

Abstract
Background
Intimal hyperplasia remains the principal lesion in the development of restenosis after vessel wall injury. Modulation of the extracellular matrix by proteases is a pivotal component of the response to injury. The aim of this study is to characterize the changes in gelatinase (MMP-2/TIMP-2 and MMP-9/TIMP-1) systems in a murine model.
Methods
The murine femoral wire injury model was employed in which a micro wire is passed through a branch of the femoral and used to denude the common femoral artery. Pluronic gel was used to apply a proteass inhibitor (GM6001) to the exterior of the vessels. Specimens were perfusion-fixed and sections were stained with H&E and Movat’s stain such that morphometry could be performed using an ImagePro system. Additional specimens of femoral artery were also harvested and snap frozen for western blotting and zymography to allow for the study of gelatinase expression and activation. Contralateral vessels were used as controls
Results
MMP-2 activity increased significantly at day 1 and peaked again at day 7 and then showed a continual decline in activity to day 56. MMP-9 activity peaked early at day 3 and declined thereafter. Protein expression for both MMP-2 and MMP-9 increased significantly after injury and were both maximal at day 14. There was an initial decrease in TIMP-1 and TIMP-2 expression and activity after injury until day 5. Both expression and activation gradually increased thereafter to level out by day 21 and correlated well with the early increases in MMP-2 and MMP-9 activity and their subsequent decline. Local application of protease inhibitor (GM6001) within a pluronic gel, decreased cell proliferation, and at 14 days there was a decrease in intimal hyperplasia.
Conclusions
These data demonstrate that femoral wire injury in the mouse is associated with a time dependent increase in gelatinase activity. Cell proliferation is associated with increased MMP-2/MMP-9 activity, and decreased TIMP-2/TIMP-1 activity, while migration is associated with increased in MMP-2 activity. Modulation of proteases and their inhibitors control the vessels’ response to injury.
Keywords: Gelatinase, Injury, vascular smooth muscle cell
INTRODUCTION
Vessel wall remodeling during atherogenesis in response to flow and after injury induces significant changes in arterial structure as a result of an alteration in the balance between medial vascular smooth muscle cell (VSMC) proliferation, apoptosis and migration and changes in the extracellular matrix (1). Regulation of these activities while mutlifactorial does involve the complex regulation of proteases, integrins and extracellular molecules (2). We recently described the changes in structure and examined the role of cell kinases in modulating the development of intimal hyperplasia in a murine model and demonstrated that femoral wire injury in the mouse induces a consistent model of intimal hyperplasia and that it is associated with a time dependent increase in signaling kinase (ERK1/2, p38MAPK and JNK) activity (3). There were also time dependent alterations in the activity and expression of the protease, urokinase (uPA) and its inhibitor, PAI-1 after injury Interruption of the MAPK kinase activity decrease the intimal hyperplastic response and interrupted the uPA/PAI-1 pathway (3). uPA activates Metalloproteinase-2 (MMP-2), a gelatinase that is implicated in cell migration and vessel remodeling (1, 4, 5). Metalloproteinase-9 (MMP-9) a second gelatinase has been associated with cell proliferation in several animal models (6, 7). Tissue inhibitors of Metalloproteinase-(TIMP)-1 and -2 control the activity of MMP-9 and MMP-2 respectively. The aim of this study is to characterize the changes in the gelatinase system (MMP-2/TIMP-2 and MMP-9/TIMP-1) in the murine model of arterial injury and determine if interruption of protease activity will alter the remodeling response.
MATERIALS AND METHODS
Experimental Design
6 week old male FVB mice underwent femoral wire injury model and were harvested at various time points over a 56 day period. Specimens (n=6 per time point) were perfusion-fixed and sections were stained for morphometry using an ImagePro system. Additional specimens of femoral artery (n=6 per time point) were also harvested and snap frozen for western blotting and zymography to allow for the study of protease expression and activation. Additional vessels (n=6 per group) were also coated in pluronic gel with and without the global MMP inhibitor (GM6001) and the plasminogen activation inhibitor inhibitor (aprotinin) and harvested at various time points to determine the impact of local inhibitor therapy. Additional vessels were also coated in pluronic gel with and without MAPK kinase inhibitors (PD98059 – ERK inhibitor; SB230580 – p38MAPK inhibitor; and SP600125 – JNK inhibitor) and harvested at various time points to determine the impact of local inhibitor therapy. Contralateral vessels were used as controls. Animal care and procedures were conducted at the University of Rochester Medical Center in accordance with state and federal laws and under protocols approved by the University of Rochester Animal Care and Use Committee. Animal care complied with the “Guide for the Care and Use of Laboratory Animals” issued by the Institute of Laboratory Animal Resources.
Femoral wire injury
Endoluminal injury to the common femoral artery was produced by 3 passages of a 0.25-mm-diameter angioplasty guidewire (Advanced Cardiovascular Systems) as previously described (3). Control sham-operated arteries underwent dissection, temporary clamping, arteriotomy, and ligature, without passage of the wire. Non-operated normal femoral arteries were used as additional controls. Based on the data derived from the control experiments, additional mice underwent femoral wire injury after being randomly allocated to receive a pluronic gel with or without GM6001 (25μM) or aprotinin (1000 units). Additional mice underwent femoral wire injury after being randomly allocated to receive a pluronic gel with or without (PD98059 [25μM], SB230580 [25μM] and SP600125 [25μM]). The vessels were harvested for western blotting to detect peak gelatinase expression and activity and final morphometry. Pluronic gel (1ml of 30% F127 Pluronic gel; Sigma, St Louis, MO) was applied to the external surface of the femoral artery at the end of the procedure as previously described (3, 8, 9). The pluronic gel remains in a liquid form at 4°C and rapidly forms a gel on exposure to body temperature. It is dissipated within 4 hrs.
Morphometry
Following perfusion fixation, each vessel was bisected and central segments from each of these halves were embedded in paraffin and cross sections were cut (10, 100μm apart). Standard morphometric measurements were performed on histological cross sections stained with Hematoxylin/Eosin and Movat’s stain using SPOT camera system. The luminal, intimal and medial areas of each vessel are determined. The volume of cells, collagen, proteoglycan and elastin were calculated using an ImagePro system. Following determination of dimensions of each vessel, a ratio of the intimal and medial areas was calculated.
Determination of in vivo proliferation and apoptosis
Labeling of proliferating VSMC in specimens is performed using the thymidine analogue 5-bromo-2í deoxyuridine (BrdU; Boehringer-Mannheim Corp., Indianapolis, IN; administered by Intraperitoneal injection 24 hours prior to sacrifice, 60 μg/g body weight). The number of BrdU-stained VSMC nuclei are counted and the BrdU labeling index (% of unlabelled cells) is calculated on 10 slides (100μm apart) per vessel (6 vessels per time point). Labeling of apoptotic VSMC on adjacent sections from each vessel specimens are evaluated using terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL; Boehringer-Mannheim Corp.) after processing the tissue according to the manufacturer’s guidelines on 10 slides (100μm apart) per vessel (6 vessels per time point). Stained VSMC nuclei are counted and the TUNEL labeling index (% of unlabelled cells) is calculated.
En Face SMC Migration Assay
Femoral arteries were harvested and were opened longitudinally and pinned down onto on agar plate with the luminal surface facing upward and fixed by immersion (3). Arteries were then stained with hematoxylin for 10 seconds and incubated in Scott’s blue for 2 minutes. Migrated VSMCs were identified as the hematoxylin-stained cells on the luminal surface of the endothelium-denuded area. The length of the femoral artery was visualized under a microscope at x100 magnification, and the total number of VSMC above and below the internal elastic lamina are counted and results expressed as % cells on the luminal surface of the artery.
Immunoblotting
At harvest, both sham and femoral arteries were rapidly retrieved at the various time points, cleaned once in ice-cold dPBS and then paced in ice-cold kinase buffer (200μl; 25mM Hepes, pH7.5, 10% glycerol, 5mM EDTA, 5mM EGTA, 150mM NaCl, 100mM benzamidine, 0.1% 2-mercaptoethanol, 1% Triton X-100, 1mM pepstatin A, 2mg/ml leupeptin and 20 kallikrein inhibitor units/ml aprotinin). Protein determinations were performed using a bicinchonic acid assay (Biorad Laboratories, Richmond, CA) with bovine serum albumen as the standard. Equal amounts of protein lysates (20μg) were loaded onto a 10% polyacrylamide gel separated by electrophoresis and then transferred to a nitrocellulose membrane (Biorad) (10). The membrane was blocked in 5% non-fat milk. The blot was then immunostained with antibodies against either phosphorylated or unphosphorylated target protein. Protein bands were visualized using an anti-mouse IgG horseradish peroxidase-conjugated antibody followed by enhanced chemiluminescence (ECL, Amersham Bioscience, Piscataway, NJ) using the manufacturer’s protocols. Band intensity was measured using Gel-Imaging software (Kodak, Rochester, NY). All experiments were performed at least three times.
Gelatin Zymography
For assay of MMP-2 and MMP-9 activity, vessels were isolated and snap frozen after rinsing in ice cold PBS. Segments were then pulverized on liquid nitrogen using a mortar and pestle and extracted with a guanidine based buffer (0.05M TRIS, pH7.5, 0.01M CaCl2, 2.0M Guanidine and 0.2% Triton X-100). The suspension was centrifuged and dialyzed against 0.05M TRIS and 0.2% Triton for 48 hours. BCA protein assay (Pierce, Rockford, IL) was performed and 10μg protein is loaded per lane. Gelatin zymography for matrix metalloproteinases (MMPs) was performed as described previously (11).
Reverse Gelatin Zymography
Gelatin Zymography was performed as described previously (12) with minor modifications. Equal amounts of proteins were mixed with SDS sample buffer without reducing agent and subjected to 7.5% SDS-PAGE containing gelatin (2.5 mg/ml) and pro-MMP-2 (1.2μg/ml) or pro-MMP-9 (1.2μg/ml). After electrophoresis, the gels were washed twice in 2.5% Triton X-100 for 30 min each at room temperature to remove SDS and then incubated for 18 hrs at 37°C in buffer containing Tris-HCl (40mM), CaCl2 (10 mM) and NaCl (50 mM). The proteins in the gel were stained with 0.2% Coomassie Brilliant Blue in 30% Methanol: 10% Acetic acid solution for 2–3 hrs and then destained in 30% Methanol: 10% acetic acid. TIMP activity was detected as blue bands against a clear digested background of MMP digestion.
Data and Statistical Analysis
All data are presented as the mean ± standard error of the mean (s.e.m.) of six observations and statistical differences between groups were tested with a Kruskal-Wallis non-parametric test with post hoc Dunn’s multiple comparison correction, where appropriate. A p-value less than 0.05 was regarded as significant.
RESULTS
Morphological Response
The injured femoral arteries developed intimal hyperplasia, which is maximal at 28 days and does not change substantially between day 28 and day 56 (Fig 1A). Sham operated on vessels did not produce such a response There were time dependent changes in the volume of cells, collagen, proteoglycan and elastin with a substantial increase in the volume of proteoglycans over time (Fig 1A). Local application of protease inhibitors against MMP and plasminogen activators (GM6001 and aprotinin) within a pluronic gel, decreased cell proliferation and cell migration respectively compared to gel treated injured vessels (Fig 1B).. Sham vessels should no significant responses at these time points (Fig 1B). At 28 days, there was a decrease in intimal hyperplasia in both the GM6001-treated and aprotinin-treated groups (Fig 1C). Application of the MMP inhibitor GM6001 and the plasminogen activation inhibitor aprotinin also reduced the composition of the intimal hyperplasia with a decrease in the percentage of proteoglycans and a rise in the percent of collagen within the intimal hyperplasia (Fig 1D).
Figure 1.
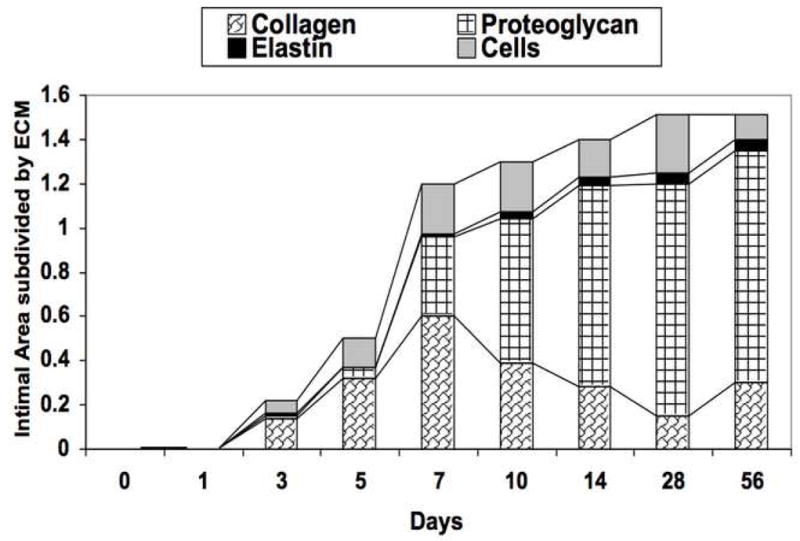
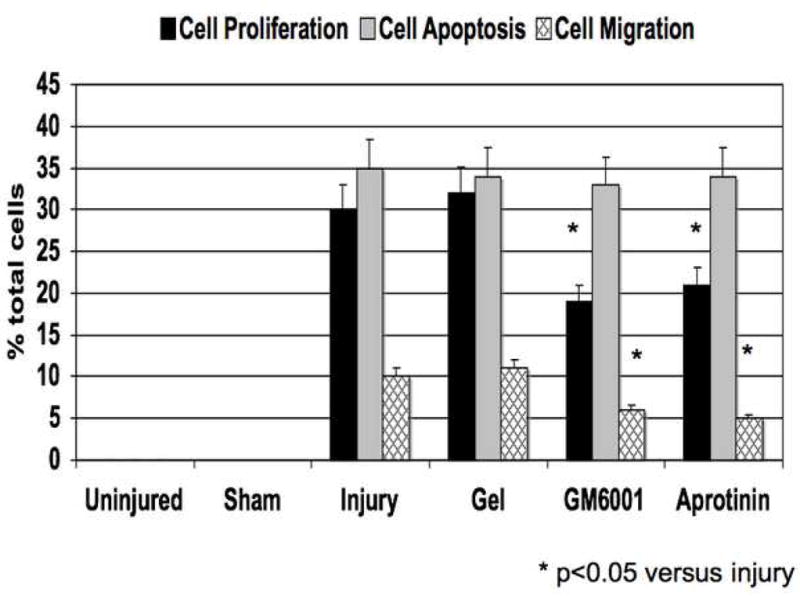
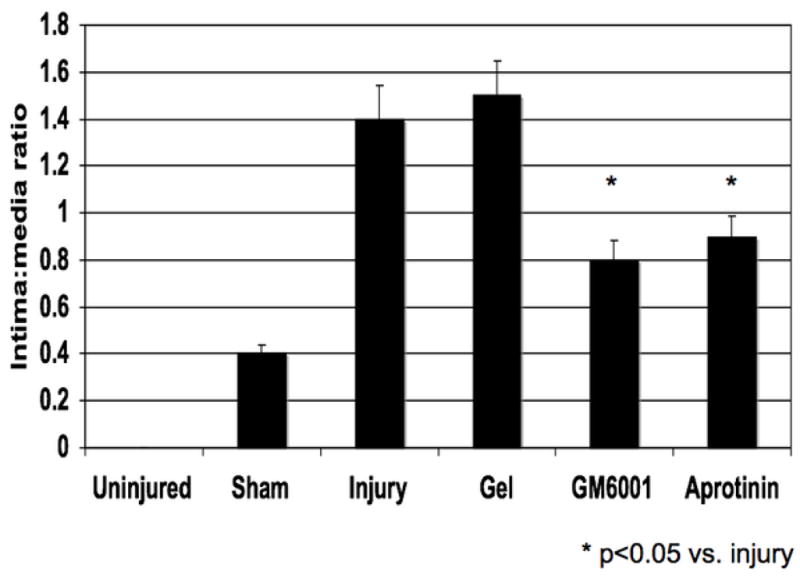
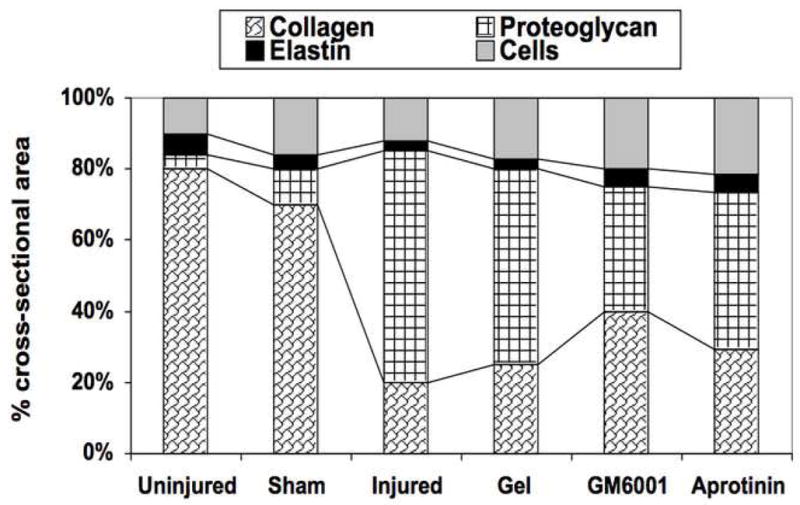
(A) Changes in the ratio of intimal to medial areas and the intimal area of cells, collagen, proteoglycan and elastin in wire injured femoral vessels over time from day 0 to day 56. (B) A comparison of the effect of pluronic gel with and without GM6001 (general MMP inhibitor) and aprotinin (plasminogen activation inhibitor) on intimal hyperplasia development at 28 days after wire injury. (C) Changes in the area of cells, collagen, proteoglycan and elastin in wire injured femoral vessels in the presence of pluronic gel with and without GM6001 and aprotinin at 28 days after wire injury. (D) Changes in cell proliferation, cell apoptosis and cell migration in wire injured femoral vessels in the presence of pluronic gel with and without GM6001 and aprotinin. Values are the mean± s.e.m. (n=6 per group or time point). Statistical differences between groups were tested with a Kruskal-Wallis non-parametric test with post hoc Dunn’s multiple comparison correction, where appropriate.
Gelatinase Activity
MMP-2 activity increased significantly at day 1 and peaked again at day 7 and then showed a continual decline in activity to day 56 (Fig 2A). MMP-9 activity peaked early at day 3 and declined thereafter (Fig 3A). Protein expression for both MMP-2 and MMP-9 increased significantly after injury and were both maximal at day 14 (Fig 2B and 3B). There was an initial decrease in TIMP-2 expression and activity after injury until Day 5 (Fig 2A and 2B), Thereafter TIMP-2 activity rose to 4-times control from day 7 onwards (Fig 2A), without a significant change in protein expression (Fig 2B). There was an increase in TIMP-1 activity without a change in TIMP-1 expression after injury until day 5 (Fig 3A and 3B) and then a gradual 2-fold increase in protein expression and activity after Day 14. Both expression and activation of TIMP-1 and TIMP-2 correlated inversely with the early increases in MMP-2 and MMP-9 activity and their subsequent decline (Fig 2 and 3). The activity of MMP-2/TIMP-2 and MMP-9/TIMP-1 in the sham vessels was similar until day 3 when the expression and activity returned to baseline. Application of the MMP inhibitor GM6001 but not the plasminogen activation inhibitor aprotinin decreased MMP-2 and MMP-9 expression; there was no change in TIMP-1 And TIMP-2 expression with local pharmacological manipulation (Fig 3A). Activity of MMP-2 and MMP-9 were decreased by GM6001. Only the activity of MMP-2 was affected by the presence of aprotinin (Fig 3B). There was no change in TIMP-1 And TIMP-2 activity (Fig 3B).
Figure 2.
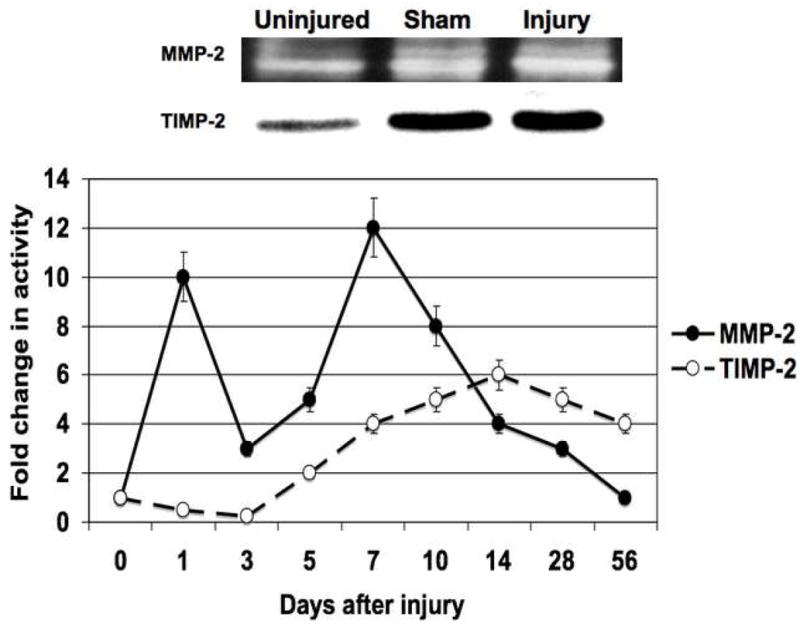
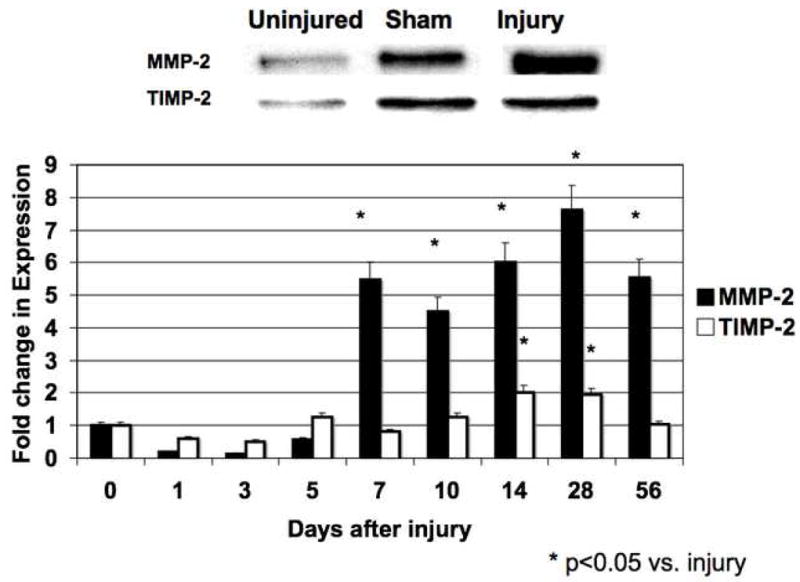
The activity (A) and expression (B) of MMP-2/TIMP-2 in the wire injured femoral vessels over time. Top panel (A) shows a representative zymograms for MMP-2/TIMP-2 at day 7. Top panel (B) shows a representative western blots for MMP-2/TIMP-2 at day 14. Bottom panels show the quantification of the activity or expression data for the injured vessels. Values are the mean± s.e.m. (n=6 per time point). Statistical differences between groups were tested with a Kruskal-Wallis non-parametric test with post hoc Dunn’s multiple comparison correction, where appropriate.
Figure 3.
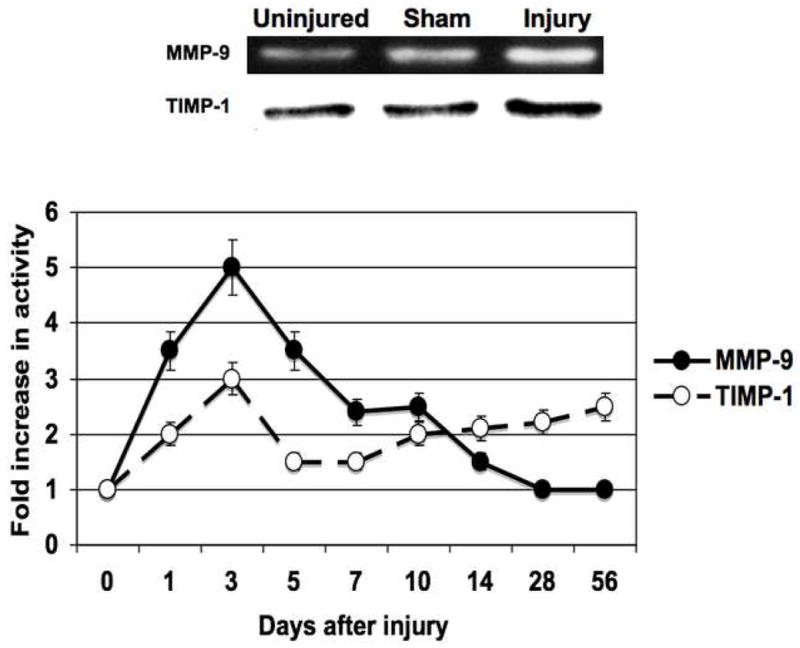
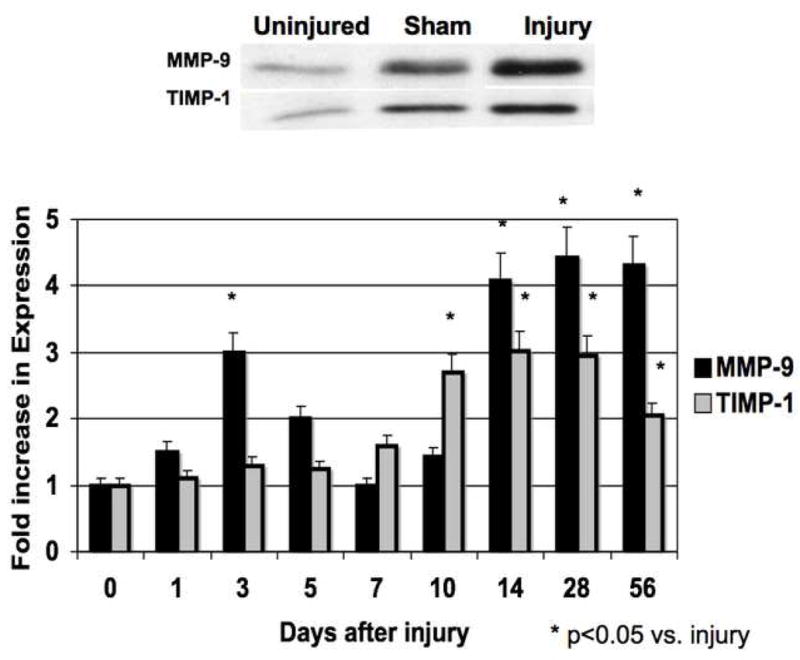
The activity (A) and expression (B) of MMP-9/TIMP-1 in the wire inured femoral vessels over time. Top panel (A) shows a representative zymograms for MMP-9/TIMP-1 at day 7. Top panel (B) shows a representative western blots for MMP-9/TIMP-1 at day 14. Bottom panels show the quantification of the activity or expression data for the injured vessels. Values are the mean± s.e.m. (n=6 per time point). Statistical differences between groups were tested with a Kruskal-Wallis non-parametric test with post hoc Dunn’s multiple comparison correction, where appropriate.
Control of Gelatinase System Expression
Application of PD98059, SB230580 and SP600125 in pluronic gel blunted MMP and TIMP activities The increases in MMP-2 expression and activity were decreased by ERK1/2 and p38MAPK inhibition and that the increases in MMP-9 expression and activity were blunted by inhibition of p38MAPK and JNK (Fig 5). TIMP-2 expression and activity was diminished by inhibition of p38MAPK and JNK inhibition and TIMP-1 expression and activity was diminished by ERK1/2 inhibition (Fig 5)
Figure 5.
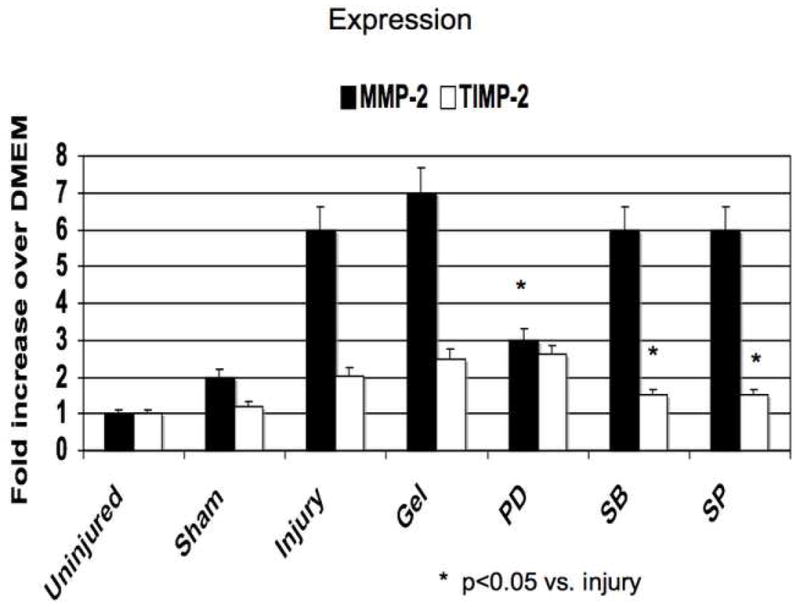
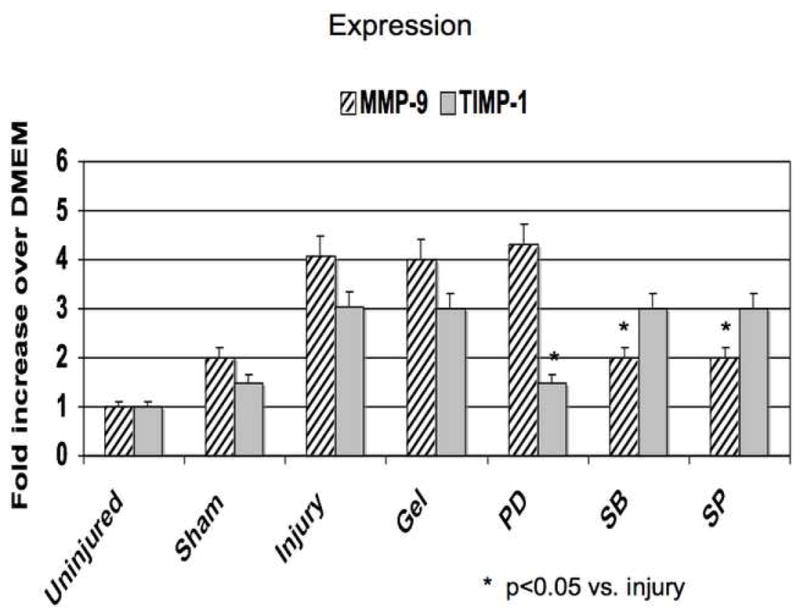
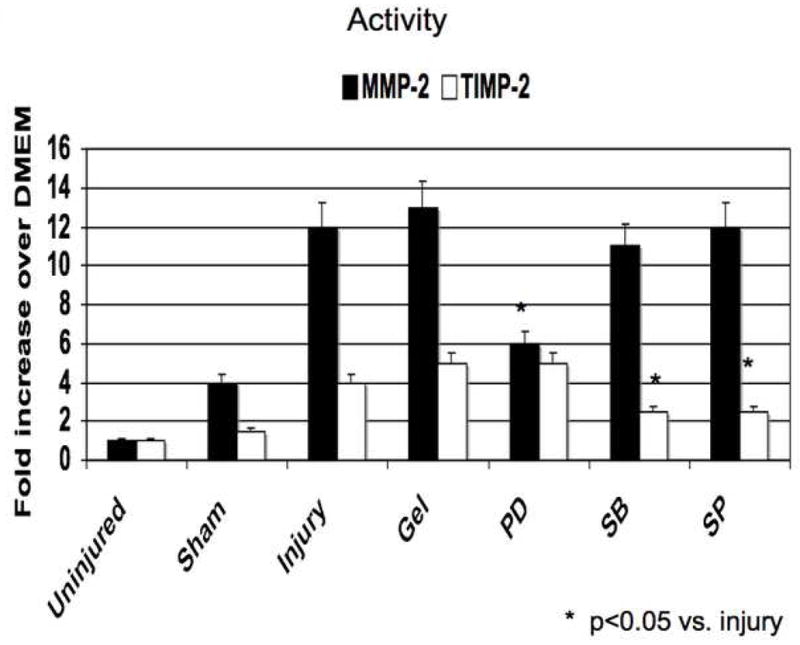
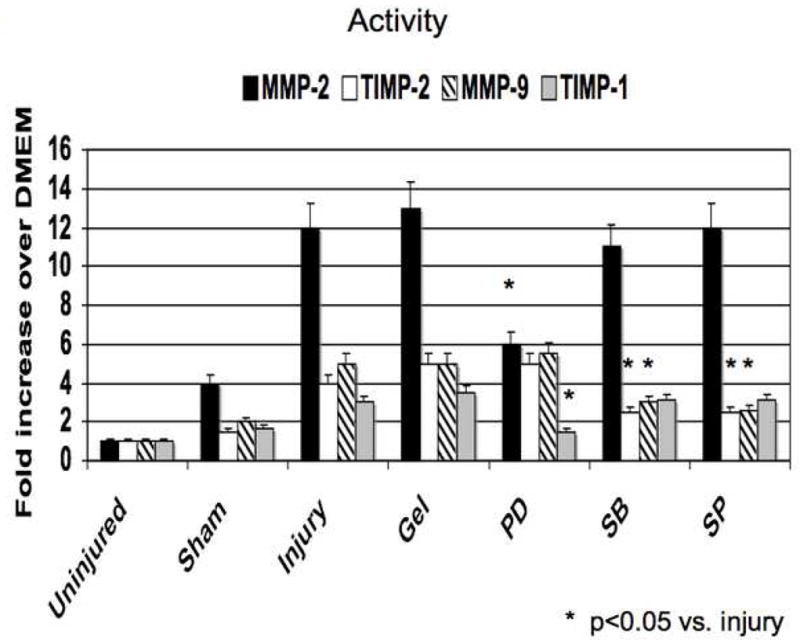
Alterations in the expression (A) and activity (B) of MMP-2/TIMP-2 and in the expression (C) and activity (D) of MMP-9/TIMP-1 in wire injured femoral vessels in the presence of pluronic gel with and without PD98059 (PD), SB230580 (SB) and SP600125 (SP). Values are the mean± s.e.m. (n=6 per group). Statistical differences between groups were tested with a Kruskal-Wallis non-parametric test with post hoc Dunn’s multiple comparison correction, where appropriate.
DISCUSSION
Response to Injury
The balance between proteases, protease inhibitors, integrins and extracellular matrix molecules constitute the proteolytic thermostat of a tissue. Proteases have been shown to play a role in hemostasis, inflammation, cell migration, cell proliferation and extracellular matrix (ECM) remodeling in many wound-healing responses (13–15). A major role of MMPs is to enable vascular remodeling through decomposition of the existing ECM scaffold, while a new ECM is synthesized and organized. MMP activity may come from leukocytes particularly in the early stages of arterial injury. In this murine model, Roque and Taubman have shown that leukocytes rapidly adhered to the injured arterial surface immediately after injury but that there is no significant macrophage infiltration into the arterial wall (16) This study demonstrates that femoral wire injury in the mouse is associated with a time dependent alterations in the gelatinase system. Cell proliferation is associated with increases in MMP-2/MMP-9 activity, and decreases TIMP-1/TIMP-2 activity, while migration is associated with increases in MMP-2 activity. The increases in MMP-2 expression and activity appear dependent ERK1/2 while increases in MMP-9 expression and activity is dependent on the stress kinases p38MAPK and JNK. Changes in TIMP-1 and TIMP-2 expression were dependent on the activity of all MAPKs. Early inhibition of gelatinase activity by GM6001 results in decreased cell proliferation, cell migration and intimal hyperplasia development.
MMP-2/MMP-9
MMP-2 is constitutively expressed in VSMC and has been shown to be required for SMC migration (17, 18). It has been demonstrated that there is a rapid upregulation of MMP-2 production after mechanical injury in the rat injury model (15, 19). The increase in activated MMP-2 levels appears to be maximal at 5 days after injury, when VSMC migration is identified (19–21). We noted a similar pattern in this model. Compared to controls, MMP-2 knock out mice show significantly reduced formation of intimal hyperplasia 28 days after carotid artery ligation, corresponding to fewer numbers of intimal smooth muscle cells suggesting a significant decrease in smooth muscle cell migration (22, 23). The data in the current study supports these findings and demonstrates that there is decrease VSMC migration when MMP activity is reduced. Activation of MMP-2 can be by several pathways. One of the activators of MMP-2, MT-MMP-1 has been shown to have enhanced expression in injured vessels and coincides with increased MMP-2 activity (24). A second activator of MMP-2 is urokinase (uPA) and we have demonstrated that this protease is also increased before peak MMP-2 activity is detected (3). Inhibition of uPA activity with aprotinin also decreases MMP-2 activity and intimal hyperplasia. Inhibition of ERK 1/2 results in decreased MMP-2 expression and subsequent detectable activity suggesting that MMP-2 expression is ERK1/2 dependent in this model. Similarly, inhibition of uPA with aprotinin decreases MMP-2 activity but not expression confirming the importance of uPA in remodeling, that uPA is necessary for MMP-2 activation and that MMP-2 is one of its downstream targets. In contrast to MMP-2, MMP-9 is not constitutively expressed in VSMC but can be induced by phorbol esters and cytokines. After arterial injury in the rat carotid, MMP-9 expression and activity increases at 24 hrs and this increase in activity coincides with the onset of cell proliferation (19–21, 25). The current data suggests that the stress kinases play a role in MMP-9 expression and that peak MMP-9 expression and activity is associated with VSMC proliferation with the vessel wall. An absence of MMP-9 in knock out mice significantly reduced the formation of intimal hyperplasia 28 days after carotid ligation (22, 23). Administration of a MMP inhibitor will decrease smooth muscle cell proliferation and migration (19, 26). In the current study, we demonstrate similar findings using local application of a MMP inhibitor but demonstrate the role of MAPKs in MMP regulation and the effect MMP inhibition has on ECM development.
TIMP-1/TIMP-2
The regulation of TIMPs is distinctly different from MMPs. The inhibitor associated with MMP-9 is TIMP-1. TIMP-1 is not constitutively expressed in VSMC but can be upregulated by the fibrogenic cytokines. TIMP-1 expression appears unchanged early after injury (27, 28). We saw a decrease in TIMP-1 expression and activity after injury. This decrease was associated with the initial peak of apoptosis (3) and the increased activity of MMP-9. TIMP-2 is more closely associated with MMP-2 and TIMP-2 secretion is constitutive expressed in VSMC and the vessel wall; this would be consistent with the theory of homeostasis as MMP-2 is also expressed with the vessel wall. It has been reported that TIMP-2 mRNA expression rises 24 hrs after injury and both protein expression and activity peak at 3 days. Expression is found in the intima and the media (28). We saw a similar pattern of response in the murine vessel. Arterial injury in TIMP-1 −/− mice demonstrates an enhanced response confirming the important role of TIMP-1 in moderating the neo-intimal response once injury has been initiated (29, 30). TIMP-1 and TIMP-2 overexpression in the injured rat carotid will induce similar results, namely decreased intimal hyperplasia formation (28, 31–33). One can induce a similar biological situation with the MMP inhibitor GM6001. It allows for enhanced MMP inhibition during the nadir in TIMP-1 and TIMP-2 activity thus blunting the remodeling response.
Plasminogen activation
Elevated uPA and decreased PAI-1 levels are predictors for angiographic coronary restenosis (34). Overexpression of uPA or a deficiency of PAI-1 in transgenic mice has been shown to promote neointimal formation in response to transmural electrical injury and to alter MMP-2/MMP-9 activity (28, 35–37). A lack of uPA leads to a reduction in neointimal hyperplasia (38). We have previously reported that we saw a steady rise in uPA protein, which stabilized at 14 days and a peak in uPA activity at 7 days in this model (3). Coupled to this change in uPA, we saw a rapid increase in PAI-1 protein early with a slow rise in PAI-1 activity over the first 3 days of injury. Both responses were blunted by the presence of MAPK inhibitors (3). Elevated PAI-1 protein expression (induced by gene therapy) in the rat carotid will also inhibit intimal thickening (39). Tranexamic acid, a plasmin inhibitor, will inhibit smooth muscle cell migration after carotid balloon injury in the rat (26). Inhibition with aprotinin has similar effects in this model. uPA inhibition also effected MMP-2 activity and may in part be the mechanism whereby uPA regulates the remodeling response. It is also of interest that application of all three MAPK inhibitors could reduce uPA expression and activity while only ERK1/2 inhibition effected MMP-2 expression suggesting separate regulation of the pathways leading to MMP-2 expression and activation.
Conclusions
This report demonstrates that femoral wire injury in the mouse induces intimal hyperplasia and that it is associated with a time dependent increase in gelatinase activity. Cell proliferation is associated with increased MMP-9 activity and diminished TIMP-1/TIMP-2 activity, while migration is associated with increased MMP-2 activity. Modulation of gelatinase activity with local application of an inhibitor may control the vessels’ response to injury.
Figure 4.
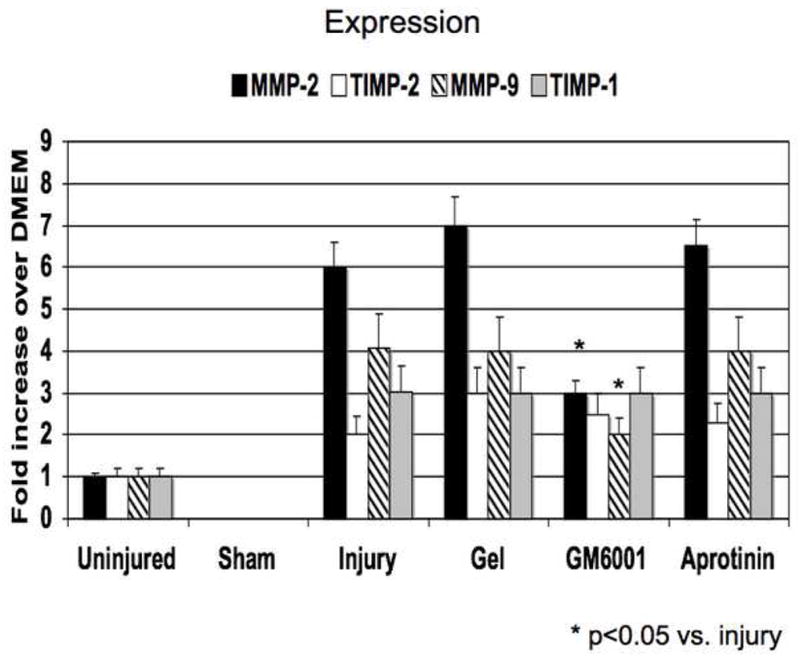
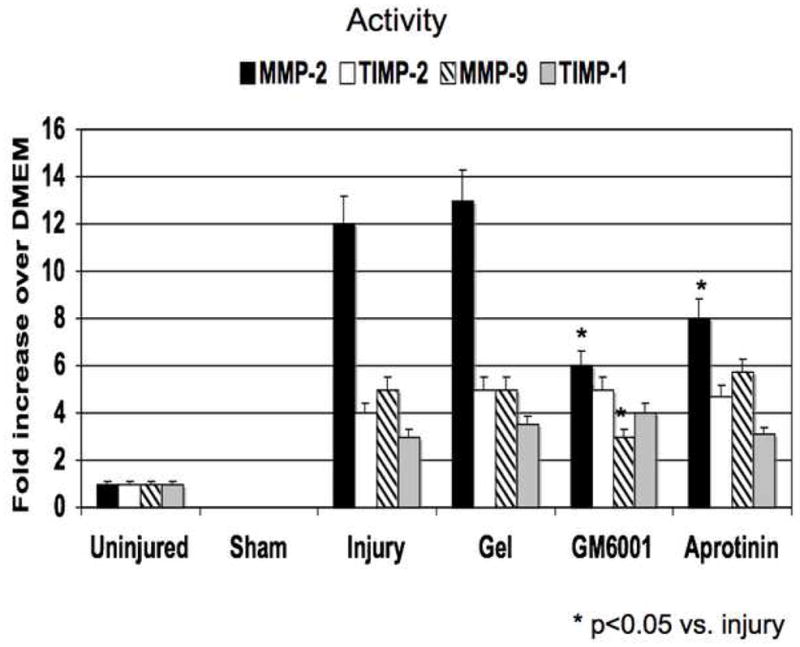
(A) A comparison of the effect of pluronic gel with and without GM6001 and aprotinin on the expression of MMP-2/TIMP-2, MMP-9/TIMP-1 in the wire injured femoral vessels over time. (B) A comparison of the effect of pluronic gel with and without GM6001 and aprotinin on the activity of MMP-2/TIMP-2, MMP-9/TIMP-1 and in the wire injured femoral vessels over time. Values are the mean± s.e.m. (n=6 per group). Statistical differences between groups were tested with a Kruskal-Wallis non-parametric test with post hoc Dunn’s multiple comparison correction, where appropriate.
Acknowledgments
This research was supported by U.S. Public Health Service HL086968 and HL067746 to Mark G. Davies, MD, PhD.
Supported by: U.S. Public Health Service HL086968 and HL067746
Footnotes
Presented at the 3rd Academic Surgical Congress (Huntington Beach, CA; Feb 2008)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Davies MG, Hagen P-O. Pathobiology of Intimal Hyperplasia. Br J Surg. 1994;81:1254–1269. doi: 10.1002/bjs.1800810904. [DOI] [PubMed] [Google Scholar]
- 2.Casscells W. Migration of smooth muscle and endothelial cells. Circulation. 1992;86:723–729. doi: 10.1161/01.cir.86.3.723. [DOI] [PubMed] [Google Scholar]
- 3.Zou Y, Qi Y, Roztocil E, Nicholl SM, Davies MG. Patterns Of Kinase Activation Induced By Injury In The Murine Femoral Artery. J Surg Res. 2007;14:1332–340. doi: 10.1016/j.jss.2007.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ip JH, Fuster V, Badimon L, Taubman MB, Chesebro JH. Syndromes of accelerated atherosclerosis: role of vascular injury and smooth muscle cell proliferation. J Am Coll Cardiol. 1990;15:1667–1687. doi: 10.1016/0735-1097(90)92845-s. [DOI] [PubMed] [Google Scholar]
- 5.Clowes AW. Intimal hyperplasia and graft failure. Cardiovasc Pathol. 1993;2 (suppl):179S–186S. [Google Scholar]
- 6.Mason DP, Kenagy RD, Hasentab D, Bowen-Pope DF, Seifert RA, Coats S, et al. Metalloproteinase-9 Overexpression Enhances Vascular Smooth Muscle Cell Migration and Alters Remodeling in the Injured Rat Carotid Artery Matrix. Circ Res. 1999;85:1179–1185. doi: 10.1161/01.res.85.12.1179. [DOI] [PubMed] [Google Scholar]
- 7.Whatling C, McPheat W, Hurt-Camejo E. Matrix Management: Assigning different roles for MMP-2 and MMP-9 in vascular remodeling. Arterioscler Thromb Vasc Biol. 2004;24:10–11. doi: 10.1161/01.ATV.0000100562.63144.C1. [DOI] [PubMed] [Google Scholar]
- 8.Simons M, Edelman ER, Dekeyser J-L, Langer R, Rosenberg RD. Antisense c-myb oligonucleotides inhibit intimal arterial smooth muscle cell accumulation in vivo. Nature. 1992;359:67–70. doi: 10.1038/359067a0. [DOI] [PubMed] [Google Scholar]
- 9.Fulton GJ, Davies MG, Koch WJ, Dalen H, Svendsen E, Hagen P-O. Antisense oligonucleotide to proto-oncogene c-myb inhibits the formation of intimal hyperplasia in experimental vein grafts. J Vasc Surg. 1997;25:453–463. doi: 10.1016/s0741-5214(97)70255-6. [DOI] [PubMed] [Google Scholar]
- 10.Tanski WJ, Fegley AJ, Roztocil E, Davies MG. Domain dependent actions of urokinase on smooth muscle cell responses. J Vasc Surg. 2004;39:214–22. doi: 10.1016/s0741-5214(03)01031-0. [DOI] [PubMed] [Google Scholar]
- 11.Kenagy RD, Nikkari ST, Welgus HG, Clowes AW. Heparin inhibits the induction of three metalloproteinases (stromelysin, 92kD gelatinase and collagenase) in primate arterial smooth muscle cells. J Clin Invest. 1994;93:1987–1993. doi: 10.1172/JCI117191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oliver GW, Leferson JD, Stetler-Stevenson WG, Kleiner DE. Quantitative reverse zymography: analysis of picogram amounts of metalloproteinase inhibitors using gelatinase A and B reverse zymogram. Anal Biochem. 1997;244(1):161–6. doi: 10.1006/abio.1996.9895. [DOI] [PubMed] [Google Scholar]
- 13.Nagase H, Woessner JF. Matrix Metalloproteinases. J Biol Chem. 1999;274:21491–4. doi: 10.1074/jbc.274.31.21491. [DOI] [PubMed] [Google Scholar]
- 14.Birkedal-Hansen H. Proteolytic remodeling of extracellular matrix. Curr Opin Cell Biol. 1995;7:728–35. doi: 10.1016/0955-0674(95)80116-2. [DOI] [PubMed] [Google Scholar]
- 15.Galis ZS, Khatri JJ. MMPs in vascular remodeling and atherogenesis: the good, the bad and the ugly. Circ Res. 2002;90:251–262. [PubMed] [Google Scholar]
- 16.Roque M, Kim WJ, Gazdoin M, Malik A, Reis ED, Fallon JT, et al. CCR2 deficiency decreases intimal hyperplasia after arterial injury. Arterioscler Thromb Vasc Biol. 2002;22(4):554–9. doi: 10.1161/hq0402.105720. [DOI] [PubMed] [Google Scholar]
- 17.Pauly RR, Passaniti A, Bilato C, Monticone R, Cheng L, Papadopoulos N, et al. Migration of cultured vascular smooth muscle cells through a basement membrane barrier requires type IV collagenase activity and is inhibited by cellular differentiation. Circ Res. 1994;75:41–54. doi: 10.1161/01.res.75.1.41. [DOI] [PubMed] [Google Scholar]
- 18.Kenagy RD, Hart CE, Stetler-Stevenson WG, Clowes AW. Primate smooth muscle cell migration from aortic explants is mediated by endogenous PDGF and bFGF acting through MMP-2 and MMP-9. Circulation. 1997;96:3555–3560. doi: 10.1161/01.cir.96.10.3555. [DOI] [PubMed] [Google Scholar]
- 19.Zempo NRDK, Au YPT, Bendeck M, Clowes MM, Reidy MA, et al. Matrix metalloproteinase of vascular wall cells are increased in balloon injured rat carotid artery. J Vasc Surg. 1994;20(2):209–17. doi: 10.1016/0741-5214(94)90008-6. [DOI] [PubMed] [Google Scholar]
- 20.Southgate KM, Fisher M, Banning A, Thurston VJ, Baker AH, Fabunmi RP, et al. Upregulation of basement membrane degrading metalloproteinase secretion after balloon injury of pig carotid arteries. Circ Res. 1996;79:1177–1187. doi: 10.1161/01.res.79.6.1177. [DOI] [PubMed] [Google Scholar]
- 21.Lijnen HR, VanHoef B, Lupu F, Moon L, Carmeliet P, Collen D. Function of the plasminogen/plasmin and MMP systems after vascular injury in mice with targeted inactivation of fibrinolytic genes. Arterioscler Thromb Vasc Biol. 1998;18(7):1035–45. doi: 10.1161/01.atv.18.7.1035. [DOI] [PubMed] [Google Scholar]
- 22.Galis Z, Johnson C, Godin D, Magid R, Shipley J, Senior R, et al. Targeted disruption of the matrix metalloproteinase-9 gene impairs smooth muscle cell migration and geometrical arterial remodeling. Circ Res. 2002;91:852–859. doi: 10.1161/01.res.0000041036.86977.14. [DOI] [PubMed] [Google Scholar]
- 23.Kuzuya M, Kanda S, Sasaki T, Tamaya-Mori N, Cheng XWC, Itoh T, et al. Deficiency of Gelatinase A Suppresses Smooth Muscle Cell Invasion and Development of Experimental Intimal Hyperplasia. Circulation. 2003;108:1375–1381. doi: 10.1161/01.CIR.0000086463.15540.3C. [DOI] [PubMed] [Google Scholar]
- 24.Jenkins GM, Crow MT, Bilato C, Gluzband Y, Ryu W-S, Li Z, et al. Increased expression of MT-MMP and preferential localization of matrix metalloproteinase-2 to the neointima of balloon injured rat carotid arteries. Circulation. 1998;97:82–90. doi: 10.1161/01.cir.97.1.82. [DOI] [PubMed] [Google Scholar]
- 25.Bassiouny HS, Song RH, Hong XF, Singh A, Glagov S. Reduced flow enhances in vivo collagenase IV transcription after arterial injury. Circulation. 1996;94(suppl):I–349. (abstract) [Google Scholar]
- 26.Bendeck MP, Zempo N, Clowes AW, Galardy RE, Reidy MA. Smooth muscle cell migration and matrix metalloproteinase expression after arterial injury in the rat. Circ Res. 1994;75:539–545. doi: 10.1161/01.res.75.3.539. [DOI] [PubMed] [Google Scholar]
- 27.Webb KE, Henney AM, Anglin S, Humphries SE, McEwan JR. Expression of MMP and their inhibitor TIMP-1 in the rat carotid artery after balloon injury. Arterioscler Thromb Vasc Biol. 1997;17:1837–1844. doi: 10.1161/01.atv.17.9.1837. [DOI] [PubMed] [Google Scholar]
- 28.Hasentaub D, Forough R, Clowes AW. Plasminogen activator inhibitor type I and tissue inhibitor of metalloproteinases-2 increase after arterial injury in rats. Circ Res. 1997;80:490–496. doi: 10.1161/01.res.80.4.490. [DOI] [PubMed] [Google Scholar]
- 29.Lijnen HR. Molecular interactions between the plasminogen/plasmin and matrix metalloproteinase systems. Fibrinolysis Proteolysis. 2000;14:175–81. [Google Scholar]
- 30.Lijnen HR, Soloway P, Collen D. TIMP-1 impairs arterial neointima formation after vascular injury. Circ Res. 1999;85:1186–1191. doi: 10.1161/01.res.85.12.1186. [DOI] [PubMed] [Google Scholar]
- 31.Forough R, Koyama N, Hasentaub D, Lea H, Clowes M, Nikkari ST, et al. Overexpression of TIMP-1 inhibits vascular smooth muscle cell formation in vitro and in vivo. Circ Res. 1996;79:812–20. doi: 10.1161/01.res.79.4.812. [DOI] [PubMed] [Google Scholar]
- 32.Forough R, Lea H, Starcher B, Koyama N, Hasentaub D, Clowes AW. Metalloproteinase blockade by local overexpression of TIMP-1 increase elastin accumulation in rat carotid artery intima. Arterioscler Thromb Vasc Biol. 1998;18:803–807. doi: 10.1161/01.atv.18.5.803. [DOI] [PubMed] [Google Scholar]
- 33.Furman C, Luo Z, Walsh K, Duverger N, Copin C, Fruchart JC, et al. Systemic tissue inhibitor of metalloproteinase-1 gene delivery reduces neointimal hyperplasia in balloon-injured rat carotid artery. FEBS Lett. 2002;531:122–126. doi: 10.1016/s0014-5793(02)03388-4. [DOI] [PubMed] [Google Scholar]
- 34.Strauss BH, Lau HK, Bowman KA, Sparkes J, Chisholm RJ, Garvey MB, et al. Plasma urokinase antigen and PAI-1 antigen levels predict angiographic coronary restenosis. Circulation. 1999;100(15):1616–22. doi: 10.1161/01.cir.100.15.1616. [DOI] [PubMed] [Google Scholar]
- 35.Carmeliet P, Moons L, Herbert J-M, Crawley J, Lupu F, Lijnen R, et al. Urokinase but not tissue plasminogen activator mediates arterial neointima formation in mice. Circ Res. 1997;81:829–839. doi: 10.1161/01.res.81.5.829. [DOI] [PubMed] [Google Scholar]
- 36.Carmeliet P, Moons L, Lijnen R, Janssens S, Lupu F, Collen D, et al. Inhibitory role of PAI-1 in arterial wound healing and neointima formation. Circulation. 1997;96:3180–3191. doi: 10.1161/01.cir.96.9.3180. [DOI] [PubMed] [Google Scholar]
- 37.Carmeliet P, Moons L, Ploplis V, Plow E, Collen D. Impaired arterial neointima formation in mice with disruption of the plasminogen gene. J Clin Invest. 1997;99(2):200–8. doi: 10.1172/JCI119148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shen GX. Vascular cell-derived fibrinolytic regulators and atherothrombotic vascular disorders. Inter J Mol Med. 1998;1(2):399–408. [PubMed] [Google Scholar]
- 39.Hasentaub D, Lea H, Clowes AW. Local plasminogen activator inhibitor Type 1 overexpression in rat carotid artery enhances thrombosis and endothelial regeneration while inhibiting intimal thickening. Arterioscler Thromb Vasc Biol. 2000;20:853–859. doi: 10.1161/01.atv.20.3.853. [DOI] [PubMed] [Google Scholar]