

Abstract
Protein Nα-terminal acetylation is one of the most common protein modifications in eukaryotic cells. In yeast, three major complexes, NatA, NatB, and NatC, catalyze nearly all N-terminal acetylation, acetylating specific subsets of protein N termini. In human cells, only the NatA and NatB complexes have been described. We here identify and characterize the human NatC (hNatC) complex, containing the catalytic subunit hMak3 and the auxiliary subunits hMak10 and hMak31. This complex associates with ribosomes, and hMak3 acetylates Met-Leu protein N termini in vitro, suggesting a model in which the human NatC complex functions in cotranslational N-terminal acetylation. Small interfering RNA-mediated knockdown of NatC subunits results in p53-dependent cell death and reduced growth of human cell lines. As a consequence of hMAK3 knockdown, p53 is stabilized and phosphorylated and there is a significant transcriptional activation of proapoptotic genes downstream of p53. Knockdown of hMAK3 alters the subcellular localization of the Arf-like GTPase hArl8b, supporting that hArl8b is a hMak3 substrate in vivo. Taken together, hNatC-mediated N-terminal acetylation is important for maintenance of protein function and cell viability in human cells.
Approximately 50% of yeast proteins and 80% of mammalian proteins are N-terminally acetylated (9). The impact of this modification on a large range of cellular processes, including links to cancer development, is being increasingly recognized. However, knowledge about the functional importance and underlying mechanisms of protein Nα-terminal acetylation is still limited.
In yeast, three NAT complexes, NatA, NatB, and NatC, account for most of the protein Nα-terminal acetylation in what is believed to be a cotranslational process. They acetylate different subsets of proteins as defined by the sequence and nature of the N-terminal amino acid residues of the substrates (18, 28, 29).
Yeast NatC contains the catalytic subunit Mak3p and the auxiliary subunits Mak10p and Mak31p, where all three subunits are necessary for NatC activity. The NatC complex potentially acetylates methionine N termini when the second residue is one of the hydrophobic amino acids leucine, isoleucine, phenylalanine, or tryptophan. The mutant yeast strains mak3-Δ, mak10-Δ, and mak31-Δ display similar phenotypes: lack of acetylation of substrates in vivo, diminished growth at 37°C in medium without fermentable carbon sources (30), defective L-A virus propagation (39), and loss of telomere elongation (11). In a screen for conserved synthetic lethal genetic interactions in Saccharomyces cerevisiae and Schizosaccharomyces pombe, MAK10 was found to be part of a core genetic interaction network. In this screen, NatC-mediated acetylation was suggested to be critical for efficient DNA replication in eukaryotes (15). Deletion of MAK10 has also been observed to increase sensitivity to rapamycin (41), indicating a link to the mTOR pathway. Identified NatC substrates include the Gag major coat protein of the L-A virus (35), the tRNA methyltransferase Trm1p-II (25), and the ARF-like GTPase Arl3p (12, 32). For all these substrates, NatC-mediated acetylation is necessary for normal protein activity. In the case of Arl3p, NatC-mediated N-terminal acetylation is important for its Golgi apparatus association (12, 32).
In humans, the NatA and NatB complexes have been identified. The human NatA complex (hNatA) is composed of the hNat1/NATH, hArd1, and hNat5/hSan subunits (2, 3). Ard1, the catalytic subunit of NatA, is essential for growth of Caenorhabditis elegans (33), the parasite Trypanosoma brucei (21), and human cells (6, 16). Human Ard1 has been suggested as a potential cancer drug target (6, 8). The hNatB complex consists of the subunits hNat3 and hMdm20. Knockdown of the hNatB subunits disturbs cell proliferation and cell cycle progression, and hNat3 has been linked to tumor growth (1, 34). A vertebrate NatC complex has been identified, consisting of Mak3, Mak31, and the Mak10p homologue embryonic growth-associated factor. Knockdown of zebrafish embryonic growth-associated factor induced embryonic growth lethality and reduced the protein level and signaling activity of zTOR protein kinase (38).
In the present communication, we describe the hNatC complex. It is evolutionarily conserved from yeast with respect to subunit composition, localization, enzymatic activity, and substrate specificity. Knockdown of individual hNatC subunits leads to cell growth defects and p53-dependent apoptosis. Knockdown of hMAK3 induces a change in the subcellular localization of hArl8b, suggesting that hNatC-mediated acetylation is necessary for hArl8b localization. Our present data suggest an important role for hNatC-mediated acetylation in various cellular processes necessary for normal cell viability in humans.
MATERIALS AND METHODS
Construction of plasmids.
Plasmids encoding Xpress and/or V5-tagged hMak10, hMak3 (Nat12), hMak31 (Lsmd1), and Lsm8 were constructed from cDNA made from total RNA isolated from human HEK293 and HeLa cells. PCR products were inserted into the TOPO TA vectors pcDNA 3.1/V5-His TOPO and pcDNA4/HisMax-TOPO (Invitrogen) according to the instruction manual. SuperScript II reverse transcriptase (Invitrogen) was used to make cDNA. Plasmid encoding Xpress-tagged hNat5 was previously described (3). Plasmid encoding MBP-hMak3 was constructed by subcloning hMak3 from pXpress-hMak3 to the pETM-41 vector. Primer sequence information is available upon request. pETM-41 was generously provided by G. Stier, EMBL, Heidelberg, Germany. hARL8b-GFP plasmid was kindly provided by S. Munro, MRC Laboratory of Molecular Biology, Cambridge, United Kingdom.
Cell culture and transfection.
HeLa cells and HEK293 cells were cultured and transfected as described previously (2). CAL-62 cells (human thyroid anaplastic carcinoma; DSMZ no. ACC 507) were cultured in Dulbecco's modified Eagle's medium with 10% fetal bovine serum. HCT116 (human colon cancer carcinoma) p53+/+ and parental p53−/− cells were kindly provided by F. Bunz, B. Vogelstein, and K. W. Kinzler (Johns Hopkins University School of Medicine and Howard Hughes Medical Institute, MD) and were routinely maintained in McCoy's 5A medium supplemented with 10% normal calf serum. To specifically inhibit caspase activities 20 μM ZVAD-fmk (R&D Systems Europe Ltd.) was added when appropriate. Small interfering RNA (siRNA)-mediated knockdown was performed using Dharmafect 1 transfection reagent (Dharmacon) according to the instruction manual. Gene-specific SMART pool siRNAs were purchased from Dharmacon and used at a final concentration of 20 to 50 nM to silence the hMAK3 (NAT12), hMAK10, and hMAK31 (LSMD1) genes (sihMAK3 [NAT12], catalog no. M-009961-02; sihMAK10, catalog no. L-021268-01; sihMAK31 [LSMD1], catalog no. L-014876-01). Nontargeting pool (catalog no. D-001810-10) and ON-TARGETplus GAPDH control pool (catalog no. D-001830) were used as controls. Where indicated, two individual siRNAs from the sihMAK3 SMART pool were used for knockdown of hMAK3 (Dharmacon catalog nos. D-009961-01 and D-009961-05).
Immunoprecipitation.
HeLa and HEK293 cells were harvested and lysed as described previously (2). The cell lysates were incubated for 2 h at 4°C with specific antibody (2 μg) before 50 μl of protein A/G-agarose was added. After incubation for 16 h, centrifugation, and washing three times in 2× phosphate-buffered saline, the samples were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting.
Isolation of polysomes.
Total ribosome isolation was performed using a modification of previously described methods (27, 37). Approximately 2 × 107 HEK293 cells were used per experiment. Prior to harvesting, cells were treated with 10 μg/ml cycloheximide for 5 min at 37°C. Cells were harvested and lysed with KCl ribosome lysis buffer (1.1% [wt/vol] KCl, 0.15% [wt/vol] triethanolamine, 0.1% [wt/vol] magnesium acetate, 8.6% [wt/vol] sucrose, 0.05% [wt/vol] Na-deoxycholate, 0.5% [vol/vol] Triton X-100, 0.25% [vol/vol] Pefabloc), and incubated on ice for 15 min. After removing nuclei and membranes by centrifugation at 400 × g for 10 min, 700 μl cell lysate was centrifuged at 436,000 × g for 25 min on a 0.4-ml cushion of 25% sucrose in KCl ribosome lysis buffer using an MLA-130 rotor (Beckman, Geneva, Switzerland). Pellets were resuspended in ribosome lysis buffer with the indicated KCl concentrations, followed by ultracentrifugation as described above. Pellets were resuspended in KCl ribosome lysis buffer and prepared for analysis by SDS-PAGE and Western blotting.
Immunofluorescence.
HeLa cells grown on coverslips were washed in phosphate-buffered saline, fixed in methanol, permeabilized in 0.1% Triton X-100, and blocked in 10% bovine serum albumin (BSA). Primary antibodies were anti-V5 or anti-Xpress (Invitrogen). Secondary antibodies were Alexa Fluor 488-, Alexa Fluor 594-, or Texas Red-conjugated immunoglobulin G (Invitrogen). Blue Hoechst 33342 or 4′,6-diamidino-2-phenylindole staining was used to stain the nucleus.
For terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assays, cells were incubated in TUNEL reaction mixture (In Situ Cell Death Detection kit with TMR red; Roche) for 1 h at 37°C. Positive and negative controls for each sample were obtained by treating samples with DNase I or incubation in TUNEL label reaction mixture without enzyme, respectively.
For investigation of hArl8b localization after hMAK3 knockdown, hMAK3 knockdown cells were transfected with hARL8b-GFP. Cells were treated with ZVAD (20 μM) after knockdown of hMAK3 to prevent induction of apoptosis. Anti-LAMP-1 (Santa Cruz Biotechnology) was used as a lysosomal marker. The microscopic recordings were processed by deconvolution (Leica 4000 software). The data for quantification of hArl8b-green fluorescent protein (GFP) localization after hMAK3 knockdown are shown as the means of at least 500 cells counted in five to six samples from three independent experiments.
Western blotting.
SDS-PAGE and Western blotting were performed as described previously (5). The following antibodies were used: anti-V5, anti-Xpress, and anti-HA (Invitrogen); anti-L26 (Novus Biologicals); anti-β-tubulin and anti-Mdm2 clone SMP14 (Sigma); anti-cytochrome c (Santa Cruz Biotechnology); anti-cleaved α-Fodrin (Asp1185), anti-cleaved poly(ADP) ribose polymerase (PARP; Asp214), anti-p53, anti-phospho-p53 (Ser15), and anti-phospho-p53 (Ser37) (Cell Signaling Technology). Anti-hMak3 (Biogenes) was generated by immunizing rabbits with purified hMak3 protein produced in Escherichia coli, followed by immunoglobulin G isolation from the resulting sera. Horseradish peroxidase-linked anti-mouse and anti-rabbit antibodies were from Amersham Biosciensce (Little Chalfont, Buckinghamshire, United Kingdom).
In vitro Nα-acetyltransferase assays.
Expression and purification of maltose binding protein (MBP)-hMak3 was carried out as previously described (7). Acetylation assays were performed with 30 μl MBP-hMak3 (2.78 μM stock), 5 μl synthetic peptide (1 mM stock of custom-made peptides from Biogenes), 5 μl [14C]acetyl coenzyme A (50 μCi, 2.07 GBq/mmol; GE Healthcare), and 250 μl acetylation buffer (50 mM Tris-HCl, pH 8.5, 1 mM dithiothreitol, 800 μM EDTA, 10 mM Na-butyrate, 10% glycerol). The mixture was incubated for 2 h at 37°C with rotation and then added to 250 μl SP Sepharose (50% slurry in 0.5 M acetic acid; Sigma) and incubated on a rotor for 5 min. The mixture was centrifuged and the pellet was washed three times with 0.5 M acetic acid and one time in methanol. Radioactivity in the peptide-containing pellet was determined by scintillation counting. All custom-made peptides contain seven unique amino acids from the N terminus, since these residues are the major determinants for Nα-terminal acetyltransferase specificity. The next 17 amino acids are identical to the ACTH peptide sequence to maintain a positive charge facilitating peptide solubility and effective isolation by cation exchange Sepharose beads. The ACTH-derived lysines were replaced by arginines to eliminate any potential interference from Nɛ acetylation. The peptide sequences were as follows: hArl8b (Q9NVJ2), [H]MLALISRRWGRPVGRRRRPVRVYP[OH]; mTOR (P42345), [H]MLGTGPARWGRPVGRRRRPVRVYP[OH]; hnRNP H (P31943), [H]MLGTEGGRWGRPVGRRRRPVRVYP[OH]; hnRNP F (P52597), [H]MLGPEGGRWGRPVGRRRRPVRVYP[OH]; NF-κB p65 (Q04206), [H]MDELFPLRWGRPVGRRRRPVRVYP[OH]; kinesin KIF4A (O95239), [H]MKEEVKGRWGRPVGRRRRPVRVYP[OH]; high-mobility group protein A1 (P17096), [H]SESSSKSRWGRPVGRRRRPVRVYP[OH].
Semiquantitative and real-time PCR.
Verification of gene-specific silencing after siRNA transfection was done by semiquantitative real-time PCR (RT-PCR) using specific primers (primer sequence information is available upon request). Relative gene expression levels of p53-induced genes in HeLa cells transfected with siRNA against hMAK3 were determined by real-time quantitative PCR. The sequences of the primers used to amplify the fragments of NOXA, KILLER/DR5, and FAS cDNA are available upon request. The primers for amplifying fragments of hMAK3 were designed to target the acetyltransferase domain. We used RPLP2 (ribosomal protein large P2) primers to amplify reference gene. Templates (equal amounts of cDNA) and primers were mixed with components from the LightCycler 480 SYBR green I Master Mix kit (Roche Applied Sciences). Reactions in triplicates were carried out in the LightCycler 480 real-time PCR machine (Roche Applied Science) under the following conditions: initial denaturation at 95°C for 5 min and then 40 cycles of denaturation at 95°C for 10 s, annealing at 54°C for 10 s, and extension at 72°C for 10 s. Melting curves were obtained to examine the purity of amplified products. CP and concentrations of amplicons were calculated with LightCycler 480 software v.1.5.0 SP1 using the second derivative max method. Normalization of obtained data was done as follows: {(concentration of gene [NOXA, KILLER/DR5, FAS, or hMAK3] in a sample)/(concentration of reference gene RPLP2 in a sample)}/{(concentration of gene [NOXA, KILLER/DR5, FAS, or hMAK3] in control)/(concentration of reference gene RPLP2 in the control)}. P values were obtained by independent t test.
Cell cycle analysis.
After harvesting of approximately 1 × 106 cells by trypsin-EDTA treatment, the cells were processed by fluorescence-activated cell sorting analysis as described in previous studies (6).
BrdU incorporation assay.
Cells growing in a 96-well plate were transfected with correspondent siRNAs. After 48 h the proliferation rate was determined by measuring the amount of bromodeoxyuridine (BrdU) incorporated into nuclear DNA using the bromodeoxyuridine incorporation assay (Cell Proliferation ELISA, BrdU, chemiluminescent; Roche Applied Science, Mannheim, Germany) according to the instructions of the manufacturer.
WST assay.
HeLa cells were seeded at 3 × 103 cells per well in 96-well plates and transfected with 50 nM correspondent siRNAs after 16 h. After 72 h water-soluble tetrazolium 1 (WST-1; Roche) was added to each well (1:10) for 1 h and absorbance at 450 nm was determined. Four replicates were used for each sample. Reduction in cell viability was expressed as the ratio of absorbance (sample) versus absorbance (control) multiplied by 100%.
RESULTS
Identification of human NatC complex components.
In the search for human homologues of the yeast NatC components Mak3p, Mak10p, and Mak31p, we used the protein sequences of these as query sequences in BLAST searches against the human proteome. We identified the putative genes encoding human homologues of Mak3p (NAT12; GeneID 122830), Mak10p (MAK10; GeneID 60560), and Mak31p (LSM8; GeneID 51691). Previously, Wenzlau and coworkers presented a vertebrate NatC complex of Mak10, Mak3, and Mak31 proteins (38). They suggested that hMAK31 is LSMD1 (GeneID 84316) and hMAK3 is NAT13/hNAT5 (GeneID 80218). However, hNat5 is the human homologue of yeast Nat5p and the Drosophila melanogaster San, and it was previously identified as the third subunit of the NatA complex (3, 18, 40). Both hNat12 and hNat13/hNat5 are predicted acetyltransferases of the GNAT superfamily. Pair-wise BLAST analysis between the different human candidates and the yeast proteins demonstrated that yeast Mak3p was more similar to hNat12 than to hNat13/hNat5/hSan (data available upon request) and that Lsm8 was more similar to yeast Mak31p than was Lsmd1 (data available upon request). Figure 1 displays alignments of putative Mak3 and Mak31 candidates from different species.
FIG. 1.

Alignment of Mak3p and Mak31p homologues. Identities are given in dark gray. Conservative substitutions are given in light gray. (A) Putative homologues of S. cerevisiae (Sc) Mak3p: human (Hs) Nat12, Mus musculus (Mm) Nat12, S. pombe (Sp) NP_596246, D. melanogaster (Dm) NP_181348, A. thaliana (At) NP_181348, C. elegans (Ce) NP_504411. (B) Putative homologues of yeast Mak31p: Hs Lsm8, Hs Lsmd1, Sp NP588509, Dm NP_647660, M. sativa (Ms) P24715, At NP_187757, Thermoplasma acidophilum (Ta) NP_394385, Ce NP_498708. The alignments were performed using JalView (14) and MAFFT sequence aligner (22).
To determine which of these proteins interacts to form the human NatC complex, the open reading frames encoding the candidate proteins were amplified from HeLa cell cDNA and used to construct plasmids expressing the candidate genes as Xpress- and/or V5-tagged proteins. For hMAK10, a splice event had taken place on the candidate retained from the PCR, resulting in the loss of nucleotides 1465 to 1704 compared to the predicted sequence (data available upon request). As this was the only observed product, possibly representing a biological feature of hMAK10 expression in HeLa cells, the product was further tested as an hNatC subunit.
We tested all components for their ability to associate with hMak10, since the sequence information identified hMak10 as the most likely hNatC subunit compared to the hMak3 and hMak31 candidates. The results demonstrated that hMak10 interacted with hNat12 and Lsmd1. We were not able to detect any interaction between hMak10 and hNat5 or Lsm8 (Fig. 2A). It was technically difficult to detect the Xpress-Lsm8 protein by Western blotting, but the significant expression of Xpress-Lsm8 in HeLa cells was confirmed by immunofluorescence microscopy (data not shown). To confirm that hMak10, hNat12, and Lsmd1 associated to form a complex, we expressed Xpress-hNat12, hMak10-V5, and Lsmd1-V5 in HEK293-cells and used an Xpress antibody for immunoprecipitation. When analyzing the complexes by SDS-PAGE and Western blotting with anti-V5, specific signals were detected for hMak10-V5 and Lsmd1-V5 compared to the control (Fig. 2B). The interaction between hNat12 and Lsmd1 was further confirmed by expressing Lsmd1-V5 in HeLa cells, using a hNat12 antibody for immunoprecipitation. When analyzing the complexes by SDS-PAGE and Western blotting as described above, specific signals for Lsmd1-V5 were detected (data not shown). Due to technical difficulties we were not able to use the hNat12-specific antibody for analyzing the interaction between hNat12 and hMak10. Based on these results, we propose that hNat12 is the hMak3, while Lsmd1 is the hMak31. The names hMak3 and hMak31 will be used in the following discussion of our results.
FIG. 2.

hMak10, hNat12/hMak3, and Lsmd1/hMak31 interact to form a stable complex. (A) HeLa cells expressing hMak10-V5 and Xpress-lacZ (negative control), Xpress-hNat12, Xpress-hNat5, Xpress-Lsm8, or Xpress-Lsmd1 were harvested and the lysates were immunoprecipitated (IP) with anti-Xpress antibody (Invitrogen). Immunoprecipitated proteins were identified by Western blotting using the indicated antibodies. (B) Cells expressing hMak10-V5, Lsmd1-V5, and Xpress-hNat12 or Xpress-lacZ (negative control) were immunoprecipitated as for panel A using an anti-Xpress antibody in the immunoprecipitation. Molecular mass (in kDa) is indicated on the left-hand side. Results shown are representative of more than three independent experiments.
The human NatC subunits are located in the cytoplasm and associated with ribosomes.
To determine the subcellular localization of the hNatC components, HeLa cells were used for immunostaining analysis. hMak3 and hMak10 localized almost exclusively to the cytoplasm while hMak31 also localized to the nucleus (Fig. 3).
FIG. 3.

Subcellular localization of hMak10-V5, Xpress-hMak3, and Xpress-hMak31. HeLa cells were transfected with hMak10-V5 (A and B), Xpress-hMak3 (C and D), or Xpress-hMak31 (E and F). Anti-V5 antibodies and Alexa-594-conjugated anti-mouse antibodies were used to visualize hMak10-V5 (A). Anti-Xpress antibodies and Alexa-488-conjugated anti-mouse antibodies were used to visualize Xpress-hMak3 (C). Anti-Xpress antibodies and Texas Red-conjugated anti-mouse antibodies were used to visualize Xpress-hMak31 (E). 4′,6-diamidino-2-phenylindole staining was used to visualize the nuclei of the cells (B, D, and F). Bar, 25 μm.
The NAT complexes are believed to cotranslationally acetylate nascent polypeptides in yeast, and it has been shown that the NatA complex associates with ribosomes via its Nat1p subunit (18). Similarly, the components hNat1 and hArd1 of hNatA form a complex cosedimenting with ribosomes (2). To investigate the ribosome-binding properties of the human NatC complex, we isolated polysomes from HeLa cells overexpressing the different hNatC subunits. Western blot analysis demonstrated that xp-hMak3, hMak10-V5, and hMak31-V5 cosediment with ribosomal pellets, suggesting that all subunits are stably associated with ribosomes (Fig. 4A). Endogenous hMak3 also cosediments with ribosomal pellets in a salt-sensitive manner (Fig. 4B). These results support the hypothesis that hMak3, hMak10, and hMak31, as a complex, are involved in cotranslational acetylation. In addition, the hNatC subunits were present in the nonribosomal fraction. Thus, they may be dynamically associated with the ribosomes, or they may have functions that are independent of cotranslational acetylation. Interestingly, there seem to be some differences in the amount of bound and unbound forms of the various subunits.
FIG. 4.

hNatC subunits cosediment with polysomal fractions in a salt-sensitive manner. (A) Polysomal pellets from HeLa cells expressing hMak10-V5, Xpress-hMak3, and Xpress-hMak3 were resuspended in buffer containing increasing concentrations of KCl. Cell lysate (L), supernatant post-first ultracentrifugation (S), and polysomal pellets after KCl treatment were analyzed by SDS-PAGE and Western blotting. The membrane was incubated with anti-V5, anti-Xpress, anti-L26 (ribosomal protein), and anti-CytC antibodies. Molecular mass markers (in kDa) are indicated on the left-hand side. (B) For endogenous hMak3, untransfected cells were treated as described for panel A and analyzed with anti-hMak3 antibody. Molecular mass (in kDa) is indicated on the left-hand side. Results shown are representative of more than three independent experiments.
Human Mak3 displays N-terminal acetyltransferase activity.
The yeast NatC complex acetylates Met-Leu/Ile/Phe/Trp N termini. To determine whether the human NatC complex expresses a similar activity, the catalytic subunit hMak3 was expressed with an MBP tag in E. coli and purified using several steps of His affinity column and gel filtration. The purified MBP-hMak3 was assayed for in vitro activity using oligopeptides with various N-terminal amino acid residues as substrates. Significant activity was observed for peptide substrates having the N termini MLALI, MLGTG, and MLGTE. Also, some activity was obtained toward the peptide starting with MLGPE (Fig. 5). The predicted NatA and NatB substrates, SESSS and MDELF, respectively, were not significantly acetylated. Thus, it seems that hMak3 is the catalytic subunit of hNatC and that its substrate specificity is highly conserved from yeast. Interestingly, it is clear from these data that the substrate specificity of NatC at least in part is contained within the catalytic subunit alone.
FIG. 5.

hMak3 displays sequence-specific N-acetyltransferase activity. E. coli-expressed and purified MBP-hMak3 was analyzed for Nα-acetyltransferase activity using peptides differing in their N-terminal residues and [1-14C]acetyl coenzyme A. The acetyl incorporation was determined by isolation of the peptides followed by scintillation counting. Experiments were performed three to five times for each peptide. Error bars indicate standard deviations.
NatC knockdown induces p53-dependent cell death and growth arrest in human cell lines.
To investigate the biological importance of the human NatC complex, we performed siRNA-mediated knockdown of the genes encoding the hNatC subunits hMak10, hMak3, and hMak31. RT-PCR demonstrated efficient knockdown of respective RNA transcripts in HeLa cells (Fig. 6A). Using a BrdU assay we detected a significantly diminished rate of DNA synthesis in cell cultures treated with siRNAs targeting hMAK3, hMAK10, or hMAK31 (Fig. 6B). The WST-1 cell proliferation/viability assay clearly demonstrated that knockdown of all three hNatC subunits reduced cell viability (Fig. 6C). By live cell microscopy, we also observed cell death after treatment with siRNAs targeting hMAK3 (Fig. 6D). To investigate whether caspases were activated as a consequence of a decrease in the hNatC complex level, we assayed for cleavage of the known caspase target PARP. Western blot analysis demonstrated that knockdown of all three hNatC subunits induced PARP cleavage (Fig. 6E). A TUNEL assay confirmed the presence of DNA breaks as a further indication of apoptosis (Fig. 6F).
FIG. 6.

hMAK3, hMAK10, or hMAK31 knockdown induces apoptosis in HeLa cells. (A) Cells cultured in six-well plates were transfected with 50 nM hMAK3, hMAK10, or hMAK31 SMART pool siRNAs or control siRNA (siGAPDH or nontargeting siRNA). After 48 h, total RNA was isolated and processed by RT-PCR with specific primers against the genes of interest. (B) At 48 h posttransfection the cell proliferation rate was determined using a BrdU assay, measuring the amount of bromodeoxyuridine incorporated into nuclear DNA. The results are given as percent mitogenic activity. Error bars (B and C) represent standard deviations. P values for independent t tests for samples versus control are indicated (*, P < 0.001). (C) At 72 h posttransfection the cell viability was measured using a WST assay. The results are given as percent cell viability. P values for independent t tests for samples versus control are indicated (*, P < 0.00001). (D) Live cell imaging of hMAK3 knockdown cells 72 h posttransfection. Arrowheads indicate apoptotic cells. Hoechst 33342 staining was used to stain the nuclei. Phase contrast (PH) was used to visualize cells. (E) PARP cleavage was observed by harvesting cells 72 h posttransfection and analyzing cell lysates by Western blotting. The membrane was incubated in anti-cleaved PARP and anti-β-tubulin (loading control). All experiments were performed a minimum of three times. (F) At 72 h posttransfection hMAK3, hMAK10, or hMAK31 knockdown cells were analyzed for DNA breaks by using a TUNEL assay. Blue Hoechst 33342 staining was used to visualize the nuclei. Cells were visualized by phase contrast (PH).
Furthermore, we performed fluorescence-activated cell sorting cell cycle analysis on hNatC knockdown cells. Here we observed an increase in the sub-G1/G0 fraction, representing nuclear fragmentation (Fig. 7). Knockdown of hMAK3, hMAK10, or hMAK31 in the colon carcinoma cells HCT116 (p53+/+) induced apoptosis (Fig. 7, top panel). However, in similar experiments using HCT116 (p53−/−) cells, apoptosis was not observed, indicating that p53 is essential for the hNatC knockdown-mediated induction of apoptosis (Fig. 7, middle panel). Accumulation of hNatC knockdown cells in the sub-G1/G0 fraction was also observed for HeLa cells (Fig. 7, lower panel). Adding the pan-caspase inhibitor ZVAD reversed the sub-G1/G0 accumulation, supporting that sihNatC-treated cells undergo caspase-mediated cell death (data not shown).
FIG. 7.

Cell cycle analysis of hMAK3, hMAK10, and hMAK31 knockdown cells. Flow cytometric cell cycle analysis results are shown for hMAK3, hMAK10, and hMAK31 knockdown cells at 72 h posttransfection. Experiments were performed three times, and representative values are given.
Western blot assays of HeLa cell extracts 72 h posttransfection demonstrated that hMAK3 knockdown by two different hMAK3-specific siRNAs induced a decrease in Mdm2 levels (Fig. 8A) and a stabilization of p53. The Mdm2 reduction may potentially contribute to the stabilization of p53. Previously, it was demonstrated that phosphorylation of p53 at Ser15 and Ser37 mediates transcriptional activation of p53 (23). The observation that p53 is phosphorylated at Ser37 in hMAK3 knockdown cells points to a possible pathway through which p53 stabilization and/or activation is affected (Fig. 8A). In contrast to daunorubicin-treated cells, Ser15 of p53 was not phosphorylated in sihMAK3-treated cells (Fig. 8A). Apoptosis was confirmed by the detection of cleaved PARP and α-Fodrin. Using anti-hMak3, we confirmed efficient hMAK3 knockdown at the endogenous protein level, and furthermore we demonstrated that daunorubicin-induced apoptosis caused significantly reduced levels of hMak3 protein (Fig. 8A). Daunorubicin treatment did not alter the mRNA levels of hMAK3 (Fig. 8B), suggesting that the daunorubicin-induced reduction of hMak3 protein occurs directly at the protein level. To further elaborate the mechanism through which p53 mediates apoptosis after hNatC mRNA knockdown, we investigated the transcriptional levels of KILLER/DR5, NOXA, and FAS by quantitative PCR (Fig. 8B). These are markers of transcriptionally mediated p53-dependent apoptosis (as reviewed by Chipuk and Green [13]). The levels of KILLER/DR5, NOXA, and FAS were all significantly increased by hMAK3 knockdown, suggesting that sihMAK3 mediates apoptosis through transcriptional regulation of apoptosis genes. We did not detect any increase in levels of mitochondria-associated p53 after hMAK3 knockdown (data not shown). Thus, apoptosis resulting from hMAK3 knockdown does not seem to be mediated through an increase in p53 association with the mitochondria. When exogenous hMak3 was expressed in hMAK3 knockdown cells, phenotype rescue was observed (Fig. 9). This verified that the observed phenotypes are indeed due to the loss of hMak3.
FIG. 8.

hMAK3 knockdown induces apoptosis via p53 stabilization and transcriptional activation. (A) HeLa cells cultured in six-well plates were transfected with 50 nM hMAK3-1 or hMAK3-2 taken from the SMART pool sihMAK3 or with control siRNA (nontargeting siRNA). Daunorubicin (Dau) treatment was used as a positive control for apoptosis. At 72 h posttransfection, cell lysates were analyzed by Western blotting. The membrane was incubated with anti-cleaved α-Fodrin (Asp1185), anti-cleaved PARP (Asp214), anti-Mdm2, anti-p53, anti-phospho-p53 (Ser15), anti-phospho-p53 (Ser37), anti-hMak3, and anti-β-tubulin (loading control). Experiments were performed a minimum of three times, and representative results are shown. (B) Cells were treated as for panel A, but after harvesting, total RNA was isolated and processed by quantitative RT-PCR with gene-specific primers against hMAK3, KILLER/DR5, FAS, and NOXA. Error bars indicate standard deviations. P values for independent t tests for samples versus control are indicated (*, P < 0.0001). Values indicated in the diagram are given in the table below.
FIG. 9.
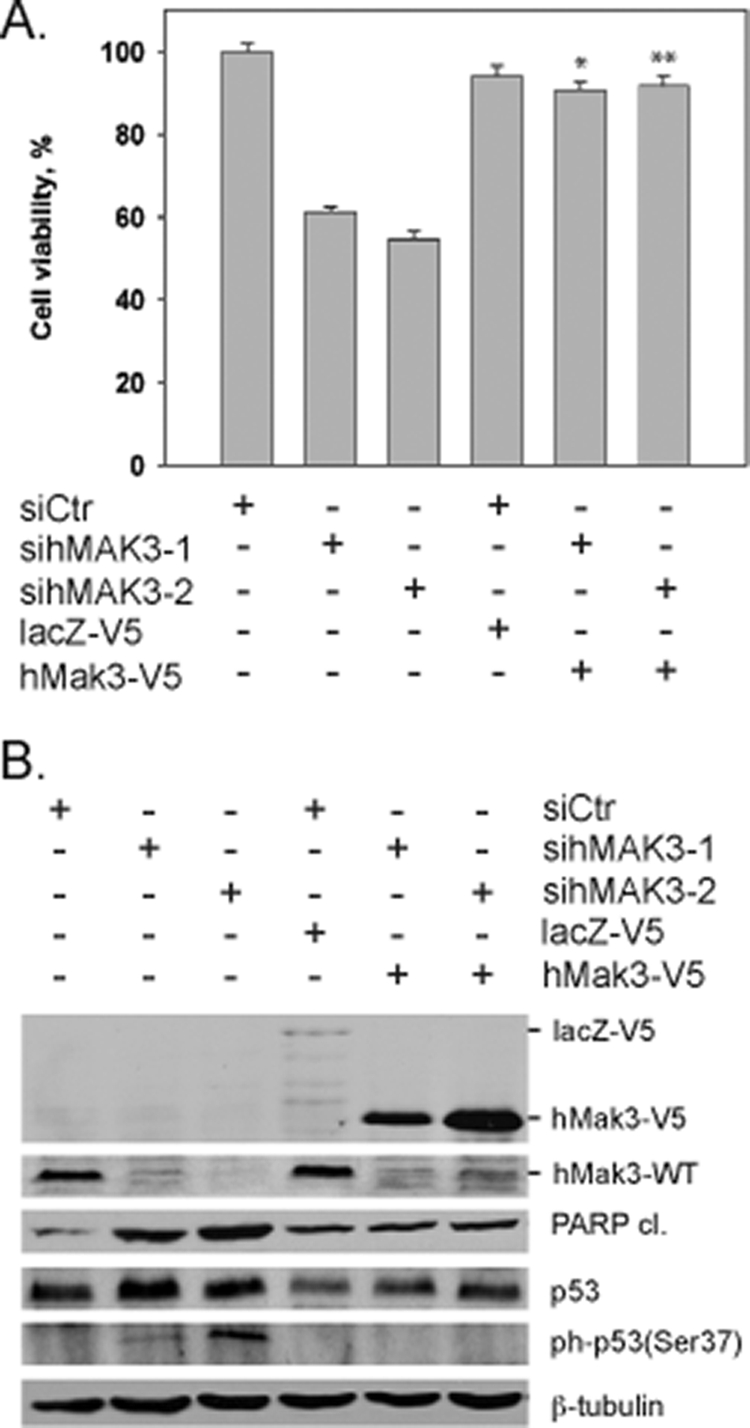
Expression of exogenous hMak3 rescues hMAK3 knockdown phenotypes. (A) The viability of hMAK3 knockdown cells expressing exogenous hMak3-V5 was compared to hMAK3 knockdown cells not expressing exogenous hMak3. Cell viability was measured 72 h posttransfection using a WST-1 assay. Results are given as percent cell viability. Experiments were performed in two parallel assays using two individual hMAK3 siRNAs. DharmaFECT Duo transfection reagent was used for cotransfection. Nontargeting SmartPool siRNA and lacZ-V5 were used as cotransfection controls. HeLa cells were used in all experiments. P values for hMak3 knockdown rescued with exogenous expression of hMak3 obtained with paired t tests are indicated: *, P < 0.004; **, P < 0.001. (B) Cells were treated as for panel A, and cell lysates were analyzed by SDS-PAGE and Western blotting. The membrane was incubated with anti-cleaved PARP (Asp214), anti-p53, anti-phospho-p53 (Ser37), anti-hMak3, anti-V5, and anti-β-tubulin (loading control). Results shown are representative of three independent experiments.
hArl8b is dependent on hMak3-mediated acetylation for lysosomal localization.
It was recently shown that the human Arf-like GTPase hArl8b was N-terminally acetylated and that the acetylated methionine was essential for its lysosomal localization (20). However, the human NAT responsible for the acetylation was not identified. As the N terminal of hArl8b is MLALI, we investigated whether hArl8b is an hMak3 substrate. Our in vitro results show that the hArl8b N-terminal sequence is indeed a substrate for hMak3-mediated acetylation (Fig. 5).
In HeLa cells subjected to knockdown of hMAK3 expression, a shift in hArl8b-GFP localization was observed (Fig. 10). In cells treated with control siRNA, hArl8b-GFP displayed a punctuate distribution, as previously published (20), overlapping the lysosomal marker LAMP-1 (Fig. 10D). When treated with sihMAK3, a significant fraction of the observed cells displayed aberrant localization of hArl8b-GFP, including loss of punctuate distribution and the formation of aggregate-like structures (Fig. 10E to H).
FIG. 10.

hMAK3 knockdown alters hArl8b localization. hArl8b-GFP was transfected into siControl cells (A to D) and sihMAK3 cells (E to H). (A and E) Localization of hArl8b-GFP (green); (B and F) the lysosomal marker LAMP-1 (red). (C and G) An overlay of hArl8b-GFP and LAMP-1, in addition to blue Hoechst 33342 staining for visualization of DNA. (D and H) Detailed view from panels C and F, respectively, as indicated by the marked areas in panels C and F. Bar, 25 μm. (I) hArl8b-GFP-transfected cells were counted (A to H), and the percentage of cells displaying a nonpunctuate distribution, as exemplified in panels E to H, was calculated. At least 500 transfected cells of each type from three independent experiments were registered. Error bars indicate the standard deviations. The P value for the two groups (indicated with an asterisk) was calculated to be <0.0143 based on a paired t test.
DISCUSSION
Protein N-terminal acetylation is a very common event in eukaryotic cells, occurring on a majority of proteins (9). However, compared to the yeast homologues, the human protein N-terminal acetyltransferases have not been fully identified or extensively characterized. In this study, we describe the human homologues of the yeast NatC complex. Our candidate subunits, hMak3, hMak10, and hMak31, stably interact with each other, localize to the cytoplasm, and associate with ribosomes. Furthermore, our semipurified hMak3 displays in vitro N-terminal acetyltransferase activity toward peptide substrates matching the observed substrate specificity of the yeast NatC complex, displaying a high degree of substrate specificity. Interestingly, Munro and colleagues showed in a previous study that hNat12/hMak3 functionally complemented yMak3p in Δmak3 yeast strains (12). Taken together, these findings strongly argue that hMak3 (hNat12) is the human homologue of yeast Mak3p. Based on these observations, we suggest that hMak3, hMak10, and hMak31 constitute the human NatC complex and that hMak3 is the catalytic subunit of the complex. Interestingly, the hMak3 protein (362 amino acids) is 176 amino acids larger than the yeast Mak3p. This is mainly due to an N-terminal region not shared by the yeast homologue. Similarly, the Arabidopsis thaliana Mak3 also contains additional residues compared to yeast Mak3, and AtMak3 displays enzymatic activity independent of AtMak10 (26). The function of the N-terminal hMak3 domain is not known. Using the ELM functional site prediction resource (http://elm.eu.org/) (31), the N-terminal region was found to contain several potential phosphorylation sites, making it a possible region for posttranslational regulation of hMak3 activity. Similarly, the catalytic subunit of hNatA, hArd1, has a C-terminal domain that is phosphorylated to different extents depending on cell culture conditions (24).
All the hNatC subunits localize to the cytoplasm and bind ribosomes. However, hMak31 also significantly localizes to the nucleus (Fig. 3). The nuclear localization of hMak31 would be expected, as its small size allows it to pass through the nuclear pore complexes. hMak31 (Lsmd1) belongs to the Sm and Sm-like proteins, which associate with RNA and are often involved in RNA processing events. Thus, hMak31 may have a nuclear role linked to RNA processing independent of the hNatC complex. hMak3, hMak10, and hMak31 are all present, both in a ribosome-bound and an unbound state. This pattern is also observed for subunits in the hNatA (T. Arnesen et al., submitted for pubication) and the hNatB complexes (34). In addition, there seems to be some difference between hMak3, hMak10, and hMak31 with respect to the ratio of ribosome-bound versus unbound protein. In particular, a larger fraction of hMak10 seems to be ribosome bound compared to hMak3 and hMak31. One could hypothesize that hMak10 may function as an anchor for ribosomal binding of the complex, similar to what is observed for hNat1 in the hNatA complex.
In a previous study, Nat13/Nat5 was suggested to be the vertebrate homologue of yMak3p (38). However, Nat13/Nat5 is the vertebrate protein most similar to yeast Nat5p. Both in humans and in yeast, Nat5 is described to be a subunit of the NatA complex (3, 18). To our knowledge, Nat5p has not been physically linked to the yeast NatC complex or functionally associated with NatC activity. In our present study we were not able to demonstrate an interaction between hNat13/hNat5 and hMak10. Whether hNat13/hNat5 is capable of binding to the human NatC is therefore doubtful, but more thorough experimentation is needed to settle this issue.
Knockdown of hNatC subunits in HeLa cells leads to cell death and growth arrest. Knockdown cells displayed reduced metabolic activity and DNA synthesis, an increase in the sub-G0/G1 fraction representing nuclear fragmentation, an increase in double-stranded DNA breaks, and PARP cleavage indicating caspase-dependent cell death. All three subunits are important for normal growth, supporting a model where the observed phenotypes are caused by lack of hNatC-mediated N-acetylation. Upon knockdown of hMAK3, hMAK10, or hMAK31 in the colon carcinoma cell lines HCT116 (p53−/−), no apoptosis was induced, in contrast to HCT116 (p53+/+) cells (Fig. 7). This difference in response between the cell variants strongly indicates that p53 is essential for induction of apoptosis in hNatC-depleted cells. Our findings suggest that this effect is mediated mainly through the transcriptional activity of p53, since hMAK3 knockdown increased the protein levels of p53, increased p53 Ser37 phosphorylation, and induced the transcription of p53 downstream death effector genes (Fig. 8).
NatC is believed to acetylate relatively few substrates compared to NatA in a manner of high amino acid sequence specificity at the N-terminal region. This could make hMak3 a potential target for drug-mediated cancer treatment, as previously discussed for hNatA (6, 8). An important goal of future studies will be to further elaborate the pathways through which hMAK3 knockdown mediates p53-dependent apoptosis.
In combination with the previous studies of the hNatA and hNatB complexes (1, 6, 34) our present results emphasize the biological importance of protein N-terminal acetylation in human cells. It should be noted that hMAK3 knockdown cells in general demonstrated a stronger phenotype than hMAK10 and hMAK31 knockdown cells. These effects may indicate that hMak3 has an additional function independent of hMak10 and hMak31. Leister and coworkers demonstrated that A. thaliana Mak3 alone is able to functionally replace the yeast NatC complex (26). Also, in contrast to AtMak3, knockout of AtMak10 did not result in any obvious defects. This indicates that Mak3 can have functions independent of the NatC complex in organisms higher than yeast.
The oligopeptides MLALI and MLGTG are identical to the N termini of the predicted human NatC substrates hArl8b and mTOR, respectively (38). hArl8b was recently described to be N-terminally acetylated, and an intact N terminus is essential for its association with lysosomes (20). mTOR was suggested to be a direct downstream target for NatC acetylation in zebrafish. Our in vitro acetyltransferase assays support our hypothesis that both hArl8b and mTOR are substrates for hNatC in vivo (Fig. 5). Knockdown of hMAK3 in HeLa cells induces a change in subcellular distribution of hArl8b-GFP. The observed change was statistically significant, but only a fraction of the cells (∼15%) display altered hArl8b-GFP localization as a consequence of hMAK3 knockdown (Fig. 10). The observed phenotype changes will to some extent be dependent on the transfection efficiency of sihMAK3: only a fraction of the hArl8b-GFP-expressing cells will have efficient knockdown of hMAK3. Among this subpopulation, one would expect a larger fraction of cells to display aberrant hArl8b-GFP localization. Furthermore, large-scale proteomics analyses of NAT knockdown in human cell lines have demonstrated only partial downstream N-terminal acetylation effects in agreement with our observations (9). Combined, these data strongly support that hArl8b indeed is an hNatC substrate in vivo. This is to our knowledge the only human NatC substrate where a clear consequence is observed when the N-terminal acetylation is lost. Also, it is the only known case with a link between a specific human NAT and a substrate of which N-terminal acetylation affects the substrate function. Further characterization of functionally important human NatC substrates will be of great importance in order to understand the overall impact of N-terminal acetylation, and in particular, N-terminal acetylation mediated by the hNatC complex.
With the present study, the human homologues of all three major yeast NAT complexes, NatA, NatB, and NatC, have been experimentally determined. In Table 1 we present an overview of the currently known human protein N-terminal acetyltransferases. The nomenclature of this enzyme class is under revision (B. Polevoda, T. Arnesen, and F. Sherman, unpublished data), and the NatC components will be denoted as follows: Naa30 (Mak3), Naa35 (Mak10), and Naa38 (Mak31).
TABLE 1.
Overview of human protein NATs
| Yeast NAT | Yeast substrates (N termini) | NAT subunit | Human homologue(s) and synonym(s)a | Reference(s) or source |
|---|---|---|---|---|
| NatA | Ser-, Thr-, Ala-, Gly- | Ard1p (catalytic subunit) | hArd1, ARD1A, TE2 | 2, 10, 36 |
| hArd2, ARD1B | 4 | |||
| Nat1p (auxiliary subunit) | NATH/hNat1, Tubedown | 2, 10, 17, 19 | ||
| Nat5pb | hNat5/hSan | 3, 18 | ||
| NatB | Met-Asp-, Met-Glu-, Met-Asn- | Nat3p (catalytic subunit) | hNat3, hNat5 | 1, 34 |
| Mdm20p (auxiliary subunit) | hMdm20 | 34 | ||
| NatC | Met-Leu-, Met-Ile-, Met-Phe-, Met-Trp- | Mak3p (catalytic subunit) | hMak3, hNat12 | This study |
| Mak10p (auxiliary subunit) | hMak10 | This study | ||
| Mak31p (auxiliary subunit) | hMak31, Lsmd1 | This study | ||
| NatD | Ser-Gly-Gly-, Ser-Gly-Arg- | Nat4p | —c | —c |
Listed homologues are based on experimental data. Potential homologues based on database predictions are not included.
Nat5p is predicted to be an acetyltransferase with specific and unknown substrates. Nat5p is not required for NatA activity.
—, no information available.
In summary, we here identify hMak3, hMak10, and hMak31 as the subunits of the human NatC complex. Knockdown of these subunits induces p53-dependent apoptosis. hMak3-mediated acetylation is necessary for the lysosomal localization and function of hArl8b (Fig. 11).
FIG. 11.

Schematic representation of functions associated with the human NatC complex. (A) The hNatC complex, composed of the subunits hMak3, hMak10, and hMak31, associates with ribosomes and acetylates nascent Met-Leu and similar polypeptides. One protein of this type, the Arf-like GTPase hArl8b, depends on the N-terminal acetylation for its localization to lysosomes. (B) Knockdown of hNatC complex subunits by siRNA most likely reduces the N-terminal acetylation of some NatC-type substrates. Directly or indirectly, this stabilizes and activates p53, which in turn induces transcription of genes leading to the induction of apoptosis. Apoptosis decreases the protein level of hMak3, while the mRNA level is unaffected.
Acknowledgments
We thank C. Hoff, J. Torsvik, E. Skjelvik, L. Vikebø, and M. Algrøy for technical assistance.
This work was supported by The Norwegian Cancer Society, The Meltzer Foundation, and Norwegian Health Region West.
Footnotes
Published ahead of print on 27 April 2009.
REFERENCES
- 1.Ametzazurra, A., E. Larrea, M. P. Civeira, J. Prieto, and R. Aldabe. 2008. Implication of human N-alpha-acetyltransferase 5 in cellular proliferation and carcinogenesis. Oncogene 277296-7306. [DOI] [PubMed] [Google Scholar]
- 2.Arnesen, T., D. Anderson, C. Baldersheim, M. Lanotte, J. E. Varhaug, and J. R. Lillehaug. 2005. Identification and characterization of the human ARD1-NATH protein acetyltransferase complex. Biochem. J. 386433-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnesen, T., D. Anderson, J. Torsvik, H. B. Halseth, J. E. Varhaug, and J. R. Lillehaug. 2006. Cloning and characterization of hNAT5/hSAN: an evolutionarily conserved component of the NatA protein N-alpha-acetyltransferase complex. Gene 371291-295. [DOI] [PubMed] [Google Scholar]
- 4.Arnesen, T., M. J. Betts, F. Pendino, D. A. Liberles, D. Anderson, J. Caro, X. Kong, J. E. Varhaug, and J. R. Lillehaug. 2006. Characterization of hARD2, a processed hARD1 gene duplicate, encoding a human protein N-alpha-acetyltransferase. BMC Biochem. 713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arnesen, T., D. Gromyko, O. Horvli, O. Fluge, J. Lillehaug, and J. E. Varhaug. 2005. Expression of N-acetyl transferase human and human Arrest defective 1 proteins in thyroid neoplasms. Thyroid 151131-1136. [DOI] [PubMed] [Google Scholar]
- 6.Arnesen, T., D. Gromyko, F. Pendino, A. Ryningen, J. E. Varhaug, and J. R. Lillehaug. 2006. Induction of apoptosis in human cells by RNAi-mediated knockdown of hARD1 and NATH, components of the protein N-alpha-acetyltransferase complex. Oncogene 254350-4360. [DOI] [PubMed] [Google Scholar]
- 7.Arnesen, T., X. Kong, R. Evjenth, D. Gromyko, J. E. Varhaug, Z. Lin, N. Sang, J. Caro, and J. R. Lillehaug. 2005. Interaction between HIF-1 alpha (ODD) and hARD1 does not induce acetylation and destabilization of HIF-1 alpha. FEBS Lett. 5796428-6432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arnesen, T., P. R. Thompson, J. E. Varhaug, and J. R. Lillehaug. 2008. The protein acetyltransferase ARD1: a novel cancer drug target? Curr. Cancer Drug Targets 8545-553. [DOI] [PubMed] [Google Scholar]
- 9.Arnesen, T., P. Van Damme, B. Polevoda, K. Helsens, R. Evjenth, N. Colaert, J. Varhaug, J. Vandekerckhove, J. R. Lillehaug, F. Sherman, and K. Gevaert. 2009. Proteomics analyses reveal the evolutionary conservation and divergence of N-terminal acetyltransferases from yeast and humans. Proc. Natl. Acad. Sci. USA 1068157-8162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Asaumi, M., K. Iijima, A. Sumioka, K. Iijima-Ando, Y. Kirino, T. Nakaya, and T. Suzuki. 2005. Interaction of N-terminal acetyltransferase with the cytoplasmic domain of beta-amyloid precursor protein and its effect on A beta secretion. J. Biochem. (Tokyo) 137147-155. [DOI] [PubMed] [Google Scholar]
- 11.Askree, S. H., T. Yehuda, S. Smolikov, R. Gurevich, J. Hawk, C. Coker, A. Krauskopf, M. Kupiec, and M. J. McEachern. 2004. A genome-wide screen for Saccharomyces cerevisiae deletion mutants that affect telomere length. Proc. Natl. Acad. Sci. USA 1018658-8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Behnia, R., B. Panic, J. R. Whyte, and S. Munro. 2004. Targeting of the Arf-like GTPase Arl3p to the Golgi requires N-terminal acetylation and the membrane protein Sys1p. Nat. Cell Biol. 6405-413. [DOI] [PubMed] [Google Scholar]
- 13.Chipuk, J. E., and D. R. Green. 2006. Dissecting p53-dependent apoptosis. Cell Death Differ. 13994-1002. [DOI] [PubMed] [Google Scholar]
- 14.Clamp, M., J. Cuff, S. M. Searle, and G. J. Barton. 2004. The Jalview Java alignment editor. Bioinformatics 20426-427. [DOI] [PubMed] [Google Scholar]
- 15.Dixon, S. J., Y. Fedyshyn, J. L. Koh, T. S. Prasad, C. Chahwan, G. Chua, K. Toufighi, A. Baryshnikova, J. Hayles, K. L. Hoe, D. U. Kim, H. O. Park, C. L. Myers, A. Pandey, D. Durocher, B. J. Andrews, and C. Boone. 2008. Significant conservation of synthetic lethal genetic interaction networks between distantly related eukaryotes. Proc. Natl. Acad. Sci. USA 10516653-16658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fisher, T. S., S. D. Etages, L. Hayes, K. Crimin, and B. Li. 2005. Analysis of ARD1 function in hypoxia response using retroviral RNA interference. J. Biol. Chem. 28017749-17757. [DOI] [PubMed] [Google Scholar]
- 17.Fluge, O., O. Bruland, L. A. Akslen, J. E. Varhaug, and J. R. Lillehaug. 2002. NATH, a novel gene overexpressed in papillary thyroid carcinomas. Oncogene 215056-5068. [DOI] [PubMed] [Google Scholar]
- 18.Gautschi, M., S. Just, A. Mun, S. Ross, P. Rucknagel, Y. Dubaquie, A. Ehrenhofer-Murray, and S. Rospert. 2003. The yeast Nα-acetyltransferase NatA is quantitatively anchored to the ribosome and interacts with nascent polypeptides. Mol. Cell. Biol. 237403-7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gendron, R. L., L. C. Adams, and H. Paradis. 2000. Tubedown-1, a novel acetyltransferase associated with blood vessel development. Dev. Dyn. 218300-315. [DOI] [PubMed] [Google Scholar]
- 20.Hofmann, I., and S. Munro. 2006. An N-terminally acetylated Arf-like GTPase is localised to lysosomes and affects their motility. J. Cell Sci. 1191494-1503. [DOI] [PubMed] [Google Scholar]
- 21.Ingram, A. K., G. A. Cross, and D. Horn. 2000. Genetic manipulation indicates that ARD1 is an essential Nα-acetyltransferase in Trypanosoma brucei. Mol. Biochem. Parasitol. 111309-317. [DOI] [PubMed] [Google Scholar]
- 22.Katoh, K., K. Kuma, T. Miyata, and H. Toh. 2005. Improvement in the accuracy of multiple sequence alignment program MAFFT. Genome Inform. 1622-33. [PubMed] [Google Scholar]
- 23.Li, D. W., J. P. Liu, P. C. Schmid, R. Schlosser, H. Feng, W. B. Liu, Q. Yan, L. Gong, S. M. Sun, M. Deng, and Y. Liu. 2006. Protein serine/threonine phosphatase-1 dephosphorylates p53 at Ser-15 and Ser-37 to modulate its transcriptional and apoptotic activities. Oncogene 253006-3022. [DOI] [PubMed] [Google Scholar]
- 24.Malen, H., J. Lillehaug, and T. Arnesen. 2009. The protein Nα-terminal acetyltransferase hNaa10p (hArd1) is phosphorylated in HEK293 cells. BMC Res. Notes 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murthi, A., and A. K. Hopper. 2005. Genome-wide screen for inner nuclear membrane protein targeting in Saccharomyces cerevisiae: roles for N-acetylation and an integral membrane protein. Genetics 1701553-1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pesaresi, P., N. A. Gardner, S. Masiero, A. Dietzmann, L. Eichacker, R. Wickner, F. Salamini, and D. Leister. 2003. Cytoplasmic N-terminal protein acetylation is required for efficient photosynthesis in Arabidopsis. Plant Cell 151817-1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pfund, C., N. Lopez-Hoyo, T. Ziegelhoffer, B. A. Schilke, P. Lopez-Buesa, W. A. Walter, M. Wiedmann, and E. A. Craig. 1998. The molecular chaperone Ssb from Saccharomyces cerevisiae is a component of the ribosome-nascent chain complex. EMBO J. 173981-3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Polevoda, B., S. Brown, T. S. Cardillo, S. Rigby, and F. Sherman. 2008. Yeast Nα-terminal acetyltransferases are associated with ribosomes. J. Cell Biochem. 103492-508. [DOI] [PubMed] [Google Scholar]
- 29.Polevoda, B., and F. Sherman. 2003. N-terminal acetyltransferases and sequence requirements for N-terminal acetylation of eukaryotic proteins. J. Mol. Biol. 325595-622. [DOI] [PubMed] [Google Scholar]
- 30.Polevoda, B., and F. Sherman. 2001. NatC Nα-terminal acetyltransferase of yeast contains three subunits, Mak3p, Mak10p, and Mak31p. J. Biol. Chem. 27620154-20159. [DOI] [PubMed] [Google Scholar]
- 31.Puntervoll, P., R. Linding, C. Gemund, S. Chabanis-Davidson, M. Mattingsdal, S. Cameron, D. M. Martin, G. Ausiello, B. Brannetti, A. Costantini, F. Ferre, V. Maselli, A. Via, G. Cesareni, F. Diella, G. Superti-Furga, L. Wyrwicz, C. Ramu, C. McGuigan, R. Gudavalli, I. Letunic, P. Bork, L. Rychlewski, B. Kuster, M. Helmer-Citterich, W. N. Hunter, R. Aasland, and T. J. Gibson. 2003. ELM server: a new resource for investigating short functional sites in modular eukaryotic proteins. Nucleic Acids Res. 313625-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Setty, S. R., T. I. Strochlic, A. H. Tong, C. Boone, and C. G. Burd. 2004. Golgi targeting of ARF-like GTPase Arl3p requires its Nα-acetylation and the integral membrane protein Sys1p. Nat. Cell Biol. 6414-419. [DOI] [PubMed] [Google Scholar]
- 33.Sonnichsen, B., L. B. Koski, A. Walsh, P. Marschall, B. Neumann, M. Brehm, A. M. Alleaume, J. Artelt, P. Bettencourt, E. Cassin, M. Hewitson, C. Holz, M. Khan, S. Lazik, C. Martin, B. Nitzsche, M. Ruer, J. Stamford, M. Winzi, R. Heinkel, M. Roder, J. Finell, H. Hantsch, S. J. Jones, M. Jones, F. Piano, K. C. Gunsalus, K. Oegema, P. Gonczy, A. Coulson, A. A. Hyman, and C. J. Echeverri. 2005. Full-genome RNAi profiling of early embryogenesis in Caenorhabditis elegans. Nature 434462-469. [DOI] [PubMed] [Google Scholar]
- 34.Starheim, K. K., T. Arnesen, D. Gromyko, A. Ryningen, J. E. Varhaug, and J. R. Lillehaug. 2008. Identification of the human Nα-acetyltransferase complex B (hNatB): a complex important for cell-cycle progression. Biochem. J. 415325-331. [DOI] [PubMed] [Google Scholar]
- 35.Tercero, J. C., and R. B. Wickner. 1992. MAK3 encodes an N-acetyltransferase whose modification of the L-A gag NH2 terminus is necessary for virus particle assembly. J. Biol. Chem. 26720277-20281. [PubMed] [Google Scholar]
- 36.Tribioli, C., M. Mancini, E. Plassart, S. Bione, S. Rivella, C. Sala, G. Torri, and D. Toniolo. 1994. Isolation of new genes in distal Xq28: transcriptional map and identification of a human homologue of the ARD1 N-acetyl transferase of Saccharomyces cerevisiae. Hum. Mol. Genet. 31061-1067. [DOI] [PubMed] [Google Scholar]
- 37.Vedeler, A., I. F. Pryme, and J. E. Hesketh. 1991. The characterization of free, cytoskeletal and membrane-bound polysomes in Krebs II ascites and 3T3 cells. Mol. Cell. Biochem. 100183-193. [DOI] [PubMed] [Google Scholar]
- 38.Wenzlau, J. M., P. J. Garl, P. Simpson, K. R. Stenmark, J. West, K. B. Artinger, R. A. Nemenoff, and M. C. Weiser-Evans. 2006. Embryonic growth-associated protein is one subunit of a novel N-terminal acetyltransferase complex essential for embryonic vascular development. Circ. Res. 98846-855. [DOI] [PubMed] [Google Scholar]
- 39.Wickner, R. B., and A. Toh-e. 1982. [HOK], a new yeast non-Mendelian trait, enables a replication-defective killer plasmid to be maintained. Genetics 100159-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Williams, B. C., C. M. Garrett-Engele, Z. Li, E. V. Williams, E. D. Rosenman, and M. L. Goldberg. 2003. Two putative acetyltransferases, san and deco, are required for establishing sister chromatid cohesion in Drosophila. Curr. Biol. 132025-2036. [DOI] [PubMed] [Google Scholar]
- 41.Xie, M. W., F. Jin, H. Hwang, S. Hwang, V. Anand, M. C. Duncan, and J. Huang. 2005. Insights into TOR function and rapamycin response: chemical genomic profiling by using a high-density cell array method. Proc. Natl. Acad. Sci. USA 1027215-7220. [DOI] [PMC free article] [PubMed] [Google Scholar]