhang et al. demonstrated that the combination of a hypomorphic ck1ɛ mutation and ectopic expression of RNAi-ck1α enhances ectopic Wg signaling during Drosophila limb formation, suggesting a synergistic interaction between the two kinases (56).
The Drosophila homologue of ck1ɛ is called doubletime (dbt) for its effect on the degradation of Period, a regulator of the circadian clock (39), or disc overgrown (dco) because of the hyperplastic growth of dco3 mutant imaginal discs (57). First, we analyzed clones of cells mutant for the dco3 allele. We observed that dco3 clones (marked by the absence of GFP) were larger than their associated wild-type sibling clones (marked by strong GFP expression) (Fig. 5). In particular, clones generated with the presumptive notum/hinge territory autonomously accumulate high levels of dMyc protein (right panel), indicating that CK1ɛ activity negatively regulates dMyc expression within this region. The same was also suggested by the behavior of clones of cells that ectopically express DCO/CK1ɛ: they were significantly smaller than control wild-type clones that ectopically expressed only GFP (see Fig. S4 in the supplemental material; Table 1), and were associated with reduced levels of endogenous dMyc protein (data not shown). Next, we generated clones of cells ectopically expressing UAS-DCO/CK1ɛ, together with UAS-dMyc, and compared them to clones ectopically expressing UAS-DCO/CK1ɛ alone or UAS-dMyc alone. The sizes of the clones were measured, and their dMyc protein levels were examined. UAS-DCO/CK1ɛ; UAS-dMyc clones were larger than UAS-DCO/CK1ɛ clones but smaller than UAS-dMyc clones. In addition, UAS-DCO/CK1ɛ; UAS-dMyc clones revealed less ectopic dMyc protein than UAS-dMyc clones (see Fig. S4 in the supplemental material; Table 1), further confirming that DCO/CK1ɛ downregulates dMyc protein expression.
FIG. 5.

dMyc is upregulated in cells with reduced levels of dco3/ck1ɛ and ck1α. (A) Expression of dMyc protein (red) in dco3/ck1ɛ mitotic clones from wing imaginal discs. Clones were induced at 65 ± 2 h AEL by mitotic recombination. dMyc protein was analyzed in dco3/ck1ɛ mutant clones (GFP negative) and compared to levels observed in sibling clones (marked with GFP2). A higher magnification of the area (on the right) shows an increase in dMyc protein level in dco3/ck1ɛ clones (marked with a white line). (B) Flp-out clones in the wing imaginal discs expressing UAS-RNAi-ck1α were induced at 52 ± 2 h AEL. Expression of endogenous dMyc in these clones (marked by GFP) was visualized by immunostaining with anti-dMyc antiserum (red). Expression of dMyc is higher in clones located in the hinge region (arrow) and lower in the ZNC (arrowhead). Discs are oriented ventral up and anterior to the left.
Finally, we generated clones of cells ectopically expressing RNAi-ck1α (40) and tested them for dMyc protein levels. UAS-RNAi-ck1α clones, when located within the presumptive notum and hinge domains, showed autonomous upregulation of dMyc protein levels (Fig. 5, right panel, arrow). In contrast, dMyc protein levels were reduced in clones that were generated with the presumptive wing pouch, in agreement with the loss of ck1α causing ectopic activation of Wg signaling and thus the repression of dmyc transcription within this region (12, 19, 22).
Taken together, our data suggest that within tissues in which Wg signaling is not involved in the repression of dmyc transcription (as in presumptive hinge and notum), both CK1ɛ and CK1α, as well as Sgg/GSK3β, are directly implicated in the downregulation of dMyc protein.
In vitro characterization of dMyc phosphorylation mutants reveals that dMyc-PII and AB mutants are resistant to CK1α and GSK3β degradation.
Analysis of the dMyc amino acid sequence reveals the presence of conserved motifs that are potential substrates for phosphorylation by CK1s and GSK3β (Fig. 1A). In order to understand the contribution of these domains to dMyc protein stability, we performed site-directed mutagenesis and replaced serine and threonine residues within the dMyc-PI, -PII, and -PV boxes with alanine. We also converted the AB glutamic acid and aspartate residues into glycine and asparagine, respectively (Table 1). All dMyc mutants were tagged with the hemagglutinin (HA) epitope at their N termini and expressed in S2 cells using the UAS/Gal4 method. The dMyc mutants' half-life was measured biochemically in Western blot experiments using the protein synthesis inhibitor CHX. Cells were transfected with dMyc-WT, the various mutants were treated with CHX, and extracts were prepared at the indicated times after treatment. The expression level of the dMyc mutants was quantified by Western blotting with anti-HA antibodies. Mutation of the residues within the dMyc-PI domain shortened dMyc half-life from 30 to 15 min (Fig. 6, upper panel). Conversely, mutations within the dMyc-PII and dMyc-PV domains lengthened dMyc half-life up to 60 min for PII and to over 180 min for PV. Similarly, substitution of acidic amino acids in the AB region increased the Myc protein half-life to more than 180 min (Fig. 6, lower panel), suggesting an important role for the acidic domain in the regulation of dMyc stability.
FIG. 6.

Stability of dMyc phosphorylation mutants. The half-life of dMyc mutant protein was determined in S2 cells transfected with the indicated constructs and treated with CHX for various lengths of time. Protein levels were quantified by Western blotting with anti-HA antibody. Actin was used as a loading control.
Next, we analyzed whether the newly identified dMyc domains associated with its protein stability were sensitive to CK1α and GSK3β kinase-induced degradation. dMyc-WT, dMyc-PI, dMyc-PII, dMyc-PV, and dMyc-AB were coexpressed in S2 cells, together with increasing concentrations of CK1α or GSK3β kinases. Expression of the kinases was induced with copper sulfate, and the dMyc protein level was analyzed by Western blotting with anti-HA antiserum. As shown in Fig. 7, dMyc-PII and dMyc-AB were resistant to degradation induced by CK1α and GSK3β kinases, while dMyc-PI and dMyc-PV presented degradation kinetics similar to those of dMyc-WT.
FIG. 7.

Stability of dMyc-WT and mutants in the presence of GSK3β and CK1α kinases. HA-dMyc-WT and mutants were transfected in S2 cells together with the indicated concentrations (in μg) of plasmids encoding HA-CK1α or HA-GSK3β kinases. Proteins expression was analyzed in the cell extracts by Western blotting with anti-HA antiserum. Actin was used as loading control.
These data suggest that dMyc-PII and the AB contain functional domains necessary for the regulation of dMyc protein stability, and this effect is regulated by CK1α and GSK3β kinases.
Increased dMyc stability inhibits ommatidial differentiation and induces cell death during eye development.
To characterize the physiological relevance of dMyc mutants in vivo, we used Gal4 promoter lines to express various UAS-dMyc transgenes in different tissues. dMyc-PI-overexpressing flies, and to lesser extent, dMyc-WT-overexpressing flies were viable with all of the tested promoters, while expression the dMyc-PII, dMyc-PV, or dMyc-AB mutants resulted in developmental lethality (see Table S2 in the supplemental material). To overcome the lethality of our transgenes, we restrict their expression to the eye, using flies with reduced dmyc levels (dmycP0), which allows for expression of the transgenes under the eyeless compartment (4).
Since expression of dMyc during compound eye formation modulates the growth and the number of ommatidia (22), we performed genetic epistasis experiments to assess the contribution of various UAS-dMyc mutants to the size and number of ommatidia. Expression of dMyc-PI increased ommatidial size, but to a lesser extent than expression of dMyc-WT (317 μm2 in ey>dmycP0/Y; dMyc-PI/+ and 341 μm2 in ey>dmycP0/Y; dMyc-WT/+) (Fig. 8B). In addition, the total number of ommatidia in ey>dmycP0/Y; dMyc-PI/+ flies (n = 756) was higher than that in ey>dmycP0/Y; dMyc-WT/+ animals (n = 633), suggesting that the expression of dMyc-PI is less effective than the expression of dMyc-WT for inducing apoptosis (33). Conversely, expression of dMyc-PII, dMyc-PV, and dMyc-AB increased the ommatidial size of ey>dmycP0/Y flies (Fig. 8B), with the noteworthy observation that the number of ommatidia was substantially reduced specifically when dMyc-PV and dMyc-AB transgenes were expressed, suggesting that the presence of the stable dMyc mutant proteins resulted in a strong eye defect accompanied by a reduction of the tissue of the head capsule (Fig. 8A). To understand, at the cellular level, the mechanism responsible for these defects, we monitored how the expression of dMyc mutants affected apoptosis (33, 47) and photoreceptor differentiation (2) during the eye development. Eye imaginal discs of the indicated genotypes were collected from third-instar larvae and stained by immunofluorescence for the neuronal markers ELAV (Fig. 9A to F), for dMyc (Fig. 9A′ to F′), and for the cleaved form of caspase-3 (A‴ to F‴). The expression of dMyc-PV or dMyc-AB significantly reduced the number of differentiated photoreceptors (ELAV positive) and increased the number of apoptotic cells (caspase-3 positive). Conversely, expression of dMyc-PI affected neither photoreceptor differentiation nor apoptosis, further confirming that dMyc-PI has a lower transcriptional activity than that of dMyc-WT.
FIG. 8.

Expression of dMyc mutants in vivo affects ommatidial size and number. (A) Representative scanning electron micrographs of adult eyes from ey>dmycP0/Y expressing the transgenes UAS-dMyc-WT, UAS-dMyc-PI, UAS-dMyc-PII, UAS-dMyc-PV, and UAS-dMyc-AB. (B) Graphics show the quantification of ommatidial size and number of the indicated genotypes. Standard deviations were calculated based on the number of animals analyzed and are indicated in parentheses.
FIG. 9.

Expression of dMyc-PV and dMyc-AB inhibits ommatidial differentiation and increases apoptosis in eye imaginal discs. Photographs of eye imaginal discs from third-instar larvae of the indicated genotypes were stained for ELAV (green) and dMyc (red) expression (A to F). Apoptosis is visualized by using anti-active caspase 3 antibodies (white) and nuclei stained with DAPI (A‴ to F‴). Photographs were taken at ×20 magnification. The morphogenetic furrow is indicated by an arrowhead.
Taken together, the in vivo data and the biochemical analysis in S2 cells indicate that dMyc-PII, dMyc-PV, and dMyc-AB mutants are more stable than dMyc-WT, and they also illustrate how the modulation of dMyc stability critically impacts patterning in vivo.
DISCUSSION
Myc is a member of a family of transcription factors and plays a pivotal role in the regulation of proliferation, growth, and apoptosis. Myc protein expression is tightly regulated, and its stability is modulated by phosphorylation events downstream of mitotic signals. Here, we showed that members of the CK1 family are novel regulatory components of Myc protein stability. Our biochemical data demonstrated that CK1α induces dMyc ubiquitination and degradation through the proteasome pathway. Further, we showed that the specific proteasome degradation pathway induced by CK1α-mediated dMyc phosphorylation is the same as the one induced by GSK3β phosphorylation (Fig. 1 and 2), a kinase that was previously identified as a key regulator of c-Myc protein degradation (46).
In vivo downregulation of GSK3β and CK1α or CK1ɛ kinases in wing imaginal discs results in the accumulation of dMyc protein, an effect particularly visible in the hinge and notum regions but not in cells adjacent to the ZNC (Fig. 4 and 5). Reduction of GSK3β and CK1α activates Wingless (Wg) signaling (28), which in turns negatively regulates dmyc RNA in the ZNC (12, 19, 22). This functional relationship might explain the lack of expression of dMyc protein in clones falling in the wing pouch area and in the ZNC. This positional effect also suggests that dMyc activity is regulated by patterning signals active during the development of the wing imaginal discs.
Our analysis of the dMyc amino acid sequence uncovered novel conserved domains, which serve as potential phosphorylation substrates for CK1s or GSK3β kinases. Biochemical characterization of these domains indicated that a combination of amino acid substitutions (S201A, S205A, and S207A) in the dMyc-PI sequence produces a protein with a shorter half-life than dMyc-WT (Table 1). In vivo expression of the dMyc-MPI mutant did not confer the typical ommatidial roughness that is induced by the expression of dMyc-WT. Moreover, expression of dMyc-PI failed to induce apoptosis in the eye imaginal discs, an effect normally associated with dMyc-WT overexpression (Fig. 9). In conclusion, our data suggest that dMyc-PI produces a protein that is less stable than dMyc-WT. In vertebrates, phosphorylation of c-Myc on Ser-62 by MAPK/ERK, JNK N-terminal kinase, or CDK4 increases its stability (18, 46). The dMyc-PI sequence does not contain a bona fide ERK phosphorylation site (PXSP). However, Ser-201 lies in a favorable context for phosphorylation by the ribosomal S6 kinase-p90 RSK (37). RSK-p90 belongs to a class of Ser/Thr kinases, activated by ERK (20) and insulin signaling (23, 43), that phosphorylates the S6 protein component of the 40S ribosomal subunit in response to mitogenic stimulation, resulting in enhanced translation (24). Interestingly, it has been reported that RSK-p90 activation by ERK is capable of switching on mTOR signaling via inactivation of the TSC1/2 complex (27), suggesting a role for this kinase in protein synthesis and mass accumulation. No evidence for this regulatory mechanism has been described thus far in Drosophila. We hypothesize that growth factors may stabilize Myc protein, possibly through phosphorylation by the RSK-p90 kinase, and promote ribosomal biogenesis, in accordance with the prominent role played by dMyc in the production of mass and growth regulation (17).
Biochemical analysis of the protein stability of dMyc-PII, dMyc-PV, and dMyc-AB showed an increased half-life of these mutants compared to dMyc-WT. The sequence within the dMyc-PII domain (S324A-T328A-S330A) contains potential targets for phosphorylation by GSK3β at Ser-324 [324-S/T-XXX-S/T-(PO4)+4], which requires a priming event of phosphorylation at the +4 position (Thr-330) (10). This phosphorylation event also acts as priming for other kinases (i.e., CK1s) and creates an optimum consensus site for phosphorylation by CK1s at Thr-330 [S/T-(PO4)-XX-330-S/T] (30). We found that alanine substitutions of amino acids 324, 328, and 330 conferred resistance to dMyc protein degradation upon phosphorylation by the CK1α and GSK3β kinases (Fig. 7). Previously, Moberg et al. proposed Ser-324 to be a phosphorylation target for GSK3β and to act as a potential binding site for the ubiquitin ligase Ago, the Drosophila homologue of the human Fbw7 ligase (32). These authors found that mutation at Ser-324 did not decrease the capacity of dMyc to bind Ago, nor did it influence dMyc protein stability (32). Our experiments show that mutation of the residues S324, T328, and S330 confers to the dMyc-PII mutant a resistance to degradation mediated by the ubiquitin ligase Ago. Moreover, we found that dMyc-MPV, which is degraded by CK1α and GSK3β kinases, is somewhat resistant to degradation by Ago (see Fig. S5 in the supplemental material), suggesting that CK1α- and GSK3β-mediated phosphorylation of dMyc is not sufficient to induce its degradation by Ago but perhaps by another unknown ubiquitin ligase.
Our data also demonstrate that the dMyc-AB plays an important role in the regulation of dMyc protein stability. Mutation of acidic amino acids imparted to dMyc resistance to degradation primed by CK1α and GSK3β kinases (Fig. 7). It was recently proposed that acidic domains act as docking sites for the CK1 and CK2, enabling proper positioning of the kinases to recognize their substrates. We speculate that the conserved acidic amino acid stretch in Myc protein helps the binding of CK1 and CK2 kinases and favors Myc phosphorylation (30). In support of this hypothesis, we found the dMyc-PV amino acid sequence (residues 405, 407, and 409), located within the AB (amino acids 404 to 414), to be highly homologous to the PEST domain of c-Myc (amino acids 226 to 270). This domain was previously demonstrated to be relevant for c-Myc stability and to act as a potential substrate for CK2 phosphorylation (29). Our biochemical data show that mutations of the dMyc-PV and the AB domains confer increased stability to dMyc protein and suggest that the acidic sequence functions similarly to the PEST domain to control dMyc stability. Notably, Ser-407 constitutes an optimum consensus site for phosphorylation by CK2 (S/T-407-XX-D/E) (37). This observation agrees with the hypothesis that in mammals CK2 is involved in the regulation of c-Myc degradation by targeting the PEST domain (29).
In vivo expression of the stable mutants dMyc-PII, dMyc-PV, and dMyc-AB resulted in a visible eye defect (Fig. 8), accompanied by a reduction of the head capsule and a diminution of the number of the ommatidia. This was particularly visible for dMyc-PV and -AB. Cellular analysis of third-instar larvae eye imaginal discs revealed that expression of these mutants induced apoptosis during disc development. Apoptosis was detected not only within the compartment of dMyc expression (cell autonomous) but also in the neighboring cells (non-cell autonomous; Fig. 9). This is a well-documented phenomenon and illustrates the role of dMyc in cell competition, where cells expressing high dMyc kill slower-proliferating neighboring cells nonautonomously through an unidentified mechanism (11, 35, 48).
In conclusion, multiple phosphorylation events may work hierarchically to prime Myc phosphoamino acids for binding by multiple kinases. We propose here that different kinases respond to a “phosphorylation code” that is required to properly control Myc protein stability. This code will depend on an upstream program that in turn activates these kinases. The identification of other phosphorylation residues in dMyc will help in drawing a complete map of phosphorylation activities and will elucidate the events necessary for robust regulation of Myc protein stability. For example, we speculate that components of growth signaling pathways, such as ras or insulin, may influence the activities of different combinations of kinases, thus affecting phosphorylation at different amino acids to control dMyc protein stability. In support of this hypothesis, we produced preliminary data showing that activation of the DILP (for Drosophila insulinlike peptides) pathway increases dMyc protein stability in vivo through the inactivation of GSK3β kinase (F. Parisi, unpublished data), suggesting that the metabolic and nutrient pathways affect growth by partially controlling dMyc protein expression.
Supplementary Material
Acknowledgments
We thank Shin-ichi Yanagawa and Kennet Moberg for plasmids; Daniel Fimiartz for confocal imaging; Jorge Morales for electron microscopy support; and Peter Gallant, Nathalie Methot, and Myriam Zecca for critical reading of the manuscript. We thank the Developmental Studies Hybridoma Bank under the auspices of NICHD for antibodies.
This study was supported by the National Center for Research Resources (NIH 5G12RR03060), by a Public Health Service grant from the NIH (5SC1DK085047-1) and the Research Foundation RF-CUNY (P.B.), and by the CaRisBo Foundation (S.R.)
Footnotes
Published ahead of print on 13 April 2009.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Amit, S., A. Hatzubai, Y. Birman, J. S. Andersen, E. Ben-Shushan, M. Mann, Y. Ben-Neriah, and I. Alkalay. 2002. Axin-mediated CKI phosphorylation of beta-catenin at Ser 45: a molecular switch for the Wnt pathway. Genes Dev. 161066-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Basler, K., and E. Hafen. 1991. Specification of cell fate in the developing eye of Drosophila. Bioessays 13621-631. [DOI] [PubMed] [Google Scholar]
- 3.Bellosta, P., M. Costa, D. A. Lin, and C. Basilico. 1995. The receptor tyrosine kinase ARK mediates cell aggregation by homophilic binding. Mol. Cell. Biol. 15614-625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bellosta, P., T. Hulf, S. Balla Diop, F. Usseglio, J. Pradel, D. Aragnol, and P. Gallant. 2005. Myc interacts genetically with Tip48/Reptin and Tip49/Pontin to control growth and proliferation during Drosophila development. Proc. Natl. Acad. Sci. USA 10211799-11804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Benassayag, C., L. Montero, N. Colombie, P. Gallant, D. Cribbs, and D. Morello. 2005. Human c-Myc isoforms differentially regulate cell growth and apoptosis in Drosophila melanogaster. Mol. Cell. Biol. 259897-9909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernard, S., and M. Eilers. 2006. Control of cell proliferation and growth by Myc proteins. Results Probl. Cell Differ. 42329-342. [DOI] [PubMed] [Google Scholar]
- 7.Brand, A. H., and N. Perrimon. 1993. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118401-415. [DOI] [PubMed] [Google Scholar]
- 8.Cohen, P., and S. Frame. 2001. The renaissance of GSK3. Nat. Rev. Mol. Cell Biol. 2769-776. [DOI] [PubMed] [Google Scholar]
- 9.Cong, F., L. Schweizer, and H. Varmus. 2004. Casein kinase Iepsilon modulates the signaling specificities of dishevelled. Mol. Cell. Biol. 242000-2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dajani, R., E. Fraser, S. M. Roe, N. Young, V. Good, T. C. Dale, and L. H. Pearl. 2001. Crystal structure of glycogen synthase kinase 3 beta: structural basis for phosphate-primed substrate specificity and autoinhibition. Cell 105721-732. [DOI] [PubMed] [Google Scholar]
- 11.de la Cova, C., M. Abril, P. Bellosta, P. Gallant, and L. A. Johnston. 2004. Drosophila myc regulates organ size by inducing cell competition. Cell 117107-116. [DOI] [PubMed] [Google Scholar]
- 12.Duman-Scheel, M., L. A. Johnston, and W. Du. 2004. Repression of dMyc expression by Wingless promotes Rbf-induced G1 arrest in the presumptive Drosophila wing margin. Proc. Natl. Acad. Sci. USA 1013857-3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gallant, P., Y. Shiio, P. F. Cheng, S. M. Parkhurst, and R. N. Eisenman. 1996. Myc and Max homologs in Drosophila. Science 2741523-1527. [DOI] [PubMed] [Google Scholar]
- 14.Gao, Z. H., J. M. Seeling, V. Hill, A. Yochum, and D. M. Virshup. 2002. Casein kinase I phosphorylates and destabilizes the beta-catenin degradation complex. Proc. Natl. Acad. Sci. USA 991182-1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Golic, K. G. 1991. Site-specific recombination between homologous chromosomes in Drosophila. Science 252958-961. [DOI] [PubMed] [Google Scholar]
- 16.Gregory, M. A., and S. R. Hann. 2000. c-Myc proteolysis by the ubiquitin-proteasome pathway: stabilization of c-Myc in Burkitt's lymphoma cells. Mol. Cell. Biol. 202423-2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grewal, S. S., L. Li, A. Orian, R. N. Eisenman, and B. A. Edgar. 2005. Myc-dependent regulation of rRNA synthesis during Drosophila development. Nat. Cell Biol. 7295-302. [DOI] [PubMed] [Google Scholar]
- 18.Hann, S. R. 2006. Role of posttranslational modifications in regulating c-Myc proteolysis, transcriptional activity and biological function. Semin. Cancer Biol. 16288-302. [DOI] [PubMed] [Google Scholar]
- 19.Herranz, H., L. Perez, F. A. Martin, and M. Milan. 2008. A Wingless and Notch double-repression mechanism regulates G1-S transition in the Drosophila wing. EMBO J. 271633-1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ikuta, M., M. Kornienko, N. Byrne, J. C. Reid, S. Mizuarai, H. Kotani, and S. K. Munshi. 2007. Crystal structures of the N-terminal kinase domain of human RSK1 bound to three different ligands: implications for the design of RSK1 specific inhibitors. Protein Sci. 162626-2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jia, J., K. Amanai, G. Wang, J. Tang, B. Wang, and J. Jiang. 2002. Shaggy/GSK3 antagonizes Hedgehog signalling by regulating Cubitus interruptus. Nature 416548-552. [DOI] [PubMed] [Google Scholar]
- 22.Johnston, L. A., D. A. Prober, B. A. Edgar, R. N. Eisenman, and P. Gallant. 1999. Drosophila myc regulates cellular growth during development. Cell 98779-790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim, M., J. H. Lee, H. Koh, S. Y. Lee, C. Jang, C. J. Chung, J. H. Sung, J. Blenis, and J. Chung. 2006. Inhibition of ERK-MAP kinase signaling by RSK during Drosophila development. EMBO J. 253056-3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim, S. G., and S. J. Lee. 2007. PI3K, RSK, and mTOR signal networks for the GST gene regulation. Toxicol. Sci. 96206-213. [DOI] [PubMed] [Google Scholar]
- 25.Kim, S. Y., A. Herbst, K. A. Tworkowski, S. E. Salghetti, and W. P. Tansey. 2003. Skp2 regulates Myc protein stability and activity. Mol. Cell 111177-1188. [DOI] [PubMed] [Google Scholar]
- 26.Knippschild, U., S. Wolff, G. Giamas, C. Brockschmidt, M. Wittau, P. U. Wurl, T. Eismann, and M. Stoter. 2005. The role of the casein kinase 1 (CK1) family in different signaling pathways linked to cancer development. Onkologie 28508-514. [DOI] [PubMed] [Google Scholar]
- 27.Kwiatkowski, D. J., and B. D. Manning. 2005. Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum. Mol. Genet. 2(14 Spec. No.)R251-R258. [DOI] [PubMed] [Google Scholar]
- 28.Liu, C., Y. Li, M. Semenov, C. Han, G. H. Baeg, Y. Tan, Z. Zhang, X. Lin, and X. He. 2002. Control of beta-catenin phosphorylation/degradation by a dual-kinase mechanism. Cell 108837-847. [DOI] [PubMed] [Google Scholar]
- 29.Luscher, B., E. A. Kuenzel, E. G. Krebs, and R. N. Eisenman. 1989. Myc oncoproteins are phosphorylated by casein kinase II. EMBO J. 81111-1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marin, O., V. H. Bustos, L. Cesaro, F. Meggio, M. A. Pagano, M. Antonelli, C. C. Allende, L. A. Pinna, and J. E. Allende. 2003. A noncanonical sequence phosphorylated by casein kinase 1 in β-catenin may play a role in casein kinase 1 targeting of important signaling proteins. Proc. Natl. Acad. Sci. USA 10010193-10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Methot, N., and K. Basler. 2001. An absolute requirement for Cubitus interruptus in Hedgehog signaling. Development 128733-742. [DOI] [PubMed] [Google Scholar]
- 32.Moberg, K. H., A. Mukherjee, A. Veraksa, S. Artavanis-Tsakonas, and I. K. Hariharan. 2004. The Drosophila F box protein archipelago regulates dMyc protein levels in vivo. Curr. Biol. 14965-974. [DOI] [PubMed] [Google Scholar]
- 33.Montero, L., N. Muller, and P. Gallant. 2008. Induction of apoptosis by Drosophila Myc. Genesis 46104-111. [DOI] [PubMed] [Google Scholar]
- 34.Morata, G., and F. A. Martin. 2007. Cell competition: the embrace of death. Dev. Cell 131-2. [DOI] [PubMed] [Google Scholar]
- 35.Moreno, E., and K. Basler. 2004. dMyc transforms cells into super-competitors. Cell 117117-129. [DOI] [PubMed] [Google Scholar]
- 36.Noguchi, K., C. Kitanaka, H. Yamana, A. Kokubu, T. Mochizuki, and Y. Kuchino. 1999. Regulation of c-Myc through phosphorylation at Ser-62 and Ser-71 by c-Jun N-terminal kinase. J. Biol. Chem. 27432580-32587. [DOI] [PubMed] [Google Scholar]
- 37.Obenauer, J. C., L. C. Cantley, and M. B. Yaffe. 2003. Scansite 2.0: proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 313635-3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peters, J. M., R. M. McKay, J. P. McKay, and J. M. Graff. 1999. Casein kinase I transduces Wnt signals. Nature 401345-350. [DOI] [PubMed] [Google Scholar]
- 39.Price, J. L., J. Blau, A. Rothenfluh, M. Abodeely, B. Kloss, and M. W. Young. 1998. double-time is a novel Drosophila clock gene that regulates PERIOD protein accumulation. Cell 9483-95. [DOI] [PubMed] [Google Scholar]
- 40.Price, M. A., and D. Kalderon. 2002. Proteolysis of the Hedgehog signaling effector Cubitus interruptus requires phosphorylation by glycogen synthase kinase 3 and casein kinase 1. Cell 108823-835. [DOI] [PubMed] [Google Scholar]
- 41.Prober, D. A., and B. A. Edgar. 2002. Interactions between Ras1, dMyc, and dPI3K signaling in the developing Drosophila wing. Genes Dev. 162286-2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prober, D. A., and B. A. Edgar. 2000. Ras1 promotes cellular growth in the Drosophila wing. Cell 100435-446. [DOI] [PubMed] [Google Scholar]
- 43.Rintelen, F., H. Stocker, G. Thomas, and E. Hafen. 2001. PDK1 regulates growth through Akt and S6K in Drosophila. Proc. Natl. Acad. Sci. USA 9815020-15025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sears, R., G. Leone, J. DeGregori, and J. R. Nevins. 1999. Ras enhances Myc protein stability. Mol. Cell 3169-179. [DOI] [PubMed] [Google Scholar]
- 45.Sears, R., F. Nuckolls, E. Haura, Y. Taya, K. Tamai, and J. R. Nevins. 2000. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 142501-2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sears, R. C. 2004. The life cycle of C-myc: from synthesis to degradation. Cell Cycle 31133-1137. [PubMed] [Google Scholar]
- 47.Secombe, J., L. Li, L. Carlos, and R. N. Eisenman. 2007. The Trithorax group protein Lid is a trimethyl histone H3K4 demethylase required for dMyc-induced cell growth. Genes Dev. 21537-551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Senoo-Matsuda, N., and L. A. Johnston. 2007. Soluble factors mediate competitive and cooperative interactions between cells expressing different levels of Drosophila Myc. Proc. Natl. Acad. Sci. USA 10418543-18548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trumpp, A., Y. Refaeli, T. Oskarsson, S. Gasser, M. Murphy, G. R. Martin, and J. M. Bishop. 2001. c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 414768-773. [DOI] [PubMed] [Google Scholar]
- 50.Vervoorts, J., J. Luscher-Firzlaff, and B. Luscher. 2006. The ins and outs of MYC regulation by posttranslational mechanisms. J. Biol. Chem. 28134725-34729. [DOI] [PubMed] [Google Scholar]
- 51.von der Lehr, N., S. Johansson, S. Wu, F. Bahram, A. Castell, C. Cetinkaya, P. Hydbring, I. Weidung, K. Nakayama, K. I. Nakayama, O. Soderberg, T. K. Kerppola, and L. G. Larsson. 2003. The F-box protein Skp2 participates in c-Myc proteosomal degradation and acts as a cofactor for c-Myc-regulated transcription. Mol. Cell 111189-1200. [DOI] [PubMed] [Google Scholar]
- 52.Xu, T., and G. M. Rubin. 1993. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development 1171223-1237. [DOI] [PubMed] [Google Scholar]
- 53.Yanagawa, S., Y. Matsuda, J. S. Lee, H. Matsubayashi, S. Sese, T. Kadowaki, and A. Ishimoto. 2002. Casein kinase I phosphorylates the Armadillo protein and induces its degradation in Drosophila. EMBO J. 211733-1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yeh, E., M. Cunningham, H. Arnold, D. Chasse, T. Monteith, G. Ivaldi, W. C. Hahn, P. T. Stukenberg, S. Shenolikar, T. Uchida, C. M. Counter, J. R. Nevins, A. R. Means, and R. Sears. 2004. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat. Cell Biol. 6308-318. [DOI] [PubMed] [Google Scholar]
- 55.Zecca, M., K. Basler, and G. Struhl. 1996. Direct and long-range action of a wingless morphogen gradient. Cell 87833-844. [DOI] [PubMed] [Google Scholar]
- 56.Zhang, L., J. Jia, B. Wang, K. Amanai, K. A. Wharton, Jr., and J. Jiang. 2006. Regulation of wingless signaling by the CKI family in Drosophila limb development. Dev. Biol. 299221-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zilian, O., E. Frei, R. Burke, D. Brentrup, T. Gutjahr, P. J. Bryant, and M. Noll. 1999. double-time is identical to discs overgrown, which is required for cell survival, proliferation and growth arrest in Drosophila imaginal discs. Development 1265409-5420. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.